 Sam Afkhami1,2,3
Sam Afkhami1,2,3 Anne Drumond Villela1,2,3
Anne Drumond Villela1,2,3 Michael R. D’Agostino1,2,3
Michael R. D’Agostino1,2,3 Mangalakumari Jeyanathan1,2,3
Mangalakumari Jeyanathan1,2,3 Amy Gillgrass1,2,3
Amy Gillgrass1,2,3 Zhou Xing1,2,3*
Zhou Xing1,2,3*- 1McMaster Immunology Research Center, McMaster University, Hamilton, ON, Canada
- 2Department of Pathology and Molecular Medicine, McMaster University, Hamilton, ON, Canada
- 3Michael G. DeGroote Institute for Infectious Disease Research, McMaster University, Hamilton, ON, Canada
Chemotherapeutic intervention remains the primary strategy in treating and controlling tuberculosis (TB). However, a complex interplay between therapeutic and patient-related factors leads to poor treatment adherence. This in turn continues to give rise to unacceptably high rates of disease relapse and the growing emergence of drug-resistant forms of TB. As such, there is considerable interest in strategies that simultaneously improve treatment outcome and shorten chemotherapy duration. Therapeutic vaccines represent one such approach which aims to accomplish this through boosting and/or priming novel anti-TB immune responses to accelerate disease resolution, shorten treatment duration, and enhance treatment success rates. Numerous therapeutic vaccine candidates are currently undergoing pre-clinical and clinical assessment, showing varying degrees of efficacy. By dissecting the underlying mechanisms/correlates of their successes and/or shortcomings, strategies can be identified to improve existing and future vaccine candidates. This mini-review will discuss the current understanding of therapeutic TB vaccine candidates, and discuss major strategies that can be implemented in advancing their development.
Introduction
In 2014, the World Health Organization (WHO) supported a post-2015 END TB Resolution that aimed to curb 90% of both cases and deaths associated with tuberculosis (TB) by 2035 (1). Although measurable progress toward these goals has been made to-date, recent reports show that many interim goals originally set for 2020 will not be met globally (2). A major proportion of these shortcomings stem from current anti-TB chemotherapy regimens including fragmented patient adherence, treatment failure, and emergence of multi- and extensively drug resistant disease (3). Novel strategies to be administered in adjunct with conventional chemotherapy, known as Host-Directed Therapies (HDT), are designed to combat such shortcomings by improving patient adherence, improving cure rates, and preventing disease relapse (4, 5). In this mini-review, we focus on therapeutic vaccination strategies as HDTs for TB. We discuss current requirements for therapeutic vaccines in accordance with WHO standards, their mechanisms of action, detail the current pipeline, and expand on strategies to improve therapeutic vaccines.
Current TB Chemotherapy Landscape and Its Shortcomings
Chemotherapy for drug-susceptible TB requires two months of continuous treatment with a cocktail of rifampicin, isoniazid, ethambutol, and pyrazinamide followed by four months of rifampicin and isoniazid. Although this regimen has remained largely unchanged for the last 35 years, numerous healthcare policies such as Direct Observed Treatment Short course have allowed for cure rates as high as 85% (2, 6). This, however, starkly contrasts the low success rate (56%) for drug-resistant disease, for which treatment can last upwards of 24 months (2). Although there are numerous factors that contribute to such low rates, many arise from, and are further exacerbated by poor patient adherence due to the duration of treatment required to successfully treat disease.
It is important to note that novel drugs and drug regimens progressing through the clinical pipeline continue to substantially improve TB treatment outcomes. Novel chemotherapeutics such as bedaquiline and pretomanid have allowed for regimens as short as nine months to be effective in curing drug-resistant forms of TB (7). However, despite such continued improvements, patient adherence remains poor. Chemotherapy durations remain longwinded and costly, and novel antibiotics pose the threat of off-target toxicities such as peripheral neuropathy and cardiac manifestations (8). Non-compliance additionally drives drug resistance which has alarmingly already been documented for bedaquiline (9). These, alongside other socioeconomic issues, are entangled with the longevity of treatment and therefore remain a major roadblock toward the success of chemotherapy against TB.
The development of novel immunotherapeutic strategies that synergize with antibiotics to further shorten the duration of chemotherapy required to successfully treat disease is an alluring platform for improving patient compliance and treatment success.
Rationale Behind Prolonged Multi-Drug Regimens
Logically, multiple drugs are required to treat TB as to avoid the selection of resistant mycobacterial subpopulations that arise from single-drug monotherapy (defined as genetic tolerance) (10). Therefore, individuals with a higher bacillary burden are statistically more likely to carry intrinsically resistant mutants. In stark contrast, the rationale behind the length of therapy is perhaps less intuitive and requires scrutinization of the host-pathogen interface.
Airway macrophages comprise the major niche for Mycobacterium tuberculosis (M.tb) infection and outgrowth (11, 12). Infected macrophages deploy numerous mechanisms to eliminate M.tb which include phagolysosome fusion, upregulation of antimicrobial peptides, and autophagy (13–15). Eventual priming and recruitment of adaptive Th1 immunity further enhances mycobacterial control in part by promoting innate-mediated killing (16). Unfortunately, innate and adaptive immune responses together are often unable to fully eliminate the pathogen. This necessitates addition of chemotherapeutic interventions to further enhance mycobacterial clearance through inhibition of cell wall synthesis (isoniazid and pyrazinamide), and transcription (rifampicin) (10).
However, M.tb is fully capable of adapting to both the immunological and pharmacological pressures placed upon it. Examples of such bacterial countermeasures include inhibiting phagosome acidification, delaying adaptive responses, and upregulation of drug efflux pumps (15–18). In addition, immunological and chemotherapeutic stresses also cause M.tb to undergo transcriptomic and metabolic shifts leading to a state of dormancy. In this non-replicative state, the antigenic profile of M.tb shifts toward expression of stress/dormancy associated genes (19–25). Collectively, this not only further circumvents adaptive immune responses established against the replication antigens, but it also thwarts the efficacy of such therapies (defined as phenotypic tolerance) since the majority of TB chemotherapeutics only target actively replicating bacilli. These phenotypically tolerant, non-replicating mycobacteria are a major and under-investigated subpopulation responsible for disease relapse and the need for prolonged antibiotic therapy (26). As such, there has been a growing interest in strategies which can address such problems. Host-directed therapies represent a promising therapeutic category designed to accomplish this goal.
Improving Treatment Outcome With Host-Directed Therapies
Host-directed therapies are administered in tandem with conventional chemotherapy to improve treatment success and reduce disease relapse. Depending on the strategy, HDTs accomplish this by (1) augmenting the host anti-TB immune response, (2) limiting lung pathology, and/or (3) enhancing mycobacterial sensitivity to chemotherapy. Numerous drug-based HDTs are currently under pre-clinical and clinical investigation. For example, the antihyperglycemic agent metformin has shown promise as it enhances macrophage intracellular mycobacterial killing by increasing phagolysosome fusion and production of reactive oxygen species (27, 28). Drug-based HDTs have been reviewed extensively elsewhere and will not be the focus of this review (4, 5, 29).
Vaccines as Host-Directed Therapies Against TB
Tuberculosis vaccination strategies encompass those administered prophylactically (to prevent infection), therapeutically (to improve treatment of active disease), and post-exposure (to prevent re-infection/re-activation) (30). Therapeutic vaccines continue to gain traction as leading candidates for TB HDTs.
Therapeutic vaccines are administered in adjunct (at the start or during) with conventional chemotherapy to accelerate treatment, shorten chemotherapy duration, and improve treatment completion rates. The WHO has generated a detailed list of parameters necessary for an ideal therapeutic vaccine candidate, of which the major targets/goals are presented in Figure 1 (31). Therapeutic TB vaccines aspire to accomplish these goals by boosting host anti-TB immunity, priming novel immune responses, and modulating the host inflammatory response. The following section briefly outlines the most recent advancements in emerging therapeutic TB vaccines, as identified by the TuBerculosis Vaccine Initiative (TBVI)1 and the 2018 WHO Global TB Report (Figure 2).
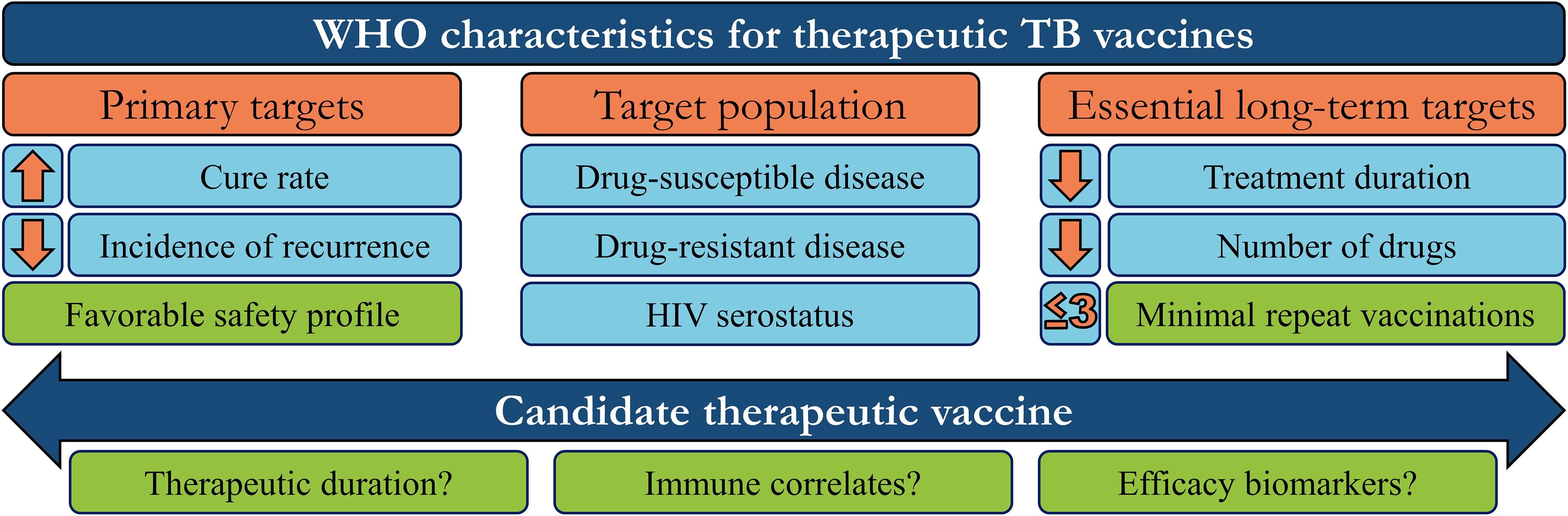
Figure 1. WHO target characteristics for therapeutic tuberculosis vaccines. In order to guide research and development, the WHO has set aspirational targets/characteristics for candidate therapeutic TB vaccines. These include three major areas to address (orange boxes): Vaccine targets that determine efficacy, the target population, and the subsequent long-term impact on the chemotherapy regimen. Candidates which meet some/all of these targets must also be safe, efficacious in a minimum number of repeated doses, and investigated to define their mechanism-of-action or potential efficacy biomarkers, to further refine/guide future vaccination strategies (green boxes).
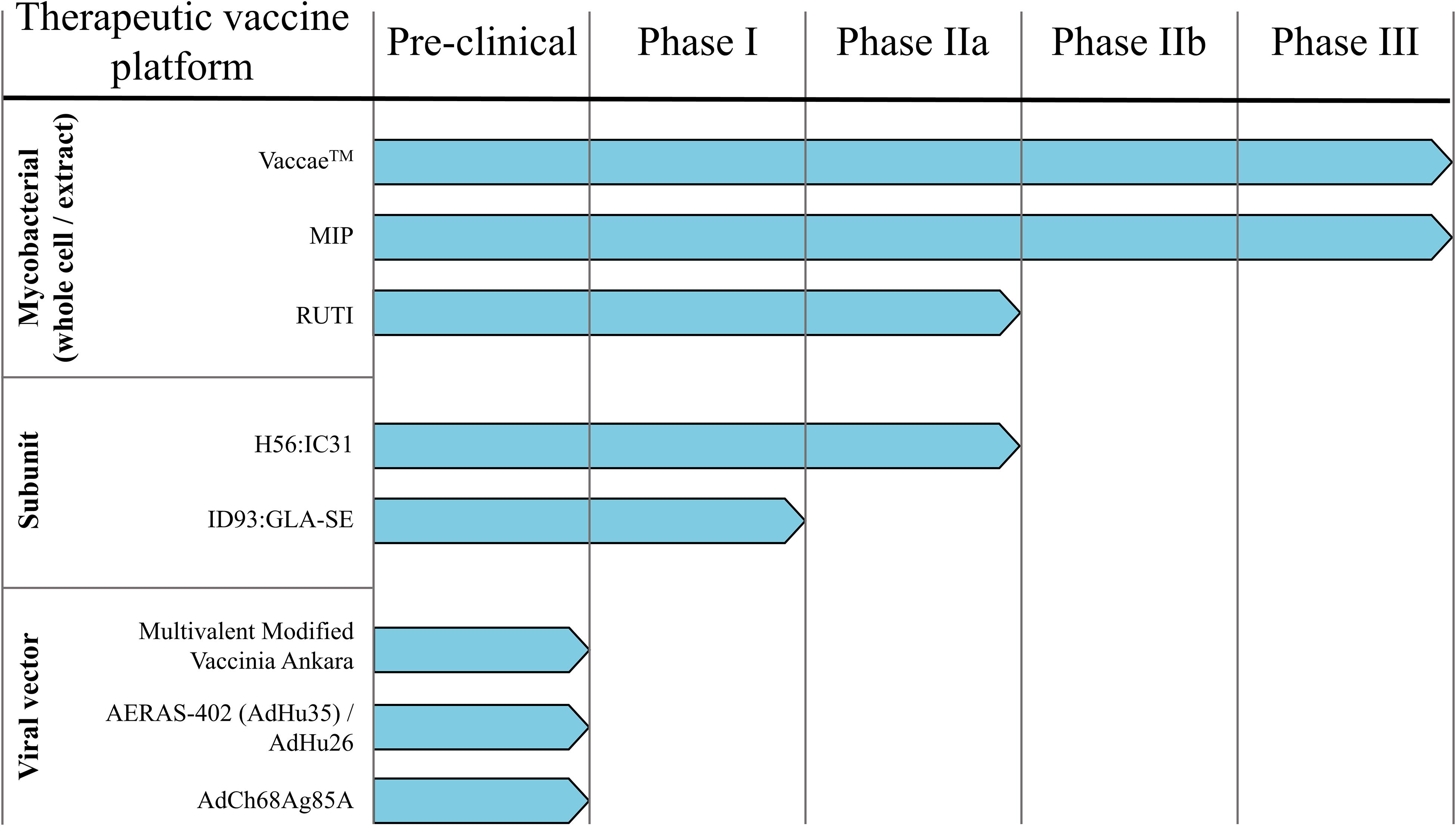
Figure 2. Therapeutic TB vaccine pipeline. Numerous TB vaccine candidates under pre-clinical and/or clinical testing for their prophylactic efficacy are also evaluated as immunotherapies.
Mycobacterial-Based Therapeutic TB Vaccines
VaccaeTM
VaccaeTM consists of a heat-killed Mycobacterium vaccae (M vaccae) variant (32). This non-tuberculous mycobacterium is currently the most advanced therapeutic TB vaccine candidate and is approved for administration alongside standard chemotherapy in patients with active TB in China (33, 34). Although clinical data pertaining to VaccaeTM efficacy is conflicting (with certain meta-analyses indicating little-to-no improvement), recent studies have highlighted its therapeutic efficacy (35, 36). In particular, such studies have highlighted the ability of orally administered VaccaeTM to accelerate negative sputum smear conversion. A large-scale phase III trial with 10,000 recruited participants is ongoing with interim results showing high retention rates (clinicaltrials.gov, NCT01979900). However immunological data pertaining to VaccaeTM mechanism of action is limited. Pre-clinical and clinical immunological studies have suggested that induction of robust Th1 (and conversely skewing away from Th2) immune responses, alongside induction of cytotoxic CD8 T lymphocytes are correlatives of efficacy (37).
Mycobacterium indicus pranii
Mycobacterium indicus pranii (MIP) is a non-pathogenic mycobacterium whose heat-killed variant has been successfully used as an immunotherapeutic for leprosy (38, 39). Characterization of the antigenic profile of MIP has shown broad antigenic congruency with M.tb, warranting further investigation toward its potential as a therapeutic vaccine. A recently published phase II trial where MIP was intradermally administered in adjunct to chemotherapy showed success in enhancing bacterial clearance, and improved lung pathology in patients with bi-lateral, drug-resistant disease (40). Pre-clinical studies in numerous animal models (including hyper-susceptible guinea pigs) have correlated the therapeutic efficacy of MIP to its ability to drive robust Th1-skewed immunity (41–43). Interestingly, these studies also compared the contribution of immunization route to efficacy, showing that respiratory mucosal vaccination was immunologically superior to the parenteral route.
RUTI®
In contrast to whole, heat-killed mycobacteria, RUTI® is composed of liposomes containing detoxified fragments of M.tb grown under hypoxic/stress conditions (44, 45). The growth of virulent M.tb under these conditions drives expression of an array of stress and latency associated antigens, thereby expanding the antigenic breadth of the RUTI® formulation, theoretically allowing for adaptive immune responses to target latent bacilli (46). The added advantage of this strategy is targeting the latent bacillary population which, as stated previously, is a major contributor to disease relapse. RUTI® is currently being investigated as a subcutaneously administered post-exposure vaccine in individuals with drug-resistant disease who have completed chemotherapy (45, 47). As such, RUTI® remains to be tested therapeutically in patients with active disease, potentially due to issues related to the Koch phenomenon (45).
Recombinant-Based Therapeutic TB Vaccines
H56:IC31
H56:IC31 is a recombinant fusion protein of three M.tb antigens: Ag85B, ESAT-6, and Rv2660c combined in a stabilizing agent containing a TLR9 agonist as an adjuvant (48). By expressing replication and pathogenicity-based antigens (Ag85B, and ESAT-6, respectively) and latency-associated antigens (Rv2660c), H56:IC31 is designed to drive multi-functional Th1 immunity against both actively replicating and dormant bacilli, and has been shown to prevent M.tb reactivation in macaques following parenteral delivery (48, 49). H56:IC31 has undergone extensive clinical testing as a prophylactic vaccine in phase 1 and 2 trials showing a favorable safety and immunogenicity profile in IGRA− and IGRA+ individuals (50). In line with its success and clinical advancement as a prophylactic vaccine, H56:IC31 is also being investigated as an intramuscularly administeredtherapeutic for TB. This includes a phase 1 combinatorial HDT regimen with a Cyclooxygenase-2 inhibitor etoricoxib (clinicaltrials.gov, NCT02503839), and a phase II efficacy trial in South Africa addressing prevention of disease reoccurrence (clinicaltrials.gov, NCT03512249).
ID93/GLA-SE
ID93/GLA-SE is a recombinant vaccine expressing three virulence (Rv2608, Rv3619, and Rv3620) and one latency-associated (Rv1913) antigen in combination with a synthetic TLR4 agonist in an oil-in-water emulsion (51, 52). Similar to the other vaccine candidates described in this review, ID93/GLA-SE has been extensively characterized in pre-clinical animal models. Such studies have shown that ID93/GLA-SE drives robust and sustained anti-TB specific CD4 and CD8 T cell responses following parenteral delivery, correlating with significantly improved prophylactic protection against pulmonary TB (53). Therapeutic vaccination by the intramuscular route with ID93/GLA-SE in non-human primate models significantly reduced pulmonary mycobacterial burden, pathology, and improved survival (53). Thus far, a phase I trial has shown that ID93/GLA-SE is safe and immunogenic in QuantiFERON positive individuals, thereby supporting its further assessment as an immunotherapeutic (54).
Viral-Vectored Therapeutic TB Vaccines
Modified Vaccinia Ankara
Modified Vaccinia Ankara (MVA)-based TB vaccines have a checkered history with a phase IIb trial of MVA85A failing to enhance prophylactic immunity following parenteral immunization in BCG-vaccinated infants (55). Regardless, MVA-vectors remain widely utilized as TB vaccines given their remarkable ability to accommodate large transgene inserts and drive long-lived Th1 immunity (56). A recently developed MVA-based vaccine expressing 10 M.tb antigens has been assessed for its therapeutic efficacy in a murine model. Interestingly, this vaccine provided minimal reduction of bacterial burden within the lungs and a modest reduction in relapse (57). This was observed following parenteral, but not respiratory mucosal immunization. Additionally, immunological studies revealed that immune responses against some antigens were not induced. These observations highlight certain key considerations for vaccine design which will be addressed further in this review.
Adenovirus
Adenoviruses represent the most widely used viral vector platform for vaccine design (58). Human adenoviruses, in particular human adenovirus serotype 5 (AdHu5), have been extensively tested as a prophylactic vaccine platform delivered both parenterally and via the respiratory mucosal route (58–62). Unfortunately, in some instances the high seroprevalence of antibodies against this common respiratory pathogen has limited its efficacy. This has prompted the development of platforms founded in serotypes exotic to humans (63).
Non-human adenoviruses, such as chimpanzee adenoviruses have gained traction as vaccine vector candidates given their robust immunogenicity and low seroprevalence (63).
We previously characterized the prophylactic potential of a chimpanzee adenoviral-vectored vaccine expressing M.tb Ag85A (AdCh68Ag85A) (64). Our study showcased the superior immunogenicity and efficacy of this vector following respiratory mucosal administration in comparison to its AdHu5 counterpart. Additionally, utilizing a clinically relevant murine model of chemotherapy treated active TB disease, our group has shown that respiratory mucosal therapeutic immunization with AdCh68Ag85A accelerated bacterial clearance, limited lung pathology, and limited disease relapse following pre-mature chemotherapy cessation (65).
Advancing Therapeutic TB Vaccine Design
Although definite protective immune correlates for TB remain elusive, insights from ongoing pre-clinical and clinical trials provide invaluable information to steer development of efficacious therapeutic TB vaccination strategies. Such insights stem from both the immunological as well as the mycobacterial areas of research. In this section we highlight three categories (Figure 3) which we believe are most important in advancing therapeutic TB vaccine design: Vector formulation and immunization route, antigen/epitope optimization, and antigen selection dictated by the mycobacterial life cycle.
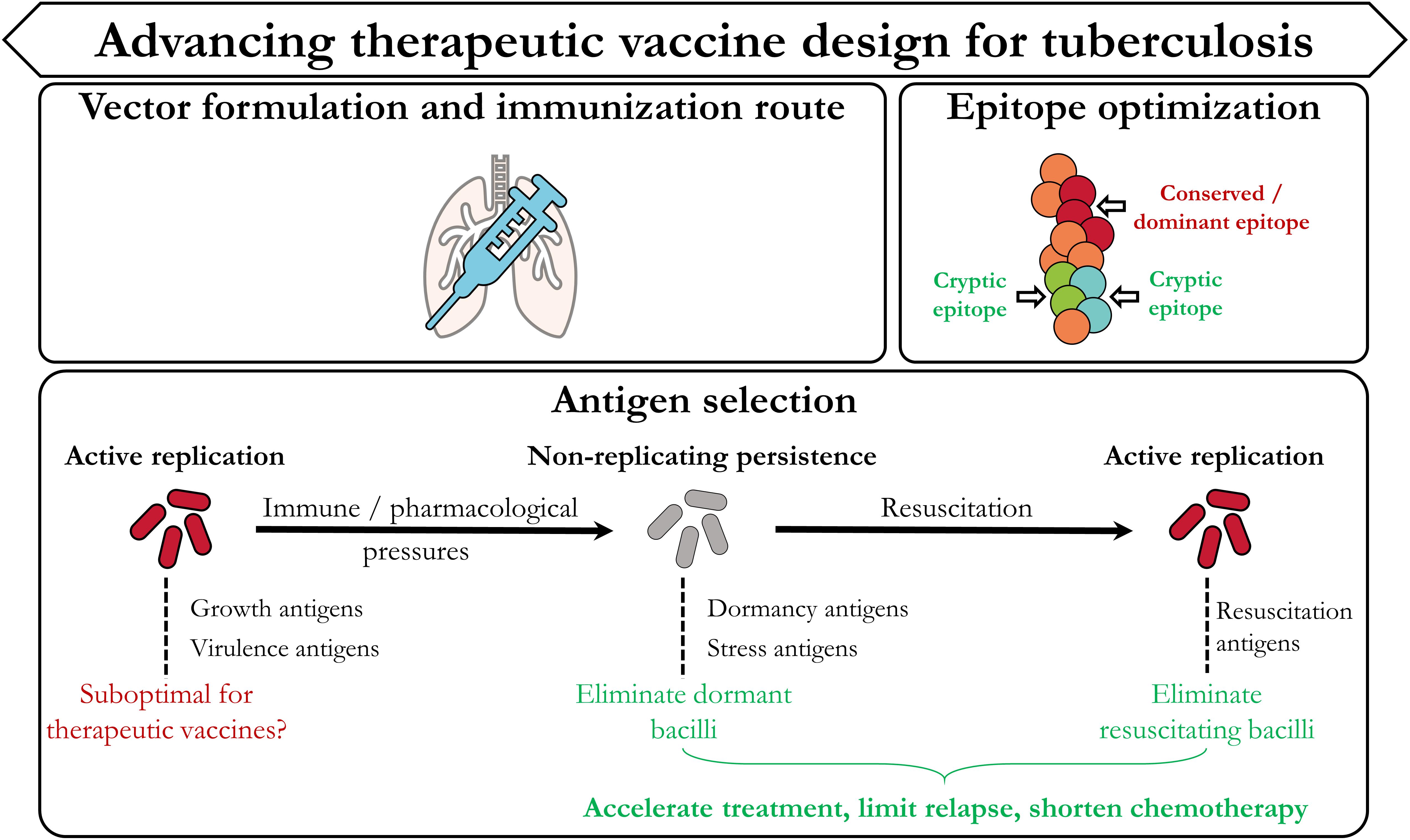
Figure 3. Designing improved therapeutic TB vaccination strategies. In accordance with Figure 1, the current TB vaccine landscape has provided invaluable evidence which can be used to improve therapeutic vaccination strategies. These include the type of vaccine vector, the route of immunization, the selection and design of the epitope(s)/antigen(s), and further focus on identifying targets from the different stages of the mycobacterial life cycle.
Vector Choice
Factors such as feasibility, safety, and immunogenicity are largely dictated by vector choice. As such, this represents a major consideration when developing novel TB therapeutic vaccine strategies. Whole organism-based vaccines (VaccaeTM and PIM) are advantageous given their antigenic breadth. These vectors however may not be amenable for delivery via the respiratory mucosal route in humans due to potential safety considerations. Additionally, utilization of whole mycobacteria/mycobacterial fragments can potentially skew and/or mask immunological responses against less immunogenic, yet protective antigens (see below). As such, utilization of recombinant molecular biology techniques allows for construction of recombinant vaccines that focus immune responses against well-recognized protective antigens. This is the case for recombinant protein and viral-vectored vaccination strategies (58, 66).
In addition, through careful adjuvant selection, antigen-specific immunity can be polarized toward certain immune profiles. Use of TLR agonists (such as TLR9 in H56:IC31and TLR4 in ID93:GLA-SE) enhance both the cytokine functionality and longevity of Th1 immune responses which are critical for anti-TB immunity (67). Identification of adjuvants capable of inducing cytotoxic CD8 T cell responses can be challenging however and may represent a roadblock in developing optimal therapeutic TB vaccines. We have recently shown that antigen-specific CD8 T cells are the major cell type involved in the therapeutic efficacy of AdCh68Ag85A, thereby providing supporting evidence of their importance in anti-TB immunity (65).
Viral vectors, particularly adenoviruses, remain the most widely utilized platforms for vaccine design given their genetic malleability, safety, and amenability for respiratory mucosal administration (58). However as echoed by us and others, viral serotype selection is a major consideration in the downstream immunogenicity and efficacy of a vaccine candidate. For example, human adenovirus serotype 35 (AdHu35) has been used in place of AdHu5 given its low seroprevalence, which allows it to circumvent anti-vector immunity (68). Thus, this vector has been extensively tested as a putative prophylactic TB vaccine candidate (AERAS-402) (69). Unfortunately, recent studies have shown that parenteral administration of this vaccine did not significantly induce antigen-specific immunity (70, 71). In line with this observation, it is well documented that AdHu35 induces robust type 1 interferon responses, leading to downstream loss of vector transgene expression, which may negatively impact T cell immunity (72). Alongside the choice of immunization route (explained below), this also may explain why AERAS-402 failed to confer efficacy when administered therapeutically (73).
Progress in the development of other novel vaccine platforms provide new opportunities in further advancing therapeutic TB vaccine development. For example, nanoparticle-based vaccines have shown tremendous progress against a plethora of infectious diseases (74). Nanoparticles not only act as antigen carriers, but also possess intrinsic immunogenic properties (often not requiring adjuvants) which can be fine-tuned by altering their physiochemical properties (75). Recent studies have shown that nanoparticle-based TB vaccines are not only amenable for respiratory mucosal delivery, but can drive robust Th1-skewed immune responses which provide similar-to-grater immunity than BCG alone (76, 77).
Immunization Route
Compelling evidence suggests that protection against mucosal pathogens such as M.tb is heavily reliant on the presence of pathogen-specific immune cells at the primary site of infection (78–80). As seen during natural M.tb infection, bacterial control is observed when anti-TB specific T cells appear in the lung (81). Specifically, it is the presence of such immune responses within the airways that is critical in anti-TB immunity. Immunization route largely determines the anatomical location of antigen-specific T cells and therefore, vaccine efficacy (82). Pre-clinical studies show that parenteral immunization with TB vaccines drives robust antigen-specific T cell immune responses. However, such cells are primarily restricted to the periphery, unable to quickly enter the lung interstitium and airway lumen, and largely fail to confer protection against pulmonary TB (83). In stark contrast, respiratory mucosal immunization generates a long-lasting population of tissue-resident polyfunctional T cells that are primed to express homing molecules to allow preferential migration and residence in the airway lumen and lungs (80, 82, 84, 85). These immune cells, located at the portal of infection, are able to rapidly respond and carry out their effector function, providing markedly enhanced protection against pulmonary M.tb infection.
Antigen/Epitope Optimization
Most TB vaccine candidates in development continue to utilize antigens that are widely characterized in M.tb-infected individuals and are proven to be immunogenic (e.g., Ag85A, ESAT6, TB10.4). Seminal studies however have shown that M.tb infection in humans is characterized by adaptive T cell responses which are skewed toward hyper conserved epitopes (86, 87). Broad conservation of immune responses against such epitopes is a non-prototypic measure of immune evasion which M.tb is speculated to have evolved to control and concentrate the host immune response against non-protective antigens. As most vaccine strategies are formulated to include such immunodominant antigens and have been ineffective, it suggests that these candidates may be suboptimal for vaccine-derived protection. Consequently, targeting non-dominant (cryptic) antigens may represent a superior avenue for designing efficacious vaccines (88).
Expanding the repertoire of T cell immunity through immunization with cryptic antigens has been evaluated in TB in pre-clinical models (89, 90). Such studies have shown that T cell responses against cryptic antigens are significantly more functional than those elicited against classical immunodominant antigens and provide enhanced protection. Importantly, T cell responses against such cryptic antigens are longer-lived, and are less prone to exhaustion, making them ideal for long-lasting immunity. Collectively, such observations warrant the inclusion of cryptic antigens in therapeutic vaccination strategies.
Antigen Selection
Following pulmonary infection, adaptive immune responses are primarily skewed toward replication and virulence-associated antigens. As previously mentioned, immunological and pharmacological stresses proceed to drive M.tb into a non-replicating, dormant state. Antigenically, this is associated with a shift from replication-associated antigens to those involved in the stress and the dormancy response. Consequently this allows M.tb to evade existing adaptive immune responses that have already been primed against such antigens. Two major conclusions can be inferred from this: Only targeting replication-associated antigens may be suboptimal for therapeutic vaccination platforms, and including dormancy/stress-associated antigens may improve therapeutic vaccine efficacy. A plethora of dormancy antigens are currently under investigation as tantalizing vaccine antigen candidates. As detailed earlier, H56:IC31 expresses the latency antigen Rv2660c and ID93:GLA-SE expresses Rv1813 (48, 53). These represent just two of many potential dormancy candidates (such as those controlled by the DosR Regulon) that warrant further investigation. In addition, it will be critical to assess whether immune responses to such antigens are able to specifically eliminate dormant bacilli. To do so will require implementation and standardization of experimental techniques that delineate between these mycobacterial subpopulations, which have been described previously (91).
In addition, targeting M.tb populations resuscitating from dormancy would also be critical in the development of therapeutic vaccines, as to eliminate/minimize reactivation. Mycobacterial resuscitation is a complex process and is regulated in a coordinated transcriptional burst preceding expression of metabolic and growth-related pathways (92). Resuscitation promoting factors (RPFs) represent a major class of antigens involved in this process and may represent promising vaccine antigen targets (92–94).
Evidence supporting further investigation of RPFs as vaccine antigens stems from clinical studies that correlated the long term maintenance of multifunctional adaptive immunity against these antigens in M.tb-infected non-progressors (95). This strongly supports the role of such immune responses in controlling reactivation from latency. Although RPFs have been included in numerous pre-clinical prophylactic vaccine candidates, no study to-date has investigated the contribution of RPF-specific immunity in restricting resuscitation (96, 97). Given the significant contribution of resuscitating M.tb populations to disease relapse, developing therapeutic vaccines to improve immune surveillance during or post-chemotherapy against such subpopulations would have substantial benefits.
Concluding Statement
Reaching the END TB set goals for 2035 remains a daunting task which we are unlikely to meet short of a revolution in current treatments. Investigation into therapeutic vaccines has expanded rapidly in the last several years offering novel insights toward vector design, and targeted antigen selection from differing stages of the bacterial lifecycle. While this field lags behind the development of prophylactic strategies, therapeutic vaccines have the potential to enhance treatment success rates, even for difficult-to-treat drug-resistant forms of TB. As this emerging field of TB vaccine development continues to flourish, there are several factors that will be critical to assessing their potential success, and eventual utilization. Namely, to what extent are these vaccines able to shorten the duration of existing chemotherapeutic regimens? How will we evaluate the efficacy of these vaccines under realistic situations such as fragmented or incomplete therapy? Can therapeutic immunization protect against recurrent infection? Intertwined with this are establishing correlates to measure efficacy, and the standardization of complex, pre-clinical models necessary to account for the myriad of moving parts at play. This includes, but is not limited to, dissecting immune responses from vaccines versus those induced from infection, the role of chemotherapy in driving dormancy/escape from immunological pressures, and safety/efficacy in individuals living with HIV.
Author Contributions
SA, AV, and ZX designed the manuscript. SA and AV wrote the manuscript. ZX approved the manuscript. All authors revised and edited the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer HM declared a past co-authorship with one of the authors ZX to the handling Editor.
Acknowledgments
We acknowledge the support from the Canadian Institutes of Health Research Foundation Program and the Natural Sciences and Engineering Research Council of Canada.
Footnotes
References
1. World Health Organization Executive Board. Global Strategy and Targets for Tuberculosis Prevention, Care and Control After 2015. Geneva: WHO (2015). p. 1–23.
3. World Health Organization. Consolidated Guidelines on Tuberculosis Treatment. Geneva: WHO (2019).
4. Wallis RS, Hafner R. Advancing host-directed therapy for tuberculosis. Nat Rev Immunol. (2015) 15:255–63. doi: 10.1038/nri3813
5. Young C, Walzl G, Du Plessis N. Therapeutic host-directed strategies to improve outcome in tuberculosis. Mucosal Immunol. (2020) 13:190–204. doi: 10.1038/s41385-019-0226-5
6. World Health Organization. What is DOTS? A Guide to Understanding the WHO-recommended TB Control Strategy Known as DOTS. Geneva: WHO (1999). p. 1–39.
7. Conradie F, Diacon AH, Ngubane N, Howell P, Everitt D, Crook AM, et al. Treatment of highly drug-resistant pulmonary tuberculosis. N Engl J Med. (2020) 382:893–902. doi: 10.1056/NEJMoa1901814
8. Diacon AH, Pym A, Grobusch MP, de los Rios JM, Gotuzzo E, Vasilyeva I, et al. Multidrug-resistant tuberculosis and culture conversion with bedaquiline. N Engl J Med. (2014) 371:723–32. doi: 10.1056/NEJMoa1313865
9. de Vos M, Ley SD, Wiggins KB, Derendinger B, Dippenaar A, Grobbelaar M, et al. Bedaquiline microheteroresistance after cessation of tuberculosis treatment. N Engl J Med. (2019) 380:2178–80. doi: 10.1056/NEJMc1815121
10. Colijn C, Cohen T, Ganesh A, Murray M. Spontaneous emergence of multiple drug resistance in tuberculosis before and during therapy. PLoS One. (2011) 6:e18327. doi: 10.1371/journal.pone.0018327
11. Cohen SB, Gern BH, Delahaye JL, Adams KN, Plumlee CR, Winkler JK, et al. Alveolar macrophages provide an early Mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe. (2018) 24:439–46.e4. doi: 10.1016/j.chom.2018.08.001
12. Huang L, Nazarova EV, Tan S, Liu Y, Russell DG. Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J Exp Med. (2018) 215:1135–52. doi: 10.1084/jem.20172020
13. Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature. (2015) 528:565–9. doi: 10.1038/nature16451
14. Songane M, Kleinnijenhuis J, Netea MG, van Crevel R. The role of autophagy in host defence against Mycobacterium tuberculosis infection. Tuberculosis. (2012) 92:388–96. doi: 10.1016/j.tube.2012.05.004
15. Levitte S, Adams KN, Berg RD, Cosma CL, Urdahl KB, Ramakrishnan L. Mycobacterial acid tolerance enables phagolysosomal survival and establishment of tuberculous infection in vivo. Cell Host Microbe. (2016) 20:250–8. doi: 10.1016/j.chom.2016.07.007
16. Lai R, Jeyanathan M, Shaler CR, Damjanovic D, Khera A, Horvath C, et al. Restoration of innate immune activation accelerates Th1-cell priming and protection following pulmonary mycobacterial infection. Eur J Immunol. (2014) 44:1375–86. doi: 10.1002/eji.201344300
17. Romagnoli A, Etna MP, Giacomini E, Pardini M, Remoli ME, Corazzari M, et al. ESX-1 dependent impairment of autophagic flux by Mycobacterium tuberculosis in human dendritic cells. Autophagy. (2012) 8:1357–70. doi: 10.4161/auto.20881
18. Lai R, Jeyanathan M, Afkhami S, Zganiacz A, Hammill JA, Yao Y, et al. CD11b + dendritic cell–mediated anti– Mycobacterium tuberculosis Th1 activation is counterregulated by CD103 + dendritic cells via IL-10. J Immunol. (2018) 200:1746–60. doi: 10.4049/jimmunol.1701109
19. Karakousis PC, Yoshimatsu T, Lamichhane G, Woolwine SC, Nuermberger EL, Grosset J, et al. Dormancy phenotype displayed by extracellular Mycobacterium tuberculosis within artificial granulomas in mice. J Exp Med. (2004) 200:647–57. doi: 10.1084/jem.20040646
20. Voskuil MI, Schnappinger D, Visconti KC, Harrell MI, Dolganov GM, Sherman DR, et al. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J Exp Med. (2003) 198:705–13. doi: 10.1084/jem.20030205
21. Gengenbacher M, Kaufmann SHE. Mycobacterium tuberculosis?: success through dormancy. FEMS Microbiol Rev. (2012) 36:514–32. doi: 10.1111/j.1574-6976.2012.00331.x
22. Raffetseder J, Pienaar E, Blomgran R, Eklund D, Patcha Brodin V, Andersson H, et al. Replication rates of Mycobacterium tuberculosis in human macrophages do not correlate with mycobacterial antibiotic susceptibility. PLoS One. (2014) 9:e112426. doi: 10.1371/journal.pone.0112426
23. Park H-D, Guinn KM, Harrell MI, Liao R, Voskuil MI, Tompa M, et al. Rv3133c/dosR is a transcription factor that mediates the hypoxic response of Mycobacterium tuberculosis. Mol Microbiol. (2003) 48:833–43. doi: 10.1046/j.1365-2958.2003.03474.x
24. Mehra S, Foreman TW, Didier PJ, Ahsan MH, Hudock TA, Kissee R, et al. The DosR regulon modulates adaptive immunity and is essential for Mycobacterium tuberculosis persistence. Am J Respir Crit Care Med. (2015) 191:1185–96. doi: 10.1164/rccm.201408-1502OC
25. Bartek IL, Rutherford R, Gruppo V, Morton RA, Morris RP, Klein MR, et al. The DosR regulon of M. tuberculosis and antibacterial tolerance. Tuberculosis. (2009) 89:310–6. doi: 10.1016/j.tube.2009.06.001
26. Hu Y, Pertinez H, Liu Y, Davies G, Coates A. Bedaquiline kills persistent Mycobacterium tuberculosis with no disease relapse: an in vivo model of a potential cure. J Antimicrob Chemother. (2019) 74:1627–33. doi: 10.1093/jac/dkz052
27. Singhal A, Jie L, Kumar P, Hong GS, Leow MK-S, Paleja B, et al. Metformin as adjunct antituberculosis therapy. Sci Transl Med. (2014) 6:263ra159. doi: 10.1126/scitranslmed.3009885
28. Lachmandas E, Eckold C, Böhme J, Koeken VACM, Marzuki MB, Blok B, et al. Metformin alters human host responses to Mycobacterium tuberculosis in healthy subjects. J Infect Dis. (2019) 220:139–50. doi: 10.1093/infdis/jiz064
29. Torfs E, Piller T, Cos P, Cappoen D. Opportunities for overcoming Mycobacterium tuberculosis drug resistance: emerging mycobacterial targets and host-directed therapy. Int J Mol Sci. (2019) 20:2868. doi: 10.3390/ijms20122868
30. Orme IM. Tuberculosis vaccine types and timings. Clin Vaccine Immunol. (2015) 22:249–57. doi: 10.1128/CVI.00718-14
31. World Health Organization. WHO Preferred Product Characteristics for Therapeutic Vaccines to Improve Tuberculosis Treatment Outcomes. Geneva: WHO (2019). doi: 10.1128/cvi.00718-14
32. Stanford JL, Rook GAW, Bahr GM, Dowlati Y, Ganapati R, Ghazi Saidi K, et al. Mycobacterium vaccae in immunoprophylaxis and immunotherapy of leprosy and tuberculosis. Vaccine. (1990) 8:525–30. doi: 10.1016/0264-410X(90)90002-4
33. Huang C, Hsieh W. Efficacy of Mycobacterium vaccae immunotherapy for patients with tuberculosis: a systematic review and meta-analysis. Hum Vaccin Immunother. (2017) 13:1960–71. doi: 10.1080/21645515.2017.1335374
34. Weng H, Huang J-Y, Meng X-Y, Li S, Zhang G-Q. Adjunctive therapy of Mycobacterium vaccae vaccine in the treatment of multidrug-resistant tuberculosis: a systematic review and meta-analysis. Biomed Rep. (2016) 4:595–600. doi: 10.3892/br.2016.624
35. de Bruyn G, Garner P. Mycobacterium vaccae immunotherapy for treating tuberculosis. Cochrane Database Syst Rev. (2003) CD001166.
36. Yang XY, Chen QF, Cui XH, Yu Y, Li YP. Mycobacterium vaccae vaccine to prevent tuberculosis in high risk people: a meta-analysis. J Infect. (2010) 60:320–30. doi: 10.1016/j.jinf.2010.02.005
37. Skinner MA, Yuan S, Prestidge R, Chuk D, Watson JD, Tan PLJ. Immunization with heat-killed Mycobacterium vaccae stimulates CD8+ cytotoxic T cells specific for macrophages infected with Mycobacterium tuberculosis. Infect Immun. (1997) 65:4525–30. doi: 10.1128/IAI.65.11.4525-4530.1997
38. Burdick AE, Ramirez CC. The role of mycophenolate mofetil in the treatment of leprosy reactions. Int J Lepr Other Mycobact Dis. (2005) 73:127–8.
39. Zaheer SA, Mukherjee R, Ramkumar D, Misra RS, Sharma AK, Kar HK, et al. Combined multidrug and Mycobacterium w vaccine therapy in patients with multibacillary leprosy. J Infect Dis. (1993) 167:401–10. doi: 10.1093/infdis/167.2.401
40. Sharma SK, Katoch K, Sarin R, Balambal R, Kumar Jain N, Patel N, et al. Efficacy and safety of Mycobacterium indicus pranii as an adjunct therapy in category II pulmonary tuberculosis in a randomized trial. Sci Rep. (2017) 7:3354. doi: 10.1038/s41598-017-03514-1
41. Guleria I, Mukherjee R, Kaufmann SE. In vivo depletion of CD4 and CD8 T lymphocytes impairs Mycobacterium w vaccine-induced protection against M. tubercolosis in mice. Med Microbiol Immunol. (1993) 182:129–35. doi: 10.1007/BF00190265
42. Gupta A, Ahmad FJ, Ahmad F, Gupta UD, Natarajan M, Katoch VM, et al. Protective efficacy of Mycobacterium indicus pranii against tuberculosis and underlying local lung immune responses in guinea pig model. Vaccine. (2012) 30:6198–209. doi: 10.1016/j.vaccine.2012.07.061
43. Das S, Chowdhury BP, Goswami A, Parveen S, Jawed J, Pal N, et al. Mycobacterium indicus pranii (MIP) mediated host protective intracellular mechanisms against tuberculosis infection: involvement of TLR-4 mediated signaling. Tuberculosis. (2016) 101:201–9. doi: 10.1016/j.tube.2016.09.027
44. Cardona P-J, Amat I, Gordillo S, Arcos V, Guirado E, Díaz J, et al. Immunotherapy with fragmented Mycobacterium tuberculosis cells increases the effectiveness of chemotherapy against a chronical infection in a murine model of tuberculosis. Vaccine. (2005) 23:1393–8. doi: 10.1016/j.vaccine.2004.09.008
45. Nell AS, D’lom E, Bouic P, Sabaté M, Bosser R, Picas J, et al. Safety, tolerability, and immunogenicity of the novel antituberculous vaccine RUTI: randomized, placebo-controlled phase II clinical trial in patients with latent tuberculosis infection. PLoS One. (2014) 9:e89612. doi: 10.1371/journal.pone.0089612
46. Guirado E, Gil O, Caìceres N, Singh M, Vilaplana C, Cardona P-J. Induction of a specific strong polyantigenic cellular immune response after short-term chemotherapy controls bacillary reactivation in murine and guinea pig experimental models of tuberculosis. Clin Vaccine Immunol. (2008) 15:1229–37. doi: 10.1128/CVI.00094-08
47. Vilaplana C, Montané E, Pinto S, Barriocanal AM, Domenech G, Torres F, et al. Double-blind, randomized, placebo-controlled Phase I clinical trial of the therapeutical antituberculous vaccine RUTI®. Vaccine. (2010) 28:1106–16. doi: 10.1016/j.vaccine.2009.09.134
48. Lin PL, Dietrich J, Tan E, Abalos RM, Burgos J, Bigbee C, et al. The multistage vaccine H56 boosts the effects of BCG to protect cynomolgus macaques against active tuberculosis and reactivation of latent Mycobacterium tuberculosis infection. J Clin Invest. (2012) 122:303–14. doi: 10.1172/JCI46252
49. Aagaard C, Hoang T, Dietrich J, Cardona P-J, Izzo A, Dolganov G, et al. multistage tuberculosis vaccine that confers efficient protection before and after exposure. Nat Med. (2011) 17:189–94. doi: 10.1038/nm.2285
50. Luabeya AKK, Kagina BMN, Tameris MD, Geldenhuys H, Hoff ST, Shi Z, et al. First-in-human trial of the post-exposure tuberculosis vaccine H56:IC31 in Mycobacterium tuberculosis infected and non-infected healthy adults. Vaccine. (2015) 33:4130–40. doi: 10.1016/j.vaccine.2015.06.051
51. Coler RN, Bertholet S, Moutaftsi M, Guderian JA, Windish HP, Baldwin SL, et al. Development and characterization of synthetic glucopyranosyl lipid adjuvant system as a vaccine adjuvant. PLoS One. (2011) 6:e16333. doi: 10.1371/journal.pone.0016333
52. Bertholet S, Ireton GC, Kahn M, Guderian J, Mohamath R, Stride N, et al. Identification of human T cell antigens for the development of vaccines against Mycobacterium tuberculosis. J Immunol. (2008) 181:7948–57. doi: 10.4049/jimmunol.181.11.7948
53. Coler RN, Bertholet S, Pine SO, Orr MT, Reese V, Windish HP, et al. Therapeutic immunization against Mycobacterium tuberculosis is an effective adjunct to antibiotic treatment. J Infect Dis. (2013) 207:1242–52. doi: 10.1093/infdis/jis425
54. Penn-Nicholson A, Tameris M, Smit E, Day TA, Musvosvi M, Jayashankar L, et al. Safety and immunogenicity of the novel tuberculosis vaccine ID93 + GLA-SE in BCG-vaccinated healthy adults in South Africa: a randomised, double-blind, placebo-controlled phase 1 trial. Lancet Respir Med. (2018) 6:287–98. doi: 10.1016/S2213-2600(18)30077-8
55. Tameris MD, Hatherill M, Landry BS, Scriba TJ, Snowden MA, Lockhart S, et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled phase 2b trial. Lancet. (2013) 381:1021–8. doi: 10.1016/S0140-6736(13)60177-4
56. Cottingham MG, Carroll MW. Recombinant MVA vaccines: dispelling the myths. Vaccine. (2013) 31:4247–51. doi: 10.1016/j.vaccine.2013.03.021
57. Leung-Theung-Long S, Coupet C, Gouanvic M, Schmitt D, Ray A, Hoffmann C, et al. A multi-antigenic MVA vaccine increases efficacy of combination chemotherapy against Mycobacterium tuberculosis. PLoS One. (2018) 13:e0196815. doi: 10.1371/journal.pone.0196815
58. Afkhami S, Yao Y, Xing Z. Methods and clinical development of adenovirus-vectored vaccines against mucosal pathogens. Mol Ther Methods Clin Dev. (2016) 3:16030. doi: 10.1038/mtm.2016.30
59. Tchilian E, Ahuja D, Hey A, Jiang S, Beverley P. Immunization with different formulations of Mycobacterium tuberculosis antigen 85A induces immune responses with different specificity and protective efficacy. Vaccine. (2013) 31:4624–31. doi: 10.1016/j.vaccine.2013.07.040
60. Smaill F, Jeyanathan M, Smieja M, Medina MF, Thanthrige-Don N, Zganiacz A, et al. A human type 5 adenovirus-based tuberculosis vaccine induces robust T cell responses in humans despite preexisting anti-adenovirus immunity. Sci Transl Med. (2013) 5:205ra134. doi: 10.1126/scitranslmed.3006843
61. Jeyanathan M, Shao Z, Yu X, Harkness R, Jiang R, Li J, et al. AdHu5Ag85A respiratory mucosal boost immunization enhances protection against pulmonary tuberculosis in BCG-primed non-human primates. PLoS One. (2015) 10:e0135009. doi: 10.1371/journal.pone.0135009
62. Yao Y, Lai R, Afkhami S, Haddadi S, Zganiacz A, Vahedi F, et al. Enhancement of antituberculosis immunity in a humanized model system by a novel virus-vectored respiratory mucosal vaccine. J Infect Dis. (2017) 216:135–45. doi: 10.1093/infdis/jix252
63. Colloca S, Barnes E, Folgori A, Ammendola V, Capone S, Cirillo A, et al. Vaccine vectors derived from a large collection of simian adenoviruses induce potent cellular immunity across multiple species. Sci Transl Med. (2012) 4:115ra2. doi: 10.1126/scitranslmed.3002925
64. Jeyanathan M, Thanthrige-Don N, Afkhami S, Lai R, Damjanovic D, Zganiacz A, et al. Novel chimpanzee adenovirus-vectored respiratory mucosal tuberculosis vaccine: overcoming local anti-human adenovirus immunity for potent TB protection. Mucosal Immunol. (2015) 8:1373–87. doi: 10.1038/mi.2015.29
65. Afkhami S, Lai R, D’Agostino MR, Vaseghi-Shanjani M, Zganiacz A, Yao Y, et al. Single-dose mucosal immunotherapy with chimpanzee adenovirus-based vaccine accelerates TB disease control and limits its rebound following antibiotic cessation. J Infect Dis. (2019) 220:1355–66. doi: 10.1093/infdis/jiz306
66. Nascimento IP, Leite LCC. Recombinant vaccines and the development of new vaccine strategies. Brazilian J Med Biol Res. (2012) 45:1102–11. doi: 10.1590/s0100-879x2012007500142
67. Schellack C, Prinz K, Egyed A, Fritz JH, Wittmann B, Ginzler M, et al. IC31, a novel adjuvant signaling via TLR9, induces potent cellular and humoral immune responses. Vaccine. (2006) 24:5461–72. doi: 10.1016/j.vaccine.2006.03.071
68. Thorner AR, Vogels R, Kaspers J, Weverling GJ, Holterman L, Lemckert A, et al. Age dependence of adenovirus-specific neutralizing antibody titers in individuals from sub-saharan Africa. J Clin Microbiol. (2006) 44:3781–3. doi: 10.1128/JCM.01249-06
69. Abel B, Tameris M, Mansoor N, Gelderbloem S, Hughes J, Abrahams D, et al. The novel tuberculosis vaccine, AERAS-402, induces robust and polyfunctional CD4 + and CD8 + T cells in adults. Am J Respir Crit Care Med. (2010) 181:1407–17. doi: 10.1164/rccm.200910-1484OC
70. van Zyl-Smit RN, Esmail A, Bateman ME, Dawson R, Goldin J, van Rikxoort E, et al. Safety and immunogenicity of adenovirus 35 tuberculosis vaccine candidate in adults with active or previous tuberculosis. A Randomized Trial. Am J Respir Crit Care Med. (2017) 195:1171–80. doi: 10.1164/rccm.201603-0654OC
71. Darrah PA, Bolton DL, Lackner AA, Kaushal D, Aye PP, Mehra S, et al. Aerosol vaccination with AERAS-402 elicits robust cellular immune responses in the lungs of rhesus macaques but fails to protect against high-dose Mycobacterium tuberculosis challenge. J Immunol. (2014) 193:1799–811. doi: 10.4049/jimmunol.1400676
72. Johnson MJ, Björkström NK, Petrovas C, Liang F, Gall JGD, Loré K, et al. Type I interferon-dependent activation of NK cells by rAd28 or rAd35, but not rAd5, leads to loss of vector-insert expression. Vaccine. (2014) 32:717–24. doi: 10.1016/j.vaccine.2013.11.055
73. Alyahya SA, Nolan ST, Smith CMR, Bishai WR, Sadoff J, Lamichhane G. Immunogenicity without efficacy of an adenoviral tuberculosis vaccine in a stringent mouse model for immunotherapy during treatment. PLoS One. (2015) 10:e0127907. doi: 10.1371/journal.pone.0127907
74. Pati R, Shevtsov M, Sonawane A. Nanoparticle vaccines against infectious diseases. Front Immunol. (2018) 9:2224. doi: 10.3389/fimmu.2018.02224
75. Gregory AE, Titball R, Williamson D. Vaccine delivery using nanoparticles. Front Cell Infect Microbiol. (2013) 3:1–13. doi: 10.3389/fcimb.2013.00013
76. Stylianou E, Diogo GR, Pepponi I, Dolleweerd C, Arias MA, Locht C, et al. Mucosal delivery of antigen−coated nanoparticles to lungs confers protective immunity against tuberculosis infection in mice. Eur J Immunol. (2014) 44:440–9. doi: 10.1002/eji.201343887
77. Ballester M, Nembrini C, Dhar N, de Titta A, de Piano C, Pasquier M, et al. Nanoparticle conjugation and pulmonary delivery enhance the protective efficacy of Ag85B and CpG against tuberculosis. Vaccine. (2011) 29:6959–66. doi: 10.1016/j.vaccine.2011.07.039
78. Jeyanathan M, Heriazon A, Xing, Z. Airway luminal T cells: a newcomer on the stage of TB vaccination strategies. Trends Immunol. (2010) 31:247–52. doi: 10.1016/j.it.2010.05.002
79. Xing Z, Jeyanathan M, Smaill F. New approaches to TB vaccination. Chest. (2014) 146:804–12. doi: 10.1378/chest.14-0439
70. Wang J, Thorson L, Stokes RW, Santosuosso M, Huygen K, Zganiacz A, et al. Single mucosal, but not parenteral, immunization with recombinant adenoviral-based vaccine provides potent protection from pulmonary tuberculosis. J Immunol. (2004) 173:6357–65. doi: 10.4049/jimmunol.173.10.6357
81. Cooper AM. Cell-mediated immune responses in tuberculosis. Annu Rev Immunol. (2009) 27:393–422. doi: 10.1146/annurev.immunol.021908.132703
82. Lai R, Afkhami S, Haddadi S, Jeyanathan M, Xing Z. Mucosal immunity and novel tuberculosis vaccine strategies: route of immunisation determined T-cell homing to restricted lung mucosal compartments. Eur Respir Rev. (2015) 24:356–60. doi: 10.1183/16000617.00002515
83. Jeyanathan M, Afkhami S, Khera A, Mandur T, Damjanovic D, Yao Y, et al. CXCR3 signaling is required for restricted homing of parenteral tuberculosis vaccine–induced T cells to both the lung parenchyma and airway. J Immunol. (2017) 199:2555–69. doi: 10.4049/jimmunol.1700382
84. Haddadi S, Thanthrige-Don N, Afkhami S, Khera A, Jeyanathan M, Xing Z. Expression and role of VLA-1 in resident memory CD8 T cell responses to respiratory mucosal viral-vectored immunization against tuberculosis. Sci Rep. (2017) 7:9525. doi: 10.1038/s41598-017-09909-4
85. Haddadi S, Vaseghi-Shanjani M, Yao Y, Afkhami S, D’Agostino MR, Zganiacz A, et al. Mucosal-pull induction of lung-resident memory CD8 T cells in parenteral TB vaccine-primed hosts requires cognate antigens and CD4 T cells. Front Immunol. (2019) 10:2075. doi: 10.3389/fimmu.2019.02075
86. Comas I, Chakravartti J, Small PM, Galagan J, Niemann S, Kremer K, et al. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat Genet. (2010) 42:498–503. doi: 10.1038/ng.590
87. Coscolla M, Copin R, Sutherland J, Gehre F, de Jong B, Owolabi O, et al. tuberculosis T cell epitope analysis reveals paucity of antigenic variation and identifies rare variable TB antigens. Cell Host Microbe. (2015) 18:538–48. doi: 10.1016/j.chom.2015.10.008
88. Woodworth JS, Andersen P. Reprogramming the T cell response to tuberculosis. Trends Immunol. (2016) 37:81–3. doi: 10.1016/j.it.2015.12.009
89. Woodworth JS, Aagaard CS, Hansen PR, Cassidy JP, Agger EM, Andersen P. Protective CD4 T cells targeting cryptic epitopes of Mycobacterium tuberculosis resist infection-driven terminal differentiation. J Immunol. (2014) 192:3247–58. doi: 10.4049/jimmunol.1300283
90. Aagaard CS, Hoang TTKT, Vingsbo-Lundberg C, Dietrich J, Andersen P. Quality and vaccine efficacy of CD4 + T cell responses directed to dominant and subdominant epitopes in ESAT-6 from Mycobacterium tuberculosis. J Immunol. (2009) 183:2659–68. doi: 10.4049/jimmunol.0900947
91. Loraine J, Pu F, Turapov O, Mukamolova GV. Development of an in vitro assay for detection of drug-induced resuscitation-promoting-factor-dependent mycobacteria. Antimicrob Agents Chemother. (2016) 60:6227–33. doi: 10.1128/AAC.00518-16
92. Salina EG, Grigorov AS, Bychenko OS, Skvortsova YV, Mamedov IZ, Azhikina TL, et al. Resuscitation of dormant “Non-culturable” Mycobacterium tuberculosis is characterized by immediate transcriptional burst. Front Cell Infect Microbiol. (2019) 9:272. doi: 10.3389/fcimb.2019.00272
93. Shleeva MO, Bagramyan K, Telkov MV, Mukamolova GV, Young M, Kell DB, et al. Formation and resuscitation of ‘non-culturable’ cells of Rhodococcus rhodochrous and Mycobacterium tuberculosis in prolonged stationary phase. Microbiology. (2002) 148:1581–91. doi: 10.1099/00221287-148-5-1581
94. Mukamolova GV, Turapov O, Malkin J, Woltmann G, Barer MR. Resuscitation-promoting factors reveal an occult population of Tubercle bacilli in sputum. Am J Respir Crit Care Med. (2010) 181:174–80. doi: 10.1164/rccm.200905-0661OC
95. Commandeur S, van Meijgaarden KE, Lin MY, Franken KLMC, Friggen AH, Drijfhout JW, et al. Identification of human T-cell responses to Mycobacterium tuberculosis resuscitation-promoting factors in long-Term latently infected individuals. Clin Vaccine Immunol. (2011) 18:676–83. doi: 10.1128/CVI.00492-10
96. Romano M, Aryan E, Korf H, Bruffaerts N, Franken CLMC, Ottenhoff THM, et al. Potential of Mycobacterium tuberculosis resuscitation-promoting factors as antigens in novel tuberculosis sub-unit vaccines. Microbes Infect. (2012) 14:86–95. doi: 10.1016/j.micinf.2011.08.011
Keywords: tuberculosis, therapeutic vaccine, chemotherapy, immunotherapy, respiratory mucosa, mycobacterial life cycle
Citation: Afkhami S, Villela AD, D’Agostino MR, Jeyanathan M, Gillgrass A and Xing Z (2020) Advancing Immunotherapeutic Vaccine Strategies Against Pulmonary Tuberculosis. Front. Immunol. 11:557809. doi: 10.3389/fimmu.2020.557809
Received: 30 April 2020; Accepted: 18 August 2020;
Published: 09 September 2020.
Edited by:
Juraj Ivanyi, King’s College London, United KingdomReviewed by:
Helen McShane, University of Oxford, United KingdomRajko Reljic, St George’s, University of London, United Kingdom
Copyright © 2020 Afkhami, Villela, D’Agostino, Jeyanathan, Gillgrass and Xing. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhou Xing, eGluZ3pAbWNtYXN0ZXIuY2E=