 Giacomo Casella1
Giacomo Casella1 Javad Rasouli1
Javad Rasouli1 Rodolfo Thome1
Rodolfo Thome1 Hélène C. Descamps2Asrita Vattikonda1Larissa Ishikawa1Alexandra Boehm1Daniel Hwang1Weifeng Zhang1Dan Xiao1
Hélène C. Descamps2Asrita Vattikonda1Larissa Ishikawa1Alexandra Boehm1Daniel Hwang1Weifeng Zhang1Dan Xiao1 Jeongho Park3,4
Jeongho Park3,4 Guang-Xian Zhang1Jorge I. Alvarez3
Guang-Xian Zhang1Jorge I. Alvarez3 Abdolmohamad Rostami1
Abdolmohamad Rostami1 Bogoljub Ciric1*
Bogoljub Ciric1*- 1Department of Neurology, Thomas Jefferson University, Philadelphia, PA, United States
- 2Department of Microbiology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, United States
- 3Department of Pathobiology, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, PA, United States
- 4College of Veterinary Medicine & Institute of Veterinary Science, Kangwon National University, Chuncheon, South Korea
Antigen (Ag)-specific tolerance induction by intravenous (i. v.) injection of high-dose auto-Ags has been explored for therapy of autoimmune diseases, including multiple sclerosis (MS). It is thought that the advantage of such Ag-specific therapy over non-specific immunomodulatory treatments would be selective suppression of a pathogenic immune response without impairing systemic immunity, thus avoiding adverse effects of immunosuppression. Auto-Ag i.v. tolerance induction has been extensively studied in experimental autoimmune encephalomyelitis (EAE), an animal model of MS, and limited clinical trials demonstrated that it is safe and beneficial to a subset of MS patients. Nonetheless, the mechanisms of i.v. tolerance induction are incompletely understood, hampering the development of better approaches and their clinical application. Here, we describe a pathway whereby auto-Ag i.v. injected into mice with ongoing clinical EAE induces interferon-gamma (IFN-γ) secretion by auto-Ag-specific CD4+ T cells, triggering interleukin (IL)-27 production by conventional dendritic cells type 1 (cDC1). IL-27 then, via signal transducer and activator of transcription 3 activation, induces programmed death ligand 1 (PD-L1) expression by monocyte-derived dendritic cells (moDCs) in the central nervous system of mice with EAE. PD-L1 interaction with programmed cell death protein 1 on pathogenic CD4+ T cells leads to their apoptosis/anergy, resulting in disease amelioration. These findings identify a key role of the IFN-γ/IL-27/PD-L1 axis, involving T cells/cDC1/moDCs in the induction of i.v. tolerance.
Introduction
Autoimmune diseases develop due to a break in immune tolerance toward certain auto-antigens (auto-Ags). It follows that the healthy state could be achieved by restoring peripheral immune tolerance toward those auto-Ags. Devising therapies based on the restoration of Ag-specific immune tolerance induction has been a long-standing goal for treatment of autoimmune diseases, including multiple sclerosis (MS) (1). One such approach relies on intravenous (i.v.) injection of free myelin-derived auto-Ags that are targets of autoimmune response. This approach, and variants thereof, have been proven beneficial in experimental autoimmune encephalomyelitis (EAE), an animal model of MS (1). Some clinical trials have confirmed that repeatedly i.v. injecting large doses of myelin auto-Ag can be safe and beneficial to a subset of MS patients (1). In comparison with non-specific immunomodulatory therapies currently in use, the principal advantage of Ag-specific therapy would be that it suppresses harmful autoimmune response while sparing the rest of the immune system. This would eliminate side effects and adverse events due to systemic immunosuppression caused by non-specific immunomodulation.
Even though certain key players in i.v. tolerance induction, such as interleukin (IL)-10, IL-27, and programmed death ligand 1 (PD-L1), have been identified (2, 3), its complete mechanisms have not been elucidated. This lack of specific knowledge also includes the cell types involved, sequence of their interactions, and relative relevance of the periphery vs. the central nervous system (CNS) in tolerance induction and maintenance over time. A more thorough understanding of these mechanisms will be helpful in developing better Ag-specific therapies for MS and possibly other autoimmune diseases.
Consistent with the suppressive role of IL-10 in EAE development (4), i.v. tolerance induction in EAE requires IL-10. Tolerization by i.v. injection of an auto-Ag elicits IL-10 production, and blockade of IL-10 signaling precludes tolerance induction (2, 5, 6).
The lack of IL-27 signaling leads to the development of more severe EAE (7), and treatment with recombinant IL-27 suppresses EAE (8–10), demonstrating its anti-inflammatory role in EAE. The anti-inflammatory effects of IL-27 encompass inhibition of Th17 cell development; suppression of granulocyte-macrophage colony-stimulating factor (GM-CSF) expression; induction of PD-L1, CD39, and IL-10 expression; and enhancement of Treg development and function (8–10). We have shown that IL-27 is necessary for induction of i.v. tolerance in EAE (3); in particular, IL-27 signaling in DCs was required for tolerance induction, whereas its signaling in T cells was not. IL-27-dependent tolerance induction relied on cooperation of distinct subsets of spleen DCs with the ability to induce T cell-derived IL-10 and interferon-gamma (IFN-γ) (3).
Programmed cell death protein 1 (PD-1) and its ligands, PD-L1 and PD-L2, regulate the balance between T cell activation and immune tolerance (11, 12). The majority of CD4+ T cells in the CNS of mice with EAE express PD-1, while PD-L1 and PD-L2 are differentially expressed by populations of Ag-presenting cells (APC) (13). PD-1−/− and PD-L1−/− mice develop atypically severe EAE, with enhanced T cell proliferation and increased production of inflammatory cytokines (14, 15). Genetic deficiency in PD-L2 did not lead to more severe EAE (14), suggesting that PD-L1 is a dominant inhibitory PD-1 ligand in EAE development. In contrast, blockade of PD-L2, but not PD-L1, in advanced EAE in C57BL/6 mice led to worsening of disease, indicating that these two PD-1 ligands, or possibly cell types that express them, have distinct roles in regulating different stages of EAE (2, 16, 17). In regard to i.v. tolerance induction in EAE, it has been shown that tolerization induces PD-L1 expression by APCs and that PD-1 blockade abrogates tolerance induction (2, 18).
IFN-γ is a cytokine released by almost all activated immune cells, with natural killer (NK) and T cells being its major sources (19). Although IFN-γ has been traditionally considered a pro-inflammatory cytokine, it is now clear that IFN-γ also has prominent anti-inflammatory roles that balance its possibly damaging inflammatory effects (20). Numerous studies have firmly established that IFN-γ suppresses EAE; mice lacking IFN-γ signaling develop severe EAE, and mouse strains resistant to EAE become susceptible (21–23). Consistent with this, IFN-γ production by myelin-specific CD4+ T cells is not required for their encephalitogenicity (24), IFN-γ-deficient CD4+ T cells could be notably more pathogenic than their IFN-γ-sufficient counterparts (25), and IFN-γ production by encephalitogenic T cells in the CNS is required for recovery from EAE (26). Further, an increase in IFN-γ levels in the CNS of mice with EAE leads to disease suppression (27, 28). Taken together, these findings indicate that IFN-γ could be important in EAE suppression by i.v. tolerance induction as well, a possibility that has not been explored.
Here we show that i.v. administration of auto-Ag (free encephalitogenic peptide) halts EAE progression by inducing PD-L1 expression in CNS monocyte-derived dendritic cells (moDCs) via an IFN-γ/IL-27-dependent mechanism. Blockade of PD-L1, but not PD-L2, or the lack of PD-1 in CD4+ T cells precluded i.v. tolerance induction. The lack of IFN-γ in CD4+ T cells, or IFN-γR in conventional DCs type 1 (cDC1), abrogated IL-27 production and PD-L1 expression by moDCs. Collectively, our data reveal a mechanism of Ag-dependent induction of PD-L1 expression in moDCs that in turn suppresses Ag-specific Th cell responses and ameliorates CNS autoimmunity.
Materials and Methods
Mice
C57BL/6, B6.Ly5.1 (CD45.1+), RAG1−/−, PD-1−/−, 2D2, Zbtb46-iDTR, IFN-γ−/−, IFN-γRα−/−, GREAT (IFN-γ reporter), Ccr2−/−, Wsx−/−, and Stat3mut mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). IL-27p28 reporter mice were a gift of Dr. Ross M. Kedl (University of Colorado). Mice were kept in specific pathogen-free conditions with a maximum of 5 mice per cage, in 12/12 h of light/dark cycles and food ad libitum throughout the experimental procedures. Every effort was made to minimize suffering of mice. Experimental protocols using mice were approved by the Institutional Animal Care and Use Committee of Thomas Jefferson University.
Generation of BMDCs
STAT1−/−, STAT3−/−, and WT BMDCs were generated according to a previously described protocol (3). Briefly, BM cells were seeded at 2 × 106 cells/mL in Petri dish in complete IMDM supplemented with 100 ng/mL of recombinant mouse Flt-3 (R&D Systems, Minneapolis, MN, USA). Culture medium was changed every 3 days. Maturation of the DCs was induced with LPS (100 ng/mL) for 16 h. At day 9 after starting the culture, DCs were enriched by anti-Flt-3-biotin Ab and anti-biotin microbeads (Miltenyi Biotec, CA, USA), and CD11c+MHCII+ cells were then FACS-sorted.
IFN-γRα−/− and WT cDCs were generated from BM cells following a protocol described in (29) with slight modifications.
Ag-presentation Assays
Naive CD4+ T cells from spleens of 2D2 mice were isolated using magnetic beads (Naive CD4+ T cell isolation kit, Miltenyi Biotec, CA, USA). 2 × 105 naive CD4+ T cells were added to each well of the cell culture plate containing moDCs, cDC1, or BMDCs (ratio of 1 DC: 10 T cells) and plates were incubated at 37°C in the presence of MOG35−55 peptide (20 μg/mL) and anti-PD-L1 MAb (1 μg/mL; clone 10F.9G2, BioXCell). Cells were collected after 72 h and analyzed by flow cytometry, while cytokine concentrations in culture supernatants were measured by ELISA.
EAE and i.v. Tolerance Induction
Anesthetized mice were subcutaneously injected with 200 μL of an emulsion containing 200 μg of MOG35−55 peptide (MEVGWYRSPFSRVVHLYRNGK, Genscript, NJ, USA) in PBS and equal volume of Complete Freund's adjuvant supplemented with 10 mg/mL of heat-killed Mycobacterium tuberculosis H37Ra. Additionally, mice were intraperitoneal (i.p.) injected with 200 ng of pertussis toxin at immunization time and 48 h later. Mice were weighed and scored for clinical signs daily. Clinical assessment of EAE was performed according to the following scoring criteria: 0, healthy; 1, limp tail; 2, ataxia and/or paresis of hindlimbs; 3, paralysis of hindlimbs and/or paresis of forelimbs; 4, tetraparalysis; and 5, moribund or death (30).
i.v. tolerance was induced in mice after onset of clinical disease by injections of 200 μg MOG35−55 in PBS every third day, 3 times in total. Control mice received PBS only (3).
Bone Marrow Chimeras
B6.Ly5.1 (CD45.1+) congenic mice were lethally irradiated with 2 × 2.5 Gy with an 8 h interval between irradiation and were then i.v. injected with 5 × 106 CD45.2+ BM cells from WT, or Zbtb46-DTR donors. Recipient mice were in other experiments reconstituted with 1:1 mixture (total 1 × 107 cells) of BM cells from Wsx−/− and Ccr2−/− mice, or with mixture of BM cells from Stat3mut and Ccr2−/− mice. Mice were allowed to reconstitute for 6–8 weeks prior to use.
DT Ablation
Diphtheria toxin (DTX; Sigma-Aldrich) was administered i.p. at 1 μg/20 g of mouse weight in 200 μl of PBS, 1 day before i.v. injection of MOG35−55. Mice received three injections of DTX once every 3 days.
PD-L1, PD-L2, and IFN-γ Blockade
Mice with EAE were i.p. injected with 200 μg/mouse of αPD-L1 MAb (clone 10F.9G2, BioXCell), or with 200 μg/mouse of αPD-L2 MAb (clone TY25, BioXCell), or with 150 μg/mouse of αIFN-γ MAb (clone R4-6A2, BioXCell) 1 day before i.v. injection of MOG35−55. Mice received two MAb injections, 3 days apart in each treatment.
Ag-Specific Recall Response
Spleens of mice with EAE were dissociated through a 70 μm strainer to prepare single-cell suspensions in complete IMDM, containing 10% heat-inactivated fetal bovine serum, penicillin (100 U), streptomycin (10 μg/mL), L-glutamine (0.3 mg/mL), and 2-mercaptoethanol (55 μM). After treatment with RBC lysis buffer (Biolegend, CA, USA), cells were extensively washed with complete IMDM by centrifugation at 1,300 rpm for 5 min at 4°C and the cell density was adjusted to 2 × 106 /mL. One hundred microliter of adjusted cell suspension was added to each well of a 96-well plate. MOG35−55 was added to a final concentration of 20 μg/mL. Cells were incubated at 37°C for 3 days. As negative control, cells were cultured without MOG35−55. Cell culture supernatants were collected and stored at −20°C until use, and cells were analyzed for proliferation and cytokine production by flow cytometry.
Reconstitution of WT and RAG1–/– Mice
WT mice with EAE received i.v. 2 × 106 FACS-sorted CD11b+ CD11c+ Ly6chigh MHCII+ cells from the CNS of WT mice with EAE previously i.v. injected with MOG35−55 or PBS. CD45.1+ mice reconstituted with CD45.2+ BM cells from WT, or Zbtb46-iDTR donors, received i.v. 2 × 106 of in vitro Flt-3-differentiated cDCs. RAG1−/− mice were i.v. reconstituted with 5 × 106 magnetic bead-isolated total CD4+ T cells from spleens of WT, PD1−/−, or IFN-γ−/− mice. After 72 h of adoptive transfer, mice were immunized for EAE induction.
Isolation of CNS Infiltrating Leukocytes
Brain and spinal cord tissues were incubated for 30 min at 37°C with 0.4 mg/mL type IV collagenase (Sigma-Aldrich) and dissociated by passing through a 19-gouge needle. Cells were enriched by centrifugation on a Percoll gradient as previously described (31).
Flow Cytometry and Cell Sorting
Flow cytometry was performed using a FACSaria II (Becton Dickinson) and analyzed with FlowJo software (Tree Star). Fluorochrome-conjugated MAbs specific for: CD45 (clone 30-F11), CD45.1 (A20), CD11b (M1/70), CD3 (17A2), CD8α (53-6.7) CD4 (RM4-5), CD19 (1D3/CD19), CD11c (N418), CD26 (H194-112), CD88 (20/70), CD172α (P84), PDCA1 (927), Ly6c (AL-21), F4/80 (MB8), Ly6g (1A8), MHC-II (M5/114.15.2), PD-1 (29F.1A12), PD-L1 (10F.9G2), PD-L2 (TY25), Caspase 3, and Annexin V were purchased either from BD Biosciences, R&D, Biolegend, Santa Cruz, or Abcam.
For intracellular staining, cells were stimulated for 4 h with phorbol 12-myristate 13-acetate (PMA; 50 ng/ml, Sigma-Aldrich) and ionomycin (500 ng/ml, Sigma-Aldrich) in the presence of GolgiPlug (1:1,000, BD Pharmigen), permeabilized using a Cytofix/Cytoperm Plus kit (BD Bioscience) and stained with the following fluochrome-conjugated MAbs: GM-CSF (MP1-22E9), IL-17A (TC11-18H10.1), and IFN-γ (XMG1.2) from Biolegend and BD Pharmingen. Dead cells were excluded using L/D stain (BD Pharmingen). Data were acquired on a FACSAria Fusion (BD Biosciences) and analyzed using FlowJo software (TreeStar).
ELISA
Supernatants from cell cultures were kept at −20°C until use. Cytokine concentrations in culture supernatants were measured with sandwich enzyme-linked immunosorbent assay (ELISA) using commercial kits, following the manufacturer's recommendation (R&D Systems, Minneapolis, MN, USA).
qPCR
Total RNA was extracted from moDCs, CD4+ T cells, and cDC1 with RNeasy Mini Kit (Qiagen), whereas from total CNS and spleen with Trizol (Invitrogen). Genomic DNA was removed by treatment with DNAse I type (Qiagen). cDNA synthesis was performed using ThermoscriptTM RT-PCR system (Invitrogen). Apoptosis (cat# 4413255), Jak/Stat signaling (cat# 4391524) arrays, Il27ra, (Mm00497259_m1), and Gapdh (4352339E). mRNA levels were measured by real-time RT-PCR (Applied Biosystems, Invitrogen). The 2–ΔΔCT method was used to calculate relative changes in gene expression (32).
Statistical Analysis
Statistical analysis was performed by GraphPad Prism 8 software. Statistical evaluations are expressed as mean ± s.d. or mean ± s.e.m., as appropriate. Results were analyzed using Two- or One-way ANOVA and posttested with Bonferroni, and with unpaired, two-tailed Student's t-test. Statistical significance was ranked *p < 0.05; **p < 0.001; ***p < 0.0001; ****p < 0.00001.
Results
Intravenous Tolerance Induction in Experimental Autoimmune Encephalomyelitis Is Dependent on Programmed Death Ligand 1 and Programmed Cell Death Protein 1, but Not on Programmed Death Ligand 2
We and others have reported that i.v. delivery of auto-Ag in mice with EAE induces expression of PD-L1 by APCs (2, 3). Given the importance of PD-1 and its ligands in immune tolerance, we investigated their role in i.v. tolerance induction in EAE. Mice were immunized with myelin oligodendrocyte glycoprotein (MOG)35−55 for EAE induction and i.v. injected with 200 μg of MOG35−55 in phosphate-buffered saline (PBS), or with PBS, at the onset of clinical disease; two more doses of MOG35−55 and PBS were injected 3 days apart (Supplementary Figures 1A,B). Mice were sacrificed at 21 days post immunization (d.p.i.), and cells isolated from their CNS were analyzed by flow cytometry. t-distributed stochastic neighbor embedding (t-SNE) analysis (33) identified eight populations of CD45+ MHCII+ cells: cDC1, cDC2, microglia, plasmacytoid DCs (pDCs), moDCs, macrophages (MΦ), neutrophils, and B cells (Figure 1A). MOG35−55-treated mice had increased numbers of PD-L1+ and PD-L2+ cells compared with PBS-treated mice. Next, we investigated which of these cells expressed PD-L1 and PD-L2. moDCs (75%) were the bulk of PD-L1+ cells, whereas in control mice, cDC1 (58%) and cDC2 (33%) were the most abundant PD-L1+ cells (Figure 1B). In mice that received MOG35−55, PD-L2 was mostly expressed by neutrophils (50%) and MΦ (35%), whereas in control mice, PD-L2 was expressed by microglia (64%) and cDC2 (34%) (Figure 1C). These data show that i.v. delivery of auto-Ag induces a robust PD-L1 expression by moDCs and PD-L2 expression by neutrophils and MΦ. We also found increased numbers of PD-L1+ and PD-L2+ cells among lymph node and splenic cells of MOG35−55-tolerized mice (data not shown).
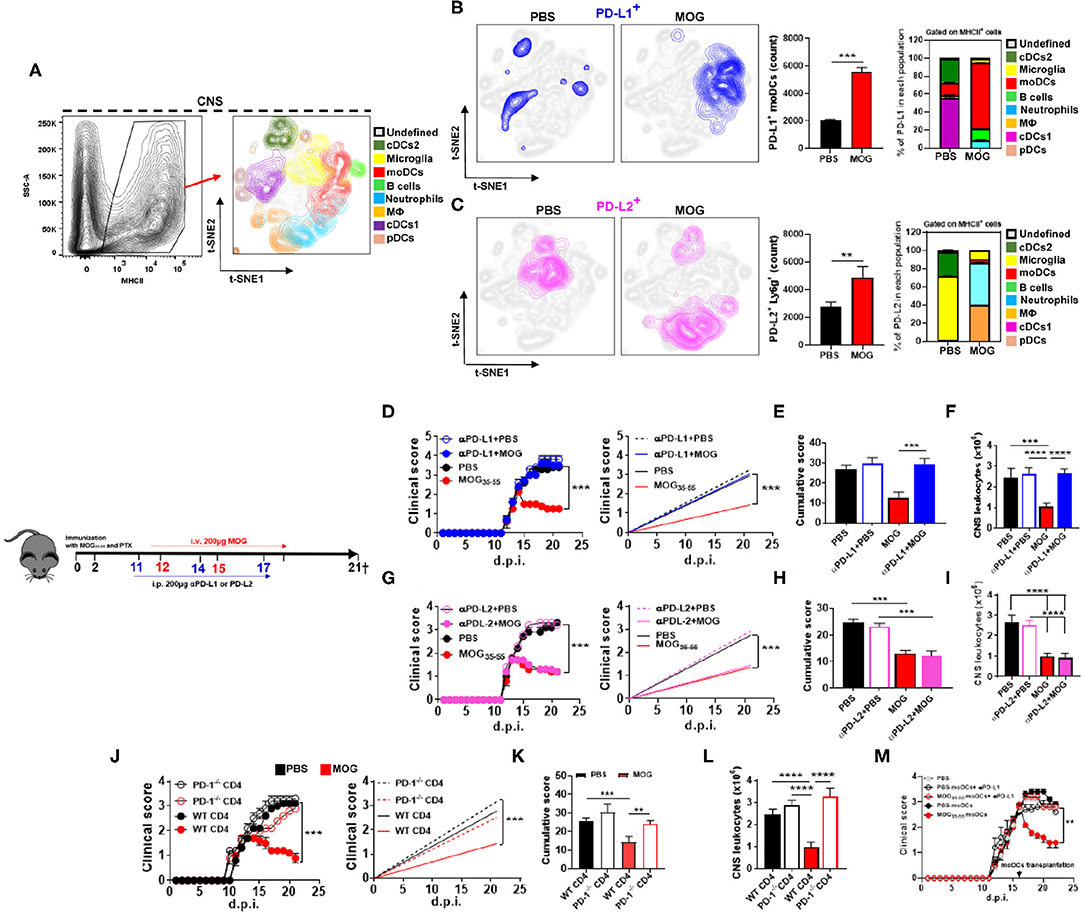
Figure 1. Intravenous administration of myelin oligodendrocyte glycoprotein (MOG)35−55 induces programmed death ligand 1 (PD-L1) expression in central nervous system (CNS) monocyte-derived dendritic cells (moDCs) and suppresses experimental autoimmune encephalomyelitis (EAE) in a programmed cell death protein 1 (PD-1)/PD-L1 manner. C57BL/6 wild-type (WT) mice (n = 10/group each experiment) were immunized with MOG35−55 for EAE induction and starting from disease onset i.v. injected with 200 μg of MOG35−55 in PBS every 3 days. Mice were sacrificed 21 days post immunization (d.p.i.) and CD45+ MHCII+ cells from the CNS were analyzed by flow cytometry. (A) Eight populations were identified: microglia (CD11b+ CD45low), monocytes (CD11b+ CD11c+ Ly6chigh Ly6g−), macrophages (CD11b+ Ly6clow Ly6g− F4/80high), cDC1 (CD11b+ CD11c+ Ly6cmed CD26+), cDC2 (CD11b+ CD11c+ Ly6cmed CD172α+), plasmacytoid DCs (pDCs; Lin− PDCA1high), and B cells (CD19+). (B,C) t-SNE graphs showing the expression and percentage of PD-L1+ (B) and PD-L2 (C) cells from the CNS of PBS- or MOG35−55-treated EAE mice. (D) Mice with EAE (n = 5–7/group in each experiment) were i.p. injected with blocking anti-PD-L1 MAb (200 μg/mouse/injection; clone 10F.9G2), or isotype control MAb, on 11, 14, and 17 d.p.i. MOG35−55 or PBS was i.v. injected on 12, 15, and 18 d.p.i. (E) Cumulative disease score for mice described in (D). (F) Mice described in (D) were sacrificed at day 21 p.i. and the numbers of CD45+ leukocytes obtained from their CNS were determined by flow cytometry. (G) Mice with EAE (n = 5–7/group in each experiment) were i.p. injected with blocking anti-PD-L2 MAb (200 μg/mouse/injection; clone TY25), or isotype control MAb, as described in (D). (H) Cumulative disease score for mice described in (G). (I) Mice described in (G) were sacrificed at day 21 p.i. and the numbers of CD45+ leukocytes obtained from their CNS were determined by flow cytometry. (J) RAG1−/− mice were reconstituted with 5 × 106 total CD4+ T cells from WT or PD-1−/− mice (n = 10/group in each experiment); 72 h post reconstitution, recipient mice were immunized for EAE induction and MOG35−55 or PBS was injected i.v. three times, starting from EAE onset. (K) Cumulative disease score for mice described in (J). (L) Mice described in (J) were sacrificed at day 21 p.i. and the numbers of CD45+ leukocytes obtained from their CNS were determined by flow cytometry. (M) Recipient WT mice with EAE were transplanted at the peak of disease with 2 × 106 moDCs from the CNS of donor mice with EAE that were previously i.v. tolerized with MOG35−55 or PBS (n = 5/group in each experiment). Groups of recipient mice with EAE received moDCs that were pretreated for 1 h with anti-PD-L1 MAb (1 μg/ml). All data are representative of at least two experiments and symbols depict mean ± SEM. Analyses between two groups were carried out by Student's t-test and between four groups by one-way ANOVA with Bonferroni post-test (E,F,H,I,K,L). EAE experiments were analyzed by two-way ANOVA with Bonferroni's multiple comparison. Values of **P < 0.001, ***P < 0.0001, and ****P < 0.00001 were considered significant.
We then tested the role of PD-L1 and PD-L2 in tolerance induction by i.p. injecting anti-PD-L1 or anti-PD-L2 MAbs 24 h before i.v. injecting MOG35−55. Treatment with anti-PD-L1 MAb precluded tolerance induction (Figures 1D,E) and led to an increase in the number of leukocytes in the CNS (Figure 1F), whereas treatment with anti-PD-L2 MAb did not have an effect (Figures 1G–I). Further, we transplanted PD-1−/− or wild-type (WT) CD4+ T cells into RAG1−/− mice and induced EAE in them. MOG35−55/i.v. treatment suppressed EAE in mice with WT CD4+ T cells, but not in mice with PD-1−/− CD4+ T cells (Figures 1J–L). These data demonstrate that PD-L1 and PD-1 are critical for i.v. tolerance induction in ongoing EAE, whereas PD-L2 does not play a role.
Monocyte-Derived Dendritic Cells From the Central Nervous System of Tolerized Mice With Experimental Autoimmune Encephalomyelitis Are Suppressive via Programmed Death Ligand 1
Given that tolerized mice had increased numbers of apoptotic annexin V+ CD4+ T cells (Supplementary Figures 1C,D) and PD-L1+ moDCs in the CNS (Figure 1B), we investigated a correlation between their numbers. There was a robust positive correlation of apoptotic CD4+ T cells with PD-L1+ moDCs (Supplementary Figure 1E), but not with PD-L2+ moDCs (Supplementary Figure 1F) or PD-L2+ neutrophils (data not shown). This suggests that PD-L1+ moDCs mediate apoptosis of T cells.
We next evaluated the effect of moDCs on myelin-specific CD4+ T cells ex vivo. MHCII+ Ly6chigh moDCs were sorted from the CNS of mice with EAE treated with MOG35−55/i.v. and co-cultured with naive CD4+ T cells expressing a transgenic T cell receptor for MOG35−55 from 2D2 mice. Anti-PD-L1 MAb was added in some co-cultures. In comparison with moDCs from control mice, moDCs from MOG35−55-tolerized mice induced lower T cell proliferation and lower GM-CSF and IL-17A production, but greater IFN-γ and PD-1 expression and annexin V staining by T cells (Supplementary Figures 1G–I). We also measured a larger quantity of regulatory cytokines, IL-27 and IL-10, in supernatants of these co-cultures compared with controls (Supplementary Figure 1J). In the presence of anti-PD-L1 MAb, moDCs did not reduce T cell proliferation or induce their apoptosis, demonstrating that PD-L1 expressed by moDCs limits myelin-specific CD4+ T cell responses in vitro.
To validate the immunosuppressive phenotype of MOG35−55/i.v.-induced moDCs in vivo, we transplanted moDCs from the CNS of tolerized mice with EAE into mice with ongoing EAE. Control mice received either moDCs from PBS/i.v. mice, PBS, or moDCs from MOG35−55/i.v. mice that were pretreated for 1 h with anti-PD-L1 MAb. The transfer of moDCs from MOG35−55-treated donors, but not from those that were PBS-treated, led to recovery from the disease, and anti-PD-L1 MAb pretreatment of moDCs from MOG35−55/i.v. donors precluded their suppressive effect on EAE in recipient mice (Figure 1M). These data show that moDCs of MOG35−55-treated mice with EAE are suppressive via PD-L1 in vitro and in vivo.
Interferon-γ Secreted by CD4+ T Cells Is Necessary for Experimental Autoimmune Encephalomyelitis Suppression Upon Myelin Oligodendrocyte Glycoprotein35−55/Intravenous Treatment
Given that IFN-γ suppresses EAE (28, 34) and induces PD-L1 expression (35), we tested whether IFN-γ plays a role in tolerance induction. We injected anti-IFN-γ MAb into mice after EAE onset, 24 h before MOG35−55/i.v. injection (Figure 2A). Anti-IFN-γ-treated mice developed a severe disease that did not respond to MOG35−55/i.v. treatment (Figures 2B,C). These mice had markedly increased numbers of leukocytes in the CNS compared with control MOG35−55/i.v.-treated mice (Figure 2D). Anti-IFN-γ-treated mice also had lower expression of PD-L1+ in moDCs, reduced numbers of apoptotic CD4+ T cells, and greater frequencies of GM-CSF+ IL-17A+ CD4+ T cells (Figures 2E–I). Consistent with this observation, we found higher concentrations of GM-CSF and IL-17A in culture supernatants from MOG35−55-stimulated splenocytes of the above anti-IFN-γ-treated mice compared with controls (Figures 2J,K); however, while MOG35−55/i.v. treatment resulted in an increase of IL-10, and especially IL-27 production, treatment with anti-IFN-γ MAb precluded these increases (Figures 2L,M). We next determined the kinetics of IFN-γ expression upon MOG35−55 injection into mice with EAE. Ifng mRNA levels, both in the CNS and spleen, were highest at 6 h after the injection and declined to base levels by 12 h post injection (Figures 3A,B). These data show that IFN-γ plays a critical role in the induction of i.v. tolerance in EAE.
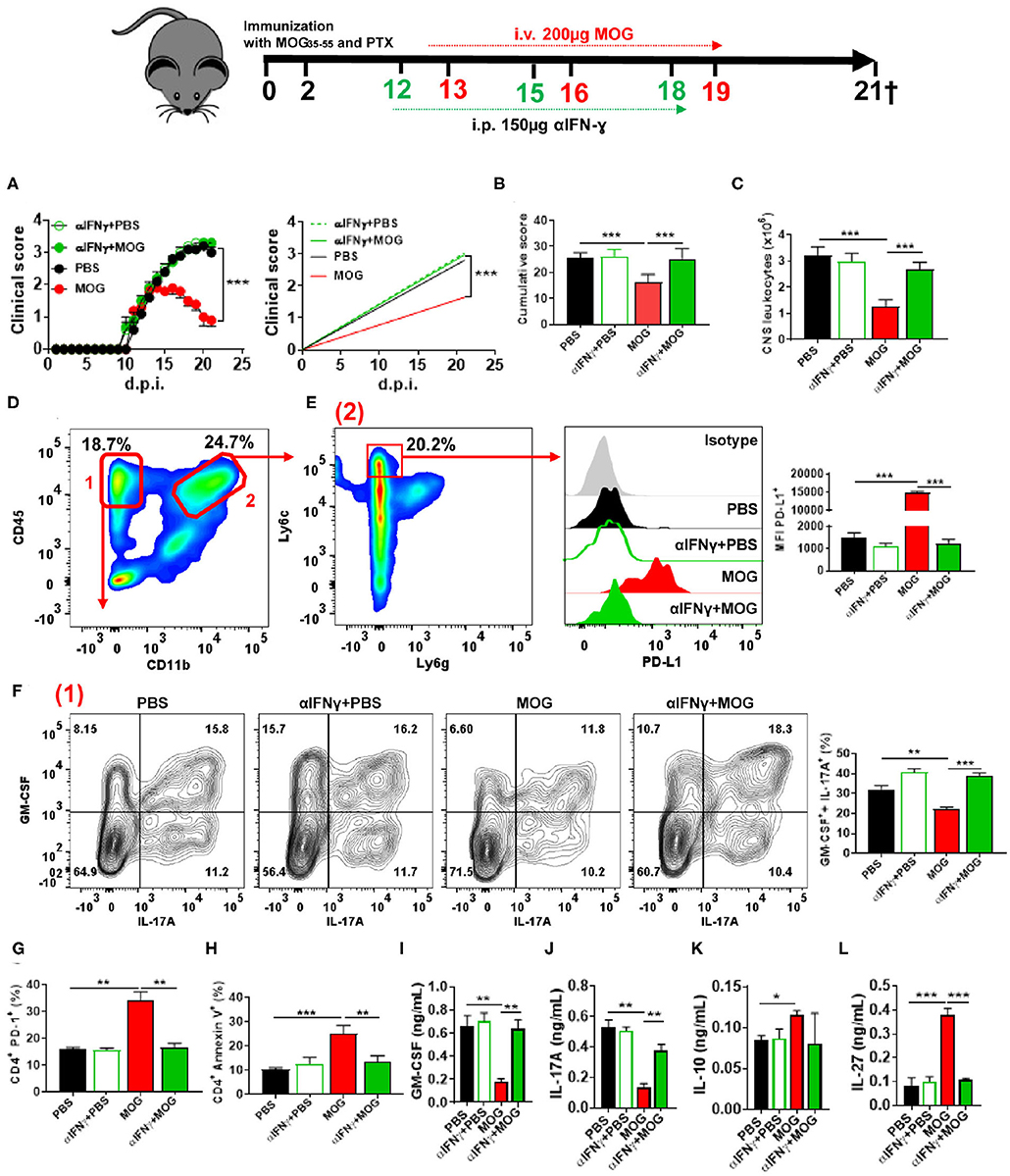
Figure 2. IFN-γ is critical for i.v. tolerance induction in EAE. (A) Mice with EAE (n = 5–7/group in each experiment) were i.p. injected with blocking anti-IFN-γ MAb (150 μg/mouse/injection; clone H22), or isotype control MAb, on 12, 15, and 18 d.p.i. MOG35−55 or PBS was i.v. given on 13, 16, and 19 d.p.i. (B) Cumulative disease score for mice described in (A). (C) Mice described in (A) were sacrificed on day 21 p.i. and the numbers of CD45+ leukocytes in their CNS were determined by flow cytometry. (D) Flow cytometry plot showing lymphoid (1) and infiltrating myeloid (2) cells from the CNS of mice shown in (A). (E) MFI of PD-L1+ moDCs from the CNS of mice described in (A). Frequencies of GM-CSF+ IL-17A+ (F), PD-1+ (G), and annexin V+ (H) CD4+ T cells from the CNS of mice described in (A). (J–M) Cytokine concentrations in supernatants from cultures of spleen cells from mice described in (A) stimulated for 72 h with MOG35−55. Cytokine concentrations were measured by ELISA. All data are representative of at least two experiments, and symbols depict mean ± SEM. Analysis between four groups was carried out by one-way ANOVA with Bonferroni post-test. EAE experiments in (A) were analyzed by two-way ANOVA with Bonferroni's multiple comparison. Values of *P < 0.05, **P < 0.001, and ***P < 0.0001 were considered significant.
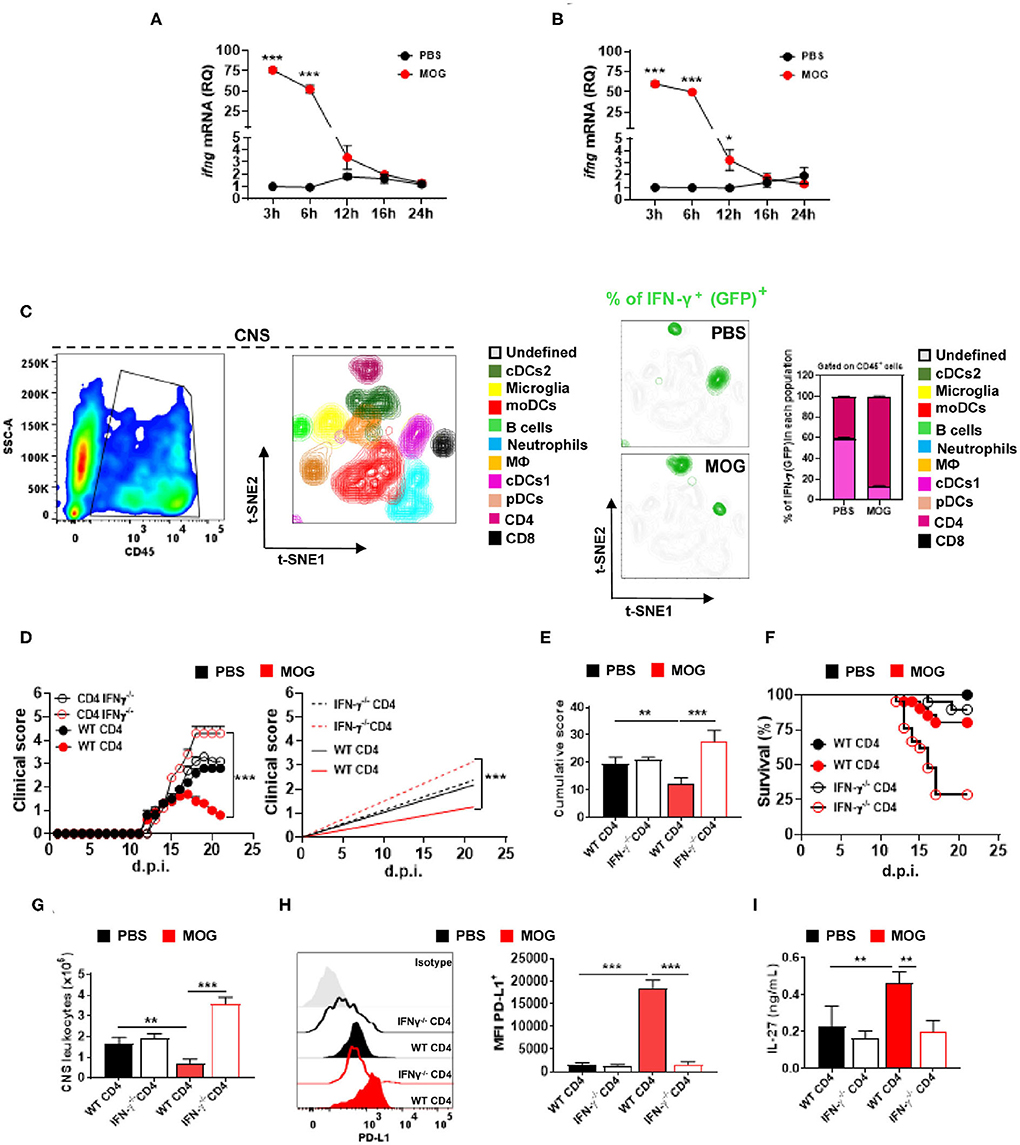
Figure 3. CD4+ T cell-derived IFN-γ is critical for i.v. tolerance induction in EAE. Time course of Ifng mRNA, analyzed by qPCR, in the spleen (A) and CNS (B) after treating mice with EAE i.v. with MOG35−55 peptide (200 μg) or PBS (n = 5/group in each experiment). (C) GFP/IFN-γ reporter mice were immunized for EAE induction and i.v. injected with MOG35−55 or PBS at disease onset (n = 5/group in each experiment). Mice were sacrificed 3 h post injection and CD45+ cells from the CNS were analyzed by flow cytometry. t-SNE graphs showing the expression and percentage of IFN-γ+ (GFP+) among these cells. Ten populations were identified: microglia (CD11b+ CD45low), monocytes (CD11b+ CD11c+ Ly6chigh MHCIIhigh Ly6g−), macrophages (CD11b+ Ly6clow Ly6g− MHCII+ F4/80high), cDC1 (CD11b+ CD11c+ Ly6cmed MHCIIhigh CD26+), cDC2 (CD11b+ CD11c+ Ly6cmed MHCII+ CD172α+), pDCs (Lin− PDCA1high), B cells (CD19+ MHCII+), CD8+ T cells (CD11b− CD3+ CD8α+), and CD4+ T cells (CD11b− CD3+ CD4+). (D) RAG1−/− mice were reconstituted with 5 × 106 total CD4+ T cells from WT or IFN-γ−/− mice (n = 10/group in each experiment); 72 h post reconstitution, recipient mice were immunized for EAE induction and MOG35−55 or PBS was given i.v. three times, starting at EAE onset. (E) Cumulative disease score for mice shown in (D). (F) Survival (%) of EAE mice treated described in (D). (G) Mice described in (D) were sacrificed 21 d.p.i. and the numbers of CD45+ leukocytes in their CNS were determined by flow cytometry. (H) MFI of PD-L1+ moDCs from the CNS of mice shown in (D). (I) Splenocytes from mice described in (D) were stimulated with MOG35−55 for 72 h. Concentrations of IL-27 in culture supernatants were measured by ELISA. All data are representative of at least two experiments, and symbols depict mean ± SEM. Analyses between two groups were carried out by Student's t-test, and analyses between four groups by one-way ANOVA with Bonferroni post-test. EAE experiments in (D) were analyzed by two-way ANOVA with Bonferroni's multiple comparison. Values of *P < 0.05, **P < 0.001, and ***P < 0.0001 were considered significant.
To identify the cellular sources of MOG35−55/i.v.-induced IFN-γ, we induced EAE in IFN-γ reporter mice (express GFP from the IFN-γ gene), injected them with MOG35−55/i.v. at disease onset, and 3 h after the injection analyzed their CNS. IFN-γ was primarily produced by CD4+ T cells, whereas in control mice injected with PBS, both cDCs and CD4+ T cells produced IFN-γ (Figure 3C). We then investigated whether CD4+ T cells are a relevant source of IFN-γ in tolerance induction. To this end, we reconstituted RAG1−/− mice with WT or IFN-γ−/− CD4+ T cells and immunized them for EAE induction; mice received PBS or MOG35−55/i.v. at disease onset. MOG35−55/i.v. significantly suppressed disease in mice with WT CD4+ T cells, whereas it exacerbated the disease in mice with IFN-γ−/− T cells (70% of mice died) (Figures 3D,E). Consistent with clinical outcome, mice with IFN-γ−/− T cells had reduced PD-L1+ expression in CNS moDCs compared with mice with WT T cells (Figure 3H). Moreover, splenocytes of mice with IFN-γ−/− T cells upon in vitro activation produced significantly less IL-27 compared with control mice (Figure 3I). These data show that IFN-γ secretion by CD4+ T cells is required for EAE suppression by MOG35−55/i.v. treatment.
The Lack of Interferon-γ Signaling in Conventional Dendritic Cells Type 1 Precludes Their Interleukin-27 Expression in Intravenous Tolerance Induction in Experimental Autoimmune Encephalomyelitis
Blocking IFN-γ resulted in reduced IL-27 production, prompting us to search for the cellular source of IL-27 upon MOG35−55/i.v. treatment. We induced EAE in IL-27 reporter mice (express GFP from the IL-27p28 gene) and injected them with MOG35−55 at disease onset. IL-27 production in the spleen and CNS peaked at 16 h after the injection (Figures 4A,B), and among cells from the CNS, the primary IL-27-producing cells were cDC1 (~80% of GFP+ cells), while in PBS-treated mice, moDCs also produced IL-27 (Figure 4C).
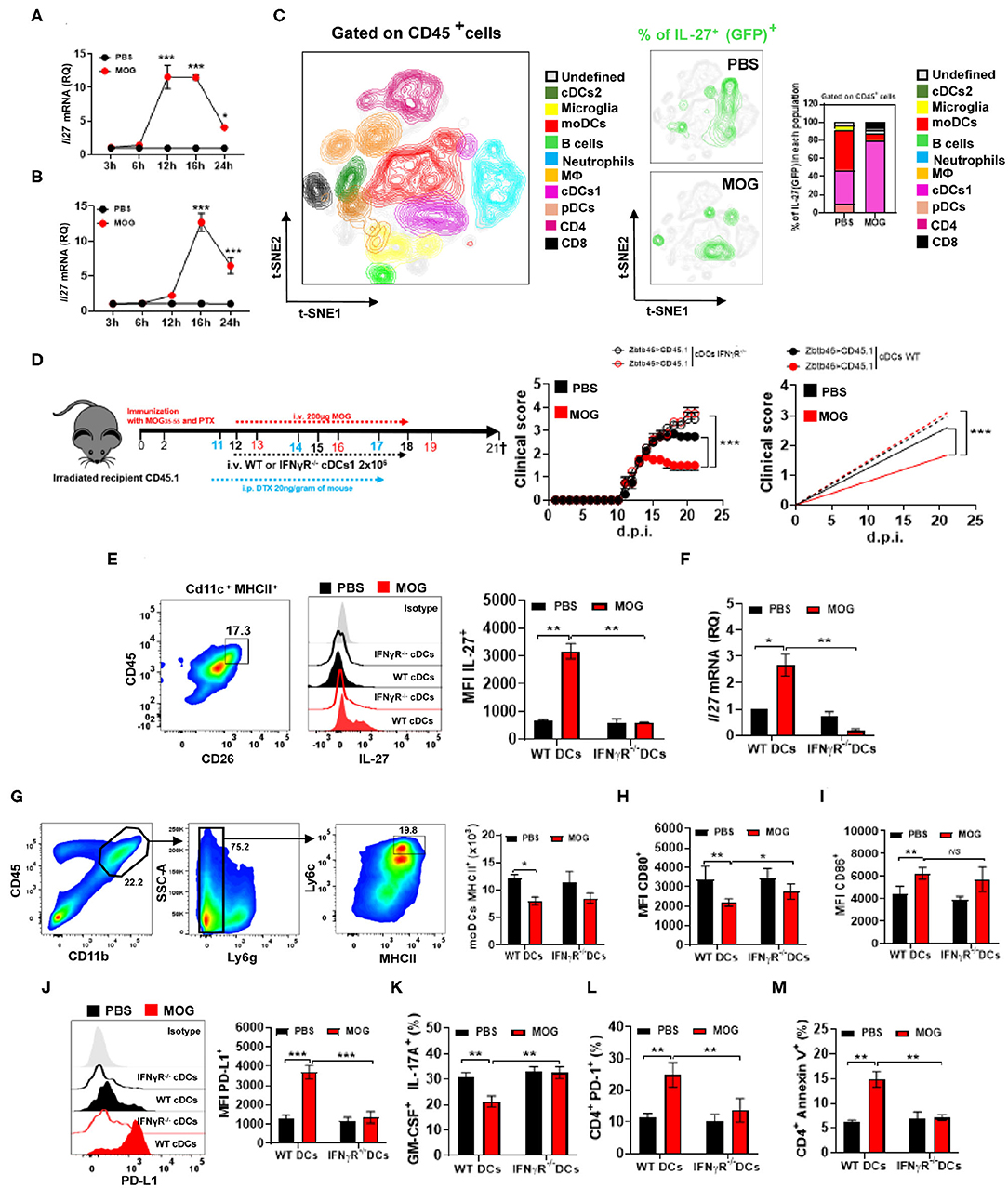
Figure 4. The lack of IFN-γ signaling in cDC1 abrogates their IL-27 production upon i.v. tolerance induction in EAE. Time course of Il27 mRNA expression analyzed by qPCR, in the spleen (A) and CNS (B) 24 h after treating mice with EAE i.v. with MOG35−55 or PBS (n = 5/group in each experiment). (C) GFP/IL-27p28 reporter mice were immunized with MOG35−55 for EAE induction and i.v. injected with MOG35−55 or PBS at disease onset (n = 5/group in each experiment). Mice were sacrificed 16 h post injection and cells from the CNS analyzed by flow cytometry. Ten CD45+ cell populations were identified: microglia (CD11b+ CD45low), monocytes (CD11b+ CD11c+ Ly6chigh MHCIIhigh Ly6g−), macrophages (CD11b+ Ly6clow Ly6g− MHCII+ F4/80high), cDC1 (CD11b+ CD11c+ Ly6cmed MHCIIhigh CD26+), cDC2 (CD11b+ CD11c+ Ly6cmed MHCII+ CD172α+), pDCs (Lin− PDCA1high), B cells (CD19+ MHCII+), CD8+ T cells (CD11b− CD3+ CD8α+), and CD4+ T cells (CD11b− CD3+ CD4+). t-SNE graphs show the expression and percentage of IL-27p28+ (GFP+) cells. (D) CD45.2+ mice were irradiated and transplanted with Zbtb46-iDTR or CD45.1+ BM and 6–8 weeks later immunized with MOG35−55 (n = 7–10/group in each experiment). cDC depletion (Zbtb46+ MHCII+ CD11c+) was accomplished by i.p. injecting DTX (20 ng/g of mouse weight) every third day after EAE onset. In vitro Flt-3-differentiated BMDCs, IFN-γRα−/−, or WT was i.v. transferred 1 day post DTX injection, in total twice. MOG35−55 or PBS was i.v. injected at 13, 16, and 19 d.p.i. (E) At 21 d.p.i, cDC1 (CD11b+ CD11c+ CD45+ Ly6cmed MHCIIhigh CD26+) were isolated from the CNS of EAE mice described in (D) and their IL-27p28 production was analyzed by flow cytometry and (F) by qPCR. The numbers of MHCII+ (G), CD80+ (H), CD86+ (I), and MFI of PD-L1+ (J) in moDCs from the CNS of mice described in (D). Frequencies of GM-CSF+ IL-17A+ (K), PD-1+ (L), and annexin V+ (M) CD4+ T cells from the CNS of mice described in (D). All data are representative of at least two experiments, and symbols depict mean ± SEM. Analysis between two groups was carried out by Student's t-test, whereas analysis between four groups was carried out by one-way ANOVA with Bonferroni post-test. EAE experiments in (D) were analyzed by two-way ANOVA with Bonferroni's multiple comparison. Values of *P < 0.05, **P < 0.001, and ***P < 0.0001 were considered significant.
We next investigated whether the lack of IFN-γ signaling in cDC1 affects IL-27 production and compromises tolerance induction. We generated a Zbtb46-DTR (CD45.2+)→ CD45.1+ bone marrow (BM) chimera mice in which cDC1 can be depleted with diphtheria toxin (DTX) (36) (Supplementary Figure 2A). WT (CD45.2+)→ CD45.1+ BM chimeras served as control. We immunized chimera mice for EAE induction and depleted cDC1 by administering DTX, starting at disease onset and then every other day. We then transplanted in vitro Flt3-differentiated WT or IFN-γR−/− cDC1 into these DTX-treated chimera mice with EAE (Figure 4D). Mice were treated with DTX and transplanted with cDC1 twice. Moreover, 24 h post cDC1 transplantation, mice were injected with PBS or MOG35−55, three times, 3 days apart. MOG35−55/i.v. treatment had no effect in mice that received IFN-γR−/− cDC1, whereas in mice with WT cDC1, it significantly suppressed disease (Figure 4D). We also found a reduced IL-27 production (both mRNA and protein) by CNS IFN-γR−/− cDC1 (Figures 4E,F), suggesting that IFN-γ induces IL-27 expression in cDC1. We next investigated whether reduced IL-27 production by IFN-γR−/− cDC1 affected PD-L1 expression by moDCs and encephalitogenic CD4+ T cells. CNS moDCs from IFN-γR−/− cDC1-transplanted mice did not differ in MHCII, CD80, and CD86 expression from moDCs of control mice, but they had significantly fewer PD-L1+ moDCs (Figures 4G–J). Failure of tolerance induction in IFN-γR−/− cDC1-transplanted mice was associated with increased frequencies of IL-17+ and GM-CSF+ CD4+ T cells in the CNS (Figure 4K) and reduced numbers of annexin V+ and PD-1+ CD4+ T cells (Figures 4L,M) compared with WT cDC1-transplanted mice.
We then investigated whether, upon MOG35−55/i.v. injection, CNS and splenic cDC1 uptake MOG35−55, enabling them to directly interact with MOG35−55-specific T cells. Two hours post MOG35−55 or PBS i.v. injection into mice with ongoing EAE, cDC1 were FACS-sorted from the CNS (Supplementary Figures 2B,C) and co-cultured with 2D2 CD4+ T cells. cDC1 from MOG35−55-injected mice induced greater T cell proliferation and IFN-γ production, compared with cDC1 from PBS-injected mice (Supplementary Figures 2D–G). Overall, these data show that cDC1 acquire i.v. injected MOG35−55, which enables them to activate T cells and induce IFN-γ secretion. IFN-γ signaling in cDC1 is required to induce their IL-27 production, PD-L1 expression by moDCs, and i.v. tolerance in EAE.
Interleukin-27 Induces Programmed Death Ligand 1 Expression in Monocyte-Derived Dendritic Cells
Given that IL-27 signaling is critical for i.v. tolerance induction (3) and that Il27ra−/− (Wsx-1−/−) mice with EAE had reduced numbers of PD-L1+ moDCs in the CNS upon MOG35−55/i.v. treatment, we investigated whether the lack of IL-27 signaling in moDCs affects their PD-L1 expression and tolerance induction. We generated a mixed BM chimeras in which recipient mice (CD45.1) received half BM from Ccr2−/− mice and another half from Il27ra−/− (CD45.2) mice. In these chimera mice, virtually all monocytes outside of the BM are Il27ra−/−Ccr2+/+, as Ccr2−/− monocytes fail to leave the BM (37). As a control, we generated mixed chimeras with BMs (1:1) from WT and Ccr2−/− mice. Chimera mice were immunized for EAE induction and treated with MOG35−55/i.v. at disease onset. Mice with Il27ra−/− BM developed severe disease, compared with mice with WT BM, and MOG35−55/i.v. treatment failed to suppress disease (Figures 5A,B). Worsening of disease and treatment failure were associated with a higher number of CNS-infiltrating leukocytes (Figure 5C) and greater frequencies of Th1 and Th17 cells, whereas the frequency of apoptotic CD4+ T cells was reduced (Figures 5D–F). Moreover, the lack of IL-27 signaling in CNS moDCs precluded upregulation of PD-L1 on them upon MOG35−55/i.v. treatment (Figure 5G). Taken together, these data show that MOG35−55/i.v.-induced IL-27 in turn induces PD-L1 expression in CNS moDCs.
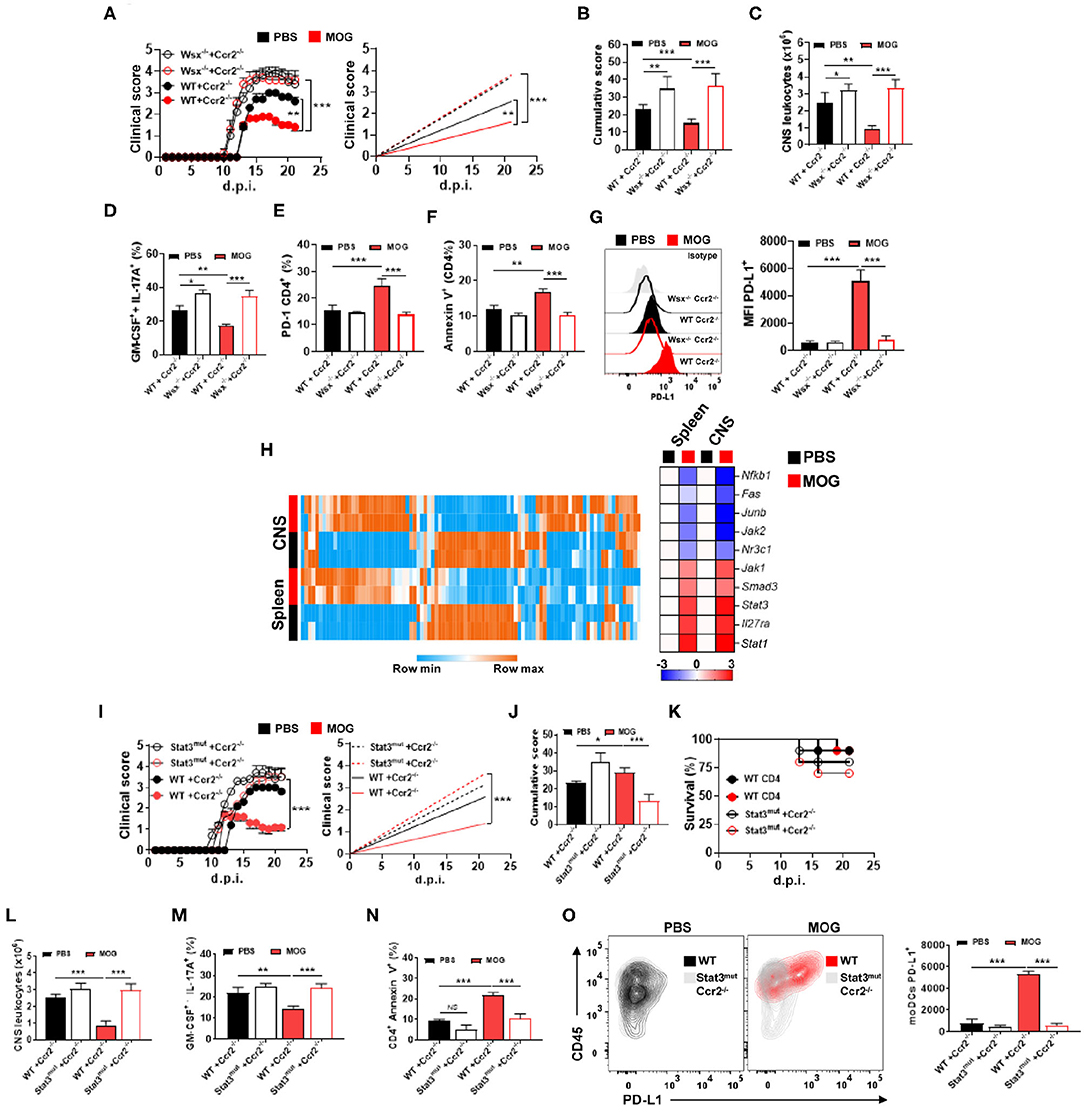
Figure 5. IL-27 induces PD-L1 expression in moDCs via STAT3. (A) CD45.1+ mice were irradiated and transplanted with 1:1 Wsx−/− and Ccr2−/− BM or WT and Ccr2−/− BM and 6–8 weeks later immunized with MOG35−55 (n = 7–10/group in each experiment). Starting from disease onset, mice were i.v. injected with 200 μg of MOG35−55 every 3 days. (B) Cumulative disease score for mice shown in (A). (C) Mice described in (A) were sacrificed at day 21 p.i. and the numbers of CD45+ leukocytes in their CNS were determined by flow cytometry. Frequencies of GM-CSF+ IL-17A+ (D), PD-1+ (E), and annexin V+ (F) CD4+ T cells from the CNS of mice shown in (A). (G) MFI of PD-L1+ moDCs from the CNS of mice shown in (A). (H) Splenic and CNS Ly6chigh MHCII+ monocytes were FACS-sorted from WT mice with EAE at 21 d.p.i. and Jak/Stat signaling gene array analysis was performed by qPCR. Heat map showing the expression levels of the top 10 genes. Gene expression levels are row centered, log2 transformed, and saturated at −3 and +3 for visualization. (I) CD45.1+ mice were irradiated and transplanted with 1:1 Stat3mut and Ccr2−/− BM, or WT and Ccr2−/− BM, and 6–8 weeks later immunized with MOG35−55 (n = 7–10/group in each experiment). Starting from disease onset, mice were i.v. injected with 200 μg of MOG35−55 every 3 days. (J) Cumulative disease score for mice shown in (I). (K) Survival (%) of mice treated as described in (I) (n = 10/group in each experiment). (L) Mice were sacrificed at day 21 p.i. and the numbers of CD45+ leukocytes from the CNS were determined by flow cytometry. Frequencies of GM-CSF+ IL-17A+ (M) and annexin V+ (N) CD4+ T cells in the CNS of mice shown in (I). (O) Numbers of PD-L1+ moDCs from the CNS of mice shown in (I). All data are representative of at least two experiments, and symbols depict mean ± SEM. Analysis between four groups was carried out by one-way ANOVA with Bonferroni post-test. EAE experiments (A,I) were analyzed by two-way ANOVA with Bonferroni's multiple comparison. Values of *P < 0.05, **P < 0.001, and ***P < 0.0001 were considered significant.
CNS and spleen moDCs upregulated PD-L1 under MOG35−55/i.v. treatment, expressing high levels of genes related to Jak–Stat signaling (Figure 5H), especially signal transducer and activator of transcription-1 (STAT1) and STAT3. We therefore investigated whether they were involved in IL-27-mediated PD-L1 induction in moDCs. WT, Stat1−/−, and Stat3mut [mutant STAT3 gene with impaired activity (38)] BM-derived DCs (BMDCs) were treated with IL-27 for 24 h, and PD-L1 expression was evaluated. While IL-27 treatment induced PD-L1 expression in WT and Stat1−/− moDCs, Stat3mut moDCs failed to upregulate PD-L1. Next, when cultured with naive 2D2 T cells, IL-27-treated Stat3mut BMDCs were less suppressive to T cell proliferation and GM-CSF production, compared with IL-27-treated WT BMDCs.
To test whether STAT3 is necessary for PD-L1 induction in moDCs in vivo, we generated mixed BM chimera mice in which recipient mice (CD45.1) received BM cells from Ccr2−/− mice and from Stat3mut mice. In these mice, virtually all monocytes and monocyte-derived cells (e.g., moDCs) outside of BM are Stat3mut, whereas other immune cells are approximately 1:1 mixture of Stat3mut and Stat3WT cells. Control chimera mice received BM cells from Ccr2−/− and WT mice; in these mice, monocytes and moDCs outside of the BM are Stat3WT. Chimera mice were immunized for EAE induction and, after onset of clinical disease, treated with MOG35−55/i.v. Mice with Stat3mut BM developed severe EAE and did not respond to MOG35−55/i.v. treatment (Figures 5I,J). Treatment failure was associated with an increased number of CNS-infiltrating leukocytes (Figure 5L), higher frequencies of CNS-infiltrating Th1 and Th17 cells (Figure 5M), reduced frequency of apoptotic CD4+ T cells (Figure 5N), and reduced PD-L1+ expression in moDCs (Figure 5O). Moreover, although Ccr2−/− Stat3mut chimera mice developed severe EAE, we did not find statistical differences in survival compared with Ccr2−/− WT mice (Figure 5K). These findings show that IL-27 induces PD-L1 expression in moDCs via the STAT3 pathway.
Discussion
Extinguishing harmful immune responses by restoring peripheral tolerance toward auto-Ags has been a long-standing goal in the search for therapies for autoimmune diseases (1, 39). Although depletion of autoreactive T cells and induction of Tregs and tolerogenic DCs are well-known mechanisms of peripheral tolerance (5, 40, 41), our study defines an interplay between molecular and cellular factors that leads to the development of tolerogenic DCs and depletion of autoreactive T cells. Auto-Ag administered i.v. is acquired by cDC1 and presented to auto-Ag-specific T cells, leading to their activation and IFN-γ secretion, which in turn induces IL-27 secretion from cDC1. IL-27 acts on moDCs to induce PD-L1 expression, which then promotes apoptosis of PD-1+ autoreactive T cells and disease amelioration.
It has been established that IFN-γ plays a protective role in EAE through multiple mechanisms (22, 25, 42, 43); it is therefore not surprising that it also mediates disease suppression in i.v. tolerance induction. We have shown that i.v. tolerized mice with EAE have higher frequencies of IFN-γ+ CD4+ T cells compared with controls (3). We show here that i.v. injection of auto-Ag in mice with EAE induces a robust and rapid production of IFN-γ by CD4+ T cells and that blockade of IFN-γ inhibits IL-27 production and PD-L1 expression in CNS moDCs and abrogates tolerance induction. This is in agreement with the findings that IFN-γ prevents accumulation of activated CD4+ T cells in response to Ag stimulation by both inhibiting proliferation and inducing apoptosis of CD4+ T cells (25). Rapid in vivo/in situ IFN-γ secretion by peptide-specific effector memory CD4+ T cells upon i.v. injection of the peptide has been demonstrated (44), a finding fully applicable to our system. Thus, our results demonstrate that IFN-γ derived from CD4+ T cells is critical for i.v. tolerance induction in ongoing EAE.
Several studies have reported that distinct DC subpopulations can uptake Ags and induce immune tolerance by the induction of IL-27 production and Tregs (3, 10, 45). We have shown that CD11b+ CD103− DCs are the major source of IL-27 in i.v. tolerance induction in EAE (3). The engagement of IFN-γR on DCs induces their expression of IL-27 and several other regulatory molecules, and IFN-γ-modified DCs modulate EAE severity in an IL-27-dependent manner (42). DCs treated with IFN-γ in vitro and injected into mice with EAE suppress disease (46). Consistent with this, we show here that mice lacking IFN-γ signaling in cDC1 fail to recover from EAE upon auto-Ag i.v. treatment. We also show that cDC1 uptake i.v. injected myelin Ag activate CD4+ T cells and their IFN-γ expression and induce tolerance.
The role of monocytes in EAE is viewed as solely pro-inflammatory (47, 48). However, there is evidence that CNS moDCs can acquire regulatory phenotype and facilitate tissue repair (49). Given that IL-27 induces PD-L1 in moDCs (8, 50) and that IL-27 signaling is beneficial in EAE (10, 51), we investigated whether the absence of IL-27 signaling in moDCs affects tolerance induction. We show that the absence of IL-27R in CNS moDCs abrogates the expression of PD-L1 and EAE recovery upon auto-Ag i.v. treatment. In contrast, neutrophils do not upregulate PD-L1 upon injection of auto-Ag; instead, they upregulate PD-L2, which is dispensable for tolerance induction.
It is well-known that IL-27 induces IL-10 expression by T cells and other types of immune cells (10). We and others have shown that i.v. tolerance induction in EAE induces IL-10 production (2, 5), which was also the case in this study. Further, the essential role of IL-10 in i.v. tolerance induction in EAE has been clearly demonstrated (2, 3, 41). We therefore did not test its role again here; however, in future studies, it would be interesting to define a pathway by which IL-10 mediates i.v. tolerance, such as its relevant cellular sources and targets, and to determine which effects of IL-27, if any, are not reliant on IL-10 induction.
Mice lacking PD-L1 develop exacerbated EAE, with PD-L1 on CD11c+ DCs playing an important role in limiting self-reactive CD4+ T cells (52). However, the lack of PD-L2, also a PD-1 ligand, did not worsen EAE (53), demonstrating that PD-L1 has a dominant role in regulating EAE severity. In agreement with these findings, our data reveal that PD-L1 is required for tolerance induction, whereas PD-L2 is dispensable. This is seemingly at odds with studies showing that blockade of PD-L1 with MAb at chronic stage EAE in C57BL/6 mice does not worsen the disease, whereas blockade of PD-L2 does (16, 53). A possible reason for this inconsistency is that we induced i.v. tolerance while clinical disease was still developing, whereas the abovementioned studies started PD-L1 and PD-L2 blockade later, in the chronic phase of disease. Taken together, these findings suggest that the relative importance of cell types and factors they express in regulating disease does change over the disease course.
Studies have shown that IL-27 induces PD-L1 expression (8) and that STAT3, which together with STAT1 mediates IL-27R signaling (3, 10), is required for PD-L1 expression (12, 54). However, the intracellular pathways downstream of IL-27 in i.v. tolerance induction in EAE are still unclear. Consistent with our previous finding that STAT1 is not necessary for IL-27-induced DC modulation (3), we show here that IL-27 from cDC1 induces PD-L1 expression in moDCs via STAT3. Indeed, BM chimera mice with impaired STAT3 signaling in moDCs failed to upregulate PD-L1 and to recover from EAE upon MOG35−55/i.v. treatment.
Our findings define the regulatory pathway that suppresses auto-Ag-specific immune response. The prerequisite for activation of this pathway is the existence of a large pool of auto-Ag-specific effector T cells that secrete IFN-γ upon activation with auto-Ag presented by APCs. Injection of a large quantity of auto-Ag induces a burst of IFN-γ secretion from auto-Ag-specific effector T cells, eliciting IL-27 and PD-L1 expression by APCs, which then in turn suppress immune response by causing anergy/apoptosis of the T cells. This is a regulatory feedback mechanism for dampening strong and possibly damaging immune responses. It is likely that this mechanism regulates myelin-specific autoimmune responses in EAE throughout its course, not only after i.v. tolerance induction. The existence of this pathway provides a unifying explanation for more severe disease in IFN-γ-, IL-27-, and PD-L1-deficient animals. It is likely that additional molecules participate in this pathway, such as IL-10, which is induced in and essential to i.v. tolerance induction in EAE (2, 3, 5, 55); upregulation of TGF-β has also been noted (55), but its significance not explored. Even though we have defined it in the context of i.v. tolerance induction in EAE, this regulatory pathway is certainly relevant in other contexts, being either beneficial or detrimental in them. In addition to CD4+ T cells, the source of IFN-γ could be, for example, pathogen-specific CD8+ T cells or NK cells as well.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by Thomas Jefferson University.
Author Contributions
GC and BC designed the concept and experiments and wrote the manuscript. GC performed most of the experiments. JR performed some of the flow cytometry experiments and revised the manuscript. RT, HD, AV, LI, and AB performed some in vivo experiments and helped with flow cytometry analysis. DH, WZ, and DX revised the manuscript. JP helped with the in vivo experiments and revised the manuscript. G-XZ revised the manuscript. JA conducted some in vivo experiments and revised the manuscript. AR and BC supervised the studies. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from the National Institutes of Health (5R01AI106026) to AR.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Katherine Regan for editing the manuscript. They would also like to thank Dr. Ross M. Kedl at the University of Colorado and Dr. Cristopher Hunter at the University of Pennsylvania for providing IL-27p28 reporter mice.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.576752/full#supplementary-material
References
1. Serra P, Santamaria P. Antigen-specific therapeutic approaches for autoimmunity. Nat Biotechnol. (2019) 37:238–51. doi: 10.1038/s41587-019-0015-4
2. Getts DR, Turley DM, Smith CE, Harp CT, McCarthy D, Feeney EM, et al. Tolerance induced by apoptotic antigen-coupled leukocytes is induced by PD-L1+ and IL-10-producing splenic macrophages and maintained by T regulatory cells. J Immunol. (2011) 187:2405–17. doi: 10.4049/jimmunol.1004175
3. Thome R, Moore JN, Mari ER, Rasouli J, Hwang D, Yoshimura S, et al. Induction of peripheral tolerance in ongoing autoimmune inflammation requires interleukin 27 signaling in dendritic cells. Front Immunol. (2017) 8:1392. doi: 10.3389/fimmu.2017.01392
4. Bettelli E, Das MP, Howard ED, Weiner HL, Sobel RA, Kuchroo VK. IL-10 is critical in the regulation of autoimmune encephalomyelitis as demonstrated by studies of IL-10- and IL-4-deficient and transgenic mice. J Immunol. (1998) 161:3299–306. doi: 10.1016/S0165-5728(98)91500-4
5. Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZN, Yap WT, et al. Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat Biotechnol. (2012) 30:1217–24. doi: 10.1038/nbt.2434
6. Wang L, Li Z, Ciric B, Safavi F, Zhang GX, Rostami A. Selective depletion of CD11c(+) CD11b(+) dendritic cells partially abrogates tolerogenic effects of intravenous MOG in murine EAE. Eur J Immunol. (2016) 46:2454–66. doi: 10.1002/eji.201546274
7. Batten M, Li J, Yi S, Kljavin NM, Danilenko DM, Lucas S, et al. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol. (2006) 7:929–36. doi: 10.1038/ni1375
8. Casella G, Finardi A, Descamps H, Colombo F, Maiorino C, Ruffini F, et al. IL-27, but not IL-35, inhibits neuroinflammation through modulating GM-CSF expression. Sci Rep. (2017) 7:16547. doi: 10.1038/s41598-017-16702-w
9. Fitzgerald DC, Ciric B, Touil T, Harle H, Grammatikopolou J, Das Sarma J, et al. Suppressive effect of IL-27 on encephalitogenic Th17 cells and the effector phase of experimental autoimmune encephalomyelitis. J Immunol. (2007) 179:3268–75. doi: 10.4049/jimmunol.179.5.3268
10. Mascanfroni ID, Yeste A, Vieira SM, Burns EJ, Patel B, Sloma I, et al. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat Immunol. (2013) 14:1054–63. doi: 10.1038/ni.2695
11. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. (2010) 236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x
12. Diskin B, Adam S, Cassini MF, Sanchez G, Liria M, Aykut B, et al. PD-L1 engagement on T cells promotes self-tolerance and suppression of neighboring macrophages and effector T cells in cancer. Nat Immunol. (2020) 21:442–54. doi: 10.1038/s41590-020-0620-x
13. Schreiner B, Bailey SL, Shin T, Chen L, Miller SD. PD-1 ligands expressed on myeloid-derived APC in the CNS regulate T-cell responses in EAE. Eur J Immunol. (2008) 38:2706–17. doi: 10.1002/eji.200838137
14. Carter LL, Leach MW, Azoitei ML, Cui J, Pelker JW, Jussif J, et al. PD-1/PD-L1, but not PD-1/PD-L2, interactions regulate the severity of experimental autoimmune encephalomyelitis. J Neuroimmunol. (2007) 182:124–34. doi: 10.1016/j.jneuroim.2006.10.006
15. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
16. Zhu B, Guleria I, Khosroshahi A, Chitnis T, Imitola J, Azuma M, et al. Differential role of programmed death-ligand 1 [corrected] and programmed death-ligand 2 [corrected] in regulating the susceptibility and chronic progression of experimental autoimmune encephalomyelitis. J Immunol. (2006) 176:3480–9. doi: 10.4049/jimmunol.176.6.3480
17. Garnier L, Laffont S, Lelu K, Yogev N, Waisman A, Guery JC. Estrogen signaling in bystander Foxp3(neg) CD4(+) T cells suppresses cognate Th17 differentiation in trans and protects from central nervous system autoimmunity. J Immunol. (2018) 201:3218–28. doi: 10.4049/jimmunol.1800417
18. McCarthy DP, Yap JW, Harp CT, Song WK, Chen J, Pearson RM, et al. An antigen-encapsulating nanoparticle platform for TH1/17 immune tolerance therapy. Nanomedicine. (2017) 13:191–200. doi: 10.1016/j.nano.2016.09.007
19. Castro F, Cardoso AP, Goncalves RM, Serre K, Oliveira MJ. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front Immunol. (2018) 9:847. doi: 10.3389/fimmu.2018.00847
20. Lees JR, Interferon gamma in autoimmunity: a complicated player on a complex stage. Cytokine. (2015) 74:18–26. doi: 10.1016/j.cyto.2014.10.014
21. Duong TT, Finkelman FD, Singh B, Strejan GH. Effect of anti-interferon-gamma monoclonal antibody treatment on the development of experimental allergic encephalomyelitis in resistant mouse strains. J Neuroimmunol. (1994) 53:101–7. doi: 10.1016/0165-5728(94)90069-8
22. Billiau A, Heremans H, Vandekerckhove F, Dijkmans R, Sobis H, Meulepas E, et al. Enhancement of experimental allergic encephalomyelitis in mice by antibodies against IFN-gamma. J Immunol. (1988) 140:1506–10.
23. Lublin FD, Knobler RL, Kalman B, Goldhaber M, Marini J, Perrault M, et al. Monoclonal anti-gamma interferon antibodies enhance experimental allergic encephalomyelitis. Autoimmunity. (1993) 16:267–74. doi: 10.3109/08916939309014645
24. Kroenke MA, Chensue SW, Segal BM. EAE mediated by a non-IFN-gamma/non-IL-17 pathway. Eur J Immunol. (2010) 40:2340–8. doi: 10.1002/eji.201040489
25. Chu CQ, Wittmer S, Dalton DK. Failure to suppress the expansion of the activated CD4 T cell population in interferon gamma-deficient mice leads to exacerbation of experimental autoimmune encephalomyelitis. J Exp Med. (2000) 192:123–8. doi: 10.1084/jem.192.1.123
26. Ponomarev ED, Novikova M, Yassai M, Szczepanik M, Gorski J, Dittel BN. Gamma delta T cell regulation of IFN-gamma production by central nervous system-infiltrating encephalitogenic T cells: correlation with recovery from experimental autoimmune encephalomyelitis. J Immunol. (2004) 173:1587–95. doi: 10.4049/jimmunol.173.3.1587
27. Voorthuis JA, Uitdehaag BM, De Groot CJ, Goede PH, P.H. van der Meide, Dijkstra CD. Suppression of experimental allergic encephalomyelitis by intraventricular administration of interferon-gamma in Lewis rats. Clin Exp Immunol. (1990) 81:183–8. doi: 10.1111/j.1365-2249.1990.tb03315.x
28. Furlan R, Brambilla E, Ruffini F, Poliani PL, Bergami A, Marconi PC, et al. Intrathecal delivery of IFN-gamma protects C57BL/6 mice from chronic-progressive experimental autoimmune encephalomyelitis by increasing apoptosis of central nervous system-infiltrating lymphocytes. J Immunol. (2001) 167:1821–9. doi: 10.4049/jimmunol.167.3.1821
29. Meredith MM, Liu K, Darrasse-Jeze G, Kamphorst AO, Schreiber HA, Guermonprez P, et al. Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J Exp Med. (2012) 209:1153–65. doi: 10.1084/jem.20112675
30. Casella G, Colombo F, Finardi A, Descamps H, Ill-Raga G, Spinelli A, et al. Extracellular vesicles containing IL-4 modulate neuroinflammation in a mouse model of multiple sclerosis. Mol Ther. (2018) 26:2107–18. doi: 10.1016/j.ymthe.2018.06.024
31. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
32. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. (2001) 25:402–8. doi: 10.1006/meth.2001.1262
33. Mundt S, Mrdjen D, Utz SG, Greter M, Schreiner B, Becher B. Conventional DCs sample and present myelin antigens in the healthy CNS and allow parenchymal T cell entry to initiate neuroinflammation. Sci Immunol. (2019) 4. doi: 10.1126/sciimmunol.aau8380
34. Naves R, Singh SP, Cashman KS, Rowse AL, Axtell RC, Steinman L, et al. The interdependent, overlapping, and differential roles of type I and II IFNs in the pathogenesis of experimental autoimmune encephalomyelitis. J Immunol. (2013) 191:2967–77. doi: 10.4049/jimmunol.1300419
35. White MPJ, Webster G, Leonard F, La Flamme A.C. Innate IFN-gamma ameliorates experimental autoimmune encephalomyelitis and promotes myeloid expansion and PDL-1 expression. Sci Rep. (2018) 8:259. doi: 10.1038/s41598-017-18543-z
36. Giles DA, Duncker PC, Wilkinson NM, Washnock-Schmid JM, Segal BM. CNS-resident classical DCs play a critical role in CNS autoimmune disease. J Clin Invest. (2018) 128:5322–34. doi: 10.1172/JCI123708
37. Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med. (2000) 192:1075–80. doi: 10.1084/jem.192.7.1075
38. Steward-Tharp SM, Laurence A, Kanno Y, Kotlyar A, Villarino AV, Sciume G, et al. A mouse model of HIES reveals pro- and anti-inflammatory functions of STAT3. Blood. (2014) 123:2978–87. doi: 10.1182/blood-2013-09-523167
39. Steinman L, Ho PP, Robinson WH, Utz PJ, Villoslada P. Antigen-specific tolerance to self-antigens in protein replacement therapy, gene therapy and autoimmunity. Curr Opin Immunol. (2019) 61:46–53. doi: 10.1016/j.coi.2019.07.011
40. Li H, Zhang GX, Chen Y, Xu H, Fitzgerald DC, Zhao Z, et al. CD11c+CD11b+ dendritic cells play an important role in intravenous tolerance and the suppression of experimental autoimmune encephalomyelitis. J Immunol. (2008) 181:2483–93. doi: 10.4049/jimmunol.181.4.2483
41. Mari ER, Moore JN, Zhang GX, Rostami A. Mechanisms of immunological tolerance in central nervous system inflammatory demyelination. Clin Exp Neuroimmunol. (2015) 6:264–74. doi: 10.1111/cen3.12196
42. Murugaiyan G, Mittal A, Weiner HL. Identification of an IL-27/osteopontin axis in dendritic cells and its modulation by IFN-gamma limits IL-17-mediated autoimmune inflammation. Proc Natl Acad Sci USA. (2010) 107:11495–500. doi: 10.1073/pnas.1002099107
43. Heremans H, Dillen C, Groenen M, Martens E, Billiau A. Chronic relapsing experimental autoimmune encephalomyelitis (CREAE) in mice: enhancement by monoclonal antibodies against interferon-gamma. Eur J Immunol. (1996) 26:2393–8. doi: 10.1002/eji.1830261019
44. Reinhardt RL, Khoruts A, Merica R, Zell T, Jenkins MK. Visualizing the generation of memory CD4 T cells in the whole body. Nature. (2001) 410:101–5. doi: 10.1038/35065111
45. Mari ER, Rasouli J, Ciric B, Moore JN, Conejo-Garcia JR, Rajasagi N, et al. Galectin-1 is essential for the induction of MOG35-55-based intravenous tolerance in experimental autoimmune encephalomyelitis. Eur J Immunol. (2016) 46:1783–96. doi: 10.1002/eji.201546212
46. Xiao BG, Wu XC, Yang JS, Xu LY, Liu X, Huang YM, et al. Therapeutic potential of IFN-gamma-modified dendritic cells in acute and chronic experimental allergic encephalomyelitis. Int Immunol. (2004) 16:13–22. doi: 10.1093/intimm/dxh003
47. Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, et al. The cytokine GM-CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity. (2015) 43:502–14. doi: 10.1016/j.immuni.2015.08.010
48. Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. (2011) 14:1142–9. doi: 10.1038/nn.2887
49. Giles DA, Washnock-Schmid JM, Duncker PC, Dahlawi S, Ponath G, Pitt D, et al. Myeloid cell plasticity in the evolution of central nervous system autoimmunity. Ann Neurol. (2018) 83:131–41. doi: 10.1002/ana.25128
50. Karakhanova S, Bedke T, Enk AH, Mahnke K. IL-27 renders DC immunosuppressive by induction of B7-H1. J Leukoc Biol. (2011) 89:837–45. doi: 10.1189/jlb.1209788
51. Do J, Kim D, Kim S, Valentin-Torres A, Dvorina N, Jang E, et al. Treg-specific IL-27Ralpha deletion uncovers a key role for IL-27 in Treg function to control autoimmunity. Proc Natl Acad Sci USA. (2017) 114:10190–5. doi: 10.1073/pnas.1703100114
52. Sage PT, Schildberg FA, Sobel RA, Kuchroo VK, Freeman GJ, Sharpe AH. Dendritic Cell PD-L1 limits autoimmunity and follicular T cell differentiation and function. J Immunol. (2018) 200:2592–602. doi: 10.4049/jimmunol.1701231
53. Salama AD, Chitnis T, Imitola J, Ansari MJ, Akiba H, Tushima F, et al. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J Exp Med. (2003) 198:71–8. doi: 10.1084/jem.20022119
54. Wolfle SJ, Strebovsky J, Bartz H, Sahr A, Arnold C, Kaiser C, et al. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur J Immunol. (2011) 41:413–24. doi: 10.1002/eji.201040979
Keywords: peripheral tolerance, monocytes, experimental autoimmune encephalitis, PD-L1, cytokines
Citation: Casella G, Rasouli J, Thome R, Descamps HC, Vattikonda A, Ishikawa L, Boehm A, Hwang D, Zhang W, Xiao D, Park J, Zhang G-X, Alvarez JI, Rostami A and Ciric B (2020) Interferon-γ/Interleukin-27 Axis Induces Programmed Death Ligand 1 Expression in Monocyte-Derived Dendritic Cells and Restores Immune Tolerance in Central Nervous System Autoimmunity. Front. Immunol. 11:576752. doi: 10.3389/fimmu.2020.576752
Received: 26 June 2020; Accepted: 02 September 2020;
Published: 26 October 2020.
Edited by:
Dipyaman Ganguly, Indian Institute of Chemical Biology, IndiaReviewed by:
Sin-Hyeog Im, Pohang University of Science and Technology, South KoreaSylvaine You, Institut National de la Santé et de la Recherche Médicale, France
Copyright © 2020 Casella, Rasouli, Thome, Descamps, Vattikonda, Ishikawa, Boehm, Hwang, Zhang, Xiao, Park, Zhang, Alvarez, Rostami and Ciric. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bogoljub Ciric, Ym9nb2xqdWIuY2lyaWNAamVmZmVyc29uLmVkdQ==