 Masakatsu Yanagimachi1,2*
Masakatsu Yanagimachi1,2* Koji Kato3,4Akihiro Iguchi5Koji Sasaki6Chikako Kiyotani7
Koji Kato3,4Akihiro Iguchi5Koji Sasaki6Chikako Kiyotani7 Katsuyoshi Koh8Takashi Koike9
Katsuyoshi Koh8Takashi Koike9 Hideki Sano10Tomonari Shigemura11Hideki Muramatsu12Keiko Okada13Masami Inoue14Ken Tabuchi15
Hideki Sano10Tomonari Shigemura11Hideki Muramatsu12Keiko Okada13Masami Inoue14Ken Tabuchi15 Toyoki Nishimura16
Toyoki Nishimura16 Tomoyuki Mizukami16,17Hiroyuki Nunoi16
Tomoyuki Mizukami16,17Hiroyuki Nunoi16 Kohsuke Imai1
Kohsuke Imai1 Masao Kobayashi18
Masao Kobayashi18 Tomohiro Morio1
Tomohiro Morio1- 1Department of Pediatrics, Tokyo Medical and Dental University, Tokyo, Japan
- 2Department of Hematology/Oncology, Kanagawa Children's Medical Center, Yokohama, Japan
- 3Department of Hematology and Oncology, Children's Medical Center, Japanese Red Cross Nagoya First Hospital, Nagoya, Japan
- 4Central Japan Cord Blood Bank, Seto, Japan
- 5Department of Pediatrics, Hokkaido University Hospital, Sapporo, Japan
- 6Department of Pediatrics, Yokohama City University, Yokohama, Japan
- 7Children's Cancer Center, National Center for Child Health and Development, Tokyo, Japan
- 8Department of Hematology/Oncology, Saitama Children's Medical Center, Saitama, Japan
- 9Department of Pediatrics, Tokai University School of Medicine, Isehara, Japan
- 10Department of Pediatric Oncology, Fukushima Medical University Hospital, Fukushima, Japan
- 11Department of Pediatrics, Shinshu University School of Medicine, Nagano, Japan
- 12Department of Pediatrics, Nagoya University Graduate School of Medicine, Nagoya, Japan
- 13Department of Pediatric Hematology/Oncology, Osaka City General Hospital, Osaka, Japan
- 14Department of Pediatric Hematology/Oncology, Osaka Women's and Children's Hospital, Osaka, Japan
- 15Division of Pediatrics, Tokyo Metropolitan Cancer and Infectious Disease Center Komagome Hospital, Tokyo, Japan
- 16Division of Pediatrics, Developmental and Urological-Reproductive Medicine Faculty of Medicine, University of Miyazaki, Miyazaki, Japan
- 17Department of Pediatrics, NHO Kumamoto Medical Center, Kumamoto, Japan
- 18Department of Pediatrics, Hiroshima University Graduate School of Biomedical & Health Sciences, Hiroshima, Japan
Hematopoietic cell transplantation (HCT) is established as a curative treatment for severe chronic granulomatous disease (CGD). However, outcomes of HCT for CGD in Japan had not been precisely reported. We evaluated the outcome of HCT for CGD in Japan by means of a nationwide survey. A total of 91 patients (86 males and 5 females) with CGD who received HCT between 1992 and 2013 was investigated. Their median age at HCT was 11 years (0–39). Sixty-four patients had X-linked CGD caused by CYBB gene mutations, 13 had autosomal recessive CGD (7 CYBA and 6 NCF2), and 14 were genetically undetermined. Seventy patients are still alive at a median follow-up of 38.9 (3.7–230) months. Three-year OS and EFS was 73.7 and 67.6%, respectively. Twenty-one patients died mainly from transplant-related mortality. The cumulative incidence of grade II to IV acute GVHD and extensive chronic GVHD was 27.2 and 17.9%, respectively. Risk factors for EFS after HCT for CGD were age >30 years (P < 0.01), non-CYBB gene mutations (P < 0.01) and CBT (P < 0.01). Regarding the reduced intensity conditioning (RIC) regimen, risk factors for EFS included anti-thymocyte globulin (P = 0.048) and not using low-dose irradiation therapy (P < 0.01), in addition to the preceding risk factors. We report outcomes of HCT for CGD in Japan. Future studies are needed to improve such outcomes, especially for patients harboring non-CYBB gene mutations and suffering from adult CGD. A RIC regimen including low-dose irradiation may be a good option to explore further.
Introduction
Chronic granulomatous disease (CGD), caused by a dysfunction of the phagocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidase system, is an inborn error of immunity (1, 2). CGD is characterized by recurrent and severe infections with bacterial and fungal pathogens and by inflammatory complications, such as granuloma and colitis. Although the natural history of CGD patients has improved because of advances in supportive care, such as anti-fungal prophylaxis, CGD patients still suffer from severe uncontrollable infections or inflammatory complications. Hematopoietic cell transplantation (HCT) is an established therapy for the curative treatment of inborn errors of immunity including severe CGD (3). Recently, improved outcomes of HCT for CGD were reported from other countries (4, 5). However, the outcome of HCT for CGD in Japan had not been precisely reported until now. There are differences in the distribution of the types of molecular defects associated with CGD between Japan and other countries, which may influence the outcome of HCT for CGD (6). With this in mind, we evaluated the outcome of HCT for CGD in Japan by means of a nationwide survey.
Patients and Methods
Patients
A total of 91 patients (86 males and 5 females) with CGD who received HCT in 37 HCT centers from 1992 to 2013 was included in this study. They received a total of 100 transplants (2 patients twice, 2 thrice and one patient four times). Analyses in this study were limited to first-time transplants. Median patient age at CGD diagnosis was 0 years (0–19) and median age at first HCT was 11 years (0–39) (Table 1). Sixty-four patients had X-linked CGD caused by CYBB gene mutations, 13 had autosomal recessive (AR) CGD (7 CYBA and 6 NCF2), and 14 were genetically undetermined. The 14 patients were diagnosed CGD only according to their clinical symptoms and low or absent NAPDH-oxidase activity, especially before 2000 or could not be genetically categorized even after genetic analysis. No CGD patient carrying NCF1 and NCF4 gene mutations was identified in this study.
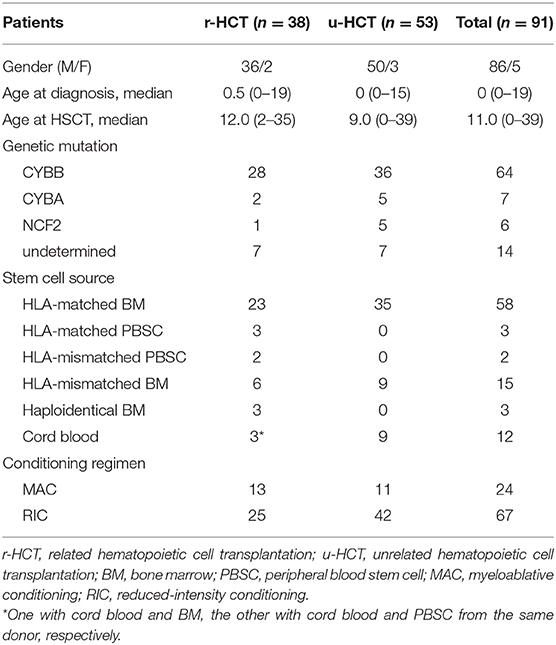
Table 1. Patients' characteristics.
Clinical data were collected by the Japan Society for Hematopoietic Cell Transplantation (JSHCT) and the Japanese Data Center for Hematopoietic Cell Transplantation (JDCHCT) using the Transplant Registry Unified Management Program (TRUMP). Genetic data were collated in the Primary Immunodeficiency database in Japan (PIDJ). This study was approved by the Institutional Review Boards at JSHCT and Tokyo Medical and Dental University. All patients and/or their guardians gave written consent to the JDCHCT and PIDJ.
Transplantation
The degree of HLA compatibility between donor and recipient was established by serotyping of HLA-A, B, and DR. Twenty-seven patients received a transplant from an HLA-matched sibling donor, of which 23 were bone marrow transplants (BMT), 3 were peripheral blood stem cell transplants (PBSCT), and 3 were cord blood transplants (CBT). Two of the CBT patients received stem cells from two sources simultaneously, namely PBSCT plus CBT, and BMT plus CBT, from HLA-matched sibling donors (Figure S1, Table 1). For statistical analyses, these two patients were assigned to the PBSCT group and BMT group, respectively. Of the patients without a matched sibling donor, 3 received a BMT from haploidentical related donors, and 8 from HLA-mismatched related donors (6 BMT and 2 PBSCT). A total of 53 patients received a transplant from an unrelated donor, including 35 HLA-matched BMT, 9 HLA-mismatched BMT and 9 CBT (1 HLA-8/8 match and 8 HLA-mismatched) (Table 1).
The choice of conditioning regimen was at the discretion of each HCT institution. Myeloablative conditioning (MAC) was defined as a regimen including total body irradiation (TBI), thoraco-abdominal irradiation (TLI) (≥8 Gy, fractionated) or busulfan (BU) (≥8 mg/kg); the other regimens were defined as reduced-intensity conditioning (RIC) [Table 1; (7)]. Six TBI/TLI and 18 BU-based regimens were included in the MAC group, and 27 fludarabine (FLU)/cyclophosphamide (CY), 14 FLU/melphalan (MEL), 19 FLU/CY/MEL and 5 FLU/BU-based regimens were included in the RIC group. For the latter, 48 low-dose TBI (mean 3.3 Gy), 3 TLI (mean 6 Gy), and 4 total abdominal irradiation (TAI) (mean 4.2 Gy) were included, as well as 39 anti-thymocyte globulin (ATG) and 4 anti-lymphocyte globulin (ALG)-treated patients. GVHD prophylaxis was almost always with cyclosporine (n = 28) or tacrolimus (n = 63) accompanied by methotrexate.
Donor chimerism was defined as >80% donor cells present in the whole white blood cell sample analyzed, whereas mixed chimerism was defined as 20–80% donor chimerism, and graft failure (GF) as <20% donor chimerism, according to the TRUMP classification. Methods of measuring chimerism status were vary, including short tandem repeat analysis and FISH analysis of X/Y chromosome.
Graft versus host disease (GVHD) was scored according to standard criteria (8, 9). The hematopoietic cell transplantation comorbidity index (HCT-CI) was also scored by standard criteria (10).
Statistical Analysis
Overall survival (OS), event-free survival (EFS), and GVHD-free, event-free survival (GEFS) were described by Kaplan-Meier estimates. Differences among clinical groups were compared using the log-rank test. An event of EFS was defined as GF or death. An event of GEFS was defined as the first event among grades III to IV acute GVHD, extended chronic GVHD, GF, and death. The probability of neutrophil engraftment (≥500/mm3), acute and chronic GVHD was evaluated by Gray's test for comparing cumulative incidence curves. For neutrophil engraftment, death before neutrophil recovery was counted as a competing event; for GVHD, death without GVHD was counted as a competing event. The influence of different factors on OS/EFS was evaluated using Cox proportional hazard regression modeling. The influence of different factors on the cumulative incidence of engraftment and GVHD was evaluated using Fine-Gray proportional hazard regression modeling. P < 0.05 were considered statistically significant. All analyses were performed with EZR (Saitama Medical Center, Jichi Medical University, Saitama, Japan), which is a graphical user interface for R (the R foundation for statistical Computing, Vienna, Austria) (11).
Results
Engraftment and Death
The cumulative incidence of neutrophil engraftment at day 100 was 80.9% for all patients, which broke down to 86.1% in BMT/PBSCT and 40.0% in CBT (Figure S2). Thus, CBT was associated with less neutrophil engraftment, but this was not quite statistically significant (P = 0.057). Thirteen GF occurred, after which 10 patients received second or more transplants and 7 survived. GF at day 100 after HCT was associated with poor OS, compared to donor or mixed chimerism (P < 0.01; Figure S3). There were two (17%) GF occurred in 13 MAC regimens, 3 (8%) in 37 FLU/CY-based regimens and 5 (31%) in 16 non-FLU/CY-based regimens at day 100 after HCT. In the aspect of stem cell sources, there were 4 (7%) GF occurred in 55 BMT and 6 (67%) in 9 CBT at day 100 after HCT.
In total, 21 patients died, 4 from graft rejection, 4 from GVHD, 2 thrombotic microangiopathy (TMA), 2 infections, 3 hemorrhages, 1 secondary malignancy (osteosarcoma), and 5 others (Table 2).
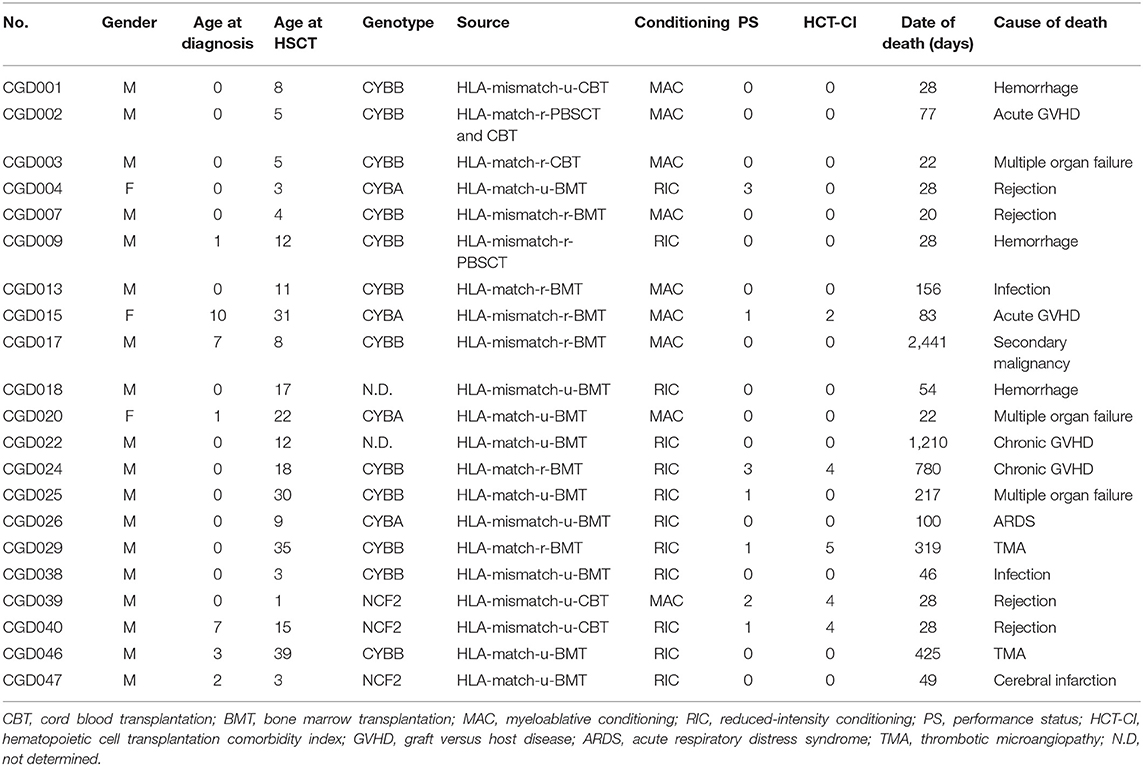
Table 2. Summary of deceased cases.
GVHD
The cumulative incidence of grade II to IV acute GVHD was 25.3% and the incidence of extensive chronic GVHD was 19.6%. ATG/ALG, donor source and HLA compatibility were not associated with the incidence of grade II to IV acute GVHD and extensive chronic GVHD.
EFS/OS and GEFS
Seventy patients are alive at a median follow-up of 38.9 (3.7–230) months. Three-year OS, EFS and GEFS was 73.7, 67.6, and 57.0%, respectively (Figure 1). Twenty-one patients died mainly from transplant-related mortality (TRM) (Table 2). Eighteen male and 3 female patients died (P = 0.08). CBT was a significant risk factor for low EFS/GEFS (P < 0.01), but not for OS (P = 0.12; Figure 2, Table 3).
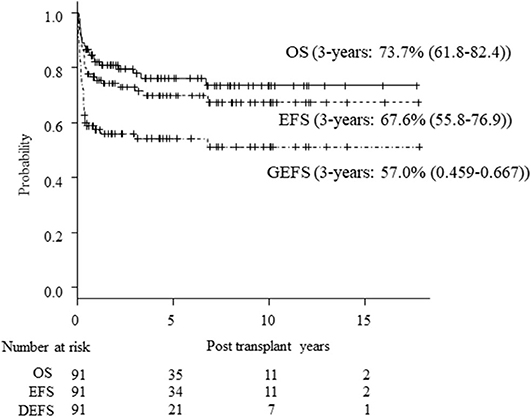
Figure 1. Kaplan-Meier estimates of OS/EFS/GEFS in all patients. Kaplan-Meier estimates of overall survival (OS), event-free survival (EFS), and GVHD-free, event-free survival (GEFS) of CGD patients undergoing HCT.
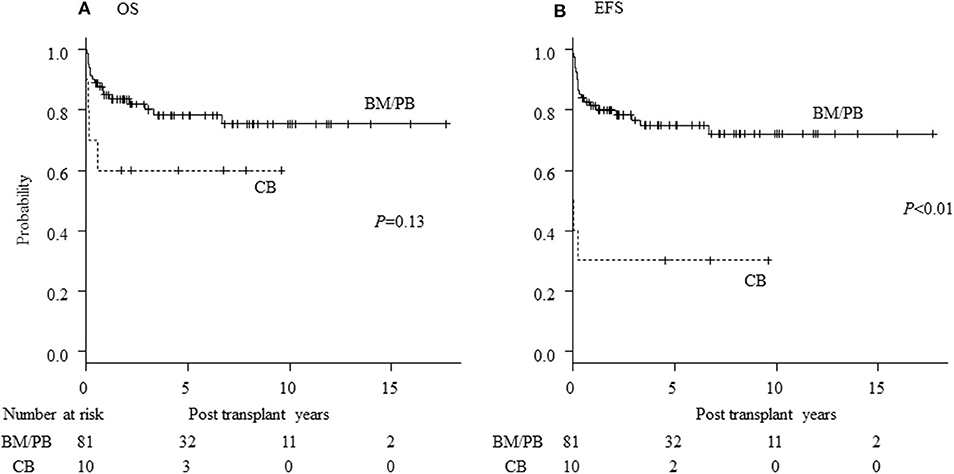
Figure 2. Influence of stem cell source for HCT of CGD. Influence of stem cell source of transplantation on (A) overall survival (OS) and (B) event-free survival (EFS) of CGD patients undergoing HCT. CB, cord blood cell transplantation; BM, bone marrow transplantation; PB, peripheral blood stem cell transplantation.
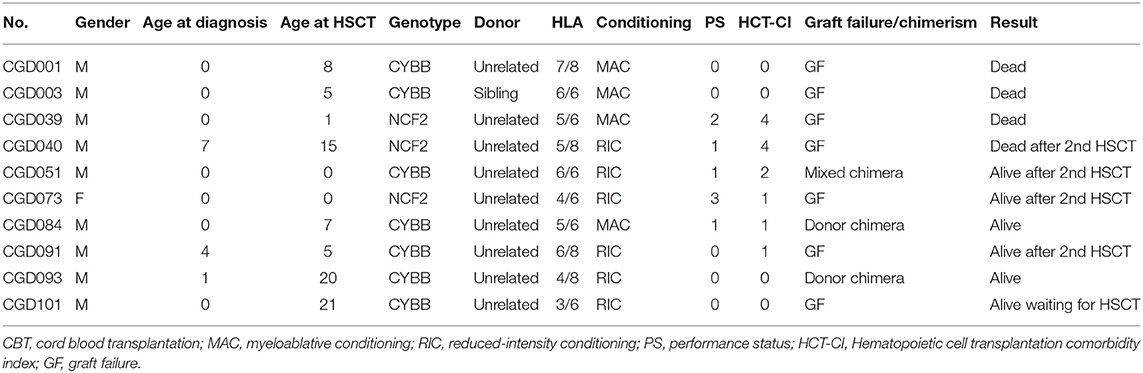
Table 3. Outcome of CBT for CGD.
There was no difference in OS/EFS/GEFS(OK) between patients transplanted 2008-2013 or earlier either for all patients or in the CBT setting only. Age >30 years at HCT resulted in poorer OS/EFS/GEFS than in younger patients (OS; P < 0.01/ EFS; P < 0.01/GEFS; P = 0.04; Figure 3). A total of 4 patients aged >30 years at HCT died from TRM (Table 2). A high HCT-CI score (over 3) also influenced the outcome of HCT in all patients (Figure S4).
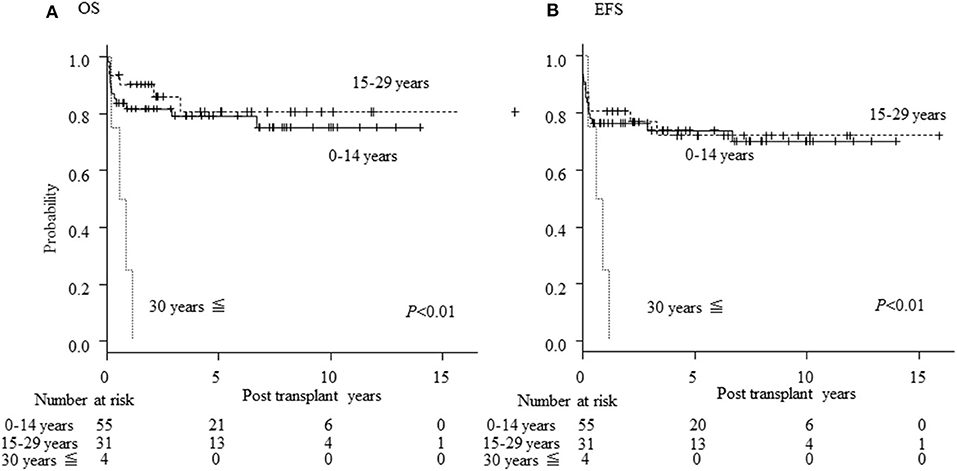
Figure 3. Influence of age on outcome of HCT. Influence of age at HCT on outcome. (A) Overall survival; (B) event-free survival of CGD patients undergoing HCT.
Patients harboring CYBA or NCF2 (non-CYBB) gene mutations had poor OS/EFS/GEFS(OK) compared to those with CYBB mutations (P < 0.01; Figure 4). There was no difference in the patients' characteristics between those with or without CYBB mutations [Table S1; (10)].
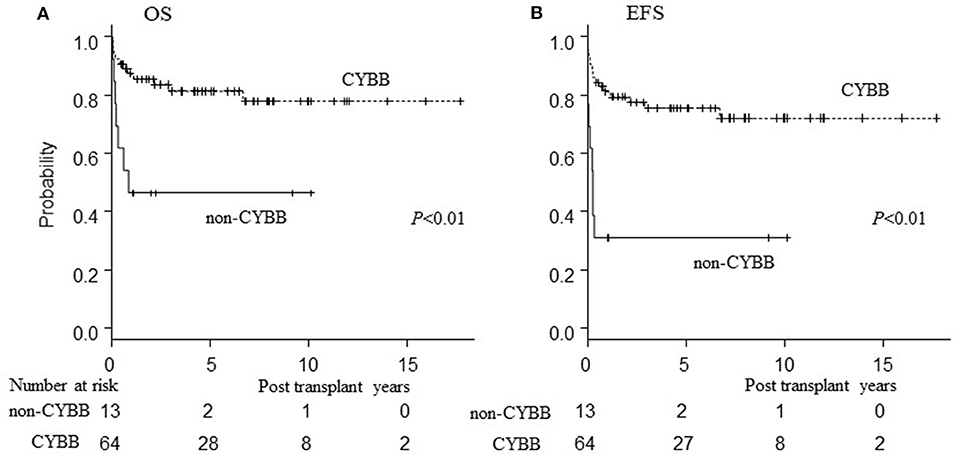
Figure 4. Influence of genetic mutation on outcome of HCT. Influence of genetic mutation on outcome of HCT. (A) Overall survival; (B) event-free survival of CGD patients undergoing HCT. CYBB, patients with CYBB gene mutation. Non-CYBB, patients with CYBA or NCF2 gene mutations.
Active bacterial or fungal infection at the time of HCT did not influence OS/EFS/GEFS in this cohort.
For BMT from HLA-matched donors, there was no difference in OS/EFS/GEFS between related (n = 27) and unrelated (n = 36) donor groups (OS; P = 0.67/EFS; P = 0.50/GEFS). Also for BMT from unrelated donors, no differences were seen in OS/EFS/GEFS; z between HLA fully matched donors (n = 19) and donors mismatched for one allele (n = 9) (OS; P = 0.64/EFS; P = 0.77/GEFS; P = 0.73). No differences were seen in OS/EFS/GEFS between HLA fully matched related donors and the other donors in all patients (OS; P = 0.58/EFS; P = 0.18/GEFS; P = 0.61) and in patients received HCT with RIC regimens (OS; P = 0.30/EFS; P = 0.07/GEFS; P = 0.10).
Conditioning Regimens
A total of 24 patients on different MAC regimens and 67 on RIC regimens had been selected at the discretion of each institution (Table 1). After the year 2000, mainly RIC regimens were employed. There were no differences in OS/EFS between patients receiving RIC or MAC regimens by univariate analysis (Figure S5). Thus, in the univariate analysis, ATG/ALG and TBI/TLI/TAI use was not associated with OS/EFS in the whole patient cohort or the RIC group (Figure S6). EFS was higher in the 47 patients receiving FLU/CY-based regimens (n = 27) and FLU/CY/MEL regimens (n = 19) than in the 19 patients not receiving FLU/CY-based regimens [FLU/BU (n = 5) and FLU/MEL (n = 14)] (p = 0.02) in the RIC group (Figure S7).
By multivariate analysis in the Cox proportional hazard regression model, risk factors for EFS of HCT for CGD in all patients were found to be age >30 years (P < 0.01), CBT (P < 0.01) and non-CYBB mutation (P < 0.01). Age >30 years (P < 0.01), MAC (P = 0.03) and non-CYBB mutation (P = 0.03) were also risk factors for OS in all patients (Table 4). Although there was no risk factor for GEFS in all patients, CBT tended to be associated with lower GEFS (P = 0.068). Risk factors for EFS of HCT for CGD in patients receiving RIC regimens were again age >30 years (P < 0.01), CBT (P < 0.01), non-CYBB mutation (P < 0.01), and also ATG/ALG (P = 0.048) and non-TBI/TLI/TAI (P < 0.01). Age over 30 years (P < 0.01), non-CYBB mutation (P < 0.01) and HCT-CI score (over 3) (P = 0.02) were also risk factors for OS in patients on RIC regimens (Table 4).
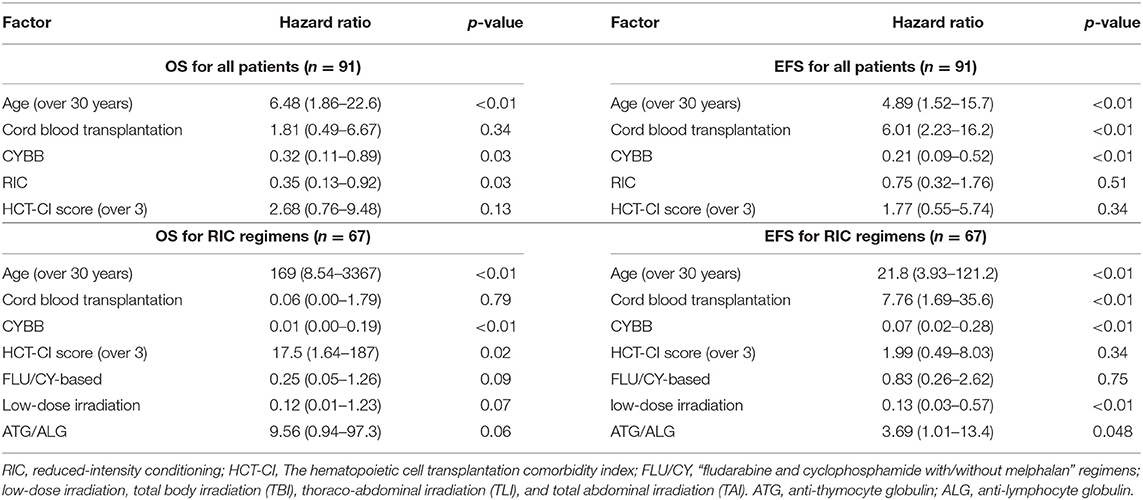
Table 4. Cox regression analysis for risk factors.
Discussion
HCT is an established curative treatment for severe CGD (4, 5, 12). However, overall results of HCT for CGD in Japan had not been precisely reported until now. Therefore, we carried out a nationwide survey of the outcome of HCT for CGD in Japan. Three-year OS (73.7%) in our cohort is very similar to that reported in previous publications, except for Güngör's study with HLA-matched donors (4, 5, 12–15). Recently, improved outcomes of HCT for CGD were reported from other countries (4, 5). HCT with “submyeloablative” conditioning regimens using low-dose or targeted busulfan administration for CGD resulted in excellent 2-year EFS (91%) and stable donor chimerism (93%) (4). Treosulfan-based conditioning resulted in 81% EFS (median follow-up, 34 months) without severe adverse events such as sinusoidal obstruction syndrome (5). These procedures will be introduced for Japanese CGD patients shortly.
This present report is the first to document poor HCT outcomes for CGD patients harboring non-CYBB gene mutations, whereas previous studies had noted that non-CYBB gene mutations were associated with better HCT outcome than CYBB mutations (4, 5, 15). In general, CGD patients harboring non-CYBB gene mutations have less severe infections and lower mortality rates than with CYBB mutations (16). Indications for HCT, especially for CGD patients with non-CYBB gene mutations, have not been established. However, patients harboring non-CYBB mutations in our cohort tended to have high HCT-CI scores (Table S1). Although there was no data of NAPDH-oxidase activity of these CGD patients harboring non-CYBB gene mutations, they might have low or absent NAPDH-oxidase activity that caused severe phenotype of these patients. These severe general conditions before and at the time of HCT might have been an influence contributing to the poor outcome of non-CYBB CGD patients in the present study. There are also possible differences in the outcomes of HCT for CGD with non-CYBB gene mutations between countries; thus, these mutations are associated with lower incidence of GF and death after HCT in other countries [Table S2; (4, 5, 15, 17, 18)]. It is necessary to evaluate the outcomes of HCT for CGD patients with non-CYBB gene mutations in detail internationally, because of rarity of these gene mutation subtypes of CGD.
Adult CGD patients are a high-risk group for HCT, because of organ dysfunction, overt infections and inflammatory and post-inflammatory complications (14, 19). Although there was no difference in the outcome of HCT between patients grouped according to the age range 0–14 years compared with 15–29 years, all 4 patients >30 years old in this study died from TRM, such as TMA and acute GVHD (Figure 3, Table 2). It was reported that donor cell infusion in CGD patients with GF or poor donor chimerism after HCT was associated with poor outcome (20). In this study, three of the four died after donor lymphoid infusion, which should therefore be used with great caution for treating patients with GF or mixed chimerism.
Mixed chimerism did not influence OS/EFS in this study (Figure S3). Older female carriers of CGD tended to suffer more complications, such as suppurative infections, gastrointestinal manifestations, and autoimmunity that CGD patients frequently experience (21, 22). Long term follow-up of CGD patients with mixed chimerism after HCT is needed for evaluating whether these complications may occur at a later time in such patients.
CBT was correlated with GF and death in this study, consistent with a previous report [Figure 2, Table 3; (23)]. It was also reported that the outcome of CBT for CGD using MAC was acceptable (24). Recently, the use of “submyeloablative” busulfan-based and treosulfan-based conditioning regimens for BMT and PBSCT has resulted in excellent engraftment and OS. These regimens are expected also to deliver promising results in unrelated CBT for CGD (4, 5). In the present study, we determined that using RIC regimens is associated with better OS than MAC regimens and that low-dose TBI/TLI/TAI resulted in higher EFS rates than RIC without TBI/TLI/TAI (Table 4). Prospective evaluations are now warranted as to whether addition of low-dose irradiation in “submyeloablative” busulfan- or treosulfan- based regimens with HLA-mismatched donor and the CBT setting will improve outcomes of HCT, according to the previous research (25). ATG/ALG was a risk factor for EFS in RIC regimens but was not associated with post-HCT viral and fungal infections (p = 0.55).
Here, we report the first nationwide study of HCT for CGD in Japan. However, this is a retrospective study using the TRUMP and PIDJ databases. Therefore, there are intrinsic limitations to the study, such as data quality regarding donor chimerism status, HLA compatibility, and CGD genotyping. The TRUMP database does not fully include CGD-related complications before HCT, such as inflammatory bowel disease. Therefore, we could not adequately evaluate the influence of the patient's condition pre-HCT on the outcome of HCT, although this is probably an important factor. Prospective studies are needed to establish the most effective HCT conditioning regimen for CGD patients. The establishment of eligibility criteria for HCT for severe CGD is also needed. Meanwhile, the outcome of HCT in adult CGD patients under 30 years old was as good as younger patients. It was reported that 3-years OS was 81.8% for adult CGD patients who had HCT [n = 11, median age at HCT; 19 (17–27)] (26). When a CGD patient with absent NAPDH-oxidase activity suffers from uncontrollable infections or severe inflammatory complications and has a suitable donor, HCT may be the treatment of choice, especially in patients younger than 30 years old, but if the patient has no available matched donor, gene therapy, or HLA-haploidentical BMT/PBSCT should be considered.
We could not adequately evaluate the late effects of HCT for CGD patients, such as issues with fertility and secondary malignancy. Anecdotally, we can say that some CGD patients who had HCT with certain RIC regimens have in the meantime born children (personal communication). Therefore, long term follow-up will be required to establish the nature of late effects of HCT and for the evaluation of the best HCT conditioning regimen and age at HCT.
We have reported on outcomes of HCT for CGD in Japan. Future studies are needed to improve these outcomes, especially for patients harboring non-CYBB gene mutations or who are adults. Improved outcomes of HCT for CGD were recently reported, and RIC regimens using low-dose irradiation therapy may be one of the best approaches.
Data Availability Statement
The datasets presented in this article are not readily available because the dataset belongs to the Japan Society for Hematopoietic Cell Transplantation (JSHCT) and the Japanese Data Center for Hematopoietic Cell Transplantation (JDCHCT). Requests to access the datasets should be directed to http://www.jdchct.or.jp/.
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional Review Boards at JSHCT and Tokyo Medical and Dental University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
MY designed the research, analyzed the data, and wrote the paper. KKa, AI, and MK analyzed the data and wrote the paper. KS, CK, KKo, TK, HS, HM, and KO performed the research and provided the clinical data. MI and TK contributed in the transplantation data management as the members of Japanese Data Center for Hematopoietic Cell Transplantation and analyzed the data. TN, TMi, HN, and KI analyzed the gene mutation causing CGD. KI and TMo designed the research and wrote the paper. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the Grant from Japan Ministry of Health, Labour and Welfare (201911006B).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors appreciate all of the staff of the JMDP and PIDJ for their assistance and all medical staff who provided medical care for CGD patients. The authors also thank Hiroaki Goto, Fumiko Tanaka, Hirokazu Kanegane, and Yoshiko Atsuta for their support for this project.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01617/full#supplementary-material
References
1. Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. (2010) 363:2600–10. doi: 10.1056/NEJMoa1007097
2. Seger RA. Advances in the diagnosis and treatment of chronic granulomatous disease. Curr Opin Hematol. (2011) 18:36–41. doi: 10.1097/MOH.0b013e32834115e7
3. Gennery AR, Albert MH, Slatter MA, Lankester A. Hematopoietic stem cell transplantation for primary immunodeficiencies. Front Pediatr. (2019) 7:445. doi: 10.3389/fped.2019.00445
4. Gungor T, Teira P, Slatter M, Stussi G, Stepensky P, Moshous D, et al. Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet. (2014) 383:436–48. doi: 10.1016/S0140-6736(13)62069-3
5. Morillo-Gutierrez B, Beier R, Rao K, Burroughs L, Schulz A, Ewins AM, et al. Treosulfan-based conditioning for allogeneic HSCT in children with chronic granulomatous disease: a multicenter experience. Blood. (2016) 128:440–8. doi: 10.1182/blood-2016-03-704015
6. Ishibashi F, Nunoi H, Endo F, Matsuda I, Kanegasaki S. Statistical and mutational analysis of chronic granulomatous disease in Japan with special reference to gp91-phox and p22-phox deficiency. Hum Genet. (2000) 106:473–81. doi: 10.1007/s004390000288
7. Bacigalupo A, Ballen K, Rizzo D, Giralt S, Lazarus H, Ho V, et al. Defining the intensity of conditioning regimens: working definitions. Biol Blood Marrow Transplant. (2009) 15:1628–33. doi: 10.1016/j.bbmt.2009.07.004
8. Filipovich AH, Weisdorf D, Pavletic S, Socie G, Wingard JR, Lee SJ, et al. National institutes of health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant. (2005) 11:945–56. doi: 10.1016/j.bbmt.2005.09.004
9. Przepiorka D, Weisdorf D, Martin P, Klingemann HG, Beatty P, Hows J, et al. 1994 Consensus conference on acute GVHD grading. Bone Marrow Transplant. (1995) 15:825–8.
10. Sorror ML, Maris MB, Storb R, Baron F, Sandmaier BM, Maloney DG, et al. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood. (2005) 106:2912–9. doi: 10.1182/blood-2005-05-2004
11. Kanda Y. Investigation of the freely available easy-to-use software 'EZR' for medical statistics. Bone Marrow Transplant. (2013) 48:452–8. doi: 10.1038/bmt.2012.244
12. Martinez CA, Shah S, Shearer WT, Rosenblatt HM, Paul ME, Chinen J, et al. Excellent survival after sibling or unrelated donor stem cell transplantation for chronic granulomatous disease. J Allergy Clin Immunol. (2012) 129:176–83. doi: 10.1016/j.jaci.2011.10.005
13. Cole T, Pearce MS, Cant AJ, Cale CM, Goldblatt D, Gennery AR. Clinical outcome in children with chronic granulomatous disease managed conservatively or with hematopoietic stem cell transplantation. J Allergy Clin Immunol. (2013) 132:1150–5. doi: 10.1016/j.jaci.2013.05.031
14. Gungor T, Halter J, Klink A, Junge S, Stumpe KD, Seger R, et al. Successful low toxicity hematopoietic stem cell transplantation for high-risk adult chronic granulomatous disease patients. Transplantation. (2005) 79:1596–606. doi: 10.1097/01.TP.0000163466.73485.5E
15. Seger RA, Gungor T, Belohradsky BH, Blanche S, Bordigoni P, Di Bartolomeo P, et al. Treatment of chronic granulomatous disease with myeloablative conditioning and an unmodified hemopoietic allograft: a survey of the European experience, 1985-2000. Blood. (2002) 100:4344–50. doi: 10.1182/blood-2002-02-0583
16. Winkelstein JA, Marino MC, Johnston RB Jr, Boyle J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore). (2000) 79:155–69. doi: 10.1097/00005792-200005000-00003
17. Horwitz ME, Barrett AJ, Brown MR, Carter CS, Childs R, Gallin JI, et al. Treatment of chronic granulomatous disease with nonmyeloablative conditioning and a T-cell-depleted hematopoietic allograft. N Engl J Med. (2001) 344:881–8. doi: 10.1056/NEJM200103223441203
18. Kutukculer N, Aykut A, Karaca NE, Durmaz A, Aksu G, Genel F, et al. Chronic granulamatous disease: two decades of experience from a paediatric immunology unit in a country with high rate of consangineous marriages. Scand J Immunol. (2019) 89:e12737. doi: 10.1111/sji.12737
19. Dunogue B, Pilmis B, Mahlaoui N, Elie C, Coignard-Biehler H, Amazzough K, et al. Chronic granulomatous disease in patients reaching adulthood: a nationwide study in France. Clin Infect Dis. (2017) 64:767–75. doi: 10.1093/cid/ciw837
20. Parta M, Kelly C, Kwatemaa N, Theobald N, Hilligoss D, Qin J, et al. Allogeneic reduced-intensity hematopoietic stem cell transplantation for chronic granulomatous disease: a single-center prospective trial. J Clin Immunol. (2017) 37:548–58. doi: 10.1007/s10875-017-0422-6
21. Battersby AC, Cale AM, Goldblatt D, Gennery AR. Clinical manifestations of disease in X-linked carriers of chronic granulomatous disease. J Clin Immunol. (2013) 33:1276–84. doi: 10.1007/s10875-013-9939-5
22. Marciano BE, Zerbe CS, Falcone EL, Ding L, DeRavin SS, Daub J, et al. X-linked carriers of chronic granulomatous disease: illness, lyonization, and stability. J Allergy Clin Immunol. (2018) 141:365–71. doi: 10.1016/j.jaci.2017.04.035
23. Morio T, Atsuta Y, Tomizawa D, Nagamura-Inoue T, Kato K, Ariga T, et al. Outcome of unrelated umbilical cord blood transplantation in 88 patients with primary immunodeficiency in Japan. Br J Haematol. (2011) 154:363–72. doi: 10.1111/j.1365-2141.2011.08735.x
24. Tewari P, Martin PL, Mendizabal A, Parikh SH, Page KM, Driscoll TA, et al. Myeloablative transplantation using either cord blood or bone marrow leads to immune recovery, high long-term donor chimerism and excellent survival in chronic granulomatous disease. Biol Blood Marrow Transplant. (2012) 18:1368–77. doi: 10.1016/j.bbmt.2012.02.002
25. Umeda K, Yabe H, Kato K, Imai K, Kobayashi M, Takahashi Y, et al. Impact of low-dose irradiation and in vivo T-cell depletion on hematopoietic stem cell transplantation for non-malignant diseases using fludarabine-based reduced-intensity conditioning. Bone Marrow Transplant. (2019) 54:1227–36. doi: 10.1038/s41409-018-0418-8
Keywords: hematopoietic cell transplantation, chronic granulomatous disease, CYBB, adult, cord blood transplantation, low-dose irradiation
Citation: Yanagimachi M, Kato K, Iguchi A, Sasaki K, Kiyotani C, Koh K, Koike T, Sano H, Shigemura T, Muramatsu H, Okada K, Inoue M, Tabuchi K, Nishimura T, Mizukami T, Nunoi H, Imai K, Kobayashi M and Morio T (2020) Hematopoietic Cell Transplantation for Chronic Granulomatous Disease in Japan. Front. Immunol. 11:1617. doi: 10.3389/fimmu.2020.01617
Received: 11 May 2020; Accepted: 17 June 2020;
Published: 29 July 2020.
Edited by:
Andrew R. Gennery, Newcastle University, United KingdomReviewed by:
Reinhard Alexander Seger, University of Zurich, SwitzerlandElizabeth Kang, National Institutes of Health (NIH), United States
Copyright © 2020 Yanagimachi, Kato, Iguchi, Sasaki, Kiyotani, Koh, Koike, Sano, Shigemura, Muramatsu, Okada, Inoue, Tabuchi, Nishimura, Mizukami, Nunoi, Imai, Kobayashi and Morio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masakatsu Yanagimachi, bXlhbmFnaW1hY2hpLnBlZEB0bWQuYWMuanA=