Abstract
Circular RNAs (circRNAs) constitute a class of covalently circular non-coding RNA molecules formed by 5′ and 3′ end back-splicing. The rapid development of bioinformatics and large-scale sequencing has led to the identification of functional circRNAs. Despite an overall upward trend, studies focusing on the roles of circRNAs in immune diseases remain relatively scarce. In the present study, we obtained a differential circRNA expression profile based on microarray analysis of peripheral blood mononuclear cells (PBMCs) in children with type 1 diabetes mellitus (T1DM). We characterized one differentially expressed circRNA back-spliced from the MYB Proto-Oncogene Like 2 (MYBL2) gene in patients with T1DM, termed as hsa_circ_0060450. Subsequent assays revealed that hsa_circ_0060450 can serve as the sponge of miR-199a-5p, release its target gene, Src homology 2 (SH2)-containing protein tyrosine phosphatase 2 (SHP2), encoded by the tyrosine-protein phosphatase non-receptor type 11 gene (PTPN11), and further suppress the JAK-STAT signaling pathway triggered by type I interferon (IFN-I) to inhibit macrophage-mediated inflammation, which indicates the important roles of circRNAs in T1DM and represents a promising therapeutic molecule in the treatment of T1DM.
Introduction
Type 1 diabetes (T1DM) is an autoimmune disease, also known as insulin-dependent diabetes mellitus, owing to the complete absence of insulin secretion. During the pathogenesis of T1DM, insulin-producing pancreatic β cells are attacked by the immune system, resulting in destruction of islet function and decreased insulin secretion (1, 2). Immune tolerance mediated by Treg cells is outweighed by immune activation involving autoreactive CD8+ T cells, Th1 cells, and B cells (3). Furthermore, the expansion of IL-17-secreting Th17 cells also contributes to pancreatic inflammation in patients with T1DM (4, 5). In addition to adaptive immune cells, innate immune cells are involved in the onset of T1DM (6–8). Emerging evidence suggests that the absence of Toll-like receptor signaling in innate immune cells, caused by TIR-domain-containing adapter-inducing interferon-β (TRIF) deficiency, has a protective effect against diabetes in the non-obese diabetic (NOD) mouse via alteration of the gut microbiota and reduced immune cell activation (9).
Macrophages, known as inflammatory mediator-secreting immune cells, play a critical role in insulitis, and support autoimmune T cells to aggravate the infiltration of inflammatory cells during T1DM (10, 11). Macrophage migration inhibitory factor (Mif) knockout (KO) mice were found to express lower levels of costimulatory molecules in the streptozocin-induced diabetic mouse model and Toll-like receptors on the surface of their macrophages, leading to functional deficit of macrophages and impaired T cell activation, thereby alleviating pancreatic injury (12). Moreover, Unanue et al. have showed that islet-resident macrophages are in a persistent inflammatory state prior to signs of β cell pathology and their depletion significantly reduced the onset of diabetes (13, 14).
In addition to inflammation caused by bacterial infection, viral infection and subsequent IFN-I production are linked to T1DM pathogenesis (15–17). Viral infections can lead to apoptosis of pancreatic β cells and pancreas damage (18). Some viruses, such as coxsackievirus B1, parainfluenza, and enteroviruses initiated a devastating immune response in pancreatic β cells and other surrounding cells, resulting in the occurrence of fulminant T1DM through the induction of IFN-I and inflammation (19, 20). In addition, 2 independent longitudinal studies have showed the expression of IFN-I–induced genes in PBMCs isolated from children at risk for T1DM preceded onset of autoimmunity (21, 22). Moreover, some studies have confirmed that abrogation of IFN-I function significantly ameliorated the incidence of diabetes in NOD mice (23, 24). Marro et al. also found that macrophages promoted autoreactive CD8+ T cell infiltration into islets in the Rip-LCMV-GP T1DM model via IFN-I signaling, and abrogation of IFN-I signaling on macrophages significantly limited the onset of T1DM (25).
Circular RNAs (circRNAs) are a class of covalently circular non-coding RNA molecules (26, 27). Existing studies have shown circRNAs are generated mainly through three distinct mechanisms: exon skipping, RNA binding protein (RBP)-driven circularization, and intron pairing-driven circularization, which bring the 5′ and 3′ termini of mRNA into spatial proximity and induce back-splicing. The length of most human exonic circRNAs is <1,500 nucleotides (nt), and the median length is 500 nt (28). Compared with their homologous linear transcripts, circRNAs are not easily degraded by RNase R (29), and highly conserved between tissues, developmental stages, and cell types (30). By integrating large-scale deep-sequencing data sets, Zheng et al. annotated 14 867 human circRNAs in deepBase v2.0, 1260 of which are orthologous to mouse circRNAs (31). Furthermore, some circRNAs have been shown to exert important physiological functions. In 2013, Memczak et al. found that there are 63 miR-7 binding sites within the circular transcript ciRS-7 sequence of the CDR1as gene (30). Hansen et al. also found that circRNA Sry can act as a “sponge” to adsorb miR-138 (32). Substantial evidence has been published describing the competitive endogenous RNA (ceRNA) mechanism, which is the most widely studied mechanism in circRNA research. Through adsorption of microRNAs, circRNA reduces the quantity of free microRNAs in the cytoplasm and releases target genes of microRNAs, leading to occurrence or inhibition of certain diseases.
Notably, despite an overall upward trend, circRNA studies focusing on immune diseases are relatively scarce. In the present study, we obtained a differential circRNA expression profile based on microarray analysis of PBMCs in children with T1DM. We characterized an upregulated circRNA, back-spliced from MYB Proto-Oncogene Like 2 (MYBL2) in patients with T1DM, termed as hsa_circ_0060450. Subsequent assays revealed that hsa_circ_0060450 can serve as the sponge of miR-199a-5p, releasing its target Src homology 2 (SH2)-containing protein tyrosine phosphatase 2 (SHP2) encoded by tyrosine-protein phosphatase non-receptor type 11 gene (PTPN11) in macrophages, and further suppress the JAK-STAT signaling pathway triggered by IFN-I to inhibit macrophage-mediated inflammation.
Materials and Methods
Human Normal and T1DM Samples
Peripheral blood samples of T1DM patients and healthy controls were collected from the Children's Hospital of Fudan University, Shanghai, China. The follow-up PBMCs were separated by Ficoll-Hypaque (GE Healthcare) within 24 h after collection. Human samples were obtained with informed consents from their parents, and the study was approved by the Research Ethics Board of the Children's Hospital of Fudan University.
Microarray Analysis
PBMCs of T1DM patients and healthy controls were separated and performed SBC Human (4*180K) ceRNA microarray analysis (Shanghai Biotechnology Corporation, China). In brief, total RNA was extracted and purified using miRNeasy Mini Kit (QIAGEN, GmBH, Germany), followed by amplification and label using Low Input Quick Amp Labeling Kit, One-Color (Agilent technologies, Santa Clara, CA, USA). Each slide was hybridized with Cy3-labeled cRNA using Gene Expression Hybridization Kit (Agilent technologies, Santa Clara, CA, USA) in Hybridization Oven. After hybridization, slides were washed in staining dishes with Gene Expression Wash Buffer Kit (Agilent technologies, Santa Clara, CA, USA), and were then scanned by Agilent Microarray Scanner (Agilent technologies, Santa Clara, CA, USA). Finally, data were extracted with Feature Extraction software 10.7 (Agilent technologies, Santa Clara, CA, USA), and raw data were normalized by Quantile algorithm. Fold change > 2.0 and P < 0.05 was the criterion to screen differentially expressed circRNAs between T1DM patients and healthy controls. Microarray data have been deposited in the Gene Expression Omnibus (GEO) under accession numbers GSE133225 (submitted in other draft).
Cell Culture and Treatment
Human THP1 and HEK-293T cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). THP1 cells were maintained in RPMI-1640 medium with inactivated 10% fetal bovine serum (FBS, Gibco, Gaithersburg, MD, USA) and 1% penicillin-streptomycin (Gibco, Gaithersburg, MD, USA). HEK-293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco, Gaithersburg, MD, USA) supplemented with 10% FBS and 1% penicillin-streptomycin. Cells were grown at 37°C in a 5% CO2 humidified incubator. THP1-derived macrophages were induced using THP1 cells treated with 50 ng/ml phorbol 12-myristate 13-acetate (PMA) for 48 h. Chemically synthesized miR-199a-5p mimics, miR-133a-3p mimics, miR-133b mimics, hsa_circ_0060450 siRNA-1 and hsa_circ_0060450 siRNA-2 (200 nM, GenePharma, Shanghai, China) were transiently transfected into macrophages using Lipofectamine RNAiMAX reagent (Invitrogen, USA), according to the standard protocol. After 48 h of transfection, macrophages were treated with type I interferon (1,000 units/ml, PBL Assay Science, USA). The sequences of siRNAs, microRNA mimics and NC are listed in Supplementary Table 1.
RNA Extraction and Reverse Transcription Quantitative PCR (RT-qPCR)
Total RNA was extracted using TRIzol (Invitrogen, USA) followed by reverse transcription using the PrimeScript II 1st Strand cDNA Synthesis Kit (Takara, Japan), according to the manufacturer's instructions. Real-time quantitative PCR reaction was performed with SYBR® Premix Ex Taq™ II (Takara, Japan) using the Roche 480 Real Time PCR System. We used β-actin as internal control and calculated fold change via the 2−ΔΔCT method. The primer pairs used for qPCR are listed in Supplementary Table 2.
Nuclear and Cytoplasmic Separation
Nuclear and cytoplasmic separation assay was conducted using the Nuclear and Cytoplasmic Extraction Kit (CWBiotech, Beijing, China) according to the manufacturer's protocol. Briefly, 1 × 107 cells were centrifuged at 3 × 103 rpm for 5 min, resuspended in 500 μl of ribonuclease inhibitor added-Nc-buffer A, and incubated on ice for 20 min. Then, 27.5 μl of Nc-Buffer B was added, and incubated on ice for 1 min. The mixtures were then centrifuged at 12,000 g, 4°C for 15 min. The supernatants (cytoplasmic component) were mixed with TRIzol reagent for RNA extraction. Next, the nuclear pellets were resuspended with 250 μl of ribonuclease inhibitor added-Nc-buffer C, and then mixed with TRIzol reagent for RNA extraction. RNA extraction and RT-qPCR were performed as previously described.
Western Blot
Cell lysates were prepared with RIPA buffer (Thermo Fisher, USA); then, total protein was quantified with the Protein BCA Assay Kit (Bio-Rad, USA), and denatured at 100°C for 10 min. Next, 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was used to separate the protein (20 μg). The proteins were transferred to polyvinylidene fluoride (PVDF) membranes (Millipore Corporation, USA) and blocked in 5% bovine serum albumin (BSA) at room temperature (RT) for 1 h, followed by incubation overnight at 4°C with the rabbit anti-β-tubulin antibody (1:5000, Abcam, USA), anti-p-Stat1 antibody (Y701, 1:1000, Cell Signaling Technology, USA), anti-p-Stat3 antibody (Y705, 1:1000, Cell Signaling Technology, USA), anti-Stat1 antibody (1:1000, Cell Signaling Technology, USA), anti-Stat3 antibody (1:1000, Cell Signaling Technology, USA) and anti-SHP2 antibody (1:500, Santa Cruz, USA). Subsequently, after washing with tris buffered saline solutions with 0.1% Tween-20 (TBST) for three times, the membranes were incubated with horseradish peroxidase (HRP)-conjugated goat-anti-rabbit antibody (1:5000, Cell Signaling Technology, USA) at RT for 1 h. Finally, after washing again with TBST solution for three times, the bands were immersed in chemiluminescent HRP substrate (Millipore Corporation) determination and imaged with a Molecular Imager® (Bio-RAD, ChemiDocTM XRS+ Imaging System).
Dual-Luciferase Reporter Assay
The wild type (WT) and mutant hsa_circ_0060450 fragments and SHP2 3′-UTR containing corresponding miRNAs binding sites were fused into Renilla luciferase gene (hRluc) included psicheck2 vector (Promega, Madison, WI, USA) using the Xho I and Not I restriction sites. The fused recombinant vectors were verified by sanger sequencing. The PCR primers for WT and mutant hsa_circ_0060450 fragments and SHP2 fragments are listed in Supplementary Table 2.
HEK-293T cells were seeded into a 96-well plate at a density of 10,000 cells/well. The recombinant vector or empty vector and miR-199a-5p, miR-133a-3p, miR-133b mimics, or NC were co-transfected simultaneously into HEK-293T cells using Lipofectamine 2000 (Invitrogen, CA, USA). Luciferase activity was determined 24 h after transfection using a dual luciferase reporter assay kit (Promega, USA).
RNase R Treatment
A total of 5 μg of RNA extracted from cells using TRIzol reagent was incubated at 37°C for 30 min with RNase R (20 U/μl, Lucigen, USA) treatment according to the manufacturer's instructions; the control group was treated under RNase R-free conditions. The remaining RNA was then purified with phenol/chloroform mixture (5:1, Sigma-Aldrich, USA). Subsequent RNA extraction and RT-qPCR was performed as previously described.
Actinomycin D Treatment
THP1 cells were seeded into a 24-well plate at a density of 120,000 cells/well with PMA (50 ng/ml) stimulation for 48 h. Then, total RNA was harvested at 0, 3, 6, 12, and 24 h, with or without actinomycin D (ActD) treatment (1 μg/ml). Subsequent RNA extraction and RT-qPCR was performed as previously described.
Statistical Analysis
Results from three independent, replicate experiments were obtained to confirm the reproducibility of each experiment. Bars represent the means ± SEM. The two-tailed Student's t-tests was used for comparisons between two groups, and one-way analysis of variance (ANOVA) was used for comparisons of multiple groups. A p-value of <0.05 was considered to indicate statistically significant data. Statistical analyses were performed with SPSS v.19.0 software and visualized data were obtained with Graphpad prism 7.0 software.
Results
Hsa_circ_0060450 Is Upregulated in PBMCs of T1DM Patients
In a pilot study, we obtained the differential circRNA expression profile based on microarray analysis of PBMCs in children with T1DM and age-matched healthy controls. We identified 15 upregulated and 17 downregulated circRNAs between T1DM patients and healthy controls (n = 3, fold change > 2, and p < 0.05) (Figure 1A). Next, we verified the expressions of the top 10 circRNAs (Figure 1B) with the highest differential expression fold in a larger population of clinical samples of PBMCs collected from 20 T1DM children and 20 healthy controls by RT-qPCR, and the results showed hsa_circ_0060450 was detectable and significantly upregulated in PBMCs of T1DM patients than controls (Figure 1C). In addition, through ROC curve analysis, we found that the expression of hsa_circ_0060450 in PBMCs could discriminate normal subjects and T1DM patients (Figure 1D). Using RT-PCR with divergent primers and DNA sequencing, we confirmed the back-splicing site sequences of hsa_circ_0060450 (Figure 1E). The full-length sequences of hsa_circ_0060450 were listed in Supplementary Figure 1. After treatment with RNase R, hsa_circ_0060450 did not degrade readily compared with its cognate linear transcript MYBL2 (Figure 1F). Actinomycin D assay also revealed that hsa_circ_0060450 is more stable than its linear transcript MYBL2, with a half-life exceeding 24 h (Figure 1G). Furthermore, to investigate the roles of hsa_circ_0060450 in T1DM pathogenesis, we separated human PBMCs to detect the expression of hsa_circ_0060450 in T cells, B cells and monocytes, respectively, and found the expression of hsa_circ_0060450 is higher in monocytes compared with T cells and B cells (Figure 1H). Given that monocytes can transform into macrophages to participate in pancreas inflammation, we next mainly explored the function of hsa_circ_0060450 in macrophages. Subsequently, we performed nuclear and cytoplasmic separation experiments in THP1-derived macrophages, and found that ~40% of hsa_circ_0060450 is located in the cytoplasm, with the remaining proportion in the nucleus (Figure 1I).
Figure 1
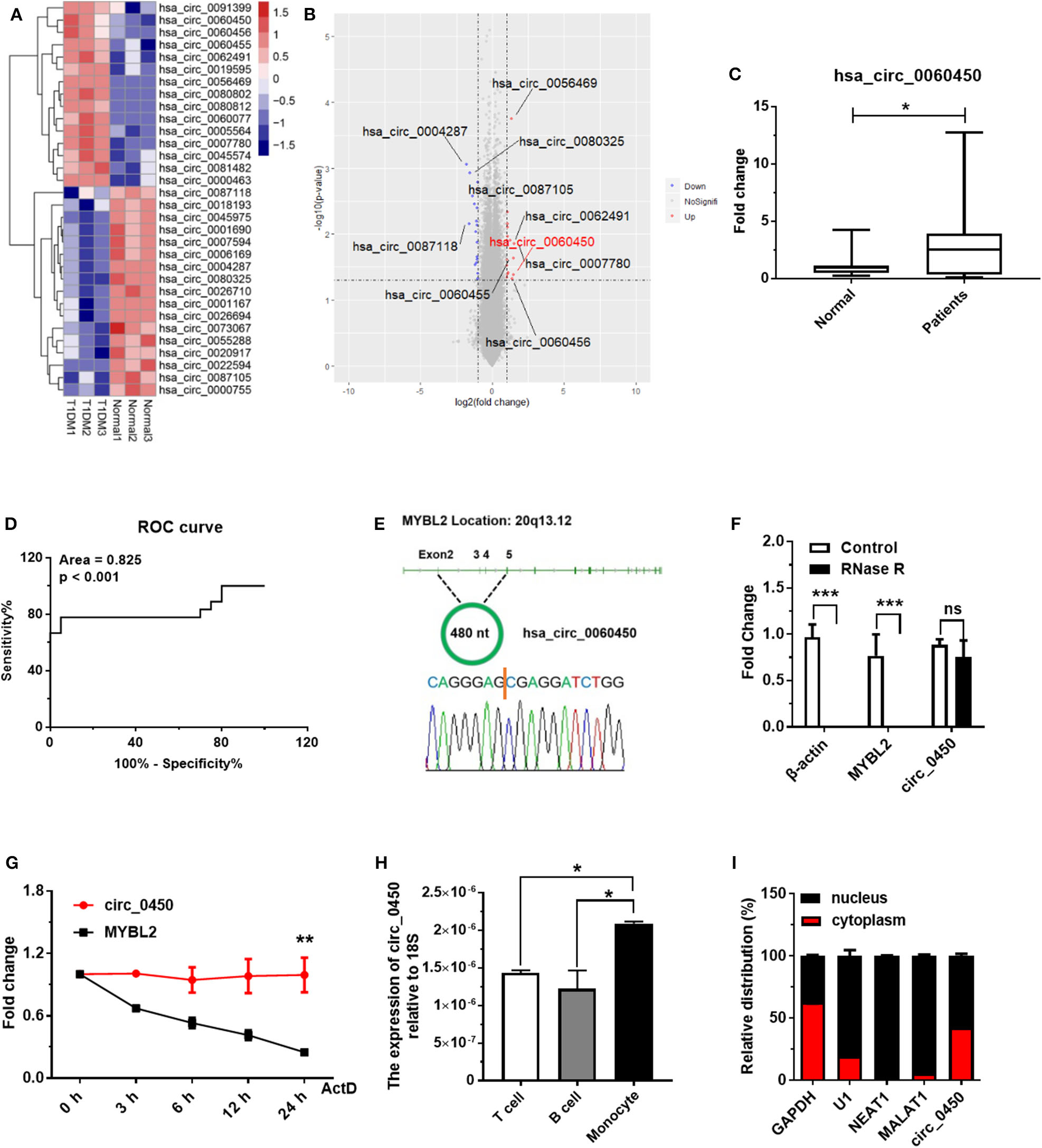
Hsa_circ_0060450 is upregulated in the PBMCs of T1DM patients. (A) Heatmap of circRNAs with significantly altered expression in the PBMCs of 3 normal controls and 3 T1DM patients. (B) Volcano plot of circRNAs with significantly altered expression in the PBMCs of 3 normal controls and 3 T1DM patients. (C) The expression of hsa_circ_0060450 in PBMCs of 20 children with T1DM and 20 healthy controls, detected by RT-qPCR. (D) Receiver operating curve (ROC) analysis of hsa_circ_0060450 levels in the study population. (E) The genomic loci of the MYBL2 and hsa_circ_0060450. The expression of hsa_circ_0060450 was detected by RT-PCR and validated by Sanger sequencing. (F) RT-qPCR analyses of β-actin, MYBL2, and hsa_circ_0060450 after treatment with RNase R in THP1-derived macrophages. (G) RT-qPCR analyses of MYBL2 and hsa_circ_0060450 after treatment with actinomycin D at the indicated time points in THP1-derived macrophages. (H) The expressions of hsa_circ_0060450 in T cells, B cells and monocytes of human PBMCs, detected by RT-qPCR. (I) The relative distribution of hsa_circ_0060450 in the nucleus and cytoplasm in THP1-derived macrophages. circ_0450 is the abbreviation of hsa_circ_0060450. ActD, actinomycin D. Data represent means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. ns, not significance.
Hsa_circ_0060450 Negatively Regulates IFN-I-Induced Inflammation Through the JAK-STAT1/3 Pathway
To further explore whether hsa_circ_0060450 plays a role in IFN-I-induced inflammation, we designed two small interfering RNAs (siRNAs) to silence hsa_circ_0060450 expression in THP1-derived macrophages (Figure 2A). Notably, the two siRNAs were targeted specifically to the back-splicing site of hsa_circ_0060450, ensuring that only hsa_circ_0060450 was knocked down and its cognate linear transcript MYBL2 was not targeted. RT-qPCR assay showed that hsa_circ_0060450 silencing resulted in significant upregulation of the expressions of IFIT1, IFIH1, CXCL10, and iNOS under IFN-I stimulation (Figure 2B). We then performed Western blotting assay to investigate the effect of hsa_circ_0060450 on the JAK-STAT1/3 pathway activated by IFN-I. Experimental results showed that hsa_circ_0060450 silencing promoted STAT1 and STAT3 protein phosphorylation after 15 min and 30 min of IFN-I stimulation (Figure 2C). This was indicative of the inhibitory action of hsa_circ_0060450 on IFN-I-induced inflammation. Collectively, these results suggest that hsa_circ_0060450 negatively regulates IFN-I-induced inflammation through the JAK-STAT1/3 pathway.
Figure 2
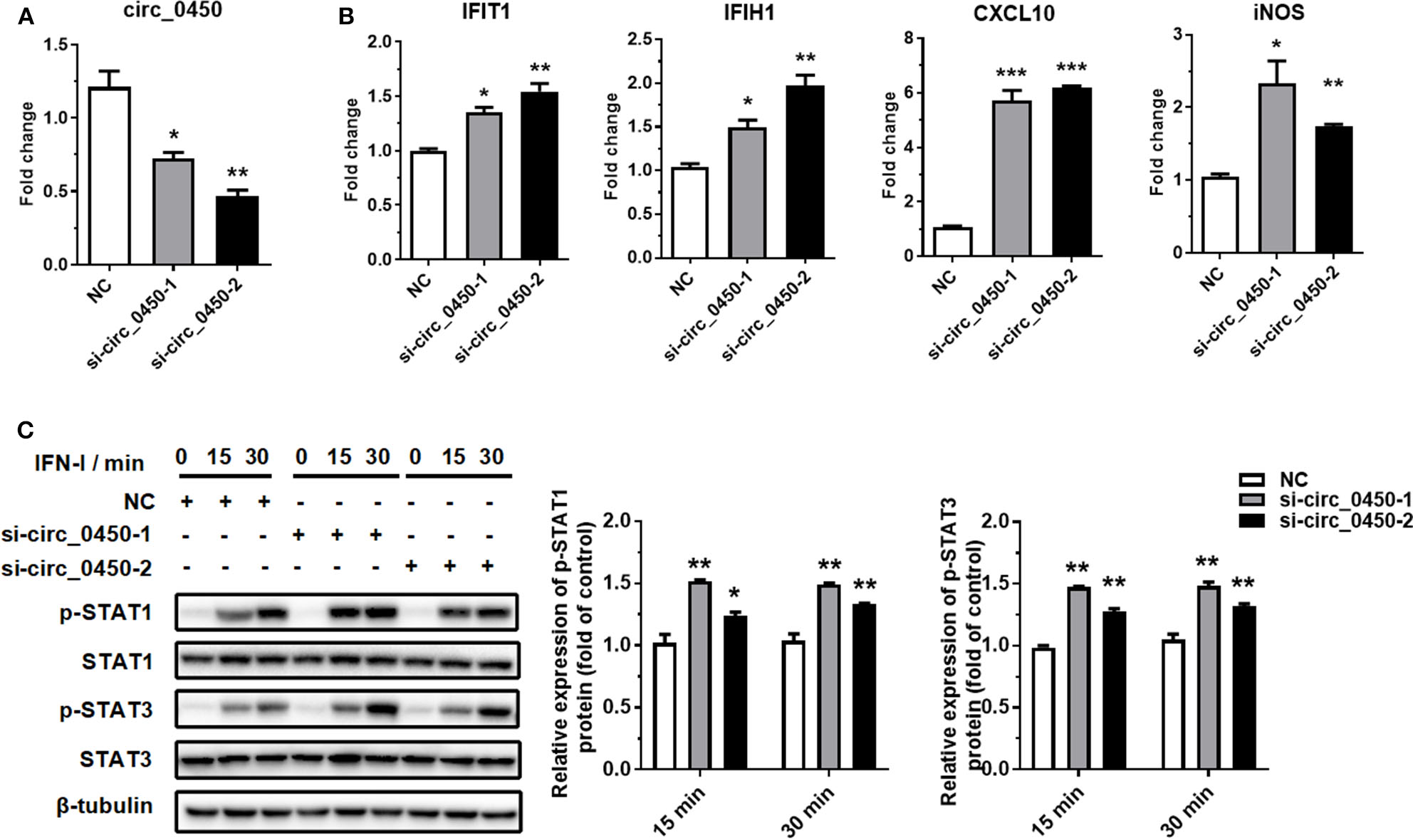
Silencing of hsa_circ_0060450 promotes IFN-I-induced inflammation through the JAK-STAT1/3 pathway. (A) RT-qPCR analyses of hsa_circ_0060450 to confirm the silencing efficiencies of two siRNAs. (B) RT-qPCR analyses of IFIT1, IFIH1, CXCL10, and iNOS under IFN-I stimulation after treatment with two hsa_circ_0060450 siRNAs. (C) STAT1 and STAT3 protein phosphorylation assessment at 15 and 30 min of IFN-I stimulation after treatment with two hsa_circ_0060450 siRNAs. NC, negative control. Data represent means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Hsa_circ_0060450 Serves as a Sponge of miR-199a-5p to Inhibit Inflammation Induced by IFN-I
Recent evidence suggests that exonic circRNAs within the cytoplasm can serve as a “sponge” of microRNAs. Therefore, we determined to investigate whether hsa_circ_0060450 adsorbs microRNAs to exert its function. We first selected several microRNAs that have been studied for their pro-inflammatory effects, and among these microRNAs, miR-199a-5p, miR-133a-3p and miR-133b were predicted to have binding sites to hsa_circ_0060450 by RNA22 database (https://cm.jefferson.edu/rna22/Interactive/) (Figure 3A). Next, we performed luciferase assay by constructing hsa_circ_0060450 fragment-containing psicheck2 recombinant vectors (Figure 3B). Results showed that the luciferase activity of the group co-transfected with miR-199a-5p mimics and psicheck2 recombinant vector containing hsa_circ_0060450 fragment, as well as that of the other two groups co-transfected with miR-133a or miR-133b mimics and psicheck2 recombinant vector containing hsa_circ_0060450 fragment, was significantly reduced (Figure 3C). We also performed luciferase assay to confirm the target sequence of miR-199a-5p, miR-133a or miR-133b in hsa_circ_0060450 by constructing mutant hsa_circ_0060450 fragment-containing psicheck2 recombinant vectors (Figure 3A). Results showed that the luciferase activity of the group co-transfected with miR-199a-5p, miR-133a or miR-133b mimics and corresponding hsa_circ_0060450-mutant vector was not changed compared with the NC group (Figure 3D and Supplementary Figure 2). Furthermore, we investigated the pro-inflammatory effects of these three microRNAs in THP1-derived macrophages. The Western blotting results suggested that miR-199a-5p overexpression promoted STAT1 and STAT3 protein phosphorylation (Figure 3E); however, the overexpression of miR-133a and miR-133b inhibited STAT1 and STAT3 protein phosphorylation after IFN-I stimulation (Supplementary Figures 3A,B). Of note, RT-qPCR assay indicated that miR-199a-5p overexpression significantly upregulated the expressions of IFIT1, IFIH1, CXCL10, and iNOS under the stimulation of IFN-I (Figure 3F), consistent with the pro-inflammatory features of miR-199a as reported previously (33, 34), and miR-133a or miR-133b overexpression significantly reduced the expressions of IFIT1, IFIH1, CXCL10, and CCL5 under the stimulation of IFN-I (Supplementary Figure 3C). To summarize, hsa_circ_0060450 may serve as a sponge of miR-199a-5p to inhibit inflammation induced by IFN-I.
Figure 3
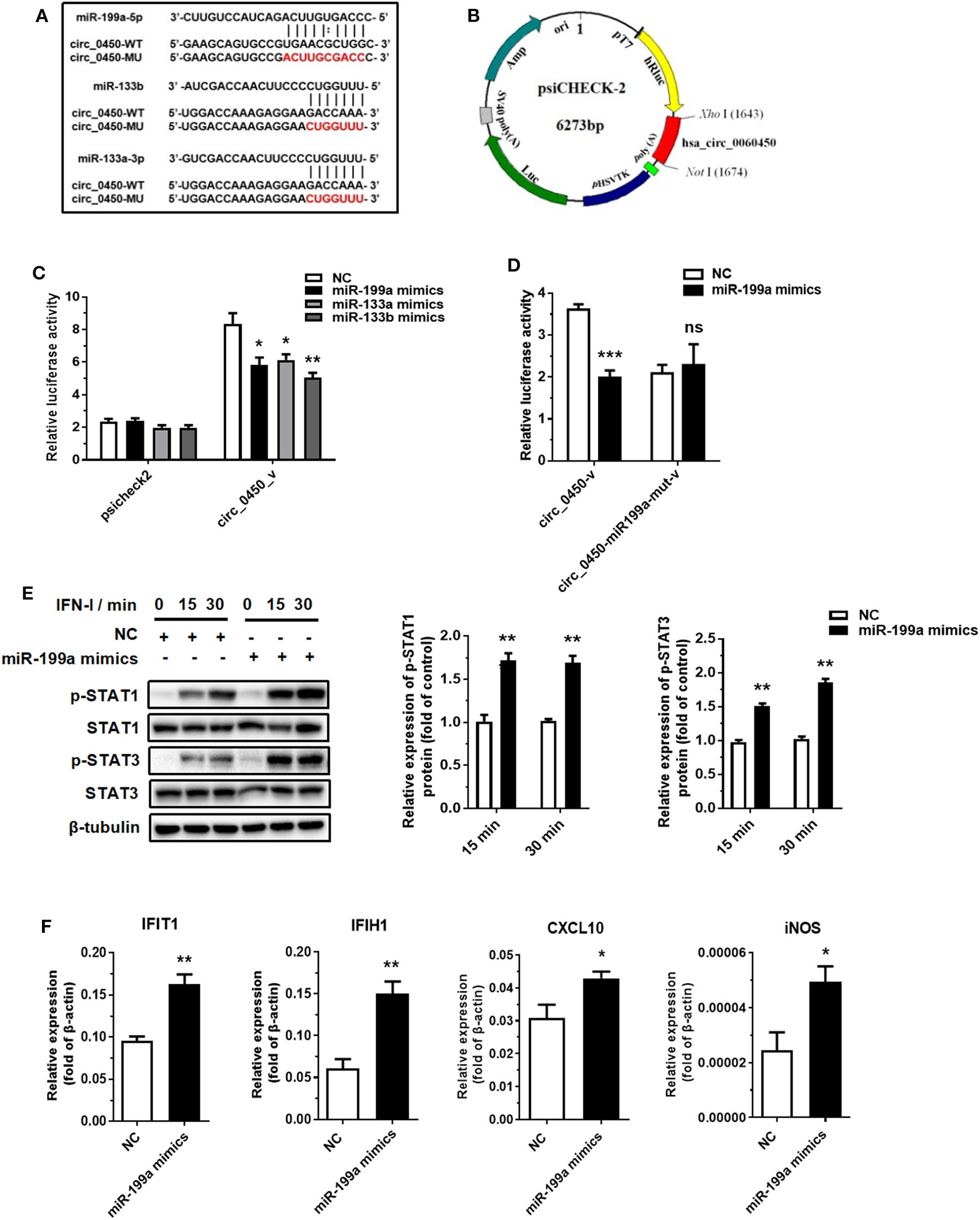
Hsa_circ_0060450 serves as a sponge of miR-199a-5p. (A) The predicted target sequence of miR-199a-5p, miR-133a or miR-133b in hsa_circ_0060450 and corresponding mutant target sequence for binding sites-mutant luciferase assay. (B) The construction of hsa_circ_0060450 fragment-containing psicheck2 luciferase vector. (C) A luciferase reporter assay was used to detect the luciferase activity of 293T cells co-transfected with blank psicheck2 vector or psicheck2 recombinant vector containing hsa_circ_0060450 fragment and miR-199a-5p, miR-133a, miR-133b mimics, or NC. (D) A luciferase reporter assay was used to detect the luciferase activity of 293T cells co-transfected with psicheck2 recombinant vector containing mutant hsa_circ_0060450 fragment and miR-199a-5p or NC. (E) STAT1 and STAT3 protein phosphorylation assessment and quantification analyses at 15 and 30 min of IFN-I stimulation after treatment with miR-199a-5p mimics. (F) RT-qPCR analyses of IFIT1, IFIH1, CXCL10, and iNOS under IFN-I stimulation after treatment with miR-199a-5p mimics. NC, negative control. circ_0450-v, psicheck2 recombinant vector containing WT hsa_circ_0060450 fragment. circ_0450-miR199a-mut-v, psicheck2 recombinant vector containing miR-199a-5p binding site-mutant hsa_circ_0060450 fragment. Data represent means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. ns, not significance.
miR-199a-5p Promotes IFN-I-Induced Inflammation by Targeting SHP2
MicroRNAs can bind to the 3′ UTR of their targets to suppress translation or accelerate mRNA degradation (35, 36). miR-199a-5p may act as a pro-inflammatory factor by binding to its target to promote JAK-STAT1/3 pathway activation. We then selected five typical negative regulators of JAK-STAT pathway through a survey of the literature: SHP2, SOCS3, SHP1, SOCS1, and PTP1B (37). RT-qPCR assay demonstrated that the overexpression of miR-199a-5p reduced the expression of SHP2 at mRNA level, but not that of the other four negative regulators (Figure 4A). Western blotting assay also showed that overexpression of miR-199a-5p reduced the expression of SHP2 at protein level (Figure 4B). SHP2 was predicted to contain three miR-199a-5p binding sites according to the starBase database (http://starbase.sysu.edu.cn/index.php) (Figure 4C), and subsequent luciferase assay showed that the luciferase activity of the group co-transfected with miR-199a-5p mimics and part SHP2 sequence-containing psicheck2 vector was significantly reduced (Figure 4D). These findings suggest that SHP2 is a target of miR-199a-5p.
Figure 4
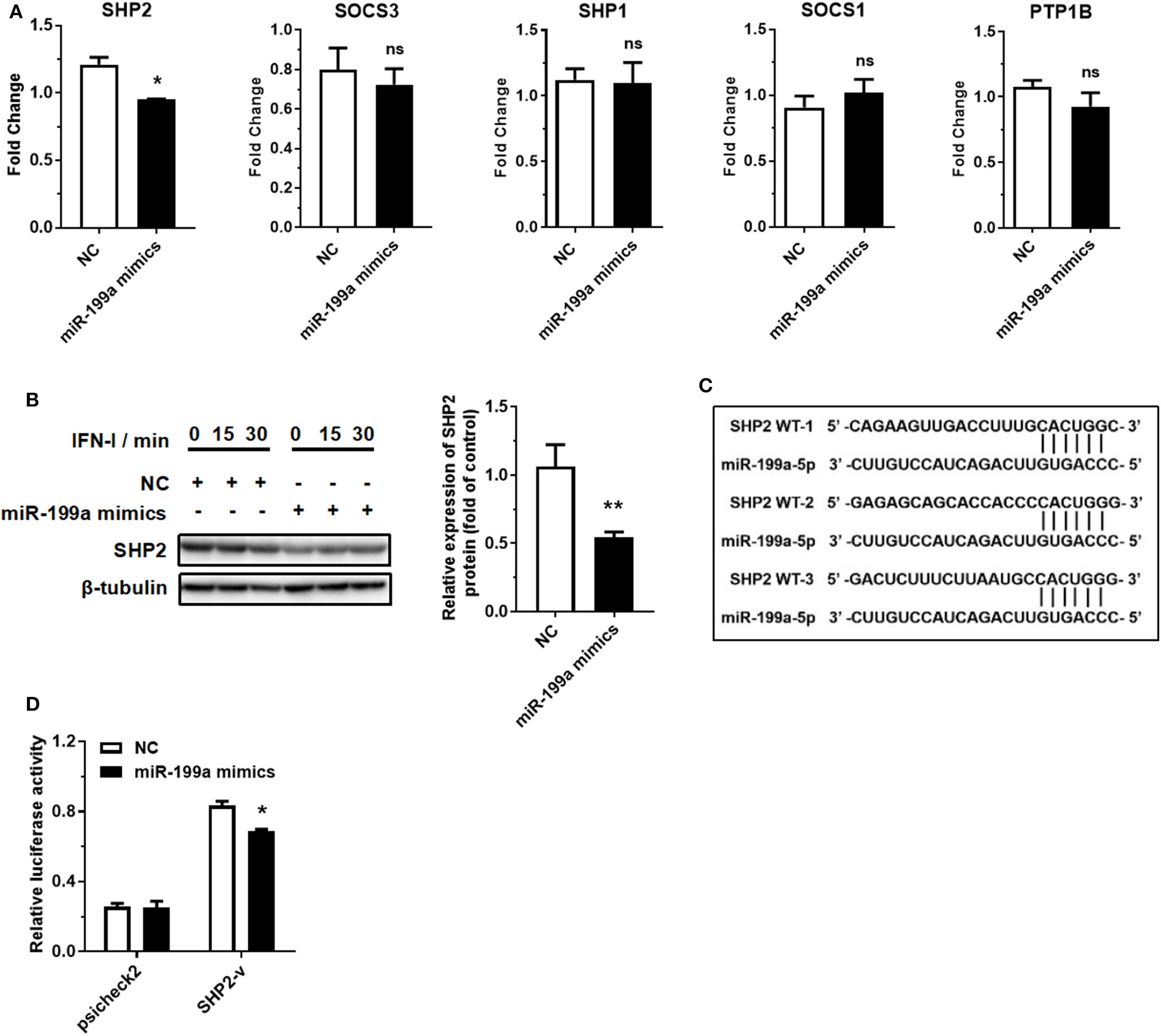
miR-199a-5p promotes IFN-I-induced inflammation by targeting SHP2. (A) RT-qPCR analyses of five predicted miR-199a-5p target genes after treatment with miR-199a-5p mimics. (B) SHP2 protein assessment at 0, 15, and 30 min of IFN-I stimulation after treatment with miR-199a-5p mimics. (C) Three miR-199a-5p binding sites within SHP2 3′ UTR predicted using the starBase database. (D) Luciferase activities of 293T cells co-transfected with blank psicheck2 vector or psicheck2 recombinant vector containing SHP2 3′ UTR fragment and miR-199a-5p mimics or NC. NC, negative control. SHP2-v, psicheck2 recombinant vector containing SHP2 3′ UTR fragment. Data represent means ± SEM. *p < 0.05, **p < 0.01. ns, not significance.
Hsa_circ_0060450 Upregulates SHP2 Expression by Adsorbing miR-199a-5p
Further, we performed an in-depth investigation of the hsa_circ_0060450-miR-199a-5p-SHP2 axis. RT-qPCR and Western blotting results showed that hsa_circ_0060450 silencing depressed SHP2 expression at both mRNA and protein levels (Figures 5A,B). Subsequently, we designed two SHP2-siRNAs to silent the expression of SHP2 (Figure 5C), and further to verify the inhibitory effect of SHP2, as a typical dephosphorylation enzyme, on IFN-I-induced JAK-STAT1/3 pathway activation. Western blotting results showed SHP2 silencing promoted STAT1 and STAT3 protein phosphorylation after IFN-I stimulation (Figure 5D). In addition, silencing SHP2 boosted the expressions of IFN-I-induced genes, including IFIT1, IFIH1, CXCL10, and iNOS (Figure 5E).
Figure 5
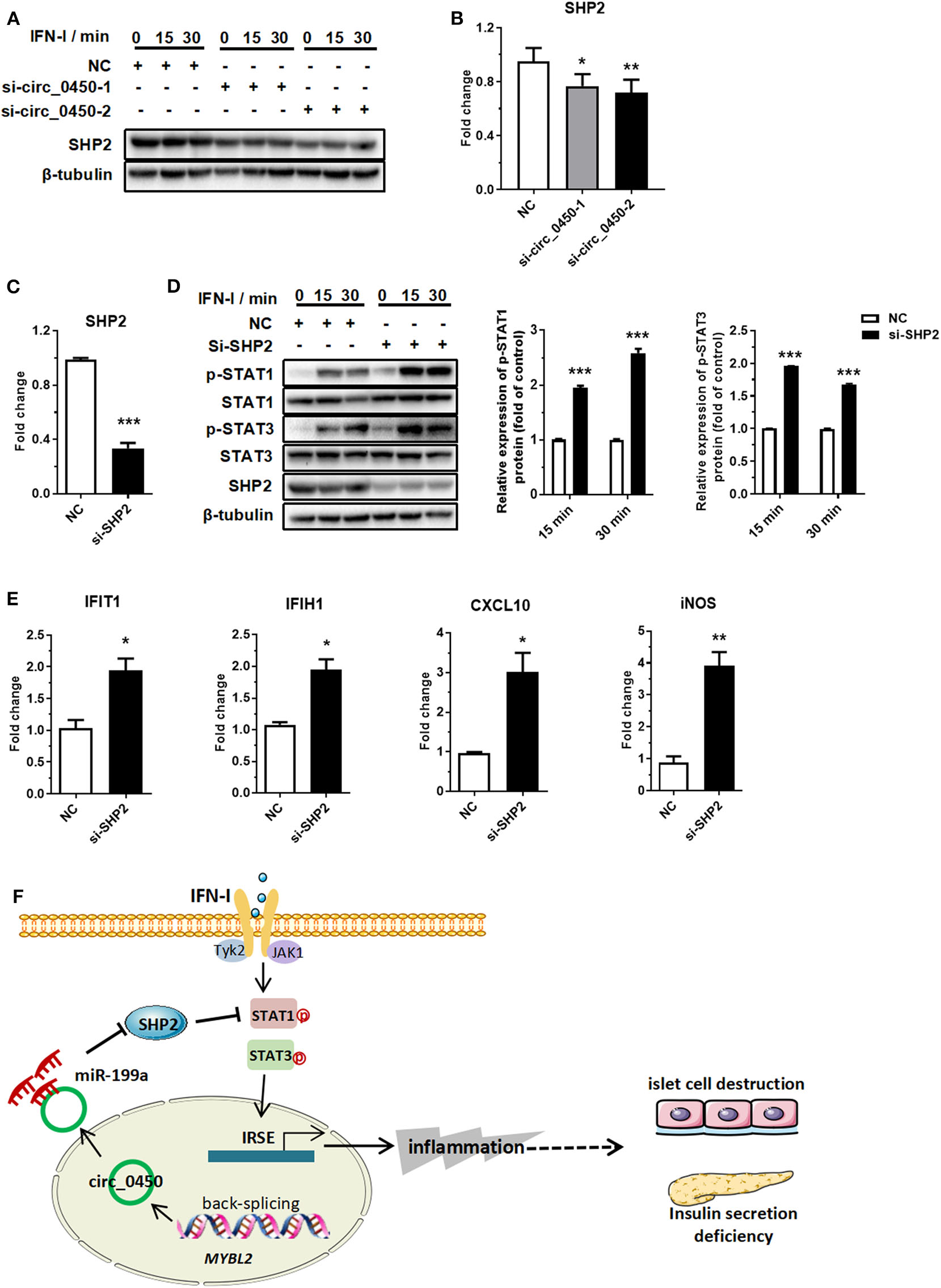
Hsa_circ_0060450 upregulates SHP2 by adsorbing miR-199a-5p. (A) SHP2 protein assessment at 0, 15, and 30 min of IFN-I stimulation after treatment with two hsa_circ_0060450 siRNAs. (B) RT-qPCR analyses of SHP2 expression after treatment with two hsa_circ_0060450 siRNAs. (C) RT-qPCR analysis of SHP2 expression to confirm the silencing efficiency of the SHP2 siRNA. (D) STAT1 and STAT3 phosphorylation level assessment at 15 and 30 min of IFN-I stimulation after treatment with the SHP2 siRNA. (E) RT-qPCR analyses of IFIT1, IFIH1, CXCL10, and iNOS under IFN-I stimulation after treatment with the SHP2 siRNA. (F) A schematic model of the function of hsa_circ_0060450 in IFN-I-stimulated macrophages and in T1DM. NC, negative control. Data represent means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
In this study, we obtained a differential circRNA expression profile based on microarray analysis of PBMCs in children with T1DM. We then characterized one circRNA, back-spliced from MYBL2, termed as hsa_circ_0060450, which exhibited upregulated expression in patients with T1DM. Subsequent assays revealed that hsa_circ_0060450 serves as a “sponge” of miR-199a-5p to inhibit IFN-I-induced inflammation (Figure 5F). miR-199a-5p was reported to exert various biological functions, and existing studies have indicated the potential roles of miR-199a-5p in the pathogenesis of diabetes. Lin et al. demonstrated that miR-199a-5p is upregulated in pancreatic β cells in response to high glucose and promotes apoptosis and ROS generation in type 2 diabetes (38). In addition, a bioinformatics analysis showed that mmu-miR-199a-5p is closely related to inflammatory response, and insulin signal pathway in the liver of diabetic mice (39). SHP2 is a widely expressed SH2 domain-containing protein tyrosine phosphatase (40). Although sufficient evidence has shown that SHP2 is involved in tumorigenesis, its roles in immune regulation have also been gradually elucidated. Shuai et al. found SHP2 negatively regulated the IFN-induced JAK1/STAT1 pathway by dephosphorylating STAT1 (37). Wu et al. also demonstrated that phosphorylation at the tyrosine residue Tyr701 of STAT1 induced by IFN-α was enhanced and prolonged in SHP2-deletion cells (41). In addition, Zhang et al. and Ke et al. observed similar negative regulation on STAT3 in SHP2 knockout mice (42, 43). More recently, Krajewska et al. showed that mice with a neuron-specific, conditional SHP2 deletion developed obesity and diabetes-associated complications, including hyperglycemia, insulin resistance, etc. This suggests a protective role of SHP2 in diabetes (44). Despite these existing studies, the present study is the first, to our knowledge, to investigate the role of hsa_circ_0060450-miR-199a-5p-SHP2 axis in IFN-I-induced inflammation in T1DM.
Unexpectedly, even if showing an upward trend in the PBMCs of T1DM patients, hsa_circ_0060450 did not prevent the occurrence of pancreatic inflammation and T1DM. Possibly, we consider that hsa_circ_0060450 is produced reactively during pancreatic inflammation, but the amount produced in the body is not sufficient to inhibit macrophage inflammation. In addition, there may be other circRNAs or other types of molecules in T1DM patients that antagonistically inhibit anti-inflammatory effect of hsa_circ_0060450. Therefore, it is necessary to administer a larger dose of exogenous hsa_circ_0060450 to achieve a certain effect of alleviating inflammation and treating T1DM.
Generally, circRNAs are classified into three types: exon circular RNA (ecircRNA), circular intronic RNA (ciRNA), and exon-intron circRNA (EIciRNA) (45). EcircRNA mainly exists in the cytoplasm, serving as a “sponge” of microRNAs, while the other two circRNAs are confined to the nucleus owing to the presence of introns (46–48). Zhang et al. demonstrated that intron-derived circular RNA ci-ankrd52 exists in the nucleus and cis-regulates transcription of its parental gene ANKRD52 via the catalytic effect of RNA Pol II (47). Li et al. subsequently found that intron-exon circRNA circEIF3J and circPAIP2 form an RNA-protein complex with U1SNP and RNA Pol II through RNA-RNA interaction to cis-regulate the transcription of their parental genes (46). The parental gene of hsa_circ_0060450, MYBL2, is a transcription factor of the MYB family, and was first studied to be a physiological regulator of cell cycle progression, cell differentiation and cell survival, but recently it was frequently found deregulation in numerous cancer entities and significantly drove cancer initiation and progression (49). Moreover, a new non-synonymous SNP R687C in exon 14 of MYBL2 was identified in a genome scan study in Ashkenazi patients with type 2 diabetes (50), suggesting a potential role of MYBL2 in diabetes. In the present study, we found MYBL2 showed an upregulated trend in T1DM patients compared with healthy controls (Supplementary Figure 4). In addition, we discovered that 40% of hsa_circ_0060450 is present in the cytoplasm, with the remaining proportion in the nucleus, and in the current study we mainly focused on the role of cytoplasmic hsa_circ_0060450; however, it remains to be elucidated whether hsa_circ_0060450 in the nucleus also functions in T1DM through the interaction with MYBL2.
In summary, in this study, we obtained a differential circRNA expression profile, and characterized a differentially expressed circRNA, hsa_circ_0060450. Subsequent experiments showed that hsa_circ_0060450 can adsorb miR-199a-5p, releasing its target SHP2, by which hsa_circ_0060450 inhibits macrophage-mediated inflammation through the suppression of JAK-STAT1/3 signaling pathway triggered by type I interferon. Overall, this study indicates the important role of hsa_circ_0060450 in T1DM and implies a promising therapeutic molecule in the treatment of T1DM in the future.
Statements
Data availability statement
All datasets generated for this study are included in the article/Supplementary Material.
Ethics statement
The studies involving human participants were reviewed and approved by Research Ethics Board of the Children's Hospital of Fudan University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author contributions
LY, XH, CZ, CS, SH, WX, YG, QL, FL, and WL designed, carried out experiments, and analyzed data. LY and YZ wrote the manuscript. YZ and JF planned, designed, supervised, and coordinated the overall research efforts. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Key R&D Program of China (2016YFC1305102 to YZ), National Natural Science Foundation of China (81671561 and 81974248 to YZ, 81900751 to XH), the International Joint Laboratory Program of National Children's Medical Center (EK1125180109 to YZ), Program for Outstanding Medical Academic Leader (2019LJ19 to YZ), and Shanghai Municipal Planning Commission of Science and Research Fund (201740065 to YZ and 20174Y0079 to XH). Shanghai Pujiang Program (16PJ1401600 to JF). Shanghai Committee of Science and Technology (19ZR1406400 to JF).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.576903/full#supplementary-material
References
1.
Pozzilli P Maddaloni E Buzzetti R . Combination immunotherapies for type 1 diabetes mellitus. Nat Rev Endocrinol. (2015) 11:289–97. 10.1038/nrendo.2015.8
2.
Ni Q Pham NB Meng WS Zhu G Chen X . Advances in immunotherapy of type I diabetes. Adv Drug Deliv Rev. (2018) 139:83–91. 10.1016/j.addr.2018.12.003
3.
Roep BO Tree TI . Immune modulation in humans: implications for type 1 diabetes mellitus. Nat Rev Endocrinol. (2014) 10:229–42. 10.1038/nrendo.2014.2
4.
Marwaha AK Crome SQ Panagiotopoulos C Berg KB Qin H Ouyang Q et al . Cutting edge: increased IL-17-secreting T cells in children with new-onset type 1 diabetes. J Immunol. (2010) 185:3814–8. 10.4049/jimmunol.1001860
5.
Ferraro A Socci C Stabilini A Valle A Monti P Piemonti L et al . Expansion of Th17 cells and functional defects in T regulatory cells are key features of the pancreatic lymph nodes in patients with type 1 diabetes. Diabetes. (2011) 60:2903–13. 10.2337/db11-0090
6.
Sanjeevi CB . Genes influencing innate and acquired immunity in type 1 diabetes and latent autoimmune diabetes in adults. Ann N Y Acad Sci. (2006) 1079:67–80. 10.1196/annals.1375.010
7.
Ting C Bansal V Batal I Mounayar M Chabtini L El Akiki G et al . Impairment of immune systems in diabetes. Adv Exp Med Biol. (2012) 771:62–75. 10.1007/978-1-4614-5441-0_8
8.
Eizirik DL Colli ML Ortis F . The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. (2009) 5:219–26. 10.1038/nrendo.2009.21
9.
Gulden E Chao C Tai N Pearson JA Peng J Majewska-Szczepanik M et al . TRIF deficiency protects non-obese diabetic mice from type 1 diabetes by modulating the gut microbiota and dendritic cells. J Autoimmun. (2018) 93:57–65. 10.1016/j.jaut.2018.06.003
10.
Jun HS Santamaria Lim HW Zhang ML Yoon JW . Absolute requirement of macrophages for the development and activation of beta-cell cytotoxic CD8+ T-cells in T-cell receptor transgenic NOD mice. Diabetes. (1999) 48:34–42. 10.2337/diabetes.48.1.34
11.
Willcox A Richardson SJ Bone AJ Foulis AK Morgan NG . Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol. (2009) 155:173–81. 10.1111/j.1365-2249.2008.03860.x
12.
Sanchez-Zamora YI Juarez-Avelar I Vazquez-Mendoza A Hiriart M Rodriguez-Sosa M . Altered macrophage and dendritic cell response in Mif -/- mice reveals a role of mif for inflammatory-Th1 response in type 1 diabetes. J Diabetes Res. (2016) 2016:7053963. 10.1155/2016/7053963
13.
Ferris ST Zakharov PN Wan X Calderon B Artyomov MN Unanue ER et al . The islet-resident macrophage is in an inflammatory state and senses microbial products in blood. J Exp Med. (2017) 214:2369–85. 10.1084/jem.20170074
14.
Carrero JA McCarthy D Ferris ST Wan X Hu H Zinselmeyer BH et al . Resident macrophages of pancreatic islets have a seminal role in the initiation of autoimmune diabetes of NOD mice. Proc Natl Acad Sci USA. (2017) 114:E10418–27. 10.1073/pnas.1713543114
15.
Drescher KM Tracy SM . The CVB and etiology of type 1 diabetes. Curr Top Microbiol Immunol. (2008) 323:259–74. 10.1007/978-3-540-75546-3_12
16.
Devendra D Jasinski J Melanitou E Nakayama M Li M Hensley B et al . Interferon-alpha as a mediator of polyinosinic : polycytidylic acid-induced type 1 diabetes. Diabetes. (2005) 54:2549–56. 10.2337/diabetes.54.9.2549
17.
Lang KS Recher M Junt T Navarini AA Harris NL Freigang S et al . Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat Med. (2005) 11:138–45. 10.1038/nm1105-1256
18.
Op de Beeck A Eizirik DL . Viral infections in type 1 diabetes mellitus–why the beta cells?Nat Rev Endocrinol. (2016) 12:263–73. 10.1038/nrendo.2016.30
19.
Oikarinen S Tauriainen S Hober D Lucas B Vazeou A Sioofy-Khojine A et al . Virus antibody survey in different European populations indicates risk association between coxsackievirus B1 and type 1 diabetes. Diabetes. (2014) 63:655–62. 10.2337/db13-0620
20.
Imagawa A Hanafusa T . Fulminant type 1 diabetes–an important subtype in East Asia. Diabetes Metab Res Rev. (2011) 27:959–64. 10.1002/dmrr.1236
21.
Ferreira RC Guo H Coulson RM Smyth DJ Pekalski ML Burren OS et al . A type I interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes. (2014) 63:2538–50. 10.2337/db13-1777
22.
Kallionpaa H Elo LL Laajala E Mykkanen J Ricano-Ponce I Vaarma M et al . Innate immune activity is detected prior to seroconversion in children with HLA-conferred type 1 diabetes susceptibility. Diabetes. (2014) 63:2402–14. 10.2337/db13-1775
23.
Li Q Xu BH Michie SA Rubins KH Schreriber RD McDevitt HO . Interferon-alpha initiates type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci USA. (2008) 105:12439–44. 10.1073/pnas.0806439105
24.
Diana J Simoni Y Furio L Beaudoin L Agerberth B Barrat F et al . Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat Med. (2013) 19:65–73. 10.1038/nm.3042
25.
Marro BS Legrain S Ware BC Oldstone MB . Macrophage IFN-I signaling promotes autoreactive T cell infiltration into islets in type 1 diabetes model. JCI Insight. (2019) 4:e125067. 10.1172/jci.insight.125067
26.
Lasda E Parker R . Circular RNAs: diversity of form and function. RNA. (2014) 20:1829–42. 10.1261/rna.047126.114
27.
Wilusz JE . A 360 degrees view of circular RNAs: from biogenesis to functions. Wiley Interdiscip Rev RNA. (2018) 9:e1478. 10.1002/wrna.1478
28.
Lyu D Huang S . The emerging role and clinical implication of human exonic circular RNA. RNA Biol. (2017) 14:1000–6. 10.1080/15476286.2016.1227904
29.
Jeck WR Sorrentino JA Wang K Slevin MK Burd CE Liu J et al . Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. (2013) 19:141–57. 10.1261/rna.035667.112
30.
Memczak S Jens M Elefsinioti A Torti F Krueger J Rybak A et al . Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. (2013) 495:333–8. 10.1038/nature11928
31.
Zheng LL Li JH Wu J Sun WJ Liu S Wang ZL et al . deepBase v2.0: identification, expression, evolution and function of small RNAs, LncRNAs and circular RNAs from deep-sequencing data. Nucleic Acids Res. (2016) 44:D196–202. 10.1093/nar/gkv1273
32.
Hansen TB Jensen TI Clausen BH Bramsen JB Finsen B Damgaard CK et al . Natural RNA circles function as efficient microRNA sponges. Nature. (2013) 495:384–8. 10.1038/nature11993
33.
Zhang PX Cheng JJ Zou SY D'Souza AD Koff JL Lu J et al . Pharmacological modulation of the AKT/microRNA-199a-5p/CAV1 pathway ameliorates cystic fibrosis lung hyper-inflammation. Nat Commun. (2015) 6:221. 10.1038/ncomms7221
34.
Liu Y Guan H Zhang JL Zheng Z Wang HT Tao K et al . Acute downregulation of miR-199a attenuates sepsis-induced acute lung injury by targeting SIRT1. Am J Physiol Cell Physiol. (2018) 314:C449–55. 10.1152/ajpcell.00173.2017
35.
Esquela-Kerscher A Slack FJ . Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. (2006) 6:259–69. 10.1038/nrc1840
36.
Jansson MD Lund AH . MicroRNA and cancer. Mol Oncol. (2012) 6:590–610. 10.1016/j.molonc.2012.09.006
37.
Shuai K Liu B . Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol. (2003) 3:900–11. 10.1038/nri1226
38.
Lin N Li XY Zhang HM Yang Z Su Q . microRNA-199a-5p mediates high glucose-induced reactive oxygen species production and apoptosis in INS-1 pancreatic beta-cells by targeting SIRT1. Eur Rev Med Pharmacol Sci. (2017) 21:1091–98.
39.
Bai Y Bao X Jiang G Ge D He W Zhao D et al . Jiang tang xiao ke granule protects hepatic tissue of diabetic mice through modulation of insulin and ras signaling - a bioinformatics analysis of micrornas and mrnas network. Front Pharmacol. (2020) 11:173. 10.3389/fphar.2020.00173
40.
Feng GS Hui CC Pawson T . SH2-containing phosphotyrosine phosphatase as a target of protein-tyrosine kinases. Science. (1993) 259:1607–11. 10.1126/science.8096088
41.
Wu TR Hong YK Wang XD Ling MY Dragoi AM Chung AS et al . SHP-2 is a dual-specificity phosphatase involved in Stat1 dephosphorylation at both tyrosine and serine residues in nuclei. J Biol Chem. (2002) 277:47572–80. 10.1074/jbc.M207536200
42.
Zhang EE Chapeau E Hagihara K Feng GS . Neuronal Shp2 tyrosine phosphatase controls energy balance and metabolism. Proc Natl Acad Sci USA. (2004) 101:16064–9. 10.1073/pnas.0405041101
43.
Ke Y Zhang EE Hagihara K Wu D Pang Y Klein R et al . Deletion of Shp2 in the brain leads to defective proliferation and differentiation in neural stem cells and early postnatal lethality. Mol Cell Biol. (2007) 27:6706–17. 10.1128/MCB.01225-07
44.
Krajewska M Banares S Zhang EE Huang X Scadeng M Jhala US et al . Development of diabesity in mice with neuronal deletion of Shp2 tyrosine phosphatase. Am J Pathol. (2008) 172:1312–24. 10.2353/ajpath.2008.070594
45.
Yang L Fu JR Zhou YF . Circular RNAs and their emerging roles in immune regulation. Front Immunol. (2018) 9:2977. 10.3389/fimmu.2018.02977
46.
Li Z Huang C Bao C Chen L Lin M Wang X et al . Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol. (2015) 22:256–64. 10.1038/nsmb.2959
47.
Zhang Y Zhang XO Chen T Xiang JF Yin QF Xing YH et al . Circular intronic long noncoding RNAs. Mol Cell. (2013) 51:792–806. 10.1016/j.molcel.2013.08.017
48.
Li Z Huang C Bao C Chen L Lin M Wang X et al . Corrigendum: exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol. (2017) 24:194. 10.1038/nsmb0217-194a
49.
Musa J Aynaud MM Mirabeau O Delattre O Grunewald TG . MYBL2 (B-Myb): a central regulator of cell proliferation, cell survival and differentiation involved in tumorigenesis. Cell Death Dis. (2017) 8:e2895. 10.1038/cddis.2017.244
50.
Permutt MA Wasson J Love-Gregory L Ma J Skolnick G Suarez B et al . Searching for type 2 diabetes genes on chromosome 20. Diabetes. (2002) 51(Suppl. 3):S308–15. 10.2337/diabetes.51.2007.S308
Summary
Keywords
type I interferon, SHP2, macrophage, type 1 diabetes mellitus, circular RNA
Citation
Yang L, Han X, Zhang C, Sun C, Huang S, Xiao W, Gao Y, Liang Q, Luo F, Lu W, Fu J and Zhou Y (2020) Hsa_circ_0060450 Negatively Regulates Type I Interferon-Induced Inflammation by Serving as miR-199a-5p Sponge in Type 1 Diabetes Mellitus. Front. Immunol. 11:576903. doi: 10.3389/fimmu.2020.576903
Received
27 June 2020
Accepted
31 August 2020
Published
29 September 2020
Volume
11 - 2020
Edited by
Menno de Winther, Amsterdam University Medical Center, Netherlands
Reviewed by
Tomohide Takaya, Shinshu University, Japan; Peixiang Lan, Houston Methodist Research Institute, United States
Updates
Copyright
© 2020 Yang, Han, Zhang, Sun, Huang, Xiao, Gao, Liang, Luo, Lu, Fu and Zhou.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yufeng Zhou yfzhou1@fudan.edu.cnJinrong Fu fujinrong@hotmail.com
This article was submitted to Molecular Innate Immunity, a section of the journal Frontiers in Immunology
†These authors have contributed equally to this work and share first authorship
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.