 Yun Ge
Yun Ge Man Huang1*
Man Huang1* Yong-ming Yao
Yong-ming Yao- 1Department of General Intensive Care Unit, The Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou, China
- 2Trauma Research Center, Fourth Medical Center and Medical Innovation Research Department of the Chinese PLA General Hospital, Beijing, China
The interleukin (IL)-17 family includes six structure-related cytokines (A–F). To date, majority of studies have focused on IL-17A. IL-17A plays a pivotal role in various infectious diseases, inflammatory and autoimmune disorders, and cancer. Several recent studies have indicated that IL-17A is a biomarker as well as a therapeutic target in sepsis. In the current review, we summarize the biological functions of IL-17, including IL-17-mediated responses and signal transduction pathways, with particular emphasis on clinical relevance to sepsis.
Introduction
The interleukin (IL)-17 family includes six structure-related cytokines (A–F; Figure 1). IL-17A, the first discovered member of the IL-17 family, was cloned in 1993 and originally termed as cytotoxic T lymphocyte antigen 8 (1). Unexpectedly, IL-17A shares sequence homology with an open reading frame in Herpesvirus saimiri (1). IL-17-binding receptor (IL-17RA) was subsequently identified in 1995 (2). Screens for homologous genes caused the discovery of other five highly conserved homologous members of the IL-17 family (IL-17B to IL-17F) (3, 4). IL-17F shows high homology with IL-17A, whereas IL-17E (also known as IL-25) shows only 16% sequence homology with IL-17A (5, 6). Analogous to platelet-derived growth factor and nerve growth factor, these molecules adopt a cysteine knot fold (3, 4). IL-17B and IL-17D have been shown to induce the secretion of chemokines and proinflammatory cytokines, but their biological actions remain scarcely explored (7–9). In contrast, the proinflammatory properties of IL-17A and IL-17F are well characterized (5, 6). Additionally, the IL-17 receptor family includes five cytokine receptors (IL-17RA to IL-17RE) characterized by a shared cytoplasmic motif named the SEF/IL-17R (SEFIR) (5).
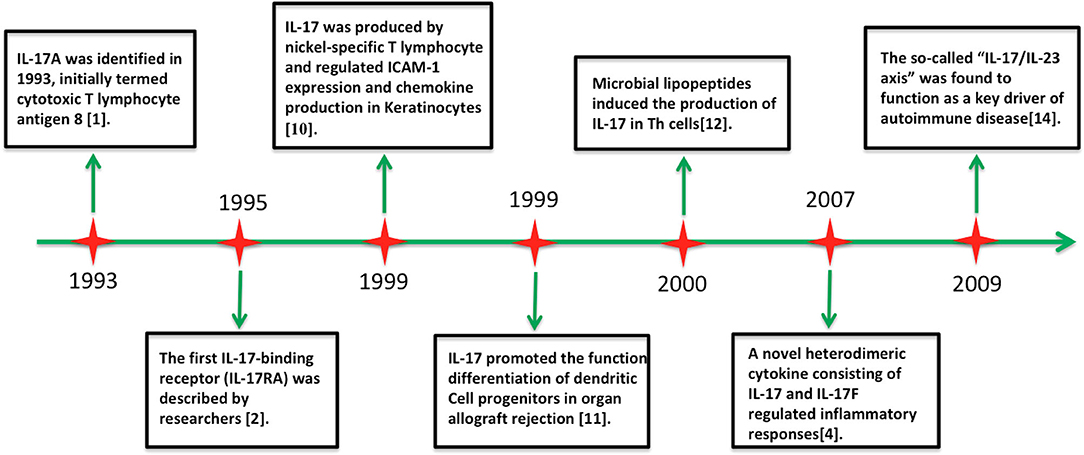
Figure 1. Timeline of advances in the research on interleukin-17 cytokine.
IL-17A expression was first reported in T helper (Th) cells (10, 11). IL-17A is recognized as a hallmark molecule of CD4+ T helper 17 (Th17) cells, which characteristically express the transcription factor RAR-related orphan receptor γ (RORγt) (12). The development of Th17 cells, including clonal expansion and phenotype stabilization of IL-17A production, is dependent on IL-23 (13). Mechanistically, IL-23 expands the Th17 cell population by upregulating signal transducers and activators of transcription (STAT)-triggered RORγt and subsequent promotion of IL-17A release (14). Thus, IL-23 is recognized as a potent inducer of IL-17A. Recent studies suggest that the so-called “IL-17/IL-23 axis” is a key element in inflammation and is involved in the immune responses to fungal and bacterial infection and autoimmune diseases (12, 13).
IL-17A can be secreted by other cell subsets, such as γδT cells, cytotoxic CD8+ T cells, innate tissue-specific cells, innate lymphoid cells (ILCs), and myeloid cells (Figure 2) (14, 15). IL-17A-mediated inflammation is required for host protection and survival against infection (8, 9, 16). IL-17A can also exacerbate fetal inflammatory responses, and has been implicated in immunopathology. IL-17A levels are elevated in various inflammatory conditions, including sepsis, pneumonia, systemic lupus erythematosus, rheumatoid arthritis, allograft rejection, and cancer (17, 18). Here we review the literature on IL-17-driven inflammatory and immune cascades during the development of sepsis.
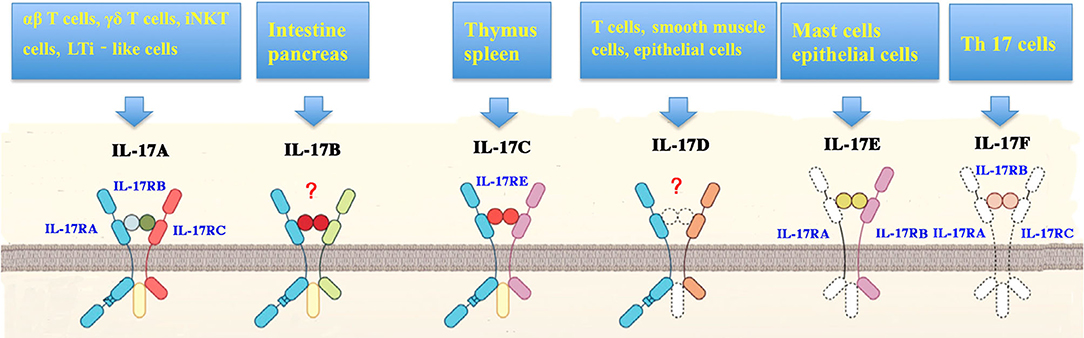
Figure 2. Cellular sources and receptors of interleukin-17. Interleukin (IL)-17 family includes isoforms A to F. IL-17 can be induced by a variety of cells including T helper (Th) 17 cells, γδT cells, natural killer T (NKT) cells, group 3 innate lymphoid cells (ILC3s), CD8+ (Tc17) cells, neutrophils, microglia, mast cells, myeloid cells in the lung and kidneys, and Paneth cells in the intestine. In addition, antigen-presenting cells (APCs) stimulate the production of IL-17in response to stress proteins, pathogen-associated molecular patterns (PAMPs), or microbial metabolites. IL-17 receptor (IL-17R) family is composed of IL-17RA, IL-17RB, IL-17RC, IL-17RE, as well as IL-17RA and C heterodimeric complex.
IL-17 Biology
Structural Features of IL-17
The IL-17 family contains six isoforms of 20–30 kDa molecular weight and is a group of secreted and glycosylated proteins. All other members of the IL-17 family show 20–55% sequence homology to IL-17A, with IL-17E exhibiting the lowest homology with other family members (8, 9). IL-17A and IL-17F could exist as heterodimers or homodimers and are co-expressed by linked genes (19). Structurally, IL-17 family proteins have a conserved C-terminus with four cysteine residues, which form intramolecular disulfide bridges (20).
Production of IL-17
IL-17 can be produced by a broad spectrum of cell populations (Figure 2), including Th17 cells, γδT cells, NKT cells, group 3 innate lymphoid cells (ILC3s), CD8+ (Tc17) cells, neutrophils, microglia, and mast cells (10, 12, 15). IL-23 and RORγt are indispensable for all IL-17-producing cell types (13).
Th17 cells were first identified in 2005 and were recognized as the primary source of IL-17 (16). In addition to IL-17, Th17 cells also produce a variety of inflammatory cytokines including granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-21, and IL-22 (16). Th17-induced responses are implicated in host defense against infections, inflammatory and autoimmune disorders, and tumorigenesis (21). Other major sources of IL-17 include myeloid cells (e.g., kidneys and lungs) and Paneth cells in the intestinal crypts (8).
In response to stress proteins, pathogen-associated molecular patterns (PAMPs), or microbial metabolites, antigen-presenting cells (APCs) produce IL-23 and IL-1β to accelerate IL-17 release (8). IL-17 levels are depended on the particular context, including the pathogen and site and severity of infection (10, 12).
IL-17 Receptor and Its Signal Transduction
IL-17 cytokines bind to five cytokine receptors (IL-17RA to IL-17RE, Figure 2) on target cells to drive their biological actions (2, 9). IL-17R is expressed in a variety of cell populations, including keratinocytes, fibroblasts, mesothelial cells, epithelial cells, and leukocytes (9). All these receptors share a SEFIR domain in the intracellular domain and a fibronectin III-like region in the extracellular region (10). IL-17RA is a shared receptor for different IL-17 isoforms. IL-17 cytokines can trigger signals via an IL-17RA/IL-17RC receptor complex. IL-17RB and IL-17RE serve as the specific receptors for IL-17B and IL-17RA/IL-17RB heterodimeric complex, respectively (9). IL-17A and IL-17F act through the same IL-17RA/IL-17RC receptor complex.
Studies suggest that IL-17RD also drives IL-17-mediated signaling, but the ligand of IL-17RD remains unknown (21).
IL-17A/IL-17C interacts with IL-17R to stimulate inflammatory reactions via activation of mitogen-activated protein kinase (MAPK), nuclear factor-kappa B (NF-κB), CCAAT/enhancer-binding protein (C/EBP), Janus kinase (JAK)/phosphatidylinostitol 3-kinase (PI3K), and JAK/STAT signaling (Figure 3) (22–25). IL-17A can also signal through Toll-IL-1 receptor (TIR)-domain-containing adapter-inducing interferon-β (TRIF), myeloid differentiation primary response gene 88 (MyD88), and the adaptor proteins (9, 21). Interestingly, IL-17A does not induce IL-17R cascades in embryonic fibroblasts of mice deficient in necrosis factor receptor-associated factor (TRAF)-6, suggesting that TRAF-6 is essential for IL-17A/IL-17R signaling (26). Furthermore, several studies have identified NF-κB activator 1 (Act1) as a key adapter molecule for TRAF-6 recruitment in the induction of IL-17R signaling (27).
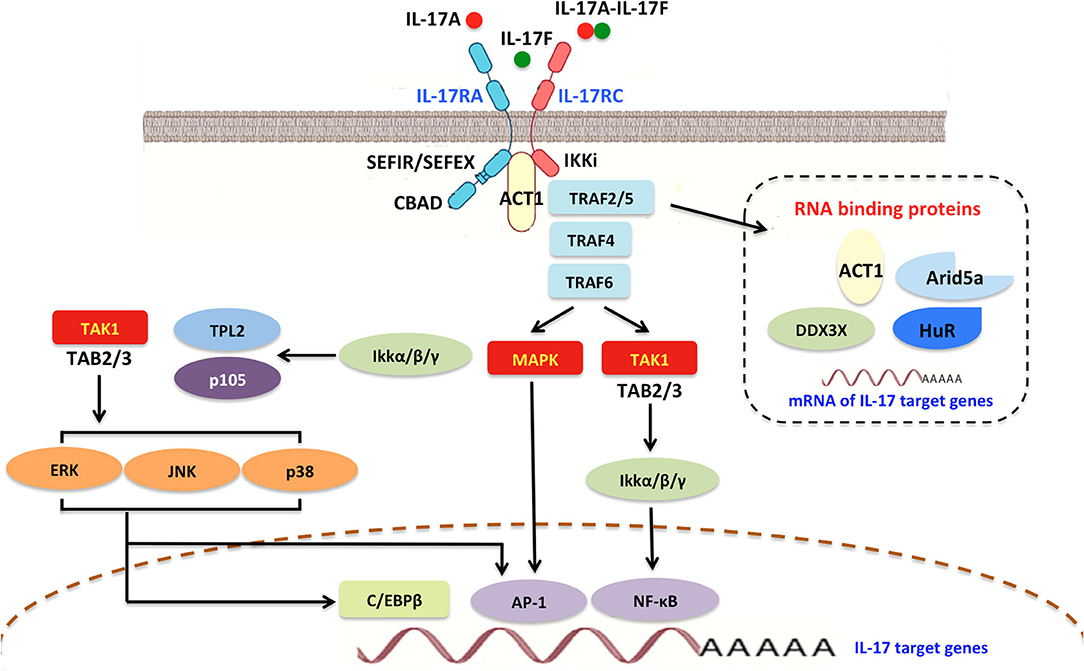
Figure 3. Interleukin-17A signaling transduction pathway. In general, all IL-17R members contain a shared and conserved cytoplasmic motif named a SEFIR/SEFEX domain similar to the TIR region in IL-1 receptors and TLRs. Initially, IL-17R signaling triggers the recruitment of Act1, which includes a SEFIR domain and is required for IL-17R-Act1 combination. Subsequently, Act1 can ubiquitinate TRAF6 and E3 ubiquitin ligase. Upon ligand binding, TRAF2/5, TRAF4, and TRAF6 can be engaged by Act1 and stabilize mRNAs by activating RNA-binding proteins such as Arid5a, HuR, and DDX3X. IL-17A promotes the activities of AP1 and MAPK, as well as δ transcription factors and C/EBPβ activation (CBAD). IL-17A also accelerates IκBα degradation and IKK activation, thereby inducing NF-κB signaling. NF-κB in turn improves IL-17A-mediated proinflammatory and anti-microbial responses. IL-17A/NF-κB signal transduction can trigger a feedback loop that controls overactivation of the NF-κB cascade. TRAF, TNF-receptor associated factor; SEFIR, similar expression of fibroblast growth factor and IL-17Rs; SEFEX, SEFIR extension; TLRs, Toll-like receptors; C/EBPβ, CCAAT/enhancer-binding protein β; CBAD, C/EBPβ activation domain; AP1, activator protein 1; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor κB; IKK, IκBα degradation and IκB kinase; JNK, Janus kinase; HuR, human antigen R.
Functions of IL-17
IL-17A
IL-17A is the most studied member of the IL-17 family. IL-17A interacts with several mediators [e.g., GM-CSF, interferon (IFN)-γ, IL-22, IL-1β, tumor necrosis factor-α (TNF-α)] to exert its proinflammatory effect (15).
In general, IL-17A-mediated downstream pathways induce the production of inflammatory molecules, chemokines, antimicrobial peptides (AMPs), and remodeling proteins (Figure 4). IL-17A elicits crucial impacts on host defense, cell trafficking, immune modulation, and tissue repair, with a key role in the induction of innate immune defenses. IL-17A stimulates non-hematopoietic cells (e.g., epithelial cells) and then acts alone or synergistically with additional proinflammatory mediators to promote chemokine production [e.g., chemokine (C-C motif) ligand (CCL)-20, granulocyte-colony stimulating factor (G-CSF), C-X-C motif ligand (CXCL)-1, CXCL-2, and CXCL-8], thereby attracting myeloid cells to inflammatory sites (28). IL-17A also stimulates the release of IL-2 from Th cells, which in turn expands regulatory T cells (5). IL-17A also promotes the secretion of AMPs (e.g., β-defensins, calgranulin, S100A8, and lipocalin-2) from macrophages and neutrophils in response to acute pathogen invasion (21). Moreover, IL-17A may induce other AMPs and proteins, including inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) (29).
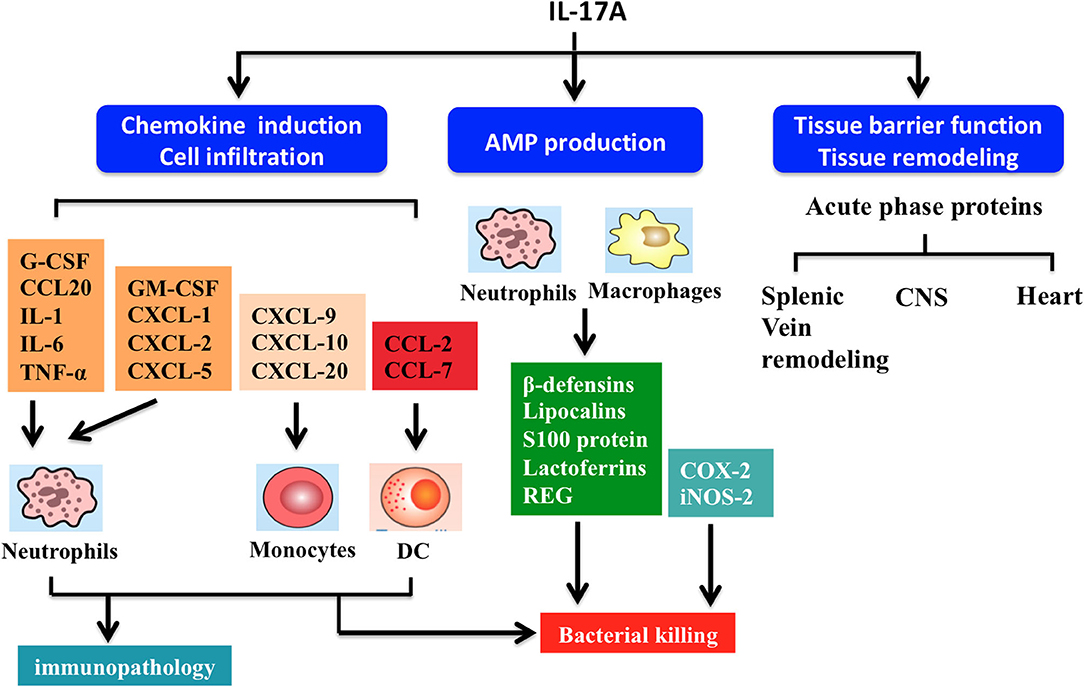
Figure 4. Summary of interleukin-17A functions. IL-17A-driven signaling drives several effector functions including chemokine induction, cell infiltration, antimicrobial peptide (AMP) production, and tissue barrier function and remodeling. Firstly, IL-17A triggers production of chemokine (C-C motif) ligand (CCL)-20 and granulocyte-colony stimulating factor (G-CSF), which act in synergy with other proinflammatory cytokines (e.g., TNF-α, IL-6, and IL-1) to induce neutrophil recruitment. IL-17A regulates the release of various chemokines. For example, chemokine (C-X-C motif) ligand (CXCL)-1, CXCL-2, CXCL-5, and granulocyte-macrophage colony-stimulating factor (GM-CSF) can improve neutrophil response. CCL7, CCL2, CXCL-9, CXCL-10, and CXCL-20 modulate the activities of monocytes and dendritic cells (DCs). Secondly, IL-17A stimulate macrophages and neutrophils to produce AMPs including β-defensins, lipocalins, S100 proteins, lactoferrins, and regenerating (REG) proteins, helping to kill bacteria. Thirdly, IL-17A signaling maintains tight junctions in mucosal tissues through formation of several acute phase proteins. IL-17A also participates in remodeling of the airway vascular, splenic vein, central nervous system (CNS), and heart tissues.
IL-17A is critical for maintaining mucosal barrier integrity and function by increasing tight junctions and inducing acute phase proteins (8, 9). IL-17A helps to orchestrate airway vascular remodeling through Th17 cell responses in pulmonary inflammation (8). Similarly, IL-17A has been shown to upregulate endothelial tissue factor and is involved in splenic vein remodeling (30). Additionally, IL-17A plays a key role in repair and remodeling of other tissues such as ventricular tissue in myocardial infarction and bone resorption (31).
Roles of IL-17B Through IL-17F
IL-17B was originally identified as a proinflammatory mediator that accelerates neutrophil recruitment and migration (7). IL-17B inhibits IL-25 signaling and attenuates mucosal inflammation (5). IL-17B promotes the proliferation and survival of cancer cells in animal models (7), and increased IL-17B levels are linked to poor outcome in patients with several types of cancers (e.g., breast, lung, and pancreatic) (5).
IL-17C can be produced by several non-immune cells (e.g., epithelial cells, cutaneous neurons, and keratinocytes) and provide host protection in the intestine, skin, and the nervous system (5). For example, IL-17C released from epithelial cells maintains barrier integrity in an autocrine manner following epithelial damage (8). Emerging evidence indicates that IL-17C is sufficient to promote growth and survival of nerves and protect peripheral neurons during reactivation of herpes simplex virus-2 (6).
IL-17D is the least studied cytokine in the IL-17 family. Similar to other isoforms, IL-17D triggers secretion of diverse inflammatory cytokines such as GM-CSF, IL-6, and IL-8. Several studies have shown markedly increased IL-17D in viral infection and tumors (7).
Similar to IL-17A, IL-17E acts as a “mucosal barrier” molecule that confers immunity against parasitic infections (8). Thymic epithelial cells promote IL-17E production and host defenses via T cell receptor development of type 2 innate-like lymphoid cells (iNKT-2) (32). IL-17E is distinct from other member of the IL-17 family. IL-17E drives stromal cells, type 2 T helper (Th2) cells, epithelial cells, and ILC2s (5). It also accelerates production of thymic stromal lymphopoietin (TSLP), IL-13, IL-5, IL-4, and IL-13, and lowers the levels of IL-23, IL-6, and IL-1 (9). IL-17E is involved in the pathogenesis of parasitic and fungal infections, allergy, and autoimmune disorders. For example, large amounts of IL-17E are produced following infection with the parasitic helminth Nippostrongylus or Aspergillus. Furthermore, IL-17E-mediated responses depend on the airway epithelium, mast cells, eosinophils, and Th2 cells, thereby contributing to the immunopathogenesis of asthma (33).
IL-17F and IL-17A share similarities in receptors, signaling, function, and cellular sources. Similar to IL-17A, IL-17F plays a critical role in inflammatory responses and mucosal barrier maintenance (4).
Roles of IL-17 in the Development of Sepsis
IL-17 in Host Defense During Sepsis
Sepsis is caused by improper inflammatory and immune responses due to the inability of the host defenses to contain infection (34). As a crucial part of host immunity, IL-17A confers powerful protective effects against infections caused by bacteria, fungi, virus, and parasites (Figure 5); (Table 1) (16, 17). Increased circulating levels of IL-17A are observed in experimental and human sepsis (16, 17). A series of clinical studies have demonstrated that high serum IL-17A levels are associated with greater risk of sepsis, suggesting that this cytokine might serve as a novel predictor of sepsis progression as well as an attractive therapeutic target (34).
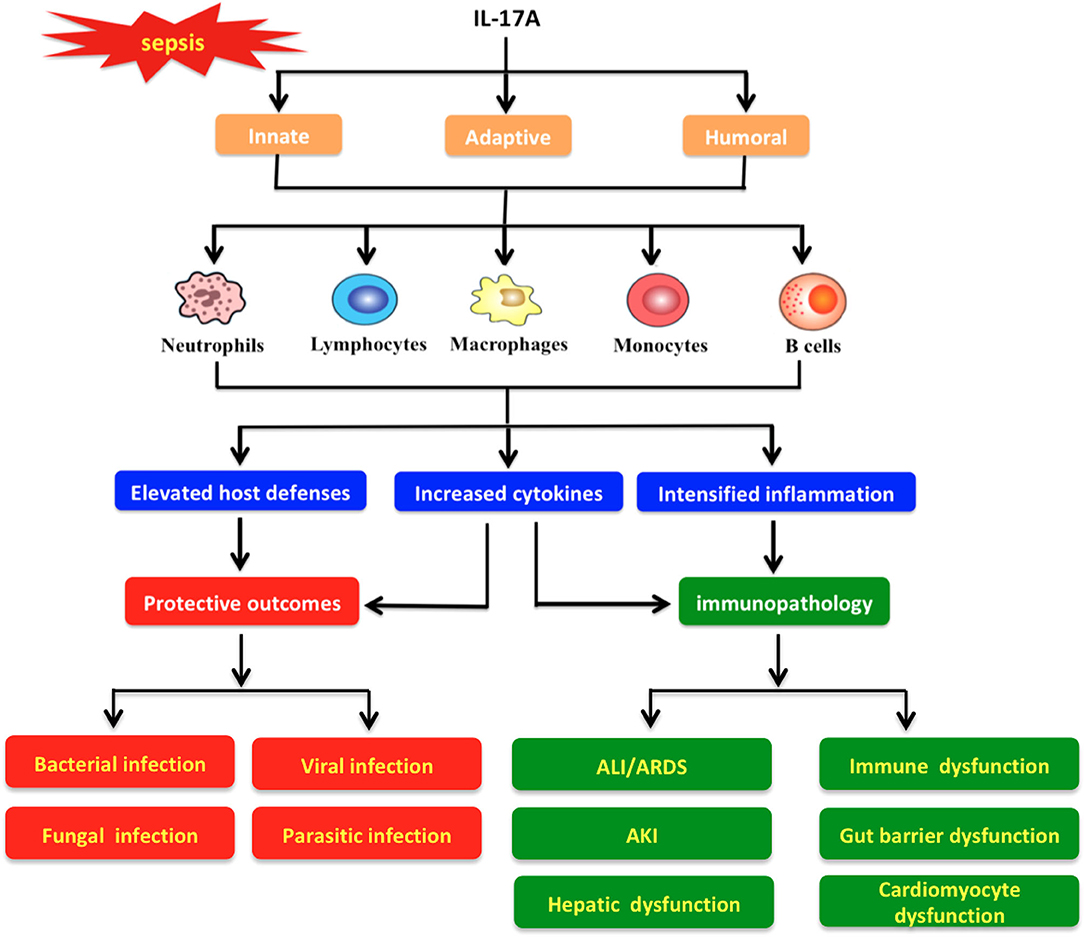
Figure 5. Protective and pathological roles of interleukin-17A in the pathogenesis of sepsis. IL-17A triggers various immune cells (e.g., neutrophils, lymphocytes, macrophages, monocytes, and B cells) to boost innate, adaptive, and humoral responses with different actions. Meanwhile, IL-17A signaling exhibits a strong protective effect against diverse infections (e.g., bacterial, fungal, viral, and parasitic infections) through promotion of host defenses and cytokine release. In addition, IL-17A may induce pathologic immune responses and then impair organ functions, which result in pathological alterations such as acute lung injury (ALI)/acute respiratory distress syndrome (ARDS), acute kidney injury (AKI), hepatic dysfunction, immune dysfunction, gut barrier dysfunction, and cardiomyocyte dysfunction.
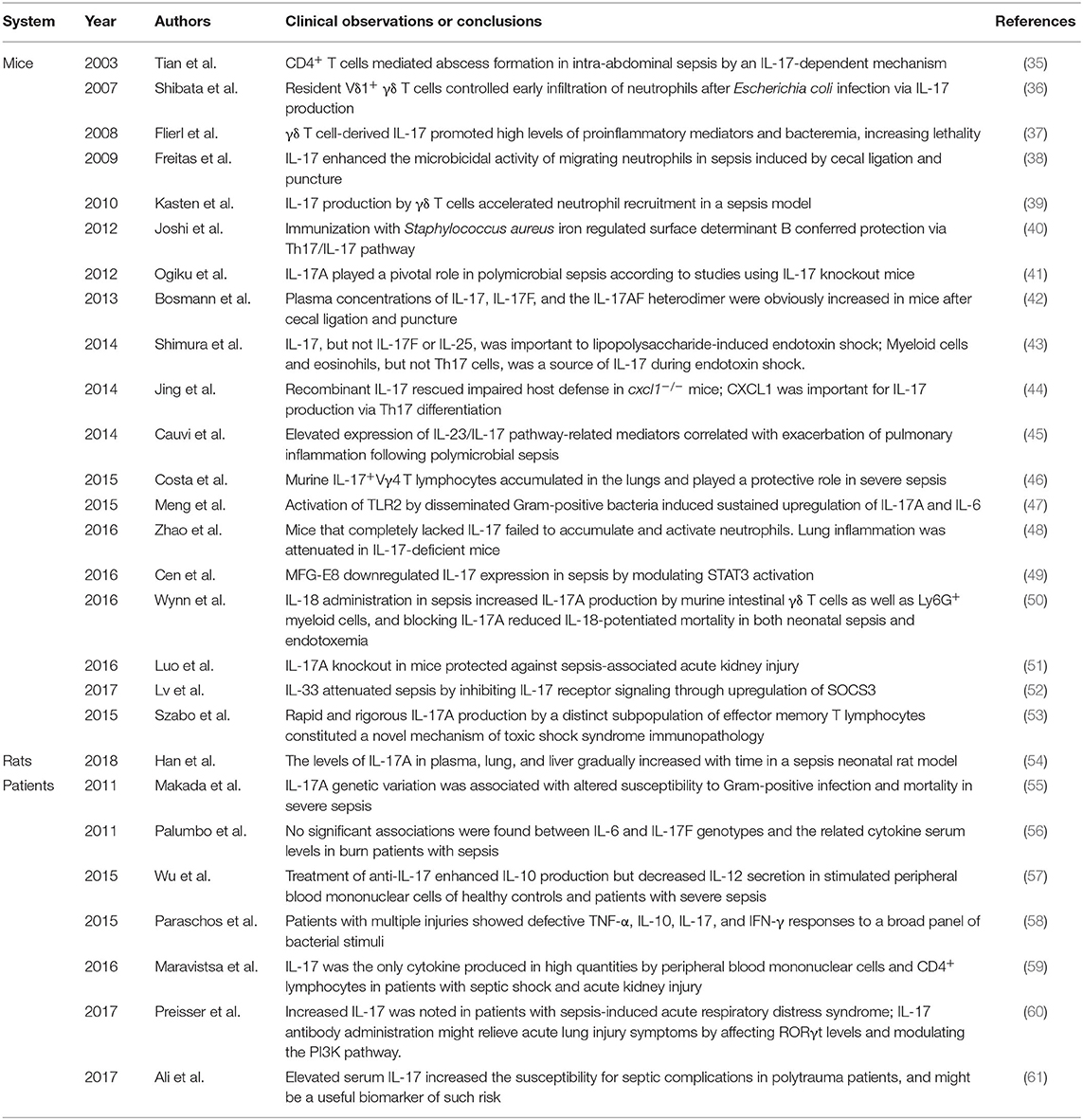
Table 1. Summary of studies on the pathophysiological significances of IL-17 in sepsis.
Bacterial Infection
It is widely accepted that bacterial infections predominantly contribute to the development of sepsis (18). Interestingly, IL-17A genetic variations influence the risk of gram-positive infection and correlate with short-term mortality in patients with severe sepsis (55). Studies suggest that IL-17A-mediated signaling orchestrates inflammatory and immune cascades by inducing proinflammatory mediators, thereby participating in preventing bacteria during sepsis (21).
IL-17A levels are positively associated with tissue bacterial loads in mice infected with Klebsiella pneumonia (62). Physiological levels of IL-17A in type 3 innate lymphoid cells (ILCs) accelerate G-GSF production and neutrophil recruitment, protecting the host against sepsis (15). Experimental and human sepsis involves impaired neutrophil recruitment and migration, the extent of which is positively associated with disease severity (63). IL-17A augments the migration and microbicidal activity of neutrophils, as well as mobilization of T lymphocytes (64). IL-17A improves neutrophil recruitment and bacterial eradication via γδT cells in a murine model of sepsis (36).
Decreased levels of IL-17A are correlated with increased risk of bacteremia (62). In IL-17R-deficient mice infected with Klebsiella pneumoniae, levels of G-CSF, macrophage inflammatory protein-2 (MIP-2), and chemokines were reduced, contributing to decreased neutrophil infiltration and microbial clearance (62). In agreement with this observation, IL-17A-deficient mice are also more susceptible to infection by other bacteria, including Bordetella pertussis, Citrobacter rodentium, Escherichia coli, and Staphylococcus aureus (65, 66). IL-17R-deficient mice showed decreased neutrophil recruitment, augmented infection spread, and exacerbated inflammatory responses following cecal ligation and puncture (CLP) (48). IL-17A also stimulates epithelial cells to trigger antimicrobial responses against intracellular bacteria such as Listeria monocytogenes, Mycobacterium bovis Bacillus-Calmette Guérin, and Salmonella typhimurium (67–69).
Fungal Infection
Invasive fungal infection is increasingly frequent in septic patients and correlates with a high risk of mortality (70). Importantly, IL-17A exerts immunoprotective effects in antifungal defense via induction of AMPs, chemokines, and proinflammatory cytokines (71). Meanwhile, IL-17A is sufficient to activate NK cells through release of GM-CSF, thereby eliciting its fungicidal activity (72). IL-17R knockdown increases susceptibility to fungal pathogens during systemic candidiasis (70, 73). Notably, low IL-17A levels are related to impaired host immunity in clinical observation (74). IL-17A responses are required for controlling infection with fungal pathogens, including Aspergillus fumigatus, Pneumocystis carinii, Cryptococcus neoformans, and Candida albicans (73–78). On the other hand, IL-17C induces lethal inflammation by exacerbating secretion of proinflammatory cytokines, contributing to the development of systemic fungal infection (78). These paradoxical effects require further investigation.
Viral Infection
Sepsis often occurs in cases of immunosuppression, when it manifests as an increased susceptibility to opportunistic infections, especially by virus (18). Several studies have shown that IL-17 promotes host immunity against influenza infection (21). In mice challenged with influenza virus, lack of IL-17 was associated with increased mortality (79). Adoptive transfer of IL-17-producing cells protected mice against a lethal dose of influenza virus (80). Also, IL-17A participates in the immunopathogenesis of influenza A (H1N1)-induced acute lung injury (80). Likely, IL-17A-mediated response is correlated with disease severity following infections by Epstein-Barr virus (EBV), herpes simplex virus (HSV), respiratory syncytial virus (RSV), vaccinia virus, and hepatitis virus (81, 82). Depletion of IL-17R mitigates inflammation and decreases neutrophil influx, hindering influenza infection (82). IL-17A neutralization could reduce viral load and mortality during herpes simplex virus infection (82). Protective or pathological roles of IL-17 during viral infections remain controversial.
Parasitic Infection
Few studies have examined the potential role of IL-17A in host defense against parasites (5). Studies suggest that Th17 cells, the major source of IL-17A, could impair host defenses against Echinococcus granulosus, Leishmania braziliensis, Toxoplasma gondii, and Trypanosoma cruzi (83–87). In patients with toxoplasmosis, CD8+ and CD4+ cells could limit parasitic replication and invasion (86). High IL-17A levels are detectable in peripheral blood mononuclear cells from patients with Leishmaniasis (87). IL-17R deficiency leads to lower expression of CXCL1 and CXCL2 in the liver and spleen as well as less neutrophil recruitment in Trypanosoma cruzi-infected mice (83). In the case of Leishmania infection, IL-17A blockade reduces disease severity in a mouse model (88). IL-17A antibody neutralization reduces inflammation and improves survival in mice following Toxoplasma gondii challenge (86). Collectively, available evidence supports a pathological role for IL-17 in parasitic infections, but further work is needed to explore whether this depends on the host, parasite, or other factors.
In summary, IL-17 contributes to host protection against diverse infectious organisms during sepsis while inducing hyperinflammation with detrimental outcomes for the host under certain conditions. Further investigation on the role of IL-17 and the interplay with other immune factors needs to be conducted in clinical settings.
IL-17 in Organ Dysfunction Resulted From Sepsis
Studies have shown that overproduction of IL-17 could exaggerate immune responses, which in turn may lead to impaired organ function (89) (Figure 5). In line with these observations, elevated levels of IL-17A have been detected in plasma and tissues during sepsis associated with multiple organ damage (37, 38, 54, 56–58, 61) (Table 1).
Acute Lung Injury or Acute Respiratory Distress Syndrome
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are early events in the course of sepsis (90). Neutrophil infiltration plays an integral part in ARDS. IL-17R signaling is proposed to regulate neutrophil trafficking into the lung to maintain host immunity (9). The addition of recombinant IL-17A into the airway causes the production of chemokines that recruit inflammatory and immune cells (48). In contrast, IL-17RA or IL-17A knockout decreases CXCL1 and G-CSF levels in the bronchoalveolar lavage fluid of mice (62). In line with this, hyperproduction of IL-17A is involved in uncontrolled pulmonary inflammation in mice infected with Haemophilus influenza (91). Interestingly, γδT lymphocytes (comprising the Vγ4 T lymphocytes) accumulate in the lungs of septic mice (46). Surprisingly, administration of anti-Vγ4 monoclonal antibody increased the mortality induced by CLP in septic mice (13, 46). In mice with lipopolysaccharide-induced ARDS, levels of IL-17A were elevated in plasma, lung tissue lysate, and bronchoalveolar lavage fluid (60). Consistently, patients with sepsis-induced ARDS show persisting high levels of IL-17A, suggesting that IL-17A is a biomarker to assess the severity and prognosis of diseases (60). Moreover, activation of the IL-23/IL-17 signaling produces harmful effects on sepsis-driven lung inflammation (45). Specifically, IL-17A expression, known to be induced by IL-23, is significantly increased in the lungs of septic mice induced by CLP (45). Neutralization of IL-17A ameliorates ALI and ARDS by modulating PI3K signaling and RORγt expression (60), suggesting that IL-17 could be explored as a therapeutic target for sepsis-associated ARDS.
Immune Dysfunction or Immunosuppression
Sepsis disrupts immune homeostasis (64). A study in septic patients showed that inhibition of IL-17A by specific antibodies could lead to immune depression by increasing IL-10 release and decreasing IL-12 production from peripheral blood mononuclear cells (57).
A previous study reported that CD4+ T cells mediated abscess formation in intra-abdominal sepsis in an IL-17A-dependent manner (35). Similarly, IL-33, a potent immunoregulator, has been shown to attenuate sepsis by suppressing IL-17A-mediated signaling via upregulation of suppressor of cytokine signaling (SOCS)-3 (52).
Toxic shock syndrome caused by Streptococcal and Staphylococcal superantigens mimicks sepsis in clinical presentation. It is characterized by overexpression of proinflammatory cytokines and pathological immune responses (53). Overproduction of IL-17A was observed in the early phase of toxic shock syndrome; in contrast, anti-IL-17A antibodies obviously reduced intestinal and hepatic impairment and mortality rate (53).
Acute Kidney Injury
Acute kidney injury (AKI) is the hallmark and a risk factor of sepsis; reversal of AKI correlates with lower risk of mortality (89). Several experimental and clinical studies support the importance of IL-17A secretion in AKI (51). Strikingly, IL-17A can function as a chemokine that recruits neutrophils to the kidneys (55).
IL-17A is associated with elevated levels of proinflammatory cytokines and accelerated tubular epithelial apoptosis in AKI (51). IL-17A is upregulated in animal models of acute tubular injury and cisplatin-induced AKI (59). In Th17 cells stimulated by heat-killed C. albicans, IL-17A levels significantly increased in patients with AKI after sepsis compared with healthy counterparts (92). In one study, Th17 cell activation was higher in patients who died from AKI following sepsis than in survivors, and the extent of activation correlated with AKI-associated inflammation (93). IL-17A knockdown can mitigate interstitial neutrophil infiltration and tubular impairment (51, 59). The intracellular receptor RORγt is required for IL-17A production, and RORγt deficiency protects against cisplatin-induced nephrotoxicity (92). IL-17A induces neutrophil migration through CXCL5, a chemokine known to be associated with higher risk of renal damage. Interestingly, high IL-17C expression has been noted in kidney epithelial cells following fungal infection (6), whereas lack of IL-17C in mice was associated with lower renal damage and improved survival (78). Altogether, the evidence supports that IL-17-induced responses potentiate AKI in septic patients.
Gut Barrier Dysfunction
Intestinal mucosa dysfunction remains a challenge in the management of sepsis (64). Sepsis-induced gut barrier dysfunction includes disrupted mucosal integrity, increased permeability of the epithelial lining, impairment of gut-blood barrier, and bacterial translocation (90).
IL-17A is important in maintaining the integrity of epithelial barriers (8). IL-17A-mediated pathological responses disrupt intestinal epithelial barrier function, increase gut permeability, and cause translocation of gut bacteria by inhibiting proliferation of enterocytes and inducing their apoptosis (8, 9). Disseminated gram-positive bacteria can upregulate IL-17A by activating Toll-like receptor (TLR)-2 on T cells and dendritic cells (47). Neutralizing IL-17 in septic mouse models protects gut barrier integrity, reduces systemic inflammation and bacterial dissemination, and lowers mortality (41).
Cardiomyocyte Dysfunction
Cardiomyocyte dysfunction is a complication that contributes to high mortality in sepsis patients (90). The molecular mechanisms underlying sepsis-induced cardiomyopathy are not well defined, but may involve a combination of endothelial disturbance, autonomic nervous system alterations, dysfunction of calcium regulation, oxidative and mitochondrial stress, extensive inflammation, and myocardial ischemia and reperfusion (I/R) injury (90). Up to now, no efficient treatment for sepsis-induced cardiomyopathy exists.
IL-17A levels are greatly increased in post-myocardial I/R injury, inflammation, and apoptotic responses (94). IL-17A accelerates the release of chemokines and proinflammatory mediators from fibroblasts, endothelial cells, leukocytes, and neutrophils (95). In viral myocarditis, neutralizing IL-17A with a monoclonal antibody attenuates myocardium inflammation, clinical symptoms, and disease progression (96). Knockdown of IL-17A in mice greatly mitigates cardiac disturbance as well as myocardial I/R injury and remodeling (97). Anti-IL-17A antibodies downregulate CCL3, CXCL1, and IL-6 in cardiomyocytes of mice with sepsis induced by CLP, suggesting that IL-17A contributes to sepsis-induced cardiomyopathy (96). Interestingly, high mobility group box-1 protein (HMGB1), a crucial late biomarker of lethal systemic inflammation in sepsis, stimulates IL-17A release during myocardial I/R injury (98). Conversely, HMGB1 inhibition remarkably lowers IL-17A levels and attenuates myocardial I/R injury (98). Therefore, the HMGB1/IL-17A axis may play a key role in myocardial dysfunction resulted from sepsis.
Other Sepsis-Related Complications
Hepatic dysfunction is a prominent feature of sepsis with crucial implications for survival, since it contributes to sepsis-induced multiple organ failure (89). IL-17 from γδ T cells protects the host from Listeria monocytogenes infection by augmenting bacterial clearance through the liver (67). Absence of IL-17A or γδ T cells in mice is associated with greater bacterial load and inflammatory lesions in the liver (67). However, another study showed that IL-22, but not IL-17A, could protect hepatocytes from acute liver inflammation (99). The precise role of IL-17 in sepsis-induced hepatic damage remains unclear and requires deeper investigation.
The stress response following sepsis involves production of glucocorticoids and catecholamines (90). Both glucocorticoids and catecholamines inhibit the expression and production of IL-17A in lipopolysaccharide-stimulated peritoneal macrophages (90). They also block c-Jun-N-terminal kinase, preventing IL-17A secretion (42).
Taken together, excessive IL-17A production disrupts immune homeostasis and contributes to the development and progression of sepsis. Nevertheless, the exact contribution of IL-17 to sepsis-induced dysfunction of the liver and other organs needs to be further explored.
Administration of Exogenous IL-17 or Targeting IL-17 in Sepsis
IL-17A is an important link between the innate and adaptive immune processes (43, 49). IL-17A-deficient mice are more susceptible to sepsis than wild-type controls (37), and IL-17A blockade impairs peritoneal eradication of E. coli, indicating that IL-17A might prevent sepsis in certain settings (63). Consistent with this idea, IL-17A administration restores impaired immunity in CXCL1-knockout mice (44). In animals infected with K. pneumoniae, treatment with IL-17A promoted bacterial clearance and neutrophil recruitment by enhancing the production of G-CSF, IL-1β, TNF-α, and CXCL-2 (36). Likewise, IL-17A administration improved anti-bacterial immunity following challenge with group B Streptococcus and Streptococcus pneumoniae in mice (34). Neutralization of IL-17A promoted resistance to intra-abdominal abscess formation in mice challenged with Bacteroides fragilis or abscess-inducing zwitterionic polysaccharides (35). Neutralizing IL-17A with antibodies could also reduce the mortality of mice immunized with S. aureus iron regulated surface determinant B after bacterial challenge (40). Thus, IL-17A plays a pivotal role in prevention of infection progression and related infectious complications.
In agreement with the findings described above, inhibition of IL-17/IL-23 significantly improved survival (from 40 to 100%) in a mouse model of endotoxic shock (39). Likely, anti-IL-17A antibody reduces systemic levels of proinflammatory cytokines/chemokines and bacteremia, improving the survival rate (50). E. coli infection results in high levels of IL-17, and blockade of IL-17 attenuates neutrophil infiltration and improves bacterial clearance (63).
In a recent study, anti-IL-17 antibody mitigated the pathology and the mortality rate of mice infected with Pseudomonas aeruginosa (100). In a colitis mouse model, IL-17 neutralization attenuated immunopathology and bacteremia (5). In a neonatal sepsis mice model, treatment with IL-17A-neutralizing antibody mitigated IL-18-related disease deterioration (57). Targeting IL-17A enhanced IL-10 production in peripheral blood mononuclear cells following sepsis (57). This evidence supports the possibility of reducing sepsis mortality through antibody-mediated blockade of IL-17R or IL-17A (Table 2). Anti-IL-17A antibodies ixekizumab and secukinumab are currently being investigated in clinical trials in septic patients (8, 9).
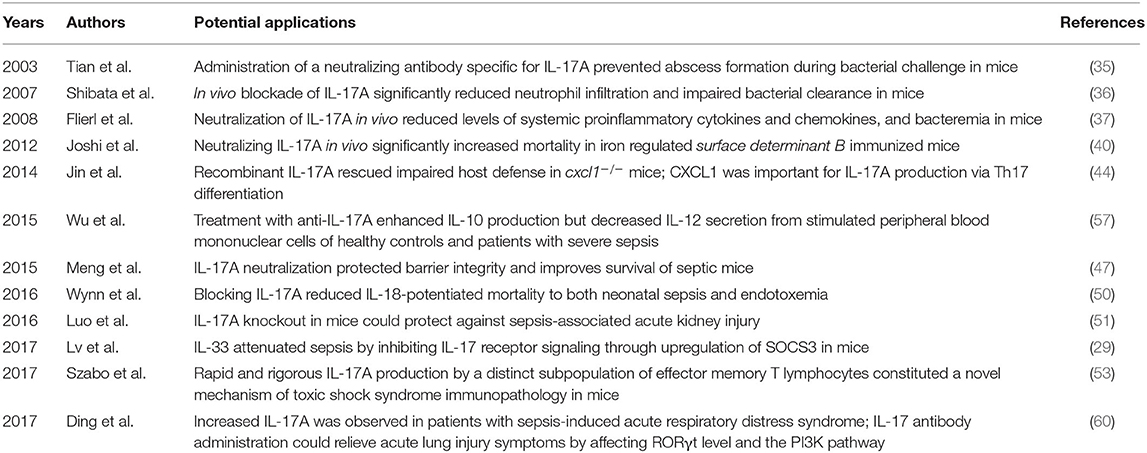
Table 2. Summary of studies on potential applications of IL-17 targeting in sepsis.
Summary and Perspectives
IL-17A can regulate host defenses against invading pathogens by producing chemokines, AMPs, and proinflammatory cytokines. In this way, IL-17A forms part of the alarm signal by sentry-like immune cells to stimulate host defenses. On one hand, IL-17A interacts with various mediators (e.g., TNF-α, IL-1, IL-6) to induce tissue-infiltrating neutrophils to eliminate the invading pathogens. On the other hand, IL-17A may interact with other proinflammatory cytokines to drive exaggerated immune response, and contribute to the development of inflammatory and autoimmune diseases. There is burgeoning evidence that IL-17A participates in the pathophysiology of sepsis, with respect to regulation of inflammatory and immune responses. Consistently, elevated level of IL-17A is apparently related with disease severity in sepsis, suggesting a potential biomarker of prognosis in the clinical setting.
Moreover, developing IL-17A as a therapeutic target must consider the fact that IL-17A might be either protective or pathogenic, depending on the specific circumstances. Future work will be needed to deeply explore the pathophysiological mechanisms by IL-17A interfere with divergent immune responses during sepsis. In addition, studies are warranted to evaluate the other forms (IL-17B-IL-17F) in clinical settings.
Author Contributions
YY conceptualized the study. YG and MH conducted the literature review and drafted the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81873946, 81974293, 81730057), the Key Project of Military Medical Innovation Program of Chinese PLA (18CXZ026), the National Key Research and Development Program of China (2017YFC1103302), and the Natural Science Foundation of Zhejiang Province (LY20H150011).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AMPs, antimicrobial peptides; APCs, antigen-presenting cells; C/EBP, CCAAT/enhancer-binding protein; CCL, chemokine (C-C motif) ligand; CXCL, C-X-C motif ligand; COX-2, cyclooxygenase-2; EBV, Epstein-Barr virus; GM-CSF, granulocyte-macrophage colony-stimulating factor; G-CSF, granulocyte-colony stimulating factor; HMGB1, high mobility group box-1 protein; IFN-γ, interferon-γ; IL, interleukin; iNOS, inducible nitric oxide synthase; iNKT-2, type 2 innate-like lymphoid cells; ILCs, innate lymphoid cells; I/R, ischemia and reperfusion; JAK, Janus kinase; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor-kappa B; NKT, natural killer T cell; PI3K, phosphatidylinostitol 3-kinase; PAMPs, pathogen-associated molecular patterns; RSV, respiratory syncytial virus; RORγt, RAR-related orphan receptor γ; SEFIR, SEF/IL-17R; STAT, signal transducers and activators of transcription; Th17, CD4+ T helper 17; TIR, Toll-IL-1 receptor; TRIF, TIR-domain-containing adapter-inducing interferon-β; TRAF, tumor necrosis factor receptor-associated factor; TNF-α, tumor necrosis factor-α; Th2, type 2 T helper; TSLP, thymic stromal lymphopoietin; MyD88, myeloid differentiation primary response gene 88.
References
1. Rouvier E, Luciani MF, Mattei MG, Denizot F, Golstein P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirussaimiri gene. J Immunol. (1993) 150:5445–56.
2. Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, et al. Herpesvirussaimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. (1995) 3:811–21. doi: 10.1016/1074-7613(95)90070-5
3. Goepfert A, Lehmann S, Wirth E, Rondeau JM. The human IL-17A/F heterodimer: a two-faced cytokine with unique receptor recognition properties. Sci Rep. (2017) 7:8906. doi: 10.1038/s41598-017-08360-9
4. Chang SH, Dong C. A novel heterodimeric cytokine consisting of IL-17 and IL-17F regulates inflammatory responses. Cell Res. (2017) 17:435–40. doi: 10.1038/cr.2007.35
5. McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 family of cytokines in health and disease. Immunity. (2019) 50:892–906. doi: 10.1016/j.immuni.2019.03.021
6. Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. (2003) 14:155–74. doi: 10.1016/S1359-6101(03)00002-9
7. Bie Q, Jin C, Zhang B, Dong H. IL-17B: a new area of study in the IL-17 family. Mol Immunol. (2017) 90:50–6. doi: 10.1016/j.molimm.2017.07.004
8. Song X, Zhu S, Shi P, Liu Y, Shi Y, Levin SD, et al. IL-17RE is the functional receptor for IL-17C and mediates mucosal immunity to infection with intestinal pathogens. Nat Immunol. (2011) 12:1151–8. doi: 10.1038/ni.2155
9. Starnes T, Broxmeyer HE, Robertson MJ, Hromas R. Cutting edge: IL-17D, a novel member of the IL-17 family, stimulates cytokine production and inhibits hemopoiesis. J Immunol. (2002) 169:642–6. doi: 10.4049/jimmunol.169.2.642
10. Albanesi C, Cavani A, Girolomoni G. IL-17 is produced by nickel-specific T lymphocytes and regulates ICAM-1 expression and chemokine production in human keratinocytes: synergistic or antagonist effects with IFN-gamma and TNF-alpha. J Immunol. (1999) 162:494–502. doi: 10.1016/S0923-1811(98)84061-9
11. Antonysamy MA, Fanslow WC, Fu F, Li W, Qian S, Troutt AB, et al. Evidence for a role of IL-17 in organ allograft rejection: IL-17 promotes the functional differentiation of dendritic cell progenitors. J Immunol. (1999) 162:577–84.
12. Infante-Duarte C, Horton HF, Byrne MC, Kamradt T. Microbial lipopeptides induce the production of IL-17 in Th cells. J Immunol. (2000) 165:6107–15. doi: 10.4049/jimmunol.165.11.6107
13. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. (2005) 6:1123–32. doi: 10.1038/ni1254
14. Lubberts E. The IL-23-IL-17 axis in inflammatory arthritis. Nat Rev Rheumatol. (2015) 11:415–29. doi: 10.1038/nrrheum.2015.53
15. Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. (2010) 10:479–89. doi: 10.1038/nri2800
16. Li Y, Wei C, Xu H, Jia J, Wei Z, Guo R, et al. The immunoregulation of Th17 in host against intracellular bacterial infection. Mediators Inflamm. (2018) 2018:6587296. doi: 10.1155/2018/6587296
17. Chamoun MN, Blumenthal A, Sullivan MJ, Schembri MA, Ulett GC. Bacterial pathogenesis and interleukin-17: interconnecting mechanisms of immune regulation, host genetics, and microbial virulence that influence severity of infection. Crit Rev Microbiol. (2018) 44:465–86. doi: 10.1080/1040841X.2018.1426556
18. Lawrence SM, Ruoss JL, Wynn JL. IL-17 in neonatal health and disease. Am J Reprod Immunol. (2018) 79:e12800. doi: 10.1111/aji.12800
19. Onishi RM, Gaffen SL. Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology. (2010) 129:311–21. doi: 10.1111/j.1365-2567.2009.03240.x
20. Kostareva OS, Gabdulkhakov AG, Kolyadenko IA, Garber MB, Tishchenko SV. Interleukin-17: functional and structural features, application as a therapeutic target. Biochemistry. (2019) 84:S193–205. doi: 10.1134/S0006297919140116
21. Amatya N, Garg AV, Gaffen SL. IL-17 signaling: the Yin and the Yang. Trends Immunol. (2017) 38:310–22. doi: 10.1016/j.it.2017.01.006
22. Ahmed M, Gaffen SL. IL-17 inhibits adipogenesis in part via C/EBPa, PPARg and Kruppel-like factors. Cytokine. (2013) 61:898–905. doi: 10.1016/j.cyto.2012.12.007
23. Wu L, Chen X, Zhao J, Martin B, Zepp JA, Ko JS, et al. A novel IL-17 signaling pathway controlling keratinocyte proliferation and tumorigenesis via the TRAF4-ERK5 axis. J Exp Med. (2015) 212:1571–87. doi: 10.1084/jem.20150204
24. Karlsen JR, Borregaard N, Cowland JB. Induction of neutrophil gelatinase-associated lipocalin expression by co-stimulation with interleukin-17 and tumor necrosis factor-alpha is controlled by IkappaB-zeta but neither by C/EBP-beta nor C/EBP-delta. J Biol Chem. (2010) 285:14088–100. doi: 10.1074/jbc.M109.017129
25. Sønder SU, Saret S, Tang W, Sturdevant DE, Porcella SF, Siebenlist U. IL-17-induced NF-kappaB activation via CIKS/Act1: physiologic significance and signaling mechanisms. J Biol Chem. (2011) 286:12881–90. doi: 10.1074/jbc.M110.199547
26. Schwandner R, Yamaguchi K, Cao Z. Requirement of tumor necrosis factor receptor-associated factor (TRAF)6 in interleukin 17 signal transduction. J Exp Med. (2000) 191:1233–40. doi: 10.1084/jem.191.7.1233
27. Herjan T, Hong L, Bubenik J, Bulek K, Qian W, Liu C, et al. IL-17-receptor-associated adaptor Act1 directly stabilizes mRNAs to mediate IL-17 inflammatory signaling. Nat Immunol. (2018) 19:354–65. doi: 10.1038/s41590-018-0071-9
28. Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. (1996) 183:2593–603. doi: 10.1084/jem.183.6.2593
29. Faour WH, Mancini A, He QW, Di Battista JA. T-cell-derived interleukin-17 regulates the level and stability of cyclooxygenase-2 (COX-2) mRNA through restricted activation of the p38 mitogen-activated protein kinase cascade: role of distal sequences in the 30-untranslated region of COX-2 mRNA. J Biol Chemy. (2003) 278:26897–907. doi: 10.1074/jbc.M212790200
30. Song X, Dai D, He X, Zhu S, Yao Y, Gao H, et al. Growth factor FGF2 cooperates with interleukin-17 to repair intestinal epithelial damage. Immunity. (2015) 43:488–501. doi: 10.1016/j.immuni.2015.06.024
31. Liu WX, Li ZJ, Niu XL, Yao Z, Deng WM. The role of T helper 17 cells and other IL-17-producing cells in bone resorption and remodeling. Int Rev Immunol. (2015) 34:332–47. doi: 10.3109/08830185.2014.952414
32. Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. (2001) 15:985–95. doi: 10.1016/S1074-7613(01)00243-6
33. Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, Gorman DM, et al. IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med. (2007) 204:161–70. doi: 10.1084/jem.20061738
34. Rendon JL, Choudhry MA. Th17 cells: critical mediators of host responses to burn injury and sepsis. J Leukoc Biol. (2012) 92:529–38. doi: 10.1189/jlb.0212083
35. Chung DR, Kasper DL, Panzo RJ, Chitnis T, Grusby MJ, Sayegh MH, et al. CD4+ T cells mediate abscess formation in intra-abdominal sepsis by an IL-17-dependent mechanism. J Immunol. (2003) 170:1958–63. doi: 10.4049/jimmunol.170.4.1958
36. Shibata K, Yamada H, Hara H, Kishihara K, Yoshikai Y. Resident Vdelta1+ gammadelta T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. J Immunol. (2007) 178:4466–72. doi: 10.4049/jimmunol.178.7.4466
37. Flierl MA, Rittirsch D, Gao H, Hoesel LM, Nadeau BA, Day DE. Adverse functions of IL-17A in experimental sepsis. FASEB J. (2008) 22:2198–205. doi: 10.1096/fj.07-105221
38. Freitas A, Alves-Filho JC, Victoni T, Secher T, Lemos HP, Sônego F, et al. IL-17 receptor signaling is required to control polymicrobial sepsis. J Immunol. (2009) 182:7846–54. doi: 10.4049/jimmunol.0803039
39. Kasten KR, Prakash PS, Unsinger J, Goetzman HS, England LG, Cave CM, et al. Interleukin-7 (IL-7) treatment accelerates neutrophil recruitment through gamma delta T-cell IL-17 production in a murine model of sepsis. Infect Immun. (2010) 78:4714–22. doi: 10.1128/IAI.00456-10
40. Joshi A, Pancari G, Cope L, Bowman EP, Cua D, Proctor RA, et al. Immunization with Staphylococcus aureus iron regulated surface determinant B (IsdB) confers protection via Th17/IL17 pathway in a murine sepsis model. Hum Vaccin Immunother. (2012) 8:336–46. doi: 10.4161/hv.18946
41. Ogiku M, Kono H, Hara M, Tsuchiya M, Fujii H. Interleukin-17A plays a pivotal role in polymicrobial sepsis according to studies using IL-17A knockout mice. J Surg Res. (2012) 174:142–9. doi: 10.1016/j.jss.2010.11.901
42. Bosmann M, Meta F, Ruemmler R, Haggadone MD, Sarma JV, Zetoune FS, et al. Regulation of IL-17 family members by adrenal hormones during experimental sepsis in mice. Am J Pathol. (2013) 182:1124–30. doi: 10.1016/j.ajpath.2013.01.005
43. Shimura E, Shibui A, Narushima S, Nambu A, Yamaguchi S, Akitsu A, et al. Potential role of myeloid cell/eosinophil-derived IL-17 in LPS-induced endotoxin shock. Biochem Biophys Res Commun. (2013) 453:1–6. doi: 10.1016/j.bbrc.2014.09.004
44. Jin L, Batra S, Douda DN, Palaniyar N, Jeyaseelan S. CXCL1 contributes to host defense in polymicrobial sepsis via modulating T cell and neutrophil functions. J Immunol. (2014) 193:3549–58. doi: 10.4049/jimmunol.1401138
45. Cauvi DM, Williams MR, Bermudez JA, Armijo G, De Maio A. Elevated expression of IL-23/IL-17 pathway-related mediators correlates with exacerbation of pulmonary inflammation during polymicrobial sepsis. Shock. (2014) 42:246–55. doi: 10.1097/SHK.0000000000000207
46. Costa MF, de Negreiros CB, Bornstein VU, Valente RH, Mengel J, Henriques MD, et al. Murine IL-17+ Vγ4 T lymphocytes accumulate in the lungs and play a protective role during severe sepsis. BMC Immunol. (2015) 16:36. doi: 10.1186/s12865-015-0098-8
47. Meng J, Banerjee S, Li D, Sindberg GM, Wang F, Ma J, et al. Opioid exacerbation of gram-positive sepsis, induced by gut microbial modulation, is rescued by IL-17A neutralization. Sci Rep. (2015) 5:10918. doi: 10.1038/srep10918
48. Zhao Y, Zhang X, Song Z, Qi D, Deng X, Xia J, et al. Rapamycin attenuates acute lung injury induced by LPS through inhibition of Th17 cell proliferation in mice. Sci Rep. (2016) 6:20156. doi: 10.1038/srep20156
49. Cen C, Aziz M, Yang WL, Nicastro J, Coppa GF, Wang P. MFG-E8 downregulates IL-17 expression in sepsis by modulating STAT3 activation. Surgery. (2016) 159:560–9. doi: 10.1016/j.surg.2015.08.011
50. Wynn JL, Wilson CS, Hawiger J, Scumpia PO, Marshall AF, Liu JH, et al. Targeting IL-17A attenuates neonatal sepsis mortality induced by IL-18. Proc Natl Acad Sci USA. (2016) 113:E2627–35. doi: 10.1073/pnas.1515793113
51. Luo CJ, Luo F, Zhang L, Xu Y, Cai GY, Fu B, et al. Knockout of interleukin-17A protects against sepsis-associated acute kidney injury. Ann Intensive Care. (2016) 6:56. doi: 10.1186/s13613-016-0157-1
52. Lv R, Zhao J, Lei M, Xiao D, Yu Y, Xie J. IL-33 attenuates sepsis by inhibiting IL-17 receptor signaling through upregulation of SOCS3. Cell Physiol Biochem. (2017) 42:1961–72. doi: 10.1159/000479836
53. Szabo PA, Goswami A, Mazzuca DM, Kim K, O'Gorman DB, Hess DA. Rapid and rigorous IL-17A production by a distinct subpopulation of effector memory T lymphocytes constitutes a novel mechanism of toxic shock syndrome immunopathology. J Immunol. (2017) 198:2805–18. doi: 10.4049/jimmunol.1601366
54. Han Y, Li X, Gao S, Liu X, Kang L, Li X, et al. Interleukin 17 is an important pathogenicity gene in pediatric sepsis. J Cell Biochem. (2019) 120:3664–71. doi: 10.1002/jcb.27644
55. Nakada TA, Russell JA, Boyd JH, Walley KR. IL-17A genetic variation is associated with altered susceptibility to gram-positive infection and mortality of severe sepsis. Crit Care. (2011) 15:R254. doi: 10.1186/cc10515
56. Accardo Palumbo A, Forte GI, Pileri D, Vaccarino L, Conte F, D'Amelio L, et al. Analysis of IL-6, IL-10 and IL-17 genetic polymorphisms as risk factors for sepsis development in burned patients. Burns. (2012) 38:208–13. doi: 10.1016/j.burns.2011.07.022
57. Wu HP, Shih CC, Chu CM, Huang CY, Hua CC, Liu YC, et al. Effect of interleukin-17 on in vitro cytokine production in healthy controls and patients with severe sepsis. J Formos Med Assoc. (2015) 114:1250–7. doi: 10.1016/j.jfma.2014.09.009
58. Paraschos MD, Patrani M, Pistiki A, Katsenos C, Tsaganos T, Netea MG, et al. Defective cytokine production early after multiple traumas: modulation in severe sepsis. Cytokine. (2015) 76:222–6. doi: 10.1016/j.cyto.2015.05.021
59. Maravitsa P, Adamopoulou M, Pistiki A, Netea MG, Louis K, Giamarellos-Bourboulis EJ. Systemic over-release of interleukin-17 in acute kidney injury after septic shock: clinical and experimental evidence. Immunol Lett. (2016) 178:68–76. doi: 10.1016/j.imlet.2016.08.002
60. Ding Q, Liu GQ, Zeng YY, Zhu JJ, Liu ZY, Zhang X, et al. Role of IL-17 in LPS-induced acute lung injury: an in vivo study. Oncotarget. (2017) 8:93704–11. doi: 10.18632/oncotarget.21474
61. Ahmed Ali M, Mikhael ES, Abdelkader A, Mansour L, EIEssawy R, EISayed R, et al. Interleukin-17 as a predictor of sepsis in polytrauma patients: a prospective cohort study. Eur J Trauma Emerg Surg. (2018) 4:621–6. doi: 10.1007/s00068-017-0841-3
62. Ye P, Garvey PB, Zhang P, Nelson S, Bagby G, Summer WR, et al. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am J Respir Cell Mol Biol. (2001) 25:335–40. doi: 10.1165/ajrcmb.25.3.4424
63. Delano MJ, Ward PA. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest. (2016) 126:23–31. doi: 10.1172/JCI82224
64. Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. (2001) 194:519–27. doi: 10.1084/jem.194.4.519
65. Cho JS, Pietras EM, Garcia NC, Ramos RI, Farzam DM, Monroe HR, et al. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J. Clin Invest. (2010) 120:1762–73. doi: 10.1172/JCI40891
66. Sivick KE, Schaller MA, Smith SN, Mobley HL. The innate immune response to uropathogenic Escherichia coli involves IL-17A in a murine model of urinary tract infection. J Immunol. (2010) 184:2065–75. doi: 10.4049/jimmunol.0902386
67. Hamada S, Umemura M, Shiono T, Tanaka K, Yahagi A, Begum MD, et al. IL-17A produced by gammadelta T cells plays a critical role in innate immunity against listeria monocytogenes infection in the liver. J Immunol. (2008) 181:3456–63. doi: 10.4049/jimmunol.181.5.3456
68. Tenland E, Håkansson G, Alaridah N, Lutay N, Rönnholm A, Hallgren O, et al. Innate immune responses after airway epithelial stimulation with Mycobacterium bovis Bacille-Calmette Guérin. PLoS ONE. (2016) 11:e0164431. doi: 10.1371/journal.pone.0164431
69. Broz P, Ohlson MB, Monack DM. Innate immune response to Salmonella typhimurium, a model enteric pathogen. Gut Microbes. (2012) 3:62–70. doi: 10.4161/gmic.19141
70. Xie GH, Fang XM, Fang Q, Wu XM, Jin YH, Wang JL, et al. Impact of invasive fungal infection on outcomes of severe sepsis: a multicenter matched cohort study in critically ill surgical patients. Crit Care. (2008) 12:R5. doi: 10.1186/cc6766
71. Gladiator A, Wangler N, Trautwein-Weidner K, Leibund Gut-Landmann S. Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J Immunol. (2013) 190:521–5. doi: 10.4049/jimmunol.1202924
72. Bär E, Whitney PG, Moor K, Reis e, Sousa C, Leibund Gut-Landmann S. IL-17 regulates systemic fungal immunity by controlling the functional competence of NK cells. Immunity. (2014) 40:117–27. doi: 10.1016/j.immuni.2013.12.002
73. Sparber F, Leibund Gut-landmann S. Interleukin 17-mediated host defense against Candida albicans. Pathogens. (2015) 4:606–19. doi: 10.3390/pathogens4030606
74. Levy R, Okada S, Beziat V, Moriya K, Liu C, Chai LYA, et al. Genetic, immunological, and clinical features of patients with bacterial and fungal infections due to inherited IL-17RA deficiency. Proc Natl Acad Sci USA. (2016) 113:E8277–85. doi: 10.1073/pnas.1618300114
75. Murdock BJ, Huffnagle GB, Olszewski MA, Osterholzer JJ. Interleukin-17A enhances host defense against cryptococcal lung infection through effects mediated by leukocyte recruitment, activation, and gamma interferon production. Infect Immun. (2014) 82:937–48. doi: 10.1128/IAI.01477-13
76. Rudner XL, Happel KI, Young EA, Shellito JE. Interleukin-23 (IL-23)-IL-17 cytokine axis in murine Pneumocystis carinii infection. Infect Immun. (2007) 75:3055–61. doi: 10.1128/IAI.01329-06
77. Guerra ES, Lee CK, Specht CA, Yadav B, Huang H, Akalin A, et al. Central role of IL-23 and IL-17 producing eosinophils as immunomodulatoryeffector cells in acute pulmonary aspergillosis and allergic asthma. PLoS Pathog. (2017) 13:e1006175. doi: 10.1371/journal.ppat.1006175
78. Huang J, Meng S, Hong S, Lin X, Jin W, Dong C. IL-17C is required for lethal inflammation during systemic fungal infection. Cell Mol Immunol. (2016) 13:474–83. doi: 10.1038/cmi.2015.56
79. Crowe CR, Chen K, Pociask DA, Alcorn JF, Krivich C, Enelow RI, et al. Critical role of IL-17RA in immunopathology of influenza infection. J Immunol. (2009) 183:5301–10. doi: 10.4049/jimmunol.0900995
80. Li C, Yang P, Sun Y, Li T, Wang C, Wang Z, et al. IL-17 response mediates acute lung injury induced by the 2009 pandemic influenza A (H1N1) virus. Cell Res. (2012) 22:528–38. doi: 10.1038/cr.2011.165
81. Mukherjee S, Lindell DM, Berlin AA, Morris SB, Shanley TP, Hershenson MB, et al. IL-17-induced pulmonary pathogenesis during respiratory viral infection and exacerbation of allergic disease. Am J Pathol. (2011) 179:248–58. doi: 10.1016/j.ajpath.2011.03.003
82. Suryawanshi A, Veiga-Parga T, Rajasagi NK, Reddy PB, Sehrawat S, Sharma S, et al. Role of IL-17 and Th17 cells in herpes simplex virus-induced corneal immunopathology. J Immunol. (2011) 187:1919–30. doi: 10.4049/jimmunol.1100736
83. Erdmann H, Roßnagel C, Böhme J, Iwakura Y, Jacobs T, Schaible UE, et al. IL-17A promotes macrophage effector mechanisms against Trypanosoma cruzi by trapping parasites in the endolysosomal compartment. Immunobiology. (2013) 218:910–23. doi: 10.1016/j.imbio.2012.10.005
84. Peckham RK, Brill R, Foster DS, Bowen AL, Leigh JA, Coffey TJ, et al. Two distinct populations of bovine IL-17+ T-cells can be induced and WC1+IL-17+γδ T-cells are effective killers of protozoan parasites. Sci Rep. (2014) 4:5431. doi: 10.1038/srep05431
85. Mezioug D, Touil-Boukoffa C. Interleukin-17A correlates with interleukin-6 production in human cystic echinococcosis: a possible involvement of IL-17A in immunoprotection against Echinococcus granulosus infection. Eur Cytokine Netw. (2012) 23:112–9. doi: 10.1684/ecn.2012.0314
86. Silva JL, Rezende-Oliveira K, da Silva MV, Gómez-Hernández C, Peghini BC, Silva NM, et al. IL-17-expressing CD4+ and CD8+ T lymphocytes in human toxoplasmosis. Mediators Inflamm. (2014) 2014:573825. doi: 10.1155/2014/573825
87. Gonzalez-Lombana C, Gimblet C, Bacellar O, Oliveira WW, Passos S, Carvalho LP, et al. IL-17 mediates immunopathology in the absence of IL-10 following Leishmania major infection. PLoS Pathog. (2013) 9:e1003243. doi: 10.1371/journal.ppat.1003243
88. Hartley MA, Bourreau E, Rossi M, Castiglioni P, Eren RO, Prevel F, et al. Leishmania virus-dependent metastatic Leishmaniasisis prevented by blocking IL-17A. PLoS Pathog. (2016) 12:e1005852. doi: 10.1371/journal.ppat.1005852
89. Pool R, Gomez H, Kellum JA. Mechanisms of organ dysfunction in sepsis. Crit Care Clin. (2018) 34:63–80. doi: 10.1016/j.ccc.2017.08.003
90. Cecconi M, Evans L, Levy M, Rhodes A. Sepsis and septic shock. Lancet. (2018) 392:75–87. doi: 10.1016/S0140-6736(18)30696-2
91. Essilfie AT, Simpson JL, Horvat JC, Preston JA, Dunkley ML, Foster PS, et al. Haemophilus influenzae infection drives IL-17-mediated neutrophilic allergic airways disease. PLoS Pathog. (2011) 7:e1002244. doi: 10.1371/journal.ppat.1002244
92. Ozkok A, Edelstein CL. Pathophysiology of cisplatin-induced acute kidney injury. Biomed Res Int. (2014) 2014:967826. doi: 10.1155/2014/967826
93. Sparber F, Leibund Gut-Landmann S. Interleukin-17 in antifungal immunity. Pathogens. (2019) 8:E54. doi: 10.3390/pathogens8020054
94. Su SA, Yang D, Zhu W, Cai Z, Zhang N, Zhao L, et al. Interleukin-17A mediates cardiomyocyte apoptosis through Stat3-iNOS pathway. Biochim Biophys Acta. (2016) 1863:2784–94. doi: 10.1016/j.bbamcr.2016.08.013
95. Tang H, Pei H, Xia Q, Tang Y, Huang J, Huang J, et al. Role of gene polymorphisms/haplotypes and serum levels of interleukin-17A in susceptibility to viral myocarditis. Exp Mol Pathol. (2018) 104:140–5. doi: 10.1016/j.yexmp.2018.03.002
96. Fan Y, Weifeng W, Yuluan Y, Qing K, Yu P, Yanlan H. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of coxsackievirus b3-induced viral myocarditis reduces myocardium inflammation. Virol J. (2011) 8:17. doi: 10.1186/1743-422X-8-17
97. Zhang Y, Zhang YY, Li TT, Wang J, Jiang Y, Zhao Y, et al. Ablation of interleukin-17 alleviated cardiac interstitial fibrosis and improved cardiac function via inhibiting long non-coding RNA-AK081284 in diabetic mice. J Mol Cell Cardiol. (2018) 115:64–72. doi: 10.1016/j.yjmcc.2018.01.001
98. Hu X, Zhang K, Chen Z, Jiang H, Xu W. The HMGB1-IL-17A axis contributes to hypoxia/reoxygenation injury via regulation of cardiomyocyte apoptosis and autophagy. Mol Med Rep. (2018) 17:336–41. doi: 10.3892/mmr.2017.7839
99. Valeri M, Raffatellu M. Cytokines IL-17 and IL-22 in the host response to infection. Pathog Dis. (2016) 74:ftw111. doi: 10.1093/femspd/ftw111
Keywords: interleukin-17, cytokine, sepsis, inflammatory response, immune response
Citation: Ge Y, Huang M and Yao Y (2020) Biology of Interleukin-17 and Its Pathophysiological Significance in Sepsis. Front. Immunol. 11:1558. doi: 10.3389/fimmu.2020.01558
Received: 02 April 2020; Accepted: 12 June 2020;
Published: 28 July 2020.
Edited by:
Laura Maggi, University of Florence, ItalyReviewed by:
Francesco Liotta, University of Florence, ItalyPeter A. Ward, University of Michigan, United States
Copyright © 2020 Ge, Huang and Yao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Man Huang, aHVhbmdtYW5Aemp1LmVkdS5jbg==; Yong-ming Yao, Y19mZkBzaW5hLmNvbQ==