 Alexander Oschwald
Alexander Oschwald Philippe Petry
Philippe Petry Katrin Kierdorf
Katrin Kierdorf Daniel Erny
Daniel Erny- 1Faculty of Medicine, Institute of Neuropathology, University of Freiburg, Freiburg, Germany
- 2Faculty of Biology, University of Freiburg, Freiburg, Germany
- 3CIBBS Centre for Integrative Biological Signalling Studies, University of Freiburg, Freiburg, Germany
- 4Center for Basics in NeuroModulation (NeuroModulBasics), Faculty of Medicine, University of Freiburg, Freiburg, Germany
The central nervous system (CNS) harbors its own immune system composed of microglia in the parenchyma and CNS-associated macrophages (CAMs) in the perivascular space, leptomeninges, dura mater, and choroid plexus. Recent advances in understanding the CNS resident immune cells gave new insights into development, maturation and function of its immune guard. Microglia and CAMs undergo essential steps of differentiation and maturation triggered by environmental factors as well as intrinsic transcriptional programs throughout embryonic and postnatal development. These shaping steps allow the macrophages to adapt to their specific physiological function as first line of defense of the CNS and its interfaces. During infancy, the CNS might be targeted by a plethora of different pathogens which can cause severe tissue damage with potentially long reaching defects. Therefore, an efficient immune response of infant CNS macrophages is required even at these early stages to clear the infections but may also lead to detrimental consequences for the developing CNS. Here, we highlight the recent knowledge of the infant CNS immune system during embryonic and postnatal infections and the consequences for the developing CNS.
Introduction
During fetal and postnatal development the central nervous system (CNS) is constantly rearranged to construct and elaborate neuronal circuits needed to fulfill complex neuronal tasks later in life (1). Even though the CNS is supposed to be immune privileged to a certain degree (2, 3), infant infections are able to reach the CNS and can cause immunopathologies, severe long-term sequelae of the CNS or even death. In 2015, 5.9 million children below the age of 5 died due to different circumstances including preterm birth complications, but also malnutrition and infections (4–6). Roughly half of these deaths are caused by infectious diseases such as pneumonia, diarrhea, neonatal sepsis and malaria, mostly in low- and middle-income countries (7, 8). During the last two decades, the numbers of child mortality due to infections were dramatically reduced by improvements in hygienic standards, vaccination programs and introduction of new antimicrobial drugs (9). However, there are still millions of children worldwide reported with prenatal and postnatal infections affecting the CNS and causing CNS pathologies (10). Therefore, fetal and early infant stages until the first years of life seem to present a vulnerable window where infections reaching the CNS can cause detrimental pathologies and malformations.
The CNS is one of the earliest formed organs during embryogenesis and needs immune cell coverage early on to assure correct CNS development and immune defense against pathogens (11). Therefore, macrophages appear in the brain very early during development. Microglia and CNS-associated macrophages (CAMs) are the tissue resident macrophages of the brain, the former residing in the CNS parenchyma and the latter inhabiting the CNS interfaces such as the perivascular space, the meninges and the choroid plexus (12–14). In mice, it was shown that microglia derive from erythro-myeloid progenitors (EMP) from the yolk sac and start to colonize the brain parenchyma at embryonic day (E) 9.5 (12, 13, 15–17). As soon as the progenitors enter the tissue they extensively expand by proliferation, distribute throughout all brain regions during development and differentiate into mature microglia (12, 18). Interestingly, microglia are not exchanged by hematopoietic stem cell (HSC)-derived circulating progenitors during later development and adulthood during steady state (19–21). Even though the HSC-independent development and endogenous maintenance is assumed for mammals during physiological conditions, it was shown in other species such as the zebrafish that microglia can be derived from different hematopoietic origins during development (22, 23). However, upon neuroinflammation in the mammalian CNS, for example during infections, recruitment of monocyte-derived macrophages is widely described due to release of chemokines and cytokines in the parenchyma and opening of the blood-brain barrier (BBB) (24).
Similar to microglia, most CAM populations also arise from EMP-derived macrophage progenitors in mammals (14, 25). However, it has been shown that subpopulations of the choroid plexus macrophages and meningeal macrophages, namely stromal and dural macrophages, are partially replaced by bone marrow-derived circulating monocytes during adulthood (14, 26). In mice it was described that CAMs start to colonize their specific niche in the CNS interfaces from E12.5 when anatomical structures of the brain interface start developing (14). However, the exact timing and distribution of the cells in the developing interfaces during development is ill-defined. In humans, there is only limited data available on the development of CNS macrophages. For microglial progenitors, it was described that they colonize the developing neuroectoderm starting around gestational week 4.5 (27–29). Post-mortem studies indicate that ionized calcium binding adaptor molecule 1 (Iba-1)+ microglial progenitors enter the brain via the meninges, choroid plexus and ventricular zone (28, 29). Human CAMs are detected shortly after in the developing CNS interfaces. Macrophages in the meninges of the human optic nerve were described as early as gestational week 8 (30). Another study describes first stromal choroid plexus macrophages around gestational week 11 (31). Future studies are needed to explore the development of human CNS macrophages in more depth.
CNS macrophage differentiation is a highly dynamic process during pre- and postnatal development controlled by both an intrinsic genetic program and extrinsic factors and is essential for CNS tissue homeostasis. Microglia differentiation depends on the transcription factors spleen focus forming virus (SFFV) proviral integration oncogene (Sfpi, encoding PU.1), interferon regulatory factor 8 (Irf8) and spalt like trancription factor 1 (Sall1) while independent of cellular myeloblastosis oncogene (c-Myb), inhibitor of DNA-binding 2 (Id2) and basic leucine zipper ATF-like transcription factor 3 (Batf3) (13–15, 26, 32, 33). Microglia and CAMs require steady colony stimulating factor 1 receptor (Csf1r) signaling (13). For microglia it was further shown that their dependence on the two ligands, colony stimulating factor 1 (CSF1) and Interleukin-34 (IL-34), seems to differ between brain regions and developmental time points (34). While CSF1 seems to be important for the whole microglial entity during embryonic and fetal development (35–37), gray matter microglia seem to depend more on IL-34 and white matter microglia on CSF1 during postnatal phases and adulthood (34, 38). It remains elusive whether a similar heterogeneity exists for CAMs in the CNS interfaces. In contrast to CAMs, microglia expansion is highly dependent on tumor growth factor β (TGF-β) signaling both during development and maintenance in the adult CNS (26, 39). Another major factor influencing microglial development, maturation and function is the endogenous gut microbiota. Absence of the host microbiota results in pre- and postnatal maturation defects in microglia and further leads to dysfunctional microglia with a hampered immune answer (18, 40). Interestingly, fetal microglia only encounters minor transcriptional changes in the absence of microbiota, whereas the effects become more pronounced in early postnatal and adult microglia (18, 41). Therefore, a detrimental contribution of microbiota to the maturation of regulator networks in microglia upon weaning is suggested. Though, the encounter with pathogenic bacteria and viruses or a dysregulated maternal microbiome during embryonic development can have effects on microglial function later in life. Furthermore, maternal immune activation due to a viral infection during pregnancy can result in microglial pre-priming and a wide spectrum of neuronal abnormalities and phenotypes (42–45).
To protect the CNS during pre- and postnatal development, CNS macrophages are equipped with a wide range of pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) or nucleotide-binding oligomerization domain-like receptors (NLRs) (46–51). Upon recognition of invading pathogens via PRRs microglia become activated and efficiently remove invading pathogens via phagocytosis (52). This process is accompanied by a release of proinflammatory cytokines and chemokines to activate neighboring microglia but also to recruit other immune cells to the CNS to resolve the infection (53–59). Microglia also upregulate genes involved in the production of inducible nitric oxide synthetase (iNOS) generating reactive oxygen species (ROS) and secrete tumor necrosis factor α (TNF-α), Interleukin-1β (IL-1β) and Interferon γ (IFN-γ) (60, 61). However, these immune defense mechanisms exist together in a fragile balance between fighting off damaging pathogens and causing tissue damage in the developing CNS. This tissue damage is either caused by the invading pathogen itself, but also to a major extent from the immune reaction against the pathogen such as release of ROS, interferons and cytokines which can be neurotoxic, but also T-cell mediated cell lysis, resulting in in neuronal apoptosis, tissue necrosis and CNS malformations (62–64).
In the following, we will explore the current knowledge and literature of different infections occurring during prenatal and early postnatal development of the brain, the responses of microglia and CAMs and their long-lasting implications on brain function.
Fetal Infections
In a healthy pregnancy the fetus is considered a sterile environment devoid of any living microorganisms under physiological conditions (65). However, upon maternal infection the maternofetal transfer of microorganisms or their by-products can have severe effects on the fetal development (Figure 1). Investigations over the last decades focused on understanding the consequences of these infections on the developing fetal immune system, but also on their effects on the developing CNS (65, 66).
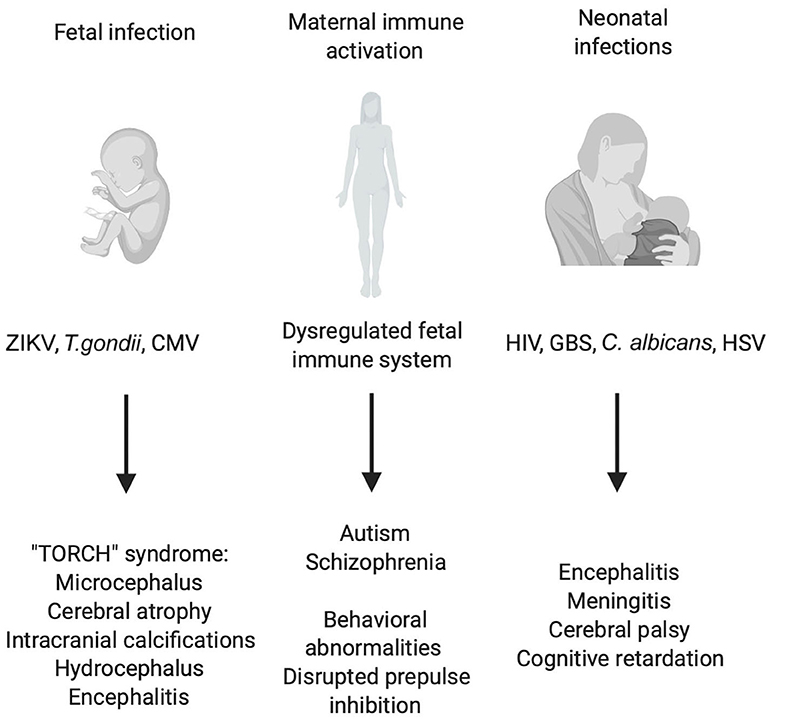
Figure 1. Consequences of early CNS infections and peripheral immune activation for CNS development. Illustration of prenatal and postnatal infections and indirect effects of maternal immune activation (MIA) on the fetus. Different viral, bacterial and parasitic infections can endanger the correct development of the CNS of the fetus leading to a plethora of symptoms grouped under the name “TORCH” syndrome. Also, maternal infection without the transmission of the pathogen to the fetus can have detrimental effects on CNS development which have robustly been linked to schizophrenia and autism spectrum disorder (ASD). Birth and the accompanying contact to environmental pathogens pose a great threat to the unchallenged immune system of the newborn in the worst cases leading to bacterial sepsis, viral encephalitis, and systemic candidiasis.
Certain layers of protection are in place to guarantee that no infectious agents are granted access to the developing fetus. The main barrier separating the maternal from the fetal blood circulation is the placenta that develops shortly after conception (67). Beside its barrier function, the placenta links the maternal blood circulation to the embryo to deliver nutrients but also antibodies that protect the developing fetus from blood-borne pathogens via passive immunization (68, 69). In most cases, viral infections of the mother are not transmitted to the fetus (70–72). Besides the physical cell-cell barrier of the placenta, the expression of antiviral interferons (IFNs) produced by the trophoblast and secretion of antimicrobial peptides further inhibits replication and pathogenesis (73, 74). However, infectious pathogens have developed different routes how they can breach this barrier and cause harmful damage to the developing fetus. Maternal infections can be transmitted vertically to the fetus either via the maternal/fetal blood interface (75), the cervical/amniotic sac interface (76, 77) or via transvaginal ascension (78, 79). Mostly at early stages of fetal development during the first and beginning of second trimester, the risk of devastating sequelae is remarkably high mainly due to the critical establishment of the placenta and the beginning of organogenesis.
The most prevalent congenital diseases are caused by a set of microorganisms grouped under the term “TORCH” who manage to overcome the placenta. This acronym stands for Toxoplasma gondii, other (Listeria monocytogenes, Treponema pallidum, varicella zoster virus, human immunodeficiency virus (HIV), parvovirus B19 and some more), rubella virus, cytomegalovirus and herpes simplex virus 1 and 2 (80). A new member added to this category is the Zika virus mostly due to its recent outbreak in South America (81). TORCH pathologies are grouped because they produce common clinical manifestations like microcephaly, hearing loss, ocular abnormalities and other congenital abnormalities leading in the worst case to fetal loss (Figure 1) (82). The severity of each of these disorders depends on the pathogen and in which gestational period the infection occurs. However, most of these TORCH infections represent with severe CNS infections and malformations (83). How TORCH pathogens are transmitted to the fetus remains predominantly unknown. Addressing the current severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic, a recent case study suggested a maternal to fetal transmission in the last trimester of pregnancy. The virus seemed to be transmitted through the placenta since the placental tissue, and both the maternal and fetal blood were positive for the SARS-CoV-2. Interestingly, magnetic resonance imaging showed bilateral gliosis of the white matter of the infant at 11 days of life, but no further long-term deficits (84).
Besides the barrier established by the placenta, the fetus itself has cellular immune protection in place to defend itself against intruders. For a long time, it was thought that the human fetal immune system was immature and will only fully develop during childhood (85, 86). This view has been challenged using new single-cell multi-omic approaches painting a more accurate picture in which both the innate and adaptive arm of the fetal immune system maintain immunity from 4 weeks post conception onwards (87–90).
The mechanism by which maternal infections translate into compromised CNS functionality, malformation and cognitive impairment of the offspring is under extensive research. In the following section we want to take a closer look on the most important TORCH pathogens, their impact on CNS development and the so far known role of the CNS immune system, especially CNS macrophages, on neuropathological outcome after congenital infection.
Zika Virus (ZIKV)
Zika virus (ZIKV) infection during pregnancy has been linked to severe congenital malformations of the CNS (91–93). ZIKV belongs to the family of Flaviviridae and was first discovered in Uganda's tropical Zika forest in 1947 (94). The virus is transmitted from mosquitoes of the Aedes species to humans which prior to 2007 spared the western hemisphere (95). A widespread outbreak occurred in 2015 in South America, most prominently in Brazil correlating with a strong increase in children born with microcephaly (Table 1) (136). The virus was shown to transmit vertically from the mother to the developing fetus and can then be detected inside the fetal brain (113). ZIKV is able to cross the placenta and infect Hofbauer and cytotrophoblast cells (114–116), however the exact mechanism of transmission is still unknown (76). Infections occurring in the first trimester of pregnancy have the worst consequences for fetal brain development leading to cerebral atrophy and resulting in microcephaly and intracranial microcalcifications (Figure 2) (113, 117, 118). Other infection-related conditions include growth restrictions and ocular abnormalities (137). These symptoms newly added ZIKV onto the list of “TORCH” diseases (76). Additionally, a paraplacental route of infection was described in which the ZIKV was shown to infect the parietal decidua and the amniochorionic membranes (115). Whereas, infections in the first two trimesters cause the above mentioned symptoms, infections during the last trimester do not pose adverse risks for abnormalities in newborns (138, 139). Currently, there are neither vaccines nor therapies against infection of this mosquito-borne disease. It was shown that ZIKV is neurotrophic, meaning that the infection mainly targets the brain (Figure 2) (113). Though some researchers also isolated viral RNA from other fetal organs such as lung, liver, muscle and spleen (114), tissue damage was only observed inside the brain parenchyma (113). Indeed, it was shown that the virus targets neuronal progenitor cells (NPCs), astrocytes and microglia, while only being cytotoxic in NPCs and causing their growth arrest and apoptosis (140, 141). Especially microglia are among the main suspects to disseminate ZIKV in the brain as the particular susceptibility between E6.5-E8.5 in mice coincides with a critical window of microglia development and the beginning of their migration via the newly formed blood circulation toward the brain (142). It was shown that an acute ablation of CSF1R resulting in a depletion of microglia results in a decreased viral load in the brain, supporting a “Trojan horse” model of ZIKV infection (142) (Figure 2). In vivo and in vitro data underlined that murine microglia progenitor cells from the yolk sac are indeed susceptible to ZIKV infection (142). In vitro data showed that infected human induced pluripotent stem cell (iPSC)- derived microglia-like cells co-cultured with neural spheroids leads to propagation of the virus to the neural tissue which supports the claim that microglia act as a viral reservoir for ZIKV and push ahead neural infection in the fetal brain (140). Infected microglia-like cells remained amoeboid with high amounts of phagocytic vesicles compared to uninfected cells taking residency inside the brain spheroid assuming a homeostatic ramified morphology (140). Furthermore, the infected amoeboid microglia only colonized the spheroid underneath the surface and signs of neurodegenerative processes and viral particle release were detected in the spheroids (140). Electrophysiological activity drastically decreased in the infected neuronal spheroids which is not reinstated over time suggesting irreversible neuronal degeneration (140). In line with these results, an in vivo study using a mouse model for adult ZIKV infection, showed the presence of the virus in neuronal stem cells in the adult hippocampus leading to the recruitment of IFN-γ producing T cells accompanied by microglial nodules and neuronophagy (143). This specific location together with a resulting reduced Homer-1 expression in synapses let to the speculation that ZIKV infection of hippocampal NPCs leads to cognitive decline in infected adult individuals (143). As already mentioned above, microglia were shown to have an activated phenotype in histological sections of human fetal brains and microglial nodules are described throughout the gray and white matter of the infected fetal CNS (Figure 2) (113). Cultured primary murine microglia showed a proinflammatory phenotype upon ZIKV infection with increased release of IL-6, IL-1β, and TNF-α correlating with an increased neurotoxicity toward fetal NPCs. Culturing of fetal murine NPCs with conditioned media from infected primary cultured microglia revealed a decrease in NPC proliferation and neuronal differentiation (144). These findings could indicate a crucial role of fetal microglial activation and microglial-mediated neurotoxicity during fetal ZIKV infection in humans. A recent study investigated the role of lipid metabolism of ZIKV infected human fetal microglia in vitro and linked a higher production of lysophosphatiylcholine (LPC) to the proinflammatory cytokine profile but also to the release of ROS by the infected cells (145). LPC is known to induce inflammasome activation and micro- and astrogliosis. Therefore, the metabolic changes observed in infected microglia could support microglial activation during the infection course (146). However, these findings need further evaluation in vivo. Another study performed ZIKV infections in adult macaques revealed a high incidence of neuroinflammation in infected animals and a disruption of the BBB (Figure 2) (147). Here, long-term increased levels of C-X-C motif chemokine ligand 12 (CXCL12), which is a chemokine involved in lymphocyte trafficking and neural repair, were described. Additionally, the authors observed inflammation in the perivascular space and meninges, accompanied by lymphocytic infiltrations (147). This was also reported in fetal human brains, where an infiltration of T and B cells was observed in the perivascular space during ZIKV infection (Figure 2) (113). Lymphocytic infiltrates are observed in neonatal and adult mouse models of ZIKV infections (143, 148), however an infiltration of monocytes or granulocytes could not be observed in infected human brain parenchyma or animal models. In the perivascular space of infected human fetuses, lymphocytic and monocytic accumulations were described and summarized as signs of meningitis. However, it was not further solved if the accumulated mononuclear cells are recruited monocytes or proliferating perivascular macrophages (149). Though, fetal mciroglia seem to play a detrimental role as a reservoir of ZIKV, the viral spread and the neurological symptoms observed, there is so far no data available on the role of fetal CAMs during ZIKV infection.
Table 1. Overview of prenatal and postnatal CNS infections.
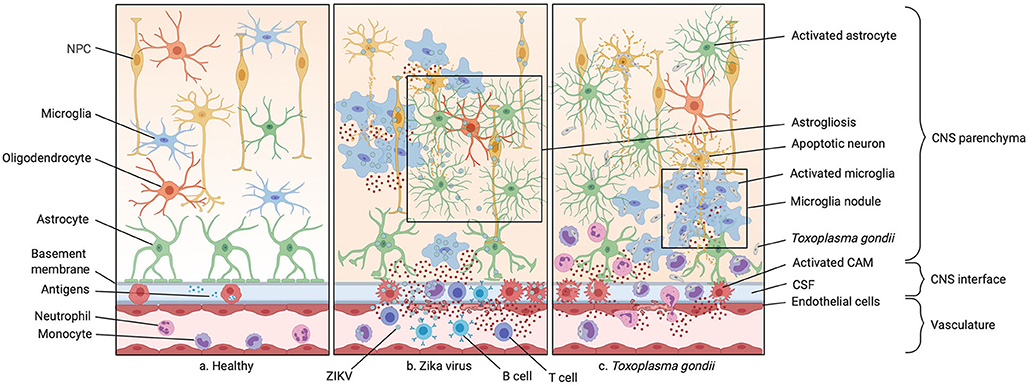
Figure 2. Infections of the prenatal CNS. (a) Illustration of the healthy central nervous system (CNS) including the vasculature, the perivascular space, and the brain parenchyma. (b) A typical ZIKV infection mostly affecting CNS macrophages inducing neuronal progenitor cell (NPC) apoptosis and recruiting peripheral monocytes and neutrophils to the site of infection via various cytokines and chemokines. The blood-brain barrier (BBB) integrity is compromised in this process. Microgliosis, astrogliosis and microglia nodules around NPC bodies are often observed. (c) A typical parasitic CNS infection by Toxoplasma gondii is shown. The pathogen gains access to the CNS with the help of circulating monocytes, carrying the parasite to through the compromised BBB, also known as a “Troian horse” infection. This causes microglia nodules, abundant cytokine release by microglia and CNS-associated macrophages (CAMs), and subsequent infiltration by monocytes and granulocytes.
Cytomegalovirus (CMV)
Congenital cytomegalovirus (CMV) infection occurs in roughly 0.2–2.2% of intrauterine infections making it the most common congenital viral infection and the main cause of long-term pediatric conditions (8, 150). CMV belongs to the betaherpesvirinae, which is a subfamily of the herpesviridae family. After primary infection of a mammal host, the virus persists in a latent state with a low virus titer periodically reactivating throughout life (96, 151). It is transmitted most frequently to pregnant women via smear infection i.e., contact with for example infectious urine and saliva of children (97). 40–50% of pregnant women infected with CMV transmit the virus to the developing fetus (8, 97). CMV is able to cross the placenta and is thus transmitted from the maternal blood to the fetus where it takes residency inside the fetal brain (112). Only 10% of congenitally infected infants are symptomatic (97). However, an infection with CMV is particularly severe during the first and second trimester of pregnancy as it may cause an infection of the trophoblast and interfere with correct development of the placenta often leading to fetal loss (77). Sequelae of congenital CMV infection mainly concern the infant brain and include microcephaly, mental and motor retardation, epilepsy and a progressive loss of vision and hearing (Table 1) (152). Other non CNS-related conditions are hepatosplenomegaly and growth retardation (8, 153, 154). Some studies also proclaimed a link between fetal CMV infection and the development of autism spectrum disorder (98). Like ZIKV, CMV can cross the human placenta thus being transmitted from the maternal to the fetal compartment (77). It is not fully understood how CMV invades the fetus through the placenta, however one study suggests maternal IgG-mediated transcytosis as a potential mechanism (155). The main cellular targets of CMV are brain macrophages, including microglia and CAMs, as well as NPCs in the cerebrum (156). Though, CMV replication has been reported in a wide range of glial cells and neurons in the CNS (157). The number of microglia, perivascular and meningeal macrophages is increased in the murine CMV-infected fetal brains which is not accounted for by proliferation, but rather a recruitment of cells from other areas of the brain as well as transvascular migration from the meninges (156). Even though, it was suggested here that these cells are recruited monocyte-derived macrophages from the fetal liver invading the infected meninges and later the parenchyma (156). Microglial cell numbers increase in infected areas but also in regions where the viral antigens are not detected (156). Interestingly, choroid plexus macrophages were also described to be elevated in numbers in this murine model for fetal CMV infection (156). As also reported during fetal ZIKV infection, microglia assemble in nodules in infected CMV foci in the brain tissue (156, 158, 159). They are located around degenerating CMV-infected neurons engaging in neuronophagy and clearance of dead cells (158, 159). In vitro studies of human fetal microglia and astrocytes showed that CMV-infected microglia can secrete TNF-α, IL-1β, and IL-6 orchestrating an antiviral response and dampening viral replication for example in infected astrocytes (55). This study suggested that CMV-infected fetal astrocytes are not competent in mounting an antiviral response but rather secrete cytokines such as CC-chemokine ligand (CCL2) to recruit microglia (55). In vitro infection of a murine microglial cell line with CMV showed extensive metabolic alteration with a shift of microglia metabolism toward glycolysis supporting the proinflammatory phenotype and activation previously reported upon CMV infection (160). Microglia and CAMs are a cellular target of CMV and are associated with neuronal degeneration and tissue damage at infectious foci in the brain. Therefore, further investigations should be performed if these CNS immune cell populations are potential therapeutic targets during fetal CMV infections. First results already suggested that targeting fetal microglia and CAMs during CMV infection in rats by intracranioventricular injection of depleting clodronate liposomes resulted in an improvement of survival, neuropathology and cognitive functions in the born litter (161).
Toxoplasma gondii (T. gondii)
The obligate intracellular protozoan parasite Toxoplasma gondii (T. gondii) causes one of the most prevalent chronic infections with around 30% of the world population being infected (162, 163). The only known sexual stages of the T. gondii life cycle take place in the family of the felidae (164). Here oocysts or sporozoites are formed and excreted in cat feces (165). Ingestion of oocysts by intermediate hosts such as rodents and birds (166) lead to a transformation into rapidly proliferating tachyzoites which are able to spread into different tissues via the blood stream, infected all nucleated resident cells and cause host cell lysis (167). Within the host cells tachyzoites replicate in specialized parasitophorous vacuoles escaping lysosomal fusion, finally induce lysis of the host cell and disseminate adjacent cells (168). Once they arrive in tissues like the CNS or muscle, they convert into bradyzoites or tissue cysts, allowing them to evade from the immune system and persist for many years (165). T. gondii can be transmitted into humans through consumption of contaminated food or water with cat fecal oocysts (169). Alternatively, oocysts can be ingested by livestock forming tissue cysts which are then transmitted into humans by consumption of uncooked contaminated tissue (165). In rarer cases T. gondii is transmitted via blood transfusion or organ transplantation (170, 171). T. gondii infection in immunocompetent adult individuals is asymptomatic or causes only mild flu-like symptoms in most cases, but opportunistic reactivation from cysts into tachyzoites can occur in immunosuppressed individuals, such as HIV patients, leading to a toxoplasmic encephalitis in adults (Table 1) (172, 173). However, vertical transmission of T. gondii from the mother to the fetus can be harmful or even fatal. Though, maternal to fetal transmission only occurs when the mother is primary infected during or up to 10 weeks before pregnancy (174). While infection in early pregnancy poses a small risk to fetal transmission (less than 6%), rates of transmission increase up to 81% in the third trimester (175). This was assumed to be due to a differential expression of placental TLRs during pregnancy (176). However, the consequences of fetal T. gondii infection are highest in early embryos and may result in severe congenital toxoplasmosis with CNS pathologies, including microcephaly, hydrocephalus, intracerebral calcifications, epilepsy, intellectual disabilities or even spontaneous abortion and fetal death (Table 1) (175, 177–181). Fetuses infected in late gestation are born normal, but can develop CNS symptoms and retinochoroiditis after birth (175). Studies suggest that maternal-fetal transmission starts with initial infection of the uterus, followed by the extravillous trophoblasts of the placenta as the parasite moves from cell to cell, and eventually lead to the infection of the fetus (182). Similar to the adult infections, it is assumed that T. gondii infects circulating cells of the developing fetus such as macrophages and use these cells as a “Trojan horse” to gain access to immune privileged sites such as the CNS or reaches the CNS via paracellular and transcellular infection routes (Figure 2) (183–187). T. gondii can infect almost any nucleated cell, but chronic infection seems to be neurotropic in a sense, since it is cleared in other tissues over time (except musculature) (172, 188). While neurons, astrocytes and microglia are infected during the acute phase by tachyzoites, it seems that bradyzoite cysts develop mostly in neurons as shown in humans and mice (172, 189, 190). In vitro studies with rat CNS cells showed that 10% of neurons and 30% of microglia were infected with T. gondii (191). However, 93% of the parasitophorous vacuoles only contained one to two degenerated parasites in microglia, while neurons contained up to 8 proliferating parasites and most of the bradyzoites four days after acute infection (189). During congenital toxoplasmosis, multifocal diffuse tissue necrosis in the developing brain is mainly caused by parasite induced lysis of infected neurons (192). Close to these lesion sites, accumulation of microglial nodules have been described (Figure 2) (192). Most of the studies investigating the role of microglia and CAMs during T. gondii infection focused on studies of the adult CNS, whereas only less information is so far available on their role during congenital toxoplasmosis. In the adult CNS, microglia and infiltrating CD11b+ CD45high monocyte-derived macrophages are one of the main source of IFN-γ during T. gondii infection, which is critical to control parasitic spread in acute but also chronic infection (193). One study showed that 37% of the IFN-γ positive cells were CD11b+CD45low microglia while 63% were CD11b+CD45high positive cells, most likely monocyte-derived macrophages (193). Recruited Ly6Chigh monocytes, but also infiltrating Ly6G+ granulocytes are a detrimental source of IFN-γ (194). Deficiency of IFN-γ upon T. gondii encephalitis in adult mice resulted in a reduced chemokine and cytokine release in the CNS parenchyma with a decrease in leukocyte infiltration to the infected CNS tissues (195). Beside IFN-γ, several more effector molecules such as nitric oxide (NO), TNF-α and IL-12 were shown to be involved in the inhibition of parasitic spread in the adult CNS (196, 197). Besides the upregulation of effector molecules, microglia exhibit a hypermigratory phenotype upon T. gondii infection (198). A recent study reported hypermigration of primary murine neonatal cortical microglia through signaling of the neurotransmitter γ-aminobutyric acid (GABA) after T. gondii infection, which was not visible after lipopolysaccharides (LPS) or heat-inactivated T. gondii stimulation, and inhibition of GABA synthesis, receptors and regulators led to an inhibition of the hypermotility of microglia (199). To this day, data about disease mechanisms of congenital T. gondii infection remains scarce and there is an urgent need to address the role of microglia and CAMs in congenital toxoplasmosis. The neurotoxic properties of T. gondii and the resulting microglial activation are likely to cause malformation of the neuronal network in the developing fetal CNS, resulting in the typical neurodegenerative symptoms of the prenatally acquired disease. Therefore, understanding microglial involvement in the disease manifestation during fetal development could be key for new treatment paradigms.
Maternal Immune Activation (MIA)
There is increasing evidence that maternal infection and consequential immune activation during pregnancy can have detrimental effects on fetal brain development and cause morbidity even without transmitting the pathogen to the fetus (200, 201). Recently, maternal immune activation (MIA) was implicated in long-term changes within the CNS which can affect CNS function much later in life. It is not entirely solved yet what the molecular mechanisms are for these long-term effects on the CNS, but the spotlight is turned on a detrimental involvement of the immune system (202). Most of all, schizophrenia and autism spectrum disorders (ASD) have been linked to a dysregulated fetal immune system which can induce changes during CNS development and maturation and many studies suggest that this is traced back to an initial activation in the womb due to non-transmitted maternal infection during pregnancy (Figure 1) (203). Elevated levels of serum proinflammatory cytokines in the infected mother may be harmful for placenta barrier integrity and lead to increased induction of cytokine levels in the fetus (204). It is thought that an overexpression of proinflammatory molecules such as IL-6, TNF-α, and IL-1β, which are required for neurodevelopment under physiological circumstances (205), can interfere with correct development of the neuronal network (206). Studies indicate that elevated maternal cytokines such as IL-8 or TNF-α correlate with a higher incidence of schizophrenia in their uninfected offspring (Figure 1) (207, 208). In many studies, absence of fetal infection is rarely confirmed, making it more difficult to assess whether long-term neurological differences are due to indirect effects of maternal inflammation or direct cytopathic effects in the fetus. To investigate the causal mechanisms behind MIA and immune dysregulation in the fetus, researchers have developed several rodent MIA models where the maternal immune system is manipulated by injections of poly-(I:C) or LPS to mimic viral or bacterial infections in the mother. Injection of poly-(I:C) into pregnant females was shown to cause disrupted prepulse inhibition in the offspring, similar to what was observed in schizophrenic patients (209). Injection of LPS in pregnant mice caused behavioral changes (210), altered synaptic pruning (211) and structural changes in the hippocampus leading to impaired memory and learning abilities (212) in their offspring (Figure 1). IL-6 was shown to be elevated after maternal immune activation both in maternal serum and fetal tissue (209). Injection of recombinant IL-6 or antibody mediated depletion of IL-6 demonstrated that elevated IL-6 levels were in part responsible for the exploratory, social, and other behavioral abnormalities observed in the offspring of poly-(I:C) injected pregnant mice (209). In line with these findings in poly-(I:C) treated pregnant mice, injection of LPS into the mother also led to increased IL-6 expression in the fetal brain (212). Beside IL-6, IL-1β, and IL-10 levels were also increased following poly-(I:C) injection into pregnant mice (213–215). Elevated CXCL-8 in maternal serum was shown to induce neuroanatomical alterations in the offspring (216). Interestingly, these regions often coincide with regions shown to be dysregulated in schizophrenia patients such as parahippocampal and superior temporal gyrus volume reductions (216). Injection of LPS into pregnant mice induced elevated levels of TNF-α in the fetal brain (217). It seems that fetal microglia are primed by MIA via potential epigenetic imprints leading to a magnified immune response later in life upon a secondary challenge (43, 218, 219). MIA offspring that received an LPS injection later in adulthood displayed an increase in IL-1β in the hippocampus (220). However, this effect was brain region specific (220). It was shown that microglia activation steadily increases throughout early life, peaking during adolescent which coincides with behavioral abnormalities also observed in schizophrenia patients (221). Many studies report an elevated number of reactive amoeboid microglia expressing high levels of major histocompatibility complex (MHC) class II and CD68 in the fetal and neonate brain indicating an activated status after injection of poly-(I:C) or LPS into pregnant mice (222–225). However, these results are highly debated as many groups fail to confirm elevated microglia numbers (226), nor do they report the presence of activated Mac-2 expressing microglia in fetal CNS parenchyma after poly-(I:C) injections (227). The same discrepancy is also present in post-mortem studies quantifying microglia numbers in the brain of diseased schizophrenia patients (228). Microglia in MIA offspring were shown to display different migratory dynamics as tangential and radial spreading to the hippocampus, corpus callosum, striatum and somatosensory cortex seems to be delayed (229). This in turn could alter developmental processes where microglia involvement is required (229). Even though epidemiological studies linking MIA during pregnancy to neuropsychiatric diseases in the offspring are still sparse, newly developed animal models start to shed light on the role of microglia in this process while the role of CAMs remains to be explored.
Postnatal Infections
Birth is the first direct contact of the newborn with the outside environment including a plethora of different microbes (230). In general, the human body lives in symbiosis with many different microbes including the species colonizing our gut and skin. The mucosa of the female genital tract is inhabited by a variety of commensal bacteria and fungi unique to every woman and vitally important for her health (231). However, beside the beneficial microbiota the human body is constantly eradicating potentially harmful pathogens which can enter the body via different routes or co-colonize microbiotic niches such as the mucosa of gut, lung or the reproductive tract. During birth, these mucosal organisms can come into contact with the newborn and its unchallenged immune system. With that said, the intrapartum period is a critical time for microbiotic colonization but also where serious infections may occur (Table 1) (232, 233). Transmission of pathogenic microorganisms from the maternal vaginal tract to the newborn can lead to dissemination to several organs of the infant, possibly resulting in long lasting morbidities or even death (Table 1) (234, 235). After birth, the infant body is immediately in contact with different microorganisms either beneficial or pathogenic. Therefore, the breastfeeding period plays a major role in preventing early diseases since it not only includes nutrients, enzymes, antimicrobial proteins/peptides, growth factors, antioxidants, anti-inflammatory elements but also transfers maternal immunoglobulins through the breast milk to compensate for the lack of immune memory in the newborn (236–239). Furthermore, studies showed that human breast milk facilitates the establishment of the neonatal microbiota indicating long term benefits for the infant beyond the breastfeeding period (239, 240). To further protect the newborn in this critical time the WHO recommended vaccination as soon as possible after birth, optimally in the first 24 h to guarantee protection against polio, hepatitis B and tuberculosis. However, vaccinations cannot establish immunity against infections occurring shortly after birth and many infections occurring in the first few weeks do not have a vaccine yet (241). Accompanied by profound changes of an immune system which needs to be fine-tuned to guarantee a balance between tolerance and immunity, the encounter of a vast assortment of antigens pose a high risk for infections to occur (Table 1) (242, 243). Studies have shown that the adaptive arm of the immune system presents certain differences compared to later time points in life (244–247). One interesting divergence is the shifted T helper (Th) cell balance in favor of a Th2 cell response during fetal and early postnatal development (248, 249). This in turn inhibits Th1 and Th17 response backing tolerance to the mother, however, making the newborn more susceptible to bacterial and fungal infections (248, 249). Furthermore, it was shown that suppressive regulatory T cells and impaired antigen-presenting cells in the fetus increased disease susceptibility at birth and may hinder effective early vaccination (244–247). As mentioned above, this was a long time assumed to point toward an immature immune system, which was recently revoked by several multi-omics studies (87–90). The maturation of the resident immune cells of the CNS was only recently started to be explored during postnatal stages using bulk and single cell RNA-sequencing (250–252). However, it is clear that CAMs and microglia play important homeostatic functions during early postnatal phases (253). In the following we will discuss how the most prevalent neonatal infections are able to establish CNS infection and explore the role of CNS macrophages in this context and the far-reaching consequences for the developing infant.
Human Immunodeficiency Virus (HIV)
Human immunodeficiency virus (HIV) is a lentivirus and belongs to the family of the retroviridae (254). Retroviruses are single-stranded RNA viruses that are able to integrate their genome after reverse transcription into the hosts DNA and thereby induce a latent infection which can persist for several decades (255). Infected cells convert the viral RNA into double-stranded DNA by using an enzyme called reverse transcriptase which is then incorporated into the hosts genome with a viral integrase and other host co-factors (256). There are two different types of HIV, HIV-1 and HIV-2, with the first being responsible for most of the global HIV infections (255). HIV is transmitted by sexual contact, exposure to infected body fluids and from mother to child during pregnancy, birth, and breastfeeding (109). The risk of vertical transmission decreased dramatically after the development of antiretroviral therapy (ART) during pregnancy and breastfeeding period for infected women in developed countries (257, 258). Primary infant infection is mostly asymptomatic, but disease progression is significantly faster than in adults (259). Before the introduction of ART, half of the infected children developed progressive HIV-1 encephalopathy (PHE) resulting in microcephaly, developmental delays, and focal white matter lesions (Table 1) (119–121). PHE further manifests in basal ganglia calcification and white matter lesions (Table 1) (260). In PHE lesions, microglial cells were often found to accumulate in nodules and seem to be the major cell type infected with the virus. Perivascular lesions with infected proliferating microglia and infiltration of CD8+ T cells and monocytes were further seen in children with HIV encephalitis (Figure 3) (261). Though, chronic neurological impairment is still obtained in HIV-1 seropositive children under ART (262–264). HIV is mostly known for infecting peripheral helper T cells via the cluster of differentiation 4 (CD4) receptor and either C-X-C chemokine receptor type 4 (CXCR4) or C-C chemokine receptor type 5 (CCR5) as a co-receptor, leading to a loss of these cells during acute infection (265). However, CD4+ T cell numbers recover after acute infection but decrease in the course of years, ultimately leading to the development of the acquired immunodeficiency syndrome (AIDS) which is characterized by low T cell counts in the blood and the occurrence of rare opportunistic diseases (266, 267). Tissue resident macrophages across different organs and circulating monocytes are also used as a viral reservoir, since they also express low levels of CD4 and the co-receptors CXCR4, CCR5 and also CCR3 in infants and adults (268). Despite CXCR4 expression macrophage-tropic (M-tropic) HIV variants mostly use CCR5 as a co-receptor to enter the cell. These viruses can bind CD4 more efficient and show enhanced interactions of their envelope glycoprotein 120 (gp120) with CCR5 (269–271). Especially yolk sac-derived tissue-resident macrophages represent an ideal viral reservoir because they are long-lived and self-renewing, making them a perfect target for latent infection (272). Yet the role of macrophages as long-term reservoirs for replication-competent viruses remained controversial. A recent study showed the presence of macrophage reservoirs in the brain of simian immunodeficiency virus (SIV) infected macaques, which is the equivalent to HIV in humans (272). Perivascular macrophages and parenchymal microglia were shown to be infected during acute SIV infection in adult macaques and acute HIV infection in adult humans (273–275). Furthermore, human fetal microglia can be efficiently infected with HIV-1 in ex vivo cultures (276). Data suggested that HIV reaches the brain via infection of bone marrow-derived monocytes which are visible in the CNS during acute infection and through the cerebrospinal fluid (CSF), using them as a “Trojan horse” similarly as described for T. gondii infection (Figure 3) (277–280). Release of proinflammatory cytokines and ROS from infected microglia, as well as synthesized viral molecules such as gp120 and trans-activator of transcription (Tat) can lead to neurotoxicity and neural injury (264, 281–283). A new mechanism of microglial activation was described for neonatal microglia in vitro where exposure to viral Tat resulted in the downregulation of microRNA-124 (miRNA-124), leading to increased methyl CpG binding protein 2 (MECP2) and signal transducer and activator of transcription 3 (STAT3) expression (284). Tat-mediated neonatal microglial activation was also induced by upregulation of miRNA-34a leading to the downregulation of NOD-like receptor C5 (NLRC5), which is a negative regulator of NF-κB signaling, and by NLR family pyrin domain containing 3 (NLRP3) inflammasome activation, both leading to enhanced cytokine release including IL-6 and IL-1β (285, 286). Primary human microglial activation was further shown by the upregulation of pro-inflammatory molecules upon HIV infection such as IL-8, IL-6 CCL2, TNF-α, CCL5 and activation of IL-1β and caspase-1 (287, 288). In adult patients but also in young infants, constant microglial activation is implicated in HIV-associated neurocognitive disorders (HAND), such as HIV-associated dementia (HAD) (289, 290). This correlates with elevated levels of TNF-α, IL-1β, caspase-1 and iNOS in microglia of HAD patients (291–293). In adult patients with HIV encephalitis, perivascular infiltration of CD14+ CD16+ HIV-infected monocytes was also correlated with the onset of dementia (290). A recent study showed that other human macrophage populations from lung and abdomen are able to restrict viral spread via sterile alpha motif and histidine/aspartic acid domain-containing protein 1 (SAMHD1) effectively compared to human microglia, which express similar levels of SAMHD1, but are more susceptible to HIV-1 infection (294). Even in the absence of neurological symptoms, HIV infected patients showed signs of immune activation in the CNS. Obtained microglial activation markers include MHC class II, CD163, IL-1β, and TNF-α levels in the cerebral cortex and white matter of seropositive patients (295–297) and TNF-α, β2-microglobulin and neopterin in the CSF, which are further markers of microglia as well as CAM activation (295). Overall, data suggest that microglia and CAMs at the CNS interfaces can serve as a potential reservoir for HIV even in infants and viral-induced constant microglial activation leading to neurodevelopmental impairment in children and potential onset of neurodegenerative disease later in age.
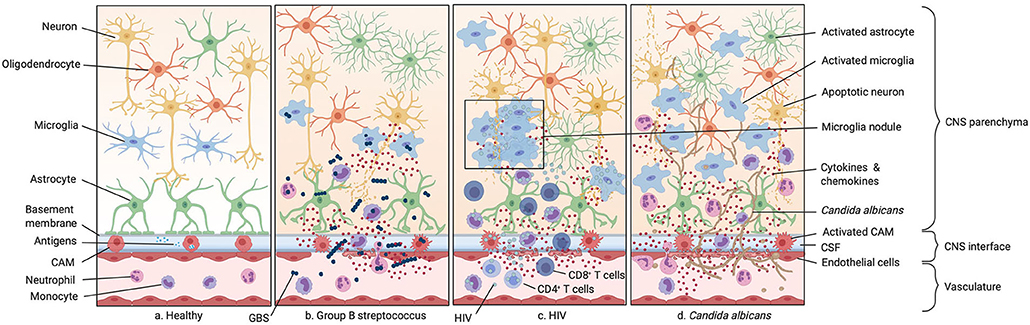
Figure 3. Infections of the neonatal CNS. (a) Illustration of the healthy CNS including the vasculature, the perivascular space, and the brain parenchyma. (b) Group B streptococcus (GBS) infection. GBS enters the CNS either paracellularly, via transcytosis or exploiting trafficking phagocytic host cells. This leads to the secretion of inflammatory cytokines by resident macrophages in the perivascular space and meninges. As GBS spreads more into the CNS this inflammatory response is also mounted by endothelial cells and microglia, leading to leakage of the BBB and therefore an increase number of monocytes and neutrophils in the CNS. (c) HIV infection. HIV infects CD4+ T cells, monocytes, and macrophages. Infection of circulating monocytes helps HIV to enter the CNS where it infects other tissue-resident macrophages. This leads to an acute immune response with secretion of proinflammatory cytokines. (d) Candida albicans infection. Candida albicans enters the CNS either para- or intracellularly or shuttled inside circulating monocytes. Infection of the CNS leads to an activation of microglia, astrocytes, and oligodendrocytes resulting in the secretion of cytokines and chemokines and subsequently to the recruitment of neutrophils and other circulating monocytes.
Herpes Simplex Virus (HSV)
Herpes simplex viruses (HSV) belong to the family of the herpesviridae and are split into two different serotypes: HSV-1 and HSV-2 (298). Like all herpesviruses they contain double-stranded DNA and are able to cause a latent infection (298). HSV infections, if symptomatic, often manifests in oral or genital lesions (298). Both of these viruses are, together with varicella zoster virus, the only herpesviruses that are neurotropic and establish a latent state in dorsal root ganglia (299). Therefore, HSV infection can also occur in the CNS, leading to more severe outcomes such as herpes simplex encephalitis (HSE) and aseptic meningitis (Table 1) (122). Interestingly, HSV-1 is the cause of 90% of HSE in adults, while HSV-2 is mostly associated with neonatal HSE and immunocompromised patients (300). Neonatal herpes is rare but fatal in 60% of cases if not treated (301). Even with treatment, neonatal infection of the CNS can still be fatal and leads to moderate-to-severe neurological abnormalities in more than 50% of the cases (Table 1) (123, 302). Human fetal astrocytes and neurons have been shown to be efficiently infected in vitro by HSV-1, but do not show an induction of cytokine and chemokine expression upon infection (303). Fetal human microglia, however, did not support viral replication but showed an extensive induction of proinflammatory factors such as CXCL10, TNF-α, CCL5, or IL-1β (303). Interestingly, CXCL10 was found to reduce viral replication in neurons, indicating a protective role of microglial-derived CXCL10 (303). Therefore, microglia could play an essential role in controlling HSV-1 replication also in the neonatal brain, as also indicated by other studies during adult encephalitis (304, 305). Upregulation of glutamate transporter 1 (GLT-1) and glutathione was found in microglia, which is an antioxidant with antiviral properties (306). Additionally, IL-6 produced by microglia was shown to protect against neuronal loss after HSV-1 infection (307) underlining the importance of microglia in this context. Purinergic receptor P2Y12+ microglia processes extended to HSV-1 positive neurons in adult HSE in humans and were shown to accumulate in an amoeboid shape around them (308). Extensive cytokine and chemokine expression of cultured human microglia such as TNF-α, IL-1β, IL-10, IFN-α/β/γ and CXCL10, CCL2, CCL4 and CCL5 was observed during HSV-1 infection and upregulation of these effector molecules is partially mediated by TLR2 (303, 309). Higher expression of CXCL10, CCL2, and CXCL9 was also found in infected BALB/c mice (310). TLR3, which recognizes double-stranded viral RNA, was also shown to be highly relevant in fighting HSV infection as two otherwise healthy children with a dominant-negative TLR3 allele were reported with HSE (311). More cases of children with HSE were found with deficiencies in proteins involved in the TLR3-signaling pathway such as TNF receptor asscociated factor 3 (TRAF3), TIR-domain-containing adapter-inducing interferon-β (TRIF) and Unc-93 homolog B (UNC93B) (312–315). Yet, an involvement of microglia and CAMs, which do express high levels of TLR3, needs to be elucidated in this context (316, 317). A recent study, however, demonstrated that adult microglial type I IFN production is dependent on stimulator of interferon genes (STING) and cyclic GMP-AMP synthase (cGAS) (305). Adult mice deficient in cGAS and STING were shown to be susceptible to HSE (305). STING deficiency in isolated neonatal microglia was shown to lead to a higher viral load in the cells and microglial type I interferons production is dependent on STING signaling (305). In conclusion, neonatal HSV infection of the CNS is still a threat and more research is needed to balance a strong immune response restricting viral spread and constant neuroinflammation damaging the developing CNS.
Candida albicans
Fungal infections pose a serious threat to nurseries and intensive care units as newborns, especially premature ones, seem to be most at risk (318). Candida albicans is the most prevalent fungus inhabiting the oropharynx and genital tract of humans and is part of the commensal gut microbiota of 60% of the population (319, 320). At the same time, it is also the major cause of fungal related pathology (Table 1) (319). Opportunistic infections can become rampant if the immune system is disturbed or suppressed for example during pharmacological treatments or immunodeficiency diseases resulting in systemic candidiasis (321). This puts mostly preterm newborns at risk, presenting with an unchallenged immune system and often undergoing heavy antibiotic or corticosteroid treatments, the latter destined to support correct development of the premature lung (322, 323). C. albicans is transmitted to the fetus in utero and to the newborn either nosocomially (in the hospital), or during birth (125, 126). Typically, mucosal membrane colonization is an important event to protect the healthy newborn from invasive candidiasis, while leaving an incidence of 0.5–20% of newborns in the US which are still developing systemic candidiasis (Table 1) (324). Another risk factor for developing candidiasis is necrotizing enterocolitis which was shown to facilitate fungal invasion into the blood stream (325). The mortality rate of newborns suffering from an acute candida infection is a staggering 35–40% despite therapy (325). Involvement of the CNS constitutes a major complication and occurs in more than half of fungal infections with conditions like brain abscess and mycotic meningoencephalitis (326, 327). Although the mechanism of how C. albicans breach the BBB is unclear, it is believed that it can either cross para- or intracellularly or shuttled inside circulating monocytes (328, 329). C. albicans produces a toxin called candidalysin which was shown to activate microglia, astrocytes and oligodendrocytes (330). Furthermore, microglia do express TLR2, Dectin-1 and complement receptor 3 which are essential to recognize invading C. albicans in the CNS (328, 330). Studies in adult mice indicate that microglia together with astrocytes and oligodendrocytes secrete CXCL1 and CXCL2 which induces neutrophil recruitment to the brain and essentially supports clearance of the pathogen from the brain (Figure 3) (330). It is not clear whether the same interplay of glial cells and neutrophils takes effect in newborns. An interesting parallel is that neutrophils isolated from newborns have decreased chemotactic capabilities compared to adults (331). Neutropenia in the CSF has been observed in certain cases of newborn CNS candidiasis (324, 326). It is unknown whether microglia display any deficiencies explaining the grim outcome of candidiasis affecting the CNS. This interplay of glial cells and neutrophils was shown to be crucial for containment and efficient clearance of the pathogen as patients with neutropenia suffered worse outcomes of the infection. This interplay is essentially dependent on the downstream pattern recognition receptor adaptor molecule C-type lectin receptor–Syk adaptor (CARD9) (330). The secreted toxin candidalysin induced microglia to release IL-1β and CXCL2 which in turn resulted in the efficient recruitment of neutrophils to the CNS (Figure 3) (330). Furthermore, microglia recruited Ly6C+ monocytes to the infected CNS which can further contribute to the recruitment of neutrophils (Figure 3). Specific deletion of Card9 in microglia resulted in an impaired neutrophil recruitment and might trigger fungal susceptibility (330). In line with these findings, it was shown that young patients with a defect in the Card9 gene cannot produce neutrophil targeted chemokines and suffer from uncontrolled fungal growth and primary immunodeficiency (330). Recruited neutrophils start secreting chemokines themselves, amplifying this positive feedback loop and driving inflammation of the CNS (332). Interestingly, Card9 deficiency did not result in candida invasion of other organs like the spleen, liver or kidney (333). It is currently unknown whether the CARD9 adaptor protein is also important for other microglia functions like phagocytosing or killing yeast cells. This detrimental role of microglia in the recruitment of neutrophils suggest a central role in the immune answer against C. albicans infection in infants and might also be a key target in understanding neonatal susceptibility to C. albicans infections. So far there is only limited data on the specific role of CAMs in neonatal C. albicans infections available.
Group B Streptococcus (GBS)
Group B streptococcus (GBS) or Streptococcus agalactiae is a gram-positive encapsulated bacterium. Infection with GBS is particularly dangerous for newborns presenting a high rate of morbidity of infected individuals who survive the primary neonatal infection (334). GBS is a major, potentially lethal cause of bacterial meningitis in newborns which is an infection of the CNS membranes covering brain and spinal cord (Table 1) (335–337). Affected newborns most frequently suffer from neurological consequences such as cerebral palsy, cognitive retardation, loss of vision and hearing and seizures (Table 1) (127). GBS naturally colonizes the genital tract of 1 in 4 healthy women, only in a minority of cases leading to an ascending in utero infection of the neonate via the amniotic fluid or during delivery via aspiration of vaginal fluids (338, 339). However, GBS is the leading cause of neonatal sepsis with an occurrence of around 42 cases per 100.000 live births and a mortality rate of around 9% (340, 341). Sepsis is divided into early onset sepsis if it occurs within the first 7 days of life and late onset sepsis if it occurs until 3 months after birth (341). Late onset sepsis often leads to meningoencephalitis which has a mortality rate currently of around 8.3% (Table 1) (340). Here, an intestinal route of GBS infection was suggested in neonates (337). Often GBS enter via the microvasculature in the choroid plexus where they adhere to endothelial cells and are able to cross the BBB either paracellularly, via transcytosis or exploiting trafficking phagocytic host cells (342, 343). In the interfaces of the brain, they encounter, besides recruited circulating monocytes, also the resident perivascular and meningeal macrophages (337). It was shown that perivascular and meningeal macrophages play an important role during bacterial infection of the CNS by secreting pro-inflammatory cytokines such as IL-1β, TNF-α, and IL-6 and phagocytose invading bacteria (Figure 3) (344). A robust inflammation is mounted by both endothelial cells outside the brain, as well as CAMs inside the brain facilitating the entry of more pathogens through the now leaky BBB and increasing the number of emigrating Ly6Chigh monocytes and neutrophils at the site of infection (Figure 3) (345). In vitro studies have shown that microglia react to GBS by TLR2/Myeloid differentiation primary response 88 (MyD88) dependent pathways leading to NO secretion specifically inducing apoptosis in neurons (346). Newborns consequently develop hydrocephalus, ischemia and increased intracerebral pressure and brain injury leading to more neuronal apoptosis (Table 1) (347). In a mouse model of vaginally colonized pregnant females, the authors were able to recapitulate most of the hallmarks of neonatal infection with GBS and following meningitis (348). Immunofluorescent imaging of infected neonatal mouse brains confirmed an activated amoeboid phenotype of microglia (348). However, in this new model of vaginal infection with GBS of pregnant mothers, the authors did not observe an increase in inflammatory cytokines when comparing infected and uninfected pups indicating that neuronal damage is rather induced by the secretion of ROS (348). In contrast, studies performed in GBS infected adult mice attribute a high importance to IL-1β signaling as they show that IL-1β deficient mice cannot keep GBS infection from disseminating to target organs such as the brain and leading to death (349). Long term sequelae of pups infected with GBS indicate a decrease in exploratory behavior in these animals impaired learning and memory and decreased glutamate levels (348). This is in line with the observation that over 30% of children that survived GBS meningitis suffer from neurodevelopmental impairment (128). As previously suggested, the exact role of microglia and CAMs in GBS mediated meningoencephalitis remains ill-defined and needs to be further explored (337).
Concluding Remarks
Besides the many improvements in hygiene and preventive treatments in recent decades, CNS infections are still a threat for the developing fetus and newborn. Information about pathogen-specific infection mechanisms remains scarce. This is due to the limited accessibility of human samples, however the refinement of in utero animal infection models will help to further investigate this issue in the future. A growing body of evidence implicates that especially microglia are an important factor for the outcome of such infections. Their crucial role in CNS development and their interplay with all different cells in the CNS, which is required for the establishment of a functional neuronal network and maintaining CNS homeostasis, may be disrupted in the presence of a pathogen. Whether tissue damage is directly caused by the necrotic properties of pathogens or through the constant activation of CNS macrophages and the subsequent secretion of proinflammatory molecules resulting in long-term complications, is dependent on the respective type of infection and needs to be studied accordingly. The embryonic origin of CAMs has only recently been revealed. Thus, prior studies did not separate long-lived CAMs from infiltrating monocytes in the context of an infection. However, CAMs should not be neglected since they are the “gatekeeper” in the interfaces of the brain and come in contact with intruders earlier than microglia. Due to the longevity of CAMs and microglia a potential role as viral reservoirs needs to be considered and was implicated in recent studies. In conclusion, understanding the impact of prenatal and neonatal CNS infection on the development and the functionality of CNS macrophages may help to comprehend the underlying mechanisms leading to pathogenesis and long-term sequelae of the infant.
Author Contributions
AO and PP wrote the manuscript and designed the figures. KK and DE conceptualized, supervised, and edited the manuscript. All authors listed approved it for publication.
Funding
KK was supported by an initial startup funding from the Neuromac SFB/TRR167 (DFG). This work was supported by a project grant of the Fritz-Thyssen Foundation to KK. DE was supported by the DFG (SFB/TRR167).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
All figures were created using Biorender.
References
1. Silbereis JC, Pochareddy S, Zhu Y, Li M, Sestan N. The cellular and molecular landscapes of the developing human central nervous system. Neuron. (2016) 89:248–68. doi: 10.1016/j.neuron.2015.12.008
2. Pachter JS, Vries de HE, Fabry Z. The blood-brain barrier and its role in immune privilege in the central nervous system. J Neuropathol Exp Neurol. (2003) 62:593–604. doi: 10.1093/jnen/62.6.593
3. Forrester JV, McMenamin PG, Dando SJ. CNS infection and immune privilege. Nat Rev Neurosci. (2018) 19:655–71. doi: 10.1038/s41583-018-0070-8
4. Ehret DY, Patterson JK, Bose CL. Improving neonatal care: a global perspective. Clin Perinatol. (2017) 44:567–82. doi: 10.1016/j.clp.2017.05.002
5. Liu L, Oza S, Hogan D, Perin J, Rudan I, Lawn JE, et al. Global, regional, and national causes of child mortality in 2000–13, with projections to inform post-2015 priorities: an updated systematic analysis. Lancet. (2015) 385:430–40. doi: 10.1016/S0140-6736(14)61698-6
6. Liu L, Oza S, Hogan D, Chu Y, Perin J, Zhu J, et al. Global, regional, and national causes of under-5 mortality in 2000–15: an updated systematic analysis with implications for the Sustainable Development Goals. Lancet. (2016) 388:3027–35. doi: 10.1016/S0140-6736(16)31593-8
7. Wang H, Bhutta ZA, Coates MM, Coggeshall M, Dandona L, Diallo K, et al. Global, regional, national, and selected subnational levels of stillbirths, neonatal, infant, and under-5 mortality, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. (2016) 388:1725–74. doi: 10.1016/S0140-6736(16)31575-6
8. Khan AM, Morris SK, Bhutta ZA. Neonatal and perinatal infections. Pediatr Clin North Am. (2017) 64:785–98. doi: 10.1016/j.pcl.2017.03.008
9. Bhutta ZA, Das JK, Rizvi A, Gaffey MF, Walker N, Horton S, et al. Evidence-based interventions for improvement of maternal and child nutrition: what can be done and at what cost? Lancet. (2013) 382:452–77. doi: 10.1016/S0140-6736(13)60996-4
10. John CC, Carabin H, Montano SM, Bangirana P, Zunt JR, Peterson PK. Global research priorities for infections that affect the nervous system. Nature. (2015) 527:S178–86. doi: 10.1038/nature16033
12. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
13. Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. (2012) 336:86–90. doi: 10.1126/science.1219179
14. Goldmann T, Wieghofer P, Jordão MJ, Prutek F, Hagemeyer N, Frenzel K, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol. (2016) 17:797–805. doi: 10.1038/ni.3423
15. Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. (2013) 16:273–80. doi: 10.1038/nn.3318
16. Palis J. Yolk-sac hematopoiesis the first blood cells of mouse and man. Exp Hematol. (2001) 29:927–36. doi: 10.1016/S0301-472X(01)00669-5
17. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. (2015) 518:547–51. doi: 10.1038/nature13989
18. Thion MS, Low D, Silvin A, Chen J, Grisel P, Schulte-Schrepping J, et al. Microbiome influences prenatal and adult microglia in a sex-specific manner. Cell. (2018) 172:500–16.e16. doi: 10.1016/j.cell.2017.11.042
19. Tay TL, Mai D, Dautzenberg J, Fernández-Klett F, Lin G, Sagar, et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat Neurosci. (2017) 20:793–803. doi: 10.1038/nn.4547
20. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
21. Goldmann T, Wieghofer P, Müller PF, Wolf Y, Varol D, Yona S, et al. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat Neurosci. (2013) 16:1618–26. doi: 10.1038/nn.3531
22. Ferrero G, Mahony CB, Dupuis E, Yvernogeau L, Di Ruggiero E, Miserocchi M, et al. Embryonic microglia derive from primitive macrophages and are replaced by cmyb-dependent definitive microglia in Zebrafish. Cell Rep. (2018) 24:130–41. doi: 10.1016/j.celrep.2018.05.066
23. Xu J, Zhu L, He S, Wu Y, Jin W, Yu T, et al. Temporal-spatial resolution fate mapping reveals distinct origins for embryonic and adult microglia in Zebrafish. Dev Cell. (2015) 34:632–41. doi: 10.1016/j.devcel.2015.08.018
24. Cain MD, Salimi H, Diamond MS, Klein RS. Mechanisms of pathogen invasion into the central nervous system. Neuron. (2019) 103:771–83. doi: 10.1016/j.neuron.2019.07.015
25. van Hove H, Martens L, Scheyltjens I, Vlaminck K de, Pombo Antunes AR, Prijck S de, et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci. (2019) 22:1021–35. doi: 10.1038/s41593-019-0393-4
26. Utz SG, See P, Mildenberger W, Thion MS, Silvin A, Lutz M, et al. Early fate defines microglia and non-parenchymal brain macrophage development. Cell. (2020) 181:557–73.e18. doi: 10.1016/j.cell.2020.03.021
27. Andjelkovic AV, Nikolic B, Pachter JS, Zecevic N. Macrophages/microglial cells in human central nervous system during development: an immunohistochemical study. Brain Res. (1998) 814:13–25. doi: 10.1016/S0006-8993(98)00830-0
28. Verney C, Monier A, Fallet-Bianco C, Gressens P. Early microglial colonization of the human forebrain and possible involvement in periventricular white-matter injury of preterm infants. J Anat. (2010) 217:436–48. doi: 10.1111/j.1469-7580.2010.01245.x
29. Monier A, Adle-Biassette H, Delezoide A-L, Evrard P, Gressens P, Verney C. Entry and distribution of microglial cells in human embryonic and fetal cerebral cortex. J Neuropathol Exp Neurol. (2007) 66:372–82. doi: 10.1097/nen.0b013e3180517b46
30. Sturrock RR. An electron microscopic study of macrophages in the meninges of the human embryonic optic nerve. J Anat. (1988) 157:145–51.
31. Mildner A, Huang H, Radke J, Stenzel W, Priller J. P2Y12 receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia. (2017) 65:375–87. doi: 10.1002/glia.23097
32. Hagemeyer N, Kierdorf K, Frenzel K, Xue J, Ringelhan M, Abdullah Z, et al. Transcriptome-based profiling of yolk sac-derived macrophages reveals a role for Irf8 in macrophage maturation. EMBO J. (2016) 35:1730–44. doi: 10.15252/embj.201693801
33. Buttgereit A, Lelios I, Yu X, Vrohlings M, Krakoski NR, Gautier EL, et al. Sall1 is a transcriptional regulator defining microglia identity and function. Nat Immunol. (2016) 17:1397–406. doi: 10.1038/ni.3585
34. Easley-Neal C, Foreman O, Sharma N, Zarrin AA, Weimer RM. CSF1R Ligands IL-34 and CSF1 are differentially required for microglia development and maintenance in white and gray matter brain regions. Front Immunol. (2019) 10:2199. doi: 10.3389/fimmu.2019.02199
35. Lin H, Lee E, Hestir K, Leo C, Huang M, Bosch E, et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science. (2008) 320:807–11. doi: 10.1126/science.1154370
36. Greter M, Lelios I, Pelczar P, Hoeffel G, Price J, Leboeuf M, et al. Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity. (2012) 37:1050–60. doi: 10.1016/j.immuni.2012.11.001
37. Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, et al. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol. (2012) 13:753–60. doi: 10.1038/ni.2360
38. Kana V, Desland FA, Casanova-Acebes M, Ayata P, Badimon A, Nabel E, et al. CSF-1 controls cerebellar microglia and is required for motor function and social interaction. J Exp Med. (2019) 216:2265–81. doi: 10.1084/jem.20182037
39. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci. (2014) 17:131–43. doi: 10.1038/nn.3599
40. Erny D, Hrabě de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. (2015) 18:965–77. doi: 10.1038/nn.4030
41. Matcovitch-Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, Sarrazin S, et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science. (2016) 353:aad8670. doi: 10.1126/science.aad8670
42. Brown AS. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev Neurobiol. (2012) 72:1272–6. doi: 10.1002/dneu.22024
43. Knuesel I, Chicha L, Britschgi M, Schobel SA, Bodmer M, Hellings JA, et al. Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol. (2014) 10:643–60. doi: 10.1038/nrneurol.2014.187
44. Cordeiro CN, Tsimis M, Burd I. Infections and brain development. Obstet Gynecol Surv. (2015) 70:644–55. doi: 10.1097/OGX.0000000000000236
45. Bale JF. Fetal infections and brain development. Clin Perinatol. (2009) 36:639–53. doi: 10.1016/j.clp.2009.06.005
46. Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. (2004) 173:3916–24. doi: 10.4049/jimmunol.173.6.3916
47. Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. (2002) 61:1013–21. doi: 10.1093/jnen/61.11.1013
48. Dalpke AH, Schäfer MK-H, Frey M, Zimmermann S, Tebbe J, Weihe E, et al. Immunostimulatory CpG-DNA activates murine microglia. J Immunol. (2002) 168:4854–63. doi: 10.4049/jimmunol.168.10.4854
49. Laflamme N, Rivest S. Toll-like receptor 4: the missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. FASEB J. (2001) 15:155–63. doi: 10.1096/fj.00-0339com
50. Zhang Y-K, Liu J-T, Peng Z-W, Fan H, Yao A-H, Cheng P, et al. Different TLR4 expression and microglia/macrophage activation induced by hemorrhage in the rat spinal cord after compressive injury. J Neuroinflammation. (2013) 10:112. doi: 10.1186/1742-2094-10-112
51. Shiau CE, Monk KR, Joo W, Talbot WS. An anti-inflammatory NOD-like receptor is required for microglia development. Cell Rep. (2013) 5:1342–52. doi: 10.1016/j.celrep.2013.11.004
52. Ribes S, Ebert S, Czesnik D, Regen T, Zeug A, Bukowski S, et al. Toll-like receptor prestimulation increases phagocytosis of Escherichia coli DH5alpha and Escherichia coli K1 strains by murine microglial cells. Infect Immun. (2009) 77:557–64. doi: 10.1128/IAI.00903-08
53. Boddeke EWGM, Meigel I, Frentzel S, Biber K, Renn LQ, Gebicke-Härter P. Functional expression of the fractalkine (CX3C) receptor and its regulation by lipopolysaccharide in rat microglia. Eur J Pharmacol. (1999) 374:309–13. doi: 10.1016/S0014-2999(99)00307-6
54. Sørensen TL, Tani M, Jensen J, Pierce V, Lucchinetti C, Folcik VA, et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest. (1999) 103:807–15. doi: 10.1172/JCI5150
55. Cheeran MC, Hu S, Yager SL, Gekker G, Peterson PK, Lokensgard JR. Cytomegalovirus induces cytokine and chemokine production differentially in microglia and astrocytes: antiviral implications. J Neurovirol. (2001) 7:135–47. doi: 10.1080/13550280152058799
56. McManus CM, Brosnan CF, Berman JW. Cytokine induction of MIP-1 alpha and MIP-1 beta in human fetal microglia. J Immunol. (1998) 160:1449–55.
57. Hanisch U-K. Microglia as a source and target of cytokines. Glia. (2002) 40:140–55. doi: 10.1002/glia.10161
58. Peterson PK, Hu S, Salak-Johnson J, Molitor TW, Chao CC. Differential production of and migratory response to beta chemokines by human microglia and astrocytes. J Infect Dis. (1997) 175:478–81. doi: 10.1093/infdis/175.2.478
59. Smith JA, Das A, Ray SK, Banik NL. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull. (2012) 87:10–20. doi: 10.1016/j.brainresbull.2011.10.004
60. Kuno R, Wang J, Kawanokuchi J, Takeuchi H, Mizuno T, Suzumura A. Autocrine activation of microglia by tumor necrosis factor-alpha. J Neuroimmunol. (2005) 162:89–96. doi: 10.1016/j.jneuroim.2005.01.015
61. Thomas DM, Francescutti-Verbeem DM, Kuhn DM. Gene expression profile of activated microglia under conditions associated with dopamine neuronal damage. FASEB J. (2006) 20:515–7. doi: 10.1096/fj.05-4873fje
62. Russo MV, McGavern DB. Immune Surveillance of the CNS following Infection and Injury. Trends Immunol. (2015) 36:637–50. doi: 10.1016/j.it.2015.08.002
63. Griffin DE. Immune responses to RNA-virus infections of the CNS. Nat Rev Immunol. (2003) 3:493–502. doi: 10.1038/nri1105
64. Hagberg H, Mallard C. Effect of inflammation on central nervous system development and vulnerability. Curr Opin Neurol. (2005) 18:117–23. doi: 10.1097/01.wco.0000162851.44897.8f
65. Park J-E, Jardine L, Gottgens B, Teichmann SA, Haniffa M. Prenatal development of human immunity. Science. (2020) 368:600–3. doi: 10.1126/science.aaz9330
66. Stinson LF. Establishment of the early-life microbiome: a DOHaD perspective. J Dev Orig Health Dis. (2019) 11:201–10. doi: 10.1017/S2040174419000588
67. Hamilton WJ, Boyd JD. Development of the human placenta in the first three months of gestation. J Anat. (1960) 94:297–328.
68. Palmeira P, Quinello C, Silveira-Lessa AL, Zago CA, Carneiro-Sampaio M. IgG placental transfer in healthy and pathological pregnancies. Clin Dev Immunol. (2012) 2012:985646. doi: 10.1155/2012/985646
69. Malek A. Role of IgG antibodies in association with placental function and immunologic diseases in human pregnancy. Expert Rev Clin Immunol. (2013) 9:235–49. doi: 10.1586/eci.12.99
70. Fowler KB, Stagno S, Pass RF. Maternal immunity and prevention of congenital cytomegalovirus infection. JAMA. (2003) 289:1008–11. doi: 10.1001/jama.289.8.1008
71. McDonagh S, Maidji E, Ma W, Chang H-T, Fisher S, Pereira L. Viral and bacterial pathogens at the maternal-fetal interface. J Infect Dis. (2004) 190:826–34. doi: 10.1086/422330
72. Delorme-Axford E, Sadovsky Y, Coyne CB. The placenta as a barrier to viral infections. Annu Rev Virol. (2014) 1:133–46. doi: 10.1146/annurev-virology-031413-085524
73. Bayer A, Lennemann NJ, Ouyang Y, Bramley JC, Morosky S, Marques ET, et al. Type III interferons produced by human placental trophoblasts confer protection against Zika Virus infection. Cell Host Microbe. (2016) 19:705–12. doi: 10.1016/j.chom.2016.03.008
74. Kwon J-Y, Aldo P, You Y, Ding J, Racicot K, Dong X, et al. Relevance of placental type I interferon beta regulation for pregnancy success. Cell Mol Immunol. (2018) 15:1010–26. doi: 10.1038/s41423-018-0050-y
75. Arora N, Sadovsky Y, Dermody TS, Coyne CB. Microbial vertical transmission during human pregnancy. Cell Host Microbe. (2017) 21:561–7. doi: 10.1016/j.chom.2017.04.007
76. Coyne CB, Lazear HM. Zika virus - reigniting the TORCH. Nat Rev Microbiol. (2016) 14:707–15. doi: 10.1038/nrmicro.2016.125
77. Yockey LJ, Lucas C, Iwasaki A. Contributions of maternal and fetal antiviral immunity in congenital disease. Science. (2020) 368:608–12. doi: 10.1126/science.aaz1960
78. Suff N, Karda R, Diaz JA, Ng J, Baruteau J, Perocheau D, et al. Ascending vaginal infection using bioluminescent bacteria evokes intrauterine inflammation, preterm birth, and neonatal brain injury in pregnant mice. Am J Pathol. (2018) 188:2164–76. doi: 10.1016/j.ajpath.2018.06.016
79. Garland SM, Ní Chuileannáin F, Satzke C, Robins-Browne R. Mechanisms, organisms and markers of infection in pregnancy. J Reprod Immunol. (2002) 57:169–83. doi: 10.1016/S0165-0378(02)00018-9
80. Nahmias AJ. The TORCH Complex. Hospital Pract. (1974) 9:65–72. doi: 10.1080/21548331.1974.11708004
81. Schwartz DA. The Origins and Emergence of Zika Virus, the newest TORCH infection: What's old is new again. Arch Pathol Lab Med. (2017) 141:18–25. doi: 10.5858/arpa.2016-0429-ED
82. Neu N, Duchon J, Zachariah P. TORCH infections. Clin Perinatol. (2015) 42:77–103. doi: 10.1016/j.clp.2014.11.001
83. Epps RE, Pittelkow MR, Su WP. TORCH syndrome. Semin Dermatol. (1995) 14:179–86. doi: 10.1016/S1085-5629(05)80016-1
84. Vivanti AJ, Vauloup-Fellous C, Prevot S, Zupan V, Suffee C, Do Cao J, et al. Transplacental transmission of SARS-CoV-2 infection. Nat Commun. (2020) 11:3572. doi: 10.1038/s41467-020-17436-6
85. Kovarik J, Siegrist C-A. Immunity in early life. Immunol Today. (1998) 19:150–2. doi: 10.1016/S0167-5699(97)01230-9
86. Hayward AR. The human fetus and newborn: development of the immune response. Birth Defects Orig Artic Ser. (1983) 19:289–94.
87. Stras SF, Werner L, Toothaker JM, Olaloye OO, Oldham AL, McCourt CC, et al. Maturation of the human intestinal immune system occurs early in fetal development. Dev Cell. (2019) 51:357–73.e5. doi: 10.1016/j.devcel.2019.09.008
88. McGovern N, Shin A, Low G, Low D, Duan K, Yao LJ, et al. Human fetal dendritic cells promote prenatal T-cell immune suppression through arginase-2. Nature. (2017) 546:662–6.
89. Zhang X, Zhivaki D, Lo-Man R. Unique aspects of the perinatal immune system. Nat Rev Immunol. (2017) 17:495–507. doi: 10.1038/nri.2017.54
90. Sagebiel AF, Steinert F, Lunemann S, Körner C, Schreurs RR, Altfeld M, et al. Tissue-resident Eomes+ NK cells are the major innate lymphoid cell population in human infant intestine. Nat Commun. (2019) 10:975. doi: 10.1038/s41467-018-08267-7
91. Schuler-Faccini L, Ribeiro EM, Feitosa IM, Horovitz DD, Cavalcanti DP, Pessoa A, et al. Possible association between Zika Virus infection and microcephaly - Brazil, 2015. MMWR Morb Mortal Wkly Rep. (2016) 65:59–62. doi: 10.15585/mmwr.mm6503e2
92. Oliveira Melo AS, Malinger G, Ximenes R, Szejnfeld PO, Alves Sampaio S, Bispo de Filippis AM. Zika virus intrauterine infection causes fetal brain abnormality and microcephaly: tip of the iceberg? Ultrasound Obstet Gynecol. (2016) 47:6–7. doi: 10.1002/uog.15831
93. Rasmussen SA, Jamieson DJ, Honein MA, Petersen LR. Zika Virus and birth defects–reviewing the evidence for causality. N Engl J Med. (2016) 374:1981–7. doi: 10.1056/NEJMsr1604338
94. Dick GWA, Kitchen SF, Haddow AJ. Zika Virus (I). Isolations and serological specificity. Trans R Soc Trop Med Hyg. (1952) 46:509–20. doi: 10.1016/0035-9203(52)90042-4
95. Lazear HM, Diamond MS. Zika Virus: new clinical syndromes and its emergence in the western hemisphere. J Virol. (2016) 90:4864–75. doi: 10.1128/JVI.00252-16
96. Wills MR, Poole E, Lau B, Krishna B, Sinclair JH. The immunology of human cytomegalovirus latency: could latent infection be cleared by novel immunotherapeutic strategies? Cell Mol Immunol. (2015) 12:128–38. doi: 10.1038/cmi.2014.75
97. Cannon MJ, Davis KF. Washing our hands of the congenital cytomegalovirus disease epidemic. BMC Public Health. (2005) 5:70. doi: 10.1186/1471-2458-5-70
98. Sweeten TL, Posey DJ, McDougle CJ. Brief report: autistic disorder in three children with cytomegalovirus infection. J Autism Dev Disord. (2004) 34:583–6. doi: 10.1007/s10803-004-2552-y
99. Wolfe B, Wiepz GJ, Schotzko M, Bondarenko GI, Durning M, Simmons HA, et al. Acute fetal demise with first trimester maternal infection resulting from listeria monocytogenes in a nonhuman primate model. mBio. (2017) 8:e01938-16. doi: 10.1128/mBio.01938-16
100. Le Monnier A, Autret N, Join-Lambert OF, Jaubert F, Charbit A, Berche P. ActA is required for crossing of the fetoplacental barrier by Listeria monocytogenes. Infect Immun. (2007) 75:950–7. doi: 10.1128/IAI.01570-06
101. Janakiraman V. Listeriosis in pregnancy: diagnosis, treatment, and prevention. Rev Obstet Gynecol. (2008) 1:179–85.
102. Laciar AL, Hasuoka RP, Correa SM, Miranda AM, Centorbi ONP. Symptomatic hydrocephalus in a newborn infected with listeria monocytogenes. Braz J Microbiol. (2000) 31:9–11. doi: 10.1590/S1517-83822000000100003
103. Cuthbertson G, Weiner CP, Giller RH, Grose C. Prenatal diagnosis of second-trimester congenital varicella syndrome by virus-specific immunoglobulin M. J Pediatr. (1987) 111:592–5. doi: 10.1016/S0022-3476(87)80128-2
104. Echevarría JM, Casas I, Martínez-Martín P. Infections of the nervous system caused by varicella-zoster virus: a review. Intervirology. (1997) 40:72–84. doi: 10.1159/000150535
105. Harding B, Baumer JA. Congenital varicella-zoster. A serologically proven case with necrotizing encephalitis and malformation. Acta Neuropathol. (1988) 76:311–5. doi: 10.1007/BF00687781
106. Isumi H, Nunoue T, Nishida A, Takashima S. Fetal brain infection with human parvovirus B19. Pediatr Neurol. (1999) 21:661–3. doi: 10.1016/S0887-8994(99)00055-7
107. Jong EP de, Walther FJ, Kroes AC, Oepkes D. Parvovirus B19 infection in pregnancy: new insights and management. Prenat Diagn. (2011) 31:419–25. doi: 10.1002/pd.2714
108. Maisonneuve E, Garel C, Friszer S, Pénager C, Carbonne B, Pernot F, et al. Fetal brain injury associated with parvovirus B19 congenital infection requiring intrauterine transfusion. Fetal Diagn Ther. (2019) 46:1–11. doi: 10.1159/000489881
109. Shaw DW, Cohen WA. Viral infections of the CNS in children: imaging features. AJR Am J Roentgenol. (1993) 160:125–33. doi: 10.2214/ajr.160.1.8416608
110. Frey TK. Neurological aspects of rubella virus infection. Intervirology. (1997) 40:167–75. doi: 10.1159/000150543
111. Waxham MN, Wolinsky JS. Rubella virus and its effects on the central nervous system. Neurol Clin. (1984) 2:367–85. doi: 10.1016/S0733-8619(18)31109-5
112. Yamamoto AY, Mussi-Pinhata MM, Boppana SB, Novak Z, Wagatsuma VM, Oliveira Pd, et al. Human cytomegalovirus reinfection is associated with intrauterine transmission in a highly cytomegalovirus-immune maternal population. Am J Obstet Gynecol. (2010) 202:297.e1-8. doi: 10.1016/j.ajog.2009.11.018
113. Mlakar J, Korva M, Tul N, Popović M, Poljšak-Prijatelj M, Mraz J, et al. Zika Virus Associated with Microcephaly. N Engl J Med. (2016) 374:951–8. doi: 10.1056/NEJMoa1600651
114. Driggers RW, Ho C-Y, Korhonen EM, Kuivanen S, Jääskeläinen AJ, Smura T, et al. Zika Virus infection with prolonged maternal viremia and fetal brain abnormalities. N Engl J Med. (2016) 374:2142–51. doi: 10.1056/NEJMoa1601824
115. Tabata T, Petitt M, Puerta-Guardo H, Michlmayr D, Wang C, Fang-Hoover J, et al. Zika Virus targets different primary human placental cells, suggesting two routes for vertical transmission. Cell Host Microbe. (2016) 20:155–66. doi: 10.1016/j.chom.2016.07.002
116. Quicke KM, Bowen JR, Johnson EL, McDonald CE, Ma H, O'Neal JT, et al. Zika Virus infects human placental macrophages. Cell Host Microbe. (2016) 20:83–90. doi: 10.1016/j.chom.2016.05.015
117. Johansson MA, Mier-y-Teran-Romero L, Reefhuis J, Gilboa SM, Hills SL. Zika and the risk of microcephaly. N Engl J Med. (2016) 375:1–4. doi: 10.1056/NEJMp1605367
118. Jaenisch T, Rosenberger KD, Brito C, Brady O, Brasil P, Marques ET. Risk of microcephaly after Zika virus infection in Brazil, 2015 to 2016. Bull World Health Organ. (2017) 95:191–8. doi: 10.2471/BLT.16.178608
119. Epstein LG, Sharer LR, Joshi VV, Fojas MM, Koenigsberger MR, Oleske JM. Progressive encephalopathy in children with acquired immune deficiency syndrome. Ann Neurol. (1985) 17:488–96. doi: 10.1002/ana.410170512
120. Epstein LG, Sharer LR, Oleske JM, Connor EM, Goudsmit J, Bagdon L, et al. Neurologic manifestations of human immunodeficiency virus infection in children. Pediatrics. (1986) 78:678–87.
121. Kauffman WM, Sivit CJ, Fitz CR, Rakusan TA, Herzog K, Chandra RS. CT and MR evaluation of intracranial involvement in pediatric HIV infection: a clinical-imaging correlation. AJNR Am J Neuroradiol. (1992) 13:949–57.
122. Tyler KL. Herpes simplex virus infections of the central nervous system: encephalitis and meningitis, including Mollaret's. Herpes. (2004) 11 Suppl 2:57A−64.
123. Kimberlin DW, Lin CY, Jacobs RF, Powell DA, Frenkel LM, Gruber WC, et al. Natural history of neonatal herpes simplex virus infections in the acyclovir era. Pediatrics. (2001) 108:223–9. doi: 10.1542/peds.108.2.223
124. Agostini S, Mancuso R, Baglio F, Cabinio M, Hernis A, Costa AS, et al. High avidity HSV-1 antibodies correlate with absence of amnestic mild cognitive impairment conversion to Alzheimer's disease. Brain Behav Immun. (2016) 58:254–60. doi: 10.1016/j.bbi.2016.07.153
125. Hammarskjöld F, Mernelius S, Andersson RE, Berg S, Hanberger H, Löfgren S, et al. Possible transmission of Candida albicans on an intensive care unit: genotype and temporal cluster analyses. J Hosp Infect. (2013) 85:60–5. doi: 10.1016/j.jhin.2013.06.002
126. Waggoner-Fountain LA, Walker MW, Hollis RJ, Pfaller MA, Ferguson JE, Wenzel RP, et al. Vertical and horizontal transmission of unique Candida species to premature newborns. Clin Infect Dis. (1996) 22:803–8. doi: 10.1093/clinids/22.5.803
127. Baker CJ. The spectrum of perinatal group B streptococcal disease. Vaccine. (2013) 31 Suppl 4:D3–6. doi: 10.1016/j.vaccine.2013.02.030
128. Libster R, Edwards KM, Levent F, Edwards MS, Rench MA, Castagnini LA, et al. Long-term outcomes of group B streptococcal meningitis. Pediatrics. (2012) 130:e8–15. doi: 10.1542/peds.2011-3453
129. Gérardin P, Barau G, Michault A, Bintner M, Randrianaivo H, Choker G, et al. Multidisciplinary prospective study of mother-to-child chikungunya virus infections on the island of La Réunion. PLoS Med. (2008) 5:e60. doi: 10.1371/journal.pmed.0050060
130. Oliveira JR, Gérardin P, Couderc T, Randrianaivo H, Fritel X, Lecuit M. Chikungunya virus-associated encephalitis: a cohort study on La Réunion Island, 2005-2009. Neurology. (2016) 86:2025–6. doi: 10.1212/WNL.0000000000002732
131. Torres JR, Falleiros-Arlant LH, Dueñas L, Pleitez-Navarrete J, Salgado DM, Castillo JB-D. Congenital and perinatal complications of Chikungunya fever: a Latin American experience. Int J Infect Dis. (2016) 51:85–8. doi: 10.1016/j.ijid.2016.09.009
132. Ramful D, Carbonnier M, Pasquet M, Bouhmani B, Ghazouani J, Noormahomed T, et al. Mother-to-child transmission of Chikungunya virus infection. Pediatr Infect Dis J. (2007) 26:811–5. doi: 10.1097/INF.0b013e3180616d4f
133. Gérardin P, Sampériz S, Ramful D, Boumahni B, Bintner M, Alessandri J-L, et al. Neurocognitive outcome of children exposed to perinatal mother-to-child Chikungunya virus infection: the CHIMERE cohort study on Reunion Island. PLoS Negl Trop Dis. (2014) 8:e2996. doi: 10.1371/journal.pntd.0002996
134. Bonacorsi S, Bingen E. Molecular epidemiology of Escherichia coli causing neonatal meningitis. Int J Med Microbiol. (2005) 295:373–81. doi: 10.1016/j.ijmm.2005.07.011
135. Kim BY, Kang J, Kim KS. Invasion processes of pathogenic Escherichia coli. Int J Med Microbiol. (2005) 295:463–70. doi: 10.1016/j.ijmm.2005.07.004
136. Campos GS, Bandeira AC, Sardi SI. Zika Virus Outbreak, Bahia, Brazil. Emerging Infect Dis. (2015) 21:1885–6. doi: 10.3201/eid2110.150847
137. Tisoncik-Go J, Gale M. Microglia in memory decline from Zika virus and west Nile virus infection. Trends Neurosci. (2019) 42:757–9. doi: 10.1016/j.tins.2019.08.009
138. Pacheco O, Beltrán M, Nelson CA, Valencia D, Tolosa N, Farr SL, et al. Zika Virus Disease in Colombia - Preliminary Report. N Engl J Med. (2020) 383:e44. doi: 10.1056/NEJMoa1604037
139. Brady OJ, Osgood-Zimmerman A, Kassebaum NJ, Ray SE, Araújo de VE, da Nóbrega AA, et al. The association between Zika virus infection and microcephaly in Brazil 2015-2017: An observational analysis of over 4 million births. PLoS Med. (2019) 16:e1002755. doi: 10.1371/journal.pmed.1002755
140. Muffat J, Li Y, Omer A, Durbin A, Bosch I, Bakiasi G, et al. Human induced pluripotent stem cell-derived glial cells and neural progenitors display divergent responses to Zika and dengue infections. Proc Natl Acad Sci USA. (2018) 115:7117–22. doi: 10.1073/pnas.1719266115
141. Tang H, Hammack C, Ogden SC, Wen Z, Qian X, Li Y, et al. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell. (2016) 18:587–90. doi: 10.1016/j.stem.2016.02.016
142. Xu P, Shan C, Dunn TJ, Xie X, Xia H, Gao J, et al. Role of microglia in the dissemination of Zika virus from mother to fetal brain. PLoS Negl Trop Dis. (2020) 14:e0008413. doi: 10.1371/journal.pntd.0008413
143. Garber C, Soung A, Vollmer LL, Kanmogne M, Last A, Brown J, et al. T cells promote microglia-mediated synaptic elimination and cognitive dysfunction during recovery from neuropathogenic flaviviruses. Nat Neurosci. (2019) 22:1276–88. doi: 10.1038/s41593-019-0427-y
144. Wang J, Liu J, Zhou R, Ding X, Zhang Q, Zhang C, et al. Zika virus infected primary microglia impairs NPCs proliferation and differentiation. Biochem Biophys Res Commun. (2018) 497:619–25. doi: 10.1016/j.bbrc.2018.02.118
145. Diop F, Vial T, Ferraris P, Wichit S, Bengue M, Hamel R, et al. Zika virus infection modulates the metabolomic profile of microglial cells. PLoS ONE. (2018) 13:e0206093. doi: 10.1371/journal.pone.0206093
146. Freeman L, Guo H, David CN, Brickey WJ, Jha S, Ting JP-Y. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med. (2017) 214:1351–70. doi: 10.1084/jem.20150237
147. Panganiban AT, Blair RV, Hattler JB, Bohannon DG, Bonaldo MC, Schouest B, et al. A zika virus primary isolate induces neuroinflammation, compromises the blood-brain barrier, and upregulates cxcl12 in adult macaques. Brain Pathol. (2020) doi: 10.1111/bpa.12873. [Epub ahead of print].
148. Manangeeswaran M, Ireland DD, Verthelyi D. Zika (PRVABC59) Infection is associated with T cell infiltration and neurodegeneration in CNS of immunocompetent neonatal C57Bl/6 Mice. PLoS Pathog. (2016) 12:e1006004. doi: 10.1371/journal.ppat.1006004
149. Azevedo RS, Sousa de JR, Araujo MT, Martins Filho AJ, Alcantara de BN, Araujo FM, et al. In situ immune response and mechanisms of cell damage in central nervous system of fatal cases microcephaly by Zika virus. Sci Rep. (2018) 8:1. doi: 10.1038/s41598-017-17765-5
150. Kenneson A, Cannon MJ. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol. (2007) 17:253–76. doi: 10.1002/rmv.535
151. Brown HL, Abernathy MP. Cytomegalovirus infection. Semin Perinatol. (1998) 22:260–6. doi: 10.1016/S0146-0005(98)80014-1
152. Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. The “silent” global burden of congenital cytomegalovirus. Clin Microbiol Rev. (2013) 26:86–102. doi: 10.1128/CMR.00062-12
153. Dreher AM, Arora N, Fowler KB, Novak Z, Britt WJ, Boppana SB, et al. Spectrum of disease and outcome in children with symptomatic congenital cytomegalovirus infection. J Pediatr. (2014) 164:855–9. doi: 10.1016/j.jpeds.2013.12.007
154. Yamamoto AY, Mussi-Pinhata MM, Isaac Md, Amaral FR, Carvalheiro CG, Aragon DC, et al. Congenital cytomegalovirus infection as a cause of sensorineural hearing loss in a highly immune population. Pediatr Infect Dis J. (2011) 30:1043–6. doi: 10.1097/INF.0b013e31822d9640
155. Maidji E, McDonagh S, Genbacev O, Tabata T, Pereira L. Maternal antibodies enhance or prevent cytomegalovirus infection in the placenta by neonatal Fc receptor-mediated transcytosis. Am J Pathol. (2006) 168:1210–26. doi: 10.2353/ajpath.2006.050482
156. Sakao-Suzuki M, Kawasaki H, Akamatsu T, Meguro S, Miyajima H, Iwashita T, et al. Aberrant fetal macrophage/microglial reactions to cytomegalovirus infection. Ann Clin Transl Neurol. (2014) 1:570–88. doi: 10.1002/acn3.88
157. van den Pol AN, Mocarski E, Saederup N, Vieira J, Meier TJ. Cytomegalovirus cell tropism, replication, and gene transfer in brain. J Neurosci.. (1999) 19:10948–65. doi: 10.1523/JNEUROSCI.19-24-10948.1999
158. Teissier N, Fallet-Bianco C, Delezoide A-L, Laquerrière A, Marcorelles P, Khung-Savatovsky S, et al. Cytomegalovirus-induced brain malformations in fetuses. J Neuropathol Exp Neurol. (2014) 73:143–58. doi: 10.1097/NEN.0000000000000038
159. Laukoter S, Rauschka H, Tröscher AR, Köck U, Saji E, Jellinger K, et al. Differences in T cell cytotoxicity and cell death mechanisms between progressive multifocal leukoencephalopathy, herpes simplex virus encephalitis and cytomegalovirus encephalitis. Acta Neuropathol. (2017) 133:613–27. doi: 10.1007/s00401-016-1642-1
160. Kučić N, Rački V, Jurdana K, Marcelić M, Grabušić K. Immunometabolic phenotype of BV-2 microglia cells upon murine cytomegalovirus infection. J Neurovirol. (2019) 25:496–507. doi: 10.1007/s13365-019-00750-1
161. Cloarec R, Bauer S, Teissier N, Schaller F, Luche H, Courtens S, et al. In utero administration of drugs targeting microglia improves the neurodevelopmental outcome following cytomegalovirus infection of the rat fetal brain. Front Cell Neurosci. (2018) 12:55. doi: 10.3389/fncel.2018.00055
162. Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet. (2004) 363:1965–76. doi: 10.1016/S0140-6736(04)16412-X
163. Boothroyd JC, Grigg ME. Population biology of Toxoplasma gondii and its relevance to human infection: do different strains cause different disease? Curr Opin Microbiol. (2002) 5:438–42. doi: 10.1016/S1369-5274(02)00349-1
164. Halonen SK, Weiss LM. Toxoplasmosis. Handb Clin Neurol. (2013) 114:125–45. doi: 10.1016/B978-0-444-53490-3.00008-X
165. Black MW, Boothroyd JC. Lytic cycle of Toxoplasma gondii. Microbiol Mol Biol Rev. (2000) 64:607–23. doi: 10.1128/MMBR.64.3.607-623.2000
166. Torrey EF, Yolken RH. Toxoplasma oocysts as a public health problem. Trends Parasitol. (2013) 29:380–4. doi: 10.1016/j.pt.2013.06.001
167. Dubey JP, Speer CA, Shen SK, Kwok OC, Blixt JA. Oocyst-induced murine toxoplasmosis: life cycle, pathogenicity, and stage conversion in mice fed Toxoplasma gondii oocysts. J Parasitol. (1997) 83:870–82. doi: 10.2307/3284282
168. Mordue DG, Sibley LD. Intracellular fate of vacuoles containing Toxoplasma gondii is determined at the time of formation and depends on the mechanism of entry. J Immunol. (1997) 159:4452–9.
169. Dubey JP, Miller NL, Frenkel JK. The Toxoplasma gondii oocyst from cat feces. J Exp Med. (1970) 132:636–62. doi: 10.1084/jem.132.4.636
170. McAuley JB. Congenital toxoplasmosis. J Pediatric Infect Dis Soc. (2014) 3 Suppl 1:S30–5. doi: 10.1093/jpids/piu077
171. Ostrander B, Bale JF. Congenital and perinatal infections. Handb Clin Neurol. (2019) 162:133–53. doi: 10.1016/B978-0-444-64029-1.00006-0
172. Schlüter D, Barragan A. Advances and challenges in understanding cerebral toxoplasmosis. Front Immunol. (2019) 10:242. doi: 10.3389/fimmu.2019.00242
173. Schmidt-Hieber M, Zweigner J, Uharek L, Blau IW, Thiel E. Central nervous system infections in immunocompromised patients: update on diagnostics and therapy. Leuk Lymphoma. (2009) 50:24–36. doi: 10.1080/10428190802517740
174. Vogel N, Kirisits M, Michael E, Bach H, Hostetter M, Boyer K, et al. Congenital toxoplasmosis transmitted from an immunologically competent mother infected before conception. Clin Infect Dis. (1996) 23:1055–60. doi: 10.1093/clinids/23.5.1055
175. Dunn D, Wallon M, Peyron F, Petersen E, Peckham C, Gilbert R. Mother-to-child transmission of toxoplasmosis: risk estimates for clinical counselling. Lancet. (1999) 353:1829–33. doi: 10.1016/S0140-6736(98)08220-8
176. Wujcicka W, Wilczyński J, Nowakowska D. Do the placental barrier, parasite genotype and Toll-like receptor polymorphisms contribute to the course of primary infection with various Toxoplasma gondii genotypes in pregnant women? Eur J Clin Microbiol Infect Dis. (2014) 33:703–9. doi: 10.1007/s10096-013-2017-3
177. Wong SY, Remington JS. Toxoplasmosis in pregnancy. Clin Infect Dis. (1994) 18:853–61; quiz 862. doi: 10.1093/clinids/18.6.853
178. McLeod R, Kieffer F, Sautter M, Hosten T, Pelloux H. Why prevent, diagnose and treat congenital toxoplasmosis? Mem Inst Oswaldo Cruz. (2009) 104:320–44. doi: 10.1590/S0074-02762009000200029
179. Oz HS, Tobin T. Atovaquone ameliorate gastrointestinal toxoplasmosis complications in a pregnancy model. Med Sci Monit. (2012) 18:BR337–45. doi: 10.12659/MSM.883342
180. Oz HS. Maternal and congenital toxoplasmosis, currently available and novel therapies in horizon. Front Microbiol. (2014) 5:385. doi: 10.3389/fmicb.2014.00385
181. Effect of timing and type of treatment on the risk of mother to child transmission of Toxoplasma gondii. BJOG. (2003) 110:112–20. doi: 10.1046/j.1471-0528.2003.02325.x
182. Robbins JR, Zeldovich VB, Poukchanski A, Boothroyd JC, Bakardjiev AI. Tissue barriers of the human placenta to infection with Toxoplasma gondii. Infect Immun. (2012) 80:418–28. doi: 10.1128/IAI.05899-11
183. Barragan A, Sibley LD. Transepithelial migration of Toxoplasma gondii is linked to parasite motility and virulence. J Exp Med. (2002) 195:1625–33. doi: 10.1084/jem.20020258
184. Da Gama LM, Ribeiro-Gomes FL, Guimarães U, Arnholdt AC. Reduction in adhesiveness to extracellular matrix components, modulation of adhesion molecules and in vivo migration of murine macrophages infected with Toxoplasma gondii. Microbes Infect. (2004) 6:1287–96. doi: 10.1016/j.micinf.2004.07.008
185. Courret N, Darche S, Sonigo P, Milon G, Buzoni-Gâtel D, Tardieux I. CD11c- and CD11b-expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood. (2006) 107:309–16. doi: 10.1182/blood-2005-02-0666
186. Baba M, Batanova T, Kitoh K, Takashima Y. Adhesion of Toxoplasma gondii tachyzoite-infected vehicle leukocytes to capillary endothelial cells triggers timely parasite egression. Sci Rep. (2017) 7:5675. doi: 10.1038/s41598-017-05956-z
187. Lachenmaier SM, Deli MA, Meissner M, Liesenfeld O. Intracellular transport of Toxoplasma gondii through the blood-brain barrier. J Neuroimmunol. (2011) 232:119–30. doi: 10.1016/j.jneuroim.2010.10.029
188. Robert-Gangneux F, Dardé M-L. Epidemiology of and diagnostic strategies for toxoplasmosis. Clin Microbiol Rev. (2012) 25:264–96. doi: 10.1128/CMR.05013-11
189. Lüder CG, Giraldo-Velásquez M, Sendtner M, Gross U. Toxoplasma gondii in primary rat CNS cells: differential contribution of neurons, astrocytes, and microglial cells for the intracerebral development and stage differentiation. Exp Parasitol. (1999) 93:23–32. doi: 10.1006/expr.1999.4421
190. Mendez OA, Koshy AA. Toxoplasma gondii: entry, association, and physiological influence on the central nervous system. PLoS Pathog. (2017) 13:e1006351. doi: 10.1371/journal.ppat.1006351
191. Arias I, Sorlozano A, Villegas E, Dios Luna de J, McKenney K, Cervilla J, et al. Infectious agents associated with schizophrenia: a meta-analysis. Schizophr Res. (2012) 136:128–36. doi: 10.1016/j.schres.2011.10.026
192. Frenkel JK. Pathology and pathogenesis of congenital toxoplasmosis. Bull N Y Acad Med. (1974) 50:182–91.
193. Suzuki Y, Claflin J, Wang X, Lengi A, Kikuchi T. Microglia and macrophages as innate producers of interferon-gamma in the brain following infection with Toxoplasma gondii. Int J Parasitol. (2005) 35:83–90. doi: 10.1016/j.ijpara.2004.10.020
194. Biswas A, French T, Düsedau HP, Mueller N, Riek-Burchardt M, Dudeck A, et al. Behavior of neutrophil granulocytes during Toxoplasma gondii infection in the central nervous system. Front Cell Infect Microbiol. (2017) 7:259. doi: 10.3389/fcimb.2017.00259
195. Strack A, Asensio VC, Campbell IL, Schlüter D, Deckert M. Chemokines are differentially expressed by astrocytes, microglia and inflammatory leukocytes in Toxoplasma encephalitis and critically regulated by interferon-gamma. Acta Neuropathol. (2002) 103:458–68. doi: 10.1007/s00401-001-0491-7
196. Sarciron ME, Gherardi A. Cytokines involved in Toxoplasmic encephalitis. Scand J Immunol. (2000) 52:534–43. doi: 10.1046/j.1365-3083.2000.00817.x
197. Hwang YS, Shin J-H, Yang J-P, Jung B-K, Lee SH, Shin E-H. Characteristics of infection immunity regulated by Toxoplasma gondii to maintain chronic infection in the brain. Front Immunol. (2018) 9:158. doi: 10.3389/fimmu.2018.00158
198. Dellacasa-Lindberg I, Fuks JM, Arrighi RB, Lambert H, Wallin RP, Chambers BJ, et al. Migratory activation of primary cortical microglia upon infection with Toxoplasma gondii. Infect Immun. (2011) 79:3046–52. doi: 10.1128/IAI.01042-10
199. Bhandage AK, Kanatani S, Barragan A. Toxoplasma-induced hypermigration of primary cortical microglia implicates GABAergic signaling. Front Cell Infect Microbiol. (2019) 9:73. doi: 10.3389/fcimb.2019.00073
200. Estes ML, McAllister AK. Maternal immune activation: implications for neuropsychiatric disorders. Science. (2016) 353:772–7. doi: 10.1126/science.aag3194
201. Meyer U, Yee BK, Feldon J. The neurodevelopmental impact of prenatal infections at different times of pregnancy: the earlier the worse? Neuroscientist. (2007) 13:241–56. doi: 10.1177/1073858406296401
202. Abu-Raya B, Kollmann TR, Marchant A, MacGillivray DM. The immune system of HIV-exposed uninfected infants. Front Immunol.. (2016) 7:383. doi: 10.3389/fimmu.2016.00383
203. Borrell J. Prenatal immune challenge disrupts sensorimotor gating in adult rats implications for the etiopathogenesis of schizophrenia. Neuropsychopharmacology. (2002) 26:204–15. doi: 10.1016/S0893-133X(01)00360-8
204. Reisinger S, Khan D, Kong E, Berger A, Pollak A, Pollak DD. The poly(I:C)-induced maternal immune activation model in preclinical neuropsychiatric drug discovery. Pharmacol Ther. (2015) 149:213–26. doi: 10.1016/j.pharmthera.2015.01.001
205. Deverman BE, Patterson PH. Cytokines and CNS development. Neuron. (2009) 64:61–78. doi: 10.1016/j.neuron.2009.09.002
206. Zhao B, Schwartz JP. Involvement of cytokines in normal CNS development and neurological diseases: recent progress and perspectives. J Neurosci Res. (1998) 52:7–16.
207. Buka SL, Tsuang MT, Torrey EF, Klebanoff MA, Wagner RL, Yolken RH. Maternal cytokine levels during pregnancy and adult psychosis. Brain Behav Immun. (2001) 15:411–20. doi: 10.1006/brbi.2001.0644
208. Brown AS. Prenatal infection as a risk factor for schizophrenia. Schizophr Bull. (2006) 32:200–2. doi: 10.1093/schbul/sbj052
209. Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci.. (2007) 27:10695–702. doi: 10.1523/JNEUROSCI.2178-07.2007
210. Sacks D, Baxter B, Campbell BC, Carpenter JS, Cognard C, Dippel D, et al. Multisociety consensus quality improvement revised consensus statement for endovascular therapy of acute ischemic stroke. Int J Stroke. (2018) 13:612–32. doi: 10.1177/1747493018778713
211. Fernández de Cossío L, Guzmán A, van der Veldt S, Luheshi GN. Prenatal infection leads to ASD-like behavior and altered synaptic pruning in the mouse offspring. Brain Behav Immun. (2017) 63:88–98. doi: 10.1016/j.bbi.2016.09.028
212. Golan HM, Lev V, Hallak M, Sorokin Y, Huleihel M. Specific neurodevelopmental damage in mice offspring following maternal inflammation during pregnancy. Neuropharmacology. (2005) 48:903–17. doi: 10.1016/j.neuropharm.2004.12.023
213. Meyer U, Feldon J, Schedlowski M, Yee BK. Immunological stress at the maternal-foetal interface: a link between neurodevelopment and adult psychopathology. Brain Behav Immun. (2006) 20:378–88. doi: 10.1016/j.bbi.2005.11.003
214. Elovitz MA, Brown AG, Breen K, Anton L, Maubert M, Burd I. Intrauterine inflammation, insufficient to induce parturition, still evokes fetal and neonatal brain injury. Int J Dev Neurosci. (2011) 29:663–71. doi: 10.1016/j.ijdevneu.2011.02.011
215. Liverman CS, Kaftan HA, Cui L, Hersperger SG, Taboada E, Klein RM, et al. Altered expression of pro-inflammatory and developmental genes in the fetal brain in a mouse model of maternal infection. Neurosci Lett. (2006) 399:220–5. doi: 10.1016/j.neulet.2006.01.064
216. Ellman LM, Deicken RF, Vinogradov S, Kremen WS, Poole JH, Kern DM, et al. Structural brain alterations in schizophrenia following fetal exposure to the inflammatory cytokine interleukin-8. Schizophr Res. (2010) 121:46–54. doi: 10.1016/j.schres.2010.05.014
217. Cai Z, Pan ZL, Pang Y, Evans OB, Rhodes PG. Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatr Res. (2000) 47:64–72. doi: 10.1203/00006450-200001000-00013
218. Giovanoli S, Engler H, Engler A, Richetto J, Voget M, Willi R, et al. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science. (2013) 339:1095–9. doi: 10.1126/science.1228261
219. Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. (2014) 10:217–24. doi: 10.1038/nrneurol.2014.38
220. Schaafsma W, Basterra LB, Jacobs S, Brouwer N, Meerlo P, Schaafsma A, et al. Maternal inflammation induces immune activation of fetal microglia and leads to disrupted microglia immune responses, behavior, and learning performance in adulthood. Neurobiol Dis. (2017) 106:291–300. doi: 10.1016/j.nbd.2017.07.017
221. van den Eynde K, Missault S, Fransen E, Raeymaekers L, Willems R, Drinkenburg W, et al. Hypolocomotive behaviour associated with increased microglia in a prenatal immune activation model with relevance to schizophrenia. Behav Brain Res. (2014) 258:179–86. doi: 10.1016/j.bbr.2013.10.005
222. Hadar R, Le Dong, Del-Valle-Anton L, Guneykaya D, Voget M, Edemann-Callesen H, et al. Deep brain stimulation during early adolescence prevents microglial alterations in a model of maternal immune activation. Brain Behav Immun. (2017) 63:71–80. doi: 10.1016/j.bbi.2016.12.003
223. Juckel G, Manitz MP, Brüne M, Friebe A, Heneka MT, Wolf RJ. Microglial activation in a neuroinflammational animal model of schizophrenia–a pilot study. Schizophr Res. (2011) 131:96–100. doi: 10.1016/j.schres.2011.06.018
224. O'Loughlin E, Pakan JM, Yilmazer-Hanke D, McDermott KW. Acute in utero exposure to lipopolysaccharide induces inflammation in the pre- and postnatal brain and alters the glial cytoarchitecture in the developing amygdala. J Neuroinflammation. (2017) 14:212. doi: 10.1186/s12974-017-0981-8
225. Wu Y, Qi F, Song D, He Z, Zuo Z, Yang Y, et al. Prenatal influenza vaccination rescues impairments of social behavior and lamination in a mouse model of autism. J Neuroinflammation. (2018) 15:228. doi: 10.1186/s12974-018-1252-z
226. Pratt L, Ni L, Ponzio NM, Jonakait GM. Maternal inflammation promotes fetal microglial activation and increased cholinergic expression in the fetal basal forebrain: role of interleukin-6. Pediatr Res. (2013) 74:393–401. doi: 10.1038/pr.2013.126
227. Smolders S, Smolders SM, Swinnen N, Gärtner A, Rigo J-M, Legendre P, et al. Maternal immune activation evoked by polyinosinic:polycytidylic acid does not evoke microglial cell activation in the embryo. Front Cell Neurosci. (2015) 9:301. doi: 10.3389/fncel.2015.00301
228. Laskaris LE, Di Biase MA, Everall I, Chana G, Christopoulos A, Skafidas E, et al. Microglial activation and progressive brain changes in schizophrenia. Br J Pharmacol. (2016) 173:666–80. doi: 10.1111/bph.13364
229. Zhang J, Jing Y, Zhang H, Bilkey DK, Liu P. Maternal immune activation altered microglial immunoreactivity in the brain of postnatal day 2 rat offspring. Synapse. (2018) 73:e22072. doi: 10.1002/syn.22072
230. Basha S, Surendran N, Pichichero M. Immune responses in neonates. Expert Rev Clin Immunol. (2014) 10:1171–84. doi: 10.1586/1744666X.2014.942288
231. Mirmonsef P, Gilbert D, Zariffard MR, Hamaker BR, Kaur A, Landay AL, et al. The effects of commensal bacteria on innate immune responses in the female genital tract. Am J Reprod Immunol. (2011) 65:190–5. doi: 10.1111/j.1600-0897.2010.00943.x
232. Wang H, Liddell CA, Coates MM, Mooney MD, Levitz CE, Schumacher AE, et al. Global, regional, and national levels of neonatal, infant, and under-5 mortality during 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. (2014) 384:957–79. doi: 10.1016/S0140-6736(14)60497-9
233. Wardlaw T, You D, Hug L, Amouzou A, Newby H. UNICEF Report: enormous progress in child survival but greater focus on newborns urgently needed. Reprod Health. (2014) 11:82. doi: 10.1186/1742-4755-11-82
234. Kimberlin DW. Neonatal herpes simplex infection. Clin Microbiol Rev. (2004) 17:1–13. doi: 10.1128/CMR.17.1.1-13.2004
235. Cortese F, Scicchitano P, Gesualdo M, Filaninno A, Giorgi de E, Schettini F, et al. Early and late infections in newborns: Where Do We Stand? A Review. Pediatr Neonatol. (2016) 57:265–73. doi: 10.1016/j.pedneo.2015.09.007
236. Mandal SM, Bharti R, Porto WF, Gauri SS, Mandal M, Franco OL, et al. Identification of multifunctional peptides from human milk. Peptides. (2014) 56:84–93. doi: 10.1016/j.peptides.2014.03.017
237. Chirico G, Marzollo R, Cortinovis S, Fonte C, Gasparoni A. Antiinfective properties of human milk. J Nutr. (2008) 138:1801S−6S. doi: 10.1093/jn/138.9.1801S
238. Turfkruyer M, Verhasselt V. Breast milk and its impact on maturation of the neonatal immune system. Curr Opin Infect Dis. (2015) 28:199–206. doi: 10.1097/QCO.0000000000000165
239. Cacho NT, Lawrence RM. Innate immunity and breast milk. Front Immunol. (2017) 8:584. doi: 10.3389/fimmu.2017.00584
240. Newburg DS, Walker WA. Protection of the neonate by the innate immune system of developing gut and of human milk. Pediatr Res. (2007) 61:2–8. doi: 10.1203/01.pdr.0000250274.68571.18
241. Kollmann TR, Marchant A, Way SS. Vaccination strategies to enhance immunity in neonates. Science. (2020) 368:612–5. doi: 10.1126/science.aaz9447
242. Kollmann TR, Kampmann B, Mazmanian SK, Marchant A, Levy O. Protecting the newborn and young infant from infectious diseases: lessons from immune ontogeny. Immunity. (2017) 46:350–63. doi: 10.1016/j.immuni.2017.03.009
243. Elahi S, Ertelt JM, Kinder JM, Jiang TT, Zhang X, Xin L, et al. Immunosuppressive CD71+ erythroid cells compromise neonatal host defence against infection. Nature. (2013) 504:158–62. doi: 10.1038/nature12675
244. Michaëlsson J, Mold JE, McCune JM, Nixon DF. Regulation of T cell responses in the developing human fetus. J Immunol. (2006) 176:5741–8. doi: 10.4049/jimmunol.176.10.5741
245. Fernandez MA, Puttur FK, Wang YM, Howden W, Alexander SI, Jones CA. T regulatory cells contribute to the attenuated primary CD8+ and CD4+ T cell responses to herpes simplex virus type 2 in neonatal mice. J Immunol. (2008) 180:1556–64. doi: 10.4049/jimmunol.180.3.1556
246. Hunt DW, Huppertz HI, Jiang HJ, Petty RE. Studies of human cord blood dendritic cells: evidence for functional immaturity. Blood. (1994) 84:4333–43. doi: 10.1182/blood.V84.12.4333.bloodjournal84124333
247. Langrish CL, Buddle JC, Thrasher AJ, Goldblatt D. Neonatal dendritic cells are intrinsically biased against Th-1 immune responses. Clin Exp Immunol. (2002) 128:118–23. doi: 10.1046/j.1365-2249.2002.01817.x
248. Lionakis MS, Iliev ID, Hohl TM. Immunity against fungi. JCI Insight. (2017) 2:e93156. doi: 10.1172/jci.insight.93156
249. Cuthbert JA, Lipsky PE. Identification of low density lipoprotein receptor abnormalities by assaying functional receptors on proliferating lymphocytes. Arteriosclerosis. (1989) 9:I43–9.
250. Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, et al. Single-Cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity. (2019) 50:253–71.e6. doi: 10.1016/j.immuni.2018.11.004
251. Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, et al. Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron. (2019) 101:207–23.e10. doi: 10.1016/j.neuron.2018.12.006
252. Masuda T, Sankowski R, Staszewski O, Böttcher C, Amann L, Sagar, et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. (2019) 566:388–92. doi: 10.1038/s41586-019-0924-x
253. Kierdorf K, Masuda T, Jordão MJ, Prinz M. Macrophages at CNS interfaces: ontogeny and function in health and disease. Nat Rev Neurosci. (2019) 20:547–62. doi: 10.1038/s41583-019-0201-x
254. Chiu IM, Yaniv A, Dahlberg JE, Gazit A, Skuntz SF, Tronick SR, et al. Nucleotide sequence evidence for relationship of AIDS retrovirus to lentiviruses. Nature. (1985) 317:366–8. doi: 10.1038/317366a0
255. Deeks SG, Overbaugh J, Phillips A, Buchbinder S. HIV infection. Nat Rev Dis Primers. (2015) 1:15035. doi: 10.1038/nrdp.2015.35
256. Smith JA, Daniel R. Following the path of the virus: the exploitation of host DNA repair mechanisms by retroviruses. ACS Chem Biol. (2006) 1:217–26. doi: 10.1021/cb600131q
257. Nielsen-Saines K. Perinatal HIV as an infectious cause of developmental regression. Neurosci Biobehav Rev. (2019) 102:417–23. doi: 10.1016/j.neubiorev.2019.05.011
258. Forbes JC, Alimenti AM, Singer J, Brophy JC, Bitnun A, Samson LM, et al. A national review of vertical HIV transmission. AIDS. (2012) 26:757–63. doi: 10.1097/QAD.0b013e328350995c
259. Chakraborty R. HIV-1 infection in children: a clinical and immunologic overview. Curr HIV Res. (2005) 3:31–41. doi: 10.2174/1570162052773022
260. DeCarli C, Civitello LA, Brouwers P, Pizzo PA. The prevalence of computed tomographic abnormalities of the cerebrum in 100 consecutive children symptomatic with the human immune deficiency virus. Ann Neurol. (1993) 34:198–205. doi: 10.1002/ana.410340216
261. Vazeux R, Lacroix-Ciaudo C, Blanche S, Cumont MC, Henin D, Gray F, et al. Low levels of human immunodeficiency virus replication in the brain tissue of children with severe acquired immunodeficiency syndrome encephalopathy. Am J Pathol. (1992) 140:137–44.
262. Franklin S, Lim HJ, Rennie KM, Eastwood D, Cuene B, Havens PL. Longitudinal Intellectual Assessment of Children With HIV Infection. J Clin Psychol Med Settings. (2005) 12:367–76. doi: 10.1007/s10880-005-7823-z
263. Paramesparan Y, Garvey LJ, Ashby J, Foster CJ, Fidler S, Winston A. High rates of asymptomatic neurocognitive impairment in vertically acquired HIV-1-infected adolescents surviving to adulthood. J Acquir Immune Defic Syndr. (2010) 55:134–6. doi: 10.1097/QAI.0b013e3181d90e8c
264. van Rie A, Harrington PR, Dow A, Robertson K. Neurologic and neurodevelopmental manifestations of pediatric HIV/AIDS: a global perspective. Eur J Paediatr Neurol. (2007) 11:1–9. doi: 10.1016/j.ejpn.2006.10.006
265. Veazey RS, DeMaria M, Chalifoux LV, Shvetz DE, Pauley DR, Knight HL, et al. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science. (1998) 280:427–31. doi: 10.1126/science.280.5362.427
266. Brenchley JM, Price DA, Douek DC. HIV disease: fallout from a mucosal catastrophe? Nat Immunol. (2006) 7:235–9. doi: 10.1038/ni1316
267. Time from HIV-1 seroconversion to AIDS and death before widespread use of highly-active antiretroviral therapy: a collaborative re-analysis. Collaborative Group on AIDS Incubation and HIV Survival including the CASCADE EU Concerted Action. Concerted Action on SeroConversion to AIDS and Death in Europe. Lancet. (2000) 355:1131–7. doi: 10.1016/S0140-6736(00)02061-4
268. He J, Chen Y, Farzan M, Choe H, Ohagen A, Gartner S, et al. CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature. (1997) 385:645–9. doi: 10.1038/385645a0
269. Arrildt KT, LaBranche CC, Joseph SB, Dukhovlinova EN, Graham WD, Ping L-H, et al. Phenotypic correlates of HIV-1 macrophage tropism. J Virol. (2015) 89:11294–311. doi: 10.1128/JVI.00946-15
270. Mefford ME, Kunstman K, Wolinsky SM, Gabuzda D. Bioinformatic analysis of neurotropic HIV envelope sequences identifies polymorphisms in the gp120 bridging sheet that increase macrophage-tropism through enhanced interactions with CCR5. Virology. (2015) 481:210–22. doi: 10.1016/j.virol.2015.01.032
271. Salimi H, Roche M, Webb N, Gray LR, Chikere K, Sterjovski J, et al. Macrophage-tropic HIV-1 variants from brain demonstrate alterations in the way gp120 engages both CD4 and CCR5. J Leukoc Biol. (2013) 93:113–26. doi: 10.1189/jlb.0612308
272. Avalos CR, Abreu CM, Queen SE, Li M, Price S, Shirk EN, et al. Brain macrophages in simian immunodeficiency virus-infected, antiretroviral-suppressed macaques: a functional latent reservoir. mBio. (2017) 8:e01186-17. doi: 10.1128/mBio.01186-17
273. Mankowski JL, Clements JE, Zink MC. Searching for clues: tracking the pathogenesis of human immunodeficiency virus central nervous system disease by use of an accelerated, consistent simian immunodeficiency virus macaque model. J Infect Dis. (2002) 186 Suppl 2:S199–208. doi: 10.1086/344938
274. Valcour V, Chalermchai T, Sailasuta N, Marovich M, Lerdlum S, Suttichom D, et al. Central nervous system viral invasion and inflammation during acute HIV infection. J Infect Dis. (2012) 206:275–82. doi: 10.1093/infdis/jis326
275. Williams KC, Corey S, Westmoreland SV, Pauley D, Knight H, deBakker C, et al. Perivascular macrophages are the primary cell type productively infected by simian immunodeficiency virus in the brains of macaques: implications for the neuropathogenesis of AIDS. J Exp Med. (2001) 193:905–15. doi: 10.1084/jem.193.8.905
276. Lee SC, Hatch WC, Liu W, Kress Y, Lyman WD, Dickson DW. Productive infection of human fetal microglia by HIV-1. Am J Pathol. (1993) 143:1032–9.
277. Ellis RJ, Moore DJ, Childers ME, Letendre S, McCutchan JA, Wolfson T, et al. Progression to neuropsychological impairment in human immunodeficiency virus infection predicted by elevated cerebrospinal fluid levels of human immunodeficiency virus RNA. Arch Neurol. (2002) 59:923–8. doi: 10.1001/archneur.59.6.923
278. Shiramizu B, Gartner S, Williams A, Shikuma C, Ratto-Kim S, Watters M, et al. Circulating proviral HIV DNA and HIV-associated dementia. AIDS. (2005) 19:45–52. doi: 10.1097/00002030-200501030-00005
279. Liu Y, Tang XP, McArthur JC, Scott J, Gartner S. Analysis of human immunodeficiency virus type 1 gp160 sequences from a patient with HIV dementia: evidence for monocyte trafficking into brain. J Neurovirol. (2000) 6 (Suppl. 1):S70–81.
280. Burdo TH, Soulas C, Orzechowski K, Button J, Krishnan A, Sugimoto C, et al. Increased monocyte turnover from bone marrow correlates with severity of SIV encephalitis and CD163 levels in plasma. PLoS Pathog. (2010) 6:e1000842. doi: 10.1371/journal.ppat.1000842
281. Nath A, Psooy K, Martin C, Knudsen B, Magnuson DS, Haughey N, et al. Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. J Virol. (1996) 70:1475–80. doi: 10.1128/JVI.70.3.1475-1480.1996
282. Bardi G, Sengupta R, Khan MZ, Patel JP, Meucci O. Human immunodeficiency virus gp120-induced apoptosis of human neuroblastoma cells in the absence of CXCR4 internalization. J Neurovirol. (2006) 12:211–8. doi: 10.1080/13550280600848373
283. Rao VR, Ruiz AP, Prasad VR. Viral and cellular factors underlying neuropathogenesis in HIV associated neurocognitive disorders (HAND). AIDS Res Ther. (2014) 11:13. doi: 10.1186/1742-6405-11-13
284. Periyasamy P, Thangaraj A, Guo M-L, Hu G, Callen S, Buch S. Epigenetic promoter DNA methylation of miR-124 promotes HIV-1 Tat-mediated microglial activation via MECP2-STAT3 Axis. J Neurosci. (2018) 38:5367–83. doi: 10.1523/JNEUROSCI.3474-17.2018
285. Periyasamy P, Thangaraj A, Bendi VS, Buch S. HIV-1 Tat-mediated microglial inflammation involves a novel miRNA-34a-NLRC5-NFκB signaling axis. Brain Behav Immun. (2019) 80:227–37. doi: 10.1016/j.bbi.2019.03.011
286. Chivero ET, Guo M-L, Periyasamy P, Liao K, Callen SE, Buch S. HIV-1 Tat primes and activates microglial NLRP3 inflammasome-mediated neuroinflammation. J Neurosci. (2017) 37:3599–609. doi: 10.1523/JNEUROSCI.3045-16.2017
287. Walsh JG, Reinke SN, Mamik MK, McKenzie BA, Maingat F, Branton WG, et al. Rapid inflammasome activation in microglia contributes to brain disease in HIV/AIDS. Retrovirology. (2014) 11:35. doi: 10.1186/1742-4690-11-35
288. Chen NC, Partridge AT, Sell C, Torres C, Martín-García J. Fate of microglia during HIV-1 infection: from activation to senescence? Glia. (2017) 65:431–46. doi: 10.1002/glia.23081
289. Tavazzi E, Morrison D, Sullivan P, Morgello S, Fischer T. Brain inflammation is a common feature of HIV-infected patients without HIV encephalitis or productive brain infection. Curr HIV Res. (2014) 12:97–110. doi: 10.2174/1570162X12666140526114956
290. Fischer-Smith T, Croul S, Sverstiuk AE, Capini C, L'Heureux D, Régulier EG, et al. CNS invasion by CD14+/CD16+ peripheral blood-derived monocytes in HIV dementia: perivascular accumulation and reservoir of HIV infection. J Neurovirol. (2001) 7:528–41. doi: 10.1080/135502801753248114
291. Sippy BD, Hofman FM, Wallach D, Hinton DR. Increased expression of tumor necrosis factor-alpha receptors in the brains of patients with AIDS. J Acquir Immune Defic Syndr Hum Retrovirol. (1995) 10:511–21. doi: 10.1097/00042560-199510050-00004
292. Zhao M-L, Kim M-O, Morgello S, Lee SC. Expression of inducible nitric oxide synthase, interleukin-1 and caspase-1 in HIV-1 encephalitis. J Neuroimmunol. (2001) 115:182–91. doi: 10.1016/S0165-5728(00)00463-X
293. Lee SC, Dickson DW, Brosnan CF. Interleukin-1, nitric oxide and reactive astrocytes. Brain Behav Immun. (1995) 9:345–54. doi: 10.1006/brbi.1995.1032
294. Cenker JJ, Stultz RD, McDonald D. Brain Microglial Cells Are Highly Susceptible to HIV-1 Infection and Spread. AIDS Res Hum Retroviruses. (2017) 33:1155–65. doi: 10.1089/aid.2017.0004
295. Tyor WR, Glass JD, Griffin JW, Becker PS, McArthur JC, Bezman L, et al. Cytokine expression in the brain during the acquired immunodeficiency syndrome. Ann Neurol. (1992) 31:349–60. doi: 10.1002/ana.410310402
296. Garvey LJ, Pavese N, Politis M, Ramlackhansingh A, Brooks DJ, Taylor-Robinson SD, et al. Increased microglia activation in neurologically asymptomatic HIV-infected patients receiving effective ART. AIDS. (2014) 28:67–72. doi: 10.1097/01.aids.0000432467.54003.f7
297. Andersson LM, Fredman P, Lekman A, Rosengren L, Gisslén M. Increased cerebrospinal fluid ganglioside GD3 concentrations as a marker of microglial activation in HIV type 1 infection. AIDS Res Hum Retroviruses. (1998) 14:1065–9. doi: 10.1089/aid.1998.14.1065
298. Gilden DH, Mahalingam R, Cohrs RJ, Tyler KL. Herpesvirus infections of the nervous system. Nat Clin Pract Neurol. (2007) 3:82–94. doi: 10.1038/ncpneuro0401
299. Kleinschmidt-DeMasters BK, Keohane C, Gray F. Herpes simplex virus infections of the CNS. In: Chrétien F, Wong KT, Sharer LR, Keohane C, Gray F, editors. Infections of the Central Nervous System: Pathology and Genetics. Hoboken, NJ: Wiley-Blackwell (2020). p. 43–54. doi: 10.1002/9781119467748.ch5
300. Kennedy PG, Chaudhuri A. Herpes simplex encephalitis. J Neurol Neurosurg Psychiatry. (2002) 73:237–8. doi: 10.1136/jnnp.73.3.237
301. Looker KJ, Magaret AS, May MT, Turner KM, Vickerman P, Newman LM, et al. First estimates of the global and regional incidence of neonatal herpes infection. Lancet Global Health. (2017) 5:e300–9. doi: 10.1016/S2214-109X(16)30362-X
302. Kimberlin DW, Lin CY, Jacobs RF, Powell DA, Corey L, Gruber WC, et al. Safety and efficacy of high-dose intravenous acyclovir in the management of neonatal herpes simplex virus infections. Pediatrics. (2001) 108:230–8. doi: 10.1542/peds.108.2.230
303. Lokensgard JR, Hu S, Sheng W, vanOijen M, Cox D, Cheeran MC, et al. Robust expression of TNF-alpha, IL-1beta, RANTES, and IP-10 by human microglial cells during nonproductive infection with herpes simplex virus. J Neurovirol. (2001) 7:208–19. doi: 10.1080/13550280152403254
304. Conrady CD, Zheng M, van Rooijen N, Drevets DA, Royer D, Alleman A, et al. Microglia and a functional type I IFN pathway are required to counter HSV-1-driven brain lateral ventricle enlargement and encephalitis. J Immunol. (2013) 190:2807–17. doi: 10.4049/jimmunol.1203265
305. Reinert LS, Lopušná K, Winther H, Sun C, Thomsen MK, Nandakumar R, et al. Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat Commun. (2016) 7:13348. doi: 10.1038/ncomms13348
306. Persson M, Brantefjord M, Liljeqvist J-A, Bergström T, Hansson E, Rönnbäck L. Microglial GLT-1 is upregulated in response to herpes simplex virus infection to provide an antiviral defence via glutathione. Glia. (2007) 55:1449–58. doi: 10.1002/glia.20560
307. Chucair-Elliott AJ, Conrady C, Zheng M, Kroll CM, Lane TE, Carr DJ. Microglia-induced IL-6 protects against neuronal loss following HSV-1 infection of neural progenitor cells. Glia. (2014) 62:1418–34. doi: 10.1002/glia.22689
308. Fekete R, Cserép C, Lénárt N, Tóth K, Orsolits B, Martinecz B, et al. Microglia control the spread of neurotropic virus infection via P2Y12 signalling and recruit monocytes through P2Y12-independent mechanisms. Acta Neuropathol. (2018) 136:461–82. doi: 10.1007/s00401-018-1885-0
309. Aravalli RN, Hu S, Rowen TN, Palmquist JM, Lokensgard JR. Cutting edge: TLR2-mediated proinflammatory cytokine and chemokine production by microglial cells in response to herpes simplex virus. J Immunol. (2005) 175:4189–93. doi: 10.4049/jimmunol.175.7.4189
310. Marques CP, Hu S, Sheng W, Lokensgard JR. Microglial cells initiate vigorous yet non-protective immune responses during HSV-1 brain infection. Virus Res. (2006) 121:1–10. doi: 10.1016/j.virusres.2006.03.009
311. Zhang S-Y, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. (2007) 317:1522–7. doi: 10.1126/science.1139522
312. Sancho-Shimizu V, Pérez de Diego R, Lorenzo L, Halwani R, Alangari A, Israelsson E, et al. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J Clin Invest. (2011) 121:4889–902. doi: 10.1172/JCI59259
313. Guo Y, Audry M, Ciancanelli M, Alsina L, Azevedo J, Herman M, et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J Exp Med. (2011) 208:2083–98. doi: 10.1084/jem.20101568
314. Pérez de Diego R, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity. (2010) 33:400–11. doi: 10.1016/j.immuni.2010.08.014
315. Casrouge A, Zhang S-Y, Eidenschenk C, Jouanguy E, Puel A, Yang K, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. (2006) 314:308–12. doi: 10.1126/science.1128346
316. Mielcarska MB, Bossowska-Nowicka M, Toka FN. Functional failure of TLR3 and its signaling components contribute to herpes simplex encephalitis. J Neuroimmunol. (2018) 316:65–73. doi: 10.1016/j.jneuroim.2017.12.011
317. Lim HK, Seppänen M, Hautala T, Ciancanelli MJ, Itan Y, Lafaille FG, et al. TLR3 deficiency in herpes simplex encephalitis: high allelic heterogeneity and recurrence risk. Neurology. (2014) 83:1888–97. doi: 10.1212/WNL.0000000000000999
318. Manzoni P, Stronati M, Jacqz-Aigrain E, Maragliano R, Ruffinazzi G, Rizzollo S, et al. Correct choices for correct treatments: key issues in the management of Candida infections in preterm neonates. Early Hum Dev. (2012) 88 Suppl 2:S98–100. doi: 10.1016/S0378-3782(12)70026-9
319. Butler KM, Baker CJ. Candida: an increasingly important pathogen in the nursery. Pediatr Clin North Am. (1988) 35:543–63. doi: 10.1016/S0031-3955(16)36471-9
320. Kullberg BJ, Arendrup MC. Invasive Candidiasis. N Engl J Med. (2015) 373:1445–56. doi: 10.1056/NEJMra1315399
321. Fishman JA. Opportunistic infections–coming to the limits of immunosuppression? Cold Spring Harb Perspect Med. (2013) 3:a015669. doi: 10.1101/cshperspect.a015669
322. Wagstaff JS, Durrant RJ, Newman MG, Eason R, Ward RM, Sherwin CM, et al. Antibiotic treatment of suspected and confirmed neonatal sepsis within 28 days of birth: a retrospective analysis. Front Pharmacol. (2019) 10:1191. doi: 10.3389/fphar.2019.01191
323. Bonanno C, Wapner RJ. Antenatal corticosteroids in the management of preterm birth: are we back where we started? Obstet Gynecol Clin North Am. (2012) 39:47–63. doi: 10.1016/j.ogc.2011.12.006
324. Fernandez M, Moylett EH, Noyola DE, Baker CJ. Candidal meningitis in neonates: a 10-year review. Clin Infect Dis. (2000) 31:458–63. doi: 10.1086/313973
325. Barton M, O'Brien K, Robinson JL, Davies DH, Simpson K, Asztalos E, et al. Invasive candidiasis in low birth weight preterm infants: risk factors, clinical course and outcome in a prospective multicenter study of cases and their matched controls. BMC Infect Dis. (2014) 14:327. doi: 10.1186/1471-2334-14-327
326. Pahud BA, Greenhow TL, Piecuch B, Weintrub PS. Preterm neonates with candidal brain microabscesses: a case series. J Perinatol. (2009) 29:323–6. doi: 10.1038/jp.2008.201
327. Park JY, Lee T, Lee H, Lim H-J, Lee J, Park JS, et al. Multivariate analysis of prognostic factors in patients with pulmonary actinomycosis. BMC Infect Dis. (2014) 14:10. doi: 10.1186/1471-2334-14-10
328. Koutsouras GW, Ramos RL, Martinez LR. Role of microglia in fungal infections of the central nervous system. Virulence. (2017) 8:705–18. doi: 10.1080/21505594.2016.1261789
329. Santiago-Tirado FH, Onken MD, Cooper JA, Klein RS, Doering TL. Trojan horse transit contributes to blood-brain barrier crossing of a eukaryotic pathogen. mBio. (2017) 8:e02183-16. doi: 10.1128/mBio.02183-16
330. Drummond RA, Swamydas M, Oikonomou V, Zhai B, Dambuza IM, Schaefer BC, et al. CARD9+ microglia promote antifungal immunity via IL-1β- and CXCL1-mediated neutrophil recruitment. Nat Immunol. (2019) 20:559–70. doi: 10.1038/s41590-019-0377-2
331. Raymond SL, Mathias BJ, Murphy TJ, Rincon JC, López MC, Ungaro R, et al. Neutrophil chemotaxis and transcriptomics in term and preterm neonates. Transl Res. (2017) 190:4–15. doi: 10.1016/j.trsl.2017.08.003
332. Drummond RA, Collar AL, Swamydas M, Rodriguez CA, Lim JK, Mendez LM, et al. CARD9-dependent neutrophil recruitment protects against fungal invasion of the central nervous system. PLoS Pathog. (2015) 11:e1005293. doi: 10.1371/journal.ppat.1005293
333. Drummond RA, Lionakis MS. Mechanistic insights into the role of C-Type Lectin Receptor/CARD9 signaling in human antifungal immunity. Front Cell Infect Microbiol. (2016) 6:39. doi: 10.3389/fcimb.2016.00039
334. Maisey HC, Doran KS, Nizet V. Recent advances in understanding the molecular basis of group B Streptococcus virulence. Expert Rev Mol Med. (2008) 10:e27. doi: 10.1017/S1462399408000914
335. Heath PT, Okike IO, Oeser C. Neonatal meningitis: can we do better? Adv Exp Med Biol. (2011) 719:11–24. doi: 10.1007/978-1-4614-0204-6_2
336. Stoll BJ, Hansen NI, Sánchez PJ, Faix RG, Poindexter BB, van Meurs KP, et al. Early onset neonatal sepsis: the burden of group B Streptococcal and E. coli disease continues. Pediatrics. (2011) 127:817–26. doi: 10.1542/peds.2010-2217
337. Gres V, Kolter J, Erny D, Henneke P. The role of CNS macrophages in streptococcal meningoencephalitis. J Leukoc Biol. (2019) 106:209–18. doi: 10.1002/JLB.4MR1118-419R
338. Hillier SL, Krohn MA, Kiviat NB, Watts DH, Eschenbach DA. Microbiologic causes and neonatal outcomes associated with chorioamnion infection. Am J Obstet Gynecol. (1991) 165:955–61. doi: 10.1016/0002-9378(91)90447-Y
339. Verani JR, McGee L, Schrag SJ. Prevention of perinatal group B streptococcal disease–revised guidelines from CDC, 2010. MMWR Recomm Rep. (2010) 59:1–36.
340. Gier de B, van Kassel MN, Sanders EA, van de Beek D, Hahné SJ, van der Ende A, et al. Disease burden of neonatal invasive Group B Streptococcus infection in the Netherlands. PLoS ONE. (2019) 14:e0216749. doi: 10.1371/journal.pone.0216749
341. Shane AL, Sánchez PJ, Stoll BJ. Neonatal sepsis. Lancet. (2017) 390:1770–80. doi: 10.1016/S0140-6736(17)31002-4
342. Tazi A, Disson O, Bellais S, Bouaboud A, Dmytruk N, Dramsi S, et al. The surface protein HvgA mediates group B streptococcus hypervirulence and meningeal tropism in neonates. J Exp Med. (2010) 207:2313–22. doi: 10.1084/jem.20092594
343. Doran KS, Liu GY, Nizet V. Group B streptococcal beta-hemolysin/cytolysin activates neutrophil signaling pathways in brain endothelium and contributes to development of meningitis. J Clin Invest. (2003) 112:736–44. doi: 10.1172/JCI200317335
344. Polfliet MM, Zwijnenburg PJ, van Furth AM, van der Poll T, Döpp EA, Renardel de Lavalette C, et al. Meningeal and perivascular macrophages of the central nervous system play a protective role during bacterial meningitis. J Immunol. (2001) 167:4644–50. doi: 10.4049/jimmunol.167.8.4644
345. Inaba Y, Ishiguro A, Shimbo T. The production of macrophage inflammatory protein-1alpha in the cerebrospinal fluid at the initial stage of meningitis in children. Pediatr Res. (1997) 42:788–93. doi: 10.1203/00006450-199712000-00012
346. Lehnardt S, Henneke P, Lien E, Kasper DL, Volpe JJ, Bechmann I, et al. A mechanism for neurodegeneration induced by group B streptococci through activation of the TLR2/MyD88 pathway in microglia. J Immunol. (2006) 177:583–92. doi: 10.4049/jimmunol.177.1.583
347. Miyairi I, Causey KT, DeVincenzo JP, Buckingham SC. Group B streptococcal ventriculitis: a report of three cases and literature review. Pediatr Neurol. (2006) 34:395–9. doi: 10.1016/j.pediatrneurol.2005.09.003
348. Andrade EB, Magalhães A, Puga A, Costa M, Bravo J, Portugal CC, et al. A mouse model reproducing the pathophysiology of neonatal group B streptococcal infection. Nat Commun. (2018) 9:3138. doi: 10.1038/s41467-018-05492-y
Keywords: microglia, CNS-associated macrophages, prenatal infections, postnatal infections, TORCH, maternal immune activation
Citation: Oschwald A, Petry P, Kierdorf K and Erny D (2020) CNS Macrophages and Infant Infections. Front. Immunol. 11:2123. doi: 10.3389/fimmu.2020.02123
Received: 05 June 2020; Accepted: 05 August 2020;
Published: 18 September 2020.
Edited by:
David J. Dowling, Boston Children's Hospital, United StatesReviewed by:
Christian Schulz, Ludwig Maximilian University of Munich, GermanyYoshiro Ohara, Kanazawa Medical University, Japan
Copyright © 2020 Oschwald, Petry, Kierdorf and Erny. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel Erny, ZGFuaWVsLmVybnlAdW5pa2xpbmlrLWZyZWlidXJnLmRl
†These authors have contributed equally to this work
‡These authors have contributed equally to this work and share last authorship