 Intan Juliana Abd Hamid1*
Intan Juliana Abd Hamid1* Nur Adila Azman2
Nur Adila Azman2 Andrew R. Gennery3
Andrew R. Gennery3 Ernest Mangantig1
Ernest Mangantig1 Ilie Fadzilah Hashim1
Ilie Fadzilah Hashim1 Zarina Thasneem Zainudeen1
Zarina Thasneem Zainudeen1- 1Primary Immunodeficiency Diseases Group, Regenerative Medicine Cluster, Institut Perubatan and Pergigian Termaju, Universiti Sains Malaysia, Kepala Batas, Malaysia
- 2Department of Biomedical Science, Universiti Islam Antarabangsa, Kuantan, Pahang, Malaysia
- 3Sir James Spence Professor of Child Health, Translational and Clinical Research Institute, Newcastle University, Great North Children's Hospital, Newcastle upon Tyne, United Kingdom
Introduction: Primary immunodeficiency diseases (PIDs) are under-reported in Malaysia. The actual disease frequency of PID in this country is unknown due to the absence of a national patient registry for PID.
Objective: This systematic review aimed to determine the prevalence rates of PID cases diagnosed and published in Malaysia from 1st of January 1979 until 1st of March 2020. It also aimed to describe the various types of PIDs reported in Malaysia.
Method: Following the development of a comprehensive search strategy, all published literature of PID cases from Malaysia was identified and collated. All cases that fulfilled the International Union of Immunological Societies (IUIS) classification diagnosis were included in the systematic review. Data were retrieved and collated into a proforma.
Results: A total of 4,838 articles were identified and screened, with 34 publications and 119 patients fulfilling the criteria and being included in the systematic review. The prevalence rate was 0.37 per 100,000 population. In accordance with the IUIS, the distribution of diagnostic classifications was immunodeficiencies affecting cellular and humoral immunities (36 patients, 30.3%), combined immunodeficiencies with associated or syndromic features (21 patients, 17.6%), predominant antibody deficiencies (24 patients, 20.2%), diseases of immune dysregulation (13 patients, 10.9%), congenital defects in phagocyte number or function (20 patients, 16.8%), defects in intrinsic and innate immunity (4 patients, 3.4%), and autoinflammatory disorders (1 patient, 0.8%). Parental consanguinity was 2.5%. Thirteen different gene mutations were available in 21.8% of the cases.
Conclusion: PIDs are underdiagnosed and under-reported in Malaysia. Developing PID healthcare and a national patient registry is much needed to enhance the outcome of PID patient care.
Introduction
Primary immunodeficiency diseases (PIDs) or inborn errors of immunity are inherited disorders that impair the immune response, leading to increased risk of infections, immune dysregulation, autoimmune phenomena, inflammation, and malignancy. They are classified as “rare diseases,” but their global incidence is more prevalent than generally thought (1). Six million people are estimated to be living with PIDs worldwide, of which only 27,000–60,000 have been diagnosed (2). Early detection and prompt action by clinicians could reduce complications and save lives by administering appropriate treatment.
Malaysia is a small country situated in Southeast Asia, with a total population of 32.6 million; the total livebirths in 2018 was 501.9 thousand, and the GDP per capita is 10,940 USD. The estimated life expectancy in Malaysia is 74.5 years, and 28% of the population is aged less than 18 years. The under-5 mortality rate was 7.8 deaths per 1,000 live births in 2018 (3).
Management and research of PIDs in Malaysia are met with many challenges. Vast gaps in knowledge of PIDs and translation of diagnosis and management into clinical practice exist. Given the scarce epidemiological PID data and the lack of patient awareness or diagnostic facilities in Malaysia, PIDs are a tremendous challenge for the biomedical community, thereby causing unnecessary suffering, and deaths. Therefore, national studies must be considered to efficiently and systematically assess the proportion of affected individuals amongst the general population (4).
Furthermore, estimates of PIDs frequency in Malaysia are needed to determine the cost-effectiveness of routine screening. Most incidence and prevalence determination is performed using data from national patient registry databases (5, 6). Given the absence of a Malaysian PID registry database, the prevalence rate was calculated in the present paper by systematically reviewing PID literature from Malaysia to provide retrospective data for the medical community, the health authorities, and PID patients and their families and improve the knowledge of PIDs. This systematic review offers a mechanism to describe the epidemiological landscape of PIDs in Malaysia and constitutes an opportunity to determine the prevalence rates of PIDs in a defined non-referred population.
Methods
In these sections, the strategy to identify relevant citations gathered for this review is described, and a summary of the results is provided. A comprehensive search of the Cochrane Library and Medline databases (via PubMed) was initially conducted from its inception to 1st of March 2020. A title/abstract keyword search was conducted, 77 candidate article abstracts were manually reviewed, and forward/backward reference searches were completed. Controlled vocabulary supplemented with keywords (which included autoimmunity) was used to search for individual primary immunodeficiencies in accordance with the IUIS 2019 classification (7). The search results and study selections are outlined in Figure 1. The PICO statement was as follows: P, patients with PID in Malaysia; I, none considering looking into the epidemiology and prevalence study; C, other neighboring Southeast Asian countries and O, prevalence and incidence rates.
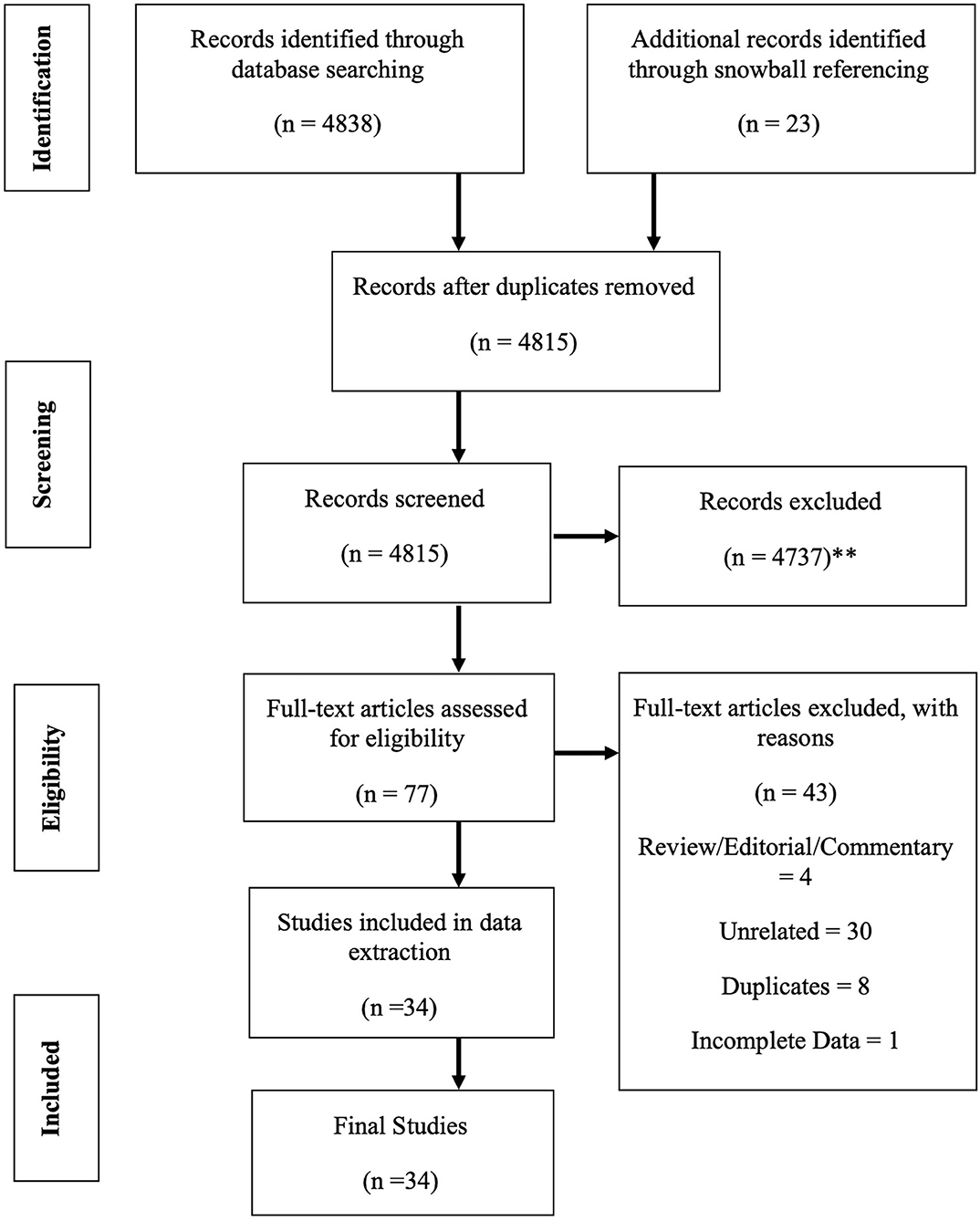
Figure 1. PRISMA flow diagram: the PRISMA (Preferred Reporting Items for Systematic Review and Meta-Analysis) diagram details our search and selection process applied during the overview. (**Reasons excluded are listed in Supplement 1).
Cases were screened using primary immunodeficiencies in accordance with the IUIS 2019 classification (7). For all published papers retrieved, the second, fifth, and sixth author reviewed the title and abstracts independently and performed the selections on the basis of pre-specified inclusion and exclusion criteria. All inclusions and exclusions were discussed and finalized with all team members. The primary author reviewed the clinical profile, laboratory parameters, other comorbid conditions, and other associated factors discussed in the papers and retrieved the relevant information. Age and year at diagnosis were retrieved from the publications. In cases where year was not listed, an assumption that the year of diagnosis was equal to the year of publication was made in specific articles/cases. Attempts were also made to contact the original authors of the included published studies via email in cases of incomplete information or for further clarification. The prevalence rates were calculated by the fourth author. The third author offer critical review on the manuscript draft.
The agreement of reviewers on the methodological quality assessment was assessed, and any disagreement was resolved by discussion or, if agreement could not be reached, by arbitration by the third author. Reviewers were not masked to study details when assessing the study quality.
The period prevalence rate per 100,000 population was calculated as the number of all existing PID cases diagnosed up to 2019, divided by the average number of the Malaysian population over a specified time period.
Results
A total of 4,838 articles were identified and screened in accordance with the pre-determined inclusion criteria. Thirty-four publications and 119 patients were included in this systematic review (Table 1). The study population was mainly Malays (67%), Chinese (14%), Indians (17%), Ibans (1%), and Melanau (1%). However, information regarding race was only available in 84 of 119 cases identified. The mean age was difficult to determine due to the nature of reporting by all the reports. Parental consanguinity was only recorded as present in three cases (2.5%). The pattern of major reporting centers, which were mainly from the public universities and the medical research team, was noted. Out of 63 cases identified with family history mentioned in the publications, only 38% had positive family history. Only two adult patients were reported to have PID (familial hemophagocycytic lymphohistiocytosis) (17), whereas the remaining aged between the 1st month of life and 17 years. However, patient age was only available in 26 publications.
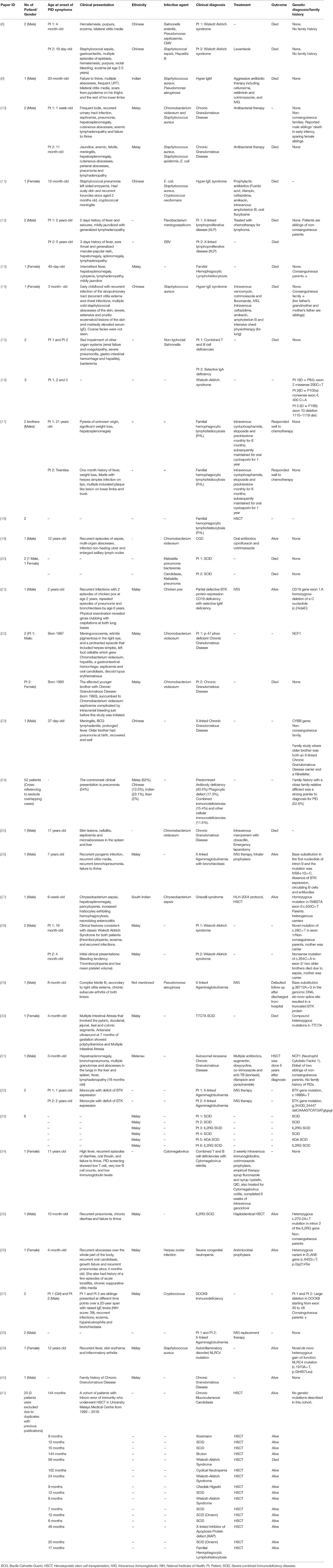
Table 1. Primary Immunodeficiency Diseases Cases Reported in Malaysia.
A total of 119 PID cases were identified, and the diagnosis and distribution are illustrated in Figure 2. The most common forms of PID were immunodeficiencies affecting cellular and humoral immunity (30.3% of the cases published, Table 2), followed by predominantly antibody deficiencies (20.2%), congenital defects in phagocyte function and number (16.8%), diseases of immune dysregulation (10.9%), defects in intrinsic and innate immunity (3.4%), and autoinflammatory disorders (0.8%). The most common PID diagnosis was severe combined immunodeficiency (SCID, 22 patients), with genetic diagnosis available in 10 patients with SCID (Table 3). The second most common PID diagnosis was X-linked agammaglobulinemia (17 cases), followed by chronic granulomatous disease (CGD), affecting 14 patients (p47-phox-deficient CGD = three patients, X-linked CGD = two patients). The calculated prevalence rate was 0.37 cases per 100,000 population (from 1979 until 2019). We did not calculate the incidence as there was limited information in the literature.
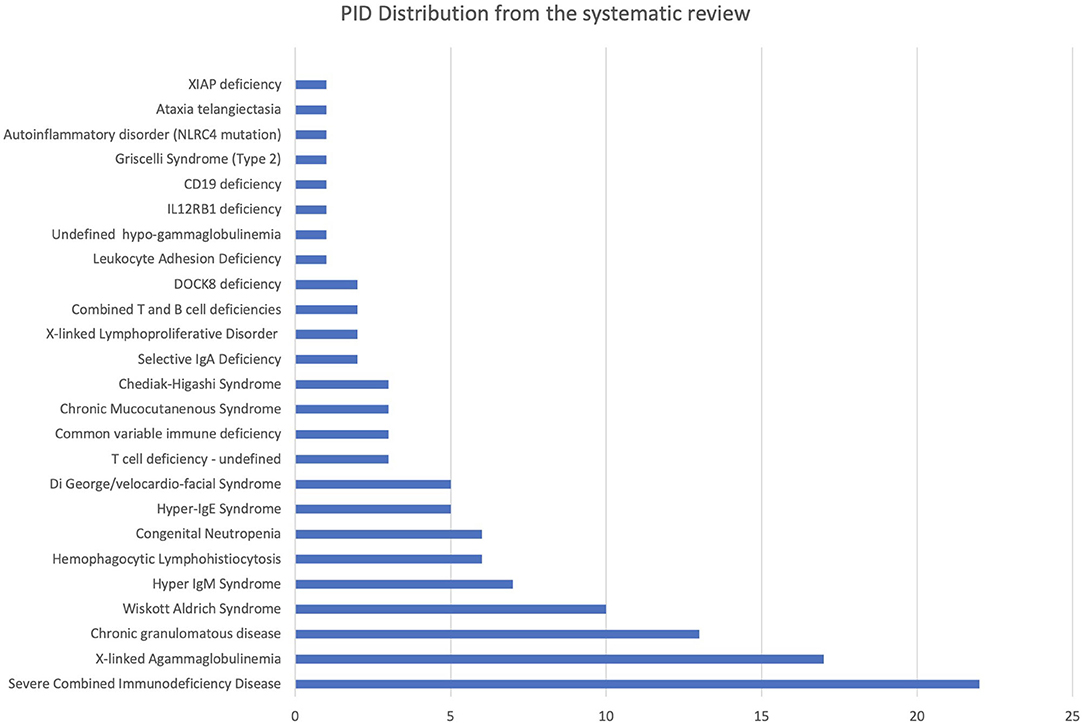
Figure 2. PID diagnosis distribution identified from systematic review.
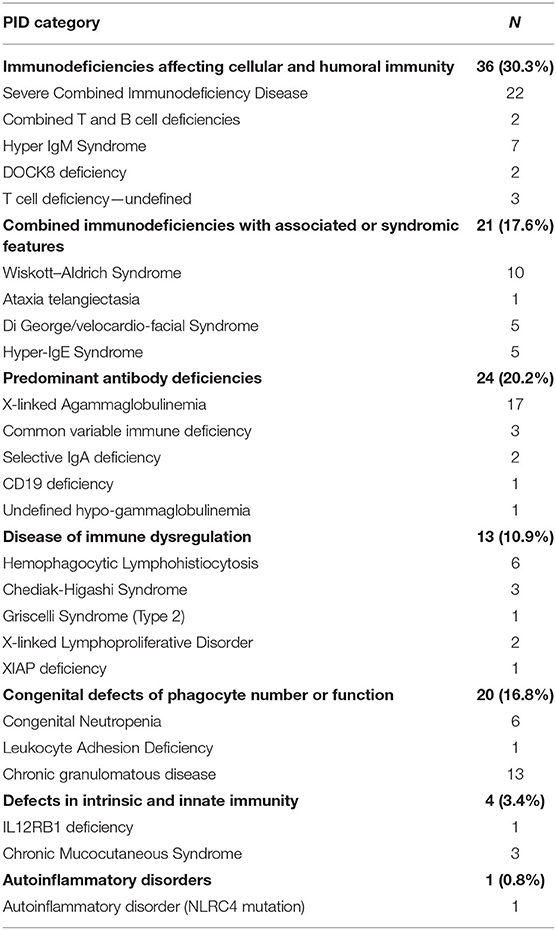
Table 2. Spectrum of PIDs, 1979–2020.

Table 3. Genetic mutations described.
Genetic tests are not universally available in Malaysia at present, and most are paid for by patients and families. Genetic mutation results were identified in 26 patients (21.8%; Table 3). Twenty-two patients underwent hematopoietic stem cell transplantation in Malaysia, whilst 19 received intravenous immunoglobulin replacement therapy. Only 28 patients were documented as alive, and 16 had died at the time of publication of the case reports. However, no data were available for the remaining publications.
In order to place results from Malaysia in context, a similar comparison of PID diseases frequencies was made, using published reports from other countries (Table 4). The prevalence of PID in Malaysia using this method is the lowest compared to reports from other nations. This low prevalence comes from a systematic review in the absence of national registry or cohort report, as reported by other countries.
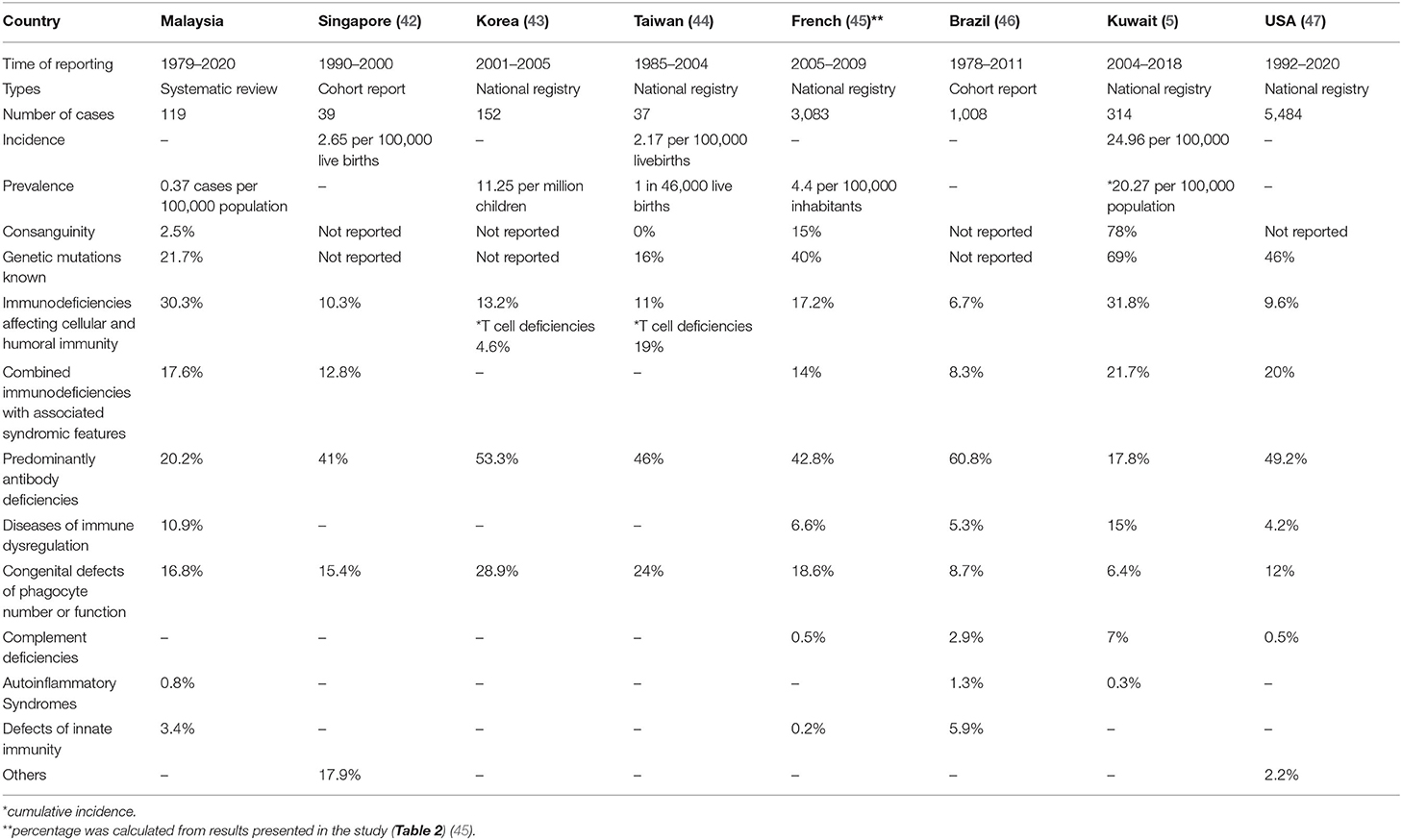
Table 4. The comparison of primary immunodeficiencies prevalence, frequencies, consanguinity, and availability of genetic results with various countries.
Discussion
To the best of the authors' knowledge, this review is the first to study the prevalence of PIDs in Malaysia, which was 0.37 per 100,000 population. These rates were comparably lower than published rates from other countries, where the prevalence rate was ~4.4–20.27 per 100,000 population (5, 42–47).
In addition, these rates were lower than those in several neighboring countries in Southeast Asia with similar geo-political environments. Singapore has an incidence rate of 2.65 per 100,000 live births and an estimated occurrence rate of one in 37,000 live births (47). Thailand reported a cohort of 67 patients with PID from 18 year-experience from a single tertiary centre (48). No national PID patient registry databases are available in Southeast Asian countries. The only countries in the Asia Pacific with national PID registries are Japan, South Korea, Taiwan, Hong Kong, and China (49). The Middle East countries with national PID registries are Kuwait, Turkey, and Iran (5, 50, 51).
There was an increase in reporting over time which may be attributed to the increase in numbers of practicing clinical immunologists, enhanced diagnostic strategies, and facilities and improved awareness amongst medical fraternities locally (52).
This systematic review found that parental consanguinity was 2.5%, which was comparable with the parental consanguinity rate of 2.4% in a Malaysian population study on birth defects (53). Consanguineous marriage is not widely practiced in Malaysia compared with other South Asian and Middle East countries but is practiced in certain ethnic groups or indigenous populations.
This study is unique because it was not based on a national registry. However, it was the best attempt in delineating the prevalence rates of PIDs in Malaysia. Systematic review offers a non-biased approach to reviewing the evidence available in the absence of a national patient registry database. Furthermore, this systematic review is a novel approach in assessing PID prevalence in a low resource country that does not have a national database and a model that could be followed by other countries, such as in South America and Africa.
The potential limitations of this study were the calculations that were based on published evidence. The prevalence rate may not reflect the actual rate, because many PID cases are undiagnosed and, even if diagnosed correctly, not published. Furthermore, reporting bias possibly existed, where certain diagnoses were reported more than others. This bias could explain why the most common PID diagnoses in this systematic review were severe combined immunodeficiencies rather than primary antibody defects, which are more prevalent in patient registry databases worldwide. Furthermore, only two adult patients were reported, and only three Common Variable Immunodeficiency cases were published, thereby emphasizing that the lack of clinical immunologists to take care of adult patients may be a major contributing factor for low adult PID cases diagnosed or not reported.
Therefore, a national PID registry database must be urgently established in Malaysia. Inter-collaboration between various agencies is the manner to move forward in terms of improving access to early diagnosis and treatment of PIDs in Malaysia.
Conclusions
The prevalence of PID is lower in Malaysia than in other geographical regions. The comparison of PID prevalence between countries may be misleading due to various case-finding strategies. Efforts should be geared toward setting up a national PID patient registry database in Malaysia.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
NA, IH, and ZZ performed the literature search, reviewed the title, and abstracts independently and conducted the stratifications. IA and NA conducted the data extraction of relevant studies. Any disagreements/clarifications were confirmed with AG. The prevalence rates were calculated by EM. IA wrote the first draft of the manuscript. All authors designed the study. All authors critically reviewed the literature search and revised the manuscript. All authors read and approved the final version of the manuscript.
Funding
Grant Agencies: Universiti Sains Malaysia Grant Name: USM 2020 Initiatives (Project 19) Grant Number: 311.CIPPT.411919.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to acknowledge USM for the financial and moral support in PID service empowerment through the USM 2020 Initiatives (Project 19) (USM: 311.CIPPT.411919), Puan Noraida Hassan (Librarian, Hamdan Tahir Library, USM) for assistance in conducting the database searches and thank all the original authors that responded and supplied the manuscript with more details.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01923/full#supplementary-material
References
1. Rezaei N, Aghamohammadi A, Notarangelo LD. Primary Immunodeficiency Diseases: Definition, Diagnosis, and Management. 2nd ed. Berlin: Springer-Verlag Berlin Heidelberg (2017).
2. Bousfiha AA, Jeddane L, Ailal F, Benhsaien I, Mahlaoui N, Casanova JL, et al. Primary immunodeficiency diseases worldwide: more common than generally thought. J Clin Immunol. (2013) 33:1–7. doi: 10.1007/s10875-012-9751-7
3. Department of Statistics Malaysia. (2019). Retrieved from: https://www.dosm.gov.my (December 7, 2019).
4. Condino-Neto A, Espinosa-Rosales FJ. Changing the lives of people with Primary Immunodeficiencies (PI) with early testing and diagnosis. Front Immunol. (2018) 9:1439. doi: 10.3389/fimmu.2018.01439
5. Waleed AH, Al-Ahmad M, Al-Khabaz A, Husain A, Sadek A, Othman Y. The Kuwait national primary immunodeficiency registry 2004-2018. Front Immunol. (2019) 10:1754. doi: 10.3389/fimmu.2019.01754
6. El-Helou SM, Biegner AK, Bode S, Ehl SR, Heeg M, Maccari ME, et al. The German National Registry of primary immunodeficiencies (2012-2017). Front Immunol. (2019) 10:1272.
7. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. (2020) 40:24–64. doi: 10.1007/s10875-019-00737-x
8. Tong YH, Sinniah D, Murugasu R, White J. (1979) Two Malaysian Chinese male children with the Wiskott-Aldrich syndrome. Singapore Med J. 20:355–9.
9. Noh LM, Low SM, Lajin I, Abdullah N. Antibody deficiency with hyper IgM-a case report. Malaysian J Pathol. (1992) 14:121–23.
10. Noh LM, Noah RM, Wu LL, Nasuruddin BA, Junaidah E, Ooi CP, et al. Chronic granulomatous disease–a report in two Malay families. Singapore Med J. (1994) 35:505–8.
11. Sendut IH, Ariffin H, Sinniah D. Cryptococcal meningitis in a patient with hyperimmunoglobulin E syndrome. Malaysian J Child Health. (1995) 7:145–7.
12. Hany A, Thong MK, Lin HP. Two brothers in a Malaysian family with X-linked lymphoproliferative disease - a case report. Singapore Med J. (1996) 37:325–7.
13. Leong CF, Cheong SK, Hamidah NH, Ainoon O, Kannaheswary Y. Serum ferritin and lactate dehydrogenase in a case of hemophagocytic lymphohistiocytosis. Malaysian J Pathol. (1998) 20:103–8.
14. Lee WS, Boey CC, Goh AY. Pulmonary nocardiosis in a child with hyperimmunoglobulin E syndrome. Singapore Med J. (1999) 40:278–80.
15. Lee WS, Puthucheary SD, Parasakthi N. Extra-intestinal non-typhoidal Salmonella infections in children. Ann Trop Paediatr. (2000) 20:125–9. doi: 10.1080/02724936.2000.11748121
16. Jin Y, Mazza C, Christie JR, Giliani S, Fiorini M, Mella P, et al. (2004) Mutations of the Wiskott-Aldrich Syndrome Protein (WASP): hotspots, effect on transcription, and translation and phenotype/genotype correlation. Blood. 104:4010–9 doi: 10.1182/blood-2003-05-1592
17. Gan GG, Eow GI, Teh A, Ng SC, Sangkar JV. Familial hemophagocytic lymphohistiocytosis in two brothers. Med J Malaysia. (2004) 59:100–2.
18. Ariffin H, Lum SH, Cheok SA, Shekhar K, Ariffin WA, Chan LP, et al. Haemophagocytic lymphohistiocytosis in children. J Paediatr Child Health. (2005) 41:136–9. doi: 10.1111/j.1440-1754.2005.00564.x
19. Sureisen M, Choon SK, Tai CC. Recurrent Chromobacterium violaceum infection in a patient with chronic granulomatous disease. Med J Malaysia. (2008) 63:346–7.
20. Lee PP, Chan K, Chen T, Jiang L, Wang X, et al. Molecular diagnosis of severe combined immunodeficiency-identification of IL2RG, JAK3, IL7R, DCLRE1C, RAG1, and RAG2 mutations in a cohort of Chinese and Southeast Asian Children. J Clin Immunol. (2010) 31:281–96. doi: 10.1007/s10875-010-9489-z
21. Noh LM, Abd Hamid I, Rus Anidah A, Hashim I, Narazah Y, Latiff AH, et al. An CD19 deficient B cell abnormality and selective isotype IgM deficiency in A Malay Child. A Case Report. In: Proceeding of the 15th Meeting of the European Society Immunodeficiencies-ESID. (2012). Retrieved from: https://www.researchgate.net/publication/236946449_An_CD19_Deficient_B_Cell_Abnormality_and_Selective_isotype_IgM_deficiency_in_A_Malay_Child_A_Case_Report
22. Gill HK, Kumar HC, Dhaliwal JS, Zabidi F, Sendut IH, Noah R, et al. Defining p47-phox deficient chronic granulomatous disease in a Malay family. Asian Pac J Allergy Immunol. (2012) 30:313–20.
23. Gill HK, Kumar HC, Cheng CK, Ming CC, Nallusamy R, Yusoff N, et al. De chronic granulomatous disease in a male child with an X-CGD carrier, Klinefelter brother. Asian Pac J Allergy Immunol. (2013) 31:167–72. doi: 10.12932/AP0274.31.2.2013
24. Noh LM, Nasuruddin BA, Abdul Latiff AH, Noah RM, Kamarul Azahar MR, Norzila M, et al. Clinical-epidemiological pattern of primary immunodeficiencies in Malaysia 1987-2006: a 20 year experience in Four Malaysian Hospitals. Med J Malaysia. (2013) 68:13–17.
25. Rashid Z, Ali U, Sulong A, Rahman R. Chromobacterium Violaceum infections; a series of case reports in a Malaysian Tertiary Hospital. Internet J Infect Dis. (2013) 11:1–6. doi: 10.5580/2cd3
26. Chear CT, Gill HK, Ramly NH, Dhaliwal JS, Bujang N, Ripen AM, et al. A novel Bruton's tyrosine kinase gene (BTK) invariant splice site mutation in a Malaysian family with X-linked agammaglobulinemia. Asian Pac J Allergy Immunol. (2013) 31:320–4. doi: 10.12932/AP0304.31.4.2013
27. Ariffin H, Geikowski A, Chin TF, Chau D, Arshad A, Abu Bakar K, et al. Griscelli syndrome. Med J Malaysia. (2014) 69:193–4.
28. Baharin MF, Begum S, Ibrahim K, Song YAP, Abdul Manaf AM, Ripen AM, et al. Molecular characterization of two Malaysian patients with Wiskott-Aldrich syndrome. Malaysian J Pathol. (2015) 37:153–8.
29. Chear CT, Ripen AM, Mohamed SAS, Dhaliwal JS. A novel BTK gene mutation creates a de-novo splice site in an X-linked agammaglobulinemia patient. Gene. (2015) 560:245–8. doi: 10.1016/j.gene.2015.02.019
30. Yang W, Lee PPW, Ramanujam TM, Shanmugam A, Koh MT, et al. (2015). Compound heterozygous mutations in TTC7A cause familial multiple intestinal atresias and severe combined immunodeficiency. Clin Genet. 88:542–9. doi: 10.1111/cge.12553
31. Ismail IH, Jamli FM, Othman IS, Noh LM, Abdul Latiff AH. Malaysia's first transplanted case of chronic granulomatous disease: the journey of overcoming obstacles. Children (Basel). (2016) 3:9. doi: 10.3390/children3020009
32. Mirsafian H, Ripen AM, Chear CT, Bin Mohamad S, et al. Transcriptome profiling of monocytes from XLA patients revealed the innate immune function dysregulation due to the BTK gene expression deficiency. Sci Rep. (2017) 7:6836. doi: 10.1038/s41598-017-06342-5
33. Luk ADW, Lee PP, Mao H, Chan K-W, Chen XY, Chen T.-X, et al. Family history of early infant death correlates with earlier age at diagnosis but not shorter time to diagnosis for severe combined immunodeficiency. Front Immunol. (2017) 8:808. doi: 10.3389/fimmu.2017.00808
34. Ngai JJ, Chong KL, Oli Mohamed S. Cytomegalovirus retinitis in primary immune deficiency disease. Case Rep Ophthalmol Med. (2018) 2018:8125806. doi: 10.1155/2018/8125806
35. Ariffin H, Chew KS, Jawin V, Thavagnanam S. T-cell replete haploidentical bone marrow transplantation for X-linked severe combined immunodeficiency. Singapore Med J. (2018) 61:285–6. doi: 10.11622/smedj.2018101
36. Mohamad SM, Abd Hamid IJ, Abdul Wahab A, Ali A, Choo CM, Bakon F, et al. Submission ID#600349: severe congenital neutropenia caused by ELANE gene mutation in a Malaysian girl. J Clin Immunol. (2019) 39(Suppl) 1–151.
37. Abd Hamid IJ, Nik Yusoff NK, Daud M, Mohamad SM, Hashim IF, Zainudeen ZT, et al. Submission ID#605833: DOCK8 immunodeficiency in a Malay family from Malaysia: a family study. J Clin Immunol. (2019) 39:1–151.
38. El-Sayed ZA, Abramova I, Aldave JC, Al-Herz W, Bezrodnik L, Boukari R, et al. X-linked agammaglobulinemia (XLA): phenotype, diagnosis, and therapeutic challenges around the world. World Allergy Organ J. (2019) 12:100018. doi: 10.1016/j.waojou.2019.100018
39. Chear CT, Nallusamy R, Scott WC, Chan KC, Baharin MF, Hishamshah M, et al. A novel de novo NLRC4 mutation reinforces the likely pathogenicity of specific LRR domain mutation. Clin Immunol. (2020) 211:108328. doi: 10.1016/j.clim.2019.108328
40. Zainudeen ZT, Hashim IF, Abd Hamid IJ. Chronic granulomatous diseases. Malaysian J Paediatr Child Health. (2020) 25:28–9.
41. Ariffin H, Ab Rahman S, Jawin V, Foo JC, Amram NF, Mahmood NM, et al. Haematopoietic stem cell transplantation for inborn errors of immunity: 25-year experience from University of Malaya Medical Centre, Malaysia. J Paediatr Child Health. (2019) 56:379–83. doi: 10.1111/jpc.14621
42. Lim DL, Thong BY, Ho SY, Shek LP, Lou J, Leong K, et al. Primary immunodeficiency diseases in Singapore-the last 11 years. Singapore Med J. (2003) 44:579–86.
43. Rhim JW, Kim KH, Kim DS, Kim BS, Kim JS, Kim CH, et al. Prevalence of primary immunodeficiency in Korea. J Korean Med Sci. (2012) 27:788–93. doi: 10.3346/jkms.2012.27.7.788
44. Lee WI, Kuo ML, Huang JL, Lin SJ, Wu CJ. Distribution and clinical aspects of primary immunodeficiencies in a Taiwan pediatric tertiary hospital during a 20-year period. J Clin Immunol. (2005) 25:162–73. doi: 10.1007/s10875-005-2822-2
45. The French national registry of primary immunodeficiency diseases. Clin Immunol. (2010) 135:264–72. doi: 10.1016/j.clim.2010.02.021
46. Carneiro-Sampaio M, Moraes-Vasconcelos D, Kokron CM, Jacob CM, Toledo-Barros M, Dorna MB, et al. Primary immunodeficiency diseases in different age groups: a report on 1,008 cases from a single Brazilian reference center. J Clin Immunol. (2013) 33:716–24. doi: 10.1007/s10875-013-9865-6
47. Total number of patients in the USIDNET registry. US Immunodeficiency Network. (2020). Available online at: http://www.usidnet.org/index.cfm (Accessed June 20, 2020).
48. Benjasupattananan P, Simasathein T, Vichyanond P, Leungwedchakarn V, Visitsunthorn N, Pacharn P, et al. Clinical characteristics and outcomes of primary immunodeficiencies in Thai children: an 18-year experience from a tertiary care center. J Clin Immunol. (2009) 29:357–64. doi: 10.1007/s10875-008-9273-5
49. Pilania RK, Chaudhary H, Jindal AK, Rawat A, Singh S. Current status and prospects of primary immunodeficiency diseases in Asia. Genes Dis. (2020) 7:3–11 doi: 10.1016/j.gendis.2019.09.004
50. Abolhassani H, Kiaee F, Tavakol M, Chavoshzadeh Z, Alireza S, Momen T, et al. Fourth update on the Iranian national registry of primary immunodeficiencies: integration of molecular diagnosis. J Clin Immunol. (2018) 38:816–32. doi: 10.1007/s10875-018-0556-1
51. Kilic SS, Ozel M, Hafizoglu D, Karaca N, Aksu G, Kutukculer N. The prevalances and patient characteristics of primary immunodeficiency diseases in Turkey-two centers study. J Clin Immunol. (2013) 33:74–83. doi: 10.1007/s10875-012-9763-3
52. Abd Hamid IJ, Zainudeen ZT, Hashim IF. Current perspectives and challenges of primary immunodeficiency diseases in Malaysia. Malaysian J Paediatr Child Health. (2020) 25:16.
Keywords: primary immunodeficiency, inborn error of immunity, epidemiology, prevalence, Malaysia
Citation: Abd Hamid IJ, Azman NA, Gennery AR, Mangantig E, Hashim IF and Zainudeen ZT (2020) Systematic Review of Primary Immunodeficiency Diseases in Malaysia: 1979–2020. Front. Immunol. 11:1923. doi: 10.3389/fimmu.2020.01923
Received: 22 May 2020; Accepted: 17 July 2020;
Published: 26 August 2020.
Edited by:
Waleed Al-Herz, Kuwait University, KuwaitReviewed by:
Antonio Condino-Neto, University of São Paulo, BrazilNizar Mahlaoui, Assistance Publique Hopitaux De Paris, France
Copyright © 2020 Abd Hamid, Azman, Gennery, Mangantig, Hashim and Zainudeen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Intan Juliana Abd Hamid, aW50YW5qQHVzbS5teQ==