 Rodrigo Sánchez-Tarjuelo1
Rodrigo Sánchez-Tarjuelo1 Isabel Cortegano1
Isabel Cortegano1 Juliana Manosalva1Mercedes Rodríguez1Carolina Ruíz1
Juliana Manosalva1Mercedes Rodríguez1Carolina Ruíz1 Mario Alía1
Mario Alía1 María Carmen Prado1Eva M. Cano2
María Carmen Prado1Eva M. Cano2 María José Ferrándiz3
María José Ferrándiz3 Adela G. de la Campa3,4
Adela G. de la Campa3,4 María Luisa Gaspar1*
María Luisa Gaspar1* Belén de Andrés1*
Belén de Andrés1*- 1Immunobiology Department, Carlos III Health Institute, Madrid, Spain
- 2Chronic Disease Programme, Carlos III Health Institute, Madrid, Spain
- 3Bacterial Genetics Department, Carlos III Health Institute, Madrid, Spain
- 4Consejo Superior de Investigaciones Científicas, Madrid, Spain
Streptococcus pneumoniae is the main cause of bacterial pneumonia, a condition that currently produces significant global morbidity and mortality. The initial immune response to this bacterium occurs when the innate system recognizes common motifs expressed by many pathogens, events driven by pattern recognition receptors like the Toll-like family receptors (TLRs). In this study, lung myeloid-cell populations responsible for the innate immune response (IIR) against S. pneumoniae, and their dependence on the TLR4-signaling axis, were analyzed in TLR4–/– and Myeloid-Differentiation factor-88 deficient (MyD88–/–) mice. Neutrophils and monocyte-derived cells were recruited in infected mice 3-days post-infection. Compared to wild-type mice, there was an increased bacterial load in both these deficient mouse strains and an altered IIR, although TLR4–/– mice were more susceptible to bacterial infection. These mice also developed fewer alveolar macrophages, weaker neutrophil infiltration, less Ly6Chigh monocyte differentiation and a disrupted classical and non-classical monocyte profile. The pro-inflammatory cytokine profile (CXCL1, TNF-α, IL-6, and IL-1β) was also severely affected by the lack of TLR4 and no induction of Th1 was observed in these mice. The respiratory burst (ROS production) after infection was profoundly dampened in TLR4–/– and MyD88–/– mice. These data demonstrate the complex dynamics of myeloid populations and a key role of the TLR4-signaling axis in the IIR to S. pneumoniae, which involves both the MyD88 and TRIF (Toll/IL-1R domain-containing adaptor-inducing IFN-β) dependent pathways.
Introduction
Streptococcus pneumoniae (pneumococcus) is a Gram-positive bacterium that colonizes and invades the respiratory tract. It is the main etiological agent of community acquired pneumonia, accounting for about 90% of all pneumonia deaths, especially in young children and the elderly (1, 2). As a consequence, it is a major cause of morbidity and mortality worldwide (2), although the present pandemic induced by SARS-Cov2 is currently changing the top list of the most dangerous respiratory pathogens (3). Globally, S. pneumoniae causes over 800,000 deaths in children and 13.8 million cases of pneumonia each year. Different vaccines have been generated against the most prevalent S. pneumoniae serotypes, of which the conjugate 13-valent (PCV13) and the polysaccharide-based 23-valent (PCV23) are often used (4). The use of antibiotics is becoming compromised by the ever-increasing appearance of antibiotic-resistant strains (5–7). Moreover, as the immune lung response underlying S. pneumoniae infection is still not fully understood, a better understanding of this response will be essential to design new effective therapeutic interventions.
It is known that S. pneumoniae recognition by lung epithelial cells and by the innate immune system (IIS) involves several pattern recognition receptors (PRRs), receptors that are expressed in different cell lineages and that recognize pathogen associated molecular patterns (PAMPs). Toll-like receptors (TLRs) constitute one of the most important PRR families, playing a critical role in initiating inflammatory responses and promoting adaptive immune responses (8, 9). Of these, both TLR2 and TLR4 may participate in the innate immune response (IIR) against S. pneumoniae. TLR2 recognizes lipoteichoic acid (LTA), peptidoglycans and lipopeptides, components of the Gram-positive cell wall (10, 11), although as it appears to play a limited role in the IIR against S. pneumoniae other TLRs are likely to be implicated in this process (12). TLR4 plays an essential role in the host’s defense against Gram-negative bacteria by recognizing lipopolysaccharide (LPS) (13), although this is not presented by S. pneumoniae. Nevertheless, it also recognizes LTA and pneumolysin (Ply), the latter a pneumococcal enzyme that is released by these bacteria. Hence, TLR4 may participate in the response against S. pneumoniae (13, 14), and the interaction between Ply and TLR4 during pneumococcal colonization of the nasopharynx may be important for protection (14). After ligand binding, TLR2 and TLR4 depend on the signaling adaptor protein MyD88 (Myeloid-Differentiation factor-88) for activation. In addition, TLR4 can signal through both MyD88- and TRIF-dependent (Toll/IL-1R domain-containing adaptor-inducing IFN-β) pathways (8, 9). Moreover, MyD88–/– mice infected with S. pneumoniae generate a stronger bacterial burden and a weaker inflammatory response in the lung (15).
Alveolar macrophages (AMs) are tissue-resident macrophages (Mϕ) that colonize lung tissues during embryogenesis and that are thereafter maintained through self-renewal (16). Alveolar macrophages initiate lung IRs by recognizing and phagocytosing S. pneumoniae, and they are essential to control bacterial numbers in the first hours after infection. However, due to the limited number of AMs in the alveoli, the efficacy of AM-mediated immunity against S. pneumoniae depends on the magnitude of the bacterial inoculum (16, 17). After antigen-activation, AMs release different cytokines (TNF-α, IFN-γ, GM-CSF, and IL-6) that attract neutrophils (Nϕ) and monocytes, promoting their infiltration into the lung parenchyma (17). Natural killer (NK) cells also play an important role in the immune response against S. pneumoniae, producing IFNγ in the early stages of lung infection, and further favoring the activation and recruitment of Nϕ to the lung (18). Nϕ are the most abundant myeloid cell population in the lung and their lung infiltration is fundamental for bacterial clearance, particularly since it is responsible for bacterial phagocytosis and complement activation (19). Furthermore, bone marrow-derived populations of Ly6Chi monocytes migrate to peripheral tissues like the lung or the spleen in response to inflammatory stimuli or to the lesions caused by S. pneumoniae, and in a CCR2-dependent manner (20). Once recruited into peripheral tissues, Ly6Chi monocytes can further differentiate into monocyte-derived dendritic cells (moDCs) and monocyte-derived Mϕ (moMϕ), two populations that participate in bacterial removal in conjunction with Nϕ (20, 21).
Mononuclear phagocytic cells are quite heterogeneous and they display significant plasticity, which allow them to acquire specialized functions. In blood, two types of monocytes have been identified with differential phenotype and functions. The classical monocytes (cMO) are CD11b+Ly6ChiCX3CR1loCD43lo, are found in tissues under homeostasis, and during inflammation they are recruited to the damaged tissues and produce proinflammatory cytokines such as TNF-α and IL6, as well as enhancing the inducible nitric oxide synthase (iNOS) available. These classical inflammatory monocytes participate in the local innate immune control of several pathogens, such as Toxoplasma gondii, Leishmania donivani, Leishmania major and Listeria monocytogenes (22–25). By contrast, the non-classical monocytes (ncMO) monocytes are CD11b+Ly6CloCX3CR1hiCD43hi, they patrol the endothelium and have been shown to have angiogenic activity by secreting vascular endothelial growth factor (VEGF) and IL-10. These ncMO monocytes are also recruited to the damaged tissues and may be involved in tissue repair and wound healing (20, 26–28). In the inflammed tissues, recruited monocytes differentiate into Mϕ. Two phenotypes of Mϕ have been proposed that are related to their functional profile (29–31). Upon activation in response to proinflammatory cytokines like TNF-α and IFN-γ, monocytes differentiate to Mϕ that adopt the M1 phenotype with enhanced microbicidal activity, and secrete pro-inflammatory cytokines (30). By contrast, M2 Mϕ are induced by the Th2-related cytokines (IL-13 and IL-4) and they secrete anti-inflammatory factors like IL-10 and TGF-β. Thus, M2 Mϕ participate in tissue repair and in the phagocytosis of apoptotic cells, limiting inflammation (32). The equilibrium between the M1 and M2 populations is essential to produce an appropriate IR, including its resolution. S. pneumoniae infection induces an inflammatory response mediated by M1 Mϕ, killing pathogens and triggering adaptive immunity. Autolysis of S. pneumoniae leads to the production of Ply and it induces the production of reactive oxygen species (ROS) by Nϕ (33). Likewise, other bacterial infections like those caused by Mycobacterium tuberculosis, induce TLR-activation of Mϕ, bacterial phagocytosis and intense ROS production as the main core of the IIR against such infection (34, 35).
Here we examined the dynamics of the lung IIR against S. pneumoniae, using a clinical isolate obtained from peripheral blood and analyzing the myeloid populations that accumulate, their markers of activation and their dependency on the TLR4-MyD88 pathway after intranasal administration. Our data show that 3 days post infection (dpi) there is a local increase in the Nϕ, monocytes, and the moMϕ and moDC monocyte-derived populations, with a prominent cMO profile. In the absence of TLR4 or MyD88, the production of these myeloid populations was dampened. The ROS produced by different myeloid cell types highlighted the importance of the TLR4-MyD88 axis in the oxidative burst within the myeloid compartment. Moreover, the lung cytokine profile and the production of ROS were less affected by infection in MyD88–/– mice than in TLR4–/– mice, although these responses were weaker than in infected WT mice. Hence, an alternative TRIF pathway appears to participate in the IIR against S. pneumoniae. Together, these data indicate that both TLR4-MyD88 and TLR4-TRIF signaling are involved in the recognition of S. pneumoniae by the cells generated through the myeloid IIR in the lung, highlighting the role of these pathways in the cytotoxic and regulatory responses that combat this bacterium.
Materials and Methods
Mice
Adult (8–10 weeks old) WT (C57BL/6 and C57BL/10), C57BL/10/TLR4–/– and C57BL/6/MyD88–/– mice were bred and maintained at the animal facility of the Centro Nacional de Microbiología-Instituto de Salud Carlos III (CNM-ISCIII, Madrid, Spain). All animal experiments were approved by the Institutional Review Board at the ISCIII, and carried out in strict accordance with EU and National Animal Care guidelines (directive 2010/63/EU and RD 53/2013). The experimental protocol was also approved by the Consejería de Medio Ambiente Comunidad de Madrid (PROEX110/15, PROEX021/18).
Induction of Infection
Mice were lightly anesthetized with an aerosol containing 4% isoflurane (Zoetis, United Kingdom) and they were inoculated intranasally with a suspension (20 μL in Phosphate Buffered Saline –PBS: Bio-Whittaker, Lonza) of the S. pneumoniae bacterial strain 1195 (serotype 3 and ST260 genotype). Strain 1195 is a clinical isolate obtained from peripheral blood that was obtained from a septic patient and submitted to the Spanish Pneumococcus Reference Laboratory. This 1195 strain was described previously (referred to as CipR1) (36) and other strains of the ST260 genotype have been used previously in a rabbit model of experimental meningitis (37). The pneumococcal strain was grown to mid-log growth phase in Todd-Hewitt broth supplemented with 0.5% yeast extract. The bacteria were then recovered by centrifugation and suspended in medium with 25% glycerol at a concentration of 108 cells/mL, and stored as aliquots at −80°C. For intranasal infection, aliquots were thawed, washed and resuspended in PBS immediately before use. We performed a dose-response study with this strain from 1 to 4 × 106 colony-forming units (CFUs) to determine the optimal concentration required to mount an immune response (Figure 1A).
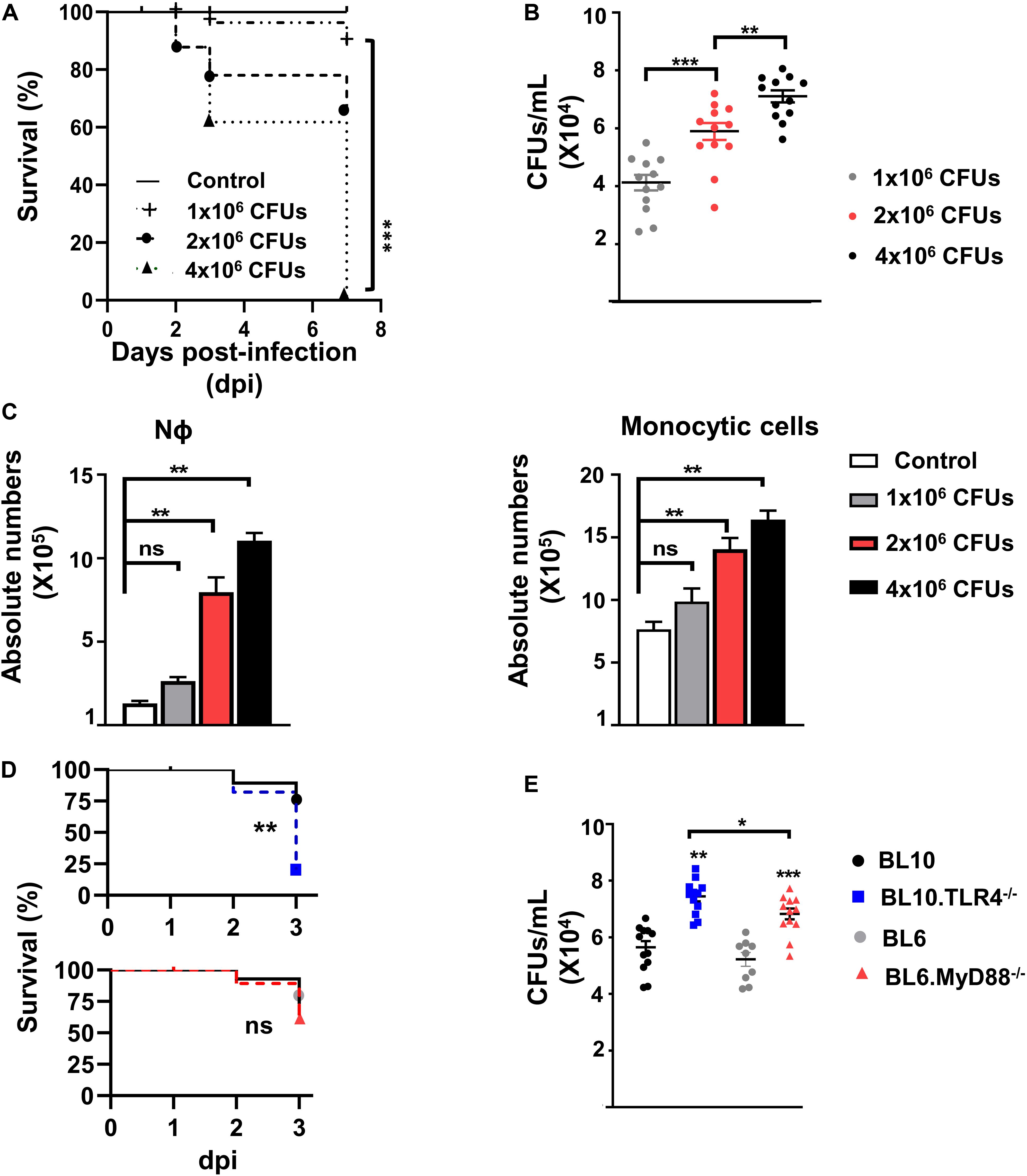
Figure 1. Dose-response study following S. pneumoniae bacterial infection of the lungs. (A) Survival of WT.BL10 mice after intranasal infection with different concentrations of S. pneumoniae CFUs. Survival was determined at 2, 3, and 7 dpi, and the differences between the groups were compared using Kaplan–Meier tests. The data represent measurements from three independent replicates: Control (n = 6); 1 × 106 CFUs (n = 8); 2 × 106 CFUs (n = 14); 4 × 106 CFUs (n = 10). Statistical analysis was performed using a log-rank Mantel–Cox test: ***p < 0.001. (B) CFUs determined in lung suspensions from infected mice at 72 h pi. Suspensions were plated on sheep blood agar plates and incubated for 24 h at 37°C in 5% CO2. The data represent the individual measurements of three independent experiments. Shown inside are means ± SEM; n = 12. (C) Quantification of Nϕ (Gr1hiCD11bhi) and monocyte cell populations (Gr1–CD11blo/hi) in whole lung cell suspensions (without ACK treatment) obtained at 3 dpi following infection with 1 × 106, 2 × 106 and 4 × 106 CFUs (as defined in Supplementary Figure 1). Bar graphs show the absolute numbers of each population calculated from the frequencies obtained by flow cytometry and the total number of cells counted in each sample in three independent experiments. Data are means ± SEM; control (n = 6); 1 × 106 CFUs (n = 8); 2 × 106 CFUs (n = 14); 4 × 106 CFUs (n = 10). (D) Survival of WT.BL10 (n = 12), TLR4–/– (n = 12), WT.BL6 (n = 10) and MyD88–/– (n = 12) mice after intranasal infection with 2 × 106 CFUs. Survival was assessed at 1, 2, and 3 dpi and the differences between the groups were compared using a log-rank Mantel–Cox test. (E) Colonies formed from the lung suspensions were counted at 3 dpi as in panel B; WT.BL10 (n = 12), TLR4–/– (n = 12), WT.BL6 (n = 9) and MyD88–/– (n = 12). The data in panels (B,C,E) were compared among multiple groups with one-way ANOVA and unpaired two tailed Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant.
Determination of the Colony-Forming Units
The lungs of the mice were dissected out at 1, 2, and 3 dpi, and cell suspensions were prepared by mechanical dissociation in 3 mL of cold (4°C) staining buffer (2.5% Fetal Calf’s Serum in PBS). No enzymatic digestion by collagenase treatment was used, since preliminary experiments showed an important reduction in the recovery of myeloid-gated cells and CD11b+ cells after such treatment (Supplementary Material). The lung tissue was cut into small pieces and cell suspensions were prepared by mechanical dissociation, disrupting the tissue and filtering through a 40 μm pore cell strainer (BD Biosciences). The filtrate was then centrifuged for 5 min at 110 g and 4°C in order to obtain the lung cells. The material recovered was analyzed by flow cytometry (see below), and the S. pneumoniae CFUs and cytokines in the supernatants were quantified. Colony-forming units were determined by growing serial dilutions of the lung supernatant on Mueller-Hinton sheep blood agar plates (BD Biosciences, San Jose, CA), counting the colonies formed after a 24 h incubation at 37°C in an atmosphere of 5% CO2. Dissemination of the bacteria to other organs was determined by CFU quantification in samples from spleen and olfactory bulbs at 3 dpi (50 ± 7 CFUs, n = 6 in splenic samples of infected BL10).
Flow Cytometry
Lung cell suspensions obtained from the entire lung were evaluated by flow cytometry, employing a multi-panel strategy to distinguish the different myeloid cell populations in the samples. Cell pellets from lung homogenates were treated for 2 min at room temperature (RT) with 1 mL ACK buffer (Potassium Bicarbonate: Life Technologies) to lyse the erythrocytes. The cells (4 × 106/200 μL) were then washed twice with staining buffer, recovered by a 5 min centrifugation at 110 g and 4°C, and then prepared in staining buffer. Cell viability was measured by Trypan blue (Merck) dye exclusion and viable cells were counted in a Neubauer’s chamber. Non-specific antibody binding was blocked by incubation for 10 min at 4°C with an anti-Fc-Block (clone 2.4G2: BD Biosciences) and the cell populations were then analyzed by Flow Cytometry after staining for 20 min at 4°C using the specific mAbs listed in Supplementary Table 1. Dead cells were discharged by staining with Fixable LIVE/DEAD violet-510 kit (Thermo Fisher Scientific) and the cells were fixed for 15 min at RT in 2% paraformaldehyde before acquisition on a LRS Fortessa X-20 cytometer (BD Biosciences). The samples were analyzed using the DIVA v8.0 software package (BD Biosciences) and the cell populations were analyzed after electronic gating on the basis of SSC-A and FSC-A, followed by a gating strategy to rule out doublets and dead cells (Supplementary Figure 1). At least 1–3 × 105 live cells were analyzed in each sample and the mAbs used in this study allowed the identification of lung myeloid populations, distinguishing the AM (Siglec-FhiCD11blo), NK (CD3–NK1.1+), Nϕ (Gr1hiCD11bhi), monocytes (Gr1– CD11bhiF4/80+CD11clo), moMϕ (Gr1–CD11bhiF4/80loCD11c+Ly6C–), and moDC (CD11bhiF4/80loCD11c+Ly6C+) populations. Finally, the differential expression of Ly6C and Ly6G was used to characterize the CD11b+ monocytic populations (22, 25, 27, 28): cMO (CD11bhiLy6ChiLy6G–) and ncMO (CD11bhiLy6CloLy6G–). Isotype fluorescence minus one (FMO) controls were performed to rule out non-specific fluorescence and to define gating boundaries (Supplementary Figure 1).
Cytometric Bead Array for Cytokines
Supernatants obtained from fresh lung suspensions (total volume 3 mL) were aliquoted and frozen at −80°C. The aliquots were thawed (only once) and the cytokines present in a 25 μL sample were analyzed (CXCL1, IL-18, IL-23, IL-12p70, IL-6, TNF-α, IL-1β, and IL-12p40) with the LEGENDplex Cytokine Analysis kit (BioLegend), according to the manufacturer’s recommendations, and using a FACS Canto I cytometer (BD Biosciences) and the BioLegend LEGENDplex software.
Quantitative Real-Time PCR
Total RNA from lung samples was extracted using NucleoZol (Macherey-Nagel) and reverse transcribed in a final volume of 25 μL with Oligo(dT)-primers, as described previously (38). The cDNA (1 μL) obtained was amplified quantitatively by real-time PCR (qPCR) on a CFX96TM Real-Time System using the SsoFastTMSupermixEvaGreen (Bio-Rad, Hercules, CA), as indicated elsewhere (39). The Bio-Rad CFX Manager software was used to calculate the CT of each reaction and the specific amount of cDNA in each sample was determined relative to the expression of the hypoxanthine phosphoribosyl transferase-1 (HPRT) gene by the 2–ΔCt method (40). The primers used in this study are listed in Supplementary Table 2.
Detection of Intracellular Reactive Oxygen Species
The ROS-sensitive probe 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA, 10 mM stock solution: Invitrogen) was used here on fresh cells surface-labeled as described in the Flow Cytometry section but without fixation procedure. The cells were then labeled for 40 min in the dark at RT with H2DCFDA (5 μM in PBS), washed twice with PBS and recovering the cells by centrifugation for 5 min at 4°C and 110 g, and resuspended them in a final volume of 1 mL. The labeled cells were incubated with 2 × 106 CFUs of heat-inactivated S. pneumoniae (60 min, 60°C) for 15, 30, and 60 min at 37°C, and they were then analyzed by flow cytometry to measure the ROS signals of the different myeloid populations. The baseline fluorescence of the cells was determined prior to their exposure to S. pneumoniae.
Statistical Analysis
The data are presented as the means ± SEM (Standard error of the mean). Statistical analyses were performed with the Prism 8.0 (Graph Pad) software, after testing the normality of the data distributions with the Kolmogorov–Smirnov and D’Agostino-Pearson tests. The data were compared with one-way ANOVA tests and two-tailed unpaired Student’s t-tests. Survival curves were obtained using Kaplan–Meier tests and analyzed using a log-rank (Mantel–Cox) test. A value of p < 0.05 was considered statistically significant: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant.
Results
Innate Immune Responses of Lung Cell After S. pneumoniae Infection
We performed initial experiments to establish the optimal dose for S. pneumoniae serotype 3 strain 1195 infection and the time required to mount an IIR, parameters that allowed us to study the myeloid cell subpopulations involved, as well as the role of TLR4 and MyD88 expression. Adult WT.BL10 mice were infected intranasally with increasing doses of bacteria (1 × 106, 2 × 106, and 4 × 106 CFUs) and their survival after infection was studied. A dose-dependent decrease in survival was found from 3 dpi, accompanied by an increase in bacterial CFUs in the lungs (Figures 1A,B). It is known that Nϕ and monocyte cell infiltration is essential for S. pneumoniae lung clearance (12, 41). Enhanced recruitment of Nϕ (Gr1hiCD11bhi) and monocytes (Gr1–CD11blo/hi) to the lung was evident at 3 dpi in mice that received 2 × 106 and 4 × 106 CFUs (Figure 1C). Thus, we chose the dose of 2 × 106 CFUs to infect and monitor the lung IIR at 3 dpi. Previous studies have shown that pneumococcal LTA and Ply were recognized by TLR2 and TLR4, as the main PRRs required to initiate the immune response (13, 14) and acting through the MyD88-signaling cascade. To analyze the contribution of TLR4 and MyD88 signaling to the IIR, TLR4–/– and MyD88–/– mice were infected intranasally with 2 × 106 CFUs S. pneumoniae (Figures 1D,E), and their survival and bacterial load was quantified at 1, 2, and 3 dpi (Figure 1E and Supplementary Figure 2). Since the different genetic backgrounds of the TLR4–/– (BL10) and MyD88–/–(BL6) mice may influence their IIR against S. pneumoniae, we used both WT.BL10 and WT.BL6 control strains, with no differences in survival and bacterial load detected between them. The TLR4–/–and MyD88–/– mice survived less time after infection than their corresponding WT animals (WT.BL10 and WT.BL6, respectively), although the TLR4–/– mice survived worse than the MyD88–/– mice at this CFU dose. Accordingly, the bacterial load was higher in the lungs of infected TLR4–/– than in the MyD88–/– mice, which both exceeded that in the WT infected mice (Figure 1E and Supplementary Figure 2).
Resident AMs (Siglec-FhiCD11blo) are essential to control bacterial numbers early after S. pneumoniae infection, driving a reduction in AM numbers by inducing their caspase-dependent apoptosis (16, 17, 42, 43). Natural killer (CD3–NK1.1+) cells also exert their effector and regulatory functions soon after infection. Accordingly, we found fewer AMs in the lungs of both infected WT strains on days 1–3 pi, along with a transient accumulation of NK cells in the lung at 1 dpi (Figures 2A, 3A). Myeloid-cells in the lung were analyzed by flow cytometry at 3 dpi (Figures 2B, 3B) and as expected, there was significant Nϕ recruitment in the infected lungs, coupled to a decrease in monocytes. Indeed, a decrease in F4/80 (a specific marker of mature Mϕs) and an increase in CD11c was observed, indicating the activation and differentiation of these mature Mϕs. When Ly6C expression was analyzed in the differentiated CD11chiF4/80lo population, an increase in the absolute numbers of both moMϕ and moDC was seen in infected mice (Figure 2B right dot plots, Figure 3C). The cMO/ncMO profile was defined on gated CD11b+ cells, detecting differential Ly6C and Ly6G expression as described previously. Monocytes in control and infected mice shifted toward a pro-inflammatory cMO/ncMO ratio (BL10 1.12 ± 0.09, n = 12; BL6 1.073 ± 0.174, n = 6) in the infected lungs when compared to the control non-infected mice (BL10 0.61 ± 0.04, n = 12; p < 0.001; BL6 0.68 ± 0.05, n = 6 p < 0.05; Figures 2C, 3D).
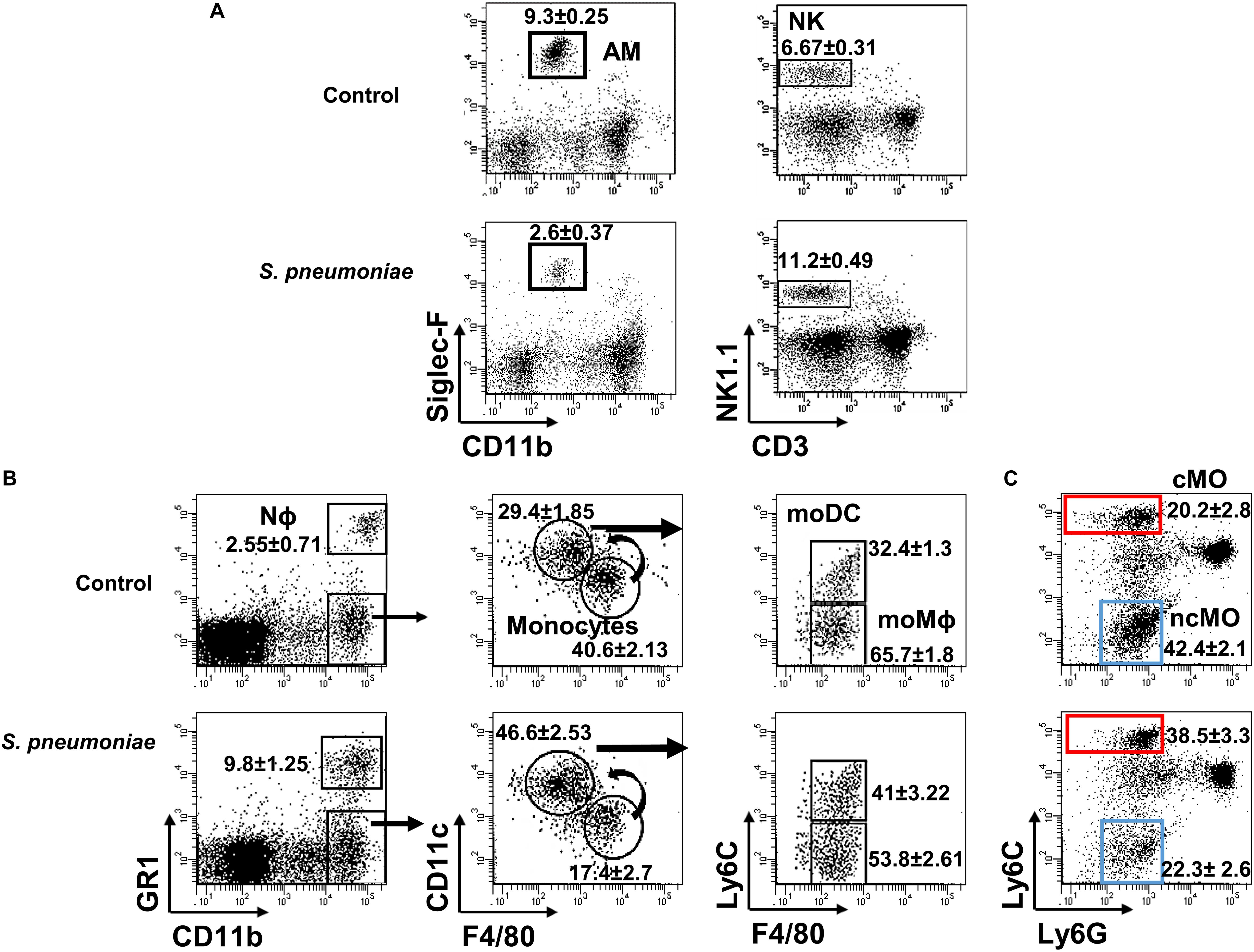
Figure 2. Lung myeloid populations after S. pneumoniae infection. Lung suspensions from infected and uninfected WT.BL10 mice were prepared at 1 and 3 dpi, and stained with the indicated mAbs (see section “Materials and Methods”) (A) Samples obtained at 1 dpi were stained with anti-CD11b and Siglec-F to identify AM (Siglec-FhiCD11blo) cells in the left dot plots. NK cells were identified using anti-NK1.1 and anti-CD3 antibodies: NK (NK1.1+CD3–) in the right dot plots. (B) At 3 dpi, myeloid and granulocyte cell populations were discriminated using the markers CD11b and Gr1, as described in Supplementary Figure 1 in which the sequential gating strategy to identify distinct monocyte populations is depicted. (C) The cMO and ncMO profile of monocytes was defined based on their Ly6C and Ly6G expression: cMO (CD11bhiLy6ChiLy6G–, red box) and ncMO (CD11bhiLy6CloLy6G–, blue box). Representative dot plots are shown and the numbers inside are the percentages mean ± SEM of four independent experiments, WT.BL10 (n = 12).
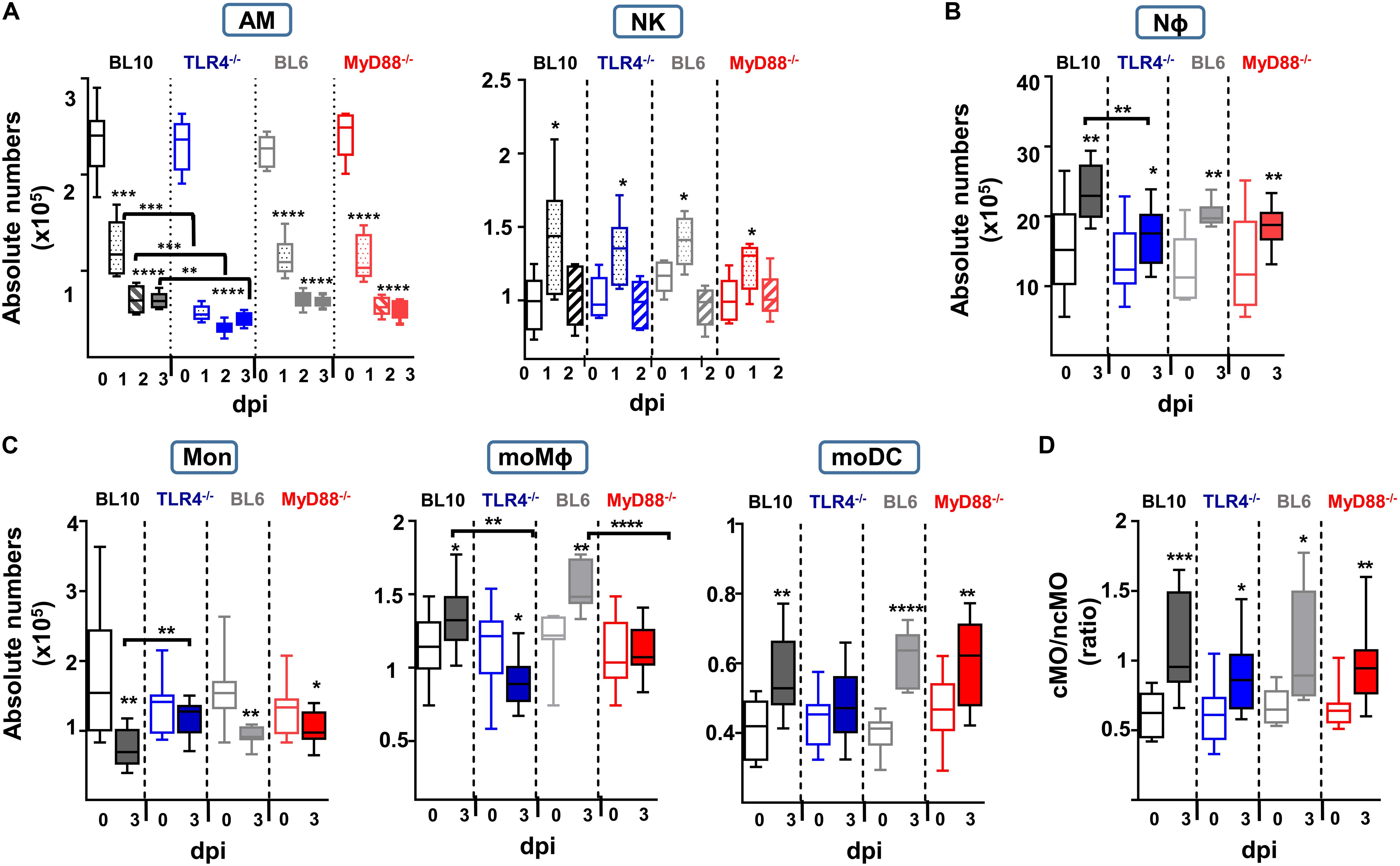
Figure 3. Quantification of the absolute numbers of different lung innate subsets after S. pneumoniae (2 × 106 CFUs) instillation to WT.BL10, TLR4–/–, WT.BL6 and MyD88–/– mice. (A) AM and NK cells were identified and counted at 0, 1, 2, and 3 dpi. Absolute numbers were calculated using the frequencies obtained by cytometry and the total number of cells counted in each sample: WT.BL10 (n = 6), TLR4–/– (n = 6), WT.BL6 (n = 6), and MyD88–/– (n = 6) at 0, 1, 2, and 3 dpi. (B–D) At 3 dpi the IIR was analyzed by quantifying the Nϕ cells, monocytes, the moDC and moMϕ cells, and the cMO and ncMO populations from control and infected mice. Box-and-whisker plots show the median values, with the bottom and top of the box indicating the first quartile to the third quartile, with the minimum and maximum values from four independent experiments: WT.BL10 (n = 12), TLR4–/– (n = 15), WT.BL6 (n = 6), and MyD88–/– (n = 15). All the populations were analyzed by one-way ANOVA and with an unpaired two-tailed Student’s t-test: *p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Together these data reveal distinct local cell dynamics in the lungs of S. pneumoniae infected mice, and both WT.BL10 and WT.BL6 control strains behave similar in terms of myeloid cell recruitment. These changes involved an initial decrease in AM cells during the first 3 dpi, along with the accumulation of NK cells on day 1. Subsequently, there was neutrophil infiltration and monocyte differentiation toward the moMϕ and moDC phenotypes at 3 dpi, with a predominant cMO profile.
Defective Innate Response to S. pneumoniae in Infected TLR4 and MyD88 Deficient Mice
The survival of TLR4–/– and MyD88–/– infected mice was worse than that of WT mice, and they had a higher bacterial load, especially the TLR4–/– mice (Figures 1D,E). As in WT.BL10 and WT.BL6 mice, S. pneumoniae infection produced an accumulation of NK cells and an important reduction in AM cells in TLR4–/– and MyD88–/– mice (Figure 3A), with the decrease in AM cells more pronounced in TLR4–/– infected mice than in either of the WT strains or the MyD88–/– mice (TLR4–/– versus MyD88–/– values p < 0.001 and p < 0.01 at days 1 and 2 pi, respectively). There was similar recruitment of NK cells to the lungs at 1 dpi in the four strains of mice. By contrast, deficient accumulation of Nϕ was detected by 3 dpi in TLR4–/– mice relative to the WT.BL10 infected mice (Figure 3B).
Although the lungs from infected WT.BL10 and WT.BL6 mice have fewer monocytes due to their differentiation toward a moMϕ and moDC populations (Figure 2B, right dot plots), there was only a minimal reduction in the monocyte populations in infected TLR4–/– mice (Figure 3C), with no increase in their moDC population and a decrease in moMϕ number. By contrast, there was a reduction in monocytes in MyD88–/– infected mice and differentiation toward the moDC lineage was observed, as occurred in WT.BL6 animals, although there was no increase in the moMϕ cell population that remained much lower than in the WT.BL6 mice (Figure 3C). Upon infection, and based on the Ly6C expression (20, 22, 26, 28) the cMO/ncMO profile shifted slightly toward a pro-inflammatory profile in TLR4–/– mice (0.64 ± 0.05 uninfected, 0.89 ± 0.079 infected mice, n = 15; p < 0.05), whereas this ratio was higher in MyD88–/– mice (0.68 ± 0.04 uninfected mice, 1.003 ± 0.087 infected, n = 15; p < 0.01), similar to that found in WT.BL10 and WT.BL6 animals. In parallel, there was an important reduction in adaptive B cell and CD4+ T cell recruitment to the lungs of TLR4–/– and MyD88–/– infected mice at 3 dpi, whereas CD8+ cells were recruited normally (manuscript in preparation).
In summary, these findings demonstrate important defects in the local lung IIR in MyD88–/– and TLR4–/– mice, with a more severe phenotype in the latter model. These changes included important defects in AM, NK, and Nϕ cell recruitment, as well as significant defects in monocyte differentiation and the adaptive response.
Differential Impairment of Inflammatory Cytokine Production in TLR4–/– and MyD88–/– Infected Mice
To further understand the susceptibility of the TLR4–/– and MyD88–/– mice to S. pneumoniae infection, we assessed the cytokines in the lung homogenates by 3 dpi (Figure 4A). There was a significant increase in pro-inflammatory cytokines like CXCL1, TNF-α, IL-6, and IL-1β in WT (BL10 and BL6) mice following S. pneumoniae infection, with CXCL1 being greater in infected WT.BL6 compared to WT.BL10 (p < 0.05), whereas no such increase in these cytokines was evident in TLR4–/– infected mice. By contrast, there was a mild increase in CXCL1, TNF-α, and IL-6 in MyD88–/– infected mice. The expression of TNF-α and IL-6 transcripts was assessed in lung homogenates from infected mice by qPCR at 3 dpi (Figure 4B), and while there was a strong increase in both cytokines in WT.BL10 and MyD88–/– infected mice, this was not the case in TLR4–/–mice. An early increase in IL-23 and IL-12p40 has been described 24 h pi with S. pneumoniae (44, 45). There was no change in IL-23, IL-18 and IL-12p70 in the lungs of WT (BL10 and BL6), TLR4–/– and MyD88–/– mice at 3 dpi, although IL-12p40 diminished in the WT mice (Figure 4A). Thus, in the absence of TLR4 S. pneumoniae impaired the pro-inflammatory cytokine cascade in the lung (CXCL1, TNF-α, IL-6, and IL-1β) at 3 dpi. Furthermore, inflammatory cytokines were moderately affected in MyD88–/– infected mice, indicating that the inflammatory response to S. pneumoniae is partially dependent on the MyD88 transduction machinery.
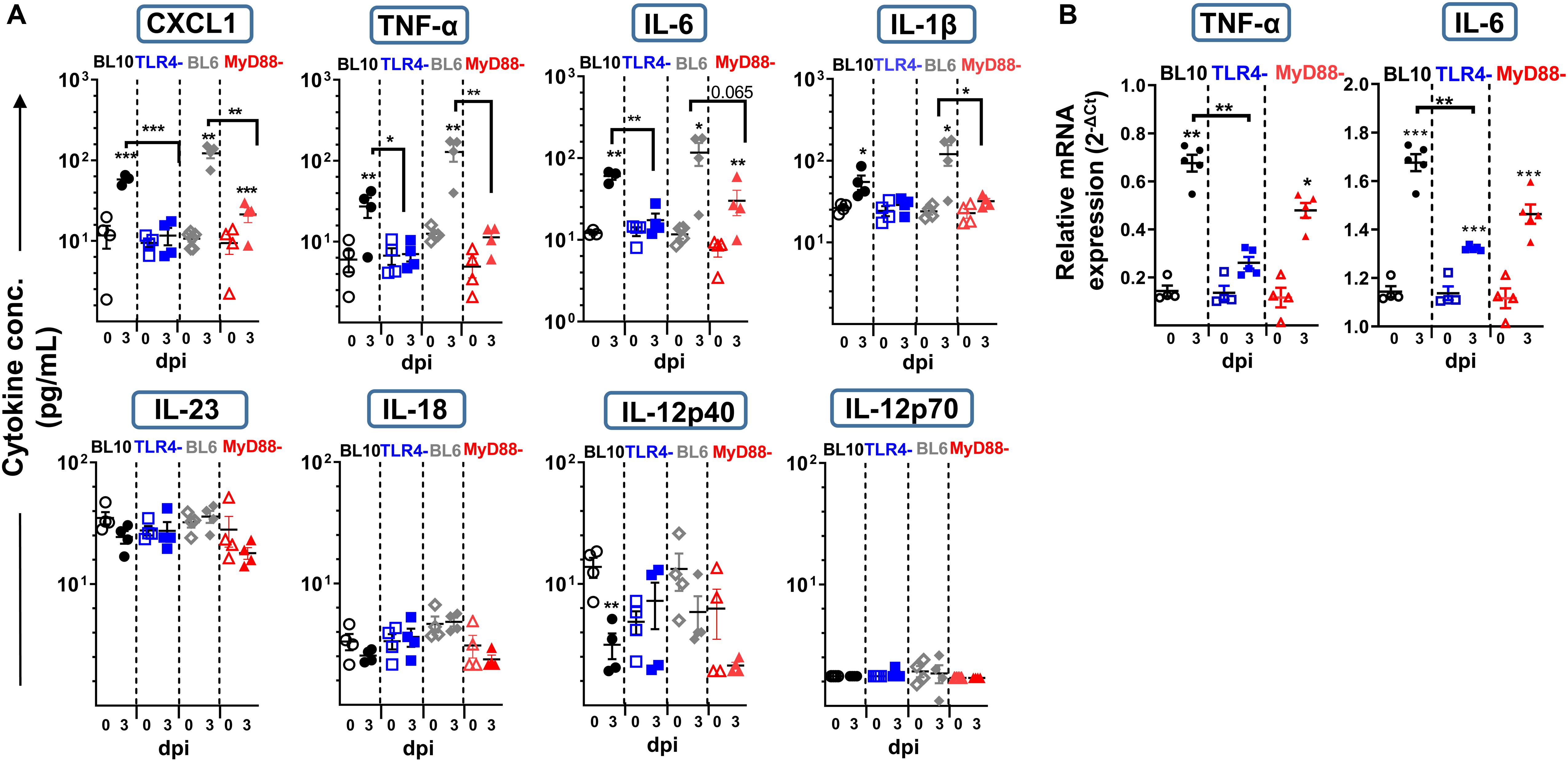
Figure 4. Lung cytokine production at 3 dpi in WT.BL10, TLR4–/–, WT.BL6 and MyD88–/– S. pneumoniae (2 × 106 CFUs) infected mice. Lung suspensions from control (empty symbols) and infected mice (filled symbols) were prepared and their supernatants analyzed. (A) CXCL1, IL-18, IL-23, IL-12p70, IL-6, TNF-α, IL-1β, and IL-12p40 protein levels were measured in LEGENDplex Cytokine Assays. (B) TNF-α and IL-6 gene expression in the lungs of WT.BL10, TLR4–/–, and MyD88–/– measured by qPCR at 3 dpi using the Bio-Rad CFX Manager software to calculate the CT for each reaction. The amount of each specific transcript in each cDNA sample was determined and expressed as the 2–ΔCT, relative to that of HPRT transcripts. The data are shown as in Figure 1B, representing 4-to-6 different samples evaluated in duplicate. The populations were compared with one-way ANOVA and with an unpaired two-tailed Student’s t-test: *p < 0.05, ** p < 0.01.
In vitro NADPH Oxidase-Activity of Distinct Myeloid Populations After S. pneumoniae Challenge
Nϕ and monocytes can activate NAPDH oxidase activity and generate ROS as part of the host’s defense against bacteria and parasites (46, 47). We studied the in vitro production of ROS in lung cell preparations after challenge with heat-killed S. pneumoniae, using the ROS-sensitive fluorescence probe H2DCFDA in Nϕ and in a variety of monocyte cell populations at different time points (Figure 5A). As expected, Nϕ rapidly produced large amounts of ROS in WT (BL10 and BL6) mice, with weaker induction and with slower kinetics in moDC, moMϕ and monocytes. The in vitro production of ROS in TLR4–/– and MyD88–/– mice differed significantly to that in WT (BL10 and BL6) mice (Figure 5B). In the case of TLR4–/– lung cell preparations, ROS production by Nϕ and by the rest of monocyte populations was inhibited after 30 and 60 minutes. Furthermore, while the production of ROS was stronger in MyD88–/– mice than in TLR4–/– mice, in all cell types analyzed except in moMϕ, it remained weaker and with slower kinetics than that detected in WT.BL6 mice.
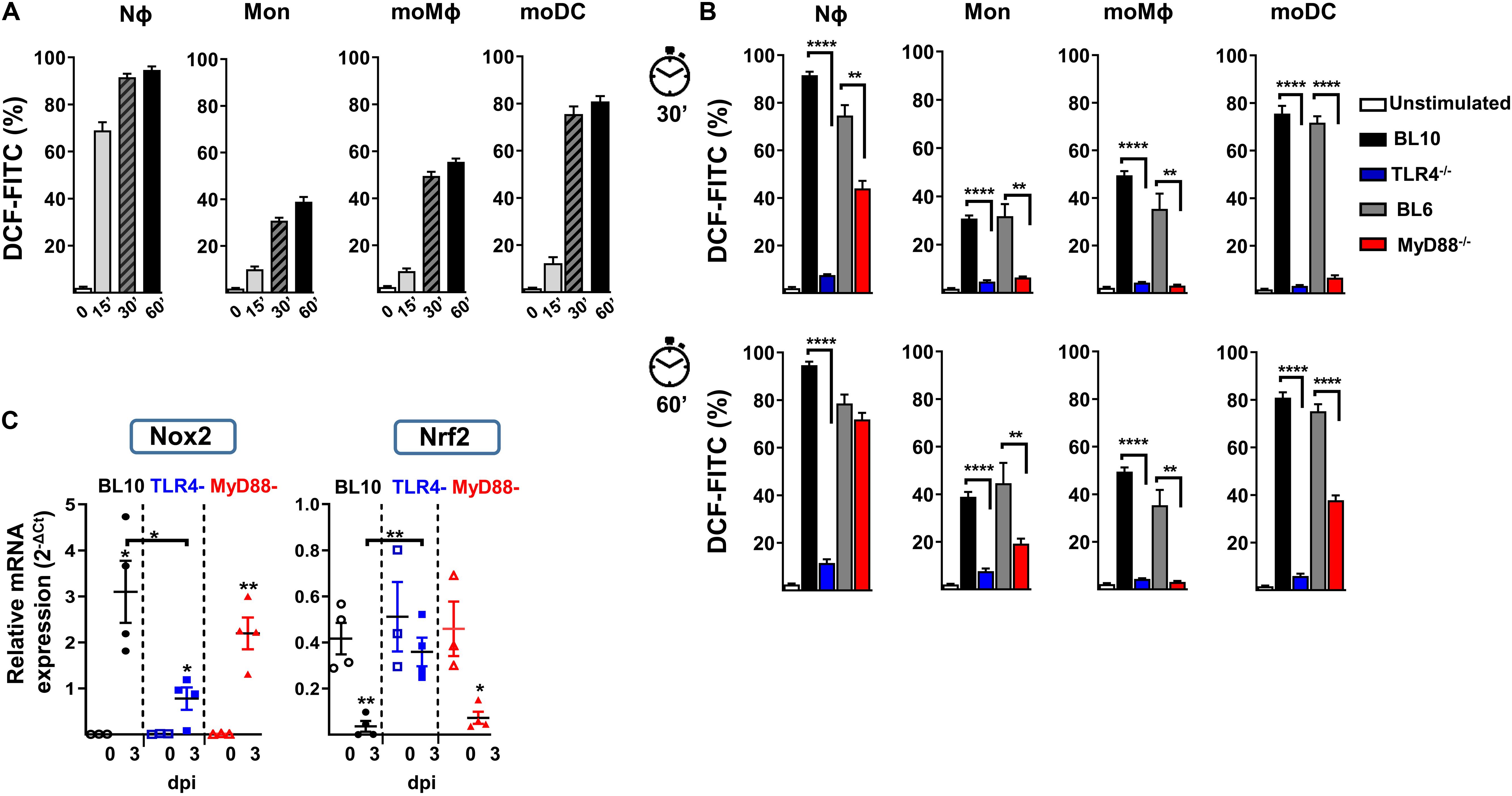
Figure 5. In vitro NADPH oxidase activity against heat-inactivated S. pneumoniae in lung cells. (A) Stained lung cells from WT.BL10 mice were labeled with the H2DCFDA-ROS sensitive probe (5 μM, DCF-FITC) and incubated with 2 × 106 CFUs heat-inactivated S. pneumoniae (60 min, 60°C) for 0, 15, 30, and 60 min at 37°C before analyzing them by flow cytometry. (B) Detection of ROS in different myeloid populations from naïve mice using heat-inactivated S. pneumoniae at 30 and 60 min as in panel (A). The bar graphs show the proportion of H2DCFDA+ cells. Unstimulated controls were shown for WT.BL10 animals. The data represents the mean ± SEM: WT.BL10 (n = 7), TLR4–/– (n = 6), WT.BL6 (n = 5), and MyD88–/– (n = 6). (C) Nox2 and Nrf2 gene expression by lung cells from WT.BL10, TLR4–/–, and MyD88–/– mice measured by qPCR at 3 dpi, as described in Figure 4. The differences between the groups were analyzed by one-way ANOVA and with an unpaired two-tailed Student’s t-test: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Regulation of the ROS-NAPDH pathway is mainly due to activation of the NADPH oxidase-2 (Nox2) transcription factor (48) and it is negatively regulated by Nrf2 (49). The transcripts of these two genes were quantified by qPCR in the three mouse models studied and Nox2 induction was evident in all infected mice, although it was weaker in the infected TLR4–/– mice (Figure 5C). Furthermore, an important reduction of Nrf2 was evident in lung samples from WT.BL10 and MyD88–/– infected mice but not in those from TLR4–/– infected mice, in which it remained unchanged. Together, maintaining Nrf2 levels and weaker Nox2 induction may contribute to the diminished ROS production in TLR4–/– infected mice.
Discussion
The airway and alveolar epithelia represent the initial barrier for the recognition of respiratory pathogens to initiate a immune response against them. Bacterial airway infections, including those caused by S. pneumoniae, are a major cause of worldwide morbidity and mortality. Indeed, pneumonia is the leading cause of mortality in children below 5 years of age, although global child mortality has decreased substantially (50, 51). In addition, the mortality of individuals over 65 years due to pneumonia remains unchanged since 1990, in part because the coverage of pneumococcal vaccines remains low (52, 53). Nasopharyngeal colonization with S. pneumoniae in healthy children is common, indicating a role of the host response in avoiding invasive pneumococcal disease.
Identifying mechanisms that can prevent bacterial lung infection is a global health priority, which requires a better understanding of the immune mechanisms involved in response to S. pneumoniae infection. The local IIR in the lung is important to combat S. pneumoniae, which involves the coordination of multiple cell populations and the activation of effector functions like phagocytosis, cytokine release, the complement cascade and antigen-presentation. Indeed, the IIR against S. pneumoniae is initiated through the recognition of PAMPs by TLRs, whose role and that of TLR-adaptor proteins and inflammasome complexes, has been studied using mouse models of infection (54–57). MyD88 and TLR4 are involved in local and systemic bacterial control, as well as in the recruitment of polymorphonuclear leukocytes in the lung (54). While TLR2 plays only a modest role in the immune response to S. pneumoniae in the respiratory tract, without affecting the overall antibacterial defense during infection (12), TLR9 has been implicated in the early clearance of bacteria, with no significant effect on local cytokine production (55). By contrast, TLR4, which signals both through MyD88-dependent and MyD88-independent (TRIF) pathways, also plays an important role by recognizing S. pneumoniae Ply and LTA (13) (Figure 6A). The impact of TLR recognition and signaling depends on the bacterial strain, and hence the pneumococcal serotype and the dose used to induce pneumonia may yield different results (54, 57). This study was carried out using a clinical serotype 3 encapsulated S. pneumoniae isolate, and by studying myeloid markers of activation and differentiation, we identified the myeloid events in the lung, local inflammatory responses and the ability to induce ROS production in response to infection of TLR4 and MyD88 deficient mice.
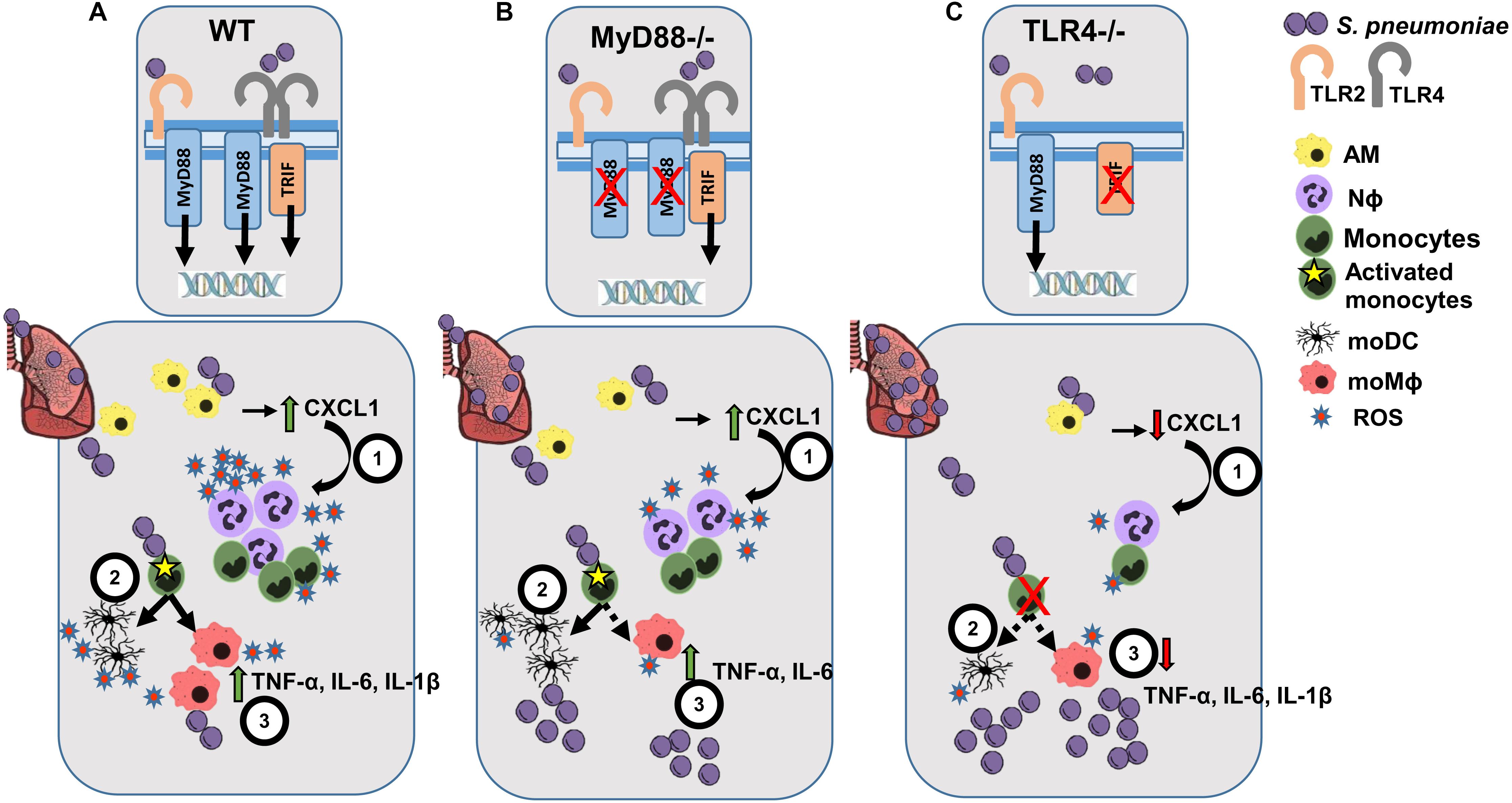
Figure 6. Scheme of the proposed lung IIR against S. pneumoniae in WT, MyD88–/– and TLR4–/– mice. (A) After infection of WT mice, TLR2 and TLR4 recognizes different PAMPs from S. pneumoniae activating the TRIF- and MyD88-dependent transduction pathways. After the initial bacterial encounter with AM cells, CXCL1 (1) is released that favors the early recruitment and activation of Nϕ cells, leading to substantial ROS production. (2) Differentiation of activated monocytes to moDCs and moMϕ helps to amplify the inflammatory response (3) through the secretion of a broad spectrum of cytokines and ROS production. (B) Infected MyD88–/– mice can still signal through the TLR4-TRIF-dependent pathway, leading to a weak IIR response while maintaining an inflammatory cytokine environment. (C) TLR4–/– infected mice experience impaired Nϕ recruitment and almost a complete blockade of monocyte differentiation, which leads to a very poor cytokine response and the development of a more intense bacterial burden.
Consistent with these previous studies (54, 57), we show here that TLR4–/– and MyD88–/– infected mice have a higher bacterial load than their corresponding WT counterparts. Interestingly, lungs from infected MyD88–/– mice had fewer bacterial CFUs than TLR4–/– mice, which may indicate that the TRIF-dependent TLR4-signaling cascade plays an important role in bacterial clearance (Figures 6B,C). It was previously shown that the interaction between Ply and TLR4 during pneumococcal colonization of the nasopharynx is important for protection (14). Indeed, our results suggest an impaired immune response to S. pneumoniae in TLR4–/– mice and to a lesser extent in MyD88–/–mice, compared to their corresponding WT genetic background (BL10 and BL6, respectively). Infected TLR4–/– mice suffer a stronger reduction in AM cells and defective Nϕ induction. By contrast, the reduction of AM and the induction of Nϕ cells was similar in MyD88–/– and WT.BL6 mice. As reported previously (20, 21), there was rapid differentiation of monocytes to moMϕ and moDC in S. pneumoniae infected WT animals. This induction of moMϕ was not observed in MyD88–/– infected mice and it was severely dampened in S. pneumoniae infected TLR4–/– mice. By contrast, moDC increased in MyD88–/– infected mice to a similar extent as in the WT.BL6 mice, whereas there was no increase in moDC number in TLR4–/– infected mice. The ratio of the cMO/ncMO populations in S. pneumoniae-infected TLR4–/– mice reflected a cMO inflammatory monocyte profile, whereas that found in MyD88–/– and in both the WT strains shifted toward a ncMO phenotype.
All these cellular defects after S. pneumoniae inoculation, mostly evident in TLR4–/– mice, may contribute to the incomplete lung cytokine profile in these mice. Firstly, CXCL1 release, together with that of IL-1β and IL-6, plays a central role in Nϕ mobilization from the bone marrow, in Nϕ activation and in the control of bacterial dissemination in the lung after pneumococcal infection (58, 59). At 3 dpi, there was more CXCL1, TNF-α, IL6, and Il-1β in the lungs of WT (BL10 and BL6) mice, while IL-18 and IL-23 were not enhanced at this time, perhaps due to an earlier action of these cytokines at 24 h (44, 45, 48). Ply is not involved in the pneumococcal induction of IL-12p40 in murine Mϕ (60) and indeed, we detected less IL-12p40 in the lungs of infected WT (BL10 and BL6) mice relative to their uninfected WT controls. These changes may be indicative of a transient Th1 response occurring in the first 24 h pi, and related to the fast action of AM and NK cells as the first innate barrier to impede bacterial growth. In TLR4-defective mice there was no increase in CXCL1, TNF-α, IL6, and Il-1β at 3 dpi, while the increase in these cytokines was only mild in MyD88-deficient mice. Whether this transient inflammatory lung environment could influence the appearance of trained Mϕ cells (resident and bone marrow-derived), with an enhanced capacity to respond to a second PAMP-associated challenge, will be addressed in the near future. As such, an in vivo model of trained immunity should be useful in designing novel “trained-induced” based vaccines.
Bacterial phagocytosis and ROS production are essential to control S. pneumoniae pathogen-dissemination and overgrowth. Nϕ cells fulfill a central role in removing invading pneumococci through the rapid production of intracellular and/or extracellular ROS in response to PAMPs (61), or after phagocytosis (58). The release of Ply after autolysis of pneumococci activates NADPH oxidase and the intracellular production of ROS by Nϕ (33). Our results following in vitro stimulation with heat killed S. pneumoniae revealed that Nϕ were the fastest producers of ROS, although moMϕ and moDC can also produce notable amounts of ROS albeit with delayed kinetics and over longer periods. These cells not only play a key role in killing ingested pathogens but also in antigen presentation, cross-presentation and the activation of cytolytic T cells (62). Reactive oxygen species production by Nϕ and by all the monocyte-derived populations was severely inhibited in TLR4–/– and to a lesser extent in MyD88–/– mice, which may at least in part explain the rapid growth of S. pneumoniae in these animals. In humans, mitochondrial ROS production by AM cells was dampened in chronic obstructive pulmonary disease (COPD) patients but not in smokers, or in control individuals after in vitro challenge with Haemophilus influenzae or S. pneumoniae (63). Moreover, NADPH defects in humans favor fungal and bacterial infection (61). Likewise, the bactericidal activity of the fluoroquinolones levofloxacin and moxifloxacin against S. pneumoniae is related to the production of ROS by the bacteria, associated with transcriptional alterations induced by these fluoroquinolones (64–66). Moreover, oxygen-derived metabolites can also damage infected tissues, regulate immune functions and induce apoptosis (58, 67). The levels of the ROS inducer Nox2 and the antioxidant Nrf2 must be counterbalanced in order to overcome lung infection by S. pneumoniae, as demonstrated in WT and MyD88–/– mice but not in TLR4–/– mice. Indeed, targeting mitochondrial regulators of NADPH-oxidase production has been evaluated in leukemia cells (68), ischemic cerebral neurons (69), and human Nϕ cells after LPS activation (70). It is tempting to speculate that novel therapies of this type might benefit COPD patients, PRR-immunodeficient subjects, aged recurrent-pneumonia and patients with sepsis as promising alternatives to conventional antibiotic treatments. Likewise, such therapies could be useful to combat an important number of multi-resistant bacteria, with important implications for treatment and national healthcare budgets.
In summary, this study involved an in-depth analysis of different resident and recruited innate cell populations in the lung, addressing their cytokine production, their differentiation into distinct phenotypes and NADPH-oxidative metabolism, which in conjunction produces efficient pneumococcal clearance. All these host-mediated responses were severely impaired when TLR4 was absent, demonstrating the critical involvement of this receptor in the defense against the Gram-positive S. pneumoniae. In MyD88 deficient mice, a partial effect on the innate cell content, cytokine profile and ROS production was evident, indicating a role for either the TRIF-dependent and MyD88-independent signaling pathways, or alternatively, the implication of other PRRs like TLR9. As a result, our data highlight the importance of the TLR4-MyD88 axis in the recognition and efficient response to S. pneumoniae infection in the lung, raising new challenging questions that warrant further attention.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding authors.
Ethics Statement
This study was carried out in accordance with EU and National Animal Care guidelines (directive 2010/63/EU and RD 53/2013). The procedures were approved by the Institutional Review Board at the ISCIII and the “Consejería de Medio Ambiente Comunidad de Madrid” (PROEX110/15 and PROEX021/18).
Author Contributions
RS-T performed the experiments, contributed substantially to the analysis and interpretation of the data, and reviewed the manuscript. IC analyzed the data and reviewed the manuscript. JM, MR, and CR performed the experiments. MP and MA helped with the flow cytometry experiments. EC reviewed the manuscript. MF prepared the bacteria and performed the CFU analysis. AC provided bacteria and reviewed the manuscript. MG designed the experimental procedures, made substantial contributions to the analysis and interpretation of the data, and critically reviewed the manuscript. BA designed the experimental procedures, made substantial contributions to the analysis and interpretation of the data, and drafted and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants of Ministerio de Ciencia SAF 2015-70880-R, RTI 2018-099114-B-100, BIO 2017-82951-R, and ISCIII PI14CIII/00049.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Julián Pardo for providing TLR defective mice (University Zaragoza, UNIZAR) and Dr. Mark Sefton medical writer at BiomedRed SL, for editing the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.02120/full#supplementary-material
References
1. Kadioglu A, Weiser JN, Paton JC, Andrew, PW. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol. (2008) 6:288–301. doi: 10.1038/nrmicro1871
2. Fitzwater SP, Chandran A, Santosham M, Johnson, HL. The worldwide impact of the seven-valent pneumococcal conjugate vaccine. Pediatr Infect Dis J. (2012) 31:501–8. doi: 10.1097/INF.0b013e31824de9f6
3. Verity R, Okell LC, Dorigatti I, Winskill P, Whittaker C, Imai N, et al. Estimates of the severity of coronavirus disease 2019: a model-based analysis. Lancet Infect Dis. (2020) 20:669–77. doi: 10.1016/S1473-3099(20)30243-7
4. Marimon JM, Ardanuy, C. Epidemiology of pneumococcal diseases in Spain after the introduction of pneumococcal conjugate vaccines. Enferm Infecc Microbiol Clin. (2020). doi: 10.1016/j.eimc.2020.02.016 [Epub ahead of print].
5. Campbell, GD Jr., Silberman, R. Drug-resistant Streptococcus pneumoniae. Clin Infect Dis. (1998) 26:1188–95. doi: 10.1086/520286
6. Jacobs MR, Felmingham D, Appelbaum PC, Gruneberg RN, Alexander Project, G. The alexander project 1998-2000: susceptibility of pathogens isolated from community-acquired respiratory tract infection to commonly used antimicrobial agents. J Antimicrob Chemother. (2003) 52:229–46. doi: 10.1093/jac/dkg321
7. van Hoek AJ, Andrews N, Waight PA, Stowe J, Gates P, George R, et al. The effect of underlying clinical conditions on the risk of developing invasive pneumococcal disease in England. J Infect. (2012) 65:17–24. doi: 10.1016/j.jinf.2012.02.017
8. Takeda K, Kaisho T, Akira, S. Toll-like receptors. Annu Rev Immunol. (2003) 21:335–76. doi: 10.1146/annurev.immunol.21.120601.141126
9. Iwasaki A, Medzhitov, R. Regulation of adaptive immunity by the innate immune system. Science. (2010) 327:291–5. doi: 10.1126/science.1183021
10. Schwandner R, Dziarski R, Wesche H, Rothe M, Kirschning, CJ. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J Biol Chem. (1999) 274:17406–9. doi: 10.1074/jbc.274.25.17406
11. Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock, D. Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J Immunol. (1999) 163:1–5.
12. Knapp S, Wieland CW, van ’t Veer C, Takeuchi O, Akira S, Florquin S, et al. Toll-like receptor 2 plays a role in the early inflammatory response to murine pneumococcal pneumonia but does not contribute to antibacterial defense. J Immunol. (2004) 172:3132–8. doi: 10.4049/jimmunol.172.5.3132
13. Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. (1999) 11:443–51. doi: 10.1016/s1074-7613(00)80119-3
14. Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, et al. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci USA. (2003) 100:1966–71. doi: 10.1073/pnas.0435928100
15. de Vos AF, Dessing MC, Lammers AJ, de Porto AP, Florquin S, de Boer OJ, et al. The polysaccharide capsule of Streptococcus pneumonia partially impedes MyD88-mediated immunity during pneumonia in mice. PLoS One. (2015) 10:e0118181. doi: 10.1371/journal.pone.0118181
16. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. (2013) 210:1977–92. doi: 10.1084/jem.20131199
17. Camberlein E, Cohen JM, Jose R, Hyams CJ, Callard R, Chimalapati S, et al. Importance of bacterial replication and alveolar macrophage-independent clearance mechanisms during early lung infection with Streptococcus pneumoniae. Infect Immun. (2015) 83:1181–9. doi: 10.1128/IAI.02788-14
18. Baranek T, Morello E, Valayer A, Aimar RF, Brea D, Henry C, et al. FHL2 regulates natural killer cell development and activation during Streptococcus pneumoniae infection. Front Immunol. (2017) 8:123. doi: 10.3389/fimmu.2017.00123
19. Menter T, Giefing-Kroell C, Grubeck-Loebenstein B, Tzankov, A. Characterization of the inflammatory infiltrate in Streptococcus pneumoniae pneumonia in young and elderly patients. Pathobiology. (2014) 81:160–7. doi: 10.1159/000360165
20. Shi C, Pamer, EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. (2011) 11:762–74. doi: 10.1038/nri3070
21. Carotta S, Dakic A, D’Amico A, Pang SH, Greig KT, Nutt SL, et al. The transcription factor PU.1 controls dendritic cell development and Flt3 cytokine receptor expression in a dose-dependent manner. Immunity. (2010) 32:628–41. doi: 10.1016/j.immuni.2010.05.005
22. Biswas A, Bruder D, Wolf SA, Jeron A, Mack M, Heimesaat MM, et al. Ly6C(high) monocytes control cerebral toxoplasmosis. J Immunol. (2015) 194:3223–35. doi: 10.4049/jimmunol.1402037
23. Glennie ND, Volk SW, Scott, P. Skin-resident CD4+ T cells protect against Leishmania major by recruiting and activating inflammatory monocytes. PLoS Pathog. (2017) 13:e1006349. doi: 10.1371/journal.ppat.1006349
24. Jia T, Leiner I, Dorothee G, Brandl K, Pamer, EG. MyD88 and type I interferon receptor-mediated chemokine induction and monocyte recruitment during Listeria monocytogenes infection. J Immunol. (2009) 183:1271–8. doi: 10.4049/jimmunol.0900460
25. Terrazas C, Varikuti S, Oghumu S, Steinkamp HM, Ardic N, Kimble J, et al. Ly6C(hi) inflammatory monocytes promote susceptibility to Leishmania donovani infection. Sci Rep. (2017) 7:14693. doi: 10.1038/s41598-017-14935-3
26. Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. (2007) 317:666–70. doi: 10.1126/science.1142883
27. Narasimhan PB, Marcovecchio P, Hamers AAJ, Hedrick, CC. Nonclassical monocytes in health and disease. Annu Rev Immunol. (2019) 37:439–56. doi: 10.1146/annurev-immunol-042617-053119
28. Guilliams M, Mildner A, Yona, S. Developmental and functional heterogeneity of monocytes. Immunity. (2018) 49:595–613. doi: 10.1016/j.immuni.2018.10.005
29. Geissmann F, Jung S, Littman, DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. (2003) 19:71–82. doi: 10.1016/s1074-7613(03)00174-2
30. Martinez FO, Gordon, S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. (2014) 6:13. doi: 10.12703/P6-13
31. Malyshev I, Malyshev, Y. Current concept and update of the macrophage plasticity concept: intracellular mechanisms of reprogramming and M3 macrophage “Switch” phenotype. Biomed Res Int. (2015) 2015:341308. doi: 10.1155/2015/341308
32. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill, AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. (2000) 164:6166–73. doi: 10.4049/jimmunol.164.12.6166
33. Martner A, Dahlgren C, Paton JC, Wold, AE. Pneumolysin released during Streptococcus pneumoniae autolysis is a potent activator of intracellular oxygen radical production in neutrophils. Infect Immun. (2008) 76:4079–87. doi: 10.1128/IAI.01747-07
34. Lambeth, JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. (2004) 4:181–9. doi: 10.1038/nri1312
35. Lv J, He X, Wang H, Wang Z, Kelly GT, Wang X, et al. TLR4-NOX2 axis regulates the phagocytosis and killing of Mycobacterium tuberculosis by macrophages. BMC Pulm Med. (2017) 17:194. doi: 10.1186/s12890-017-0517-0
36. de la Campa AG, Balsalobre L, Ardanuy C, Fenoll A, Perez-Trallero E, Linares J, et al. Fluoroquinolone resistance in penicillin-resistant Streptococcus pneumoniae clones, Spain. Emerg Infect Dis. (2004) 10:1751–9. doi: 10.3201/eid1010.040382
37. Ribes S, Taberner F, Cabellos C, Tubau F, Ardanuy C, Gerber J, et al. Contribution of capsular and clonal types and beta-lactam resistance to the severity of experimental pneumococcal meningitis. Microbes Infect. (2008) 10:129–34. doi: 10.1016/j.micinf.2007.10.014
38. Martinez-M JA, Minguet S, Gonzalo P, Soro PG, de Andres B, Izcue A, et al. Long-lived polyclonal B-cell lines derived from midgestation mouse embryo lymphohematopoietic progenitors reconstitute adult immunodeficient mice. Blood. (2001) 98:1862–71. doi: 10.1182/blood.V98.6.1862
39. Gozalbo-Lopez B, Andrade P, Terrados G, de Andres B, Serrano N, Cortegano I, et al. A role for DNA polymerase mu in the emerging DJH rearrangements of the postgastrulation mouse embryo. Mol Cell Biol. (2009) 29:1266–75. doi: 10.1128/MCB.01518-08
40. De Andres B, Cortegano I, Prado C, Palacios B, Alia M, Ruiz C, et al. Innate CD19(+) CD45Rlo cell population exhibit high proliferation rates in homeostatic conditions and is able to respond after TLR stimulation. J. Immunology. (2012) 137:334–5. doi: 10.4049/jimmunol.1200224
41. Fillion I, Ouellet N, Simard M, Bergeron Y, Sato S, Bergeron, MG. Role of chemokines and formyl peptides in pneumococcal pneumonia-induced monocyte/macrophage recruitment. J Immunol. (2001) 166:7353–61. doi: 10.4049/jimmunol.166.12.7353
42. Dockrell DH, Marriott HM, Prince LR, Ridger VC, Ince PG, Hellewell PG, et al. Alveolar macrophage apoptosis contributes to pneumococcal clearance in a resolving model of pulmonary infection. J Immunol. (2003) 171:5380–8. doi: 10.4049/jimmunol.171.10.5380
43. Taut K, Winter C, Briles DE, Paton JC, Christman JW, Maus R, et al. Macrophage turnover kinetics in the lungs of mice infected with Streptococcus pneumoniae. Am J Respir Cell Mol Biol. (2008) 38:105–13. doi: 10.1165/rcmb.2007-0132OC
44. Kim BJ, Lee S, Berg RE, Simecka JW, Jones, HP. Interleukin-23 (IL-23) deficiency disrupts Th17 and Th1-related defenses against Streptococcus pneumoniae infection. Cytokine. (2013) 64:375–81. doi: 10.1016/j.cyto.2013.05.013
45. Olliver M, Hiew J, Mellroth P, Henriques-Normark B, Bergman, P. Human monocytes promote Th1 and Th17 responses to Streptococcus pneumoniae. Infect Immun. (2011) 79:4210–7. doi: 10.1128/IAI.05286-11
46. Korir ML, Flaherty RA, Rogers LM, Gaddy JA, Aronoff DM, Manning, SD. Investigation of the role that NADH peroxidase plays in oxidative stress survival in group B Streptococcus. Front Microbiol. (2018) 9:2786. doi: 10.3389/fmicb.2018.02786
47. Nguyen GT, Green ER, Mecsas, J. Neutrophils to the ROScue: mechanisms of NADPH Oxidase activation and bacterial resistance. Front Cell Infect Microbiol. (2017) 7:373. doi: 10.3389/fcimb.2017.00373
48. Kim YM, Kim SJ, Tatsunami R, Yamamura H, Fukai T, Ushio-Fukai, M. ROS-induced ROS release orchestrated by Nox4, Nox2, mitochondria in VEGF signaling and angiogenesis. Am J Physiol Cell Physiol. (2017) 312:C749–64. doi: 10.1152/ajpcell.00346.2016
49. Kovac S, Angelova PR, Holmstrom KM, Zhang Y, Dinkova-Kostova AT, Abramov, AY. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim Biophys Acta. (2015) 1850:794–801. doi: 10.1016/j.bbagen.2014.11.021
50. McAllister DA, Liu L, Shi T, Chu Y, Reed C, Burrows J, et al. Global regional, and national estimates of pneumonia morbidity and mortality in children younger than 5 years between 2000 and 2015: a systematic analysis. Lancet Glob Health. (2019) 7:e47–57. doi: 10.1016/S2214-109X(18)30408-X
51. Wahl B, O’Brien KL, Greenbaum A, Majumder A, Liu L, Chu Y, et al. Burden of Streptococcus pneumoniae and Haemophilus influenzae type b disease in children in the era of conjugate vaccines: global, regional, and national estimates for 2000-15. Lancet Glob Health. (2018) 6:e744–57. doi: 10.1016/S2214-109X(18)30247-X
52. GBD 2016 Lower Respiratory Infections Collaborators Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory infections in 195 countries, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect Dis. (2018) 18:1191–210. doi: 10.1016/S1473-3099(18)30310-4
53. Watkins K, Sridhar, D. Pneumonia: a global cause without champions. Lancet. (2018) 392:718–9. doi: 10.1016/S0140-6736(18)31666-0
54. Albiger B, Sandgren A, Katsuragi H, Meyer-Hoffert U, Beiter K, Wartha F, et al. Myeloid differentiation factor 88-dependent signalling controls bacterial growth during colonization and systemic pneumococcal disease in mice. Cell Microbiol. (2005) 7:1603–15. doi: 10.1111/j.1462-5822.2005.00578.x
55. Albiger B, Dahlberg S, Sandgren A, Wartha F, Beiter K, Katsuragi H, et al. Toll-like receptor 9 acts at an early stage in host defence against pneumococcal infection. Cell Microbiol. (2007) 9:633–44. doi: 10.1111/j.1462-5822.2006.00814.x
56. Branger J, Leemans JC, Florquin S, Weijer S, Speelman P, Van Der Poll, T. Toll-like receptor 4 plays a protective role in pulmonary tuberculosis in mice. Int Immunol. (2004) 16:509–16. doi: 10.1093/intimm/dxh052
57. van Lieshout MHP, de Vos AF, Dessing MC, de Porto A, de Boer OJ, de Beer R, et al. ASC and NLRP3 impair host defense during lethal pneumonia caused by serotype 3 Streptococcus pneumoniae in mice. Eur J Immunol. (2018) 48:66–79. doi: 10.1002/eji.201646554
58. Warnatsch A, Tsourouktsoglou TD, Branzk N, Wang Q, Reincke S, Herbst S, et al. Reactive oxygen species localization programs inflammation to clear microbes of different size. Immunity. (2017) 46:421–32. doi: 10.1016/j.immuni.2017.02.013
59. Paudel S, Baral P, Ghimire L, Bergeron S, Jin L, DeCorte JA, et al. CXCL1 regulates neutrophil homeostasis in pneumonia-derived sepsis caused by Streptococcus pneumoniae serotype 3. Blood. (2019) 133:1335–45. doi: 10.1182/blood-2018-10-878082
60. Shoma S, Tsuchiya K, Kawamura I, Nomura T, Hara H, Uchiyama R, et al. Critical involvement of pneumolysin in production of interleukin-1alpha and caspase-1-dependent cytokines in infection with Streptococcus pneumoniae in vitro: a novel function of pneumolysin in caspase-1 activation. Infect Immun. (2008) 76:1547–57. doi: 10.1128/IAI.01269-07
61. Segal, AW. How neutrophils kill microbes. Annu Rev Immunol. (2005) 23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653
62. Paardekooper LM, Dingjan I, Linders PTA, Staal AHJ, Cristescu SM, Verberk W, et al. Human monocyte-derived dendritic cells produce millimolar concentrations of ROS in phagosomes per second. Front Immunol. (2019) 10:1216. doi: 10.3389/fimmu.2019.01216
63. Belchamber KBR, Singh R, Batista CM, Whyte MK, Dockrell DH, Kilty I, et al. Defective bacterial phagocytosis is associated with dysfunctional mitochondria in COPD macrophages. Eur Respir J. (2019) 54:1802244. doi: 10.1183/13993003.02244-2018
64. Garcia MT, Valenzuela MV, Ferrandiz MJ, de la Campa, AG. Reactive oxygen species production is a major factor directing the postantibiotic effect of fluoroquinolones in Streptococcus pneumoniae. Antimicrob Agents Chemother. (2019) 63:e00737-19. doi: 10.1128/AAC.00737-19
65. Ferrandiz MJ, Martin-Galiano AJ, Arnanz C, Zimmerman T, de la Campa, AG. Reactive oxygen species contribute to the bactericidal effects of the fluoroquinolone moxifloxacin in Streptococcus pneumoniae. Antimicrob Agents Chemother. (2016) 60:409–17. doi: 10.1128/AAC.02299-15
66. Ferrandiz MJ, de la Campa, AG. The fluoroquinolone levofloxacin triggers the transcriptional activation of iron transport genes that contribute to cell death in Streptococcus pneumoniae. Antimicrob Agents Chemother. (2014) 58:247–57. doi: 10.1128/AAC.01706-13
67. Lundqvist-Gustafsson H, Bengtsson, T. Activation of the granule pool of the NADPH oxidase accelerates apoptosis in human neutrophils. J Leukoc Biol. (1999) 65:196–204. doi: 10.1002/jlb.65.2.196
68. Robinson AJ, Hopkins GL, Rastogi N, Hodges M, Doyle M, Davies S, et al. Reactive oxygen species drive proliferation in acute myeloid leukemia via the glycolytic regulator PFKFB3. Cancer Res. (2019) 80:937–49. doi: 10.1158/0008-5472.CAN-19-1920
69. Shen W, Lu Y, Hu J, Le H, Yu W, Xu W, et al. Mechanism of miR-320 in regulating biological characteristics of ischemic cerebral neuron by mediating Nox2/ROS pathway. J Mol Neurosci. (2019) 70:449–57. doi: 10.1007/s12031-019-01434-5
Keywords: TLR4, MyD88, S. pneumoniae, monocyte differentiation, innate immune responses, ROS
Citation: Sánchez-Tarjuelo R, Cortegano I, Manosalva J, Rodríguez M, Ruíz C, Alía M, Prado MC, Cano EM, Ferrándiz MJ, de la Campa AG, Gaspar ML and de Andrés B (2020) The TLR4-MyD88 Signaling Axis Regulates Lung Monocyte Differentiation Pathways in Response to Streptococcus pneumoniae. Front. Immunol. 11:2120. doi: 10.3389/fimmu.2020.02120
Received: 13 May 2020; Accepted: 05 August 2020;
Published: 16 September 2020.
Edited by:
Silvia Sánchez-Ramón, Complutense University of Madrid, SpainReviewed by:
Alex De Vos, Amsterdam University Medical Center (UMC), NetherlandsAnja Andrea Kühl, Charité – Universitätsmedizin Berlin, Germany
Copyright © 2020 Sánchez-Tarjuelo, Cortegano, Manosalva, Rodríguez, Ruíz, Alía, Prado, Cano, Ferrándiz, de la Campa, Gaspar and de Andrés. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María Luisa Gaspar, bWxnYXNwYXJAaXNjaWlpLmVz; Belén de Andrés, YmRhbmRyZXNAaXNjaWlpLmVz