 Robert J. Hayashi
Robert J. Hayashi- Division of Pediatric Hematology/Oncology, Washington University School of Medicine, St. Louis, MO, United States
The variables that influence the selection of a preparative regimen for a pediatric hematopoietic stem cell transplant procedure encompasses many issues. When one considers this procedure for non-malignant diseases, components in a preparative regimen that were historically developed to reduce malignant tumor burden may be unnecessary. The primary goal of the procedure in this instance becomes engraftment with the establishment of normal hematopoiesis and a normal immune system. Overcoming rejection becomes the primary priority, but pursuit of this goal cannot neglect organ toxicity, or post-transplant morbidity such as graft-versus-host disease or life threatening infections. With the improvements in supportive care, newborn screening techniques for early disease detection, and the expansion of viable donor sources, we have reached a stage where hematopoietic stem cell transplantation can be considered for virtually any patient with a hematopoietic based disease. Advancing preparative regiments that minimize rejection and transplant related toxicity will thus dictate to what extent this medical technology is fully utilized. This mini-review will provide an overview of the origins of conditioning regimens for transplantation and how agents and techniques have evolved to make hematopoietic stem cell transplantation a viable option for children with non-malignant diseases of the hematopoietic system. We will summarize the current state of this facet of the transplant procedure and describe the considerations that come into play in selecting a particular preparative regimen. Decisions within this realm must tailor the treatment to the primary disease condition to ideally achieve an optimal outcome. Finally, we will project forward where advances are needed to overcome the persistent engraftment obstacles that currently limit the utilization of transplantation for haematopoietically based diseases in children.
Introduction
Since its first attempts in the 1950s, allogeneic hematopoietic stem cell transplantation (HSCT) has rapidly evolved over time (1). Initially used for the most desperate of situations, it has now become a standard of care for many disease conditions. This transformation is a product of many advancements including: (1) Improving our understanding of hematopoiesis and immune reconstitution. (2) Improvements in supportive care, (3) Improvements in the prevention of graft-versus-host disease (GVHD), (4) Expansion of donor pools, (5) Refinements in preparative regimen selection and design. These advancements have produced a steady decline in transplant related mortality rates which now approach 10% in some instances. Thus, HSCT is now viewed as a viable option for virtually any disease that originates from the hematopoietic system. Continued improvements must now take into account not only mortality, but also minimizing the long-term toxicities that a surviving patient must confront after achieving cure of their primary disease.
Long term toxicities can be a consequence of several variables. 1.) Organ damage from the preparative regimen, 2.) Sequelae from the transplant course such as mucositis, infection, or excessive bleeding, 3.) Chronic GVHD, 4.) Toxicity from other medications administered (calcineurin inhibitors, steroids, etc.) (2). Although some of these complications may be unpredictable, the choice of the preparative regimen can have a significant impact. For non-malignant conditions, the primary goal of the transplant procedure is to achieve stable engraftment that is sufficient to rectify the underlying disease yet minimize long term toxicity (3). In its simplest view, the primary obstacle of HSCT is rejection of the graft. Thus, the choice of preparative regimen should focus on its immunosuppressive properties, optimizing engraftment yet avoiding an excessive immunocompromised state leading to life threatening infections (4). This “balance” can be difficult to achieve, and the optimal regimen, which varies with the primary disease, has not been established for any condition.
This mini-review will summarize both the history and current state of the repertoire of preparative regimens that have been utilized for HSCT for non-malignant conditions. We will discuss the variables which should be considered in choosing the appropriate preparative regimen and how different conditions may warrant different approaches. Finally, we will discuss future directions where advances in preparative regimen design may improve the outcome for these patients.
Individual Agents Utilized for Preparative Regimen Design
Established preparative regimens have historically been developed utilizing standard phase I designs which advance dose intensity until a dose limiting toxicity was encountered. Hematologic toxicity was disregarded due to its reversal with the infusion of hematopoietic stem cells of the graft. Thus, doses and schedules of individual agents were limited by toxicities outside the hematopoietic system.
Modern day regimens are typically classified into three categories (3, 5, 6). Myeloablative regimens typically requires a stem cell graft infusion to reconstitute hematopoiesis. Non-myeloablative regimens, as the name implies, are less intensive and, even in the absence of a stem cell infusion, spontaneous hematopoietic recovery is expected. Reduced intensity regimens, whose definition has not been rigorously defined, falls somewhere in-between the two extremes, and is an acknowledgement that non-myeloablative regimens are associated, by their nature, with an increased risk of rejection. Reduced intensity regimens thus, fall short of full myeloablative dosing, but may achieve engraftment with less toxicity. Regardless of the type of preparative regimen, below are the components which constitute most modern day therapies.
Total Body Irradiation (TBI)
One of the first modalities developed, TBI was the primary modality utilized in early transplant studies in animals because of its known immunosuppressive and myeloablative properties (7, 8). Clinical experience in humans quickly raised awareness of TBI’s effects on the lungs and strategies that fractionated doses and shielded the lung fields led to improvements in survival (9). TBI’s toxicity unfortunately does not spare any tissue, often leading to irreversible damage to exposed organs making it less attractive for non-malignant diseases. Subsequent investigations have strived to reduce the dose and presumably the toxicity to exposed organ systems because of its usefulness in overcoming rejection particularly in mismatched donors. Long term studies have failed to identify doses that are free of significant rates of infertility, thyroid disease, and growth hormone deficiency making the use of this modality problematic.
Cyclophosphamide
A well-established alkylating agent, cyclophosphamide has maintained its role in HSCT due to its highly immunosuppressive properties and the relative resistance of hematopoietic stem cells to this agent even the highest doses (8, 10, 11). Recent studies have utilized cyclophosphamide post graft infusion to improve the outcomes of haploidentical transplant procedures (12–14). The success of this strategy has probably entrenched this agent as a major element of transplant therapy. Acute toxicities including hemorrhagic cystitis, and cardiac toxicity have been reduced with improved supportive care, with persistent long term toxicities that include sterility and secondary malignancies.
Busulfan
One of the first agents to be utilized in non-TBI containing preparative regimens, the establishment of pharmacokinetic modeling to project optimal dosing for this drug has reduced rejections and hepatotoxicity (8, 10, 11, 15). Seizures, a common complication of this agent has been minimized with prophylactic anti-epileptic drugs. Sinusoidal obstruction syndrome, (SOS) continues to be a clinical problem, but pharmacokinetic dose adjustments have reduced its risk.
Treosulfan
A structural analog of busulfan, its use is increasing with its potent immunosuppressive properties and favorable toxicity profile (16–19). Future trials will determine whether it supplants busulfan as a primary agent for preparative regimens.
Thiotepa
An alkylating agent, thiotepa has gained increasing popularity due to its immunosuppressive effects and its ability to lower rejection rates in reduced intensity preparative regimens (8, 20, 21). Its toxicity profile is comparable to other alkylating agents although it does have unique properties that lead to significant cutaneous toxicity which is typically managed with supportive care.
Melphalan
Another popular alkylating agent, its use has increased over the years as its toxicity is limited outside of the hematopoietic system particularly at doses used in modern reduced intensity regimens (22).
Etoposide
A phase specific, topoisomerase II inhibitor, etoposide has continued to be a common component of modern day preparative regimens due to its predictable toxicity profile and its ability to be combined with alkylating agents without adding excessive side effects (8). Most short term toxicities outside of myelosuppression has been restricted to gastrointestinal and dermatologic which can be typically managed, and severe liver toxicity is observed only with high doses (23). Etoposide’s association with an increased risk of secondary leukemia limits its use and makes it a somewhat less attractive agent for transplantation in non-malignant conditions.
Fludarabine
A purine analog, fludarabine’s popularity in its incorporation into more modern day preparative regimens is due to its relatively potent immunosuppressive properties without significant organ toxicity (10, 11, 24). Early use of this agent was associated with neurologic toxicity which has been overcome with dosing adjustments. Its successful incorporation into several reduced intensity preparative regimens for non-malignant diseases would indicate that it will a remain central element in HSCT for the foreseeable future.
Antibody Agents
Antibodies directed at the lymphoid compartment have an inherent attractiveness due to their lack of toxicities on other organ systems (3). Such agents can help overcome rejection. In addition, their typical long half-life allows for its persistence in the recipient where it can potentially impact GVHD, depleting T cells from the infused donor product. Appropriate premedication can overcome most infusion reactions. The greatest challenge is to tailor the dosing and schedule of administration to minimize rejection yet avoid sustained suppression of the T cell compartment that would lead to excessive opportunistic infections. Although many agents have been utilized over the years, only a few have maintained a stable presence in this field.
Anti-Thymocyte Globulin (ATG)
Two sources of anti-thymocyte globulin encompass most of its use: 1) ATGAM (horse polysera) 2. Thymoglobulin (rabbit polysera). ATGAM has been utilized for many more years than the rabbit formulation (25), but the latter is a more potent agent (26, 27). Studies with ATGAM have demonstrated that its use reduces the duration of other immunosuppressive agents (28). Both have been shown to improve engraftment rates when added to conventional preparative regimens and given their retained presence in the host, their use has reduced rates of both acute and chronic GVHD to varying degrees (29–32).
Anti-T Lymphocytes Globulin (ATLG)
Anti-T lymphocytes globulin, derived from rabbit polysera from immunization with a Jurkat T cell leukemia line, is also gaining in popularity (27, 33, 34). Most trials comparing the efficacy between ATG and ATLG have been performed in patients with malignant disease where more effective lymphodepletion and subsequent reductions in GVHD have been offset by increased rates of relapse of the primary cancer (35). More robust trials in non-malignant diseases are needed.
Alemtuzumab
A humanized monoclonal antibody against CD52, alemtuzumab has been shown to target T and B cells, NK cells, and antigen-presenting cells. It has been incorporated into several reduced intensity preparative regimens and has been used successfully for immunodeficiencies, hemophagocytic lymphohistiocytosis, lysosomal storages disease, thalassemia and sickle cell disease. Like other anti-lymphocyte products, it is associated with an increased risk for infections (36). However, since it is a monoclonal product, the clinical responses may be less variable from patient to patient in comparison to the polyclonal products listed above.
Co-Stimulation Blockade
Recent investigations have begun to examine T cell co-stimulation blockade as an additional means of immunosuppression to both reduce the risk of rejection and GVHD. Abatacept, a CTLA4-Ig agents can block the CD28-CD80/86 interactions needed for T cell activation has been incorporated into newer preparative regimens (37). Preliminary studies have demonstrated low rates of GVHD with an acceptable toxicity profile. Further trials are needed to further define its role.
Agents Less Commonly Used in Preparative Regimens for Non-Malignant Disease
Other chemotherapy agents which were initially advanced into preparative regimens have not sustained their presence in modern day treatments for non-malignant diseases due to their inherent toxicities and the lack of a need for their anti-neoplastic activity. Platinum agents, other alkylating agents, anthracyclines, are examples of agents that have not sustained their presence in modern day regimens (8).
Strategies in Preparative Regimen Selection for Non-Malignant Diseases
Lacking the necessity of eradicating malignant cells, the transplant physician contemplating HSCT for a patient with a non-malignant disease must take several considerations into account which may or may not be specific to the patient’s disease state. These include: 1) What are the specific vulnerabilities of a particular disease population that lead to transplant related complications from the preparative regimen selection? 2) How has the patient’s primary disease and the corresponding treatment to treat that disease impacted the patient’s vital organs? 3) What are the barriers to achieve engraftment which would guide minimizing the intensity of the preparative regimen? 4.) What are other immunological features beyond rejection that influence transplant outcome? Thoughtful consideration for each of these variables will optimize the course of the patient.
Specific Vulnerabilities of a Particular Disease Population
The different diseases which are considered for HSCT have different clinical phenotypes which are linked to problems, some which are severe. Although a successful HSCT procedure may ultimately alleviate the condition, specific elements of a particular preparative regimen may exacerbate a patient’s clinical condition to serious levels. An appreciation of the specific vulnerabilities for a particular disease will provide insight for thoughtful decision making to select a preparative regimen (Table 1). Given the diversity of clinical difficulties that each disease possesses and given the expected patient to patient variability in clinical courses, having a transplant team with sufficient experience for a particular disease will ensure optimal management of the unique complications that a patient may experience.
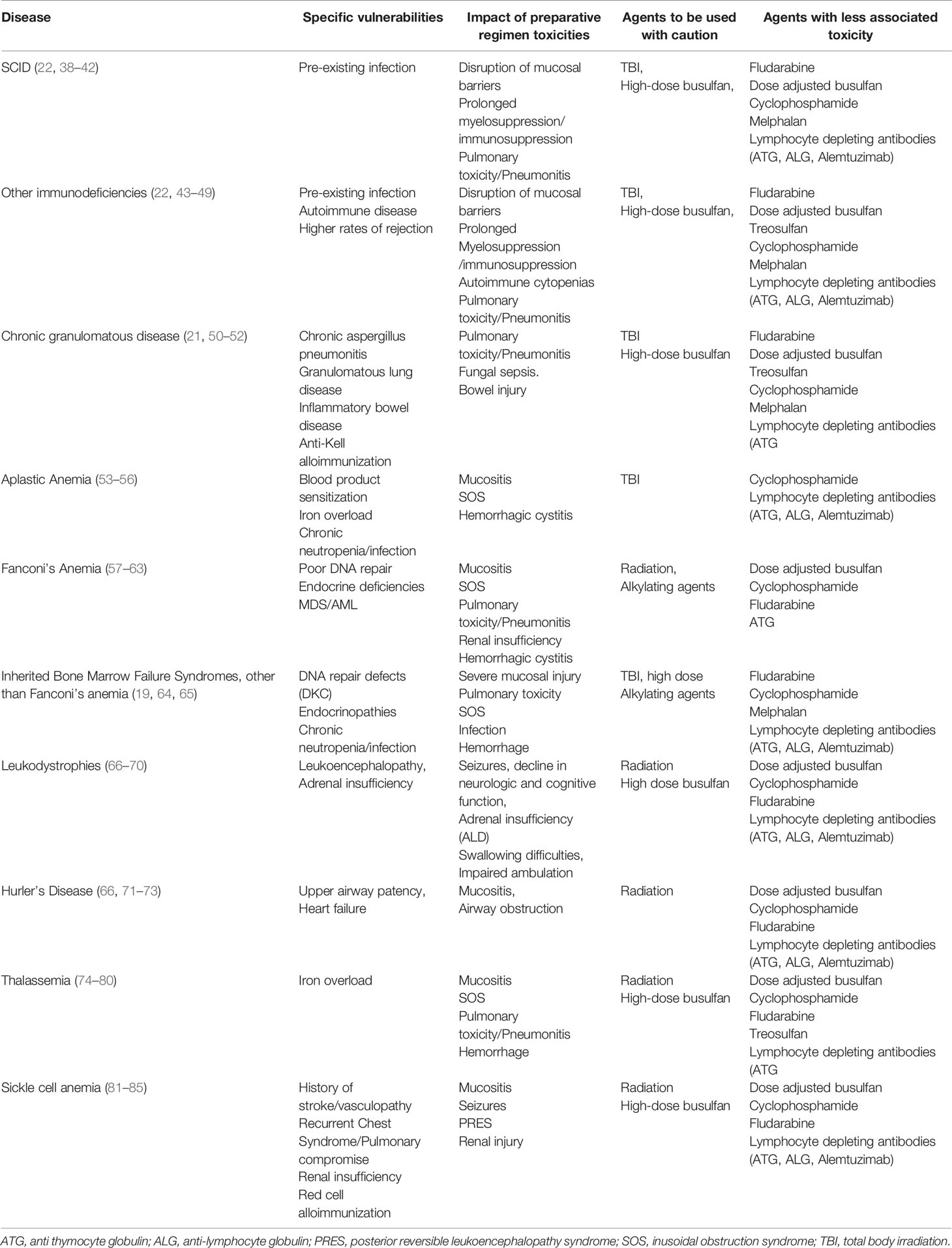
Table 1 Disease-specific vulnerabilities and the influence of preparative regimens on HSCT course.
How Has the Patient’s Primary Disease and the Corresponding Treatment to Treat That Disease Impacted the Patient’s Vital Organs?
The natural history of a particular disease may lead to organ compromise that may make the patient less tolerant to preparative regimens with specific toxicities. For instance, patients with leukodystrophies with substantial demyelination of the CNS may not tolerate TBI or high doses of neurotoxic chemotherapy such as busulfan (66, 67). A patient with sickle cell disease who has acquired substantial renal injury may handle agents cleared by the kidney poorly leading to heightened toxicity (81, 82). Alternatively, a patient with an immune compromised state such as chronic granulomatous disease may have incomplete clearance of infections which may worsen and progress once the full immunosuppressive effects of the preparative regimen have taken hold (50, 51). Thus, not only must the clinician be sufficiently familiar with the inherent vulnerabilities of the patient’s disease state, but an evaluation that sufficiently characterizes an individual’s susceptibilities to the procedure is a critical facet of the process. Preparative regimen selection and agent dosing may need to be individualized for a patient to minimize the toxicities while still striving toward a successful procedure. A sensitivity to these issues will minimize the transplant related morbidity and mortality for the patient, who could otherwise survive for a substantial number of years in the absence of the transplant procedure.
What Are the Barriers to Achieve Engraftment Which Would Guide Minimizing the Intensity of the Preparative Regimen?
The barriers to engraftment are primarily immunologic, with its magnitude dictated by the patient’s underlying disease and past treatment history (54, 57, 71). Certainly immunodeficiencies are presumed to be less capable of rejecting infused grafts, but there is wide variability in the immune competence between primary diagnoses and even for patients with the same disease. This may not necessarily be reflective in obvious differences in phenotype, but it will manifest itself in rejection (43–45). There is a tendency to provide as minimal intensity as possible for patients with immunodeficiencies to try and reduce toxicities, particularly if the patient presents with a preexisting infection. However, rejections from an inadequate preparative regimen will invariably lead to a need to repeated procedures of increasing preparative regimen intensity to avoid another rejection. Such escalation will invariably result in the accumulation of toxicities potentially leading to an unsatisfactory result.
Other disease states that are amenable to HSCT may in fact have intact immune systems. In contrast to patients with malignancies in which prior chemotherapy exposure may reduce the likelihood for rejection, non-malignant diseases, such as lysosomal storage diseases, leukodytrophies, and hemoglobinopathies may require preparative regimens with substantial immunosuppressive properties, perhaps even requiring fully myeloablative regimens (20, 66, 71, 72, 82). Such transplant procedures will lead to more severe long term toxicities.
Conditions of bone marrow failure further illustrate the complexities of choosing the right preparative regimen. Aplastic anemia, typically a disease of T cell mediated destruction of the hematopoietic system, is a condition where prior blood product exposure may sensitize the donor to an even greater risk of rejection (55). Alternatively, other conditions such as Fanconi’s Anemia or Dyskeratosis Congenita, possess difficulties in DNA repair with intolerance to the even most modest doses of radiation or alkylating agents (57–60, 64). Thus, even conditions of poor marrow function present with a wide array of clinical challenges.
What Are Other Immunological Features Beyond Rejection That Influence Transplant Outcome?
Beyond rejection, the immune system plays a central role in the clinical course of the transplanted patient. The expansion of alloreactive T cells will ultimately result in varying degrees of GVHD, and will have a substantial impact on both long term toxicity and treatment related mortality. Simultaneously, the newly reconstituting immune system is striving to achieve a protective state against infections, building new B and T cell repertoires while priming to new antigens (38, 86–90). Further complicating this process is the impact specific preparative regimen agents may have on the newly emerging lymphocyte population. Antibodies with specificity to different lymphocyte populations (ATG, ATLG, alemtuzumab etc.) will linger in the body many days after their infusion and impact not only the infused lymphocyte populations of the graft but also the newly emerging populations. The amount of antibody present as the engrafting lymphocytes develop varies with the agent, dose administered, and between patients. Thus, the transplant physician must use information from past clinical trials in selecting the appropriate regimen for an individual patient in contrast to making empiric decisions. A reduced effect on the emerging immune system may lead to extensive GVHD, while an excessive one may lead to life threatening infections (91). The inability to “fine tune” this effect is a limiting feature of the use of antibody agents.
Thoughtful Use of Preparative Regimens in HSCT in Non-Malignant Diseases
It is apparent from this review that many challenges confront the clinician when choosing a preparative regimen for a transplant candidate. Over the past several decades, investigators have reported their successes and challenges exploring different strategies (Table 2). It is apparent that virtually every element of the transplant course from rejection risk to overall survival vary tremendously from report to report. Furthermore, variables such as donor source, age of the patient, and disease status prior to the transplant procedure can influence the transplant outcome further obscuring the impact of the preparative regimen. This variability is in part due to differences in the condition of the patient population transplanted, the agents used to formulate the preparative regimen, the graft selection, (matched sibling, matched or mismatched unrelated donor, cord blood, peripheral blood verses bone marrow), and graft manipulation (T cell depletion) which will result in varying outcomes. Furthermore, many reports merge outcomes of several different preparative regimens or combine multiple diseases together, sometimes making it impossible to link specific outcomes from a preparative regimen to a specific disease. Thus, comparisons between reports can be difficult. Programs and groups that commit to a specific preparative regimen “backbone,” and then refine elements from this backbone in well-defined cohorts will provide the most useful information on how to select a preparative regimen for a patient.
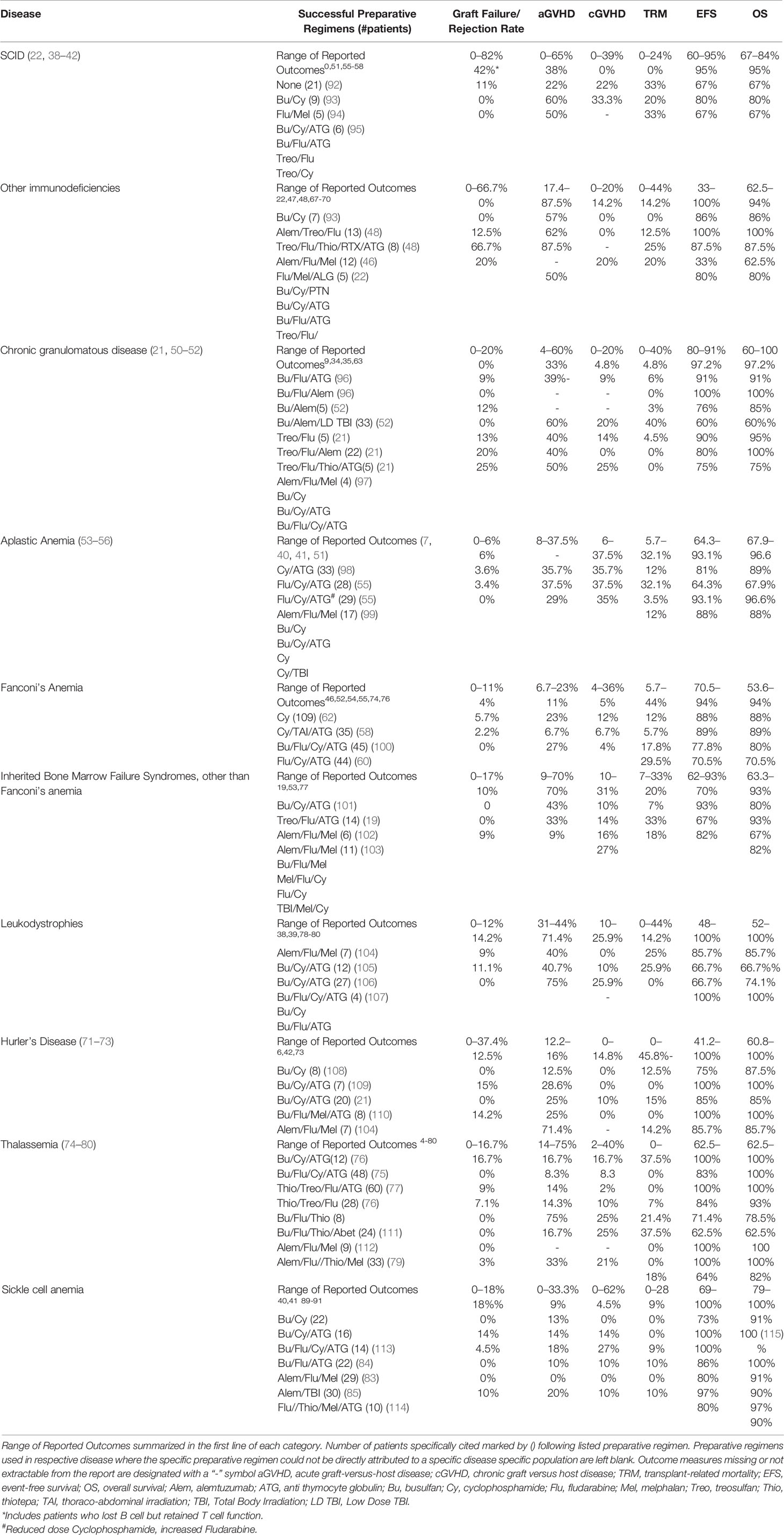
Table 2 Variation of HSCT outcomes.
Considerations of the vulnerabilities of the primary disease, the clinical status of the individualized patient, the essential needs of overcoming rejection yet temporizing GVHD and life threatening infections must all be weighed in making the appropriate decision for the patient. Unfortunately, despite over three decades of experience, there is no “formula” that can be utilized to assemble a combination of agents that will give a predictable outcome fulfilling the needs of both the clinician and the patient. Large scale studies with detailed reports of outcomes and toxicities provide our only resource to guide the clinician to make thoughtful decisions for their patient. Further research with well-designed clinical trials with full characterization of outcomes are needed to enhance our understanding of this topic.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Henig I, Zuckerman T. Hematopoietic stem cell transplantation-50 years of evolution and future perspectives. Rambam Maimonides Med J (2014) 5:e0028–8. doi: 10.5041/RMMJ.10162
2. Majhail NS. Long-term complications after hematopoietic cell transplantation. Hematol Oncol Stem Cell Ther (2017) 10:220–7. doi: 10.1016/j.hemonc.2017.05.009
3. Shaw P, Shizuru J, Hoenig M, Veys P. Conditioning Perspectives for Primary Immunodeficiency Stem Cell Transplants. Front Pediatr (2019) 7:434. doi: 10.3389/fped.2019.00434
4. Mehta RS, Rezvani K. Immune reconstitution post allogeneic transplant and the impact of immune recovery on the risk of infection. Virulence (2016) 7:901–16. doi: 10.1080/21505594.2016.1208866
5. Bacigalupo A, Ballen K, Rizzo D, Giralt S, Lazarus H, Ho V, et al. Defining the intensity of conditioning regimens: working definitions. Biol Blood Marrow Transplant (2009) 15:1628–33. doi: 10.1016/j.bbmt.2009.07.004
6. Gyurkocza B, Sandmaier BM. Conditioning regimens for hematopoietic cell transplantation: one size does not fit all. Blood (2014) 124:344–53. doi: 10.1182/blood-2014-02-514778
7. Monaco F, Scott BL, Chauncey TR, Petersen FB, Storer BE, Baron F, et al. Total body irradiation dose escalation decreases risk of progression and graft rejection after hematopoietic cell transplantation for myelodysplastic syndromes or myeloproliferative neoplasms. Haematologica (2019) 104:1221–9. doi: 10.3324/haematol.2018.199398
8. Sanders JE. Stem-cell transplant preparative regimens. Pediatr Transplant (1999) 3(Suppl 1):23–34. doi: 10.1034/j.1399-3046.1999.00073.x
9. Paix A, Antoni D, Waissi W, Ledoux MP, Bilger K, Fornecker L, et al. Total body irradiation in allogeneic bone marrow transplantation conditioning regimens: A review. Crit Rev Oncol Hematol (2018) 123:138–48. doi: 10.1016/j.critrevonc.2018.01.011
10. Harris AC, Boelens JJ, Ahn KW, Fei M, Abraham A, Artz A, et al. Comparison of pediatric allogeneic transplant outcomes using myeloablative busulfan with cyclophosphamide or fludarabine. Blood Adv (2018) 2:1198–206. doi: 10.1182/bloodadvances.2018016956
11. Gupta A, Downey M, Shanley R, Jennissen C, Miller WP, Lund TC, et al. Reduced-Toxicity (BuFlu) Conditioning Is Better Tolerated but Has a Higher Second Transplantation Rate Compared to Myeloablative Conditioning (BuCy) in Children with Inherited Metabolic Disorders. Biol Blood Marrow Transplant (2020) 26:486–92. doi: 10.1016/j.bbmt.2019.11.014
12. Oevermann L, Schulte JH, Hundsdörfer P, Hakimeh D, Kogel F, Lang P, et al. HLA-haploidentical hematopoietic stem cell transplantation in pediatric patients with hemoglobinopathies: current practice and new approaches. Bone Marrow Transplant (2019) 54:743–8. doi: 10.1038/s41409-019-0598-x
13. ElGohary G, El Fakih R, de Latour R, Risitano A, Marsh J, Schrezenmeier H, et al. Haploidentical hematopoietic stem cell transplantation in aplastic anemia: a systematic review and meta-analysis of clinical outcome on behalf of the severe aplastic anemia working party of the European group for blood and marrow transplantation (SAAWP of EBMT). Bone Marrow Transplant (2020) 55(10):906–17. doi: 10.1038/s41409-020-0897-2
14. Mallhi KK, Srikanthan MA, Baker KK, Frangoul HA, Torgerson TR, Petrovic A, et al. HLA-Haploidentical Hematopoietic Cell Transplantation for Treatment of Nonmalignant Diseases Using Nonmyeloablative Conditioning and Post-Transplant Cyclophosphamide. Biol Blood Marrow Transplant (2020) 26(7):1332–41. doi: 10.1016/j.bbmt.2020.03.018
15. McCune JS, Gooley T, Gibbs JP, Sanders JE, Petersdorf EW, Appelbaum FR, et al. Busulfan concentration and graft rejection in pediatric patients undergoing hematopoietic stem cell transplantation. Bone Marrow Transplant (2002) 30:167–73. doi: 10.1038/sj.bmt.1703612
16. van der Stoep M, Bertaina A, Ten Brink MH, Bredius RG, Smiers FJ, Wanders DCM, et al. High interpatient variability of treosulfan exposure is associated with early toxicity in paediatric HSCT: a prospective multicentre study. Br J Haematol (2017) 179:772–80. doi: 10.1111/bjh.14960
17. Slatter MA, Rao K, Abd Hamid IJ, Nademi Z, Chiesa R, Elfeky R, et al. Treosulfan and Fludarabine Conditioning for Hematopoietic Stem Cell Transplantation in Children with Primary Immunodeficiency: UK Experience. Biol Blood Marrow Transplant (2018) 24:529–36. doi: 10.1016/j.bbmt.2017.11.009
18. Huttunen P, Taskinen M, Vettenranta K. Acute toxicity and outcome among pediatric allogeneic hematopoietic transplant patients conditioned with treosulfan-based regimens. Pediatr Hematol Oncol (2020) 37(5):1–10. doi: 10.1080/08880018.2020.1738604
19. Burroughs LM, Shimamura A, Talano JA, Domm JA, Baker KK, Delaney C, et al. Allogeneic Hematopoietic Cell Transplantation Using Treosulfan-Based Conditioning for Treatment of Marrow Failure Disorders. Biol Blood Marrow Transplant (2017) 23:1669–77. doi: 10.1016/j.bbmt.2017.06.002
20. Rosales F, Peylan-Ramu N, Cividalli G, Varadi G, Or R, Naparstek E, et al. The role of thiotepa in allogeneic bone marrow transplantation for genetic diseases. Bone Marrow Transplant (1999) 23:861–5. doi: 10.1038/sj.bmt.1701758
21. Morillo-Gutierrez B, Beier R, Rao K, Burroughs L, Schulz A, Ewins AM, et al. Treosulfan-based conditioning for allogeneic HSCT in children with chronic granulomatous disease: a multicenter experience. Blood (2016) 128(3):440–8. doi: 10.1182/blood-2016-03-704015
22. Triplett BM, Wang C, Yang J, Dallas M, Hartford C, Howard V, et al. Effects of conditioning regimens and T cell depletion in hematopoietic cell transplantation for primary immune deficiency. Biol Blood Marrow Transplant (2012) 18:1911–20. doi: 10.1016/j.bbmt.2012.07.014
23. Schmitz N, Gassmann W, Rister M, Johannson W, Suttorp M, Brix F, et al. Fractionated total body irradiation and high-dose VP 16-213 followed by allogeneic bone marrow transplantation in advanced leukemias. Blood (1988) 72:1567–73. doi: 10.1182/blood.V72.5.1567.1567
24. Langenhorst JB, van Kesteren C, van Maarseveen EM, Dorlo TPC, Nierkens S, Lindemans CA, et al. Fludarabine exposure in the conditioning prior to allogeneic hematopoietic cell transplantation predicts outcomes. Blood Adv (2019) 3:2179–87. doi: 10.1182/bloodadvances.2018029421
25. Hagen P, Wagner JE, DeFor TE, Dolan M, Arora M, Warlick E, et al. The effect of equine antithymocyte globulin on the outcomes of reduced intensity conditioning for AML. Bone Marrow Transplant (2014) 49:1498–504. doi: 10.1038/bmt.2014.183
26. Yuan J, Pei R, Su W, Cao J, Lu Y. Meta-analysis of the actions of antithymocyte globulin in patients undergoing allogeneic hematopoietic cell transplantation. Oncotarget (2017) 8:10871–82. doi: 10.18632/oncotarget.14719
27. Bonifazi F, Rubio MT, Bacigalupo A, Boelens JJ, Finke J, Greinix H, et al. Rabbit ATG/ATLG in preventing graft-versus-host disease after allogeneic stem cell transplantation: consensus-based recommendations by an international expert panel. Bone Marrow Transplant (2020) 55(6):1093–102. doi: 10.1038/s41409-020-0792-x
28. Walker I, Panzarella T, Couban S, Couture F, Devins G, Elemary M, et al. Pretreatment with anti-thymocyte globulin versus no anti-thymocyte globulin in patients with haematological malignancies undergoing haemopoietic cell transplantation from unrelated donors: a randomised, controlled, open-label, phase 3, multicentre trial. Lancet Oncol (2016) 17:164–73. doi: 10.1016/S1470-2045(15)00462-3
29. Kröger N, Solano C, Wolschke C, Bandini G, Patriarca F, Pini M, et al. Antilymphocyte Globulin for Prevention of Chronic Graft-versus-Host Disease. N Engl J Med (2016) 374:43–53. doi: 10.1056/NEJMc1601364
30. Chang YJ, Wu DP, Lai YR, Liu QF, Sun YQ, Hu J, et al. Antithymocyte Globulin for Matched Sibling Donor Transplantation in Patients With Hematologic Malignancies: A Multicenter, Open-Label, Randomized Controlled Study. J Clin Oncol (2020), Jco2000150. doi: 10.1200/JCO.20.00150
31. Shiratori S, Sugita J, Ota S, Kasahara S, Ishikawa J, Tachibana T, et al. Low-dose anti-thymocyte globulin for GVHD prophylaxis in HLA-matched allogeneic peripheral blood stem cell transplantation. Bone Marrow Transplant (2020). doi: 10.1038/s41409-020-0985-3
32. Luo Y, Jin M, Tan Y, Zhao Y, Shi J, Zhu Y, et al. Antithymocyte globulin improves GVHD-free and relapse-free survival in unrelated hematopoietic stem cell transplantation. Bone Marrow Transplant (2019) 54:1668–75. doi: 10.1038/s41409-019-0502-8
33. Turki AT, Klisanin V, Bayraktar E, Kordelas L, Trenschel R, Ottinger H, et al. Optimizing anti-T-lymphocyte globulin dosing to improve long-term outcome after unrelated hematopoietic cell transplantation for hematologic malignancies. Am J Transplant (2020) 20:677–88. doi: 10.1111/ajt.15642
34. Gooptu M, Kim HT, Chen YB, Rybka W, Artz A, Boyer M, et al. Effect of Antihuman T Lymphocyte Globulin on Immune Recovery after Myeloablative Allogeneic Stem Cell Transplantation with Matched Unrelated Donors: Analysis of Immune Reconstitution in a Double-Blind Randomized Controlled Trial. Biol Blood Marrow Transplant (2018) 24:2216–23. doi: 10.1016/j.bbmt.2018.07.002
35. Soiffer RJ, Kim HT, McGuirk J, Horwitz ME, Johnston L, Patnaik MM, et al. Prospective, Randomized, Double-Blind, Phase III Clinical Trial of Anti-T-Lymphocyte Globulin to Assess Impact on Chronic Graft-Versus-Host Disease-Free Survival in Patients Undergoing HLA-Matched Unrelated Myeloablative Hematopoietic Cell Transplantation. J Clin Oncol (2017) 35:4003–11. doi: 10.1200/JCO.2017.75.8177
36. Bhatt ST, Bednarski JJ, Berg J, Trinkaus K, Murray L, Hayashi R, et al. Immune Reconstitution and Infection Patterns after Early Alemtuzumab and Reduced Intensity Transplantation for Nonmalignant Disorders in Pediatric Patients. Biol Blood Marrow Transplant (2019) 25:556–61. doi: 10.1016/j.bbmt.2018.10.008
37. Koura DT, Horan JT, Langston AA, Qayed M, Mehta A, Khoury HJ, et al. In vivo T cell costimulation blockade with abatacept for acute graft-versus-host disease prevention: a first-in-disease trial. Biol Blood Marrow Transplant (2013) 19:1638–49. doi: 10.1016/j.bbmt.2013.09.003
38. Manor U, Lev A, Simon AJ, Hutt D, Toren A, Bielorai B, et al. Immune reconstitution after HSCT in SCID-a cohort of conditioned and unconditioned patients. Immunol Res (2019) 67:166–75. doi: 10.1007/s12026-019-09081-z
39. Heimall J, Logan BR, Cowan MJ, Notarangelo LD, Griffith LM, Puck JM, et al. Immune reconstitution and survival of 100 SCID patients post-hematopoietic cell transplant: a PIDTC natural history study. Blood (2017) 130:2718–27. doi: 10.1182/blood-2017-05-781849
40. Abd Hamid IJ, Slatter MA, McKendrick F, Pearce MS, Gennery AR. Long-term outcome of hematopoietic stem cell transplantation for IL2RG/JAK3 SCID: a cohort report. Blood (2017) 129:2198–201. doi: 10.1182/blood-2016-11-748616
41. Hassan A, Booth C, Brightwell A, Allwood Z, Veys P, Rao K, et al. Outcome of hematopoietic stem cell transplantation for adenosine deaminase-deficient severe combined immunodeficiency. Blood (2012) 120:3615–24; quiz 3626. doi: 10.1182/blood-2011-12-396879
42. Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation outcomes for severe combined immunodeficiency, 2000-2009. N Engl J Med (2014) 371:434–46. doi: 10.1056/NEJMoa1401177
43. Burroughs L, Petrovic A, Brazauskas R, Liu X, Griffith LM, Ochs HD, et al. Excellent Outcomes Following Hematopoietic Cell Transplantation for Wiskott-Aldrich Syndrome: A PIDTC Report. Blood (2020) 135(23):2094–105. doi: 10.1182/blood.2019002939
44. Chan AY, Leiding JW, Liu X, Veys P, Bertrand Y, Souillet G, et al. Hematopoietic Cell Transplantation in Patients With Primary Immune Regulatory Disorders (PIRD): A Primary Immune Deficiency Treatment Consortium (PIDTC) Survey. Front Immunol (2020) 11:239. doi: 10.3389/fimmu.2020.00239
45. Dimitrova D, Gea-Banacloche J, Steinberg SM, Sadler JL, Hicks SN, Carroll E, et al. Prospective Study of a Novel, Radiation-Free, Reduced-Intensity Bone Marrow Transplantation Platform for Primary Immunodeficiency Diseases. Biol Blood Marrow Transplant (2020) 26:94–106. doi: 10.1016/j.bbmt.2019.08.018
46. Allen CE, Marsh R, Dawson P, Bollard CM, Shenoy S, Roehrs P, et al. Reduced-intensity conditioning for hematopoietic cell transplant for HLH and primary immune deficiencies. Blood (2018) 132:1438–51. doi: 10.1182/blood-2018-01-828277
47. Lum SH, Hoenig M, Gennery AR, Slatter MA. Conditioning Regimens for Hematopoietic Cell Transplantation in Primary Immunodeficiency. Curr Allergy Asthma Rep (2019) 19:52. doi: 10.1007/s11882-019-0883-1
48. Lum SH, Anderson C, McNaughton P, Engelhardt KR, MacKenzie B, Watson H, et al. Improved transplant survival and long-term disease outcome in children with MHC class II deficiency. Blood (2020) 135:954–73. doi: 10.1182/blood.2019002690
49. Wehr C, Gennery AR, Lindemans C, Schulz A, Hoenig M, Marks R, et al. Multicenter experience in hematopoietic stem cell transplantation for serious complications of common variable immunodeficiency. J Allergy Clin Immunol (2015) 135:988–97.e6. doi: 10.1016/j.jaci.2014.11.029
50. Tang X, Zhang Y, Jing Y, Lu W, Xu S, Cao X, et al. Allogeneic hematopoietic stem cell transplantation using unrelated cord blood or unmanipulated haploidentical donors is effective in pediatric chronic granulomatous disease with inflammatory complications and severe infection. Bone Marrow Transplant (2020) 55(9):1875–8. doi: 10.1038/s41409-020-0864-y
51. Connelly JA, Marsh R, Parikh S, Talano JA. Allogeneic Hematopoietic Cell Transplantation for Chronic Granulomatous Disease: Controversies and State of the Art. J Pediatr Infect Dis Soc (2018) 7:S31–s39. doi: 10.1093/jpids/piy015
52. Parta M, Kelly C, Kwatemaa N, Theobald N, Hilligoss D, Qin J, et al. Allogeneic Reduced-Intensity Hematopoietic Stem Cell Transplantation for Chronic Granulomatous Disease: a Single-Center Prospective Trial. J Clin Immunol (2017) 37:548–58. doi: 10.1007/s10875-017-0422-6
53. Darrigo LG Jr., Colturato V, de Souza MP, Loth G, Calixto R, Seber A, et al. Allogeneic Bone Marrow Transplants for Pediatric Severe Aplastic Anemia: Real-world Data comparing Matched Related and Unrelated Donors in a Developing Country. Retrospective study on behalf of the Pediatric Hematopoietic Stem Cell Transplant Working Group of the Brazilian Bone Marrow Transplantation Society (SBTMO) and the Brazil-Seattle Consortium (Gedeco). Pediatr Transplant (2019) 23:e13552. doi: 10.1111/petr.13552
54. Dufour C, Pillon M, Sociè G, Rovò A, Carraro E, Bacigalupo A, et al. Outcome of aplastic anaemia in children. A study by the severe aplastic anaemia and paediatric disease working parties of the European group blood and bone marrow transplant. Br J Haematol (2015) 169:565–73. doi: 10.1111/bjh.13297
55. Kang HJ, Hong KT, Lee JW, Kim H, Park KD, Shin HY, et al. Improved Outcome of a Reduced Toxicity-Fludarabine, Cyclophosphamide, plus Antithymocyte Globulin Conditioning Regimen for Unrelated Donor Transplantation in Severe Aplastic Anemia: Comparison of 2 Multicenter Prospective Studies. Biol Blood Marrow Transplant (2016) 22:1455–9. doi: 10.1016/j.bbmt.2016.04.003
56. Yoshida N, Kobayashi R, Yabe H, Kosaka Y, Yagasaki H, Watanabe K, et al. First-line treatment for severe aplastic anemia in children: bone marrow transplantation from a matched family donor versus immunosuppressive therapy. Haematologica (2014) 99:1784–91. doi: 10.3324/haematol.2014.109355
57. Ebens CL, DeFor TE, Tryon R, Wagner JE, MacMillan ML. Comparable Outcomes after HLA-Matched Sibling and Alternative Donor Hematopoietic Cell Transplantation for Children with Fanconi Anemia and Severe Aplastic Anemia. Biol Blood Marrow Transplant (2018) 24:765–71. doi: 10.1016/j.bbmt.2017.11.031
58. Farzin A, Davies SM, Smith FO, Filipovich A, Hansen M, Auerbach AD, et al. Matched sibling donor haematopoietic stem cell transplantation in Fanconi anaemia: an update of the Cincinnati Children’s experience. Br J Haematol (2007) 136:633–40. doi: 10.1111/j.1365-2141.2006.06460.x
59. Mehta PA, Davies SM, Leemhuis T, Myers K, Kernan NA, Prockop SE, et al. Radiation-free, alternative-donor HCT for Fanconi anemia patients: results from a prospective multi-institutional study. Blood (2017) 129:2308–15. doi: 10.1182/blood-2016-09-743112
60. Tuysuz G, Guler E, Ozel D, Kupesiz A. Results of Allogenic Hematopoietic Stem Cell Transplantation in Fanconi Anemia Caused by Bone Marrow Failure: Single-Regimen, Single-Center Experience of 14 Years. Biol Blood Marrow Transplant (2019) 25:2017–23. doi: 10.1016/j.bbmt.2019.05.039
61. AlDawsari G, Elhaddad A, El Fakih R, Ben Othman T, Ahmed P, Ghavamzadeh A, et al. Outcome of hematopoietic stem cell transplantation (HCT) from HLA-matched related donor for Fanconi anemia (FA) in adolescents and adults: a retrospective study by Eastern Mediterranean Blood and Marrow Transplantation Group (EMBMT). Bone Marrow Transplant (2020) 55(7):1485–90. doi: 10.1038/s41409-020-0809-5
62. Bonfim C, Ribeiro L, Nichele S, Bitencourt M, Loth G, Koliski A, et al. Long-term Survival, Organ Function, and Malignancy after Hematopoietic Stem Cell Transplantation for Fanconi Anemia. Biol Blood Marrow Transplant (2016) 22:1257–63. doi: 10.1016/j.bbmt.2016.03.007
63. Svahn J, Bagnasco F, Cappelli E, Onofrillo D, Caruso S, Corsolini F, et al. Somatic, hematologic phenotype, long-term outcome, and effect of hematopoietic stem cell transplantation. An analysis of 97 Fanconi anemia patients from the Italian national database on behalf of the Marrow Failure Study Group of the AIEOP (Italian Association of Pediatric Hematology-Oncology). Am J Hematol (2016) 91:666–71. doi: 10.1002/ajh.24373
64. Fioredda F, Iacobelli S, Korthof ET, Knol C, van Biezen A, Bresters D, et al. Outcome of haematopoietic stem cell transplantation in dyskeratosis congenita. Br J Haematol (2018) 183:110–8. doi: 10.1111/bjh.15495
65. Cesaro S, Pillon M, Sauer M, Smiers F, Faraci M, de Heredia CD, et al. Long-term outcome after allogeneic hematopoietic stem cell transplantation for Shwachman-Diamond syndrome: a retrospective analysis and a review of the literature by the Severe Aplastic Anemia Working Party of the European Society for Blood and Marrow Transplantation (SAAWP-EBMT). Bone Marrow Transplant (2020) 55(9):1796–809. doi: 10.1038/s41409-020-0863-z
66. Page KM, Stenger EO, Connelly JA, Shyr D, West T, Wood S, et al. Hematopoietic Stem Cell Transplantation to Treat Leukodystrophies: Clinical Practice Guidelines from the Hunter’s Hope Leukodystrophy Care Network. Biol Blood Marrow Transplant (2019) 25:e363–74. doi: 10.1016/j.bbmt.2019.09.003
67. van den Broek BTA, Page K, Paviglianiti A, Hol J, Allewelt H, Volt F, et al. Early and late outcomes after cord blood transplantation for pediatric patients with inherited leukodystrophies. Blood Adv (2018) 2:49–60. doi: 10.1182/bloodadvances.2017010645
68. Boucher AA, Miller W, Shanley R, Ziegler R, Lund T, Raymond G, et al. Long-term outcomes after allogeneic hematopoietic stem cell transplantation for metachromatic leukodystrophy: the largest single-institution cohort report. Orphanet J Rare Dis (2015) 10:94. doi: 10.1186/s13023-015-0313-y
69. Raymond GV, Aubourg P, Paker A, Escolar M, Fischer A, Blanche S, et al. Survival and Functional Outcomes in Boys with Cerebral Adrenoleukodystrophy with and without Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant (2019) 25:538–48. doi: 10.1016/j.bbmt.2018.09.036
70. Orchard PJ, Nascene DR, Miller WP, Gupta A, Kenney-Jung D, Lund TC, et al. Successful donor engraftment and repair of the blood-brain barrier in cerebral adrenoleukodystrophy. Blood (2019) 133:1378–81. doi: 10.1182/blood-2018-11-887240
71. Boelens JJ, Wynn RF, O’Meara A, Veys P, Bertrand Y, Souillet G, et al. Outcomes of hematopoietic stem cell transplantation for Hurler’s syndrome in Europe: a risk factor analysis for graft failure. Bone Marrow Transplant (2007) 40:225–33. doi: 10.1038/sj.bmt.1705718
72. Lum SH, Miller WP, Jones S, Poulton K, Ogden W, Lee H, et al. Changes in the incidence, patterns and outcomes of graft failure following hematopoietic stem cell transplantation for Hurler syndrome. Bone Marrow Transplant (2017) 52:846–53. doi: 10.1038/bmt.2017.5
73. Aldenhoven M, Jones SA, Bonney D, Borrill RE, Coussons M, Mercer J, et al. Hematopoietic cell transplantation for mucopolysaccharidosis patients is safe and effective: results after implementation of international guidelines. Biol Blood Marrow Transplant (2015) 21:1106–9. doi: 10.1016/j.bbmt.2015.02.011
74. Anurathapan U, Hongeng S, Pakakasama S, Songdej D, Sirachainan N, Pongphitcha P, et al. Hematopoietic Stem Cell Transplantation for Severe Thalassemia Patients from Haploidentical Donors Using a Novel Conditioning Regimen. Biol Blood Marrow Transplant (2020) 26(6):1106–12. doi: 10.1016/j.bbmt.2020.01.002
75. Sun L, Wang N, Chen Y, Tang L, Xing C, Lu N, et al. Unrelated Donor Peripheral Blood Stem Cell Transplantation for Patients with β-Thalassemia Major Based on a Novel Conditioning Regimen. Biol Blood Marrow Transplant (2019) 25:1592–6. doi: 10.1016/j.bbmt.2019.03.028
76. Choudhary D, Doval D, Sharma SK, Khandelwal V, Setia R, Handoo AA, et al. Allogenic Hematopoietic Cell Transplantation in Thalassemia Major: A Single-center Retrospective Analysis From India. J Pediatr Hematol Oncol (2019) 41:e296–301. doi: 10.1097/MPH.0000000000001475
77. Bernardo ME, Piras E, Vacca A, Giorgiani G, Zecca M, Bertaina A, et al. Allogeneic hematopoietic stem cell transplantation in thalassemia major: results of a reduced-toxicity conditioning regimen based on the use of treosulfan. Blood (2012) 120:473–6. doi: 10.1182/blood-2012-04-423822
78. Sabloff M, Chandy M, Wang Z, Logan BR, Ghavamzadeh A, Li CK, et al. HLA-matched sibling bone marrow transplantation for β-thalassemia major. Blood (2011) 117:1745–50. doi: 10.1182/blood-2010-09-306829
79. Shenoy S, Walters MC, Ngwube A, Soni S, Jacobsohn D, Chaudhury S, et al. Unrelated Donor Transplantation in Children with Thalassemia using Reduced-Intensity Conditioning: The URTH Trial. Biol Blood Marrow Transplant (2018) 24:1216–22. doi: 10.1016/j.bbmt.2018.01.023
80. Shenoy S, Thompson AA. Unrelated donor stem cell transplantation for transfusion-dependent thalassemia. Ann N Y Acad Sci (2016) 1368:122–6. doi: 10.1111/nyas.13019
81. Cappelli B, Volt F, Tozatto-Maio K, Scigliuolo GM, Ferster A, Dupont S, et al. Risk factors and outcomes according to age at transplantation with an HLA-identical sibling for sickle cell disease. Haematologica (2019) 104:e543–6. doi: 10.3324/haematol.2019.216788
82. Eapen M, Brazauskas R, Walters MC, Bernaudin F, Bo-Subait K, Fitzhugh CD, et al. Effect of donor type and conditioning regimen intensity on allogeneic transplantation outcomes in patients with sickle cell disease: a retrospective multicentre, cohort study. Lancet Haematol (2019) 6:e585–96. doi: 10.1016/S2352-3026(19)30154-1
83. Shenoy S, Eapen M, Panepinto JA, Logan BR, Wu J, Abraham A, et al. A trial of unrelated donor marrow transplantation for children with severe sickle cell disease. Blood (2016) 128:2561–7. doi: 10.1182/blood-2016-05-715870
84. Krishnamurti L, Neuberg DS, Sullivan KM, Kamani NR, Abraham A, Campigotto F, et al. Bone marrow transplantation for adolescents and young adults with sickle cell disease: Results of a prospective multicenter pilot study. Am J Hematol (2019) 94:446–54. doi: 10.1002/ajh.25401
85. Hsieh MM, Fitzhugh CD, Weitzel RP, Link ME, Coles WA, Zhao X, et al. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA (2014) 312:48–56. doi: 10.1001/jama.2014.7192
86. Mellgren K, Nierop AFM, Abrahamsson J. Use of Multivariate Immune Reconstitution Patterns to Describe Immune Reconstitution after Allogeneic Stem Cell Transplantation in Children. Biol Blood Marrow Transplant (2019) 25:2045–53. doi: 10.1016/j.bbmt.2019.06.018
87. Simons L, Cavazzana M, André I. Concise Review: Boosting T-Cell Reconstitution Following Allogeneic Transplantation-Current Concepts and Future Perspectives. Stem Cells Transl Med (2019) 8:650–7. doi: 10.1002/sctm.18-0248
88. Mohty M, Gaugler B, Faucher C, Sainty D, Lafage-Pochitaloff M, Vey N, et al. Recovery of lymphocyte and dendritic cell subsets following reduced intensity allogeneic bone marrow transplantation. Hematology (2002) 7:157–64. doi: 10.1080/10245330210000013898
89. Bae KW, Kim BE, Koh KN, Im HJ, Seo JJ, et al. Factors influencing lymphocyte reconstitution after allogeneic hematopoietic stem cell transplantation in children. Korean J Hematol (2012) 47:44–52. doi: 10.5045/kjh.2012.47.1.44
90. Morecki S, Gelfand Y, Nagler A, Or R, Naparstek E, Varadi G, et al. Immune reconstitution following allogeneic stem cell transplantation in recipients conditioned by low intensity vs myeloablative regimen. Bone Marrow Transplant (2001) 28:243–9. doi: 10.1038/sj.bmt.1703118
91. Goussetis E, Efstathiou E, Paisiou A, Avgerinou G, Zisaki K, Giamouris VJ, et al. Infectious complications following allogeneic stem cell transplantation by using anti-thymocyte globulin-based myeloablative conditioning regimens in children with hemoglobinopathies. Transpl Infect Dis (2015) 17:201–7. doi: 10.1111/tid.12358
92. Myers LA, Patel DD, Puck JM, Buckley RH. Hematopoietic stem cell transplantation for severe combined immunodeficiency in the neonatal period leads to superior thymic output and improved survival. Blood (2002) 99:872–8. doi: 10.1182/blood.V99.3.872
93. Dalal I, Reid B, Doyle J, Freedman M, Calderwood S, Saunders F, et al. Matched unrelated bone marrow transplantation for combined immunodeficiency. Bone Marrow Transplant (2000) 25:613–21. doi: 10.1038/sj.bmt.1702215
94. Kumaki S, Sasahara Y, Kamachi Y, Muramatsu H, Morio T, Goi K, et al. B-cell function after unrelated umbilical cord blood transplantation using a minimal-intensity conditioning regimen in patients with X-SCID. Int J Hematol (2013) 98:355–60. doi: 10.1007/s12185-013-1408-7
95. Díaz de Heredia C, Ortega JJ, Díaz MA, Olivé T, Badell I, González-Vicent M, et al. Unrelated cord blood transplantation for severe combined immunodeficiency and other primary immunodeficiencies. Bone Marrow Transplant (2008) 41:627–33. doi: 10.1038/sj.bmt.1705946
96. Güngör T, Teira P, Slatter M, Stussi G, Stepensky P, Moshous D, et al. Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet (2014) 383:436–48. doi: 10.1016/S0140-6736(13)62069-3
97. Bhatt ST, Schulz G, Hente M, Slater A, Murray L, Shenoy S, et al. A single-center experience using alemtuzumab, fludarabine, melphalan, and thiotepa as conditioning for transplantation in pediatric patients with chronic granulomatous disease. Pediatr Blood Cancer (2020) 67:e28030. doi: 10.1002/pbc.28030
98. Szpecht D, Gorczyńska E, Kałwak K, Owoc-Lempach J, Choma M, Styczyński J, et al. Matched sibling versus matched unrelated allogeneic hematopoietic stem cell transplantation in children with severe acquired aplastic anemia: experience of the polish pediatric group for hematopoietic stem cell transplantation. Arch Immunol Ther Exp (Warsz) (2012) 60:225–33. doi: 10.1007/s00005-012-0174-1
99. Ngwube A, Hayashi RJ, Murray L, Loechelt B, Dalal J, Jaroscak J, et al. Alemtuzumab based reduced intensity transplantation for pediatric severe aplastic anemia. Pediatr Blood Cancer (2015) 62:1270–6. doi: 10.1002/pbc.25458
100. Tan PL, Wagner JE, Auerbach AD, Defor TE, Slungaard A, Macmillan ML, et al. Successful engraftment without radiation after fludarabine-based regimen in Fanconi anemia patients undergoing genotypically identical donor hematopoietic cell transplantation. Pediatr Blood Cancer (2006) 46:630–6. doi: 10.1002/pbc.20538
101. Behfar M, Koochakzadeh L, Yazdanian N, Salajegheh P, Rostami T, Khodayari-Namini N, et al. Outcome of allogeneic Hematopoietic Stem Cell Transplantation on Diamond-Blackfan anemia using busulfan-based myeloablative regimen. Turk J Pediatr (2019) 61:407–12. doi: 10.24953/turkjped.2019.03.013
102. Nelson AS, Marsh RA, Myers KC, Davies SM, Jodele S, O'Brien TA, et al. A Reduced-Intensity Conditioning Regimen for Patients with Dyskeratosis Congenita Undergoing Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant (2016) 22:884–8. doi: 10.1016/j.bbmt.2016.01.026
103. Kothari A, Ngwube A, Hayashi R, Murray L, Davis J, Haut P, et al. Hematopoietic Cell Transplantation Using Reduced-Intensity Conditioning Is Successful in Children with Hematologic Cytopenias of Genetic Origin. Biol Blood Marrow Transplant (2015) 21:1321–5. doi: 10.1016/j.bbmt.2015.03.019
104. Hansen MD, Filipovich AH, Davies SM, Mehta P, Bleesing J, Jodele S, et al. Allogeneic hematopoietic cell transplantation (HCT) in Hurler’s syndrome using a reduced intensity preparative regimen. Bone Marrow Transplant (2008) 41:349–53. doi: 10.1038/sj.bmt.1705926
105. Beam D, Poe MD, Provenzale JM, Szabolcs P, Martin PL, Prasad V, et al. Outcomes of unrelated umbilical cord blood transplantation for X-linked adrenoleukodystrophy. Biol Blood Marrow Transplant (2007) 13:665–74. doi: 10.1016/j.bbmt.2007.01.082
106. Martin HR, Poe MD, Provenzale JM, Kurtzberg J, Mendizabal A, Escolar ML, et al. Neurodevelopmental outcomes of umbilical cord blood transplantation in metachromatic leukodystrophy. Biol Blood Marrow Transplant (2013) 19:616–24. doi: 10.1016/j.bbmt.2013.01.010
107. Chen Y, Xu LP, Zhang XH, Chen H, Wang FR, Liu KY, et al. Busulfan, Fludarabine, and Cyclophosphamide (BFC) conditioning allowed stable engraftment after haplo-identical allogeneic stem cell transplantation in children with adrenoleukodystrophy and mucopolysaccharidosis. Bone Marrow Transplant (2018) 53:770–3. doi: 10.1038/s41409-018-0175-8
108. Souillet G, Guffon N, Maire I, Pujol M, Taylor P, Sevin F, et al. Outcome of 27 patients with Hurler’s syndrome transplanted from either related or unrelated haematopoietic stem cell sources. Bone Marrow Transplant (2003) 31:1105–17. doi: 10.1038/sj.bmt.1704105
109. Tolar J, Grewal SS, Bjoraker KJ, Whitley CB, Shapiro EG, Charnas L, et al. Combination of enzyme replacement and hematopoietic stem cell transplantation as therapy for Hurler syndrome. Bone Marrow Transplant (2008) 41:531–5. doi: 10.1038/sj.bmt.1705934
110. Sauer M, Meissner B, Fuchs D, Gruhn B, Kabisch H, Erttmann R, et al. Allogeneic blood SCT for children with Hurler’s syndrome: results from the German multicenter approach MPS-HCT 2005. Bone Marrow Transplant (2009) 43:375–81. doi: 10.1038/bmt.2008.328
111. Khandelwal P, Yeh RF, Yu L, Lane A, Dandoy CE, El-Bietar J, et al. Graft Versus Host Disease Prophylaxis With Abatacept Reduces Severe Acute Graft Versus Host Disease in Allogeneic Hematopoietic Stem Cell Transplant for Beta Thalassemia Major with Busulfan, Fludarabine, and Thiotepa. Transplantation (2020). doi: 10.1097/TP.0000000000003327
112. King AA, Kamani N, Bunin N, Sahdev I, Brochstein J, Hayashi RJ, et al. Successful matched sibling donor marrow transplantation following reduced intensity conditioning in children with hemoglobinopathies. Am J Hematol (2015) 90:1093–8. doi: 10.1002/ajh.24183
113. Horan JT, Haight A, Dioguardi JL, Brown C, Grizzle A, Shelman C, et al. Using fludarabine to reduce exposure to alkylating agents in children with sickle cell disease receiving busulfan, cyclophosphamide, and antithymocyte globulin transplant conditioning: results of a dose de-escalation trial. Biol Blood Marrow Transplant (2015) 21:900–5. doi: 10.1016/j.bbmt.2015.01.015
114. Gilman AL, Eckrich MJ, Epstein S, Barnhart C, Cannon M, Fukes T, et al. Alternative donor hematopoietic stem cell transplantation for sickle cell disease. Blood Adv (2017) 1:1215–23. doi: 10.1182/bloodadvances.2017005462
115. Maheshwari S, Kassim A, Yeh RF, Domm J, Calder C, Evans M, et al. Targeted Busulfan therapy with a steady-state concentration of 600-700 ng/mL in patients with sickle cell disease receiving HLA-identical sibling bone marrow transplant. Bone Marrow Transplant (2014) 49:366–9. doi: 10.1038/bmt.2013.188
Keywords: transplantation, preparative, childhood and adolescence, hematopoeific stem cells, engrafted survival outcomes
Citation: Hayashi RJ (2020) Considerations in Preparative Regimen Selection to Minimize Rejection in Pediatric Hematopoietic Transplantation in Non-Malignant Diseases. Front. Immunol. 11:567423. doi: 10.3389/fimmu.2020.567423
Received: 29 May 2020; Accepted: 25 September 2020;
Published: 19 October 2020.
Edited by:
Christian Chabannon, Aix-Marseille Université, FranceReviewed by:
Benny Chen, Duke University, United StatesYing-Jun Chang, Peking University People’s Hospital, China
Copyright © 2020 Hayashi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert J. Hayashi, aGF5YXNoaV9yQHd1c3RsLmVkdQ==