Abstract
A recently developed humanized mouse has been used to assess the immune response evoked against the isolated attenuated C9 parasite clone (C9-M; carrying a single insertion disrupting the open reading frame (ORF) of PF3D7_1305500) of Plasmodium falciparum. Significant human RBC engraftment was achieved by ameliorating the residual non-adaptive immune response using clodronate-loaded liposome treatment. Controlled reactive professional phagocytic leukocytes in immunodeficient mice allowed for sizeable human blood chimerism and injected huRBCs acted as bona fide host cells for P. falciparum. huRBC-reconstituted immunodeficient mice received infectious challenge with attenuated P. falciparum C9 parasite mutants (C9-M), complemented (C9-C), and wild type (NF54) progenitors to study the role of immune effectors in the clearance of the parasite from mouse circulation. C9-M and NF54 parasites grew and developed in the huRBC-reconstituted humanized NSG mice. Further, the presence of mutant parasites in deep-seated tissues suggests the escape of parasites from the host's immune responses and thus extended the survival of the parasite. Our results suggest an evasion mechanism that may have been employed by the parasite to survive the mouse's residual non-adaptive immune responses. Collectively, our data suggest that huRBCs reconstituted NSG mice infected with attenuated P. falciparum is a valuable tool to explore the role of C9 mutation in the growth and survival of parasite mutants and their response to the host's immune responses. This mouse might help in identifying novel chemotherapeutic targets to develop new anti-malarial drugs.
Introduction
The human malaria parasite was accountable for 4,45,000 deaths in the 2016 (1). The in vitro findings do not replicate the in vivo findings and therefore a laboratory animal model is indeed needed. However, the study of human malaria parasites in animal models is severely limited by ethical and technical constraints, since only a few primate species have been found to be receptive to P. falciparum infection (2–4). Currently, the majority of in vivo investigations to understand malaria biology are dependent upon rodent malaria species (P. berghei and P. yoelii) which are used as surrogates to study human malaria (3–6). Therefore, humanized mice capable of harboring the human malaria infection are urgently needed to understand the parasite biology. A human blood chimeric mouse could serve to harmonize in vitro P. falciparum cultivation and in vivo studies carried out in rodent animal models. Introduction of several mouse strains with genetic immune deficiencies has greatly benefited the development of a small laboratory animal model (7–15) to study the asexual blood stage infection of P. falciparum. Recently, an immunodeficient mouse (16) was reconstituted with uninfected and infected huRBCs. This NSG mouse, depleted with γ-chain of the IL-2 receptor, has been shown to better tolerate a variety of human transplanted cells (17–24). The reduction in the residual innate immune effectors (mainly cells of monocytes and macrophages lineage) and co-administration of huRBCs supplied with decomplemented human serum through an intravenous route led to the development of a reproducible humanized mouse. The disruption of PF3D7_1305500 in C9-M parasites showed 50% attenuation as compared to the wild type parasites (NF54) (25). Therefore, C9-M and C9-C (Rescued phenotype of wild-type growth by genetic complementation) parasites (26) showed the attenuation in PF13_0027 knock-out parasites (C9-M) growth, which in turn resulted in the irregular cell cycle. The late entry into the S/M phase coincides with the timing for the peak expression of PF13_0027, suggesting that the deficiency in the mutant cycle can be correlated with the gene expression pattern (26).
Low parasitemia of the C9-M parasite in mouse circulation and extended survival of the C9-M parasite in deep-seated tissues suggests that the parasite may have employed a mechanism to escape the host's residual immunity. This parasitologically altered behavior of C9-M parasites was confirmed by the serum estimation of pro-inflammatory cytokines from the C9-M infected mice when compared to the NF54 and C9-C parasites.
Thus, the present study was designed to create an improved understanding of host–parasite interactions to bridge the gap between in vivo and clinical studies. The growth mutant (C9-M) parasites grafted in humanized mice showed nearly similar parasitemia patterns to that of NF54. Further, the C9-M parasite seems to have employed a mechanism to evade the host's immune responses and resides/sequesters in the deep-seated tissues. Our study showed the importance of human RBC reconstituted NSG mouse model to study the behavior of generated attenuated and complement parasites (25, 27). This humanized mouse may prove to be an important tool to study the immune mechanism(s) employed by the parasite to develop an indepth understanding on the sequestration-like phenomenon of human malaria parasite, P. falciparum. In brief, the present study showed the variation in the cytokines and parasitologically altered behavior of C9-M parasite upon engrafted in huRBC reconstituted NSG mice for its extended survival.
Materials and Methods
Animal Ethics Committee Approval
All animal procedures were carried out in compliance with The Second Hospital of Jilin University, Changchun. The procedures were reviewed and approved by The Second Hospital of Jilin University, Changchun, China.
Mice
Four- to 6-week-old male and female NOD/SCIDIL-2Rγ−/− (NSG/NOG) mice were procured from Jackson Laboratory, USA. The transgenic thymidine kinase-NOG (TK/NOG) strain was procured from Taconic, USA. The immunodeficient/transgenic mice were housed in sterile isolators and supplied with autoclaved tap water with a γ-irradiated pelleted diet ad libitum. Animals were manipulated under pathogen free conditions using a laminar flow-hood cabinet. One hundred and ten animals were used in the study. Six animals were allocated for each experimental group, except for the giemsa staining assay wherein four animals were used. Animal allocation was randomized, and mice receiving treatment were monitored daily and weighed three times (Monday, Wednesday, Fridays) per week. P. falciparum infected mice were treated with the analgesic ketoprofen to alleviate pain caused by the malaria infection.
Human Erythrocytes
The huRBCs are the bona fide host cells for the development of P. falciparum in in vitro cultivation and huRBCs reconstituted mice. The packed huRBCs were provided by the Interstate Blood Bank (Chinese blood bank). Blood was taken from donors with no history of malaria. The huRBCs were suspended in SAGM (Saline, Adenine, Glucose, Mannitol solution) and stored at 4°C for a maximum of 2 weeks. Before injection, huRBCs were washed three times in RPMI-1640 medium (Gibco-BRL, Grand Island, New York), supplemented with 1 mg of hypoxanthine per liter (Sigma-Aldrich, St. Louis, Missouri), and warmed for 10 min at 37°C. Blood samples drawn from mice were used to determine the percentage of huRBC in mice's peripheral blood at regular intervals by flow cytometer (Accuri cytometers) using FITC labeled anti-human glycophorin antibody (ebiosciences, USA).
P. falciparum Parasites Culture
C9 mutant, complement parasites (Gene complementation of the C9 mutant to rescue the wild-type phenotype) and wild type P. falciparum line (NF54) were employed in the study. C9-M and C9-C parasites were generated by the interstional mutagenesis by others (26). P. falciparum strains were cryopreserved using the glycerol/sorbitol method as described elsewhere (28). The parasites were cultured in vitro at 5% hematocrit, at 37°C with 5% CO2, using RPMI-1640 medium (Gibco/BRL), 35 mM HEPES (Sigma), 0.25% NaHCO3, 0.5% albumax II (Gibco/BRL), and 0.01 mg/ml gentamicin.
In vivo Replication of P. falciparum in the NSG-IV Model
P. falciparum was maintained in huRBCs reconstituted NSG (immunocompromised) mice undergoing additional modulation of innate immune-defenses using the clodronate-containing liposomes, as described earlier (16). huRBC administered and intravenously infected humanized mouse is referred to “NSG-IV model.” The proportion of huRBCs in the mouse's blood was measured at 3-day intervals until the end of the study by the flow cytometer (Accuri C6 flow cytometry, BD Biosciences, USA) using FITC labeled anti-human glycophorin monoclonal antibody (ebiosciences, CA, USA). Seventy to Ninety percentage of circulating huRBCs were quantified in the mice circulation. Mice were intravenously inoculated with 300 μl asynchronous P. falciparum culture maintaining 1% parasitemia. Following P. falciparum infection, thin blood films were drawn daily from the tail vein on infected humanized mice. Parasitemia has been expressed as a percentage of all erythrocytes found in mouse periphery; the real percentage of parasitized huRBCs is higher in humanized mice, proportional to the level of chimerism, since murine erythrocytes do not receive infection but were included in counts.
Estimates of the total parasite biomass in each mouse were calculated based on the mean corpuscular volume (MCV) of mouse erythrocytes (45fL), MCV of human erythrocytes (86fL), hematocrit in the mice of 0.7, weight of NSG mice (25 grams), and a conservative estimate of 5.5 ml of blood per 100 grams of mouse weight using the following equation:
Number of infected RBCs = (0.055 ml/g) (25 g) (0.7) × (huRBC parasitemia)
[86fL + (mouse Chimerism/human Chimerism) 45fL]
Sorbitol Synchronization of Plasmodium falciparum
Parasite cultures at 5% parasitemia, predominantly ring stages, were synchronized to remove late stage parasites using pre-warmed (37°C) 5% sorbitol. After harvesting culture at 1600 RPM for 6 min, medium was removed and replaced with 10 volumes of pre-warmed 5% sorbitol (29). The re-suspended culture was incubated at 37°C for 15 min, and parasites were pelleted down by the centrifugation; sorbitol was then removed and replaced with fresh complete medium.
Giemsa Staining and Parasite Count
P. falciparum infected mice were euthanized to extract organs (Kidney, liver, spleen, lung, and brain). These extracted organs were perfused, and cells collected from the organs were placed on the glass slides. Briefly, cells were fixed with methanol and stained with Giemsa, and examined at 100X magnification to perform differential counts of each stage (200 parasites from each organ counted). The analysis of deep-seated organs for parasite count in NSG mice infected with C9-M and C9-C parasites was carried out.
Genotyping of Growth Attenuated (Mutant) and Complemented P. falciparum Parasites
The original parasites C9-M and C9-C were harvested from in vitro cultures as well as from humanized mouse (huRBCNSG-IV) infected with C9-M, C9-C, and wild type parasites. The specific primers (F5′ATGGTTGGTTCGCTAAACTG3′, R5′TTAATCATTCTTCTCATATACTTCAAA3′) and (F:CTTCACTATCGCTTTGATCC, RTCGCTATCCCATAAATTACAA) were used to identify the presence of hDHFR and BSD from in vitro cultures and P. falciparum-harboring humanized mice. The genomic DNA was extracted from parasite cultures and tail snips of mice infected with P. falciparum strains using a DNA mini-kit (Qiagen). DNA was amplified in 20 μl reaction mixture by adopting the following PCR conditions: 1 cycle at 45°C for 30 min and 94°C for 2 min, followed by 35 cycles of 94°C for 15 s, 45°C for 30 s, and 65°C for 3 min. The amplified DNA showed the product band of 563 and 393 bp on 0.8% agarose gel of hDHFR and BSD, respectively.
Plasmodium falciparum Growth Assay
Growth assays were carried out by maintaining asynchronous cultures of P. falciparum wild-type and mutant parasites at 0.5–2% parasitemia in 96-well plates and diluting every 48 h for 168 h. The parasite cultures were plated in triplicate at time zero and end point of the assay, and for each time point samples were taken at every 24 h for 7 days and fixed with 0.05% glutaraldehyde after the removal of culture medium. Flow cytometer was used to estimate the parasitemia as described elsewhere (30, 31) and parasites were stained with ethidium bromide. The stained parasites were analyzed through Accuri C6 flow cytometry system (Accuri, USA). A total of 100,000 cells were counted for each sample and data were analyzed using C Flow Plus software (Accuri). Growth rate (defined as the change in parasite numbers every 24 h over a period of 7 days) analyses were performed using Microsoft Excel and (Microsoft) and SAS 9.3.
Serum Estimation of Cytokines and Chemokines
Hundred microliter blood samples were collected through the submandibular puncture of mice, and sera were stored at−80°C. Cytokines and chemokines (IL-6, MCP-1, IFNγ, TNFα, IL-12p70, and IL-10) were quantified using the BDTM Cytometric Bead Array mouse inflammatory kit (BD biosciences) following the manufacturer's recommendations.
Genotyping of TK/NOG Mice by Diagnostic PCR
Transgenic offspring were genotyped and identified by PCR (annealing temperature 59°C) using HTKF1 forward primer, 5′-CACGTCTTTATCCTGGATTACG-3′ and hGHR1 reverse primer, 5′-CACTGGAGTGGCAACTTCCA-3′. The genomic DNA was extracted from tail snips by DNA mini-kit (Qiagen), and amplified in a 20 μl reaction mixture using the PCR conditions: 2 min at 94°C, then 30 cycles of 30 s at 94°C, 30 s at 59°C, and 30 s at 72°C, and finally 3 min at 72°C. The transgene DNA showed an amplified product band of 236 bp on 1% agarose gel (32).
Statistical Analysis
Each growth assay data was analyzed using C Flow Plus software (Accuri). Growth rate and statistical analysis was carried out by Student's t-test using Prism software (Graph Pad 5 Demo) and data was expressed as the mean ± standard deviation (S.D.) of the mean (*p < 0.05, **p < 0.01, and ***p < 0.001). A value of P < 0.05 was considered statistically significant. The evaluation of inflammatory mediators' results are presented as mean SEM from the experiment performed using three mice per group.
Results
Growth and Replication of P. falciparum in PfhuRBC-NSG-IV Mice
In agreement with the previous findings (16), we used a slightly modified protocol for reconstituting NSG immunodeficient mice with huRBCs (PfhuRBC-NSG) for sustained P. falciparum growth. Experiments were performed in NSG mice using 650 μl of huRBC pellets mixed with 25% de-complemented human serum intravenously injected three times every week, and reconstituted NSG mice were infected with P. falciparum through an intravenous route. Codronate-loaded liposome (clo-lip) suspension was injected intraperitoneally, and clo-lip and huRBCs were administered the same day. This mouse model is called “Pf-NSG IV” model. With this IV protocol 100% of NSG mice, as reported earlier (16), were seen parasitized by day 1 post-inoculation. As shown in Figure 1, low to moderate levels of parasitemia were obtained. In fact, parasites could persist as long as un-infected huRBC were co-administered with human serum along with the immunomodulatory agent, clo-lip. Furthermore, despite individual variations in the maximal parasitemia reached, the parasitemia observed were found stable to determine in vivo effect of C9-M P. falciparum parasites. Thus, we sought to better understand the individual variations in the parasitemia in mouse circulation which could be explained due to the varying levels of blood chimerism. However, significant (p < 0.05) numbers of huRBCs in mouse periphery supported rapid and optimum growth of P. falciparum (Figure 1) with abundant and healthy-looking parasites showing frequent poly-parasitism of huRBC (Figure 2A).
Figure 1
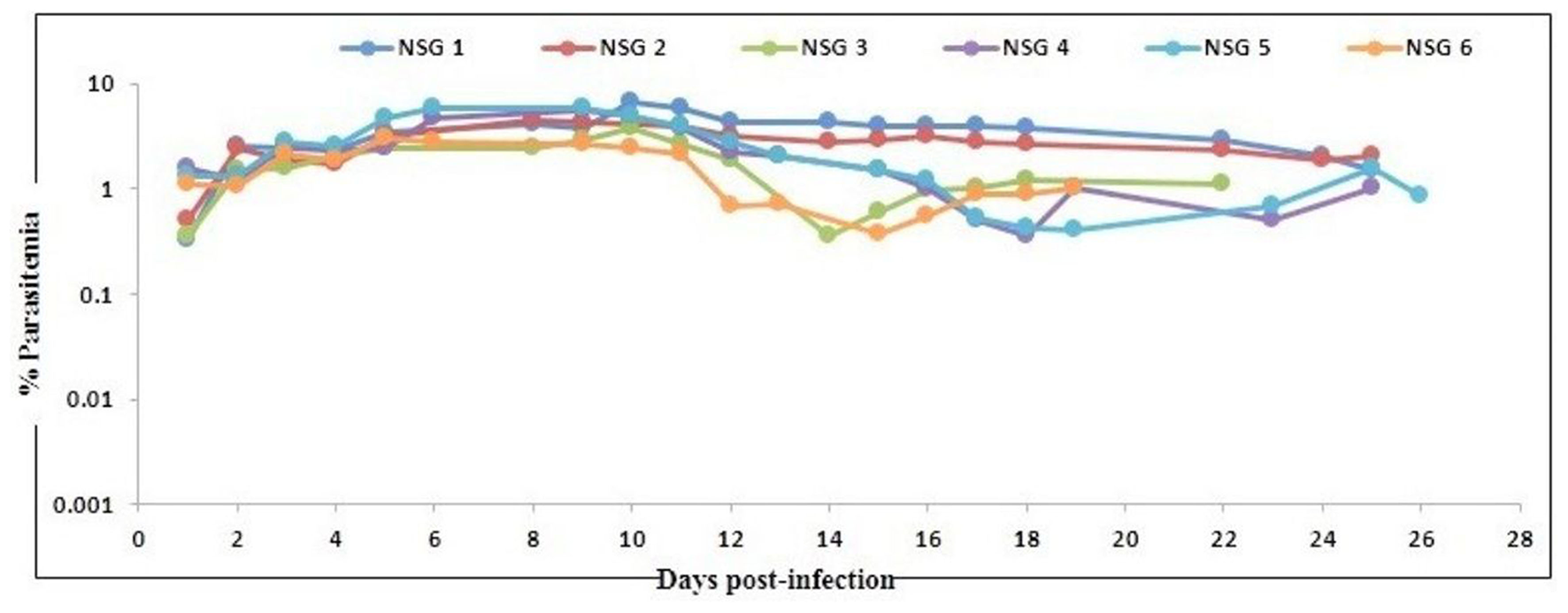
Evolution of P. falciparum (NF54) parasitemia in six untreated mice. Mice were intravenously inoculated with 0.3 ml of asynchronous cultures at 1% parasitemia of P. falciparum on day 0. Mice were supplied with huRBC every 3 days. Parasitemia in mice was expressed as the percentage of P. falciparum-huRBC in the total RBC observed on thin blood smears. Data shown are from the first day of detectable parasitemia up to the day the mice were used for other malaria studies.
Figure 2
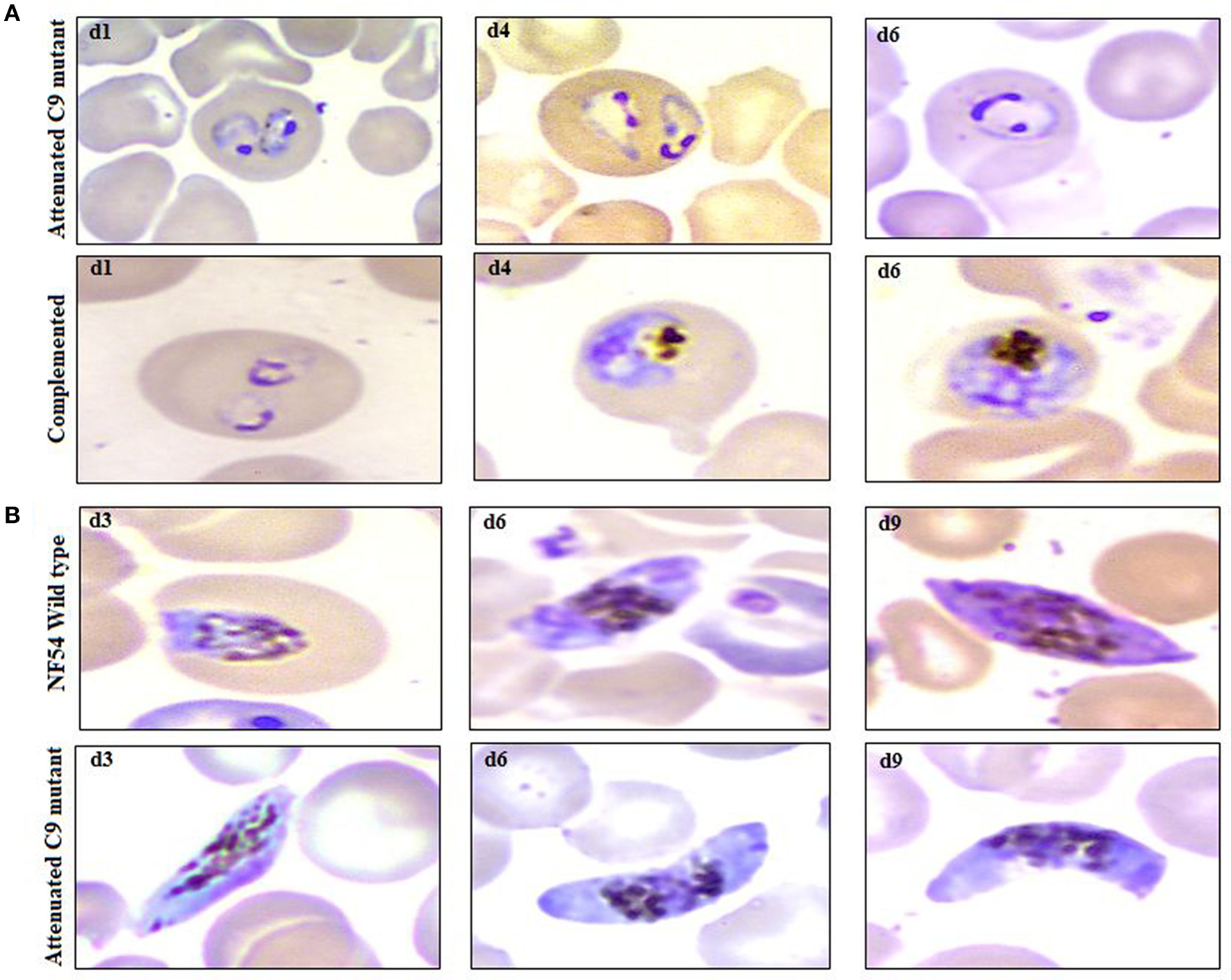
Example of parasitemia obtained from highly attenuated C9-M and complemented C9-C parasites in humanized mice (NSG-IV), (A) upper row (attenuated C9 mutant) and lower row (complemented) depicts the dominance of ring stage parasites along with the presence of trophozoites with frequent poly-parasitism seen on thin blood smears drawn from NSG mice infected with C9-M and C9-Cparasites, respectively, on day 1, 4, and 6 post-infection, (B) occurrence of mature gametocytes (stage V) in peripheral blood of NSG mice infected with NF54 (upper row) and attenuated C9 mutant (lower row) P. falciparum.
Three mice for each group were infected with NF54 (Figure 3A), C9-M (Figure 3B), and C9-C (Figure 3C) parasites. Interestingly, all NSG-IV mice showed 100% infection and supported the replication of all P. falciparum strains employed (Figure 3). However, we did not see significant (p < 0.05) differences in parasite growth in NF54, C9-M, and C9-C parasites in these humanized NSG mice. The short-term human blood chimerism in mouse circulation led to the clearance of NF54 infected erythrocytes on day 13 and 14 post-infection (Figure 3A). However, sufficient numbers of circulating huRBCs allowed C9-M (Figure 3B) and C9-C parasites (Figure 3C) to stay in the periphery until day 22 post-infection. Low standing parasitemia seen with C9-M is attributed to the knock-out of PF3D7_1305500, which may have modulated the host immune response due to the higher secretion of immunoregulatory and anti-inflammatory cytokine, IL-10.
Figure 3
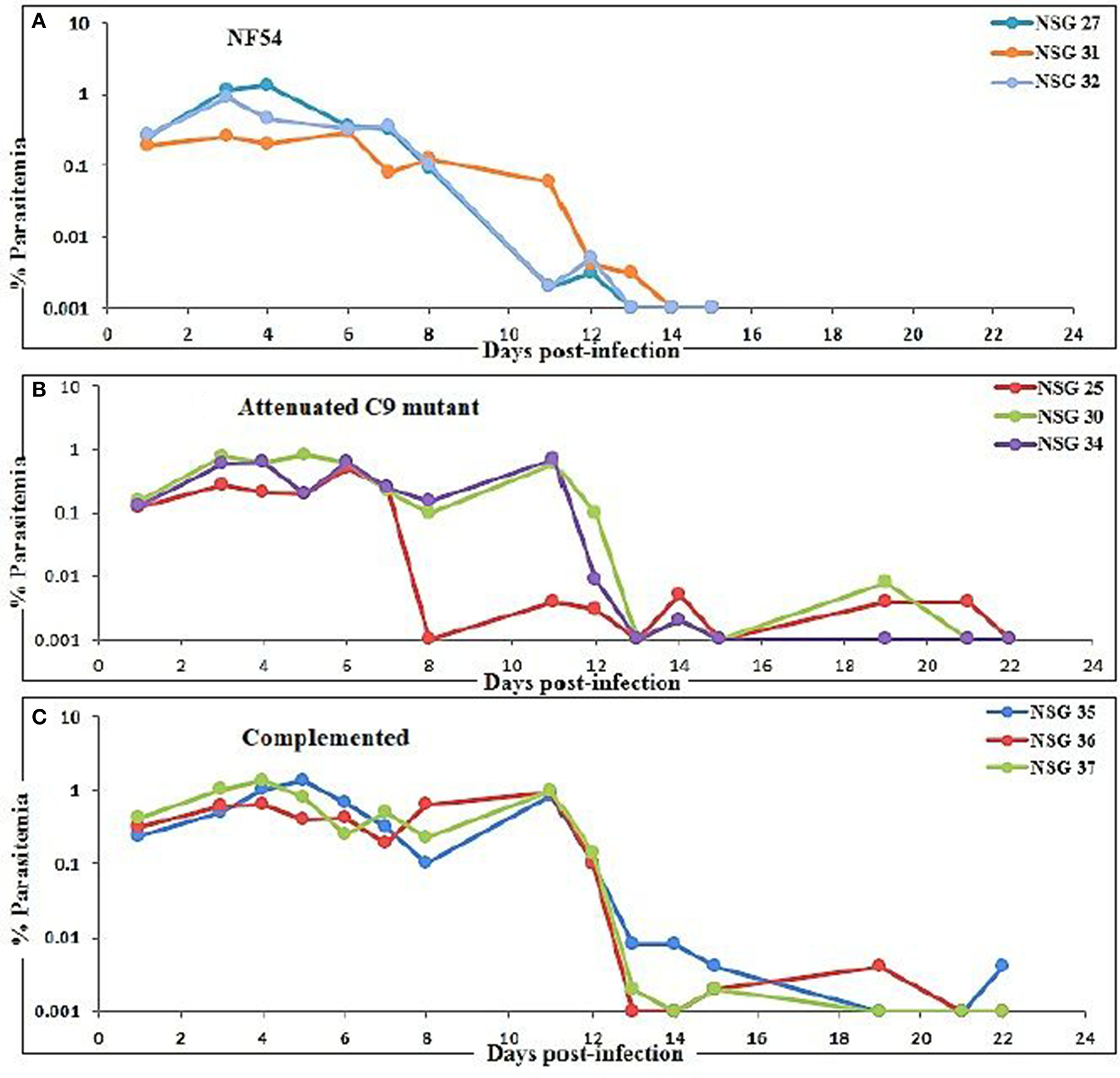
Evolution of P. falciparum parasitemia of mice treated with (A) NF54 (wild type), (B) attenuated C9 mutant, and (C) complemented P. falciparum. All mice were infected through an intravenous route with 0.3 ml of asynchronous cultures at 1% parasitemia of NF54, C9 mutant (C9-M), and complemented parasite (C9-C) of P. falciparum. The PfNSG-IV mice supported the grafting of highly attenuated parasites without needing the adaptation of the parasite to the host.
Humanized Mice Support the Development of Attenuated C9-M P. falciparum
Based on the successful development and replication of different strains of P. falciparum (NF54, C9-M, and C9-C) (Figure 3), we next carried out the growth assays to detect the effect of engraftment on the attenuation attributes of mutant parasites. Therefore, a library of unique mutant clones was created from a laboratory line of P. falciparum (NF54) using random insertional mutagenesis with a piggyBac transposon (33). Fifty percentage attenuation in the in vitro culture of intra-erythrocytic C9-M mutant parasites was seen and compared with the wild type (NF54) parasites (Figure 4A). The mutation responsible for attenuation in the growth of C9-M parasite in vitro slowed down the parasite growth and replication. As with others (26), the mean number of calculated merozoites remained the same as those seen with the parent parasites (NF54). Interestingly, C9-M parasite did not retain its phenotype when grafted in huRBC-reconstituted NSG mice. Also, we did not see significant differences in the parasitemia pattern of C9-M and wild type (Figures 3A,B) parasites, or in their morphology (Figure 2A). Growth assays were carried out on the blood drawn from the mice harboring C9-M and NF54 parasites (Figure 4B). The growth phenotype (50% growth attenuation) of C9-M parasite in parasite culture and grafted in huRBC-reconstituted humanized mice exhibited a different pattern. The humanized NSG mice supported P. falciparum infection and allowed for replication of the C9-M parasite. These attenuated growth mutants survived the residual innate immune responses of the host. Therefore, humanized mice will be useful to study the genotypic and phenotypic characteristics of mutant parasites.
Figure 4
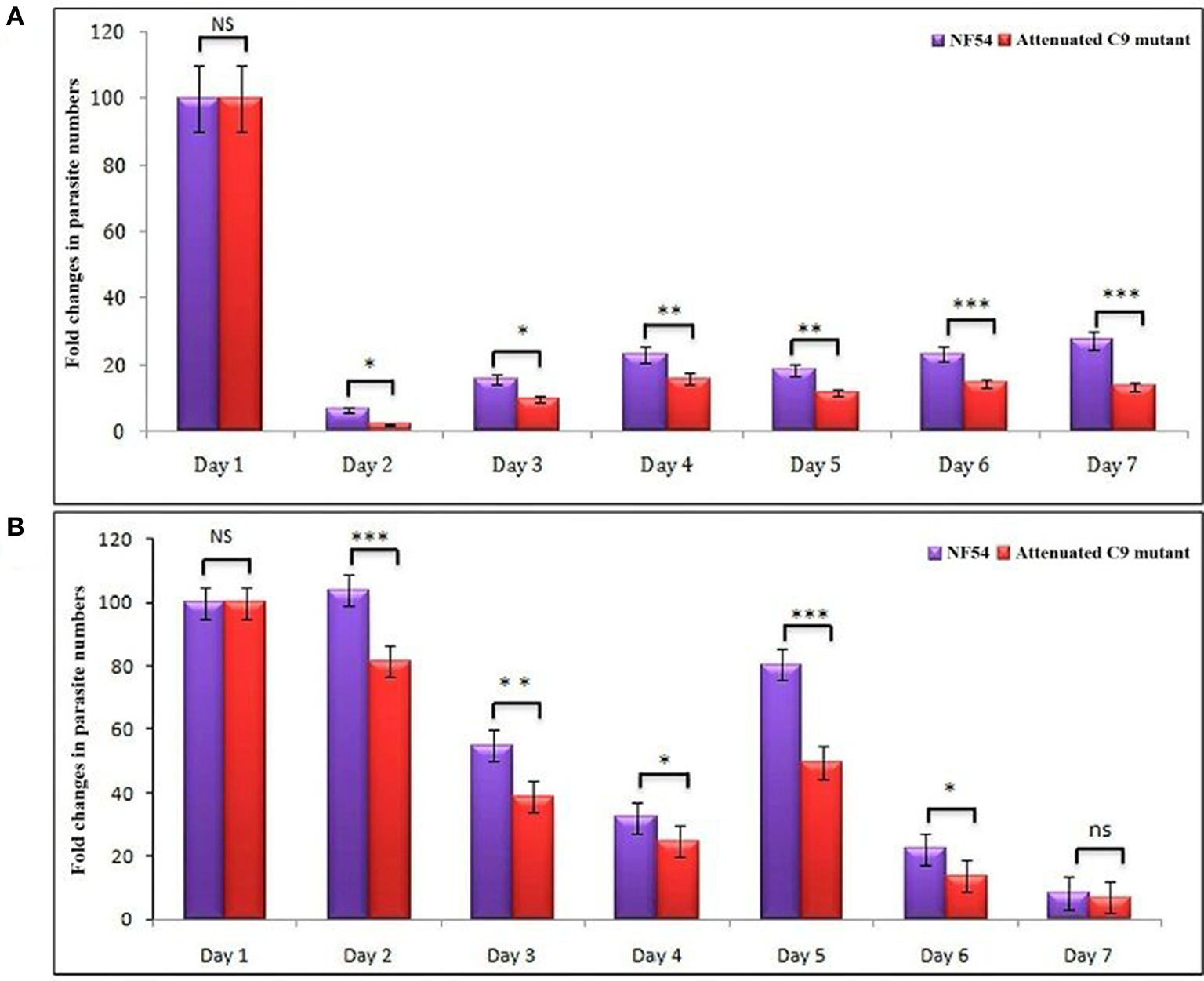
(A)In vitro and, (B)in vivo phenotypic characterization of attenuated growth mutants. A bar graph of fold changes in parasite numbers for 7 days of growth revealed a spectrum of attenuated growth phenotypes in C9-MP. falciparum parasite until day 6 in comparison to their wild type progenitors. The growth assays were carried-out on the samples collected from in vitro cultures and mice infected with wild type and C9-M P. falciparum. *p < 0.05; **p < 0.01; ***p < 0.001.
In vitro and in vivo Typing of C9-Mutant and C9-Complement Parasite
We did not see much difference in the growth pattern of wild type and mutant (C9-M) parasite when growth assays were conducted on parasites from culture and grafted humanized mice. Therefore, we decided to characterize both mutant and complement parasite strains. Parasite strains grafted in humanized mice (Pf-huRBC/NSG-IV) were typed for the attenuated C9-M and C9-C phenotype by PCR (Figure 5). Both strains were thawed, cultured, and kept under Blasticidin (BSD) pressure in vitro (Figure 5B), and the presence of human-DHFR and BSD selection markers were confirmed with the genomic DNA extracted from C9-M and C9-C parasites from culture and P. falciparum infected humanized NSG mice (Figures 5A,B). hDHFR (Figure 5A) and BSD (Figure 5B) showed an amplified product band of 536 and 393 bp of DNA extracted from C9-M and C9-C parasites from culture and mice harboring the parasite, respectively.
Figure 5
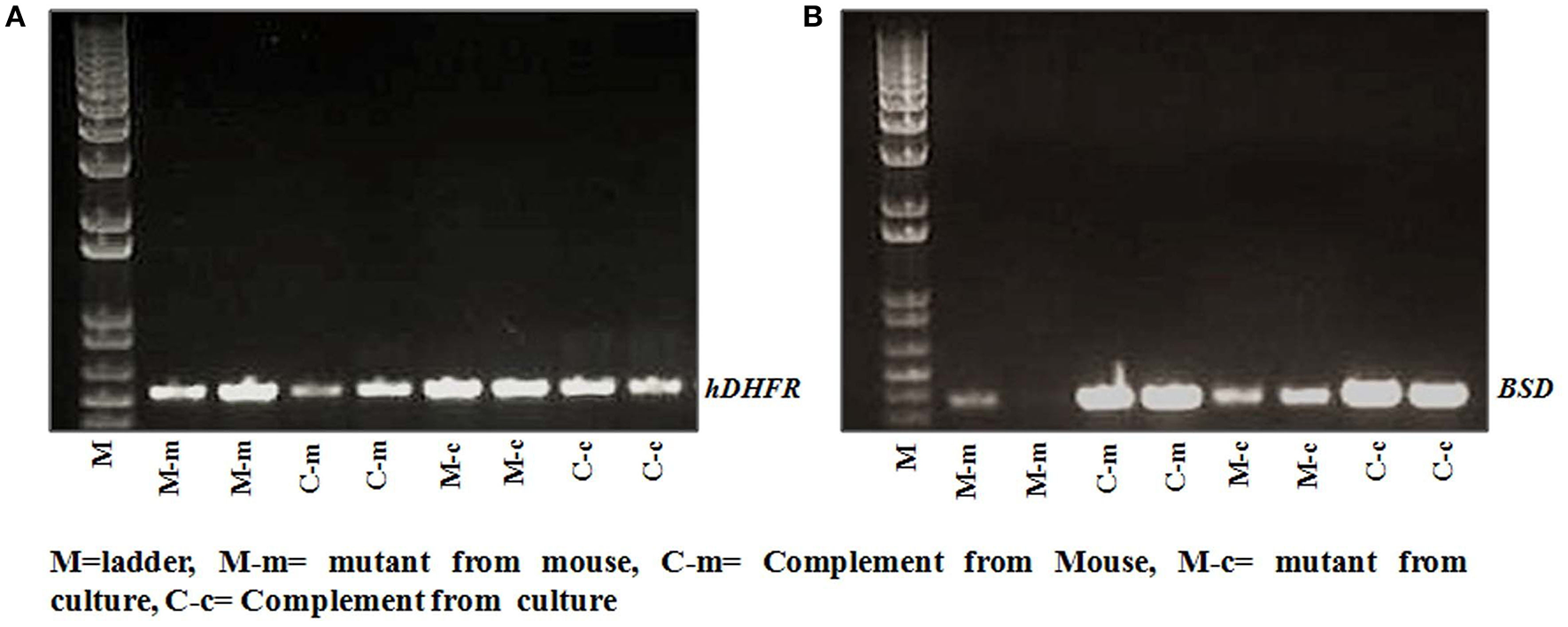
Genotyping of parasite strain(s) used in the study. The presence of (A) hDHFR, and (B) BSD cassette in integrated parasite genome from cultures, and NSG mice infected with C9-M and C9-C parasites was confirmed in both the strains tested. Lane 2 and 3: C9-M parasites from mouse; lane 4 and 5: C9-C parasite from mouse; lane 6 and 7: C9-M parasite from culture, and lane 8 and 9: C9-C parasite from culture.
Putative Phosphatase (PF3D7_1305500) Knock-Down Is Correlated With Cytokine Variations in P. falciparum Infected Humanized NSG Mice
The idea of the present work is to validate the role of knock-down effect of ribosomal binding protein (PF3D7_1305500) in huRBCs reconstituted humanized mice. Following the confirmation of the development and replication of typed mutant (PF3D7_1305500) parasite in humanized mice, upon injection with 650 μl of huRBC, our results showed the significant (p < 0.05) human blood chimerism (proportion of huRBC in mouse's periphery) ranging from 60 to 70% of total erythrocytes. This blood-chimerism was stable over a month and supported rapid and optimal growth of P. falciparum (Figures 1, 3) with abundant and very healthy-looking parasites (Figure 2A). Total hematocrit in mouse's periphery showed an average of 55–60% of huRBCs in this protocol (data not shown).
Furthermore, the use of a lower dose (450 μl) of huRBC showed an initial establishment of human blood chimerism followed by a decrease, which is most likely due to the inflammation induced by parasites (16). Therefore, we used human RBCs reconstituted NSG-IV mice to analyze inflammatory markers to investigate the role of PF3D7_1305500 RNA binding protein in the intra-erythrocytic development of the parasite. Three mice in each group were reconstituted with huRBCs, given the infectious challenge with NF54, C9-M, and C9-C parasites, and sera were collected at different time-points. The raised levels of IL-6 (632 ± 526Vs 17 ± 8 pg/ml, day 15 post-infection) and IL-10 (88 ± 60 vs. 30 ± 27.41 pg/ml, day 12 post-infection) were quantified in the mice infected with C9-M parasites and compared with NF54 wild type P. falciparum (Figure 6). The higher inflammation mounted by the host against the parasite infection is correlated with the clearance of infected and uninfected huRBC from the mouse's periphery. A greater increase in the chemokine MCP-1 (1129 ± 435 vs. 277 ± 16 pg/ml day 9 PI) in NF54 infected mice and the level of IFN-γ (158 ± 73 vs. 1.65 ± 5 pg/ml day 15 PI) in C9-M parasites than NF54 was found to be significant (p > 0.05). Interestingly, a greater increase in TNF-α (422 ± 50.63 vs. 49 ± 21.76 pg/ml day, 21 PI) and IL-12 (128 ± 69 vs. 110 ± 56 pg/ml, day 15 PI) was seen in the mice infected with C9-C parasites than those seen with NF54 infected mice (Figure 6). The sub-optimal dose of huRBC grafting resulted in the reduced human blood chimerism and resulting parasitemia. Consistent with other's findings (16), the initial rise of IL6, followed by an increase of IL-12p70 and moderate changes in TNF-α and MCP-1 that are temporally associated with anemia, may play a role in its etiology.
Figure 6

Putative phosphatase (PF3D7_1305500) knock-down is correlated with the reduction in the inflammation in P. falciparum infected humanized mice. The determination of cytokines (IL-6, IL-10, MCP-1, TNF-α, IFN-γ, and IL-12) in the serum collected from mice infected with wild type NF54, attenuated C9 mutant, complemented parasites. Results represent the mean of experiment performed using three mice per group, and the standard error was calculated.
C9 Mutant P. falciparum Partially Escapes the Residual Innate Immunity of Host
Cells of monocyte-macrophage lineage are continuously recruited and play an important role in the clearance of the parasite from mouse circulation, but mutant parasites reside for long periods of time. Therefore, we next decided to confirm the importance of monocyte/macrophage in the clearance of parasites. The macrophages are the main subsets recruited all over the course of infection, and found to be more active than other phagocytes at ingesting infected and uninfected huRBCs in the peritoneum (9, 34–36) (Supplementary Figure 1). C9-M parasite showed 100% infectivity and slow growth upon being grafted in humanized NSG mice, and parasitemia was seen to decrease by day 12 post-infection (Figure 3B). These mutant parasites were found sequestered in the liver, kidney, and brain to survive the host's residual innate immune responses (Figures 7A,B). The phagocytosis of uninfected and infected huRBCs and subsequent release of pigment suggested that complete escape of parasite from host's non-adaptive immune response was not possible (Figure 7A). Furthermore, our results suggest that the C9-M parasite may have employed evasion mechanism(s) to survive the residual innate immune response of the host. Our findings describe the role of macrophages (MP) in the rejection of both uninfected and infected huRBC by two mechanisms: the release of inflammatory mediators for the activation of monocytes, that then result in increased erythro-phagocytosis. The role of macrophages in xenograft rejection is shown by the infiltration of leukocytes during the rejection of pig-to-primate xenografts (37, 38). Furthermore, selective macrophage depletion in immunocompetent rodents showed significant delays in cellular infiltration and xenograft rejection (39, 40).
Figure 7
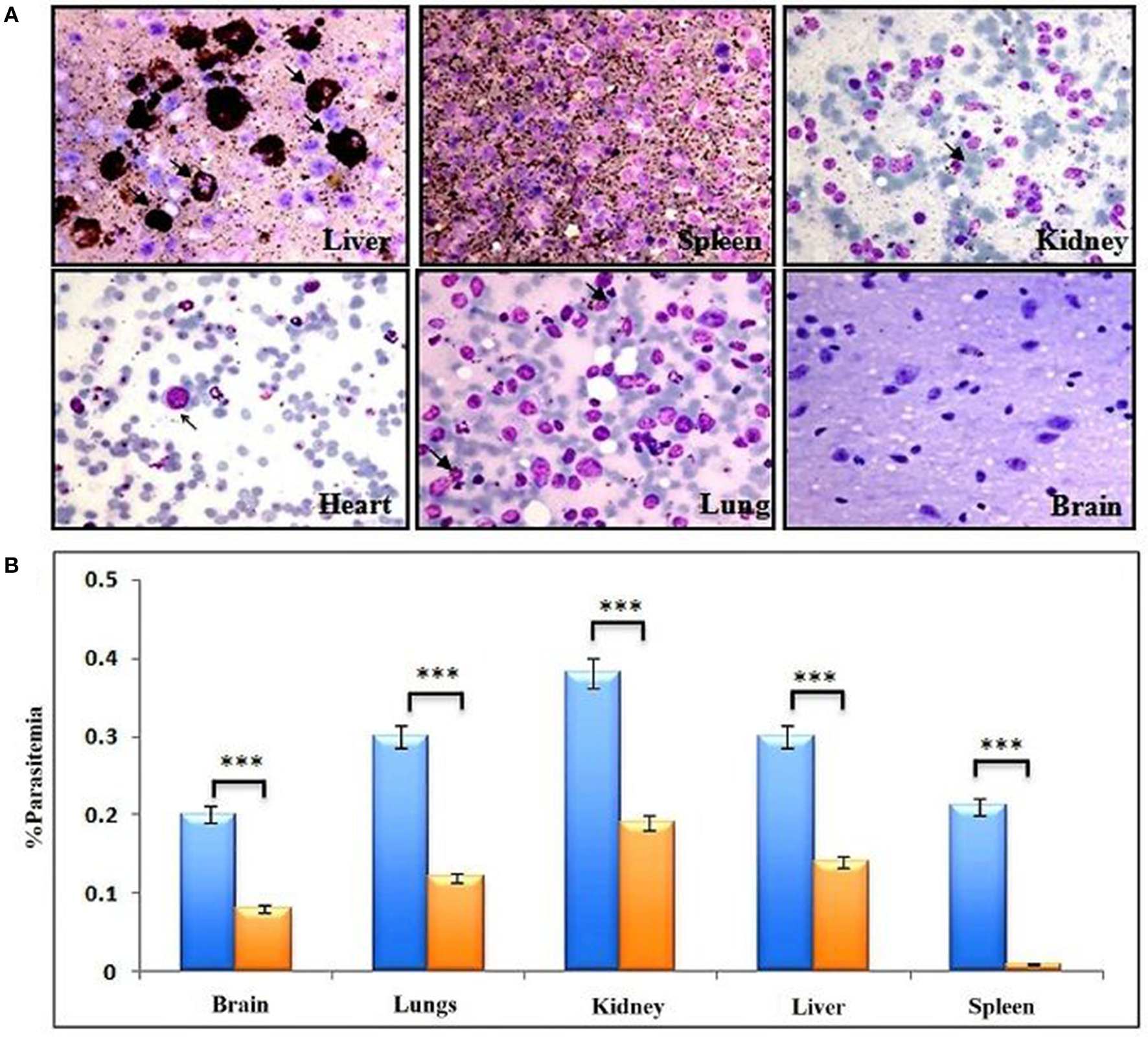
Complete escape from mouse's residual non-adaptive immunity is not possible by C9-M parasite. (A) Representative Giemsa staining of smears drawn from various organs 11 days post-infection from three different NSG mice (upper panels: liver, spleen, and kidney, lower panels: heart, lungs, and brain). Arrows indicate C9-M parasitized huRBCs. The presence of parasites in different organs, their phagocytosis, and subsequent release of pigment from active phagocytes, mainly macrophages, is an example of active residual innate immune effectors, (B) parasite count in different organs, blue bars; C9-M, red bars; C9-C parasites. ***p < 0.001.
NSG-IV Mice Supported the C9-M P. falciparum for the Development of Gametocytes (Propagation Carriers)
The propagation of the parasite from one host to another is important to confirm the replication of the parasite. NF54-wild type strain is known to develop gametocyte in vitro culture, which was supported by the humanized mice to induce the production of gametocytes (Figure 2B, lower panel, 1st row; d3, d6, d9). In addition to the remarkable growth and development of C9-M parasites seen in NSG-IV mice, gametocytes were developed in humanized mice (Figure 2B, lower panel, 2nd row; d3, d6, d9). These sexual forms (Figure 2B, lower panel) were frequently seen up to stage V. The observation of stage V gametocytes implies that infected huRBC are able to survive in mouse's circulation for at least 9 days without suffering any immune-mediated insult when NSG-IV protocol was used. We tried to feed mosquitoes on the developed gametocytes to demonstrate their infectivity, but failed to see the development of oocyte on day 11 followed by the development of sporozoites on day 16 or 17. We think more efforts are needed to demonstrate the infectivity of gametocytes developed in Pf-NSG-IV mice.
huRBC Reconstituted TK/NOG Mouse: A Better Strain for Humanization
We wanted to study the transgenic strain (32, 41–54) (expressing the thymidine kinase transgene on mouse's hepatocyte) primarily dedicated for developing the human liver-chimeric mice to examine the inflammatory disorders and liver metabolism of drugs. Since this strain is of NOG background, we thought it could support the reconstitution of huRBCs and P. falciparum infection. Thus, this mouse strain was deployed in the present study, set to breed, and littermates were genotyped by the established PCR method (32). DNA transgene showed an amplified band of thymidine kinase (Figure 8A) on the agarose gel. The absolute numbers of innate immune effectors (Leukocytes, monocytes, and polymorphonuclears) in TK/NOG were observed to be lesser than in NSG mice (Figure 8B). Next, we wanted to assess the importance of thymidine kinase transgene expression in TK/NOG mice. Therefore, TK/NOG and NSG mice were treated with clodronate-loaded liposomes to check the production of excessively recruited monocytes/macrophages, and animals were intravenously reconstituted with huRBCs. TK/NOG mice supported the significant and long-term huRBC chimerization as compared to NSG mice (Figure 8C). We went on to estimate the half-life (4–5 days) of huRBC to determine the survival of huRBC in mouse periphery (Figure 9). The human blood chimerism was found better (75%) in TK/NOG mice than NSG mice (15–20%) by day 12 post-huRBCs injections (Figure 8C).
Figure 8
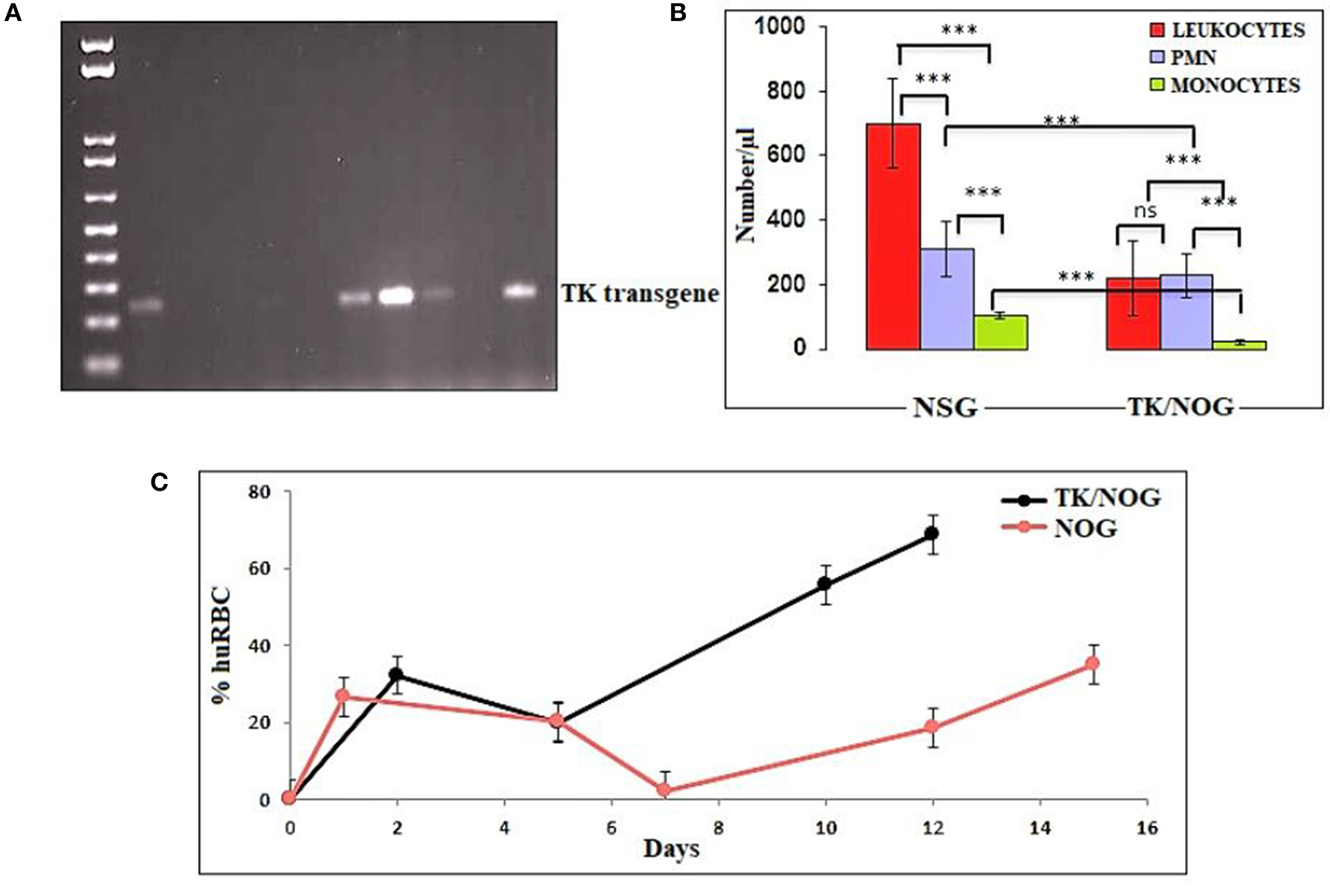
Thymidine kinase transgene expression and enhanced huRBC grafting. (A) All litters were genotyped with gDNA samples extracted from tail snips by PCR, and amplified transgene DNA (Thymidine Kinase, 236 bp) was visualized at 1% agarose, (B) The reduction in the number of innate immune cells of TK/NOG and NSG mice was compared (C) the greater human blood chimerization (huRBC grafting) is seen in TK/NOG mice than NSG mice. ***p < 0.001.
Figure 9
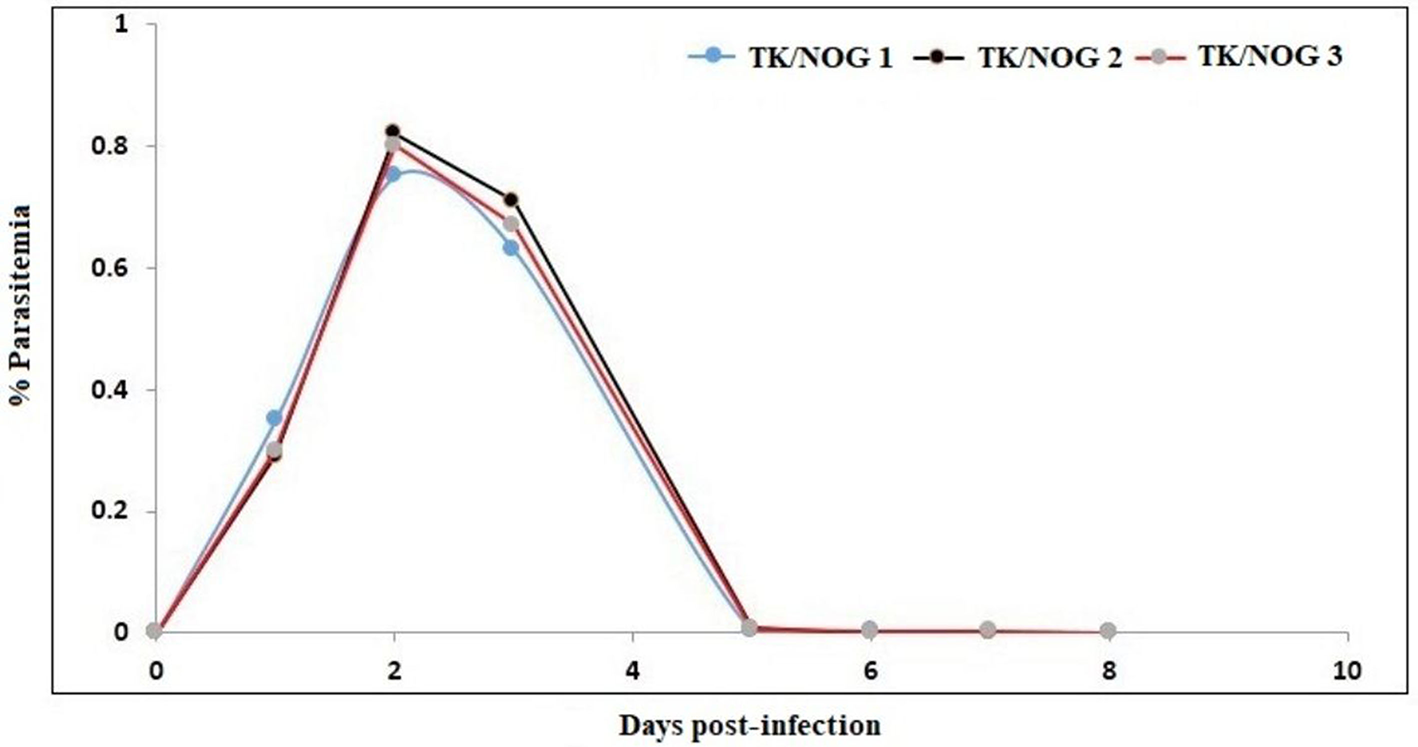
Determination of the half-life of huRBC in mouse's circulation. Mice were intravenously inoculated with 0.3 ml of asynchronous cultures at 1% parasitemia of NF54 strain of P. falciparum on day 0. The parasitemia in mice was expressed as the percentage of P. falciparum-huRBC in the total RBC observed on thin smears. The data shown is from the first day of detectable parasitemia up to the day the mice had huRBCs in their circulation.
huRBC reconstituted TK/NOG received infection with parental NF54, C9-M, C9-C, and 3D7 (three mice each) strain of P. falciparum (Figure 10). All mice receiving infection were seen parasitized by day 1 post-inoculation using IV protocol.
Figure 10
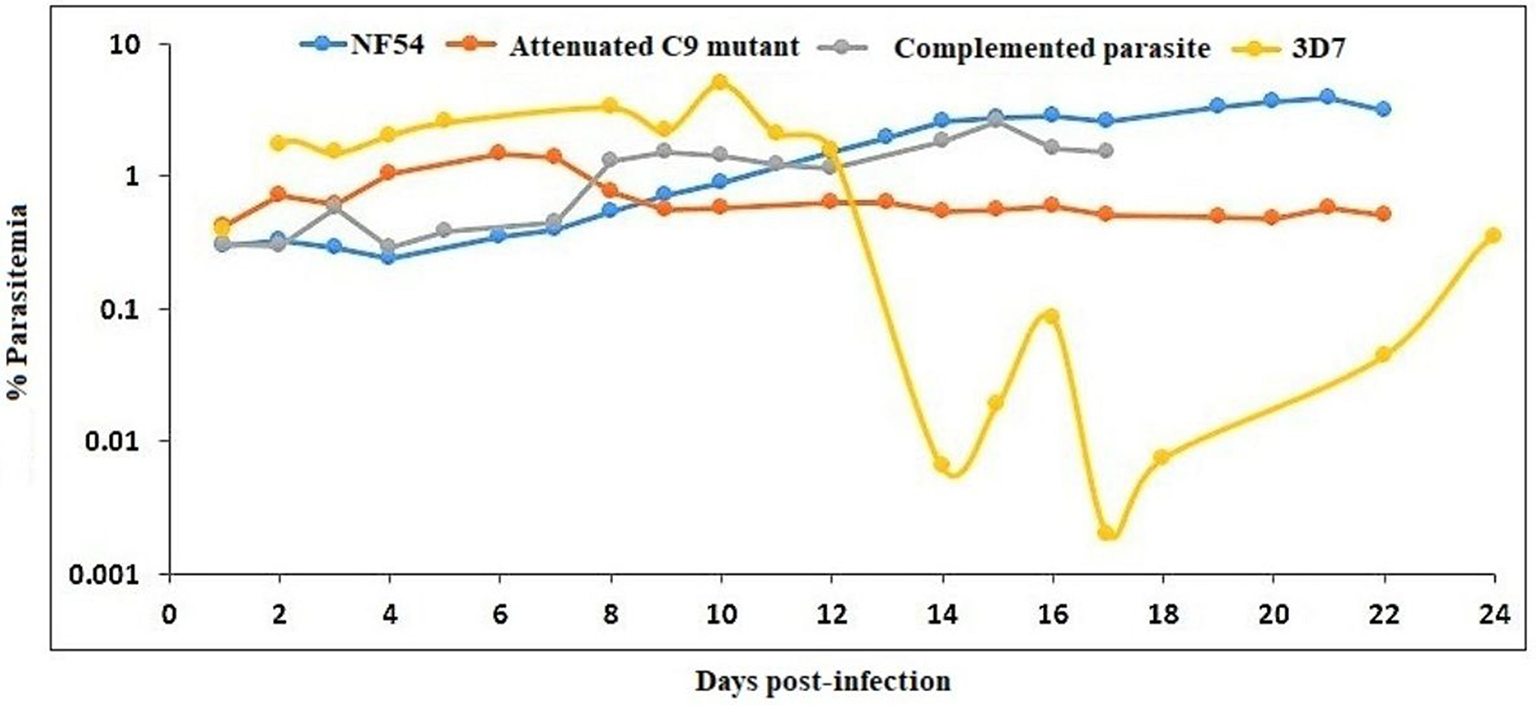
NF54 (wild type), attenuated C9-M, complemented C9-C, and 3D7 parasite strains are supported by TK/NOG mice. The trend of parasitemia is the mean of three mice each of all parasite strains tested. Mice were intravenously inoculated with 0.3 ml of asynchronous cultures at 1% P. falciparum parasitemia on day 0. Mice were supplied with huRBC every 3 days. Parasitemia in the mice was expressed as the percentage of P. falciparum parasitized huRBCs in total RBC counted on thin blood smears. The presented data is from the first day of detectable parasitemia.
The stable blood-chimerism was seen with huRBs reconstituted TK/NOG (Figure 8C). These mice supported rapid and optimal growth of all P. falciparum strains tested (Figure 10). Except for 3D7, all strains (NF54, C9-M, C9-C) showed consistency in parasitemia pattern. Lack in the adaptation of the parasite to the host was attributed to variations seen in the pattern of 3D7 parasitemia. The 3D7 strain made attempts to accommodate with the host's immune system in TK/NOG mice for nearly 2 weeks, and adapted parasites exhibited normal growth and development of 3D7 by day 17 post-infection (Figure 10).
Discussion
This study is part of our efforts to develop understanding on experimental mouse model(s) to study unknown asexual blood stage genes/RNA binding proteins of P. falciparum. As with others (25), our in vitro study showed that attenuated C9-M did not lead the normal cell cycle due to knock-down of PF3D7_1305500. This study suggests the importance of this atypical phosphatase in the regulation of the P. falciparum cell cycle. The attenuated C9 knock-out created by random insertional mutagenesis was used in this study to attest the in vitro studies (26). Moreover, the importance of PF3D7_1305500 playing a crucial role in the growth and development of asexual blood stage P. falciparum was confirmed in humanized mice (Figure 11).
Figure 11
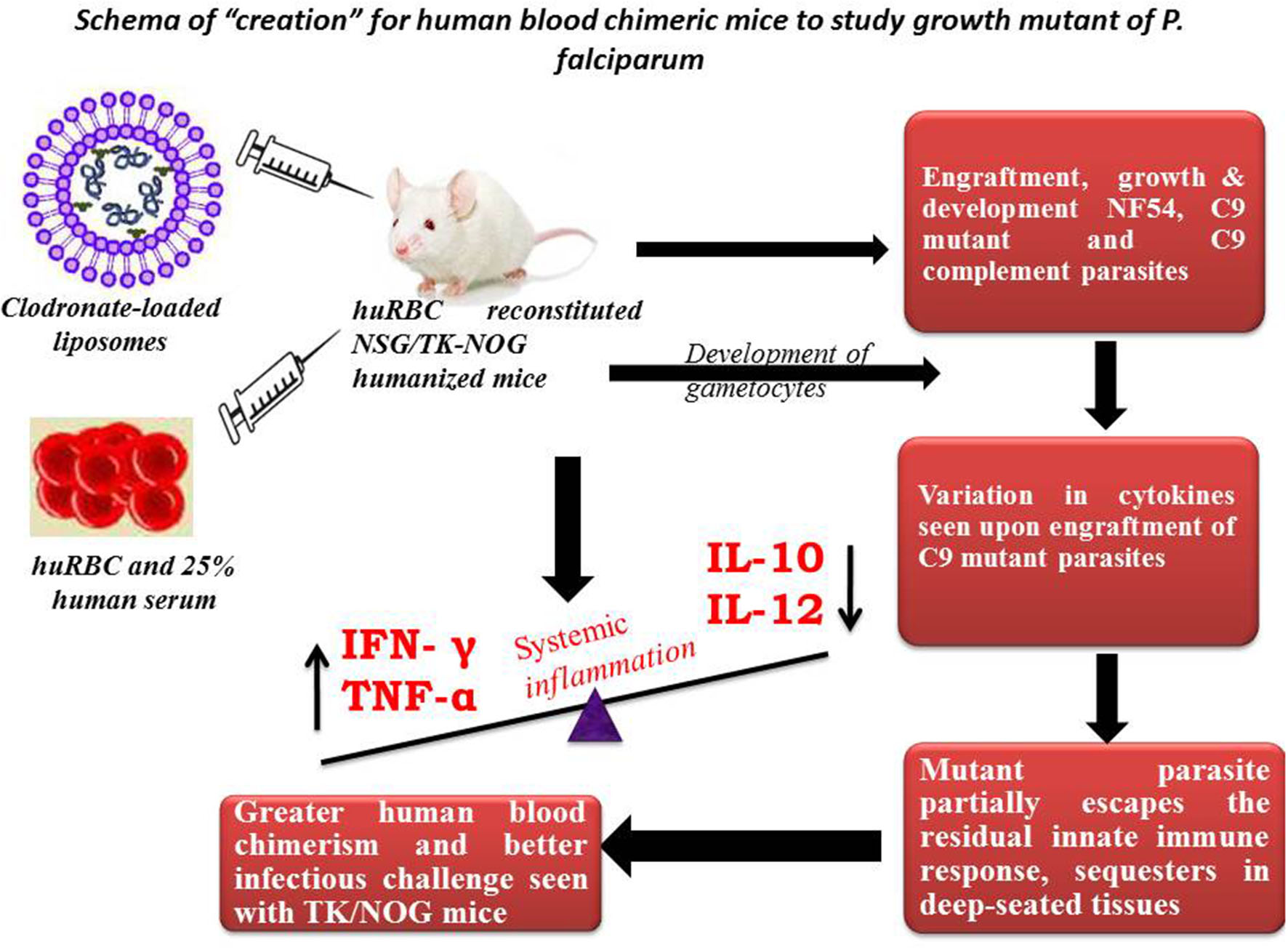
Schema of “creation” for human blood chimeric mice to study the growth mutant of P. falciparum. huRBCs reconstituted NSG mice showing variation in the cytokine levels of C9-mutant and C9-complement parasites as compared to wild type NF54. This model indicates the escape-mechanism-like phenomenon since mutant parasites reside for longer in the deep-seated tissues and evade the residual innate immunity of the host.
We succeeded to implement a reproducible infection by using different P. falciparum strains in a PfhuRBC-NSG-IV humanized mouse (16) to understand the development and replication of attenuated C9-M parasites. Consistent with the previously published findings (16), we confirm that employing an intravenous route for both parasite and huRBC delivery, clodronate-loaded liposomal suspension for macrophage suppression, and the IL-2R-γ mutation on NOD/SCID genetic background, helped in achieving a model with greater reproducibility which supported the infection with different strains, including the severely attenuated C9-M.
The present study shows that P. falciparum huRBC/NSG-IV model (huRBC-reconstituted NSG mice infected with NF54, C9-M, and C9-C parasites) may be useful to study the immune responses evoked against the grafted parasite, and possible survival mechanisms employed by the parasites. The additional defects in the innate immune system of NSG mice are most likely related to the defective activation of phagocytes which led to the reduced P. falciparum selection pressure (11).
Our results show that: (1) P. falciparum induces strain dependent moderate inflammation characterized by the release of inflammatory cytokines in the serum; (2) evolution of parasitemia in mouse's periphery remained stable but short-term blood chimerization (insufficient huRBCs) in the circulation leads to the clearance of parasites driven by the pro-inflammatory cytokines; (3) growth and development of all P. falciparum strains was supported by the developed humanized mice, indicating that, in contrast to Aotus, Saimiri, and previous mouse models, there is no requirement for the preliminary adaptation of the parasite to PfhuRBC-NSG-IV host. The slower growth of C9-M indicated that the parasite could retain the mutant phenotype upon being grafted in human RBCs reconstituted humanized mice. However, there was no difference seen in the parasitemia patterns of grafted parental NF54, C9-M, and C9-C parasites. This shows the relevance of this mouse as it helps in understanding the behavior and relevance of C9 mutation in the growth and replication of the asexual blood stage infection of P. falciparum without tampering its genotype and phenotype; (4) migration of parasites from the mouse's periphery to deep-seated organs with the extended residence indicated that C9-M parasites may have employed molecular mechanism(s) to partially evade the host's innate immune response; (5) humanized mouse supported the development of the sexual stage of P. falciparum. Stage III, IV, and V gametocytes were seen in the peripheral blood but their infectivity could not be demonstrated; and (6) transgenic/immunodeficient (TK/NOG) mice were shown to better control the non-adaptive immune response as compared to NSG mice, and therefore higher human blood chimerism was seen when reconstituted with huRBCs in the mouse prepared by clo-lip treatment. huRBCs grafted TK/NOG mice showed greater susceptibility toward infection with stable parasitemia in all P. falciparum strains tested (Figure 10).
Results obtained with the humanized mouse model, as reported earlier (9, 13, 16), concur to suggest that the model is closer to events recorded in humans (for instance, the receptivity shown toward non-adapted parasites and gametocyte production).
The disruption of any biochemical process affects the normal pattern of metabolic events which leads to the completion of the asexual blood stage development of P. falciparum. The in vitro findings (26) showed the significance of PF3D7_1305500 (C9-M) in the regulation of the asexual blood stage cell cycle of P. falciparum. The delayed transition of pre-S trophozoite to S/M schizonts showed the checkpoint of the parasite growth cycle. The rescue of C9-M phenotype by the genetic complementation confirmed the requirement of PF3D7_1305500 for normal growth and development of P. falciparum in vitro (26). Further, the in vivo selection process led to the variants of P. falciparum carrying potential rearrangements to variable molecules encoding antigenic determinants (55). The engraftment of C9-M in humanized mice shows the elicitation of active residual innate immune responses. The detailed analysis better defined the role of each cytokines/chemokines produced against the grafted C9-M and C9-C P. falciparum. And, our findings contribute to understanding the delicate balance between inflammation control and P. falciparum survival in humanized mice (Supplementary Figure 1).
The relationship between the knock-out effect of putative phosphatase and the mouse's ability to both tolerate the graft and produce fewer inflammatory mediators is an interesting finding. A marginal increase in the inflammatory mediator (IL-6) and immune-suppressor (IL-10) than their wild type progenitors was seen. We believe that improved survival of infected and uninfected huRBCs is associated with a decrease in the inflammatory cytokine and a slight increase in the production of IL-10 by the cells of myeloid origin in C9-M harboring mice. Therefore, controlled pro-inflammatory cytokines allowing the sustained parasite development in deep-seated organs indicate that P. falciparum might be employing the escape mechanisms to survive the host's residual innate immunity (55). We measured increased levels of TNF-α and IL-12 in C9-M and C9-C parasites in infected humanized mice. In addition, we saw an increase in serum levels of MCP-1 in NF54 infected humanized mice. These results are in agreement with earlier findings (16). The increase of IL-12p70, TNF-α, and MCP-1 are temporally associated with anemia which might play a role in its etiology and inflammation. Lastly, we introduced a very promising transgenic/immunodeficient strain (32, 41–45, 47–51, 56, 57), primarily dedicated for developing human liver chimeric mice to study human liver physiology, drug metabolism, and liver pathogenesis of viral etiology or liver regeneration. However, we reconstituted TK/NOG mice with huRBC and saw better blood chimerism to allow P. falciparum replication. The better control of residual non-adaptive immune effectors such as leukocytes, PMNs, and monocytes/macrophages (49, 58) also contributed to the parasite's growth and development. The monocytes/macrophage bears their most critical function in the clearance of parasitized or un-parasitized huRBCs from mouse's periphery (9). The significant human blood chimerization supported by the TK/NOG mice when grafted with huRBC advocates for better replication of P. falciparum (49).
Conclusion and Future Perspectives
The emergence of resistance to frontline drugs and the lack of diverse therapeutic agents necessitates the finding of effective anti-malarial drugs and the identification of new drug targets (7, 59–65). Our P. falciparum-NSG-IV model may allow the study of asexual blood stage growth mutant(s) and their effect on the human system. This mouse shows reproducibility of both huRBC grafting and parasite survival, less day to day variation in parasitemia, does not require preliminary adaptation of parasite strains to the mouse, and supports the development of even attenuated P. falciparum. The evasion mechanisms employed by the parasites help survive against the host's residual immune responses and raises the possibility of partial sequestration of parasites in deep-seated organs, which are key findings of the present work. The delineation of in vivo function and behavior of attenuated C9-M parasites gene (PF3D7_130550) in a humanized mouse might give insights into this unknown protein, important for the growth and development of the asexual blood stage of P. falciparum.
Statements
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by The Second Hospital of Jilin University, Changchun, China.
Author contributions
LZ and MT conceived and designed the experiments, analyzed the data, and wrote the paper. LZ, J-LL, M-XJ, DT, L-YW, and CC performed the experiments and contributed materials/analysis tools. All authors contributed to the article and approved the submitted version.
Funding
Authors express their gratitude toward The Second Hospital of Jilin University, Changchun, China for funding the proposed study.
Acknowledgments
The instrumentation facility of The Second Hospital of Jilin University, Changchun, China is duly acknowledged.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01801/full#supplementary-material
Supplementary Figure 1(a) HuRBCs are massively engulfed by macrophages in the peritoneum of mice administered with human RBCs. (b) Sizable rafting of huRBCs was seen when co-injected with clodronate loaded liposomes (9). Black Square-HuRBC, Plain circle- HuRBC+ Clo-lip, Open circle- HuRBC+ NIMP.
- ORF
open reading frame
- Clo-lip
Clodronate-loaded liposomes
- NSG
NOD/SCID/IL-2rg−/
- huRBC
Human Red Blood Cells
- TK/NOG
Thymidine kinase-NOD/SCID/IL-2rg−/
- C9 mutant
PF3D7_1305500
- MCV
mean corpuscular volume
- hDHFR
human dihydrofolate reeducates
- BSD
Blasticidin.
Abbreviations
References
1.
WHO. World Malaria Report. World Health Organization (2017). p. 1–196.
2.
MurrayCJRosenfeldLCLimSSAndrewsKGForemanKJHaringDet al. Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet. (2012) 379:413–31. 10.1016/S0140-6736(12)60034-8
3.
DruilhePHaganPRookGA. The importance of models of infection in the study of disease resistance. Trends Microbiol. (2002) 10(Suppl. 10):S38–46. 10.1016/S0966-842X(02)02437-X
4.
WhiteNJTurnerGDMedanaIMDondorpAMDayNP. The murine cerebral malaria phenomenon. Trends Parasitol. (2010) 26:11–15. 10.1016/j.pt.2009.10.007
5.
DruilhePBarnwellJW. Pre-erythrocytic stage malaria vaccines: time for a change in path. Current Opin Microbiol. (2007) 10:371–8. 10.1016/j.mib.2007.07.009
6.
SiuEPlossA. Modeling malaria in humanized mice: opportunities and challenges. Ann N Y Acad Sci. (2015) 1342:29–36. 10.1111/nyas.12618
7.
DondorpAMYeungSWhiteLNguonCDayNPSocheatDet al. Artemisinin resistance: current status and scenarios for containment. Nat Rev Microbiol. (2010) 8:272–80. 10.1038/nrmicro2331
8.
ShanksGD. Treatment of falciparum malaria in the age of drug resistance. J Postgrad Med. (2006) 52:277–80.
9.
ArnoldLTyagiRKMejiaPVan RooijenNPérignonJLDruilheP. Analysis of innate defences against Plasmodium falciparum in immunodeficient mice. Malaria J. (2010) 9:197. 10.1186/1475-2875-9-197
10.
BadellEOeuvrayCMorenoASoeSvan RooijenNBouzidiAet al. Human malaria in immunocompromised mice: an in vivo model to study defense mechanisms against Plasmodium falciparum. J Exp Med. (2000) 192:1653–660. 10.1084/jem.192.11.1653
11.
Angulo-BarturenIJiménez-DíazMBMuletTRullasJHerrerosEFerrerSet al. A murine model of falciparum-malaria by in vivo selection of competent strains in non-myelodepleted mice engrafted with human erythrocytes. PloS ONE. (2008) 3:e2252. 10.1371/journal.pone.0002252
12.
DruilhePSpertiniFSoesoeDCorradinGMejiaPSinghSet al. A malaria vaccine that elicits in humans antibodies able to kill Plasmodium falciparum. PLoS Med. (2005) 2:e344. 10.1371/journal.pmed.0020344
13.
MorenoABadellEVan RooijenNDruilheP. Human malaria in immunocompromised mice: new in vivo model for chemotherapy studies. Antimicrob Agents Chemother. (2001) 45:1847–53. 10.1128/AAC.45.6.1847-1853.2001
14.
VaughanAMKappeSHPlossAMikolajczakSA. Development of humanized mouse models to study human malaria parasite infection. Fut Microbiol. (2012) 7:657–65. 10.2217/fmb.12.27
15.
McCarthyJSMarquartLSekuloskiSTrenholmeKElliottSGriffinPet al. Linking murine and human Plasmodium falciparum challenge models in a translational path for antimalarial drug development. Antimicrob Agents Chemother. (2016) 60:3669–75. 10.1128/AAC.02883-15
16.
ArnoldLTyagiRKMeijaPSwetmanCGleesonJGPérignonJLet al. Further improvements of the P. falciparum humanized mouse model. PloS ONE. (2011) 6:e18045. 10.1371/journal.pone.0018045
17.
KingMPearsonTShultzLDLeifJBottinoRTruccoMet al. Development of new-generation HU-PBMC-NOD/SCID mice to study human islet alloreactivity. Ann N Y Acad Sci. (2007) 1103:90–93. 10.1196/annals.1394.011
18.
WatanabeSTerashimaKOhtaSHoribataSYajimaMShiozawaYet al. Hematopoietic stem cell-engrafted NOD/SCID/IL2Rgamma null mice develop human lymphoid systems and induce long-lasting HIV-1 infection with specific humoral immune responses. Blood. (2007) 109:212–8. 10.1182/blood-2006-04-017681
19.
ItoMHiramatsuHKobayashiKSuzueKKawahataMHiokiKet al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. (2002) 100:3175–82. 10.1182/blood-2001-12-0207
20.
ItoMKobayashiKNakahataT. NOD/Shi-scid IL2rgamma(null) (NOG) mice more appropriate for humanized mouse models. Curr Top Microbiol Immunol. (2008) 324:53–76. 10.1007/978-3-540-75647-7_3
21.
KenneyLLShultzLDGreinerDLBrehmMA. Humanized mice and tissue transplantation. Am J Transpl. (2016) 16:389. 10.1111/ajt.13520
22.
AudigéARochatMALiDIvicSFahrnyAMullerCKSet al. Long-term leukocyte reconstitution in NSG mice transplanted with human cord blood hematopoietic stem and progenitor cells. BMC Immunol. (2017) 18:28. 10.1186/s12865-017-0209-9
23.
Herndler-BrandstetterDShanLYaoYStecherCPlajerVLietzenmayerMet al. Humanized mouse model supports development, function, and tissue residency of human natural killer cells. Proc Natl Acad Sci USA. (2017) 114:E9626–34. 10.1073/pnas.1705301114
24.
BrehmMBortellRVermaMShultzLGreinerD. Humanized mice in translational immunology. Transl Immunol Mech Pharmacol Approach. (2016) 2016:285–326. 10.1016/B978-0-12-801577-3.00012-5
25.
BaluBSinghNMaherSPAdamsJH. A genetic screen for attenuated growth identifies genes crucial for intraerythrocytic development of Plasmodium falciparum. PloS ONE. (2010) 5:e13282. 10.1371/journal.pone.0013282
26.
BaluBCampbellCSedilloJMaherSSinghNThomasPet al. Atypical mitogen-activated protein kinase phosphatase implicated in regulating transition from pre-S-Phase asexual intraerythrocytic development of Plasmodium falciparum. Eukaryot Cell. (2013) 12:1171–178. 10.1128/EC.00028-13
27.
BaluBChauhanCMaherSPShoueDAKissingerJCFraserMJet al. piggyBac is an effective tool for functional analysis of the Plasmodium falciparum genome. BMC Microbiol. (2009) 9:83. 10.1186/1471-2180-9-83
28.
RoweAWEysterEKellnerA. Liquid nitrogen preservation of red blood cells for transfusion; a low glycerol-rapid freeze procedure. Cryobiology. (1968) 5:119–28. 10.1016/S0011-2240(68)80154-3
29.
LambrosCVanderbergJP. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol. (1979) 65:418–20. 10.2307/3280287
30.
LiQGerenaLXieLZhangJKyleDMilhousW. Development and validation of flow cytometric measurement for parasitemia in cultures of P. falciparum vitally stained with YOYO-1. Cytometry Part A. (2007) 71:297–307. 10.1002/cyto.a.20380
31.
PerssonKELeeCTMarshKBeesonJG. Development and optimization of high-throughput methods to measure Plasmodium falciparum-specific growth inhibitory antibodies. J Clin Microbiol. (2006) 44:1665–73. 10.1128/JCM.44.5.1665-1673.2006
32.
HasegawaMKawaiKMitsuiTTaniguchiKMonnaiMWakuiMet al. The reconstituted ‘humanized liver' in TK-NOG mice is mature and functional. Biochem Biophys Res Commun. (2011) 405:405–10. 10.1016/j.bbrc.2011.01.042
33.
BaluBShoueDAFraserMJAdamsJH. High-efficiency transformation of Plasmodium falciparum by the lepidopteran transposable element piggyBac. Proc Natl Acad Sci USA. (2005) 102:16391–16396. 10.1073/pnas.0504679102
34.
SilvaMTCorreia-NevesM. Neutrophils and macrophages: the main partners of phagocyte cell systems. Front Immunol. (2012) 3:174. 10.3389/fimmu.2012.00174
35.
Cassado AdosAD'ImpérioMRBortoluciKR. Revisiting mouse peritoneal macrophages: heterogeneity, development, and function. Front Immunol. (2015) 6:225. 10.3389/fimmu.2015.00225
36.
de BackDZKostovaEBvanMvan den BergTKVan BruggenR. Of macrophages and red blood cells; a complex love story. Front Physiol. (2014) 5:9. 10.3389/fphys.2014.00009
37.
ItescuSKwiatkowskiPArtripJHWangSFAnkersmitJMinanovOPet al. Role of natural killer cells, macrophages, and accessory molecule interactions in the rejection of pig-to-primate xenografts beyond the hyperacute period. Hum Immunol. (1998) 59:275–86. 10.1016/S0198-8859(98)00026-3
38.
LinYVandeputteMWaerM. Natural killer cell- and macrophage-mediated rejection of concordant xenografts in the absence of T and B cell responses. J Immunol. (1997) 158:5658–67.
39.
FoxAKoulmandaMMandelTEvan RooijenNHarrisonLC. Evidence that macrophages are required for T-cell infiltration and rejection of fetal pig pancreas xenografts in nonobese diabetic mice. Transplantation. (1998) 66:1407–16. 10.1097/00007890-199812150-00002
40.
WuGSKorsgrenOZhangJGSongZSVan RooijenNTibellA. Role of macrophages and natural killer cells in the rejection of pig islet xenografts in mice. Transplant Proc. (2000) 32:1069. 10.1016/S0041-1345(00)01127-1
41.
KosakaKHiragaNImamuraMYoshimiSMurakamiENakaharaTet al. A novel TK-NOG based humanized mouse model for the study of HBV and HCV infections. Biochem Biophys Res Commun. (2013) 441:230–5. 10.1016/j.bbrc.2013.10.040
42.
HiguchiYKawaiKYamazakiHNakamuraMBreeFGuguen-GuillouzoCet al. The human hepatic cell line HepaRG as a possible cell source for the generation of humanized liver TK-NOG mice. Xenobiotica. (2014) 44:146–53. 10.3109/00498254.2013.836257
43.
TsukadaASuemizuHMurayamaNTakanoRShimizuMNakamuraMet al. Plasma concentrations of melengestrol acetate in humans extrapolated from the pharmacokinetics established in in vivo experiments with rats and chimeric mice with humanized liver and physiologically based pharmacokinetic modeling. Regul Toxicol Pharmacol. (2013) 65:316–24. 10.1016/j.yrtph.2013.01.008
44.
YamazakiHSuemizuHMurayamaNUtohMShibataNNakamuraMet al. In vivo drug interactions of the teratogen thalidomide with midazolam: heterotropic cooperativity of human cytochrome P450 in humanized TK-NOG mice. Chem Res Toxicol. (2013) 26:486–9. 10.1021/tx400008g
45.
KimMChoiBJooSYLeeHLeeJHLeeKWet al. Generation of humanized liver mouse model by transplant of patient-derived fresh human hepatocytes. Transplant Proc. (2014) 46:1186–90. 10.1016/j.transproceed.2013.11.098
46.
XuDWuMNishimuraSNishimuraTMichieSAZhengMet al. Chimeric TK-NOG mice: a predictive model for cholestatic human liver toxicity. J Pharmacol Exp Ther. (2015) 352:274–80. 10.1124/jpet.114.220798
47.
KamimuraHItoSNozawaKNakamuraSChijiwaHNagatsukaSet al. Formation of the accumulative human metabolite and human-specific glutathione conjugate of diclofenac in TK-NOG chimeric mice with humanized livers. Drug Metab Dispos. (2015) 43:309–16. 10.1124/dmd.114.061689
48.
XuDMichieSAZhengMTakedaSWuMPeltzG. Humanized thymidine kinase-NOG mice can be used to identify drugs that cause animal-specific hepatotoxicity: a case study with furosemide. J Pharmacol Exp Ther. (2015) 354:73–8. 10.1124/jpet.115.224493
49.
SoulardVBosson-VangaHLorthioisARoucherCFranetichJFZanghiGet al. Plasmodium falciparum full life cycle and Plasmodium ovale liver stages in humanized mice. Nat Commun. (2015) 6:7690. 10.1038/ncomms8690
50.
KaiYHikitaHTatsumiTNakaboriTSaitoYMorishitaNet al. Emergence of hepatitis C virus NS5A L31V plus Y93H variant upon treatment failure of daclatasvir and asunaprevir is relatively resistant to ledipasvir and NS5B polymerase nucleotide inhibitor GS-558093 in human hepatocyte chimeric mice. J Gastroenterol. (2015) 50:1145–51. 10.1007/s00535-015-1108-6
51.
UchidaTHiragaNImamuraMTsugeMAbeHHayesCNet al. Human cytotoxic T lymphocyte-mediated acute liver failure and rescue by immunoglobulin in human hepatocyte transplant TK-NOG mice. J Virol. (2015) 89:10087–96. 10.1128/JVI.01126-15
52.
FominMEBeyerAIMuenchMO. Human fetal liver cultures support multiple cell lineages that can engraft immunodeficient mice. Open Biol. (2017) 7:170108. 10.1098/rsob.170108
53.
SuzukiEKoyamaKNakaiDGodaRKugaHChibaK. Observation of clinically relevant drug interaction in chimeric mice with humanized livers: the case of valproic acid and carbapenem antibiotics. Eur J Drug Metab Pharmacokinet. (2017) 42:965–72. 10.1007/s13318-017-0413-2
54.
ShimizuMSuemizuHMitsuiMShibataNGuengerichFPYamazakiH. Metabolic profiles of pomalidomide in human plasma simulated with pharmacokinetic data in control and humanized-liver mice. Xenobiotica. (2017) 47:844–8. 10.1080/00498254.2016.1247218
55.
LavazecCSanyalSTempletonTJ. Hypervariability within the Rifin, Stevor and Pfmc-2TM superfamilies in Plasmodium falciparum. Nucleic Acids Res. (2006) 34:6696–707. 10.1093/nar/gkl942
56.
XuDNishimuraTNishimuraSZhangHZhengMGuoYYet al. Fialuridine induces acute liver failure in chimeric TK-NOG mice: a model for detecting hepatic drug toxicity prior to human testing. PLoS Med. (2014) 11:e1001628. 10.1371/journal.pmed.1001628
57.
XuDHuangPYuZXingDHOuyangSXingG. Efficacy and safety of panax notoginseng saponin therapy for acute intracerebral hemorrhage, meta-analysis, and mini review of potential mechanisms of action. Front. Neurol. (2015) 5:274. 10.3389/fneur.2014.00274
58.
Moreno-SabaterAPérignonJLMazierDLavazecCSoulardV. Humanized mouse models infected with human Plasmodium species for antimalarial drug discovery. Expert Opin Drug Discov. (2018) 13:131–40. 10.1080/17460441.2018.1410136
59.
HydeJE. Drug-resistant malaria. Trends Parasitol. (2005) 21:494–8. 10.1016/j.pt.2005.08.020
60.
HaldarKBhattacharjeeSSafeukuiI. Drug resistance in Plasmodium. Nat Rev Microbiol. (2018) 16:156–70. 10.1038/nrmicro.2017.161
61.
ThuAMPhyoAPLandierJParkerDMNostenFH. Combating multi-drug resistant Plasmodium falciparum malaria. FEBS J. (2017) 284:2569–78. 10.1111/febs.14127
62.
MenardDDondorpA. Antimalarial drug resistance: a threat to malaria elimination. Cold Spring Harbor Perspect Med. (2017) 7:a025619. 10.1101/cshperspect.a025619
63.
ArieS. Researchers and WHO clash over global threat of drug resistant malaria. BMJ. (2017) 359:j5127. 10.1136/bmj.j5127
64.
CuiLMharakurwaSNdiayeDRathodPKRosenthalPJ. Antimalarial drug resistance: literature review and activities and findings of the ICEMR network. Am J Trop Med Hyg. (2015) 93:57–68. 10.4269/ajtmh.15-0007
65.
AmolegbeSAHiranoYAdebayoJOAdemowoOGBalogunEAObaleyeJAet al. Mesoporous silica nanocarriers encapsulated antimalarials with high therapeutic performance. Sci Rep. (2018) 8:3078. 10.1038/s41598-018-21351-8
Summary
Keywords
humanized mice, clodronate-loaded liposomes, NOD/SCID/IL-2rg−/ (NSG) growth mutants, TK/NOG, cytokine, PF3D7_1305500, C9 parasite mutants (C9-M), complemented parasites (C9-C)
Citation
Zhang L-l, Li J-L, Ji M-X, Tian D, Wang L-Y, Chen C and Tian M (2020) Attenuated P. falciparum Parasite Shows Cytokine Variations in Humanized Mice. Front. Immunol. 11:1801. doi: 10.3389/fimmu.2020.01801
Received
24 April 2020
Accepted
06 July 2020
Published
11 September 2020
Volume
11 - 2020
Edited by
Xiao-Nong Zhou, National Institute of Parasitic Diseases, China
Reviewed by
Gaurav Gupta, NIIT University, India; Sunil Joshi, University of Miami, United States
Updates
Copyright
© 2020 Zhang, Li, Ji, Tian, Wang, Chen and Tian.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miao Tian tmiaolook@zoho.com.cn
This article was submitted to Microbial Immunology, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.