 Souwelimatou Amadou Amani
Souwelimatou Amadou Amani Mark L. Lang
Mark L. Lang- Department of Microbiology and Immunology, University of Oklahoma Health Sciences Center, Oklahoma City, OK, United States
Bacterial enteric pathogens individually and collectively represent a serious global health burden. Humoral immune responses following natural or experimentally-induced infections are broadly appreciated to contribute to pathogen clearance and prevention of disease recurrence. Herein, we have compared observations on humoral immune mechanisms following infection with Citrobacter rodentium, the model for enteropathogenic Escherichia coli, Vibrio cholerae, Shigella species, Salmonella enterica species, and Clostridioides difficile. A comparison of what is known about the humoral immune responses to these pathogens reveals considerable variance in specific features of humoral immunity including establishment of high affinity, IgG class-switched memory B cell and long-lived plasma cell compartments. This article suggests that such variance could be contributory to persistent and recurrent disease.
Introduction
Enteric pathogens rapidly activate host innate and adaptive defense mechanisms upon infection. These mechanisms include activation of innate immune cells, their production of cytokines and chemokines, and antigen presentation necessary for the recruitment of inflammatory cells (1), and initiation of the adaptive immune response (2).
Bacteria such as Vibrio cholerae and Clostridioides difficile secrete enterotoxins that mediate the pathogenesis and the inflammatory responses which often leads to tissue injury and loss of intestinal barrier integrity. Other bacteria such as Salmonella and Shigella overcome the intestinal barrier through invasion via the microfold cells (M cells) which are specialized epithelial cells that overlie Peyer's Patches of the intestine (3–5). Transcytosis of the bacteria by M cells facilitate their colonization of the gut mucosa and promote the induction immune responses in the Peyer's patches (6) (Figure 1). Depending of the efficiency of the immune response and the pathogenicity of the bacteria, the infection can be cleared after an inflammatory response and with limited intestinal tissue damage. A localized inflammatory response may include recruitment and activation of dendritic cells (DCs). The activated DCs migrate within the Peyer's patches and to the draining mesenteric lymph nodes to initiate T and B cell responses. These responses lead to the production of significant amounts of mucosal IgA and some systemic IgG that can traffic back to the gut (7, 8). However, with certain bacteria, a loss of intestinal barrier integrity may be needed to facilitate bacterial dissemination through the bloodstream resulting in a systemic infection. The damage may allow the secreted virulence factors and other bacterial antigens to reach distal lymphoid organs via the lymphatic system (Figure 1). These actions may result in systemic B cell and T cell responses. This extra-intestinal adaptive response summarized in Figure 2, is necessary for the production of high affinity, antigen-specific IgG antibodies in the germinal center (GC) (9).
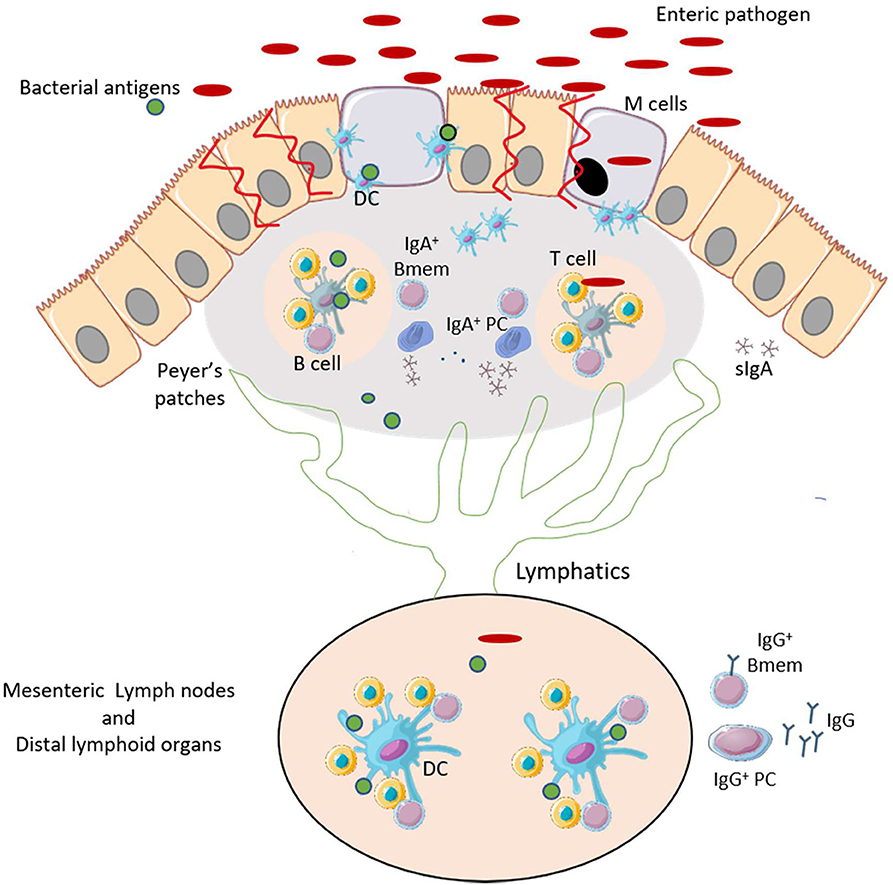
Figure 1. Enteric pathogens promote an inflammatory response and can induce gut-associated mucosal humoral immunity. After infection by enteric pathogens, bacteria and bacterial antigens may be uptaken by M cells. This causes an inflammatory response that promotes recruitment and maturation of DCs. The primed DCs activate T-helper cells in the lymphoid follicles of the Peyer's Patches. T cells interact with B cells to induce development of IgA+ memory B cells (Bmem) and plasma cells (PCs), and production of antigen-specific IgA. The bacterial antigens may reach the mesenteric and distal lymphoid organs via the lymphatic system. This leads to the production IgG antibodies encoded by Bmem cells and PCs. This figure was prepared by modifying Servier Medical Art, licensed under a Creative Common Attribution 3.0 Generic License. http://smart.servier.com/.
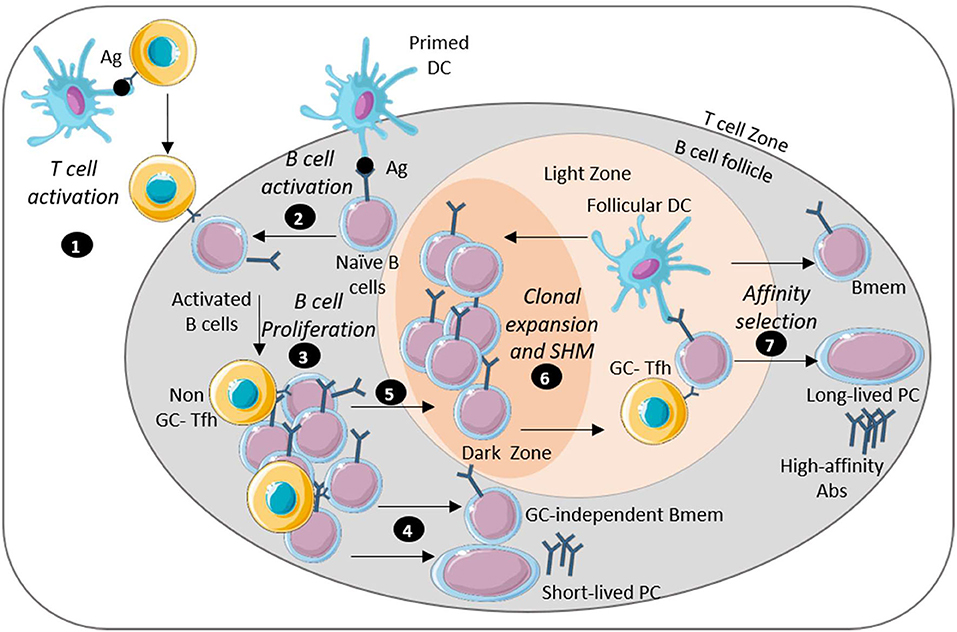
Figure 2. Long-lasting humoral immunity is mediated by long-lived plasma cells and memory B cells. Image depicts key steps in the generation of sustained humoral immunity: (1) APC-activated antigen-specific T cells migrate near the B cell follicle. (2) Naïve B cells encounter antigen through interaction with DCs and move by the T cell border. (3) B cells interact with the T cells differentiate into Tfh cells and migrate into the B cell follicle where activated B cells undergo proliferation. (4) Some extra-follicular B cells differentiate into antibody-secreting short lived plasma cells or GC-independent memory B cells. (5) Some B cells also enter the germinal center (GC). (6) The reaction in the GC starts with a rapid proliferation of the B cell leading to isotype-switch and BCR affinity maturation. (7) The B cells then undergo survival selection based on their affinity for antigen. Some post-GC B cells emerge as memory B cells or long-lived plasma cells that encode high affinity antibodies. This figure was prepared by modifying Servier Medical Art, licensed under a Creative Common Attribution 3.0 Generic License. http://smart.servier.com/.
In this article, we present an overview of what is known about the host immune responses to five enteric pathogens: C. rodentium, V. cholerae, Shigella spp., Salmonella spp., and C. Difficile, with an emphasis on humoral immunity and B cell memory. We discuss the humoral immune responses to those pathogens and the extent to which infection may induce a protective response, as well as gaps in our understanding of these processes. This information may be beneficial for understanding the course of disease.
Citrobacter rodentium
Citrobacter rodentium is a murine-specific Gram-negative extracellular bacterial pathogen that is related to the human enteropathogenic and enterohemorrhagic Escherichia coli (EPEC and EHEC, respectively). C. rodentium infection is a well characterized murine model of infectious colitis (10, 11). The disease induced by C. rodentium is dependent on the strain and the age of mice. In most strains, including Swiss Webster and C57BL/6, adult mice develop a mild self-limiting enteric disease. However, younger mice and strains such as C3H/HeOu, FVB, and C3H/HeN have a more severe disease characterized by diarrhea and severe colitis associated with weight loss, rectal prolapse, and death due to dehydration (12–14). Colonization and disease severity are also dependent on the intestinal microbial population. The variation in production of short-chain fatty acids (SCFA) determines the susceptibility to infection. A microbiome rich in butyrate-producing bacteria impairs growth of C. rodentium and protect against infection and disease (15, 16).
C. rodentium is transmitted through the fecal-oral route. After inoculation of mice, the bacteria first establish themselves in the caecal lymphoid patch and disrupt the commensal flora. Then the bacteria expand throughout the gastrointestinal tract to colonize the distal colon and the rectum (17–19). C. rodentium colonizes the gastrointestinal tract of mice via the mechanism of attaching and effacing (A/E) similarly to EPEC and EHEC (20). The pathogens adhere and bind intimately to epithelial cells through the adhesin intimin and induce pedestal-like protrusions at the site of attachment. The type III secretion system (TTSS) of the bacteria injects several virulence effectors into the host cells to interfere with signal transduction, modify actin cytoskeleton, and inhibit microtubule function. A/E bacteria also secrete virulence factors that promote junctional disruption of the epithelial cells and loss of intestinal barrier (21). These actions lead to damage of the brush border microvilli, intestinal inflammation and formation of plaques of cytoskeletal filaments underneath the adhering bacteria (19, 20).
Upon infection, C. rodentium activates host innate receptor TLR4 which then promotes recruitment of inflammatory cells including neutrophils. Neutrophil infiltration in the infected gut significantly contributes to the formation of crypt abscesses. TLR4-deficient mice have decreased bacterial colonization and tissue damage (22, 23). In contrast, infection with the EPEC and EHEC in humans is associated with a weaker inflammatory response despite the ability of the bacteria to cause disruption of epithelial barrier integrity. This can be explained by the bacteria ability to inhibit various MAP kinase pathways associated with NF-κB and induction of innate immune responses (24, 25). Although the initial host response to the infection contributes to C. rodentium pathology, this immune response also plays a protective role. The myeloid differentiation primary response protein (MyD88) is necessary for limiting bacterial colonization and promoting clearance. MyD88-dependent Toll-like receptor 2 and 4 (TLR2 and TLR4) signaling mediates production of pro-inflammatory cytokines such as Tumor necrosis factors (TNF), and recruitment of neutrophils, macrophage and innate lymphoid cells (ILCs) dependent on Interleukin 6 and 23 (IL-6 and IL-23) (26–28). At the early stage of the infection, Type 3 ILCs (ILC3) produce IL-22 which promotes maintenance of the epithelial barrier and control of bacterial burden by inducing production of antimicrobial peptides (29–31). This rapid production of cytokines and chemokines, and the recruitment of leukocytes required for the induction of a protective immune response is mediated through the activation of the intestinal G-protein couple receptors by the SCFAs.
The protective adaptive immune response against C. rodentium is mediated by CD4+ T cell and B cell responses (32, 33). Infection with C. rodentium induces a strong mucosal IgA response but also systemic IgM and IgG responses specific to several antigens such as the adhesin intimin, a TTSS effector protein (34, 35). However while serum IgG antibodies are required for protection against disease and bacterial clearance, IgA and IgM are dispensable (36). Patients with EHEC have also been shown to mount a strong IgA, IgG and IgM responses to E. coli O157:H7 intimin (37).
The neonatal Fc Receptor (FcRn) has been shown to mediate transport of IgG from the blood to the intestinal lumen to mediate defense against C. rodentium. Infection induced IgG antibodies, mostly IgG1, mediates control of the bacteria by activating complement and inducing engulfment of the bacteria by neutrophils (38). The FcRn is also involved in the transport of antigen out of the lumen to the local lymphoid organs where it initiates a systemic immune response (39–41). This protective antibody-mediated immune response against C. rodentium is predominantly regulated by T follicular helper (Tfh) cells. Following C. rodentium infection, Tfh cells expand in the mesenteric lymph nodes and spleen, and secrete IL-21 and IL-4 (41). Infection also promotes rapid germinal center responses (42). Other CD4+ T cells such as Th1 and Th17 cells are also involved in the host response against the bacteria through production of IL-17, IL-22, and IFN-γ. T cell activation promotes protective intestinal IgA and serum IgG responses (41, 43–46). The activation and recruitment of effector T cells also requires production of SCFAs. These metabolites promote intestinal antibody responses in mice infected with C. rodentium (47, 48). CD4+ T cells are also involved in the cellular response to EHEC in cattle. Following experimental colonization of cattle with EHEC 0157:H7, bacteria-specific T cells infiltrate the calves' rectal mucosa, secrete IFN-γ and demonstrate antigen-specific proliferation to TTSS effectors (49).
Vibrio cholerae
Cholera is an acute, severe diarrheal disease that remains an important global public health problem with up to 4 million cases and 143,000 associated deaths annually. The disease is caused by Vibrio cholerae, a highly motile Gram-negative facultative bacteria (50, 51).
V. cholerae is transmitted via the fecal-oral route following ingestion of contaminated water or food. After ingestion, most bacteria die because of the acidic environment of the stomach. The surviving bacteria adhere to and colonize the small intestine. The diarrhea is due the secretion of a very potent enterotoxin (cholera toxin- CT) which consists of two different subunits (A and B). Cholera toxin B binds to intestinal brush border cells leading to the endocytosis of subunit A which targets adenylyl cyclase and increases cyclic adenosine monophosphate (cAMP) levels. The increase in cAMP causes excessive secretion of chloride ions which results in the accumulation of water in the gut (52). Beside the toxin, the toxin co-regulated pilus (TCP) is also an important virulence factor. TCP is required for bacteria colonization. It provides a matrix that allows the bacteria to aggregate protecting them from host immune response (53).
V. cholerae is a non-invasive pathogen and doesn't induce a strong inflammatory response. Nonetheless, the bacteria penetrate the mucosa of the small intestine and attach to its surface to the small intestinal epithelial cells. Colonization of the intestine leads to structural changes to the epithelium including the widening of the intracellular spaces and alterations of the apical junctions (54). Those changes trigger rapid infiltration of inflammatory cells mainly neutrophils, macrophages, and dendritic cells. V. cholerae upregulates mucosal innate defense factors including TLR8, NLRP3 inflammasomes, NF-κB and MAPK signaling pathways (55–59). However, cholera toxin dampens innate immune response by inhibiting macrophage production of pro-inflammatory effectors such TNF, nitric oxide (NO) and IL-12 and by increasing secretion of the anti-inflammatory cytokine IL-10 (60). Also, V. cholerae has developed resistance against antimicrobial peptides by reducing their levels using its outer membrane vesicles (OMVs) and efflux pumps, and by inhibiting their binding (61–63).
Natural V. cholerae infection induces a protective adaptive immune response that is initiated by the activation of both B and T cells in the Peyer's patches of the intestinal mucosa and their subsequent migration into the mesenteric lymph nodes (64). Shortly after infection, bacterial antigen-specific lymphocytes are detected in the circulation. The lymphocytes express gut homing chemokine-receptors that allow homing in the intestinal mucosa where they lead mucosal immune responses. Infection induces mainly an IL-13 secreting Th2-mediated response, and some IFN-γ - secreting CD4+ and CD8+ T cell responses (65). V. cholerae infection also induces the development of circulating Tfh cells. These Tfh cells provide help to activated B cell leading to secretion class-switched antibodies by plasma cells and development of antigen-specific memory B cells (66).
The infection induced plasma cell response is characterized by the development of both systemic and gut derived V. cholerae-specific antibody secreting cells (67–69). Consequently, patients have high levels of anti-cholera toxin as well as anti-LPS IgG and sIgA antibodies. Anti-LPS sIgA in the fecal samples of patients correlates with protection against disease. However, the plasma cell response is short-lived and both systemic and mucosal antibodies significantly decrease within a few months post infection (70, 71). Natural infection by V. cholerae also induces memory B cell responses specific to various antigens including the cholera toxin, the subunit A of TCP, LPS, and the O-specific polysaccharides (OSP). These memory B cells induce a robust and rapid recall response upon reinfection and play a significant role in a longer-lasting protection against cholera (72–74). This long-lasting immune response is mediated specifically by LPS and OSP-specific IgG memory B cells. Anti-toxin IgG or anti-toxin and anti-LPS IgA memory B cells do not correlate with the long-term immunity against V. cholerae (75, 76).
The microbiome also plays an important role in protection and recovery against the infection. In children with cholera, clinical recovery correlate with a diverse and rich microbial community and a high concentration of SCFAs (77). SCFAs included acetate, propionate and butyrate inhibit the action of CT on the colon and prevent loss of fluid and electrolytes (78). The metabolites also facilitate anti-CT antibody responses by promoting dendritic cell functions and expression of plasma cell differentiation genes (48).
Salmonella enterica
Salmonellae are Gram-negative, flagellated, facultative anaerobic bacteria that cause disease in various hosts. The clinical disease in humans is typically caused by different serovars of Salmonella enterica subspecies (79). Typhoidal Salmonella serovars, including Typhi, Sendai, and Paratyphi A, B, or C, exclusively infect humans and cause a life-threatening enteric fever. Non-typhoidal Salmonella (NTS) serovars, such as S. Typhimurium, S Enteritidis, and S. Dublin cause a gastroenteritis which can manifest with acute diarrhea, abdominal pain, fever and vomiting. In some cases, largely in infants, older individuals and the immunocompromised, NTS can cause an invasive infection including bacteremia meningitis and septic arthritis (80).
Salmonellae are transmitted via the fecal-oral route through contaminated food or water, person-to-person contact, or contact with animals (81, 82). After ingestion, the bacteria survive the acidic environment of the stomach using a pH homeostasis mechanism. This mechanism is triggered by the low pH of the stomach. It allows the bacteria to maintain an internal pH above 5 and prevent severe acid stress (83). Then, the bacteria migrate into the small intestine and invade intestinal epithelial cells. Salmonellae preferably adhere to and translocate into the Microfold cells (M cells) of the small intestinal epithelium that overlie the Peyer's patches (4, 84). Invasion of the intestinal epithelial cells is mediated by the Salmonella Pathogenicity Island (SPI)-encoded virulence factors including two distinct TTSSs. The TTSSs transport their effectors in the host cells where they disrupt the actin and microtubule cytoskeletons and the cell membrane to form a membrane ruffle that facilitates engulfment of the bacteria. In the cytoplasm, the bacteria replicate in a vacuole, termed the Salmonella containing vacuole (SCV), that transcytoses to the basolateral membrane and allows invasion of the submucosa (85–88). The bacteria cells are subsequently phagocytosed by macrophages in the intestinal submucosa where they replicate within the SCVs (89). Migration of the infected macrophages facilitates dissemination of the bacteria, which in some cases allows for the establishment of a systemic infection (90). The infection to the pathogens is also modulated by the microbial community of the gut. A microbiome rich in SCFAs-producing bacteria inhibit motility, biofilm formation and gene expression of S. enterica (91).
The early host response to infection by NTS is triggered by the detection of TLRs. Activation of TLRs leads to production of proinflammatory cytokines such as TNF, IL-1β, and IL-6 which promote the production of anti-microbial peptides and the recruitment of neutrophils and macrophages in the mucosa (92–94). During NTS infection, neutrophils play an important role in the early host defense but also in the development of gastroenteritis. While neutrophils prevent bacterial replication and limit their dissemination (95), they also contribute to intestinal tissue damage and loss of barrier integrity, leading to increased inflammation and diarrhea (96).
The adaptive immune response to infection by NTS such S. Typhimirium starts with an early DC-mediated activation of pathogen-specific CD4 T cells limited to the Peyer's patches and the mesenteric lymph nodes (97, 98). This CD4 T cell response is mainly mediated by Tbet-expressing, IFNγ-secreting Th1 cells, and it is necessary for resistance to the infection and final clearance of the bacteria in the tissues (99, 100). However, in the early stage of the infection and in the absence of CD4 T cells, CD8 T cells and NK cells secrete IFNγ and are able to control the bacterial load (101, 102). Salmonella enterica serovars including Typhimirium have evolved mechanisms to limit DC function and evade T cell immunity. This evasion mechanism is mediated by Salmonella TTSS and effector proteins (103). Passive immunization against NTS has been shown to be protective, and IgA is not essential for this protection. Also, it has been shown that B cells are required for resistance against secondary infection. However, the mechanism of protection is independent of secreted antibodies (104–107).
During the course of infection, S. typhi induces an early extra-follicular, low affinity antibody response, consisting largely of non-class-switched IgM. The germinal center reaction and the production of high affinity antibodies are significantly delayed but this does not prevent clearance of the bacteria (108). Both typhoidal and NTS have been shown to impair plasma cell responses. An S. typhi adhesin protein, SiiE has been shown to reduce IgG-secreting cells in the bone marrow suggesting that the plasma cell response following Salmonella infection may not be long-lived. Also S. Typhi infection may abrogate established long-lived plasma cell response against previous infections (109). Very little is known about the Salmonella specific memory B cell response and the role it plays during infection. However, it has been shown that the outer membrane proteins of S. Typhi induces the development of IFNγ-secreting T follicular cells and IgM+ memory B cells (110).
Shigella
Shigella species are Gram-negative, non-spore forming, facultative anaerobic bacteria, including Shigella sonnei, S. boydii, S. flexneri, and S. dysenteriae that cause shigellosis. Shigellosis is an acute mucosal inflammation that leads to symptoms ranging from abdominal pain and mild diarrhea to severe dysentery, and sepsis (111). There are about 125 million cases of Shigella-associated diarrhea each year, resulting in more than 160 thousand deaths worldwide particularly in young children (112).
Shigella is transmitted via the fecal-oral route directly from one person to another or through contaminated water or food, and is highly infectious (113). After ingestion, the bacteria survive the acidic environment of the stomach and migrate to the colon and the rectum (114). The bacteria are non-motile and do not adhere to the colon but are able to invade and colonize the colonic epithelia (5). The bacteria break the intestinal epithelial barrier by invading the M cells that overlie the Peyer's patches (115, 116). They are translocated by M cells and are phagocytosed by macrophages. The bacteria escape the phagosome and quickly induce macrophage cell death by apoptosis. The bacteria are released and they propagate in the intestinal submucosa (117–119). Then, they enter the basolateral side of epithelial cells by micropinocytosis (120). The Shigella Type III secretion system (TTSS) secretes various effectors encoded in the Shigella virulence plasmid (121). Along with the TTSS, the invasion plasmid antigens (IpaB, C, and D) are the major virulence factors of Shigella. They are essential for bacterial invasion and intracellular survival, mediate the secretions of other effectors, and are dominant immunogenic antigens (122–124). Other TTSS effectors mediate polymerization of actin filaments and reorganization of the cytoskeleton by activating small GTPases. The restructuring of the cytoskeleton facilitates uptake of bacteria by epithelial cells and invasion of host cells (125–128). Some Shigella strains secrete toxins including the cytotoxic Stx1 and Stx2, and two other enterotoxins that may play a role in the early phases of Shigella-associated diarrhea (129–131).
The initial host immune response against Shigella is characterized by a severe and acute inflammation mediated by Caspase-1 activation of IL-1β and IL-18 following infection of macrophages and epithelial cells (117, 132). Invasion of Shigella can also lead to IL-8 secretion triggering massive neutrophil infiltration (133). The effect of IL-8 limits bacterial translocation but also worsens neutrophil-mediated intestinal epithelial damage (134). Besides inducing macrophage death, Shigella utilizes various mechanisms to counteract the host innate immune response. Through the TTSS and its effectors, Shigella dampens inflammatory responses to facilitate bacterial colonization by inhibiting ATP-dependent endogenous danger signaling, manipulating NF-κB pathways, and regulating expression of some pro-inflammatory cytokines and anti-microbial peptides (135–138). The administration of SCFAs may counteract this action in patients with shigellosis as the SCFAs modulate inflammation and increase the production of the anti-microbial peptides LL-37 (139). IFN-γ is crucial for the control of bacterial colonization and recovery from acute infection, however studies suggest that its levels are downregulated during shigellosis (140, 141).
Adaptive immunity, including antibody-mediated responses, have also been shown to play an important role in protection against Shigella infection. Following infection, the antibody-mediated immune responses are characterized by mucosal and systemic responses specific to Shigella lipopolysaccharide (LPS) antigens, the invasion plasmid antigens (Ipa), shiga toxins, and other TTSS effectors. These responses are Shigella serotype-specific and there is no cross-protection against infection from different strains (142, 143). Systemic Shigella LPS-specific IgG antibodies appear to be a good correlate of protection against shigellosis (144). Natural infection elicits the development of Shigella-specific IgA plasma cells detectable in the peripheral blood of patients. Contrary to immunization with live attenuated Shigella which elicits protective antigen-specific IgG and IgA Bmem cells, initial challenge with Shigella does not appear to induce development of antigen-specific IgA or IgG Bmem cells. However, there is an increase in the frequency of circulating LPS-specific IgA Bmem cells following a secondary Shigella challenge that negatively correlates with disease severity (145–148). Also, Shigella outer membrane protein A has been shown to enhance the germinal center reaction and antibody affinity maturation (149). Moreover, Shigella infection induces both primary and recall T cell expansion, predominantly of Th17 cells. Th1 cells are induced at a low frequency following reinfection, and Shigella-specific Th2 and CD8+ T cells are not detectable (150). T cells are essential for clearance of bacteria during primary infection, and Shigella-specific IL-17A secreting T cells are crucial for limiting bacterial growth during reinfection (150).
There is evidence of natural Shigella infections inducing serotype specific adaptive immune responses against recurrent infection. However, protection appears to only occur after multiple episodes, and the responses are slow and short-lived especially in young patients who have had less exposure to the infection (146, 151, 152). This can be attributed to the fact that Shigella manipulates the host adaptive immune response by targeting DCs, T and B cells. Shigella downregulates DC recruitment during infection by decreasing production of the chemokines and cytokines such as CCL20 (138). Shigella effectors also mediate apoptotic death of DCs (153). Moreover, studies have shown that Shigella invades T cells and impacts their function and dynamics in the lymph nodes by inhibiting chemokine-mediated migration (154, 155). Lastly, Shigella suppresses or evades the adaptive immune response by inducing B cell apoptosis both in vitro and in vivo during shigellosis through interaction between the TTSS effector IpaD and TLR2 (156).
Clostridioides difficile
Clostridioides difficile is a Gram-positive, spore-forming, obligate anaerobe and the main causative agent of hospital-acquired diarrhea (157, 158). The Centers for Disease Control and Prevention (CDC) has classified C. difficile as an urgent threat. The CDC reported that in 2017, there was an estimated 223,900 cases of C. difficile infection (CDI) in hospitalized patients resulting in 12,800 deaths and costing the healthcare system more than one billion dollars (159). Although healthcare-associated cases have been declining, there is a growing number of community-acquired cases which represent 41% of CDIs (160). The C. difficile-associated gastrointestinal disease (CDAD) ranges from mild diarrhea to pseudomembranous colitis, toxic megacolon, or sepsis. CDI is further complicated by a high and increasing frequency of recurrence; up to 35% of patients will relapse with often more severe disease (161).
Intestinal homeostasis, the diversity in the microbial community and the abundance of health-associated metabolites constitute an effective mechanism of resistance to C. difficile colonization. The disruption of the microbiome often due to prolonged use of broad-spectrum antibiotics is a major risk factor for infection. CDI is characterized by the depletion of essential bacteria that produce SCFAs and butyrate which normally alleviate the pathogenicity of infection (162, 163). C. difficile is transmitted via the fecal-oral route. After ingestion and upon loss of colonization resistance factors, the spores resist the acidity of the stomach, germinate upon exposure to bile acids in the small intestine and adhere and colonize the large intestine (164). CDAD is mainly caused by the two secreted toxins, toxin A (TcdA) and toxin B (TcdB). The toxins target the colonic epithelium by inducing cytoskeleton condensation leading to cell death and loss of intestinal barrier integrity (165). Besides TcdA and TcdB, C. difficile can produce other virulence factors that contribute to motility, adherence and colonization including binary toxin (CDT), flagella, adhesins such the surface layer proteins (SLPs), and hydrolytic enzymes (166).
The host innate immune response is thought to play both pathogenic and protective roles during CDI. Toxins induce the production of pro-inflammatory cytokines and other immune mediators such as IFN-γ, IL-1β, TNF, and leptin that may contribute to inflammation and intestinal damage (167–171). Conversely, the toxins and other virulence factors including the flagella and SLPs activate NOD-1 and TLR5. This in turn promotes recruitment of neutrophils which play an important role in early defense against CDAD. C. difficile antigens induce secretion of anti-inflammatory cytokines that mediate protective repair mechanisms (172). The damaging action of the toxins can be inhibited by SCFAs. Butyrate has been shown to stabilize the transcription factor HIF-1 which protects against colitis, to increase expression of epithelial tight junctions and prevent bacterial translocation (163). Also, anti-microbial peptides such as cathelicidin, NVB-302, surotomycin have been shown to be protective against C. difficile-mediated colitis by decreasing expression of pro-inflammatory cytokines, inhibiting toxin production, and facilitating bacterial killing of antibiotics (173–175).
The adaptive immune response does not play a direct role in the protection against CDI and disease (176). However, studies have demonstrated that an antibody-mediated immune response is important for limiting pathogenesis during the initial C. difficile infection, and in recurrent disease in patients. Animal models of CDI have been used to explore ways to induce protective immune responses through both passive immunization with toxin-specific antibodies and active immunization with C. difficile toxoids (177–180). Mucosal IgA responses may contribute to protection against CDI and associated disease, but it is dispensable. An initial CDI induces antigen-specific, mostly anti-toxin, IgA responses in patients and in animal models but the responses do not appear to correlate with protection against disease (181–185). High levels of fecal IgA are associated with protection against C. difficile colonization, whereas infection has been shown to significantly reduce mucosal IgA producing cells in patients (186, 187). In contrast, systemic toxin–specific IgG appears to be a better determinant of clinical outcome. IgG has been shown to protect against CDAD in both patients and animal models by decreasing gastrointestinal symptoms and mortality (188–190). C. difficile flagellar proteins (FliC and FliD), the surface layer proteins (SLPs), and the adhesins (Cwp66 and Cwp84) have also been shown to induce antibody responses in some patients. However, whether they play a role in the defense against recurrent disease has not been examined (191, 192).
Although the importance of antibody responses against CDI and disease has been well documented through assessment of antibody titers in patients, the cellular mechanisms required for protective B and T cell responses are not well defined. CDI is characterized by a poor initial T-cell response which can also be a marker for recurrence in CDI patients (193). Our group has recently shown that C. difficile infection in mice poorly activate CD4 T cells and induces a limited Tfh cell response (183).
Studies have shown that toxin A and B specific memory B cells may significantly contribute to protection against C. difficile associated disease. However, the high frequency of disease recurrence in C. difficile patients suggest that infection may not always induce a good memory response. Immunization with the inactive toxin induces toxin neutralizing antibodies and development of toxin-specific memory B cells, which confers protection against disease associated with CDI (194, 195, 197). Yet, primary C. difficile infection in mice induces a poor expansion of the memory B cell compartment (183). In CDI patients, anti-toxin memory B cells are detected but at a much lower frequency than in asymptomatic carriers (196).
Conclusions and Future Directions
Humoral immune responses make a critical but incompletely understood contribution to protection against bacterial enteric pathogens. Long-lived humoral immunity, required for sustained protection relies on the orchestrated activation of professional antigen-presenting cells, T helper and T follicular helper cells, and B cells capable of differentiation into long-lived plasma cells and memory B cells. Secretion of high affinity antibodies from long-lived plasma cells and those newly differentiated from memory B cells are essential for complement-mediated bacterial clearance, and for neutralization of bacterial toxins and other virulence factors (Figure 2). As we have discussed in this article and summarized in Table 1, there are many differences in the humoral immune responses to different bacterial enteric pathogens.
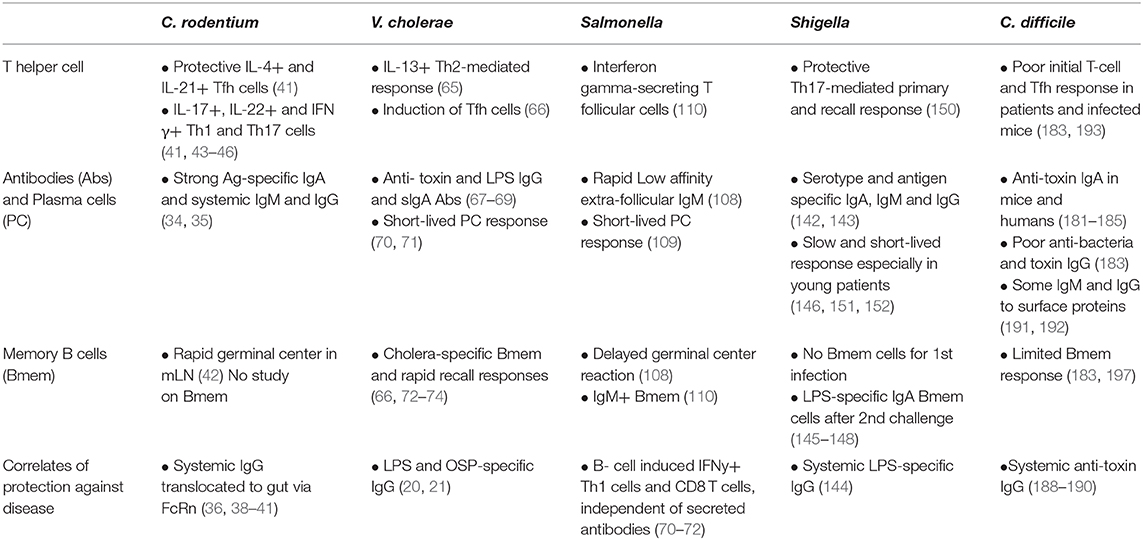
Table 1. Overview of humoral immune responses to enteric pathogens.
C. rodentium, a murine model for the attaching and effacing bacteria EPEC and EHEC, has been shown to induce a long-lasting immunity humoral immune response that is not always observed with other enteric pathogens. The plasma cell responses in V. cholerae and Salmonella infection are short-lived. The humoral response following C. difficile infection appears to be severely limited. Tfh cells and germinal center responses regulate humoral immune responses and are necessary for the induction of protective isotype-switched antibody as with C. rodentium. However, enteric pathogens such as C. difficile and Salmonella do not appear to induce a robust response. This limits not only the plasma cell response but also the Bmem compartment. Also, both pathogens mainly induce low affinity IgM responses.
Infections with C. rodentium, C. difficile, V. cholerae, and Shigella, promptly activate mucosal T and B cell responses leading to the production of IgA antibodies. Yet, secreted IgA appears to be dispensable for protection and bacteria-specific systemic IgG appears to be the best correlate of protection. This suggests that enteric pathogens or their antigens need to reach the extra-intestinal lymphoid organs to induce a systemic antibody response. Also, it suggests that there must be mechanisms by which IgG is translocated to the intestinal lumen to limit disease and bacterial dissemination. The neonatal Fc receptor (FcRn) mediates this IgG transport in C. rodentium (31), but little is known about the mechanism in the other pathogens.
As regards humoral immunity to enteric bacterial pathogens, future directions may include:
• More complete investigation of the Th/Tfh, Bmem cell, and plasma cell responses to infection in both animal models and human patients.
• Determining the mechanisms of antigen presentation in the gut, and how this influences activation and programming of B cell and Th/Tfh responses.
• Determining whether secreted toxins subvert humoral immunity and the mechanisms by which this is achieved.
• A systematic genomic approach to obtain in-depth immune profiles of the humoral response to infection and immunization.
• Single cell BCR sequencing to determine the plasma cell and memory B cell repertoires.
The application of newer technologies to the humoral response to enteric pathogens may be particularly important for infections with C. difficile and Salmonella that are characterized by persistent and/or recurrent disease. This may lead to a better understanding of the mechanisms by which memory B cell and plasma cell responses are limited, and could contribute to determining causes of persistent or recurrent disease.
Author Contributions
SA and ML wrote and edited the article. SA devised and produced the illustrations. All authors contributed to the article and approved the submitted version.
Funding
The ideas developed in this article were derived from work supported by NIH award AI134719 to ML.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Mrs. Gillian Lang (OUHSC) for assistance with editing. We also thank Ms. Nicole Giordano (OUHSC) for helpful comments on Salmonella enterica.
References
1. Bergstrom KS, Sham HP, Zarepour M, Vallance BA. Innate host responses to enteric bacterial pathogens: a balancing act between resistance and tolerance. Cell Microbiol. (2012) 14:475–84. doi: 10.1111/j.1462-5822.2012.01750.x
2. Janeway CA Jr, Travers P, Walport M, Shlomchik MJ. Principles of Innate and Adaptive Immunity. Immunobiology: The Immune System in Health and Disease. 5th ed. New York, NY: Garland Science (2001).
3. Gebert A, Rothkötter H-J, Pabst R. M cells in Peyer's patches of the intestine. Int Rev Cytol. (1996) 167:91–159. doi: 10.1016/S0074-7696(08)61346-7
4. Jones BD, Ghori N, Falkow S. Salmonella typhimurium initiates murine infection by penetrating and destroying the specialized epithelial M cells of the Peyer's patches. J Exp Med. (1994) 180:15–23. doi: 10.1084/jem.180.1.15
5. Labrec EH, Schneider H, Magnani TJ, Formal SB. Epithelial cell penetration as an essential step in the pathogenesis of bacillary dysentery. J Bacteriol. (1964) 88:1503–18. doi: 10.1128/JB.88.5.1503-1518.1964
6. Mabbott NA, Donaldson DS, Ohno H, Williams IR, Mahajan A. Microfold (M) cells: important immunosurveillance posts in the intestinal epithelium. Mucosal Immunol. (2013) 6:666–77. doi: 10.1038/mi.2013.30
7. Sansonetti PJ, Di Santo JP. Debugging how bacteria manipulate the immune response. Immunity. (2007) 26:149–61. doi: 10.1016/j.immuni.2007.02.004
8. Gutzeit C, Magri G, Cerutti A. Intestinal IgA production and its role in host-microbe interaction. Immunol Rev. (2014) 260:76–85. doi: 10.1111/imr.12189
9. Zimmermann K, Haas A, Oxenius A. Systemic antibody responses to gut microbes in health and disease. Gut Microbes. (2012) 3:42–7. doi: 10.4161/gmic.19344
10. Luperchio SA, Newman JV, Dangler CA, Schrenzel MD, Brenner DJ, Steigerwalt AG, et al. Citrobacter rodentium, the causative agent of transmissible murine colonic hyperplasia, exhibits clonality: synonymy of C. rodentium and mouse-pathogenic Escherichia coli. J Clin Microbiol. (2000) 38:4343–50. doi: 10.1128/JCM.38.12.4343-4350.2000
11. Petty NK, Bulgin R, Crepin VF, Cerdeño-Tárraga AM, Schroeder GN, Quail MA, et al. The Citrobacter rodentium genome sequence reveals convergent evolution with human pathogenic Escherichia coli. J Bacteriol. (2010) 192:525–38. doi: 10.1128/JB.01144-09
12. Papapietro O, Teatero S, Thanabalasuriar A, Yuki KE, Diez E, Zhu L, et al. R-spondin 2 signalling mediates susceptibility to fatal infectious diarrhoea. Nat Commun. (2013) 4:1–9. doi: 10.1038/ncomms2816
13. Barthold SW, Coleman G, Jacoby R, Livstone E, Jonas A. Transmissible murine colonic hyperplasia. Vet Pathol. (1978) 15:223–36. doi: 10.1177/030098587801500209
14. Vallance BA, Deng W, Jacobson K, Finlay BB. Host susceptibility to the attaching and effacing bacterial pathogen Citrobacter rodentium. Infect Immun. (2003) 71:3443–53. doi: 10.1128/IAI.71.6.3443-3453.2003
15. Osbelt L, Thiemann S, Smit N, Lesker TR, Schröter M, Gálvez EJ, et al. Variations in microbiota composition of laboratory mice influence Citrobacter rodentium infection via variable short-chain fatty acid production. PLoS Pathogens. (2020) 16:e1008448. doi: 10.1371/journal.ppat.1008448
16. Jiminez JA, Uwiera TC, Abbott DW, Uwiera RR, Inglis GD. Butyrate supplementation at high concentrations alters enteric bacterial communities and reduces intestinal inflammation in mice infected with Citrobacter rodentium. MSphere. (2017) 2:e00243-17. doi: 10.1128/mSphere.00243-17
17. Wiles S, Pickard KM, Peng K, MacDonald TT, Frankel G. In vivo bioluminescence imaging of the murine pathogen Citrobacter rodentium. Infect Immun. (2006) 74:5391–6. doi: 10.1128/IAI.00848-06
18. Wiles S, Clare S, Harker J, Huett A, Young D, Dougan G, et al. Organ specificity, colonization and clearance dynamics in vivo following oral challenges with the murine pathogen Citrobacter rodentium. Cell Microbiol. (2004) 6:963–72. doi: 10.1111/j.1462-5822.2004.00414.x
19. Luperchio SA, Schauer DB. Molecular pathogenesis of Citrobacter rodentium and transmissible murine colonic hyperplasia. Microbes Infect. (2001) 3:333–40. doi: 10.1016/S1286-4579(01)01387-9
20. Gaytán MO, Martínez-Santos VI, Soto E, González-Pedrajo B. Type three secretion system in attaching and effacing pathogens. Front Cell Infect Microbiol. (2016) 6:129. doi: 10.3389/fcimb.2016.00129
21. Garber JJ, Mallick EM, Scanlon KM, Turner JR, Donnenberg MS, Leong JM, et al. Attaching-and-effacing pathogens exploit junction regulatory activities of N-WASP and SNX9 to disrupt the intestinal barrier. Cell Mol Gastroenterol Hepatol. (2018) 5:273–88. doi: 10.1016/j.jcmgh.2017.11.015
22. Khan MA, Ma C, Knodler LA, Valdez Y, Rosenberger CM, Deng W, et al. Toll-like receptor 4 contributes to colitis development but not to host defense during Citrobacter rodentium infection in mice. Infect Immun. (2006) 74:2522–36. doi: 10.1128/IAI.74.5.2522-2536.2006
23. Higgins LM, Frankel G, Douce G, Dougan G, MacDonald TT. Citrobacter rodentium infection in mice elicits a mucosal Th1 cytokine response and lesions similar to those in murine inflammatory bowel disease. Infect Immun. (1999) 67:3031–9. doi: 10.1128/IAI.67.6.3031-3039.1999
24. Ruchaud-Sparagano MH, Maresca M, Kenny B. Enteropathogenic Escherichia coli (EPEC) inactivate innate immune responses prior to compromising epithelial barrier function. Cell Microbiol. (2007) 9:1909–21. doi: 10.1111/j.1462-5822.2007.00923.x
25. Vanaja SK, Jandhyala DM, Mallick EM, Leong JM, Balasubramanian S. Enterohemorrhagic and other Shigatoxin-Producing Escherichia coli. San Diego, CA: Elsevier (2013).
26. Lebeis SL, Bommarius B, Parkos CA, Sherman MA, Kalman D. TLR signaling mediated by MyD88 is required for a protective innate immune response by neutrophils to Citrobacter rodentium. J Immunol. (2007) 179:566–77. doi: 10.4049/jimmunol.179.1.566
27. Kum WW, Lo BC, Deng W, Ziltener HJ, Finlay BB. Impaired innate immune response and enhanced pathology during Citrobacter rodentium infection in mice lacking functional P-selectin. Cell Microbiol. (2010) 12:1250–71. doi: 10.1111/j.1462-5822.2010.01466.x
28. Gibson D, Ma C, Bergstrom K, Huang J, Man C, Vallance B. MyD88 signalling plays a critical role in host defence by controlling pathogen burden and promoting epithelial cell homeostasis during Citrobacter rodentium-induced colitis. Cell Microbiol. (2008) 10:618–31. doi: 10.1111/j.1462-5822.2007.01071.x
29. Giacomin PR, Moy RH, Noti M, Osborne LC, Siracusa MC, Alenghat T, et al. Epithelial-intrinsic IKKα expression regulates group 3 innate lymphoid cell responses and antibacterial immunity. J Exp Med. (2015) 212:1513–28. doi: 10.1084/jem.20141831
30. Guo X, Qiu J, Tu T, Yang X, Deng L, Anders RA, et al. Induction of innate lymphoid cell-derived interleukin-22 by the transcription factor STAT3 mediates protection against intestinal infection. Immunity. (2014) 40:25–39. doi: 10.1016/j.immuni.2013.10.021
31. Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. (2008) 14:282–9. doi: 10.1038/nm1720
32. Simmons CP, Clare S, Ghaem-Maghami M, Uren TK, Rankin J, Huett A, et al. Central role for B lymphocytes and CD4+ T cells in immunity to infection by the attaching and effacing pathogen Citrobacter rodentium. Infect Immun. (2003) 71:5077–86. doi: 10.1128/IAI.71.9.5077-5086.2003
33. Vallance BA, Deng W, Knodler LA, Finlay BB. Mice lacking T and B lymphocytes develop transient colitis and crypt hyperplasia yet suffer impaired bacterial clearance during Citrobacter rodentium infection. Infect Immun. (2002) 70:2070–81. doi: 10.1128/IAI.70.4.2070-2081.2002
34. Frankel G, Phillips AD, Novakova M, Field H, Candy D, Schauer DB, et al. Intimin from enteropathogenic Escherichia coli restores murine virulence to a Citrobacter rodentium eaeA mutant: induction of an immunoglobulin A response to intimin and EspB. Infect Immun. (1996) 64:5315–25. doi: 10.1128/IAI.64.12.5315-5325.1996
35. Ghaem-Maghami M, Simmons CP, Daniell S, Pizza M, Lewis D, Frankel G, et al. Intimin-specific immune responses prevent bacterial colonization by the attaching-effacing pathogen Citrobacter rodentium. Infect Immun. (2001) 69:5597–605. doi: 10.1128/IAI.69.9.5597-5605.2001
36. Maaser C, Housley MP, Iimura M, Smith JR, Vallance BA, Finlay BB, et al. Clearance of Citrobacter rodentium requires B cells but not secretory immunoglobulin A (IgA) or IgM antibodies. Infect Immun. (2004) 72:3315–24. doi: 10.1128/IAI.72.6.3315-3324.2004
37. Karpman D, Békássy ZD, Sjögren A-C, Dubois MS, Karmali MA, Mascarenhas M, et al. Antibodies to intimin and Escherichia coli secreted proteins A and B in patients with enterohemorrhagic Escherichia coli infections. Pediatr Nephrol. (2002) 17:201–11. doi: 10.1007/s00467-001-0792-z
38. Kamada N, Sakamoto K, Seo S-U, Zeng MY, Kim Y-G, Cascalho M, et al. Humoral immunity in the gut selectively targets phenotypically virulent attaching-and-effacing bacteria for intraluminal elimination. Cell Host Microbe. (2015) 17:617–27. doi: 10.1016/j.chom.2015.04.001
39. Yoshida M, Kobayashi K, Kuo TT, Bry L, Glickman JN, Claypool SM, et al. Neonatal Fc receptor for IgG regulates mucosal immune responses to luminal bacteria. J Clin Invest. (2006) 116:2142–51. doi: 10.1172/JCI27821
40. Belzer C, Liu Q, Carroll M, Bry L. The role of specific IgG and complement in combating aprimary mucosal infection of the gut epithelium. Eur J Microbiol Immunol. (2011) 1:311–8. doi: 10.1556/EuJMI.1.2011.4.7
41. Bai X, Chi X, Qiao Q, Xie S, Wan S, Ni L, et al. T follicular helper cells regulate humoral response for host protection against intestinal Citrobacter rodentium infection. J Immunol. 2020. doi: 10.4049/jimmunol.2000046
42. Clare S, John V, Walker AW, Hill JL, Abreu-Goodger C, Hale C, et al. Enhanced susceptibility to Citrobacter rodentium infection in microRNA-155-deficient mice. Infect Immun. (2013) 81:723–32. doi: 10.1128/IAI.00969-12
43. Huang X, Sun M, Chen F, Xiao Y, Chen L, Yao S, et al. IL-17 promotes intestinal IgA response to intestinal infection but does not affect memory B cell development. Am Assoc Immnol. (2017) 18:8.
44. Spahn T, Ross M, Von Eiff C, Maaser C, Spieker T, Kannengiesser K, et al. CD4+ T cells transfer resistance against Citrobacter rodentium-induced infectious colitis by induction of Th 1 immunity. Scand J Immunol. (2008) 67:238–44. doi: 10.1111/j.1365-3083.2007.02063.x
45. Simmons CP, Goncalves NS, Ghaem-Maghami M, Bajaj-Elliott M, Clare S, Neves B, et al. Impaired resistance and enhanced pathology during infection with a noninvasive, attaching-effacing enteric bacterial pathogen, Citrobacter rodentium, in mice lacking IL-12 or IFN-γ. J Immunol. (2002) 168:1804–12. doi: 10.4049/jimmunol.168.4.1804
46. Linterman MA, Denton AE, Divekar DP, Zvetkova I, Kane L, Ferreira C, et al. CD28 expression is required after T cell priming for helper T cell responses and protective immunity to infection. Elife. (2014) 3:e03180. doi: 10.7554/eLife.03180
47. Kim MH, Kang SG, Park JH, Yanagisawa M, Kim CH. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology. (2013) 145:396–406.e10. doi: 10.1053/j.gastro.2013.04.056
48. Yang W, Xiao Y, Huang X, Chen F, Sun M, Bilotta AJ, et al. Microbiota metabolite short-chain fatty acids facilitate mucosal adjuvant activity of cholera toxin through GPR43. J Immunol. (2019) 203:282–92. doi: 10.4049/jimmunol.1801068
49. Corbishley A, Ahmad NI, Hughes K, Hutchings MR, McAteer SP, Connelley TK, et al. Strain-dependent cellular immune responses in cattle following Escherichia coli O157: H7 colonization. Infect Immun. (2014) 82:5117–31. doi: 10.1128/IAI.02462-14
50. Ali M, Nelson AR, Lopez AL, Sack DA. Updated global burden of cholera in endemic countries. PLoS Neglect Trop Dis. (2015) 9:e0003832. doi: 10.1371/journal.pntd.0003832
51. Azman AS, Rudolph KE, Cummings DA, Lessler J. The incubation period of cholera: a systematic review. J Infect. (2013) 66:432–8. doi: 10.1016/j.jinf.2012.11.013
52. Finkelstein RA. Cholera, Vibrio cholerae O1 and O139, and other pathogenic vibrios. In: Baron S, editor. Medical Microbiology. Vol. 66. 4th ed. Galveston, TX: University of Texas Medical Branch at Galveston (1996). p. 432–8.
53. Taylor RK, Miller VL, Furlong DB, Mekalanos JJ. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc Natl Acad Sci USA. (1987) 84:2833–7. doi: 10.1073/pnas.84.9.2833
54. Mathan MM, Chandy G, Mathan V. Ultrastructural changes in the upper small intestinal mucosa in patients with cholera. Gastroenterology. (1995) 109:422–30. doi: 10.1016/0016-5085(95)90329-1
55. Flach C-F, Qadri F, Bhuiyan TR, Alam NH, Jennische E, Lönnroth I, et al. Broad up-regulation of innate defense factors during acute cholera. Infect Immun. (2007) 75:2343–50. doi: 10.1128/IAI.01900-06
56. Pulimood AB, Ramakrishna BS, Rita AB, Srinivasan P, Mohan V, Gupta S, et al. Early activation of mucosal dendritic cells and macrophages in acute Campylobacter colitis and cholera: an in vivo study. J Gastroenterol Hepatol. (2008) 23:752–8. doi: 10.1111/j.1440-1746.2008.05325.x
57. Qadri F, Bhuiyan T, Dutta K, Raqib R, Alam M, Alam N, et al. Acute dehydrating disease caused by Vibrio cholerae serogroups O1 and O139 induce increases in innate cells and inflammatory mediators at the mucosal surface of the gut. Gut. (2004) 53:62–9. doi: 10.1136/gut.53.1.62
58. Ellis CN, LaRocque RC, Uddin T, Krastins B, Mayo-Smith LM, Sarracino D, et al. Comparative proteomic analysis reveals activation of mucosal innate immune signaling pathways during cholera. Infect Immun. (2015) 83:1089–103. doi: 10.1128/IAI.02765-14
59. Bourque DL, Bhuiyan TR, Genereux DP, Rashu R, Ellis CN, Chowdhury F, et al. Analysis of the human mucosal response to cholera reveals sustained activation of innate immune signaling pathways. Infect Immun. (2018) 86:e00594-17. doi: 10.1128/IAI.00594-17
60. Satchell KJF. Activation and suppression of the proinflammatory immune response by Vibrio cholerae toxins. Microbes Infect. (2003) 5:1241–7. doi: 10.1016/j.micinf.2003.08.007
61. Saul-McBeth J, Matson JS. A periplasmic antimicrobial peptide-binding protein is required for stress survival in Vibrio cholerae. Front Microbiol. (2019) 10:161. doi: 10.3389/fmicb.2019.00161
62. Nizet V. Antimicrobial peptide resistance mechanisms of human bacterial pathogens. Curr Issues Mol Biol. (2006) 8:11.
63. McBroom AJ, Kuehn MJ. Release of outer membrane vesicles by Gram-negative bacteria is a novel envelope stress response. Mol Microbiol. (2007) 63:545–58. doi: 10.1111/j.1365-2958.2006.05522.x
64. Qadri F, Wennerås C, Albert MJ, Hossain J, Mannoor K, Begum YA, et al. Comparison of immune responses in patients infected with Vibrio cholerae O139 and O1. Infect Immun. (1997) 65:3571–6. doi: 10.1128/IAI.65.9.3571-3576.1997
65. Bhuiyan TR, Lundin SB, Khan AI, Lundgren A, Harris JB, Calderwood SB, et al. Cholera caused by Vibrio cholerae O1 induces T-cell responses in the circulation. Infect Immun. (2009) 77:1888–93. doi: 10.1128/IAI.01101-08
66. Rashu R, Bhuiyan TR, Hoq MR, Hossain L, Paul A, Khan AI, et al. Cognate T and B cell interaction and association of follicular helper T cells with B cell responses in Vibrio cholerae O1 infected Bangladeshi adults. Microbes Infect. (2019) 21:176–83. doi: 10.1016/j.micinf.2018.12.002
67. Rahman A, Rashu R, Bhuiyan TR, Chowdhury F, Khan AI, Islam K, et al. Antibody-secreting cell responses after Vibrio cholerae O1 infection and oral cholera vaccination in adults in Bangladesh. Clin Vaccine Immunol. (2013) 20:1592–8. doi: 10.1128/CVI.00347-13
68. Qadri F, Ryan ET, Faruque A, Ahmed F, Khan AI, Islam MM, et al. Antigen-specific immunoglobulin A antibodies secreted from circulating B cells are an effective marker for recent local immune responses in patients with cholera: comparison to antibody-secreting cell responses and other immunological markers. Infect Immun. (2003) 71:4808–14. doi: 10.1128/IAI.71.8.4808-4814.2003
69. Leung DT, Rahman MA, Mohasin M, Riyadh MA, Patel SM, Alam MM, et al. Comparison of memory B cell, antibody-secreting cell, and plasma antibody responses in young children, older children, and adults with infection caused by Vibrio cholerae O1 El Tor Ogawa in Bangladesh. Clin Vaccine Immunol. (2011) 18:1317–25. doi: 10.1128/CVI.05124-11
70. Alam MM, Riyadh MA, Fatema K, Rahman MA, Akhtar N, Ahmed T, et al. Antigen-specific memory B-cell responses in Bangladeshi adults after one-or two-dose oral killed cholera vaccination and comparison with responses in patients with naturally acquired cholera. Clin Vaccine Immunol. (2011) 18:844–50. doi: 10.1128/CVI.00562-10
71. Nelson EJ, Harris JB, Morris JG, Calderwood SB, Camilli A. Cholera transmission: the host, pathogen and bacteriophage dynamic. Nat Rev Microbiol. (2009) 7:693–702. doi: 10.1038/nrmicro2204
72. Harris AM, Bhuiyan MS, Chowdhury F, Khan AI, Hossain A, Kendall EA, et al. Antigen-specific memory B-cell responses to Vibrio cholerae O1 infection in Bangladesh. Infect Immun. (2009) 77:3850–6. doi: 10.1128/IAI.00369-09
73. Jayasekera CR, Harris JB, Bhuiyan S, Chowdhury F, Khan AI, Faruque AS, et al. Cholera toxin-specific memory B cell responses are included in patients with dehydrating diarrhea caused by Vibrio cholerae O1. J Infect Dis. (2008) 198:1055–61. doi: 10.1086/591500
74. Aktar A, Rahman MA, Afrin S, Faruk MO, Uddin T, Akter A, et al. O-specific polysaccharide-specific memory B cell responses in young children, older children, and adults infected with Vibrio cholerae O1 Ogawa in Bangladesh. Clin Vaccine Immunol. (2016) 23:427–35. doi: 10.1128/CVI.00647-15
75. Patel SM, Rahman MA, Mohasin M, Riyadh MA, Leung DT, Alam MM, et al. Memory B cell responses to Vibrio cholerae O1 lipopolysaccharide are associated with protection against infection from household contacts of patients with cholera in Bangladesh. Clin Vaccine Immunol. (2012) 19:842–8. doi: 10.1128/CVI.00037-12
76. Aktar A, Rahman MA, Afrin S, Akter A, Uddin T, Yasmin T, et al. Plasma and memory B cell responses targeting O-specific polysaccharide (OSP) are associated with protection against Vibrio cholerae O1 infection among household contacts of cholera patients in Bangladesh. PLoS Neglect Trop Dis. (2018) 12:e0006399. doi: 10.1371/journal.pntd.0006399
77. Monira S, Hoq MM, Chowdhury A, Suau A, Magne F, Endtz H, et al. Short-chain fatty acids and commensal microbiota in the faeces of severely malnourished children with cholera rehydrated with three different carbohydrates. Eur J Clin Nutr. (2010) 64:1116–24. doi: 10.1038/ejcn.2010.123
78. Rabbani G, Albert MJ, Rahman H, Chowdhury AK. Short-chain fatty acids inhibit fluid and electrolyte loss induced by cholera toxin in proximal colon of rabbit in vivo. Digest Dis Sci. (1999) 44:1547–53. doi: 10.1023/A:1026650624193
79. D'Aoust J-Y, Maurer J. Salmonella species. In: Food Microbiology: Fundamentals and Frontiers, 3rd ed. American Society of Microbiology (2007). p. 187–236. doi: 10.1128/9781555815912.ch10
80. Gal-Mor O, Boyle EC, Grassl GA. Same species, different diseases: how and why typhoidal and non-typhoidal Salmonella enterica serovars differ. Front Microbiol. (2014) 5:391. doi: 10.3389/fmicb.2014.00391
81. Heyman D. Control of Communicable Diseases Manual. 18th ed. Washington, DC: American Public Health Association.
82. Gopinath S, Carden S, Monack D. Shedding light on Salmonella carriers. Trends Microbiol. (2012) 20:320–7. doi: 10.1016/j.tim.2012.04.004
83. Foster JW, Hall HK. Inducible pH homeostasis and the acid tolerance response of Salmonella typhimurium. J Bacteriol. (1991) 173:5129–35. doi: 10.1128/JB.173.16.5129-5135.1991
84. Takeuchi A. Electron microscope studies of experimental Salmonella infection. I. Penetration into the intestinal epithelium by Salmonella typhimurium. Am J Pathol. (1967) 50:109.
85. Ohl ME, Miller SI. Salmonella: a model for bacterial pathogenesis. Annu Rev Med. (2001) 52:259–74. doi: 10.1146/annurev.med.52.1.259
86. Finlay BB, Ruschkowski S, Dedhar S. Cytoskeletal rearrangements accompanying Salmonella entry into epithelial cells. J Cell Sci. (1991) 99:283–96.
87. Francis CL, Ryan TA, Jones BD, Smith SJ, Falkow S. Ruffles induced by Salmonella and other stimuli direct macropinocytosis of bacteria. Nature. (1993) 364:639–42. doi: 10.1038/364639a0
88. Garcia-del Portillo F, Zwick MB, Leung KY, Finlay BB. Salmonella induces the formation of filamentous structures containing lysosomal membrane glycoproteins in epithelial cells. Proc Natl Acad Sci USA. (1993) 90:10544–8. doi: 10.1073/pnas.90.22.10544
89. Alpuche-Aranda CM, Racoosin EL, Swanson JA, Miller SI. Salmonella stimulate macrophage macropinocytosis and persist within spacious phagosomes. J Exp Med. (1994) 179:601–8. doi: 10.1084/jem.179.2.601
90. Worley MJ, Nieman GS, Geddes K, Heffron F. Salmonella typhimurium disseminates within its host by manipulating the motility of infected cells. Proc Natl Acad Sci USA. (2006) 103:17915–20. doi: 10.1073/pnas.0604054103
91. Lamas A, Regal P, Vázquez B, Cepeda A, Franco CM. Short chain fatty acids commonly produced by gut microbiota influence Salmonella enterica motility, biofilm formation, and gene expression. Antibiotics. (2019) 8:265. doi: 10.3390/antibiotics8040265
92. Galyov EE, Wood MW, Rosqvist R, Mullan PB, Watson PR, Hedges S, et al. A secreted effector protein of Salmonella dublin is translocated into eukaryotic cells and mediates inflammation and fluid secretion in infected ileal mucosa. Mol Microbiol. (1997) 25:903–12. doi: 10.1111/j.1365-2958.1997.mmi525.x
93. Rydström A, Wick MJ. Monocyte recruitment, activation, and function in the gut-associated lymphoid tissue during oral Salmonella infection. J Immunol. (2007) 178:5789–801. doi: 10.4049/jimmunol.178.9.5789
94. Srinivasan A, Salazar-Gonzalez R-M, Jarcho M, Sandau MM, Lefrancois L, McSorley SJ. Innate immune activation of CD4 T cells in Salmonella-infected mice is dependent on IL-18. J Immunol. (2007) 178:6342–9. doi: 10.4049/jimmunol.178.10.6342
95. Conlan JW. Neutrophils prevent extracellular colonization of the liver microvasculature by Salmonella typhimurium. Infect Immun. (1996) 64:1043–7. doi: 10.1128/IAI.64.3.1043-1047.1996
96. Tsolis RM, Adams LG, Ficht TA, Bäumler AJ. Contribution of Salmonella typhimuriumvirulence factors to diarrheal disease in calves. Infect Immun. (1999) 67:4879–85. doi: 10.1128/IAI.67.9.4879-4885.1999
97. Salazar-Gonzalez RM, Niess JH, Zammit DJ, Ravindran R, Srinivasan A, Maxwell JR, et al. CCR6-mediated dendritic cell activation of pathogen-specific T cells in Peyer's patches. Immunity. (2006) 24:623–32. doi: 10.1016/j.immuni.2006.02.015
98. McSorley SJ, Asch S, Costalonga M, Reinhardt RL, Jenkins MK. Tracking Salmonella-specific CD4 T cells in vivo reveals a local mucosal response to a disseminated infection. Immunity. (2002) 16:365–77. doi: 10.1016/S1074-7613(02)00289-3
99. Hess J, Ladel C, Miko D, Kaufmann S. Salmonella typhimurium aroA-infection in gene-targeted immunodeficient mice: major role of CD4+ TCR-alpha beta cells and IFN-gamma in bacterial clearance independent of intracellular location. J Immunol. (1996) 156:3321–6.
100. Ravindran R, Foley J, Stoklasek T, Glimcher LH, McSorley SJ. Expression of T-bet by CD4 T cells is essential for resistance to Salmonella infection. J Immunol. (2005) 175:4603–10. doi: 10.4049/jimmunol.175.7.4603
101. Lee S-J, Dunmire S, McSorley SJ. MHC class-I-restricted CD8 T cells play a protective role during primary Salmonella infection. Immunol Lett. (2012) 148:138–43. doi: 10.1016/j.imlet.2012.10.009
102. Kupz A, Bedoui S, Strugnell RA. Cellular requirements for systemic control of Salmonella enterica serovar Typhimurium infections in mice. Infect Immun. (2014) 82:4997–5004. doi: 10.1128/IAI.02192-14
103. Tobar JA, Carreno LJ, Bueno SM, González PA, Mora JE, Quezada SA, et al. Virulent Salmonella enterica serovar typhimurium evades adaptive immunity by preventing dendritic cells from activating T cells. Infect Immun. (2006) 74:6438–48. doi: 10.1128/IAI.00063-06
104. Mastroeni P, Simmons C, Fowler R, Hormaeche C, Dougan G. Igh-6–/–(B-cell-deficient) mice fail to mount solid acquired resistance to oral challenge with virulent Salmonella enterica serovar typhimurium and show impaired Th1 T-cell responses to Salmonella antigens. Infect Immun. (2000) 68:46–53. doi: 10.1128/IAI.68.1.46-53.2000
105. McSorley SJ, Jenkins MK. Antibody is required for protection against virulent but not attenuated Salmonella entericaserovar typhimurium. Infect Immun. (2000) 68:3344–8. doi: 10.1128/IAI.68.6.3344-3348.2000
106. Mittrücker H-W, Raupach B, Köhler A, Kaufmann SH. Cutting edge: role of B lymphocytes in protective immunity against Salmonella typhimurium infection. J Immunol. (2000) 164:1648–52. doi: 10.4049/jimmunol.164.4.1648
107. MacLennan CA, Gondwe EN, Msefula CL, Kingsley RA, Thomson NR, White SA, et al. The neglected role of antibody in protection against bacteremia caused by nontyphoidal strains of Salmonella in African children. J Clin Invest. (2008) 118:1553–62. doi: 10.1172/JCI33998
108. Cunningham AF, Gaspal F, Serre K, Mohr E, Henderson IR, Scott-Tucker A, et al. Salmonella induces a switched antibody response without germinal centers that impedes the extracellular spread of infection. J Immunol. (2007) 178:6200–7. doi: 10.4049/jimmunol.178.10.6200
109. Männe C, Takaya A, Yamasaki Y, Mursell M, Hojyo S, Wu T-Y, et al. Salmonella SiiE prevents an efficient humoral immune memory by interfering with IgG+ plasma cell persistence in the bone marrow. Proc Natl Acad Sci USA. (2019) 116:7425–30. doi: 10.1073/pnas.1818242116
110. Perez-Shibayama C, Gil-Cruz C, Pastelin-Palacios R, Cervantes-Barragan L, Hisaki E, Chai Q, et al. IFN-γ-producing CD4+ T cells promote generation of protective germinal center–derived IgM+ B cell memory against Salmonella Typhi. J Immunol. (2014) 192:5192–200. doi: 10.4049/jimmunol.1302526
111. Maurelli AT, Lampel KA. Shigella. In : Hui YH, Pierson MD, Gorman JR, editors. Foodborne Disease Handbook: Vol. I. Second Edition. New York, NY: Marcel Dekker, Inc. (2001). p. 323-43.
112. Bardhan P, Faruque A, Naheed A, Sack DA. Decreasing shigellosis-related deaths without Shigella spp.–specific interventions, Asia. Emerg Infect Dis. (2010) 16:1718. doi: 10.3201/eid1611.090934
113. DuPont HL, Levine MM, Hornick RB, Formal SB. Inoculum size in shigellosis and implications for expected mode of transmission. J Infect Dis. (1989) 159:1126–8. doi: 10.1093/infdis/159.6.1126
114. Gorden J, Small P. Acid resistance in enteric bacteria. Infect Immun. (1993) 61:364–7. doi: 10.1128/IAI.61.1.364-367.1993
115. Sansonetti PJ, Arondel J, Cantey JR, Prévost M-C, Huerre M. Infection of rabbit Peyer's patches by Shigella flexneri: effect of adhesive or invasive bacterial phenotypes on follicle-associated epithelium. Infect Immun. (1996) 64:2752–64. doi: 10.1128/IAI.64.7.2752-2764.1996
116. Wassef JS, Keren DF, Mailloux JL. Role of M cells in initial antigen uptake and in ulcer formation in the rabbit intestinal loop model of shigellosis. Infect Immun. (1989) 57:858–63. doi: 10.1128/IAI.57.3.858-863.1989
117. Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. (1992) 358:167–9. doi: 10.1038/358167a0
118. Zychlinsky A, Thirumalai K, Arondel J, Cantey JR, Aliprantis AO, Sansonetti PJ. In vivo apoptosis in Shigella flexneri infections. Infect Immun. (1996) 64:5357–65. doi: 10.1128/IAI.64.12.5357-5365.1996
119. Islam D, Veress B, Bardhan PK, Lindberg AA, Christensson B. In situ characterization of inflammatory responses in the rectal mucosae of patients with shigellosis. Infect Immun. (1997) 65:739–49. doi: 10.1128/IAI.65.2.739-749.1997
120. Cossart P, Sansonetti PJ. Bacterial invasion: the paradigms of enteroinvasive pathogens. Science. (2004) 304:242–8. doi: 10.1126/science.1090124
121. Van Nhieu GT, Enninga J, Sansonetti P, Grompone G. Tyrosine kinase signaling and type III effectors orchestrating Shigella invasion. Curr Opin Microbiol. (2005) 8:16–20. doi: 10.1016/j.mib.2004.12.006
122. Buysse JM, Stover CK, Oaks EV, Venkatesan M, Kopecko DJ. Molecular cloning of invasion plasmid antigen (ipa) genes from Shigella flexneri: analysis of ipa gene products and genetic mapping. J Bacteriol. (1987) 169:2561–9. doi: 10.1128/JB.169.6.2561-2569.1987
123. Blocker A, Gounon P, Larquet E, Niebuhr K, Cabiaux V, Parsot C, et al. The tripartite type III secreton of Shigella flexneri inserts IpaB and IpaC into host membranes. J Cell Biol. (1999) 147:683–93. doi: 10.1083/jcb.147.3.683
124. Oaks EV, Hale T, Formal S. Serum immune response to Shigella protein antigens in rhesus monkeys and humans infected with Shigella spp. Infect Immun. (1986) 53:57–63. doi: 10.1128/IAI.53.1.57-63.1986
125. Bourdet-Sicard R, Rüdiger M, Jockusch BM, Gounon P, Sansonetti PJ, Van Nhieu GT. Binding of the Shigella protein IpaA to vinculin induces F-actin depolymerization. EMBO J. (1999) 18:5853–62. doi: 10.1093/emboj/18.21.5853
126. Bernardini ML, Mounier J, d'Hauteville H, Coquis-Rondon M, Sansonetti PJ. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra-and intercellular spread through interaction with F-actin. Proc Natl Acad Sci USA. (1989) 86:3867–71. doi: 10.1073/pnas.86.10.3867
127. Bougnères L, Girardin SE, Weed SA, Karginov AV, Olivo-Marin J-C, Parsons JT, et al. Cortactin and Crk cooperate to trigger actin polymerization during Shigella invasion of epithelial cells. J Cell Biol. (2004) 166:225–35. doi: 10.1083/jcb.200402073
128. Van Nhieu GT, Caron E, Hall A, Sansonetti PJ. IpaC induces actin polymerization and filopodia formation during Shigella entry into epithelial cells. EMBO J. (1999) 18:3249–62. doi: 10.1093/emboj/18.12.3249
129. Tesh VL, Samoel JE, Perera LP, Sharefkin JB, O'Brien AD. Evaluation of the role of Shiga and Shiga-like toxins in mediating direct damage to human vascular endothelial cells. J Infect Dis. (1991) 164:344–52. doi: 10.1093/infdis/164.2.344
130. Bergan J, Lingelem ABD, Simm R, Skotland T, Sandvig K. Shiga toxins. Toxicon. (2012) 60:1085–107. doi: 10.1016/j.toxicon.2012.07.016
131. Fasano A, Noriega FR, Maneval DR, Chanasongcram S, Russell R, Guandalini S, et al. Shigella enterotoxin 1: an enterotoxin of Shigella flexneri 2a active in rabbit small intestine in vivo and in vitro. J Clin Invest. (1995) 95:2853–61. doi: 10.1172/JCI117991
132. Sansonetti PJ, Phalipon A, Arondel J, Thirumalai K, Banerjee S, Akira S, et al. Caspase-1 activation of IL-1β and IL-18 are essential for Shigella flexneri–induced inflammation. Immunity. (2000) 12:581–90. doi: 10.1016/S1074-7613(00)80209-5
133. Philpott DJ, Yamaoka S, Israël A, Sansonetti PJ. Invasive Shigella flexneri activates NF-κB through a lipopolysaccharide-dependent innate intracellular response and leads to IL-8 expression in epithelial cells. J Immunol. (2000) 165:903–14. doi: 10.4049/jimmunol.165.2.903
134. Sansonetti P, Arondel J, Huerre M, Harada A, Matsushima K. Interleukin-8 controls bacterial transepithelial translocation at the cost of epithelial destruction in experimental shigellosis. Infect Immun. (1999) 67:1471–80. doi: 10.1128/IAI.67.3.1471-1480.1999
135. Puhar A, Tronchère H, Payrastre B, Van Nhieu GT, Sansonetti PJ. A Shigella effector dampens inflammation by regulating epithelial release of danger signal ATP through production of the lipid mediator PtdIns5P. Immunity. (2013) 39:1121–31. doi: 10.1016/j.immuni.2013.11.013
136. Sanada T, Kim M, Mimuro H, Suzuki M, Ogawa M, Oyama A, et al. The Shigella flexneri effector OspI deamidates UBC13 to dampen the inflammatory response. Nature. (2012) 483:623–6. doi: 10.1038/nature10894
137. Okuda J, Toyotome T, Kataoka N, Ohno M, Abe H, Shimura Y, et al. Shigella effector IpaH9. 8 binds to a splicing factor U2AF35 to modulate host immune responses. Biochem Biophys Res Commun. (2005) 333:531–9. doi: 10.1016/j.bbrc.2005.05.145
138. Sperandio B, Regnault B, Guo J, Zhang Z, Stanley SL Jr, Sansonetti PJ, et al. Virulent Shigella flexneri subverts the host innate immune response through manipulation of antimicrobial peptide gene expression. J Exp Med. (2008) 205:1121–32. doi: 10.1084/jem.20071698
139. Raqib R, Sarker P, Mily A, Alam NH, Arifuzzaman ASM, Rekha RS, et al. Efficacy of sodium butyrate adjunct therapy in shigellosis: a randomized, double-blind, placebo-controlled clinical trial. BMC Infect Dis. (2012) 12:111. doi: 10.1186/1471-2334-12-111
140. Raqib R, Gustafsson A, Andersson J, Bakhiet M. A systemic downregulation of gamma interferon production is associated with acute shigellosis. Infect Immun. (1997) 65:5338–41. doi: 10.1128/IAI.65.12.5338-5341.1997
141. Way SS, Borczuk AC, Dominitz R, Goldberg MB. An essential role for gamma interferon in innate resistance to Shigella flexneri infection. Infect Immun. (1998) 66:1342–8. doi: 10.1128/IAI.66.4.1342-1348.1998
142. Cohen D, Block C, Green M, Lowell G, Ofek I. Immunoglobulin M, A, and G antibody response to lipopolysaccharide O antigen in symptomatic and asymptomatic Shigella infections. J Clin Microbiol. (1989) 27:162–7. doi: 10.1128/JCM.27.1.162-167.1989
143. Formal SB, Oaks EV, Olsen RE, Wingfield-Eggleston M, Snoy PJ, Cogan JP. Effect of prior infection with virulent Shigella flexneri 2a on the resistance of monkeys to subsequent infection with Shigella sonnei. J Infect Dis. (1991) 164:533–7. doi: 10.1093/infdis/164.3.533
144. Cohen D, Meron-Sudai S, Bialik A, Asato V, Goren S, Ariel-Cohen O, et al. Serum IgG antibodies to Shigella lipopolysaccharide antigens–a correlate of protection against shigellosis. Hum Vaccin Immunother. (2019) 15:1401–8. doi: 10.1080/21645515.2019.1606971
145. Orr N, Robin G, Lowell G, Cohen D. Presence of specific immunoglobulin A-secreting cells in peripheral blood after natural infection with Shigella sonnei. J Clin Microbiol. (1992) 30:2165–8. doi: 10.1128/JCM.30.8.2165-2168.1992
146. Wahid R, Simon JK, Picking WL, Kotloff KL, Levine MM, Sztein MB. Shigella antigen-specific B memory cells are associated with decreased disease severity in subjects challenged with wild-type Shigella flexneri 2a. Clin Immunol. (2013) 148:35–43. doi: 10.1016/j.clim.2013.03.009
147. Simon J, Wahid R, Maciel M Jr, Picking W, Kotloff K, Levine M, et al. Antigen-specific B memory cell responses to lipopolysaccharide (LPS) and invasion plasmid antigen (Ipa) B elicited in volunteers vaccinated with live-attenuated Shigella flexneri 2a vaccine candidates. Vaccine. (2009) 27:565–72. doi: 10.1016/j.vaccine.2008.10.081
148. Simon J, Maciel M Jr, Weld E, Wahid R, Pasetti M, Picking W, et al. Antigen-specific IgA B memory cell responses to Shigella antigens elicited in volunteers immunized with live attenuated Shigella flexneri 2a oral vaccine candidates. Clin Immunol. (2011) 139:185–92. doi: 10.1016/j.clim.2011.02.003
149. Bhowmick R, Pore D, Chakrabarti MK. Outer membrane protein A (OmpA) of Shigella flexneri 2a induces TLR2-mediated activation of B cells: involvement of protein tyrosine kinase, ERK and NF-κB. PLoS ONE. (2014) 9: e109107. doi: 10.1371/journal.pone.0109107
150. Sellge G, Magalhaes JG, Konradt C, Fritz JH, Salgado-Pabon W, Eberl G, et al. Th17 cells are the dominant T cell subtype primed by Shigella flexneri mediating protective immunity. J Immunol. (2010) 184:2076–85. doi: 10.4049/jimmunol.0900978
151. Raqib R, Qadri F, SarkEr P, Mia S, Sansonnetti P, Albert M, et al. Delayed and reduced adaptive humoral immune responses in children with shigellosis compared with in adults. Scand J Immunol. (2002) 55:414–23. doi: 10.1046/j.1365-3083.2002.01079.x
152. Cohen D, Green M, Block C, Slepon R, Ofek I. Prospective study of the association between serum antibodies to lipopolysaccharide O antigen and the attack rate of shigellosis. J Clin Microbiol. (1991) 29:386–9. doi: 10.1128/JCM.29.2.386-389.1991
153. Kim DW, Chu H, Joo DH, Jang MS, Choi JH, Park S-M, et al. OspF directly attenuates the activity of extracellular signal-regulated kinase during invasion by Shigella flexneri in human dendritic cells. Mol Immunol. (2008) 45:3295–301. doi: 10.1016/j.molimm.2008.02.013
154. Konradt C, Frigimelica E, Nothelfer K, Puhar A, Salgado-Pabon W, Di Bartolo V, et al. The Shigella flexneri type three secretion system effector IpgD inhibits T cell migration by manipulating host phosphoinositide metabolism. Cell Host Microbe. (2011) 9:263–72. doi: 10.1016/j.chom.2011.03.010
155. Salgado-Pabón W, Celli S, Arena ET, Nothelfer K, Roux P, Sellge G, et al. Shigella impairs T lymphocyte dynamics in vivo. Proc Natl Acad Sci USA. (2013) 110:4458–63. doi: 10.1073/pnas.1300981110
156. Nothelfer K, Arena ET, Pinaud L, Neunlist M, Mozeleski B, Belotserkovsky I, et al. B lymphocytes undergo TLR2-dependent apoptosis upon Shigella infection. J Exp Med. (2014) 211:1215–29. doi: 10.1084/jem.20130914
157. Hall IC, O'TOOLE EJAjodoc. Intestinal flora in new-born infants: with a description of a new pathogenic anaerobe. Bacillus Diff . (1935) 49:390–402. doi: 10.1001/archpedi.1935.01970020105010
158. Cunha BA. Nosocomial diarrhea. Crit Care Clin. (1998) 14:329–38. doi: 10.1016/S0749-0704(05)70398-5
159. CDC. Antibiotic Resistance Threats in the United States. US Department of Health and Human Services. Atlanta, GA: CDC (2019).
160. Khanna S, Pardi DS, Aronson SL, Kammer PP, Orenstein R, Sauver JLS, et al. The epidemiology of community-acquired Clostridium difficile infection: a population-based study. Am J Gastroenterol. (2012) 107:89. doi: 10.1038/ajg.2011.398
161. Hopkins RJ, Wilson RBJGr. Treatment of recurrent Clostridium difficile colitis: a narrative review. Gastroenterol Rep. (2018) 6:21–8. doi: 10.1093/gastro/gox041
162. Antharam VC, Li EC, Ishmael A, Sharma A, Mai V, Rand KH, et al. Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J Clin Microbiol. (2013) 51:2884–92. doi: 10.1128/JCM.00845-13
163. Fachi JL, de Souza Felipe J, Pral LP, da Silva BK, Corrêa RO, de Andrade MCP, et al. Butyrate protects mice from Clostridium difficile-induced colitis through an HIF-1-dependent mechanism. Cell Rep. (2019) 27:750–61. e7. doi: 10.1016/j.celrep.2019.03.054
164. Wilson KH, Sheagren JN, Freter RJJoID. Population dynamics of ingested Clostridium difficile in the gastrointestinal tract of the Syrian hamster. J Infect Dis. (1985) 151:355–61. doi: 10.1093/infdis/151.2.355
165. Mitchell M, Laughon B, Lin S. Biochemical studies on the effect of Clostridium difficile toxin B on actin in vivo and in vitro. Infect Immun. (1987) 55:1610–5. doi: 10.1128/IAI.55.7.1610-1615.1987
166. Janoir C. Virulence factors of Clostridium difficile and their role during infection. Anaerobe. (2016) 37:13–24. doi: 10.1016/j.anaerobe.2015.10.009
167. Warny M, Keates AC, Keates S, Castagliuolo I, Zacks JK, Aboudola S, et al. p38 MAP kinase activation by Clostridium difficile toxin A mediates monocyte necrosis, IL-8 production, and enteritis. J Clin Invest. (2000) 105:1147–56. doi: 10.1172/JCI7545
168. Savidge TC, Pan W-h, Newman P, O'Brien M, Anton PM, Pothoulakis C. Clostridium difficile toxin B is an inflammatory enterotoxin in human intestine. Gastroenterology. (2003) 125:413–20. doi: 10.1016/S0016-5085(03)00902-8
169. Kelly CP, Becker S, Linevsky JK, Joshi MA, O'Keane JC, Dickey BF, et al. Neutrophil recruitment in Clostridium difficile toxin A enteritis in the rabbit. J Clin Invest. (1994) 93:1257–65. doi: 10.1172/JCI117080
170. Ishida Y, Maegawa T, Kondo T, Kimura A, Iwakura Y, Nakamura S, et al. Essential involvement of IFN-γ in Clostridium difficile toxin A-induced enteritis. J Immunol. (2004) 172:3018–25. doi: 10.4049/jimmunol.172.5.3018
171. Mykoniatis A, Anton PM, Wlk M, Wang CC, Ungsunan L, Blüher S, et al. Leptin mediates Clostridium difficile toxin A–induced enteritis in mice. Gastroenterology. (2003) 124:683–91. doi: 10.1053/gast.2003.50101
172. Jafari NV, Kuehne SA, Bryant CE, Elawad M, Wren BW, Minton NP, et al. Clostridium difficile modulates host innate immunity via toxin-independent and dependent mechanism (s). PLoS ONE. (2013) 8:e69846. doi: 10.1371/journal.pone.0069846
173. Hing TC, Ho S, Shih DQ, Ichikawa R, Cheng M, Chen J, et al. The antimicrobial peptide cathelicidin modulates Clostridium difficile-associated colitis and toxin A-mediated enteritis in mice. Gut. (2013) 62:1295–305. doi: 10.1136/gutjnl-2012-302180
174. Nuding S, Frasch T, Schaller M, Stange EF, Zabel LT. Synergistic effects of antimicrobial peptides and antibiotics against Clostridium difficile. Antimicrob Agents Chemother. (2014) 58:5719–25. doi: 10.1128/AAC.02542-14
175. Bouillaut L, McBride S, Sorg JA, Schmidt DJ, Suarez JM, Tzipori S, et al. Effects of surotomycin on Clostridium difficile viability and toxin production in vitro. Antimicrob Agents Chemother. (2015) 59:4199–205. doi: 10.1128/AAC.00275-15
176. Abt MC, Lewis BB, Caballero S, Xiong H, Carter RA, Sušac B, et al. Innate immune defenses mediated by two ILC subsets are critical for protection against acute Clostridium difficile infection. Cell Host Microbe. (2015) 18:27–37. doi: 10.1016/j.chom.2015.06.011
177. Hussack G, Tanha J. Toxin-specific antibodies for the treatment of Clostridium difficile: current status and future perspectives. Toxins. (2010) 2:998–1018. doi: 10.3390/toxins2050998
178. Kelly C. Can we identify patients at high risk of recurrent Clostridium difficile infection? Clin Microbiol Infect. (2012) 18:21–7. doi: 10.1111/1469-0691.12046
179. Kelly CP, Kyne L. The host immune response to Clostridium difficile. J Med Microbiol. (2011) 60:1070–9. doi: 10.1099/jmm.0.030015-0
180. Kelly CP, Pothoulakis C, Vavva F, Castagliuolo I, Bostwick EF, O'Keane JC, et al. Anti-Clostridium difficile bovine immunoglobulin concentrate inhibits cytotoxicity and enterotoxicity of C. difficile toxins. Antimicrob Agents Chemother. (1996) 40:373–9. doi: 10.1128/AAC.40.2.373
181. Warny M, Vaerman J-P, Avesani V, Delmée M. Human antibody response to Clostridium difficile toxin A in relation to clinical course of infection. Infect Immun. (1994) 62:384–9. doi: 10.1128/IAI.62.2.384-389.1994
182. Azrad M, Hamo Z, Tkhawkho L, Peretz A. Elevated serum immunoglobulin A levels in patients with Clostridium difficile infection are associated with mortality. Pathog Dis. (2018) 76:fty066. doi: 10.1093/femspd/fty066
183. Amani SA, Shadid T, Ballard JD, Lang ML. C. difficile infection induces an inferior IgG response to that induced by immunization and is associated with a lack of T follicular helper cell and memory B cell expansion. Infect Immun. (2019). 88:e00829-19. doi: 10.1128/IAI.00829-19
184. Johnson S, Gerding DN, Janoff EN. Systemic and mucosal antibody responses to toxin A in patients infected with Clostridium difficile. J Infect Dis. (1992) 166:1287–94. doi: 10.1093/infdis/166.6.1287
185. Johnston PF, Gerding DN, Knight KL. Protection from Clostridium difficile infection in CD4 T cell-and polymeric immunoglobulin receptor-deficient mice. Infect Immun. (2014) 82:522–31. doi: 10.1128/IAI.01273-13
186. Bridgman SL, Konya T, Azad MB, Guttman DS, Sears MR, Becker AB, et al. High fecal IgA is associated with reduced Clostridium difficile colonization in infants. Microbes Infect. (2016) 18:543–9. doi: 10.1016/j.micinf.2016.05.001
187. Johal S, Lambert C, Hammond J, James P, Borriello S, Mahida Y. Colonic IgA producing cells and macrophages are reduced in recurrent and non-recurrent Clostridiumdifficile associated diarrhoea. J Clin Pathol. (2004) 57:973–9. doi: 10.1136/jcp.2003.015875
188. Solomon K. The host immune response to Clostridium difficile infection. Ther Adv Infect Dis. (2013) 1:19–35. doi: 10.1177/2049936112472173
189. Aronsson B, Granström M, Möllby R, Nord C. Serum antibody response to Clostridium difficile toxins in patients with Clostridium difficile diarrhoea. Infection. (1985) 13:97–101. doi: 10.1007/BF01642866
190. Bauer M, Nibbering P, Poxton I, Kuijper E, Dissel J. Humoral immune response as predictor of recurrence in Clostridium difficile infection. Clin Microbiol Infect. (2014) 20:1323–8. doi: 10.1111/1469-0691.12769
191. Drudy D, Calabi E, Kyne L, Sougioultzis S, Kelly E, Fairweather N, et al. Human antibody response to surface layer proteins in Clostridium difficile infection. FEMS Immunol Med Microbiol. (2004) 41:237–42. doi: 10.1016/j.femsim.2004.03.007
192. Sanchez-Hurtado K, Corretge M, Mutlu E, McIlhagger R, Starr JM, Poxton IR. Systemic antibody response to Clostridium difficile in colonized patients with and without symptoms and matched controls. J Med Microbiol. (2008) 57:717–24. doi: 10.1099/jmm.0.47713-0
193. Lavergne V, Beauséjour Y, Pichette G, Ghannoum M, Su SH. Lymphopenia as a novel marker of Clostridium difficile infection recurrence. J Infect. (2013) 66:129–35. doi: 10.1016/j.jinf.2012.11.001
194. Devera TS, Lang GA, Lanis JM, Rampuria P, Gilmore CL, James JA, et al. Memory B cells encode neutralizing antibody specific for toxin B from the Clostridium difficile strains VPI 10463 and NAP1/BI/027 but with superior neutralization of VPI 10463 toxin B. Infect Immun. (2016) 84:194–204. doi: 10.1128/IAI.00011-15
195. Liu Y-W, Chen Y-H, Chen J-W, Tsai P-J, Huang I. Immunization with recombinant TcdB-encapsulated nanocomplex induces protection against Clostridium difficile challenge in a mouse model. Front Microbiol. (2017) 8:1411. doi: 10.3389/fmicb.2017.01411
196. Shah HB, Smith K, Scott EJ II, Larabee JL, James JA, Ballard JD, et al. Human C. difficile toxin-specific memory B cell repertoires encode poorly neutralizing antibodies. JCI Insight. (2020) 16:138137. doi: 10.1172/jci.insight.138137
Keywords: humoral immune response, plasma cell, memory B cell, antibodies, enteric bacteria
Citation: Amadou Amani S and Lang ML (2020) Bacteria That Cause Enteric Diseases Stimulate Distinct Humoral Immune Responses. Front. Immunol. 11:565648. doi: 10.3389/fimmu.2020.565648
Received: 02 June 2020; Accepted: 18 August 2020;
Published: 16 September 2020.
Edited by:
Markus M. Heimesaat, Charité—Universitätsmedizin Berlin, GermanyReviewed by:
Allen D. Smith, Beltsville Human Nutrition Center (USDA-ARS), United StatesJaclyn Suzanne Pearson, Hudson Institute of Medical Research, Australia
Copyright © 2020 Amadou Amani and Lang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark L. Lang, bWFyay1sYW5nQG91aHNjLmVkdQ==