 Giuliana Giardino
Giuliana Giardino Carla Borzacchiello†
Carla Borzacchiello† Martina De Luca†Roberta Romano
Martina De Luca†Roberta Romano Rosaria Prencipe
Rosaria Prencipe Emilia Cirillo
Emilia Cirillo Claudio Pignata*
Claudio Pignata*- Department of Translational Medical Sciences, Pediatric Section, Federico II University of Naples, Naples, Italy
Combined Immunodeficiencies (CID) are rare congenital disorders characterized by defective T-cell development that may be associated with B- and NK-cell deficiency. They are usually due to alterations in genes expressed in hematopoietic precursors but in few cases, they are caused by impaired thymic development. Athymia was classically associated with DiGeorge Syndrome due to TBX1 gene haploinsufficiency. Other genes, implicated in thymic organogenesis include FOXN1, associated with Nude SCID syndrome, PAX1, associated with Otofaciocervical Syndrome type 2, and CHD7, one of the genes implicated in CHARGE syndrome. More recently, chromosome 2p11.2 microdeletion, causing FOXI3 haploinsufficiency, has been identified in 5 families with impaired thymus development. In this review, we will summarize the main genetic, clinical, and immunological features related to the abovementioned gene mutations. We will also focus on different therapeutic approaches to treat SCID in these patients.
Introduction
The thymus is a primary lymphoid organ which plays a pivotal role in the development of mature T cells from immature bone marrow CD34+ precursors. Together with the parathyroid glands, it develops from the 3 pharyngeal pouch (PP) (1). The epithelial components of the thymus derive from the endothelial layer, while the mesenchymal capsule derives from neural crest, originated from ectoderma (1, 2). The first stage of thymic development is independent of the transcription factor forkhead box N1 (Foxn1) expression (3). In this phase, Paired box 1 (Pax1), Eyes absent homolog 1 (Eya1), sine oculis homeobox (Six), homeobox A3 (Hoxa3), and T-box 1 (Tbx1) drive the outgrowth of the thymic epithelial anlage from the 3rd PP (1, 4, 5). Hoxa3 and Eya1 are also implicated in the development of neural crest derived mesenchymal cells (6–8). Studies suggest that chromodomain helicase DNA-binding 7 (Chd7) might be implicated in the development of both neural crest cell-derived mesenchyme and pharyngeal endoderm-derived thymic epithelial cells (TECs). Pax3 and Hoxa3 expression in mesenchymal cells allows the detachment of the thymic lobes from the pharynx (9, 10). Thymus development in early and late stages is regulated by the interactions among various cell types. The thymus three-dimensional (3D) architecture allows a proper intercellular cross talk (3). In the first phase of organogenesis, mesenchymal cells release bone morphogenetic protein 4 (Bmp4), Bmp2, fibroblast and insulin growth factors (Fgf, Igf), Wnt proteins, and retinoic acid supporting the differentiation of TECs into cortical (cTECs) and medullary (mTECs) subsets (7, 8, 11–14). In the second phase, Foxn1 induces the expression of chemokine (C-C motif) ligand 25 (CCL25), delta like canonical Notch ligand 4 (Dll4), and Hoxa3, allowing thymocyte recruitment and TECs differentiation in cTECs and mTECs. Mesenchymal cells are implicated in the recruitment of hematopoietic thymic seeding progenitors, as well (15, 16). Thymocytes participate to TECs differentiation process through the release of epidermal growth factor (Egf) and lymphotoxin factors (17–19). In support of this, mice with defective T-cell development show defective organization of the thymic medulla (20, 21), that is restored after stem cell transplantation (21, 22).
Bone marrow derived hematopoietic stem cells (HSCs) enter the thymus through cortico-medullary junction, where they proliferate (23). The V(D)J rearrangement of the double negative (DN) thymocytes T-cell receptor β (TCRβ) gene takes place in the thymic cortex (24, 25). Membrane expression of pre-TCR complex is necessary for the expression of the co-receptors CD4 and CD8, as well as V-J rearrangement of the TCRα genomic region (26). Double positive thymocytes with a functional TCR-αβ receptor capable of binding to self-MHC ligands are positively selected (27–29). This process is regulated by Prss16 and β5t, which are expressed in cTECs (30–34). Into medulla, self-reactive thymocytes are deleted through the negative selection, a process mediated by dendritic cells and Aire-expressing mTECs (35, 36). Mutations in genes implicated in different steps of thymic development, including FOXN1, PAX1, TBX1, CDH7 impair T-cell development in humans (Figure 1). Alterations of the immune system in these conditions range from an isolated reduction of T-cell count to severe combined immunodeficiency (SCID). This review is focused on definition of the role of different genes implicated in thymus development and of primary immunodeficiencies (PIDs) due to their deficiency. Moreover, therapeutic options for PIDs with congenital athymia are discussed.
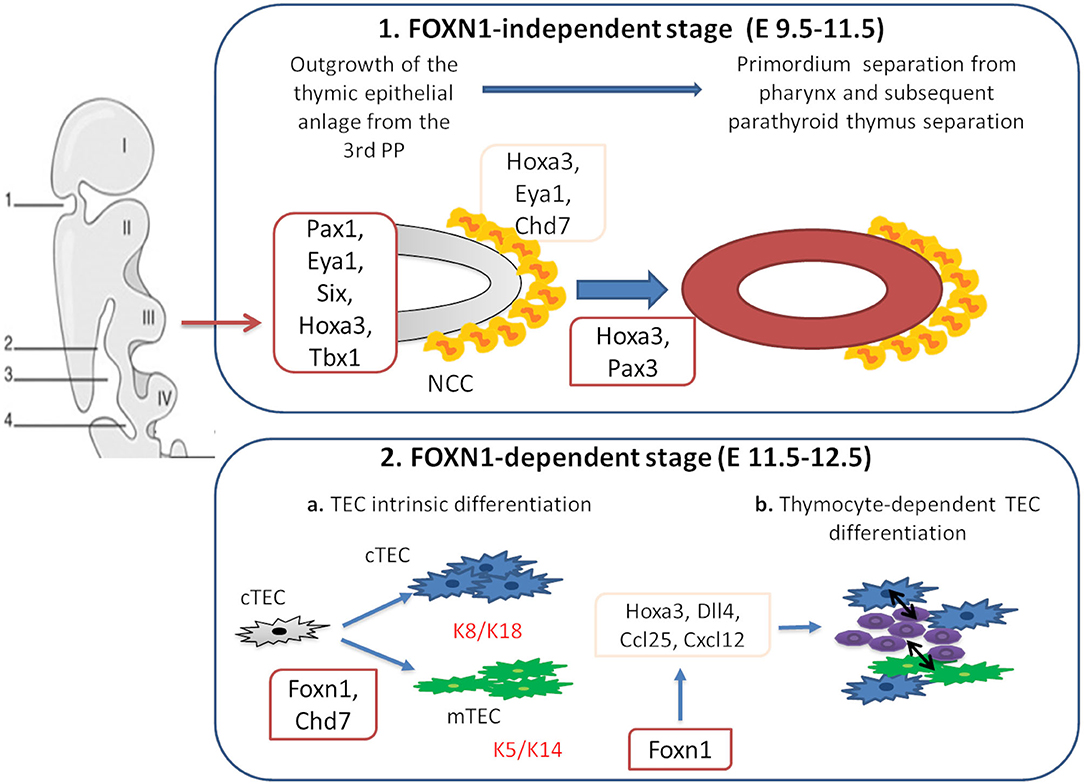
Figure 1. Genes involved in thymus organogenesis. Eya1, Six, Hoxa3, Tbx1, and Chd7 take part to the first stages of thymus development from neural crest cells (NCCs) in the posterior part of the 3rd pharyngeal pouch (PP). This step is independent from Foxn1 expression. In the second stage, Foxn1 regulates the expression of Hoxa3, Dll4, Ccl25, and Cxcl12, necessary for the thymic epithelial cells (TECs) differentiation. During this phase, cTECs (expressing K8 and K18 keratin type) and mTECs (expressing K5 and K14 keratin type) originate from the same bi-potential TECs progenitor. Chd7 is also critical for the development of cortical and medullary TECs from pharyngeal endoderm. The crosstalk between TECs and developing thymocytes is required to generate mature TECs and functional T cells.
Gene Function and Related Syndrome
FOXN1 Deficiency and Nude-SCID Syndrome
FOXN1 is located on chromosome 17q11.2 and it is composed of eight exons (30 kb) (37, 38). This gene is a member of the Fox gene family that comprises different Winged helix transcription factors implicated in development, metabolism, cancer, and aging (39). During fetal life, FOXN1 is expressed in mesenchymal and epithelial cells including liver, lung, intestine, kidney, and urinary tract. In postnatal life, its expression is restricted to keratinocytes and TECs (40, 41). FOXN1 is involved in development, function and maintenance of hair follicles, and TECs (Figure 1) (42–44). The expression of FOXN1 in TECs leads to the production of chemokines, such as C-X-C motif chemokine ligand 12 (CXCL12), implicated in the recruitment of hematopoietic progenitors and of molecules, as DLL4 notch ligand, implicated in maturation of the progenitors toward the T-cell line (Figure 1) (45–47). Studies in mice showed that Foxn1 plays a pivotal role in morphogenesis of the 3D architecture (48, 49) by inhibiting a basic morphogenetic pattern of tubulogenesis and inducing the expression of genes that drive TECs differentiation (50). FOXN1 also regulates Prss16 and β5t gene expression, implicated in positive selection (45, 51). Knockout of Hoxa1, Pax1, Pax9, Eya1, or Six1 affects Foxn1 expression resulting in impaired thymic function (52). Foxn1 expression is also regulated by signals from the surrounding endothelium and mesenchyme, including Bmp4 and Wnt (53, 54).
In epidermis, FOXN1 is expressed in keratinocytes of suprabasal layer, where these cells stop proliferating and start the terminal differentiation (55). In keratinocytes, FOXN1 regulates the transcription of more than 50 genes, including protein kinase B and C (PKB and PKC). PKC is an inhibitor of human hair follicle growth in vitro (56–58). It is up-regulated in Foxn1–/– mice while its activity is suppressed in mice overexpressing Foxn1 gene in which differentiation is inhibited. Studies conducted on human keratinocytes have confirmed the role of the gene in the differentiation of the epidermis (58).
Homozyous FOXN1 mutations cause Nude SCID syndrome, first observed in two Italian sisters presenting with congenital universal alopecia, nail dystrophy, and severe T-cell immunodeficiency with rudimentary thymus (59). In the last few years, neonatal screening and next generation sequencing techniques have led to the identification of subjects with novel homozygous, compound heterozygous, and heterozygous mutations (60). Homozygous patients suffer from immunodeficiency with susceptibility to pneumonia, chronic diarrhea, candidiasis or mycobacterial infections, and Omenn syndrome (59, 61–64). Most of the heterozygous patients show nail dystrophy, usually presenting as leukonychia and minor immunological changes (60, 65, 66). Recurrent infections and atopic dermatitis are observed only in a minority of the patients (60).
Immunological features in patients with homozygous FOXN1 mutations so far reported include reduction of T lymphocytes, particularly CD4+ T cells (61–64), reduction of T-cell receptor excision circles (TRECs) and naive T CD45RA+ lymphocytes, with increase of T memory CD45RO+ lymphocytes (62–64). In addition, an increase in the DN CD4-CD8- lymphocytes in the periphery (61–63), a reduction in CD31+ cells, recently emigrated from the thymus was observed in few cases (63). Proliferative response to phytohemagglutinin (PHA) is usually poor or absent and TCR repertoire is oligoclonal (59, 61–63). Although natural killer (NK) and B cells are usually numerically normal, they may be functionally compromised, with impaired production of specific antibodies. Recent studies proved that FOXN1 haploinsufficiency, caused by FOXN1 heterozygous mutations, may be associated with T cell-lymphopenia in infants showing low TRECs at newborn screening (60). A progressive normalization of CD4 count is observed in adulthood in heterozygous subjects while CD8 are usually persistently low (60). The increase in CD4 levels is associated to persistently low CD45RA levels suggesting a mechanism of homeostatic proliferation. A more significant homeostatic expansion in CD4 than CD8 might explain the difference in CD4 and CD8 T cells. Alternatively, another hypothesis is that FOXN1 dosage plays a stricter role in the expression of CD8 development genes (60).
PAX1 Deficiency and Otofaciocervical Syndrome Type 2
PAX1 is a member of a family of genes that encodes transcription factors implicated in embryogenesis in vertebrates (67). It is located on chromosome 20p11.22 and contains 5 exons (10 Kb) (67, 68). This gene is expressed in fetal mesenchymal cells in the body of intervertebral disks and plays an important role in formation of the segmented vertebral column in humans (69). In mice, it is also expressed in cochlea and has a role in hearing process (70). PAX1 is also implicated in thymus organogenesis by contributing to the outgrowth of the thymic epithelial anlage from the 3rd PP and to the regulation of TECs differentiation/survival balance (Figure 1) (71, 72). During pre-natal life, it is expressed in the 3rd PP from E9.5, while in the post-natal thymus it is only expressed in cTECs (72) (Figure 1). In mice, Pax1 deficiency is associated with moderate thymic hypoplasia (72) that is more severe only when it is associated with Hoxa3 haploinsufficiency (73). On the contrary, in humans, PAX1 deficiency is associated with severe thymic hypoplasia, leading to SCID (74). The difference between human and mouse phenotype may be due to a compensatory contribution by murine Pax9 when Pax1 is lacking (75).
Otofaciocervical syndrome (OTFCS) is an autosomal dominant disorder characterized by short stature, facial dysmorphism (long face, narrow mandible), shoulder girdle abnormalities, hearing loss, and mild intellectual disability (76). Two different forms of OTFCS have been described but thymus development is only affected in OTFCS2, caused by PAX1 mutations. Different biallelic deleterious PAX1 variants cause OTFCS2 and SCID, characterized by absent thymic shadow, chronic diarrhea, recurrent respiratory infections, pneumonia, and also Omenn Syndrome (74). Different severity of OTFCS2 phenotype is described in different families (70, 71, 77).
The immunological phenotype of patients with OTFC2 was described very recently in six patients who showed T-cell lymphopenia, impaired proliferative response to mitogens, and normal levels of B and NK cells. In one of them, 98% of CD4 and CD8 T cells were CD45RO+ cells, while CD45RA+CD31+ cells and TRECs were undetectable. TCRVβ analysis showed an oligoclonal spectrum. Lymph node biopsy of another patient showed absence of germinal centers and almost total absence of CD3+ T cells. Patients with Omenn Syndrome also showed eosinophilia and increased IgE levels (74).
Digeorge Syndrome and 22q11.2 Deletion
DiGeorge syndrome (DGS) is usually associated with 3 or a 1.5 Mb de novo microdeletion of 22q11.2 (78, 79). In the central region of deletion maps TBX1 gene containing 9 exons (80) and encodes a Tbx transcription factor, implicated in the regulation of nearly 2,000 genes (81, 82). It is strongly expressed in the 3rd and 4th PP endoderm and in the 4th pharyngeal arch arteries (83, 84), in the otic vesicle, vertebral column, later in tooth bud and, at a lower extent, in the brain (85). It is implicated in pharyngeal arch segmentation and outgrowth of the TECs from the 3rd PP (Figure 1). However, it should be noted that Tbx1 is not expressed in the thymic anlage and thus it is not directly implicated in TECs development (86). On the contrary, Tbx1 enforced expression within the 3rd PP represses TECs development (86). Reduced levels of Tbx1 in 22q11.2 deletion syndrome (22q11.2DS) impair the development of neural crest-derived mesenchymal cells that surround the 3rd PP, leading to thymic hypoplasia (87). TBX1 regulates the expression of secreted Fgfs molecules, namely Fgf8 and Fgf10, implicated in the control of TECs proliferation, differentiation, migration, and survival (88). Ffg receptor IIIb (FgfR2-IIIb) regulates the cascade of Hox3 paralogs transcription factors, Pax1/Pax9, and winged helix nude (Whn) (Figure 1) (89). Moreover, TBX1 interferes with the ability of small mother against decapentaplegic 1 (SMAD1) to bind SMAD4, preventing effective Bmp4 signaling (82). Bmp4 also contributes to early thymus and parathyroid morphogenesis (90). Isolated TBX1 mutations may be rarely reported in patients with DGS (91) and TBX1 gain-of-function mutations can result in the same phenotypic spectrum of loss-of-function mutations (92).
An embryonic phenocopy of DGS with impaired thymus development can be observed because of the lack of retinoic acid during gestation (79, 93, 94). Retinoic acid is able to regulate the expression of Tbx1 and other molecules implicated in thymus organogenesis including Pax1, Pax9, Hoxa3, Fgf8, and Bmp4 (95). Other epigenetic factors, including maternal diabetes (93) or prenatal exposure to retinoic acid or alcohol may also explain the alteration of thymus development in DGS (96–101). Recently, we reported on a 7-year-old DGS patient born to a mother with gestational diabetes mellitus in whom a 371 Kb-interstitial deletion of 3p12.3, involving the Zinc Finger Protein 717 (ZNF717), MicroRNA-1243 (MIR-1243), and MIR-4273 genes was identified (102). MiRNAs are small, non-coding RNAs involved in the modulation of gene expression by targeting messenger RNAs for degradation, translational repression, or both (103). miRNA-4273 regulates the expression of Bmp3, a member of the transforming growth factor β superfamily, involved in thymus and kidney development (104). The expression of other miRNAs, including MIR-185, and MIR-150 can be impaired in 22q11.2DS patients (105). MIR-185 reduction increases Bruton's tyrosine kinase (Btk) expression leading to autoantibody production while MIR-185 increase leads to dose-dependent T-cell lymphopenia (105, 106). Reduced MIR-150 expression contributes to the reduction of T and B cells (107). Dysregulation of miRNA biogenesis, due to DiGeorge Critical Region Gene 8 (DGCR8) haploinsufficiency, is implicated in the pathogenesis of immunological, cardiac, endocrinological, and neurological phenotype (105, 108). Other genes implicated in the immune response are included in the deleted region. Alterations of CrK-like (CRKL), a gene encoding a 39-kDa adapter protein belonging to the Crk family, implicated in many cellular functions, including cell migration and adhesion, are associated with impaired T-cell proliferation in response to TCR triggering (109).
DGS has a prevalence of 1:4,000 newborns (79, 110). Most of these patients present with thymic and parathyroid hypoplasia, congenital heart defects, and craniofacial dysmorphisms (78, 79). Thymic development ranges from athymia in complete DGS (cDGS) to a completely normal thymus development in partial DGS (pDGS), resulting in a variable spectrum of T-cell deficiency (78, 79, 111). cDGS is reported in about 1.5% of the patients (111). Patients with DGS show a wide spectrum of T-cell alterations ranging from completely normal T-cell development to cDGS with absent thymic development (112, 113). In 22q11.2DS patients, thymus is usually small or hypoplastic. The size of the thymus does not predict the levels circulating T cells. In fact, microscopic rests of TECs may be present at aberrant locations (114). Low CD3+ T-cell percentage is the most common T-cell defect, followed by low CD3 number (78). CD4 and CD8 compartments are similarly affected (78). T-cell number and percentage tend to increase during the follow up starting from the first year of life (78). Naive CD4 and CD8 lymphocytes are lower in pDGS patients compared to controls independently of age and they decline more rapidly with age (78). The improvement of the lymphopenia with age is not due to a recovery of the thymic function but to the peripheral homeostatic expansion of the available T cells, as suggested by the evidence that T cells are predominantly or almost exclusively of a memory phenotype, TRECs are low and the repertoire is oligoclonal (78, 115, 116). T-cell proliferation, total immunoglobulins, and specific antibody response to vaccines are typically normal. IgM levels are often low, and some patients may show selective IgA deficiency (78). The study of the thymic architecture and thymocyte development in thymi obtained from pediatric pDGS patients revealed a reduction of mature CD4+ and CD8+ T cell frequency, associated with reduced proportion and function of T regulatory cells (Tregs) (117).
The majority of DGS patients suffer from chronic otitis media, which correlates with primarily conductive hearing impairment (78). Most patients have increased susceptibility to mild infections and only rarely to atypical or severe infections (78). Abnormalities of PP derivatives, predisposing to bacterial colonization, more than immune defects are implicated in this susceptibility (78). Autoimmune diseases, mainly presenting as rheumatoid diseases and idiopathic thrombocytopenia purpura, are reported in ~10% of DGS patients (113, 116, 118). Predisposition to autoimmunity in DGS patients is partially explained by the mechanism of lymphopenia induced T-cell homeostatic proliferation together with the reduction of natural Tregs (nTregs) (117, 119).
CHD7 Haploinsufficiency and Charge Syndrome
CHARGE (coloboma, heart defects, atresia choanae, growth retardation, genital abnormalities, and ear abnormalities) syndrome is associated to haploinsufficiency in CHD7 gene, located on chromosome 8q12 (120). CHD7 is implicated in chromatin organization of mesenchymal cells, derived from the cephalic neural crest (121). Impaired CHD7 expression correlates with defects in neural crest cells, cephalic mesenchyme, pharyngeal arches, brain, otic vesicle and, in the mesoderm of the developing heart, especially in the outflow tract of the heart (122, 123). CHD7 is critical for the development of cTECs and mTECs from pharyngeal endoderm by regulating Bmp4, which in turn regulate Foxn1 expression (Figure 1) (124). Chd7 deficiency is also associated with down-regulation of Ikaros, Interelukin 7 receptor (Il7r), recombinase activating gene 1 (Rag1) (124). Studies suggest that CHD7 can also regulate TBX1 expression (74, 125).
CHARGE syndrome is characterized by different degrees of thymic alterations and even by complete thymic aplasia, resulting in combined immune deficiency (126). A wide spectrum of T-cell deficiency and isolated humoral immune deficiency may be observed in patients with CHARGE syndrome (126). They may show severe T-cell deficiency resembling SCID or, similarly to DGS, they may present transient lymphopenia, that usually normalizes over time. A close association between lymphopenia and hypocalcemia has been identified (115). Severity of the T-cell lymphopenia relates to the degree of thymic hypoplasia (127). CHARGE patients also show decreased naive CD4 and CD8 T-cells, peripheral T cells, and TRECs. Peripheral B-cell differentiation and immunoglobulin production are normal but in a 3rd of the patients an abnormal expression of IgM on class-switched memory B cells and diminished production of specific antibodies may be observed (127). Selective antibody deficiency tends to resolve spontaneously over time (127). In a recent paper, immunological features were compared between CHARGE and DGS patients. Total lymphocyte count was slightly lower in DGS patients compared to CHARGE patients and persistent lymphopenia was more common in DGS patients than in CHARGE. IgM levels were significantly lower in DGS patients compared to CHARGE (128).
Most of the patients have increased risk of recurrent infections, including recurrent otitis media, sinusitis, conjunctivitis, dermatitis, respiratory tract infections, pneumonia, and sepsis (127). Severe or atypical infections may also be reported including recurrent oral candidiasis, recurrent severe infections, septic shock, and chronic viral infections (129). As for partial DGS also in CHARGE syndrome the frequent need of invasive operative procedures and anatomical alterations, including extensive ear, sinus, nasal and palatal malformations, altered Eustachian tube anatomy, gastroesophageal reflux, or neurological abnormalities with compromised drainage and aspiration may help explain the susceptibility to infections (127). Immune defects including impaired thymus development and subsequent humoral deficiency may also contribute to the susceptibility to infections but at a lower extent (128). Patients with CHARGE syndrome also show increased risk of atopy, reported in 65% of the patients, usually presenting as food allergy (128). Increased susceptibility to autoimmune disorders may also feature this syndrome (129). Patients with complete athymia may present with atypical SCID (Omenn-like) phenotype (130).
2p11.2 Microdeletion and FOXI3 Haploinsufficiency
A microdeletion at 2p11.2 has been identified in five families presenting with DGS features, including hypocalcaemia, asymmetric crying face, low TRECs and T-cell lymphopenia, without typical facial dysmorphism, and heart abnormalities. FOXI3, a member of FOX family transcription factors, implicated in development of brachial arch-derived structures was considered the candidate gene for this phenotype (131). Early in embryonic development Foxi3 is broadly expressed in the pre-placodal ectoderm surrounding the neural plate, from which all craniofacial sensory organs derive (132, 133). Subsequently, its expression is restricted to the region from which otic and epibranchial placodes derive and finally to the ectoderm and endoderm of the pharyngeal arches. A deletion of FOXI3 gene has been recently identified in a patient with left congenital aural atresia and ipsilateral internal carotid artery agenesis (134). Foxi3 is also implicated in the differentiation of the epithelial cells within the epidermis as suggested by the identification of Foxi3 heterozygous mutations in several hairless dog breeds with hair follicle and teeth defects (135) and in thymic cortico-medullary differentiation. Foxi3 is implicated in segmentation of the pharyngeal apparatus and LOF of Foxi3 alone or in combination with Tbx1 LOF, leads to failure of the pharyngeal arch segmentation due to the inability of the epithelia to properly invaginate with subsequent thymic hypoplasia/aplasia (136).
The main clinical features of the different syndromes are compared in Table 1.
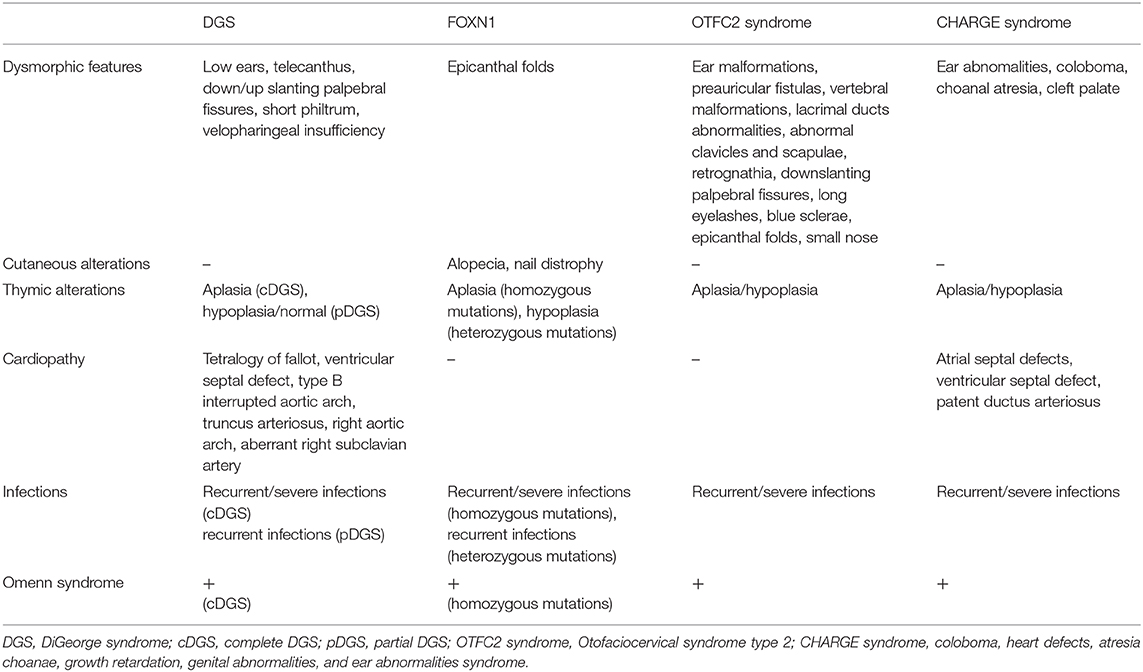
Table 1. Comparison of the main clinical features among different congenital disorders of thymic development.
Treatment Options for Athymic Conditions
Hematopoietic stem cell transplantation (HSCT) represents the cornerstone for the treatment of SCID. However, in SCID due to genetic defects that impair development and function of the thymic epithelium, theoretically thymus transplantation would represent the most appropriate therapy. Thymic tissue is obtained from infants undergoing to median sternotomy for open heart surgery. Cultured postnatal human thymic tissue is then transplanted in thin slices into the quadriceps muscle (137). In cases with successful transplant, few months after the transplant the graft is colonized by host stem cells and is able to support normal thymopoiesis (138) leading to the development of mature naive T-cell with diverse TCRVβ repertoire and able to proliferate in response to mitogens. T-cell levels in surviving patients are usually low for age but are sufficient to respond to viral, disseminated, and other infections leading to a resolution of the immunodeficiency (139). In some cases, reduced thymic output may be explained by other comorbidities, such as heart failure and hypoxia stress, that may cause hypoperfusion of the graft (139). The success of the transplant may be also limited in patients with active viral infections since the virus itself, its treatment, or both might inhibit the thymopoiesis (139). In patients with successful transplant, naive T-cells are usually detected within 6 months after the transplant (139, 140). Antibody responses and immunoglobulin levels normalize (62) even though numbers of class-switched memory B cells may remain relatively low. The success of the transplant is not correlated with the amount of tissue transplanted, HLA matching, culture conditions or immunosuppression of the recipient (141). Immunosuppression can be used to delete reactive oligoclonal T cells and mature T cells responsible of graft-versus-host disease and graft rejection (137, 139). Overall survival in DGS is 75% and mortality is usually related to pre-transplantation morbidity, mainly viral infections, and chronic lung disease (115, 140, 142). Thymus transplantation has been recently used to treat 2 patients with FOXN1 deficiency and both survived (62, 143, 144) while its use in athymic CHARGE and OTFC2 patients has never been reported. Autoimmune disorders, including thyroiditis and severe cytopenia, represent the most common complication after thymus transplant (139, 140).
Adoptive transfer of mature T cells from human leukocyte antigen identical siblings through bone marrow transplantation represents an alternative to thymus transplant to treat SCID in athymic patients (145). However, only post-thymic T cells engraft in this case and naive T cells do not develop. Survival after matched unrelated donor and matched sibling transplantations in cDGS were reported as being 33 and 60%, respectively (145) while in CHARGE out of six patients treated with HSCT three had graft vs. host disease and three died post-transplant (125, 130, 146, 147). Long-lasting survival patients after matched sibling donor transplantation are reported (148). Four FOXN1 deficient patients underwent HSCT and two survived (59, 63, 64, 149). One of them is currently well at 22 years of age (unpublished data). The study of the T cell compartment in this patient, 5 years after HSCT showed a marked reduction of CD4CD45Ra levels with normal CD8CD45Ra levels. However, TCRVβ repertoire, was largely impaired in the CD8 subset (150). Six patients with OTFC2 were treated with allogeneic HSCT. T-cell reconstitution was not observed in any of the patients, despite successful engraftment in three of them. In one of the cases with successful engraftment all the T cells showed a memory (CD45R0+) phenotype, but no de novo generation of a polyclonal repertoire of naive T cells was observed. The remaining 2 patients showed persistent T cell lymphopenia leading to severe and recurrent infections, and death for septic shock in one patient and to severe autoimmune hemolytic anemia in the other (74). Together, these data indicate that HSCT may be unable to correct the profound T cell immunodeficiency of this disease.
Conclusions
In conclusion, in this review we described different pathways involved in thymus development and the clinical phenotypes associated with their impairment. We also summarized the outcome related to different therapeutic approaches to these disorders. We highlighted the clinical importance of the early detection of a defect in the pathways involved in T-cell development. With the recent introduction of newborn screening programs a timely identification of patients affected with defects of the T-cell development before the onset of the symptoms is now possible, and this prompted the definitive treatment with HSCT. This has been proven effective in improving the prognosis. However, in some cases HSCT is not required for the management of infants with T-cell lymphopenia at birth, since the T-cell development tend to improve with age (prematurity, FOXN1 haploinsufficiency). In other cases, HSCT may not be curative since the defect involves thymus development. However, genome-wide association studies have shown that a large proportion of variants likely to cause human disease are located outside of the protein-coding domains, so whole genome wide approaches might lead to proper identification and thus correct treatment plans for immunodeficiency disorders resulting from aberrant expression of some of the genes discussed in the present review.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication. GG, CB, MD, EC, and RR reviewed the literature, organized and wrote the manuscript. CP wrote the manuscript and supervised all the work. RP did the picture. All authors reviewed and approved the manuscript.
Funding
This work was partially supported by a public grant overseen by Italian Ministry of Health as part of the project ‘Ricerca Finalizzata’ (reference: RF-2016-02364303).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Gordon J, Manley NR. Mechanisms of thymus organogenesis and morphogenesis. Development. (2011) 138:3865–78. doi: 10.1242/dev.059998
2. Gordon J. Hox genes in the pharyngeal region: how Hoxa3 controls early embryonic development of the pharyngeal organs. Int J Dev Biol. (2018) 62:775–83. doi: 10.1387/ijdb.180284jg
3. Anderson G, Jenkinson EJ. Lymphostromal interactions in thymic development and function. Nature Rev Immunol. (2001) 1:31–40. doi: 10.1038/35095500
4. Xu PX, Zheng W, Laclef C, Maire P, Maas RL, Peters H, et al. Eya1 is required for the morphogenesis of mammalian thymus, parathyroid and thyroid. Development. (2002) 129:3033–44.
5. Farley AM, Morris LX, Vroegindeweij E, Depreter ML, Vaidya H, Stenhouse FH, et al. Dynamics of thymus organogenesis and colonization in early human development. Development. (2013) 140:2015–26. doi: 10.1242/dev.087320
6. Auerbach R. Morphogenetic interactions in the development of the mouse thymus gland. Dev Biol. (1960) 2:271–84. doi: 10.1016/0012-1606(60)90009-9
7. Jenkinson WE, Jenkinson EJ, Anderson G. Differential requirement for mesenchyme in the proliferation and maturation of thymic epithelial progenitors. J Exp Med. (2003) 198:325–32. doi: 10.1084/jem.20022135
8. Jenkinson WE, Rossi SW, Parnell SM, Jenkinson EJ, Anderson G. PDGFRalpha-expressing mesenchyme regulates thymus growth and the availability of intrathymic niches. Blood. (2007) 109:954–60. doi: 10.1182/blood-2006-05-023143
9. Chen L, Zhao P, Wells L, Amemiya CT, Condie BG, Manley NR. Mouse and zebrafish hoxa3 orthologues have nonequivalent in vivo protein function. Proc Natl Acad Sci USA. (2010) 107:10555–60. doi: 10.1073/pnas.1005129107
10. Griffith AV, Cardenas K, Carter C, Gordon J, Iberg A, Engleka K, et al. Increased thymus- and decreased parathyroid-fated organ domains in Splotch mutant embryos. Dev Biol. (2009) 327:216–27. doi: 10.1016/j.ydbio.2008.12.019
11. Frank DU, Fotheringham LK, Brewer JA, Muglia LJ, Tristani-Firouzi M, Capecchi MR, et al. An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development. (2002) 129:4591–603.
12. Itoi M, Tsukamoto N, Yoshida H, Amagai T. Mesenchymal cells are required for functional development of thymic epithelial cells. Int Immunol. (2007) 19:95364. doi: 10.1093/intimm/dxm060
13. Sitnik KM, Kotarsky K, White AJ, Jenkinson WE, Anderson G, Agace WW. Mesenchymal cells regulate retinoic acid receptor-dependent cortical thymic epithelial cell homeostasis. J Immunol. (2012) 188:4801–9. doi: 10.4049/jimmunol.1200358
14. Bleul CC, Boehm T. BMP signaling is required for normal thymus development. J Immunol. (2005) 175:5213–21. doi: 10.4049/jimmunol.175.8.5213
15. Itoi M, Kawamoto H, Katsura Y, Amagai T. Two distinct steps of immigration of hematopoietic progenitors into the early thymus anlage. Int Immunol. (2001) 13:1203–11. doi: 10.1093/intimm/13.9.1203
16. Jenkinson WE, Bacon A, White AJ, Anderson G, Jenkinson EJ. An epithelial progenitor pool regulates thymus growth. J Immunol. (2008) 181:6101–8. doi: 10.4049/jimmunol.181.9.6101
17. Hikosaka Y, Nitta T, Ohigashi I, Yano K, Ishimaru N, Hayashi Y, et al. The cytokine RANKL produced by positively selected thymocytes fosters medullary thymic epithelial cells that express autoimmune regulator. Immunity. (2008) 29:438–50. doi: 10.1016/j.immuni.2008.06.018
18. Satoh R, Kakugawa K, Yasuda T, Yoshida H, Sibilia M, Katsura Y, et al. Requirement of stat3 signaling in the postnatal development of thymic medullary epithelial cells. PLoS Genet. (2016) 12:e1005776. doi: 10.1371/journal.pgen.1005776
19. Wu W, Shi Y, Xia H, Chai Q, Jin C, Ren B, et al. Epithelial LTβR signaling controls the population size of the progenitors of medullary thymic epithelial cells in neonatal mice. Sci Rep. (2017) 7:44481. doi: 10.1038/srep44481
20. Palmer DB, Viney JL, Ritter MA, Hayday AC, Owen MJ. Expression of the alpha beta T-cell receptor is necessary for the generation of the thymic medulla. Dev Immunol. (1993) 3:175–79. doi: 10.1155/1993/56290
21. Shores EW, van Ewijk W, Singer A. Maturation of medullary thymic epithelium requires thymocytes expressing fully assembled CD3-TCR complexes. Int Immunol. (1994) 6:1393–402. doi: 10.1093/intimm/6.9.1393
22. Surh CD, Sprent J. T-cell apoptosis detected in situ during positive and negative selection in the thymus. Nature. (1994) 372:100–3. doi: 10.1038/372100a0
23. Lind EF, Prockop SE, Porritt HE, Petrie HT. Mapping precursor movement through the postnatal thymus reveals specific microenvironments supporting defined stages of early lymphoid development. J Exp Med. (2001) 194:127–34. doi: 10.1084/jem.194.2.127
24. Raulet DH, Garman RD, Saito H, Tonegawa S. Developmental regulation of T-cell receptor gene expression. Nature. (1985) 314:103–7. doi: 10.1038/314103a0
25. von Boehmer H, Fehling HJ. Structure and function of the pre-T cell receptor. Annu Rev Immunol. (1997) 15:433–52. doi: 10.1146/annurev.immunol.15.1.433
26. Yamasaki S, Saito T. Molecular basis for pre-TCR-mediated autonomous signaling. Trends Immunol. (2007) 28:39–43. doi: 10.1016/j.it.2006.11.006
27. Goldrath AW, Bevan MJ. Selecting and maintaining a diverse T-cell repertoire. Nature. (1999) 402:255–62. doi: 10.1038/46218
28. Palmer E. Negative selection–clearing out the bad apples from the T-cell repertoire. Nat Rev Immunol. (2003) 3:383–91. doi: 10.1038/nri1085
29. Laufer TM, DeKoning J, Markowitz JS, Lo D, Glimcher LH. Unopposed positive selection and autoreactivity in mice expressing class II MHC only on thymic cortex. Nature. (1996) 383:81–5. doi: 10.1038/383081a0
30. Murata S, Sasaki K, Kishimoto T, Niwa S, Hayashi H, Takahama Y, et al. Regulation of CD8+ T cell development by thymus-specific proteasomes. Science. (2007) 316:1349–53. doi: 10.1126/science.1141915
31. Ripen AM, Nitta T, Murata S, Tanaka K, Takahama Y. Ontogeny of thymic cortical epithelial cells expressing the thymoproteasome subunit β5t. Eur J Immunol. (2011) 41:1278–87. doi: 10.1002/eji.201041375
32. Nitta T, Murata S, Sasaki K, Fujii H, Ripen AM, Ishimaru N, et al. Thymoproteasome shapes immunocompetent repertoire of CD8+ T cells. Immunity. (2010) 32:29–40. doi: 10.1016/j.immuni.2009.10.009
33. Gommeaux J, Grégoire C, Nguessan P, Richelme M, Malissen M, Guerder S, et al. Thymus-specific serine protease regulates positive selection of a subset of CD4+ thymocytes. Eur J Immunol. (2009) 39:956–64. doi: 10.1002/eji.200839175
34. Viret C, Lamare C, Guiraud M, Fazilleau N, Bour A, Malissen B, et al. Thymus-specific serine protease contributes to the diversification of the functional endogenous CD4 T cell receptor repertoire. J Exp Med. (2011) 208:3–11. doi: 10.1084/jem.20100027
35. Mathis D, Benoist C. Aire. Annu Rev Immunol. (2009) 27:287–312. doi: 10.1146/annurev.immunol.25.022106.141532
36. Hubert FX, Kinkel SA, Davey GM, Phipson B, Mueller SN, Liston A, et al. Aire regulates the transfer of antigen from mTECs to dendritic cells for induction of thymic tolerance. Blood. (2011) 118:2462–72. doi: 10.1182/blood-2010-06-286393
37. Schorpp M, Hoffmann M, Dear TN, Boehm T. Characterization of mouse and human nude genes. Immunogenetics. (1997) 46:509–15. doi: 10.1007/s002510050312
38. Schlake T. The nude gene and the skin. Exp Dermatol. (2001) 10:293–304. doi: 10.1034/j.1600-0625.2001.100501.x
39. Kaufmann E, Knochel W. Five years on the wings of fork head. Mech Dev. (1996) 57:3–20. doi: 10.1016/0925-4773(96)00539-4
40. Ye H, Kelly TF, Samadani U, Lim L, Rubio S, Overdier DG, et al. Hepatocyte nuclear factor 3/fork head homolog 11 is expressed in proliferating epithelial and mesenchymal cells of embryonic and adult tissues. Mol Cell Biol. (1997) 17:1626–41. doi: 10.1128/MCB.17.3.1626
41. Brissette JL, Li J, Kamimura J, Lee D, Dotto GP. The product of the mouse nude locus, whn, regulates the balance between epithelial cell growth and differentiation. Genes Dev. (1996) 10:2212–21. doi: 10.1101/gad.10.17.2212
42. Bleul CC, Corbeaux T, Reuter A, Fisch P, Schulte Monting J, Boehm T. Formation of a functional thymus initiated by a postnatal epithelial progenitor cell. Nature. (2006) 441:992–6. doi: 10.1038/nature04850
43. Cheng L, Guo J, Sun L, Fu J, Barnes PF, Metzger D, et al. Postnatal tissue-specific disruption of transcription factor FoxN1 triggers acute thymic atrophy. J Biol Chem. (2010) 285:5836–47. doi: 10.1074/jbc.M109.072124
44. Rode I, Martins VC, Küblbeck G, Maltry N, Tessmer C, Rodewald HR. Foxn1 protein expression in the developing, aging, and regenerating thymus. J Immunol. (2015) 195:5678–87. doi: 10.4049/jimmunol.1502010
45. Zuklys S, Handel A, Zhanybekova S, Govani F, Keller M, Maio S, et al. Foxn1 regulates key targets genes essential for T cell development in postnatal thymic epithelial cells. Nat Immunol. (2016) 17:1206–15. doi: 10.1038/ni.3537
46. Tsukamoto N, Itoi M, Nishikawa M, Amagai T. Lack of Delta like 1 and 4 expressions in nude thymus anlages. Cell Immunol. (2005) 234:77–80. doi: 10.1016/j.cellimm.2005.06.009
47. Hozumi K, Mailhos C, Negishi N, Hirano K, Yahata T, Ando K, et al. Delta-like 4 is indispensable in thymic environment specific for T cell development. J Exp Med. (2008) 205:2507–13. doi: 10.1084/jem.20080134
48. Mohtashami M, Zúñiga-Pflücker JC. Cutting edge: three-dimensional architecture of the thymus is required to maintain delta-like expression necessary for inducing T cell development. J Immunol. (2006) 176:730–4. doi: 10.4049/jimmunol.176.2.730
49. Zuniga-Pflucker JC. T-cell development made simple. Nat Immunol. (2004) 4:67–72. doi: 10.1038/nri1257
50. Muñoz JJ, Tobajas E, Juara S, Montero S, Zapata AG. FoxN1 mediates thymic cortex-medulla differentiation through modifying a developmental pattern based on epithelial tubulogenesis. Histochem Cell Biol. (2019) 152:397–413. doi: 10.1007/s00418-019-01818-z
51. Uddin MM, Ohigashi I, Motosugi R, Nakayama T, Sakata M, Hamazaki J, et al. Foxn1-b5t transcriptional axis controls CD8+ T-cell production in the thymus. Nat Commun. (2017) 8:14419. doi: 10.1038/ncomms14419
52. Larsen BM, Cowan JE, Wang Y, Tanaka Y, Zhao Y, Voisin B, et al. Identification of an intronic regulatory element necessary for tissue-specific expression of foxn1 in thymic epithelial cells. J Immunol. (2019) 203:686–95. doi: 10.4049/jimmunol.1801540
53. Balciunaite G, Keller MP, Balciunaite E, Piali L, Zuklys S, Mathieu YD, et al. Wnt glycoproteins regulate the expression of FoxN1, the gene defective in nude mice. Nat immunol. (2002) 3:1102–8. doi: 10.1038/ni850
54. Wertheimer T, Velardi E, Tsai J, Cooper K, Xiao S, Kloss CC, et al. Production of BMP4 by endothelial cells is crucial for endogenous thymic regeneration. Sci Immunol. (2018) 3:eaal2736. doi: 10.1126/sciimmunol.aal2736
55. Lee D, Prowse DM, Brissette JL. Association between mouse nude gene expression and the initiation of epithelial terminal differentiation. Dev Biol. (1999) 208:362–74. doi: 10.1006/dbio.1999.9221
56. Xiong Y, Harmon CS. 1beta is differentially expressed by human dermal papilla cells in response to PKC activation and is apotent inhibitor of human hair follicle growth in organ culture. J Interferon Cytokine Res. (1997) 17:151–7. doi: 10.1089/jir.1997.17.151
57. Takahashi T, Kamimura A, Shirai A, Yokoo Y. Several selective protein kinase C inhibitors including procyanidins promote hair growth. Skin Pharmacol Appl Skin Phisiol. (2000) 13:133–42. doi: 10.1159/000029918
58. Li J, Baxter RM, Weiner L, Goetinck PF, Calautti E, Brissette JL. Foxn1 promotes keratinocyte differentiation by regulating the activity of protein kinase C. Differentiation. (2007) 75:694–701. doi: 10.1111/j.1432-0436.2007.00176.x
59. Pignata C, Fiore M, Guzzetta V, Castaldo A, Sebastio G, Porta F, et al. Congenital alopecia and nail dystrophy associated with severe functional T-cell immunodeficiency in two sibs. Am J Med Genet. (1996) 65:167–70. doi: 10.1002/(SICI)1096-8628(19961016)65:2<167::AID-AJMG17>3.0.CO
60. Bosticardo M, Yamazaki Y, Cowan J, Giardino G, Corsino C, Scalia G, et al. Heterozygous FOXN1 variants cause low trecs and severe t cell lymphopenia, revealing a crucial role of foxn1 in supporting early thymopoiesis. Am J Hum Genet. (2019) 105:549–61. doi: 10.1016/j.ajhg.2019.07.014
61. Albar R, Mahdi M, Alkeraithe F, Almufarriji KN. Epstein-Barr virus associated with high-grade B-cell lymphoma in nude severe combined immunodeficiency. BMJ Case Rep. (2019) 12:e227715. doi: 10.1136/bcr-2018-227715
62. Markert ML, Marques J, Neven B, Devlin B, McCarthy E, Chinn I, et al. First use of thymus transplantation therapy for Foxn1 deficiency (nude/SCID): a report of two cases. Blood. (2011) 117:688–96. doi: 10.1182/blood-2010-06-292490
63. Chou J, Massaad MJ, Wakim RH, Bainter W, Dbaibo G, Geha R. A novel mutation in FOXN1 resultingin SCID: A case report and literature review. Clin Immunol. (2014) 155:30–2. doi: 10.1016/j.clim.2014.08.005
64. Radha Rama Devi A, Panday NN, Naushad SM. FOXN1 Italian founder mutation in indian family: implications in prenatal diagnosis. Gene. (2017) 627:222–5. doi: 10.1016/j.gene.2017.06.033
65. Adriani M, Martinez-Mir A, Fusco F, Busiello R, Frank J, Telese S, et al. Ancestral founder mutation of the nude (FOXN1) gene in congenital severe combined immunodeficiency associated with alopecia in southern Italy population. Ann Hum Genet. (2004) 68:265–8. doi: 10.1046/j.1529-8817.2004.00091.x
66. Auricchio L, Adriani M, Frank J, Busiello R, Christiano A, Pignata C. Nail distrophy associated with a heterozygous mutation of the Nude/SCID human FOXN1 (WHN) gene. Arch Dermatol. (2005) 141:647–8. doi: 10.1001/archderm.141.5.647
67. McGaughran JM, Oates A, Donnai D, Read AP, Tassabehji M. Mutations in PAX1 may be associated with Klippel-Feil syndrome. Eur J Hum Genet. (2003) 11:468–474. doi: 10.1038/sj.ejhg.5200987
68. Adham IM, Gille M, Gamel AJ, Reis A, Dressel R, Steding G, et al. The scoliosis (sco) mouse: a new allele of Pax1. Cytogenet Genome Res. (2005) 111:16–26. doi: 10.1159/000085665
69. Smith CA, Tuan RS. Human PAX gene expression and development of the vertebral column. Clin Orthop Relat Res. (1994) 302:241–50. doi: 10.1097/00003086-199405000-00038
70. Pohl E, Aykut A, Beleggia F, Karaca E, Durmaz B, Keupp K, et al. A hypofunctional PAX1 mutation causes autosomal recessively inherited otofaciocervical syndrome. Hum Genet. (2013) 132:1311–20. doi: 10.1007/s00439-013-1337-9
71. Paganini I, Sestini R, Capone GL, Putignano AL, Contini E, Giotti I, et al. A novel PAX1 null homozygous mutation in autosomal recessive otofaciocervical syndrome associated with severe combined immunodeficiency. Clin Genet. (2017) 92:664–8. doi: 10.1111/cge.13085
72. Wallin J, Eibel H, Neubuser A, Wilting J, Koseki H, Balling R. Pax1 is expressed during development of the thymus epithelium and is required for normal T-cell maturation. Development. (1996) 122:23–30.
73. Su DM, Manley NR. Hoxa3 and pax1 transcription factors regulate the ability of fetal thymic epithelial cells to promote thymocyte development. J Immunol. (2000) 164:5753–60. doi: 10.4049/jimmunol.164.11.5753
74. Yamazaki Y, Urrutia R, Franco LM, Giliani S, Zhang K, Alazami AM, et al. PAX1 is essential for development and function of the human thymus. Sci Immunol. (2020) 5:eaax1036. doi: 10.1126/sciimmunol.aax1036
75. Peters H, Wilm B, Sakai N, Imai K, Maas R, Balling R. Pax1 and Pax9 synergistically regulate vertebral column development. Development. (1999) 126:5399–408.
76. Fara M, Chlupackova V, Hrivnakova J. Familial oto-facio-cervical dysmorphia. Acta Chir Orthop Traumatol Cech. (1967) 34:511–20.
77. Patil SJ, Das Bhowmik A, Bhat V, Satidevi Vineeth V, Vasudevamurthy R, Dalal A. Autosomal recessive otofaciocervical syndrome type 2 with novel homozygous small insertion in PAX1 gene. Am J Med Genet A. (2018) 176:1200–16. doi: 10.1002/ajmg.a.38659
78. Giardino G, Radwan N, Koletsi P, Morrogh DM, Adams S, Ip W, et al. Clinical and immunological features in a cohort of patients with partial DiGeorge syndrome followed at a single center. Blood. (2019) 133:2586–96. doi: 10.1182/blood.2018885244
79. Davies EG. Immunodeficiency in digeorg syndrome and option for treating cases with completeathymia. Front Immunol. (2013) 4:322. doi: 10.3389/fimmu.2013.00322
80. Zweier C, Sticht H, Aydin-Yaylagul I, Campbell CE, Rauch A. Human TBX1 missense mutations cause gain of function resulting in the same phenotype as 22q11.2 deletions. Am J Hum Genet. (2007) 80:510–7. doi: 10.1086/511993
81. Xu H, Morishima M, Wylie JN, Schwartz RJ, Bruneau BG, Lindsay EA, et al. Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development. (2004) 131:3217–27. doi: 10.1242/dev.01174
82. Fulcoli FG, Huynh T, Scambler PJ, Baldini A. Tbx1 regulates the BMP-Smad1 pathway in a transcription independent manner. PLoS ONE. (2009) 4:e6049. doi: 10.1371/journal.pone.0006049
83. Lindsay EA. Chromosomal microdeletions: dissecting del22q11 syndrome. Nat Rev Genet. (2001) 2:858–68. doi: 10.1038/35098574
84. Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. (2001) 27:286–91. doi: 10.1038/88233
85. Chieffo C, Garvey N, Gong W, Roe B, Zhang G, Silver L, et al. Isolation and characterization of a gene from the digeorge chromosomal region homologous to the mouse Tbx1 gene. Genomics. (1997) 43:267–77. doi: 10.1006/geno.1997.4829
86. Reeh KA, Cardenas KT, Bain VE, Liu Z, Laurent M, Manley NR, et al. Ectopic TBX1 suppresses thymic epithelial cell differentiation and proliferation during thymus organogenesis. Development. (2014) 141:2950–8. doi: 10.1242/dev.111641
87. Bhalla P, Wysocki CA, van Oers NSC. Molecular insights into the causes of human thymic hypoplasia with animal models. Front Immunol. (2020) 11:830. doi: 10.3389/fimmu.2020.00830
88. Shinohara T, Honjo T. Studies in vitro on the mechanism of the epithelial/mesenchymal interaction in the early fetal thymus. Eur J Immunol. (1997) 27:522–9. doi: 10.1002/eji.1830270225
89. Revest JM, Suniara RK, Kerr K, Owen JJ, Dickson C. Development of the thymus requires signaling through the fibroblast growth factor receptor R2-IIIb. J Immunol. (2001) 167:1954–61. doi: 10.4049/jimmunol.167.4.1954
90. Li Y, Gordon J, Manley NR, Litingtung Y, Chiang C. Bmp4 is required for tracheal formation: a novel mouse model for tracheal agenesis. Dev Biol. (2008) 322:145–55. doi: 10.1016/j.ydbio.2008.07.021
91. Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. (2003) 362:1366–73. doi: 10.1016/S0140-6736(03)14632-6
92. Scambler PJ. 22q11 deletion syndrome: a role for TBX1 in pharyngeal and cardiovascular development. Pediatr Cardiol. (2010) 31:378–90. doi: 10.1007/s00246-009-9613-0
93. Wilson TA, Blethen SL, Vallone A, Alenick DS, Nolan P, Katz A, et al. DiGeorge anomaly with renal agenesis in infants of mothers with diabetes. Am J Med Genet. (1993) 47:1078–82. doi: 10.1002/ajmg.1320470729
94. Vermot J, Niederreither K, Garnier JM, Chambon P, Dollé P. Decreased embryonic retinoic acid synthesis results in a DiGeorge syndrome phenotype in newborn mice. Proc Natl Acad Sci USA. (2003) 100:1763–8. doi: 10.1073/pnas.0437920100
95. Roberts C, Ivins SM, James CT, Scambler PJ. Retinoic acid down-regulates Tbx1 expression in vivo and in vitro. Dev Dyn. (2005) 232:928–38. doi: 10.1002/dvdy.20268
96. Ammann AJ, Wara DW, Cowan MJ, Barrett DJ, Stiehm ER. The digeorge syndrome and the fetal alcohol syndrome. Am J Dis Child. (1982) 136:906–8. doi: 10.1001/archpedi.1982.03970460036008
97. Cavdar AO. DiGeorge's syndrome and fetal alcohol syndrome. Am J Dis Child. (1983) 137:806–7. doi: 10.1001/archpedi.1983.02140340086027
98. Sulik KK, Johnston MC, Daft PA, Russell WE, Dehart DB. Fetal alcohol syndrome and Digeorge anomaly: critical ethanol exposure periods for craniofacial malformations as illustrated in an animal model. Am J Med Genet Suppl. (1986) 2:97–112. doi: 10.1002/ajmg.1320250614
99. Novak RW, Robinson HB. Coincident digeorge anomaly and renal agenesis and its relation to maternal diabetes. Am J Med Genet. (1994) 50:311–2. doi: 10.1002/ajmg.1320500402
100. Gosseye S, Golaire MC, Verellen G, Van Lierde M, Claus D. Association of bilateral renal agenesis and Di george syndrome in an infant of a diabetic mother. Helv Paediatr Acta. (1982) 37:471–4.
101. Digilio MC, Marino B, Formigari R, Giannotti A. Maternal diabetes causing DiGeorge anomaly and renal agenesis. Am J Med Genet. (1995) 55:513–4. doi: 10.1002/ajmg.1320550427
102. Cirillo E, Giardino G, Gallo V, Galasso G, Romano R, D'Assante R, et al. DiGeorge-like syndrome in a child with a 3p12.3 deletion involving MIR4273 gene born to a mother with gestational diabetes mellitus. Am J Med Genet A. (2017) 7:1913–8. doi: 10.1002/ajmg.a.38242
103. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. (2009) 136:215–33. doi: 10.1016/j.cell.2009.01.002
104. Takahashi H, Ikeda T. Trascripts for two members of the trasforming growth factor-beta superfamily BMP-3 and BMP-7 are expressed in developing rat embryos. Dev Dyn. (1996) 207:439–49. doi: 10.1002/(SICI)1097-0177(199612)207:4<439::AID-AJA8>3.0.CO
105. de la Morena MT, Eitson JL, Dozmorov IM, Belkaya S, Hoover AR, Anguiano E, et al. Signature MicroRNA expression patterns identified in humans with 22q11.2 deletion DiGeorge syndrome. Clin Immunol. (2013) 147:11–22. doi: 10.1016/j.clim.2013.01.011
106. Belver L, de Yébenes VG, Ramiro AR. MicroRNAs prevent the generation of autoreactive antibodies. Immunity. (2010) 33:713–22. doi: 10.1016/j.immuni.2010.11.010
107. O'Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol. (2010) 10:111–22. doi: 10.1038/nri2708
108. Sellier C, Hwang VJ, Dandekar R, Durbin-Johnson B, Charlet-Berguerand N, Ander BP, et al. Decreased DGCR8 expression and miRNA dysregulation in individuals with 22q11.2 deletion syndrome. PLoS ONE. (2014) 9:e103884. doi: 10.1371/journal.pone.0103884
109. Giacomelli M, Kumar R, Soresina A, Tamassia N, Lorenzini T, Moratto D, et al. Reduction of CRKL expression in patients with partial DiGeorge syndrome is associated with impairment of T-cell functions. J Allergy Clin Immunol. (2016) 138:229–40.e223. doi: 10.1016/j.jaci.2015.10.051
110. Bassett AS, McDonald-McGinn DM, Devriendt K, Digilio MC, Goldenberg P, Habel A, et al. Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediatr. (2011) 159:332–9.e331. doi: 10.1016/j.jpeds.2011.02.039
111. Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. (1997) 34:798–804. doi: 10.1136/jmg.34.10.798
112. Jyonouchi S, McDonald-McGinn DM, Bale S, Zackai EH, Sullivan KE. CHARGE (coloboma, heart defect, atresia choanae, retarded growth and development, genital hypoplasia, ear anomalies/deafness) syndrome and chromosome 22q11.2 deletion syndrome: a comparison of immunologic and nonimmunologic phenotypic features. Pediatrics. (2009) 123:e871–7. doi: 10.1542/peds.2008-3400
113. Cancrini C, Puliafito P, Digilio MC, Soresina A, Martino S, Rondelli R, et al. Clinical features and follow-up in patients with 22q11.2 deletion syndrome. J Pediatr. (2014) 164:1475–80.e1472. doi: 10.1016/j.jpeds.2014.01.056
114. Sullivan KE, McDonald-McGinn D, Driscoll DA, Emanuel BS, Zackai EH, Jawad AF. Longitudinal analysis of lymphocyte function and numbers in the first year of life in chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Clin Diagn Lab Immunol. (1999) 6:906–11. doi: 10.1128/CDLI.6.6.906-911.1999
115. Mehr S, Hsu P, Campbell D. Immunodeficiency in CHARGE syndrome. Am J Med Genet C Semin Med Genet. (2017) 175:516–23. doi: 10.1002/ajmg.c.31594
116. Jawad AF, McDonald-McGinn DM, Zackai E, Sullivan KE. Immunologic features of chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). J Pediatr. (2001) 139:715–23. doi: 10.1067/mpd.2001.118534
117. Marcovecchio GE, Bortolomai I, Ferrua F, Fontana E, Imberti L, Conforti E, et al. Thymic epithelium abnormalities in digeorge and down syndrome patients contribute to dysregulation in t cell development. Front Immunol. (2019) 10:447. doi: 10.3389/fimmu.2019.00447
118. Montin D, Marolda A, Licciardi F, Robasto F, Di Cesare S, Ricotti E, et al. Immunophenotype anomalies predict the development of autoimmune cytopenia in 22q11.2 deletion syndrome. J Allergy Clin Immunol Pract. (2019) 7:2369–76. doi: 10.1016/j.jaip.2019.03.014
119. McLean-Tooke A, Spickett GP, Gennery AR. Immunodeficiency and autoimmunity in 22q11.2 deletion syndrome. Scand J Immunol. (2007) 66:1–7. doi: 10.1111/j.1365-3083.2007.01949.x
120. Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. (2004) 36:955–7. doi: 10.1038/ng1407
121. Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, et al. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature. (2010) 463:958–62. doi: 10.1038/nature08733
122. Pagon RA, Graham JM Jr, Zonana J, Yong SL. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr. (1981) 99:223–7. doi: 10.1016/S0022-3476(81)80454-4
123. Meisner JK, Martin DM. Congenital heart defects in CHARGE: The molecular role of CHD7 and effects on cardiac phenotype and clinical outcomes. Am J Med Genet C Semin Med Genet. (2020) 184:81–9. doi: 10.1002/ajmg.c.31761
124. Liu ZZ, Wang ZL, Choi TI, Huang WT, Wang HT, Han YY, et al. Chd7 is critical for early T-cell development and thymus organogenesis in zebrafish. Am J Pathol. (2018) 188:1043–58. doi: 10.1016/j.ajpath.2017.12.005
125. Inoue H, Takada H, Kusuda T, Goto T, Ochiai M, Kinjo T, et al. Successful cord blood transplantation for a CHARGE syndrome with CHD7 mutation showing DiGeorge sequence including hypoparathyroidism. Eur J Pediatr. (2010) 169:839–44. doi: 10.1007/s00431-009-1126-6
126. Writzl K, Cale CM, Pierce CM, Wilson LC, Hennekam RC. Immunological abnormalities in CHARGE syndrome. Eur J Med Genet. (2007) 50:338–45. doi: 10.1016/j.ejmg.2007.05.002
127. Wong MT, Lambeck AJ, van der Burg M, la Bastide-van Gemert S, Hogendorf LA, van Ravenswaaij-Arts CM, et al. Immune dysfunction in children with charge syndrome: a cross-sectional study. PLoS ONE. (2015) 10:e0142350. doi: 10.1371/journal.pone.0142350
128. Hsu P, Ma A, Barnes EH, Wilson M, Hoefsloot LH, Rinne T, et al. The immune phenotype of patients with charge syndrome. J Allergy Clin Immunol Pract. (2016) 4:96–103.e102. doi: 10.1016/j.jaip.2015.09.004
129. Wong MT, Schölvinck EH, Lambeck AJ, van Ravenswaaij-Arts CM. CHARGE syndrome: a review of the immunological aspects. Eur J Hum Genet. (2015) 23:1451–9. doi: 10.1038/ejhg.2015.7
130. Gennery AR, Slatter MA, Rice J, Hoefsloot LH, Barge D, McLean-Tooke A, et al. Mutations in CHD7 in patients with CHARGE syndrome cause T-B + natural killer cell + severe combined immune deficiency and may cause Omenn-like syndrome. Clin Exp Immunol. (2008) 153:75–80. doi: 10.1111/j.1365-2249.2008.03681.x
131. Bernstock JD, Totten AH, Elkahloun AG, Johnson KR, Hurst AC, Goldman F, et al. Recurrent microdeletions at chromosome 2p11.2 are associated with thymic hypoplasia and features resembling DiGeorge syndrome. J Allergy Clin Immunol. (2020) 145:358–67. doi: 10.1016/j.jaci.2019.09.020
132. Birol O, Ohyama T, Edlund RK, Drakou K, Georgiades P, Groves AK. The mouse Foxi3 transcription factor is necessary for the development of posterior placodes. Dev Biol. (2016) 409:139–51. doi: 10.1016/j.ydbio.2015.09.022
133. Khatri SB, Groves AK. Expression of the Foxi2 and Foxi3 transcription factors during development of chicken sensory placodes and pharyngeal arches. Gene Expr Patterns. (2013) 13:38–42. doi: 10.1016/j.gep.2012.10.001
134. Tassano E, Jagannathan V, Drögemüller C, Leoni M, Hytönen MK, Severino M, et al. Congenital aural atresia associated with agenesis of internal carotid artery in a girl with a FOXI3 deletion. Am J Med Genet A. (2015) 167a:537–44. doi: 10.1002/ajmg.a.36895
135. Shirokova V, Biggs LC, Jussila M, Ohyama T, Groves AK, Mikkola ML. Foxi3 deficiency compromises hair follicle stem cell specification and activation. Stem Cells. (2016) 34:1896–908. doi: 10.1002/stem.2363
136. Hasten E, Morrow BE. Tbx1 and Foxi3 genetically interact in the pharyngeal pouch endoderm in a mouse model for 22q11.2 deletion syndrome. PLoS Genet. (2019) 15:e1008301. doi: 10.1371/journal.pgen.1008301
137. McDonald-McGinn DM, Sullivan KE. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine. (2011) 90:1–18. doi: 10.1097/MD.0b013e3182060469
138. Markert ML, Kostyu DD, Ward FE, McLaughlin TM, Watson TJ, Buckley RH, et al. Successful formation of a chimeric human thymus allograft following transplantation of cultured postnatal human thymus. J Immunol. (1997) 158:998–1005.
139. Davies E, Cheung M, Gilmour K, Maimaris J, Curry J, Furmanski A, et al. Thymus transplantation for complete digeorge syndrome: European experience. J Allergy Clin Immunol. (2017) 6749:30576–86. doi: 10.1016/j.jaci.2017.03.020
140. Markert ML, Devlin BH, Alexieff MJ, Li J, McCarthy EA, Gupton SE, et al. Review of 54 patients with complete Digeorge anomaly enrolled in protocols for thymus transplantation: outcome of 44 consecutive transplants. Blood. (2007) 109:4539–47. doi: 10.1182/blood-2006-10-048652
141. Markert ML, Devlin BH, Chinn IK, McCarthy EA, Li YJ. Factors affecting success of thymus transplantation for complete DiGeorge anomaly. Am J Transplant. (2008) 8:1729–36. doi: 10.1111/j.1600-6143.2008.02301.x
142. Markert ML, Devlin BH, McCarthy EA. Thymus transplantation. Clin Immunol. (2010) 135:236–46. doi: 10.1016/j.clim.2010.02.007
143. Albuquerque AS, Marques JG, Silva SL, Ligeiro D, Devlin BH, Dutrieux J, et al. Human FOXN1-deficiency is associated with αβ double-negative and FoxP3+ T-cell expansions that are distinctly modulated upon thymic transplantation. PLoS ONE. (2012) 7:e37042. doi: 10.1371/journal.pone.0037042
144. Levy E, Neven B, Entz-Werle N, Cribier B, Lipsker D. [Post-thymus transplant vitiligo in a child with Foxn1 deficiency]. Ann Dermatol Venereol. (2012) 139:468–71. doi: 10.1016/j.annder.2012.03.014
145. Janda A, Sedlacek P, Hönig M, Friedrich W, Champagne M, Matsumoto T, et al. Multicenter survey on the outcome of transplantation of hematopoietic cells in patients with the complete form of digeorge anomaly. Blood. (2010) 116:2229–36. doi: 10.1182/blood-2010-03-275966
146. Hoover-Fong J, Savage WJ, Lisi E, Winkelstein J, Thomas GH, Hoefsloot LH, et al. Congenital T cell deficiency in a patient with charge syndrome. J Pediatr. (2009) 154:140–2. doi: 10.1016/j.jpeds.2008.07.049
147. Janda A, Sedlacek P, Mejstrikova E, Zdrahalova K, Hrusak O, Kalina T, et al. Unrelated partially matched lymphocyte infusions in a patient with complete digeorge/charge syndrome. Pediatr Transplant. (2007) 11:441–7. doi: 10.1111/j.1399-3046.2007.00702.x
148. Land MH, Garcia-Lloret MI, Borzy MS, Rao PN, Aziz N, McGhee SA, et al. Long-term results of bone marrow transplantation in complete DiGeorge syndrome. J Allergy Clin Immunol. (2007) 120:908–15. doi: 10.1016/j.jaci.2007.08.048
149. Firtina S, Cipe F, Ng YY, Kiykim A, Ng OH, Sudutan T, et al. A novel FOXN1 variant is identified in two siblings with nude severe combined immunodeficiency. J Clin Immunol. (2019) 39:144–7. doi: 10.1007/s10875-019-00615-6
Keywords: Thymus, FOXN1 gene, PAX1 gene, Pax 1/9, CHARGE, CHD7 gene, TBX1 gene, DiGeorge anomaly
Citation: Giardino G, Borzacchiello C, De Luca M, Romano R, Prencipe R, Cirillo E and Pignata C (2020) T-Cell Immunodeficiencies With Congenital Alterations of Thymic Development: Genes Implicated and Differential Immunological and Clinical Features. Front. Immunol. 11:1837. doi: 10.3389/fimmu.2020.01837
Received: 14 April 2020; Accepted: 08 July 2020;
Published: 14 August 2020.
Edited by:
Nicolai Stanislas Van Oers, University of Texas Southwestern Medical Center, United StatesReviewed by:
Jennifer Elizabeth Cowan, National Institutes of Health (NIH), United StatesNaomi Taylor, National Institutes of Health (NIH), United States
Copyright © 2020 Giardino, Borzacchiello, De Luca, Romano, Prencipe, Cirillo and Pignata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudio Pignata, cGlnbmF0YUB1bmluYS5pdA==
†These authors have contributed equally to this work