 Oluwabukola T. Gbotosho1,2
Oluwabukola T. Gbotosho1,2 Maria G. Kapetanaki2,3
Maria G. Kapetanaki2,3 Samit Ghosh1,4Flordeliza S. Villanueva5Solomon F. Ofori-Acquah1,2,4,6
Samit Ghosh1,4Flordeliza S. Villanueva5Solomon F. Ofori-Acquah1,2,4,6 Gregory J. Kato1,2*
Gregory J. Kato1,2*- 1Division of Hematology-Oncology, Department of Medicine, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
- 2Department of Medicine, Pittsburgh Heart, Lung, Blood, and Vascular Medicine Institute, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
- 3Division of Pulmonary, Allergy and Critical Care Medicine, Department of Medicine, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
- 4Department of Medicine, Center for Translational and International Hematology, School of Medicine, University of Pittsburgh, Pittsburgh, PA, United States
- 5Center for Ultrasound Molecular Imaging and Therapeutics, Heart and Vascular Institute, Pittsburgh Heart, Lung, Blood and Vascular Medicine Institute, University of Pittsburgh, Pittsburgh, PA, United States
- 6School of Biomedical and Allied Health Sciences, University of Ghana, Accra, Ghana
Emerging data indicate that free heme promotes inflammation in many different disease settings, including in sickle cell disease (SCD). Although free heme, proinflammatory cytokines, and cardiac hypertrophy are co-existing features of SCD, no mechanistic links between these features have been demonstrated. We now report significantly higher levels of IL-6 mRNA and protein in hearts of the Townes sickle cell disease (SS) mice (2.9-fold, p ≤ 0.05) than control mice expressing normal human hemoglobin (AA). We find that experimental administration of heme 50 μmoles/kg body weight induces IL-6 expression directly in vivo and induces gene expression markers of cardiac hypertrophy in SS mice. We administered heme intravenously and found that within three hours plasma IL-6 protein significantly increased in SS mice compared to AA mice (3248 ± 275 vs. 2384 ± 255 pg/ml, p ≤ 0.05). In the heart, heme induced a 15-fold increase in IL-6 transcript in SS mice heart compared to controls. Heme simultaneously induced other markers of cardiac stress and hypertrophy, including atrial natriuretic factor (Nppa; 14-fold, p ≤ 0.05) and beta myosin heavy chain (Myh7; 8-fold, p ≤ 0.05) in SS mice. Our experiments in Nrf2-deficient mice indicate that the cardiac IL-6 response to heme does not require Nrf2, the usual mediator of transcriptional response to heme for heme detoxification by heme oxygenase-1. These data are the first to show heme-induced IL-6 expression in vivo, suggesting that hemolysis may play a role in the elevated IL-6 and cardiac hypertrophy seen in patients and mice with SCD. Our results align with published evidence from rodents and humans without SCD that suggest a causal relationship between IL-6 and cardiac hypertrophy.
Introduction
Sickle cell disease (SCD) is a complex hematological disorder that affects ~100,000 Americans and millions of people worldwide, especially in sub-Saharan Africa and India (1). Hemolysis and chronic inflammation are major components of the pathophysiology of SCD. Hemolysis is caused by erythrocyte injury due to secondary defects in erythrocyte fragility, deformability and increased endothelial adhesion resulting in release of hemoglobin and heme (2, 3). Chronic inflammation in SCD is partly due to leukocytosis with the abnormally high leukocytes and monocytes that secrete proinflammatory cytokines. In addition, products of hemolysis act as damage-associated molecular patterns (DAMPs) potentiating activation of many inflammatory mechanisms (4). Additionally, products of intravascular hemolysis such as free hemoglobin and arginase-1 impair nitric oxide bioavailability, endothelial function and organ function in SCD (2). Limited data indicate that heme can induce production of proinflammatory cytokines such as interleukin-6 (IL-6) by stimulating immune responses and inflammatory reactions (5). Hemolysis and inflammation are components of a wide spectrum of other clinical conditions including sepsis (6, 7), malaria (8, 9), and preeclampsia (10, 11). Common to patients with all these syndromes is an increased risk of cardiac dysfunction (12–15). Notable among proinflammatory cytokines elevated in SCD is IL-6. Serum levels of IL-6 are elevated both at steady state and during vaso occlusive crisis in both children and adults with SCD (16–20), concurrently with severe anemia and increased markers of hemolysis. Furthermore, IL-6 is associated with cardiomyopathies such as cardiac hypertrophy and fibrosis in experimental animals (21, 22) and in the general human population (23, 24). Importantly, cardiopulmonary complications are one of the leading causes of death in SCD (25, 26). This accounts for about 26% of deaths in adults with SCD (27), with left ventricular hypertrophy (LVH) found in over 60% of children and 37% in adults with SCD (28, 29). No prior publications have investigated any potential linkage between IL-6 and hemolysis in mice and patients with SCD, particularly in cardiac disease. In this study, we assess direct heme induction in vivo of IL-6 and genes relevant to cardiac hypertrophy in the heart of sickle cell mice. Our study shows that IL-6 is highly expressed in the circulation and in the heart of sickle cell mice at steady state. Furthermore, administration of extracellular heme further increased IL-6 and cardiac hypertrophy genes expression. To gain insight of the mechanism by which heme induces IL-6, we investigated the role of nuclear factor (erythroid derived 2)-like 2 (Nfe2L2 or Nrf2). Nrf2 is the master regulator of the cellular oxidative defense system and plays a significant role in the regulation of multiple heme-induced genes (30, 31).
Materials and Methods
Mouse Strains and Treatment
Male and female Townes' knocked-in transgenic sickle mouse (SS) and strain controls expressing normal human Hb (AA mice), C57BL/6J (Nrf2+/+) and Nrf2−/− mice were used. C57BL/6 mice were obtained from the Jackson Laboratory (stock #000664) while SS, AA and Nrf2−/− mice were obtained from a colony maintained by Dr. Solomon Ofori-Acquah's laboratory in our institution. Mouse genotypes were confirmed by PCR. Hemin [Fe(III)PPIX, Sigma-Aldrich, St. Louis, MO] was prepared as described elsewhere (32, 33). Freshly prepared hemin solution was protected from light and injected into 12–16 week old mice. A range of doses and times were tested and 3 h after injection produced consistent survival with no adverse effects on all strains of mice in this study. The mice were injected in the tail vein with a hemin dose of 50 μmoles/kg body weight for SS and AA mice, and 120 μmoles/kg body weight for Nrf2+/+ and Nrf2−/− mice. Higher doses were needed for Nrf2+/+ and Nrf2−/− mice to be able to neutralize the endogenous hemopexin and other heme-binding proteins and mimic the increase in circulating heme in chronic hemolysis. This allows comparable assessment of the transcriptional response. Control mice received sterile vehicle containing 0.25M NaOH adjusted to pH 7.5 with HCl used in preparation of hemin.
Plasma Analysis
Freshly collected blood samples were centrifuged at 1,200 × g for 15 min to separate blood plasma. Plasma IL-6 concentration was measured using the mouse IL-6 ELISA kit (Sigma-Aldrich) following the manufacturer's instructions.
Real-Time PCR
Whole organs were harvested from mice 3 h after hemin injection. Freshly isolated organs (300 mg) were snap-frozen and kept at −80°C until use. Organs were homogenized in Qiazol lysis reagent using the Next Advance Bullet Blender (Next Advance, Inc. Troy, NY). Clear lysates were obtained by centrifuging homogenized samples at 18,800 × g for 10 min. All tissue processing was carried out at 4°C. Total RNA was extracted from the tissue lysates using the miRNeasy Mini Kit (#217004, QIAGEN, Germantown, MD) and quantified using the Nanodrop 8000 microvolume spectrophotometer (ThermoFisher Scientific). Real-time PCR reactions were set-up in duplicates using 50 ng of RNA. Genes of interest were evaluated using the TaqMan® Gene expression assay (ThermoFisher) and the TaqMan® RNA-to-Ct™ 1-Step Kit (ThermoFisher) according to the manufacturer's instructions. Relative quantification was calculated with the standard ΔΔCt method; amplification signals from target gene transcripts were normalized to those from beta-glucuronidase (Gusb) transcripts. Relative fold induction was calculated by further normalization to gene transcripts from vehicle treated animals. Gusb gene expressions were similar across all mouse strains used and across all organs within a given mouse strain. Gusb gene expression in organs from control mice was similar to that from the corresponding organs from hemin-injected mice. We have previously published this in different organs from SS mice here (32) and in the heart of AA control mice in Supplementary Figure 1 in this study.
IL-6 Protein Quantification
Mice were perfused with phosphate-buffered saline under anesthesia. Harvested organs (300 mg) were homogenized in RIPA buffer using the Next Advance Bullet Blender (Next Advance, Inc. Troy, NY). Homogenized samples were centrifuged at 18,800 x g for 10 min to obtain clear lysates. All tissue processing was carried out at 4°C. Heart IL-6 concentration was measured using the mouse IL-6 ELISA kit (Sigma-Aldrich) following the manufacturer's instructions. Total protein was quantified in the lysates using the BCA assay kit (ThermoFisher Scientific, #23225).
Statistical Analysis
GraphPad Prism 7 software was used for all statistical analyses. Results are reported as mean ± SEM. Group means were compared using parametric tests, such as t-test (for 2 groups) and One-way ANOVA for more than two conditions. Statistical significance was set at p values of < 0.05.
Results
High Basal Expression and Heme-Induced Cardiac IL-6 in Sickle Cell Mice
We investigated the basal expression of IL-6 transcripts in the heart of Townes SS mice and AA control mice. IL-6 expression was 2.9-fold higher in the heart of Townes SS mice compared to AA controls (Figure 1A, p ≤ 0.05). Hmox1 expression was significantly elevated in the heart (2.1-fold) of untreated Townes SS mice compared to AA controls (Figure 1B, p ≤ 0.01). We tested the hypothesis that products of hemolysis, specifically heme, would promote IL-6 expression in the AA heart to mimic the SS steady state expression. Injection of heme as previously described (32, 34) increased cardiac IL-6 transcript expression to levels comparable to vehicle-treated SS mice (Figure 1C, p < 0.05). This suggests that heme release in SS mice may be the critical factor that stimulates high SS basal IL-6 expression. SS mice were even more responsive to heme injection, with cardiac IL-6 transcripts rising 15.4-fold higher in heme-treated SS mice compared to vehicle controls (Figure 1D, p ≤ 0.05) and by about 53% in SS mice compared to AA mice controls (Figure 1C, p ≤ 0.001). We confirmed these mRNA results with analysis of IL-6 protein, which documented a 34% increase in IL-6 protein in the heart of SS mice injected with heme compared to vehicle controls (Figure 2A, p ≤ 0.05). These data support a role of heme in cardiac IL-6 regulation at steady-state and during acute heme increase in sickle cell disease.
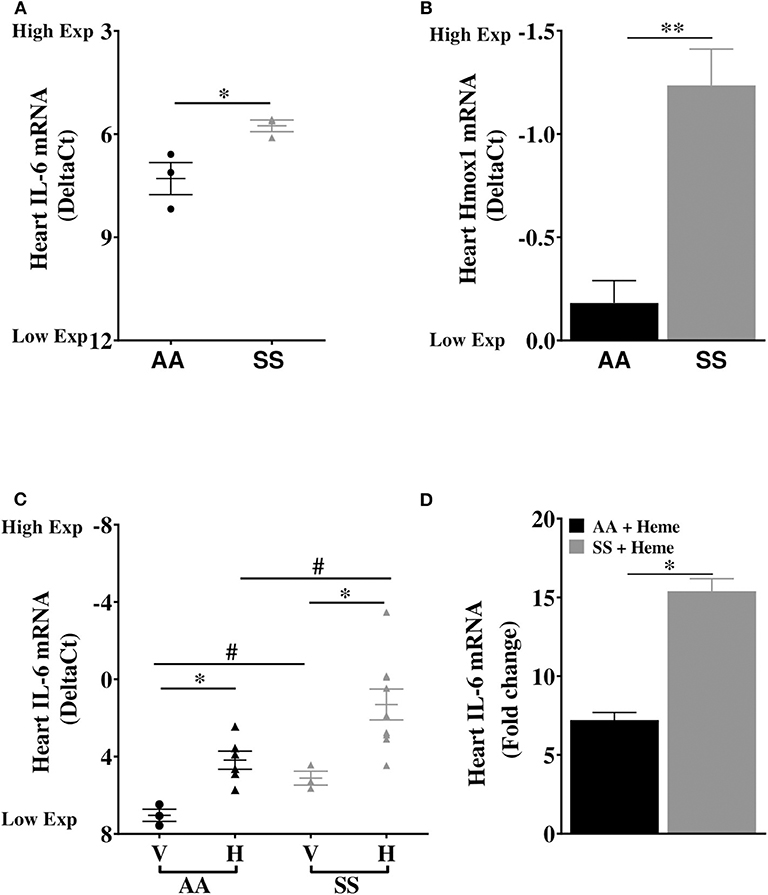
Figure 1. Cardiac expression of IL-6 and Hmox1 in SS mice. Heart mRNA expression of (A) IL-6 and (B) Hmox1 in naïve in 14 week old SS mice compared to age-matched AA mice (n = 3). The SS mice Hmox1 data was published here (32). (C) Heme induced cardiac IL-6 mRNA in SS and AA mice (n = 3–10). (D) Relative fold change in heme-induced cardiac IL-6 mRNA expression in heme treated AA and SS mice (n = 6–10). For DeltaCt, lowest value = highest expression and highest value = lowest expression. Target gene transcripts were normalized to Gusb for all mRNA expression levels. Gusb expression was similar in all mice strains used and in all of these organs in animals injected with either vehicle or hemin. For relative fold change, samples were further normalized to vehicle control gene transcripts. Unpaired Student's t-test or one-way ANOVA. Error bars indicate SEM. *p ≤ 0.05; **p ≤ 0.01. #p < 0.05 AA vs. SS. V, Vehicle and H, Heme.
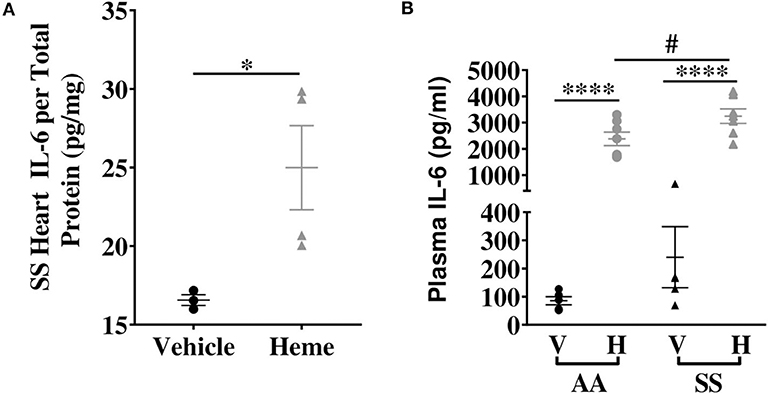
Figure 2. Heme induces IL-6 protein with a more pronounced effect in SS mice compared to AA controls. (A) Heme induced cardiac IL-6 protein in SS mice (n = 3–4). (B) Plasma IL-6 in SS and AA mice injected (IV) with 50 μmoles/kg body weight heme or vehicle (n = 5–7). Unpaired Student's t-test or one-way ANOVA. Error bars indicate SEM. *p < 0.05, ****p < 0.0001. #p < 0.05 AA vs. SS. V, Vehicle and H, Heme.
Increased Levels of Circulating IL-6 in Heme-Treated Sickle Mice
Elevated heme (3, 35) and IL-6 (16, 18) have been individually reported in the serum of SCD patients. We hypothesized that increase in extracellular heme rapidly upregulates IL-6 in the plasma of SS and AA mice. Heme treatment significantly increased plasma IL-6 protein levels about 25-fold in both AA and SS mice 3 h after heme injections. The heme-induced IL-6 level was significantly higher in SS mice than AA mice (3249 ± 276 vs. 2385 ± 256 pg/ml, p ≤ 0.05, Figure 2B). These data indicate a role for free heme in systemic regulation of IL-6.
Heme-Induced Cardiac IL-6 Expression Is Negatively Regulated via Nrf2 Pathway
Our recent studies confirmed that the Nrf2 pathway mediates heme induction of cardiac Hmox-1 expression in SS mice (32), human monocytes (36), and keratinocytes (37). This led us to investigate whether the same pathway regulates cardiac IL-6 expression and its response to heme. We find no significant differences in cardiac IL-6 mRNA expression in vehicle-treated Nrf2+/+ and Nrf2−/− mice (Figure 3A). Treatment with heme significantly augmented cardiac IL-6 mRNA levels in both strains (Figure 3A, p ≤ 0.001). Unexpectedly, IL-6 mRNA rose significantly higher in the hearts of the heme-treated Nrf2-deficient mice compared to the heme-treated Nrf2+/+ control mice. Confirming this mRNA finding, cardiac IL-6 protein was about 51% higher in heme-treated Nrf2−/− mice compared to heme-treated Nrf2+/+ mice (p < 0.01, Figure 3B). The result shows that Nrf2 is not required for heme induction of cardiac IL-6 expression.
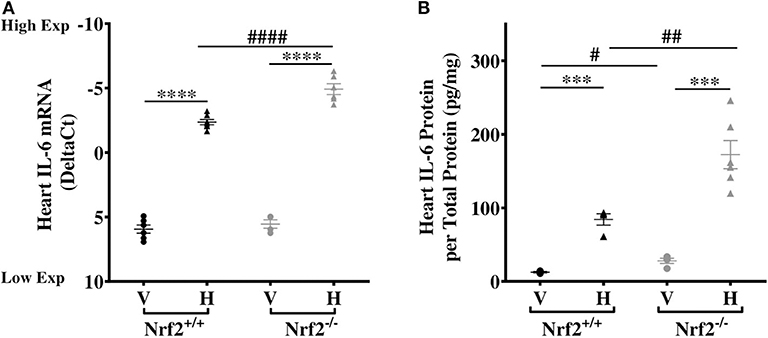
Figure 3. Nrf2 is not required for heme-induced cardiac IL-6. Heme-induced cardiac IL-6 in Nrf2+/+ and Nrf2−/− mice. (A) RNA (B) Protein. Nrf2+/+ and Nrf2−/− mice were injected with vehicle or heme (120 μmoles/Kg body weight). For DeltaCt, lowest value = highest expression and highest value = lowest expression. Target gene transcripts were normalized to Gusb for all mRNA expression level. Gusb expression was similar in all mice strains used and in all of these organs in animals injected with either vehicle or hemin. This dose was selected after standardization for producing consistent survival with no adverse effects on both strains of mice. One-way ANOVA. Error bars are SEM. ***p ≤ 0.001; ****p ≤ 0.0001, vehicle vs. heme within strain. #p ≤ 0.05 ##p ≤ 0.01 Nrf2+/+ vs. Nrf2−/− (n = 3–7; 14–16 weeks old). V, Vehicle and H, Heme.
Heme Upregulates Markers of Cardiac Hypertrophy in Sickle Mice
Our results above showed elevated basal expression of cardiac IL-6 in SS mice, which was further elevated by increase in extracellular heme. Elevated IL-6 is associated with higher risk of left ventricular dysfunction and progression to heart failure in humans (38), and hypertrophy in rodents (39). Furthermore, in SCD patients LV dysfunction was an independent risk factor for death (40, 41), while diastolic dysfunction and myocardial fibrosis were reported in sickle cell mouse model (42). Therefore, we hypothesized that heme might induce cardiac hypertrophy genes in sickle cell mice. We evaluated expression of atrial natriuretic factor (Nppa) and β-Myosin heavy chain 7 (Myh7), known to be associated with cardiac hypertrophy (43). Baseline expression of both Nppa and myh7 were similar in the hearts of AA and SS mice (Supplementary Figure 2). Heme treatment resulted in a 14.8-fold increase in Nppa transcripts and 8.1-fold increase in Myh7 transcripts in the heart of SS mice 3 h after injection of heme (Figures 4A–C), but not AA control mice (Figure 4C). The heart in SCD appears to be more sensitive to heme induction of these two cardiac hypertrophy genes.
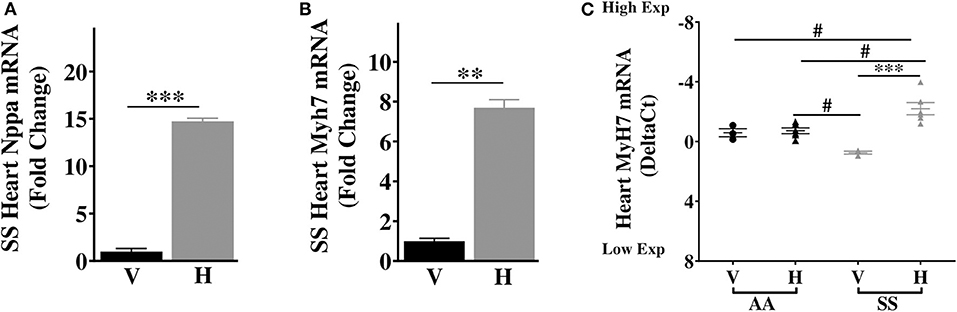
Figure 4. Heme induces high expression of transcripts of cardiac hypertrophy markers in SS mice. Heme induced the expression of (A) Nppa (B) Myh7 in the heart of SS mice 3 h after heme or vehicle injection (C) Myh7 in the heart of SS and AA mice 3 h after heme (50 μmoles/kg body weight) or vehicle injection. For DeltaCt, lowest value = highest expression and highest value = lowest expression. Target gene transcripts were normalized to Gusb for all mRNA expression level. Gusb expression was similar in all mice strains used and in all of these organs in animals injected with either vehicle or hemin. For relative fold change, samples were further normalized to vehicle control gene transcripts. Unpaired student's t-test and one-way ANOVA. Error bars indicate SEM. ***p ≤ 0.001; **p ≤ 0.001, vehicle vs. heme within strain. #p ≤ 0.05 AA vs. SS (n = 3–8; 14–16 weeks old). V, Vehicle and H, Heme.
Discussion
Intravascular hemolysis is an important modifier of outcome and pathogenesis of SCD (2). The plasma cell-free hemoglobin and heme are elevated at steady state in SCD and are associated with disease severity and end organ damage (2, 44). Cardiac-related complications represent a leading cause of death in SCD (26). In SCD patients, there is also a dysregulated expression of IL-6 and other inflammatory cytokines linked to vaso-occlusive crisis and other complications (18, 20). In this study, we report for the first time elevated basal cardiac IL-6 mRNA and protein levels in SS mice compared to AA controls. We also showed that experimental increase in circulating heme further elevates cardiac and plasma IL-6 expression in control mice and even more so in SS mice. Our result is consistent with earlier studies that reported elevated serum IL-6 in SCD patients (16, 18), but those studies did not investigate the heart. It is possible that cardiomyocytes and non-cardiomyocytes including fibroblasts and macrophages in the heart as well as cells in other organs may be contributing to the elevated plasma IL-6 after heme injection.
Hemopexin, the endogenous scavenger of free heme, is depleted from the serum of both human (45, 46) and mice (34, 47) with SCD, making them more susceptible to acute increases in heme concentration. This promotes the elevated circulating heme levels reported in human (35) and mice with SCD (34). Once the plasma heme scavenging system is saturated, circulating heme can generate reactive oxygen species, resulting in tissue injury. Our results indicate that heme also upregulates inflammatory cytokine IL-6. Our finding of heme induction of cardiac IL-6 complements a recent report of higher cardiac IL-6 transcripts in hemopexin deficient mice compared to wildtype control (48). The protective effect of hemopexin in heme injection experiments in sickle cell and hemopexin deficient mice has previously been published by our group (34, 49) and other authors (47, 50).
Our findings show that Nrf2 is dispensable for heme induction of IL-6 expression. Further investigation of this pathway was beyond the scope of this current investigation, with several possible mechanisms that might be involved in this heme response. Multiple regulatory elements in the promoter region of the IL-6 gene may contribute to its regulation in a cell type-specific manner (51). Nrf2 might act as a transcriptional repressor or it might regulate another transcriptional inhibitor of IL-6 expression (52). Additionally, reduced inducibility in Nrf2−/− mice of cardiac Hmox1, the principal enzyme in heme catabolism (32), could result in slower degradation of heme leading to prolonged heme-induced activation of IL-6 through an Nrf2-independent pathway. Chronic long-term signaling of IL-6 induces inflammation and promotes cardiac hypertrophy in other models (53). Both features are risk factors for morbidity and mortality in SCD (29, 40, 54), although their relationship to each other has not been investigated in SCD.
Our pilot analysis of other cardiac mRNAs shows a concurrent induction of transcripts of cardiac hypertrophy genes Nppa and MyH7 by heme in the heart of SS mice. Additional inflammatory cytokines such as PlGF contributes to cardiac hypertrophy through IL-6 signaling (55) and it is a predictor of increased left ventricular mass in non-hemolytic diseases such as chronic kidney disease (56). We recently published evidence of elevated basal cardiac expression of PlGF in SS mice with further inducibility by heme (32). These results support a hypothetical model that chronic hemolysis induces expression of both PlGF and IL-6, and this elevation of inflammatory cytokines might contribute to the development of LVH in SCD through a yet to be experimentally identified mechanism. The association of hemolysis, IL-6 induction and organ damage in SCD is supported by previous published research. A recent report showed an association between a polymorphism in the IL-6 gene and development of leg ulcer in SCD patients (57), while previous publications showed hemolysis as a risk factor for leg ulcer in these patients (2, 58). Taken together, these suggest that increased hemolysis and inflammatory cytokines including IL-6 may play an important role in organ injury and pathophysiology of SCD.
Our study has several limitations. We did not evaluate the hemopexin levels in plasma or the heart and we did not test whether hemopexin would be protective against heme induction of IL-6. We did not determine a specific cell type in the heart in which heme activates IL-6 expression, and this is a future goal. The mechanism of heme-induced IL-6 expression remains to be determined, although our present evidence unequivocally demonstrates that Nrf2 is not required. An alternative mechanism might involve the activation of the Toll-like receptor 4 (TLR4) induction of MyD88, activator protein-1 (AP-1) and nuclear factor–κB (NF-κB) pathways (59–61). The specific mechanisms by which hemolysis-induced IL-6 contributes to the development of cardiac hypertrophy in SCD warrants future investigation. Despite these limitations, the findings of this study are novel and set the stage for detailed mechanistic studies of heme induction of cardiac IL-6 in SCD. This may lead to the development of novel therapeutic targets for ameliorating or preventing heme-induced IL-6 and cardiac dysfunction in SCD.
In conclusion, we show for the first time direct induction of IL-6 by heme in the plasma and heart of SS mice, in a mechanism that does not require Nrf2. We also show for the first time that heme induces cardiac expression of genes associated with cardiac hypertrophy, a clinically significant complication found in SCD patients with especially severe chronic hemolysis. These new observations provide the basis for a previously unknown heme/IL-6 axis in the development of cardiac disease in patients with SCD. This new model provides potential therapeutic targets for intervention in the heme response and IL-6 pathways to prevent cardiac disease in SCD that merit additional investigation.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by University of Pittsburgh Institutional Animal Care and Use Committee (IACUC).
Author Contributions
OG and GK designed the research project, analyzed, interpreted data, and drafted the manuscript. OG performed the experiments. MK and SG assisted with the experiments. FV and SO-A supervised experiments, and interpreted data. All authors critically reviewed and approved the final version.
Funding
GK received support from NIH grants HL133864, MD009162 and from the Institute for Transfusion Medicine Hemostasis and Vascular Biology Research Institute at the University of Pittsburgh School of Medicine. SO-A is supported by NIH grants R01HL106192, U01HL117721, and U54HL141011. FV is supported by NIH grants R01EB026966, R01HL146465, UG3HL143192, and R01HL125777.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Diane Lenhart, Bethany Flage, and Danielle Crosby for technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01910/full#supplementary-material
Supplementary Figure 1. GUSB expression in vehicle and haem-injected mice. Endogenous GUSB expression in vehicle and hemin-treated AA mice (n = 3–6). For Ct, lowest value = highest expression and highest value = lowest expression. All values are mean ± SEM. Exp, Expression. There were no significant differences between vehicle control and hemin treated values.
Supplementary Figure 2. Similarity in baseline transcripts of markers of cardiac hypertrophy in AA and SS mice. Baseline expression of Nppa and Myh7 in the heart of AA and SS mice. For DeltaCt, lowest value = highest expression and highest value = lowest expression. Target gene transcripts were normalized to Gusb for all mRNA expression level. All values are mean ± SEM. Exp, Expression.
References
1. Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anaemia in children under five, 2010-2050: modelling based on demographics, excess mortality, and interventions. PLoS Med. (2013) 10:e1001484. doi: 10.1371/journal.pmed.1001484
2. Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. (2007) 21:37–47. doi: 10.1016/j.blre.2006.07.001
3. Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest. (2017) 127:750–60. doi: 10.1172/JCI89741
4. Gladwin MT, Ofori-Acquah SF. Erythroid DAMPs drive inflammation in SCD. Blood. (2014) 123:3689–90. doi: 10.1182/blood-2014-03-563874
5. Simoes RL, Arruda MA, Canetti C, Serezani CH, Fierro IM, Barja-Fidalgo C. Proinflammatory responses of heme in alveolar macrophages: repercussion in lung hemorrhagic episodes. Mediators Inflamm. (2013) 2013:946878. doi: 10.1155/2013/946878
6. Adamzik M, Hamburger T, Petrat F, Peters J, De Groot H, Hartmann M. Free hemoglobin concentration in severe sepsis: methods of measurement and prediction of outcome. Crit Care. (2012) 16:R125. doi: 10.1186/cc11425
7. Ekregbesi P, Shankar-Hari M, Bottomley C, Riley EM, Mooney JP. Relationship between anaemia, haemolysis, inflammation and haem oxygenase-1 at admission with sepsis: a pilot study. Sci Rep. (2018) 8:11198. doi: 10.1038/s41598-018-29558-5
8. Soares MP, Bach FH. Heme oxygenase-1: from biology to therapeutic potential. Trends Mol Med. (2009) 15:50–58. doi: 10.1016/j.molmed.2008.12.004
9. Yeo TW, Lampah DA, Tjitra E, Gitawati R, Kenangalem E, Piera K, et al. Relationship of cell-free hemoglobin to impaired endothelial nitric oxide bioavailability and perfusion in severe falciparum malaria. J Infect Dis. (2009) 200:1522–29. doi: 10.1086/644641
10. Abildgaard U, Heimdal K. Pathogenesis of the syndrome of hemolysis, elevated liver enzymes, and low platelet count (HELLP): a review. Eur J Obstet Gynecol Reprod Biol. (2013) 166:117–23. doi: 10.1016/j.ejogrb.2012.09.026
11. Burwick RM, Rincon M, Beeraka SS, Gupta M, Feinberg BB. Evaluation of hemolysis as a severe feature of preeclampsia. Hypertension. (2018) 72:460–5. doi: 10.1161/HYPERTENSIONAHA.118.11211
12. Pathan N, Hemingway CA, Alizadeh AA, Stephens AC, Boldrick JC, Oragui EE, et al. Role of interleukin 6 in myocardial dysfunction of meningococcal septic shock. Lancet. (2004) 363:203–9. doi: 10.1016/S0140-6736(03)15326-3
13. Nayak KC, Meena SL, Gupta BK, Kumar S, Pareek V. Cardiovascular involvement in severe vivax and falciparum malaria. J Vector Borne Dis. (2013) 50:285–91.
14. Melchiorre K, Sharma R, Thilaganathan B. Cardiovascular implications in preeclampsia: an overview. Circulation. (2014) 130:703–14. doi: 10.1161/CIRCULATIONAHA.113.003664
15. Greer J. Pathophysiology of cardiovascular dysfunction in sepsis. British Journal of Anaesthesia Education. (2015) 15:316–21. doi: 10.1093/bjaceaccp/mkv003
16. Taylor SC, Shacks SJ, Mitchell RA, Banks A. Serum interleukin-6 levels in the steady state of sickle cell disease. J Interferon Cytokine Res. (1995) 15:1061–4. doi: 10.1089/jir.1995.15.1061
17. Duits AJ, Rodriguez T, Schnog JJ. Serum levels of angiogenic factors indicate a pro-angiogenic state in adults with sickle cell disease. Br J Haematol. (2006) 134:116–9. doi: 10.1111/j.1365-2141.2006.06103.x
18. Sarray S, Saleh LR, Lisa Saldanha F, Al-Habboubi HH, Mahdi N, Almawi WY. Serum IL-6, IL-10, and TNFalpha levels in pediatric sickle cell disease patients during vasoocclusive crisis and steady state condition. Cytokine. (2015) 72:43–7. doi: 10.1016/j.cyto.2014.11.030
19. Kelaidi C, Kattamis A, Apostolakou F, Poziopoulos C, Lazaropoulou C, Delaporta P, et al. PlGF and sFlt-1 levels in patients with non-transfusion-dependent thalassemia: correlations with markers of iron burden and endothelial dysfunction. Eur J Haematol. (2018) 100:630–5. doi: 10.1111/ejh.13061
20. Barbu EA, Mendelsohn L, Samsel L, Thein SL. Pro-inflammatory cytokines associate with NETosis during sickle cell vaso-occlusive crises. Cytokine. (2019) 127:154933. doi: 10.1016/j.cyto.2019.154933
21. Dinh W, Futh R, Nickl W, Krahn T, Ellinghaus P, Scheffold T, et al. Elevated plasma levels of TNF-alpha and interleukin-6 in patients with diastolic dysfunction and glucose metabolism disorders. Cardiovasc Diabetol. (2009) 8:58. doi: 10.1186/1475-2840-8-58
22. Melendez GC, Mclarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension. (2010) 56:225–231. doi: 10.1161/HYPERTENSIONAHA.109.148635
23. Fredj S, Bescond J, Louault C, Delwail A, Lecron JC, Potreau D. Role of interleukin-6 in cardiomyocyte/cardiac fibroblast interactions during myocyte hypertrophy and fibroblast proliferation. J Cell Physiol. (2005) 204:428–36. doi: 10.1002/jcp.20307
24. Kumar S, Wang G, Zheng N, Cheng W, Ouyang K, Lin H, et al. HIMF (Hypoxia-Induced mitogenic factor)-IL (interleukin)-6 signaling mediates cardiomyocyte-fibroblast crosstalk to promote cardiac hypertrophy and fibrosis. Hypertension. (2019) 73:1058–70. doi: 10.1161/HYPERTENSIONAHA.118.12267
25. Gladwin MT, Barst RJ, Gibbs JS, Hildesheim M, Sachdev V, Nouraie M, et al. Patients risk factors for death in 632 patients with sickle cell disease in the United States and United Kingdom. PLoS ONE. (2014) 9:e99489. doi: 10.1371/journal.pone.0099489
26. Gladwin MT. Cardiovascular complications and risk of death in sickle-cell disease. Lancet. (2016) 387:2565–74. doi: 10.1016/S0140-6736(16)00647-4
27. Crocker P, Werb Z, Gordon S, Bainton D. Ultrastructural localization of a macrophage-restricted sialic acid binding hemagglutinin, SER. in macrophage-hematopoietic cell clusters. Blood. (1990) 76:1131–8. doi: 10.1182/blood.V76.6.1131.1131
28. Lester LA, Sodt PC, Hutcheon N, Arcilla RA. Cardiac abnormalities in children with sickle cell anemia. Chest. (1990) 98:1169–74. doi: 10.1378/chest.98.5.1169
29. Faro GB, Menezes-Neto OA, Batista GS, Silva-Neto AP, Cipolotti R. Left ventricular hypertrophy in children, adolescents and young adults with sickle cell anemia. Rev Bras Hematol Hemoter. (2015) 37:324–8. doi: 10.1016/j.bjhh.2015.07.001
30. Alam J, Cook JL. Transcriptional regulation of the heme oxygenase-1 gene via the stress response element pathway. Curr Pharm Des. (2003) 9:2499–511. doi: 10.2174/1381612033453730
31. Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. (2004) 10:549–57. doi: 10.1016/j.molmed.2004.09.003
32. Gbotosho OT, Ghosh S, Kapetanaki MG, Lin Y, Weidert F, Bullock GC, et al. Cardiac expression of HMOX1 and PGF in sickle cell mice and haem-treated wild type mice dominates organ expression profiles via Nrf2 (Nfe2l2). Br J Haematol. (2019) 187:666–75. doi: 10.1111/bjh.16129
33. Kapetanaki MG, Gbotosho OT, Sharma D, Weidert F, Ofori-Acquah SF, Kato GJ. Free heme regulates placenta growth factor through NRF2-antioxidant response signaling. Free Radic Biol Med. (2019) 143:300–8. doi: 10.1016/j.freeradbiomed.2019.08.009
34. Ghosh S, Adisa OA, Chappa P, Tan F, Jackson KA, Archer DR, et al. Extracellular hemin crisis triggers acute chest syndrome in sickle mice. J Clin Invest. (2013) 123:4809–20. doi: 10.1172/JCI64578
35. Oh JY, Hamm J, Xu X, Genschmer K, Zhong M, Lebensburger J, et al. Absorbance and redox based approaches for measuring free heme and free hemoglobin in biological matrices. Redox Biol. (2016) 9:167–77. doi: 10.1016/j.redox.2016.08.003
36. Boyle JJ, Johns M, Lo J, Chiodini A, Ambrose N, Evans PC, et al. Heme induces heme oxygenase 1 via Nrf2: role in the homeostatic macrophage response to intraplaque hemorrhage. Arterioscler Thromb Vasc Biol. (2011) 31:2685–91. doi: 10.1161/ATVBAHA.111.225813
37. Reichard JF, Motz GT, Puga A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res. (2007) 35:7074–86. doi: 10.1093/nar/gkm638
38. Yan AT, Yan RT, Cushman M, Redheuil A, Tracy RP, Arnett DK, et al. Relationship of interleukin-6 with regional and global left-ventricular function in asymptomatic individuals without clinical cardiovascular disease: insights from the multi-ethnic study of atherosclerosis. Eur Heart J. (2010) 31:875–82. doi: 10.1093/eurheartj/ehp454
39. Sano M, Fukuda K, Kodama H, Pan J, Saito M, Matsuzaki J, et al. Interleukin-6 family of cytokines mediate angiotensin II-induced cardiac hypertrophy in rodent cardiomyocytes. J Biol Chem. (2000) 275:29717–23. doi: 10.1074/jbc.M003128200
40. Sachdev V, Machado RF, Shizukuda Y, Rao YN, Sidenko S, Ernst I, et al. Diastolic dysfunction is an independent risk factor for death in patients with sickle cell disease. J Am Coll Cardiol. (2007) 49:472–9. doi: 10.1016/j.jacc.2006.09.038
41. Niss O, Fleck R, Makue F, Alsaied T, Desai P, Towbin JA, et al. Association between diffuse myocardial fibrosis and diastolic dysfunction in sickle cell anemia. Blood. (2017) 130:205–13. doi: 10.1182/blood-2017-02-767624
42. Bakeer N, James J, Roy S, Wansapura J, Shanmukhappa SK, Lorenz JN, et al. Sickle cell anemia mice develop a unique cardiomyopathy with restrictive physiology. Proc Natl Acad Sci USA. (2016) 113:E5182–91. doi: 10.1073/pnas.1600311113
43. Marian AJ. Genetic determinants of cardiac hypertrophy. Curr Opin Cardiol. (2008) 23:199–205. doi: 10.1097/HCO.0b013e3282fc27d9
44. Vichinsky E. Chronic organ failure in adult sickle cell disease. Hematology Am Soc Hematol Educ Program. (2017) 2017:435–9. doi: 10.1182/asheducation-2017.1.435
45. Santiago RP, Guarda CC, Figueiredo CVB, Fiuza LM, Aleluia MM, Adanho CSA, et al. Serum haptoglobin and hemopexin levels are depleted in pediatric sickle cell disease patients. Blood Cells Mol Dis. (2018) 72:34–6. doi: 10.1016/j.bcmd.2018.07.002
46. Yalamanoglu A, Deuel JW, Hunt RC, Baek JH, Hassell K, Redinius K, et al. Depletion of haptoglobin and hemopexin promote hemoglobin-mediated lipoprotein oxidation in sickle cell disease. Am J Physiol Lung Cell Mol Physiol. (2018) 315:L765–L74. doi: 10.1152/ajplung.00269.2018
47. Vercellotti GM, Zhang P, Nguyen J, Abdulla F, Chen C, Nguyen P, et al. Hepatic overexpression of hemopexin inhibits inflammation and vascular stasis in murine models of sickle cell disease. Mol Med. (2016) 22:63. doi: 10.2119/molmed.2016.00063
48. Ingoglia G, Sag CM, Rex N, De Franceschi L, Vinchi F, Cimino J, et al. Data demonstrating the anti-oxidant role of hemopexin in the heart. Data Brief. (2017) 13:69–76. doi: 10.1016/j.dib.2017.05.026
49. Ofori-Acquah SF, Hazra R, Orikogbo OO, Crosby D, Flage B, Ackah EB, et al. Hemopexin deficiency promotes acute kidney injury in sickle cell disease. Blood. (2020) 135:1044–8. doi: 10.1182/blood.2019002653
50. Belcher JD, Chen C, Nguyen J, Abdulla F, Zhang P, Nguyen H, et al. Haptoglobin and hemopexin inhibit vaso-occlusion and inflammation in murine sickle cell disease: Role of heme oxygenase-1 induction. PLoS ONE. (2018) 13:e0196455. doi: 10.1371/journal.pone.0196455
51. Wruck CJ, Streetz K, Pavic G, Gotz ME, Tohidnezhad M, Brandenburg LO, et al. Nrf2 induces interleukin-6 (IL-6) expression via an antioxidant response element within the IL-6 promoter. J Biol Chem. (2011) 286:4493–9. doi: 10.1074/jbc.M110.162008
52. Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun. (2016) 7:11624. doi: 10.1038/ncomms11624
53. Fontes JA, Rose NR, Cihakova D. The varying faces of IL-6: from cardiac protection to cardiac failure. Cytokine. (2015) 74:62–68. doi: 10.1016/j.cyto.2014.12.024
54. Bandeira IC, Rocha LB, Barbosa MC, Elias DB, Querioz JA, Freitas MV, et al. Chronic inflammatory state in sickle cell anemia patients is associated with HBB(*)S haplotype. Cytokine. (2014) 65:217–21. doi: 10.1016/j.cyto.2013.10.009
55. Jaba IM, Zhuang ZW, Li N, Jiang Y, Martin KA, Sinusas AJ, et al. NO triggers RGS4 degradation to coordinate angiogenesis and cardiomyocyte growth. J Clin Invest. (2013) 123:1718–31. doi: 10.1172/JCI65112
56. Peiskerova M, Kalousova M, Danzig V, Mikova B, Hodkova M, Nemecek E, et al. Placental growth factor may predict increased left ventricular mass index in patients with mild to moderate chronic kidney disease–a prospective observational study. BMC Nephrol. (2013) 14:142. doi: 10.1186/1471-2369-14-142
57. Vicari P, Adegoke SA, Mazzotti DR, Cancado RD, Nogutti MA, Figueiredo MS. Interleukin-1beta and interleukin-6 gene polymorphisms are associated with manifestations of sickle cell anemia. Blood Cells Mol Dis. (2015) 54:244–9. doi: 10.1016/j.bcmd.2014.12.004
58. Nolan VG, Adewoye A, Baldwin C, Wang L, Ma Q, Wyszynski DF, et al. Sickle cell leg ulcers: associations with haemolysis and SNPs in Klotho, TEK and genes of the TGF-beta/BMP pathway. Br J Haematol. (2006) 133:570–8. doi: 10.1111/j.1365-2141.2006.06074.x
59. Lavrovsky Y, Schwartzman ML, Levere RD, Kappas A, Abraham NG. Identification of binding sites for transcription factors NF-kappa B and AP-2 in the promoter region of the human heme oxygenase 1 gene. Proc Natl Acad Sci USA. (1994) 91:5987–91. doi: 10.1073/pnas.91.13.5987
60. Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, et al. Characterization of heme as activator of Toll-like receptor 4. J Biol Chem. (2007) 282:20221–9. doi: 10.1074/jbc.M610737200
Keywords: heme, hemolysis, sickle cell disease, IL-6, inflammation, cardiac hypertrophy genes
Citation: Gbotosho OT, Kapetanaki MG, Ghosh S, Villanueva FS, Ofori-Acquah SF and Kato GJ (2020) Heme Induces IL-6 and Cardiac Hypertrophy Genes Transcripts in Sickle Cell Mice. Front. Immunol. 11:1910. doi: 10.3389/fimmu.2020.01910
Received: 03 March 2020; Accepted: 16 July 2020;
Published: 21 August 2020.
Edited by:
Renata Sesti-Costa, Campinas State University, BrazilReviewed by:
John D. Belcher, University of Minnesota Twin Cities, United StatesAnn Smith, University of Missouri–Kansas City, United States
Copyright © 2020 Gbotosho, Kapetanaki, Ghosh, Villanueva, Ofori-Acquah and Kato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gregory J. Kato, Z3JlZ29yeWthdG9tZEBnbWFpbC5jb20=