 Chloe H. Lee1,2,3
Chloe H. Lee1,2,3 Mariana Pereira Pinho1,2
Mariana Pereira Pinho1,2 Paul R. Buckley1,2,3
Paul R. Buckley1,2,3 Isaac B. Woodhouse1,2,3
Isaac B. Woodhouse1,2,3 Graham Ogg1,2,5
Graham Ogg1,2,5 Alison Simmons1,4,5Giorgio Napolitani1,2
Alison Simmons1,4,5Giorgio Napolitani1,2 Hashem Koohy1,2,3*
Hashem Koohy1,2,3*- 1MRC Human Immunology Unit, Medical Research Council (MRC) Weatherall Institute of Molecular Medicine (WIMM), John Radcliffe Hospital, University of Oxford, Oxford, United Kingdom
- 2Radcliffe Department of Medicine, University of Oxford, Oxford, United Kingdom
- 3MRC WIMM Centre For Computational Biology, MRC Weatherall Institute of Molecular Medicine, John Radcliffe Hospital, University of Oxford, Oxford, United Kingdom
- 4Translational Gastroenterology Unit, John Radcliffe Hospital, Oxford, United Kingdom
- 5NIHR Oxford Biomedical Research Centre, Oxford, United Kingdom
While individuals infected with coronavirus disease 2019 (COVID-19) manifested a broad range in susceptibility and severity to the disease, the pre-existing immune memory to related pathogens cross-reactive against SARS-CoV-2 can influence the disease outcome in COVID-19. Here, we investigated the potential extent of T cell cross-reactivity against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that can be conferred by other coronaviruses and influenza virus, and generated an in silico map of public and private CD8+ T cell epitopes between coronaviruses. We observed 794 predicted SARS-CoV-2 epitopes of which 52% were private and 48% were public. Ninety-nine percent of the public epitopes were shared with SARS-CoV and 5.4% were shared with either one of four common coronaviruses, 229E, HKU1, NL63, and OC43. Moreover, to assess the potential risk of self-reactivity and/or diminished T cell response for peptides identical or highly similar to the host, we identified predicted epitopes with high sequence similarity with human proteome. Lastly, we compared predicted epitopes from coronaviruses with epitopes from influenza virus deposited in IEDB, and found only a small number of peptides with limited potential for cross-reactivity between the two virus families. We believe our comprehensive in silico profile of private and public epitopes across coronaviruses would facilitate design of vaccines, and provide insights into the presence of pre-existing coronavirus-specific memory CD8+ T cells that may influence immune responses against SARS-CoV-2.
Introduction
Faced by unprecedent health and economic crisis from the coronavirus disease 2019 (COVID-19), the scientific community is pushing forward with efforts to develop vaccines and treatments to mitigate its impact. While the severity of symptoms have been reported to be associated with age, gender, and comorbidities such as cardiovascular diseases and chronic respiratory diseases (1), the underlying mechanism of broad variation in susceptibility and severity to COVID-19 is not fully understood (2). It is however accepted that an altered immune response is a key contributor to pathology (3, 4), and the balance between generation of protective and pathological immune responses by the host may be a vital factor governing the disease outcome.
As immune memory by related pathogens has shown to help reduce severity and spread of the diseases (5–7), pre-existing immunity through cross-reactivity to familial coronavirus strains may provide individuals with protection or enhanced susceptibility against the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) without prior exposure (8–10). Therefore, we aim to characterize the potential for the existing immune memory by other coronaviruses and influenza virus to fight against SARS-CoV-2 and further identify targets for developing a “universal” vaccine against coronaviruses.
The strains infecting humans belong to either alpha and beta genera of coronavirus. The alphacoronavirus contains human coronavirus 229E (HCoV-229E) and HCoV-NL63, while the betacoronavirus contains HCoV-OC43, and HCoV-HKU1, Middle East respiratory syndrome coronavirus (MERS-CoV), SARS-CoV, and SARS-CoV-2 (11). It is known that NL63, 229E, OC43, and HKU1 usually cause only mild to moderate symptoms such as cough, runny nose, fever, and sore throat like the common cold (12), whereas MERS-CoV and SARS-CoV cause more severe symptoms including respiratory tract disease.
In this study, we investigated the level of T cell antigen cross-reactivity across the seven alpha and betacoronavirus strains, evaluated the risk of self-reactivity from SARS-CoV-2 predicted epitopes, and identified targets for vaccine developments against coronavirus and influenza virus. We first predicted the potential of peptides to be presented by 10 prevalent HLA alleles and eliciting CD8+ T cell responses, and generated a comprehensive in silico profile of public and private predicted epitopes. We also expanded the map of cross-reactivity from exact matching peptides to those with a high sequence similarity, resulting in addition of 264 and 283 public SARS-CoV-2 predicted epitopes by allowing one and two amino acid mismatches, respectively. Moreover, to assess the risk of self-reactivity and immunopathology, we compared SARS-CoV-2 predicted epitopes with human proteome sequences and detected 10 predicted epitopes that are single amino acid variant from their counterparts in the human proteome. Lastly, to explore the potential for development of vaccines against coronavirus and influenza virus, we compared our list of predicted epitopes from coronaviruses with epitopes from influenza virus deposited in Immune Epitope Database and Analysis Resource (IEDB) and detected only a limited number of epitopes with a modest sequence similarity that are shared across multiple coronavirus strains and influenza.
Methods
Retrieval of Coronavirus Proteome Sequences
The proteome sequences of coronavirus strains were obtained from NCBI. The reference numbers for these sequences are NC_002645.1 (229E), NC_006577.2 (HKU1), NC_019843.3 (MERS-CoV), NC_005831.2 (NL63), NC_006213.1 (OC43), NC_004718.3 (SARS-CoV) and NC_045512.2 (SARS-CoV-2).
Sequence Alignment and Phylogenetic Tree of Encoded Proteins in Coronaviruses
For multiple sequence alignment and phylogenetic tree generation, the open reading frames were grouped by their encoded proteins—spike protein (S), envelope protein (E), membrane protein (M), nucleocapsid protein (N), replicase polyprotein (Orf1ab), and other encoded regions (Other)—prior to analysis. The multiple sequence alignment was conducted using “msa” function from R msa v1.4.3 package and visualized by “msaPrettyPrint” function. The phylogenetic tree was produced by plotting “identity” distance generated by “dist.alignment” function from R seqinr v3.6.1 package.
Peptide Generation From Proteome Sequences
Each encoded proteins were fragmented into 9-mer peptides by scanning the proteome with a window of nine amino acids and step length of one amino acid. For strains containing two open reading frames annotated with the same functional protein e.g. 229E, MERS-CoV, SARS-CoV, and SARS-CoV-2 having two open reading frames annotated for Orf1ab, 9-mer peptides were generated from both encoded proteins and unique set of peptides were selected for subsequent analysis.
MHC Presentation Prediction
The antigen presentation of MHC was predicted using NetMHCpan v4.0 (13), a model trained on binding affinity and eluted ligand data, against HLA-A*0101, 0201, 0301, 2402, HLA-B*0702, 4001, 0801, and HLA-C*0702, 0401, 0701 alleles. Peptides with rank score <=2.0 were categorized as positive HLA-binder.
Immunogenicity Prediction
The immunogenicity potential was predicted by R package Repitope for peptides that passed NetMHCpan filtering i.e. those predicted to bind at least one HLA allele. The Repitope utilizes amino acid descriptors and TCR-peptide contact potential profiling (CPP)-based features to label immunogenicity. The Repitope package was retrieved from GitHub repository (https://github.com/masato-ogishi/Repitope.git).
After feature computation and feature selection, we utilized the published Repitope “MHCI_Human” model to extrapolate probabilistic immunogenicity scores for our dataset and made binary classification of immunogenicity. This classification was based on a threshold computed from the ROC curves of the MHCI_Human immunogenicity prediction model and was calculated using the youden index that maximizes 1-sensitivity+specificity. Probabilistic scores were extracted from the original model for a subset of peptides that were identical to peptides in the model’s training dataset.
Visualization of Private and Public Peptides
The conservation of peptides across coronavirus strains before and after MHC binding and immunogenicity prediction were visualized by “venn” function from R venn v1.9 package and upset function from R UpSetR v1.4.0 package. To identify shared peptides with up to two amino acid tolerance, the best matching peptides were identified by “pairwiseAlignment” from Biostrings v2.40.2 package using BLOSUM62 matrix, gapOpening of 100, and gapExtension of 100, followed by hamming distance to filter only peptide pairs with less than or equal to two amino acid difference.
Sequence Similarity With Human Proteome and Epitopes Deposited in IEDB
The sequence similarity of peptides derived from SARS-CoV-2 with human proteome counterparts was computed by first identifying best global-local alignment by “pairwiseAlignment” function from Biostrings v2.40.2 package with a high gap penalty, gapOpening of 100 and gapExtension of 100. The number of mismatches between best aligned pair was computed by hamming distance using “stringdist” function from R stringdist v0.9.5.5 package. Similarly, the sequence similarity between coronavirus peptides and epitopes from IEDB was compared by global-local alignment following by computing hamming distance to find the best matching pairs. In evaluating the accuracy of predicted epitopes with epitopes deposited in IEDB, the epitopes with matching sequence of predicted epitopes were retrieved.
Results
Shared Predicted Epitopes Among Coronavirus Strains
To first evaluate the homology of proteome sequences among alpha- and betacoronavirus strains, we conducted sequence alignment and generated a phylogenetic tree of encoded proteins between NL63, 229E, OC43, HKU1, MERS-CoV, SARS-CoV, and SARS-CoV-2 (see Methods). Based on sequence alignments (example alignment of spike protein illustrated in Supplementary Figure 1) and phylogenetic trees of encoded proteins, the alpha- and betacoronavirus strains shared a high sequence similarity within their own genera, i.e. higher similarity between NL63 and 229E than with betacoronaviruses. In particular, for betacoronavirus strains, phylogenetic analysis showed higher similarity between OC43 and HKU1, and between SARS-CoV and SARS-CoV-2. Notably, MERS-CoV showed relatively distinct proteome sequences to all other coronavirus strains included in this study (Figure 1A for spike protein and Supplementary Figure 2 for other encoded proteins).
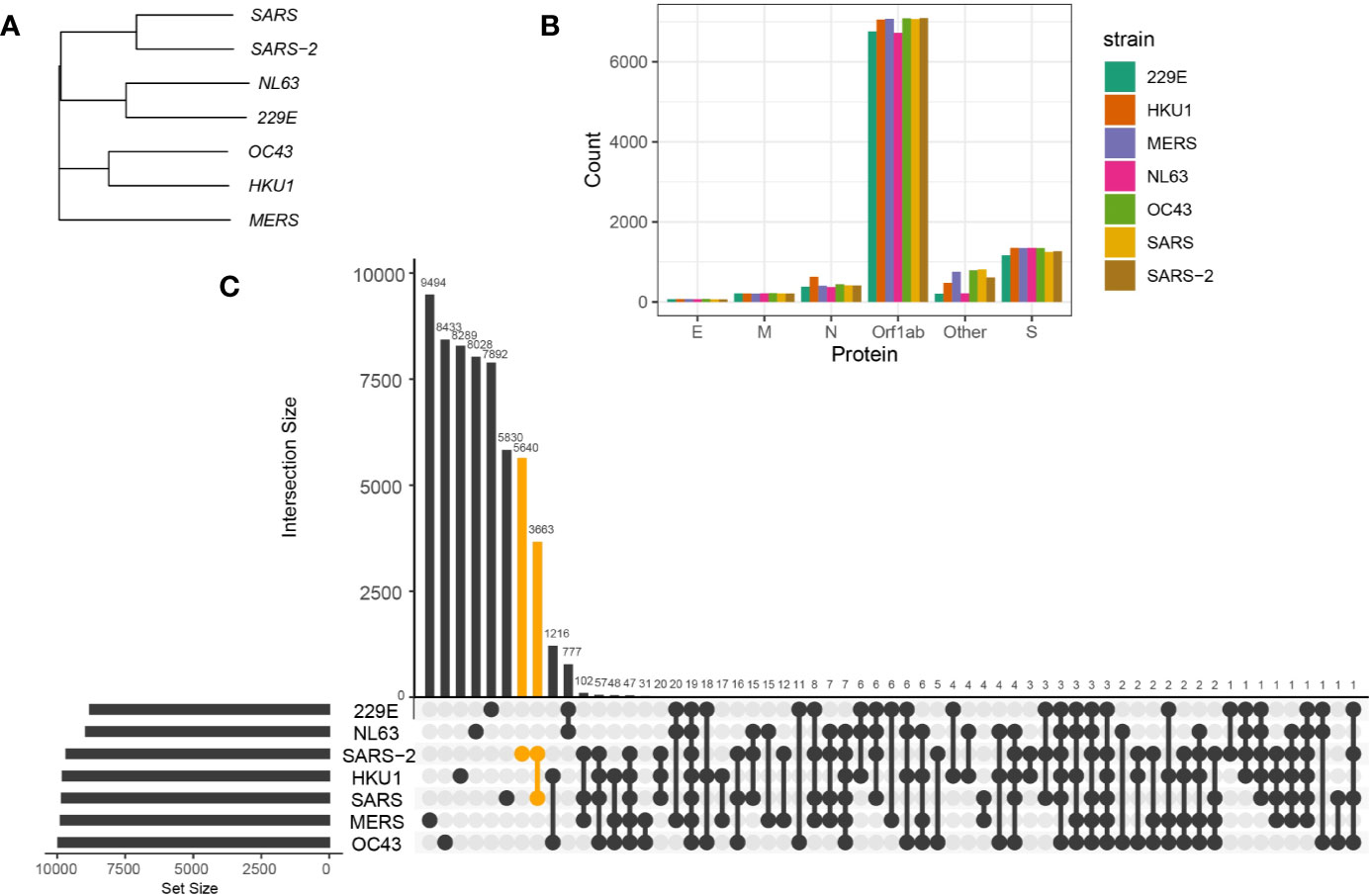
Figure 1 Sequence homology between coronavirus strains. (A) Phylogenetic tree of spike protein sequences between NL63, 229E, OC43, HKU1, MERS-CoV, SARS-CoV, and SARS-CoV-2 strains. (B) Number of 9-mer peptides generated from each coronavirus strains grouped by functional proteins, envelope protein (E), membrane protein (M), nucleocapsid protein (N) and replicase polyprotein (Orf1ab), spike protein (S), and other encoded proteins (Other). (C) Number of shared and private 9-mer peptides between coronavirus strains. The UpSet plot illustrates interaction of 9-mer peptides between seven coronavirus strains. The number of 9-mer peptides unique from SARS-CoV-2 or shared with SARS-CoV are colored in orange.
To identify the conserved 9-mer peptides across coronavirus strains, we first generated 9-mer peptides from encoded proteins of coronavirus strains (Figure 1B) and detected public peptides with identical matches (Figure 1C). Notably, there were 3,663 shared 9-mer peptides between SARS-CoV and SARS-CoV-2. Given the longest open reading frame of replicase polyproteins (Orf1ab) with an average of 7,002 amino acids across coronavirus strains, many public peptides were derived from Orf1ab, with 19 peptides shared across all studied strains (Figure 3A, first panel).
We then investigated antigen presentation potential of 9-mer peptides for 10 most prevalent HLA alleles corresponding to MHC class I (HLA-A, HLA-B, and HLA-C alleles). These include HLA-A*0101, 0201, 0301, 2402, HLA-B*0702, 4001, 0801, and HLA-C*0702, 0401, 0701 (14). Prevalence of these HLA alleles in the UK and US population has been referenced in Supplementary Figure 3A. The MHC presentation was predicted by NetMHCpan v4.0 (13) (see Methods). Generally, there was a relatively high number of peptides predicted to bind HLA-C alleles while HLA-B alleles had the lowest number of predicted binders (Figure 2A). We identified on average 2,559 (SD = 120) peptides predicted to bind at least one HLA allele (Figure 2B), which is ~21% of total number of 9-mer peptides across different strains. Of interest, there were 66 peptides from seven strains predicted to bind at least seven different HLA alleles, of which nine peptides were derived from SARS-CoV-2. The peptides predicted to bind 8 HLA alleles and their derived proteins are listed in Supplementary Figure 3B.
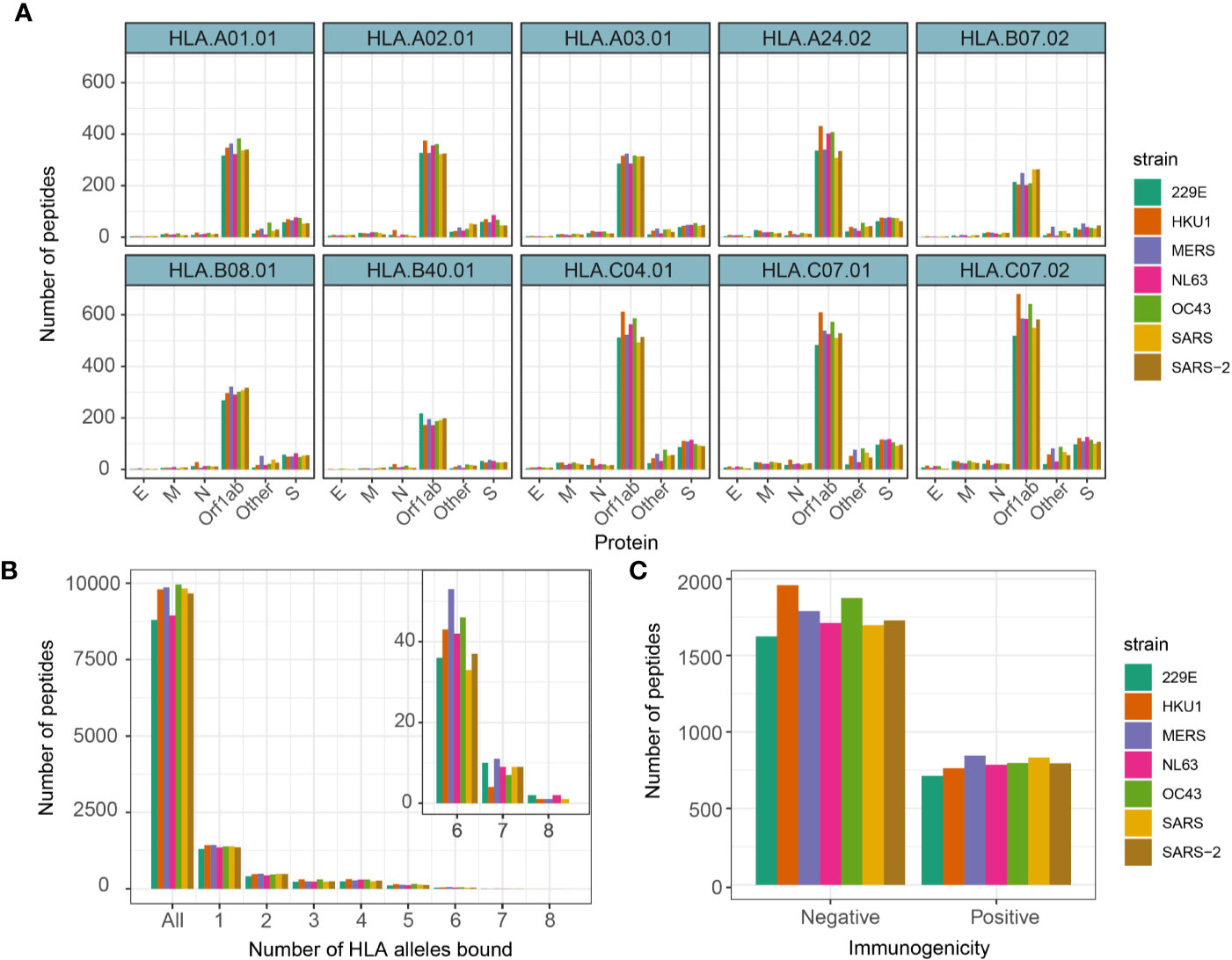
Figure 2 Number of 9-mer peptides predicted to bind HLA alleles and immunogenic. (A) Number of 9-mer peptides from each coronavirus strains predicted to bind annotated HLA alleles. (B) Number of peptides predicted to bind equal to specified number of HLA alleles. (C) Number of peptides predicted to productively interact with TCRs by Repitope prediction.
Although viral antigen presentation is a vital step in triggering immune responses, not all MHC binding peptides are immunogenic. We therefore set out to predict T cell immunogenicity, i.e. the ability of a peptide presented by an MHC molecule to productively interact with a T cell receptor, of all peptides predicted to bind at least one HLA allele. In an ongoing unpublished study, we have benchmarked the existing immunogenicity predicting models and as a result recognized a recently published model called Repitope (15) as the best performing existing model to predict immunogenicity of viral epitopes.
We therefore utilized Repitope (see Methods) and identified in total 4,894 out of 16,096 (~30%) unique predicted binders to be immunogenic (subsequently referred to as predicted epitopes), and the proportion of such predicted epitopes was comparable across different strains (Figure 2C, Supplementary Figure 4). On average, we detected 429 (SD = 26) epitopes predicted to bind at least one HLA type and be immunogenic. The full list of predicted immunogenic and nonimmunogenic HLA-binders are provided in Supplementary Table 1.
With the pool of predicted epitopes, we generated Venn diagrams to illustrate private and public epitopes across different coronavirus strains (Figure 3, Supplementary Figure 5). From a total of 794 predicted epitopes from SARS-CoV-2, 411 (/794, ~52%) were private while the remaining 383 (/794, ~48%) were public of which 379 (/383, ~99%) were shared with SARS-CoV and 21 (/383, ~5.4%) were shared with either one of four common coronaviruses, 229E, HKU1, NL63, and OC43. We detected only one predicted epitope—SLAIDAYPL derived from Orf1ab—public across all strains. Given the long sequence of Orf1ab protein, many of the public epitopes were derived from Orf1ab. This analysis suggests a large extent of CD8+ T cell cross-reactivity between SARS-CoV-2 and SARS-CoV but limited number of targets with other common coronaviruses.
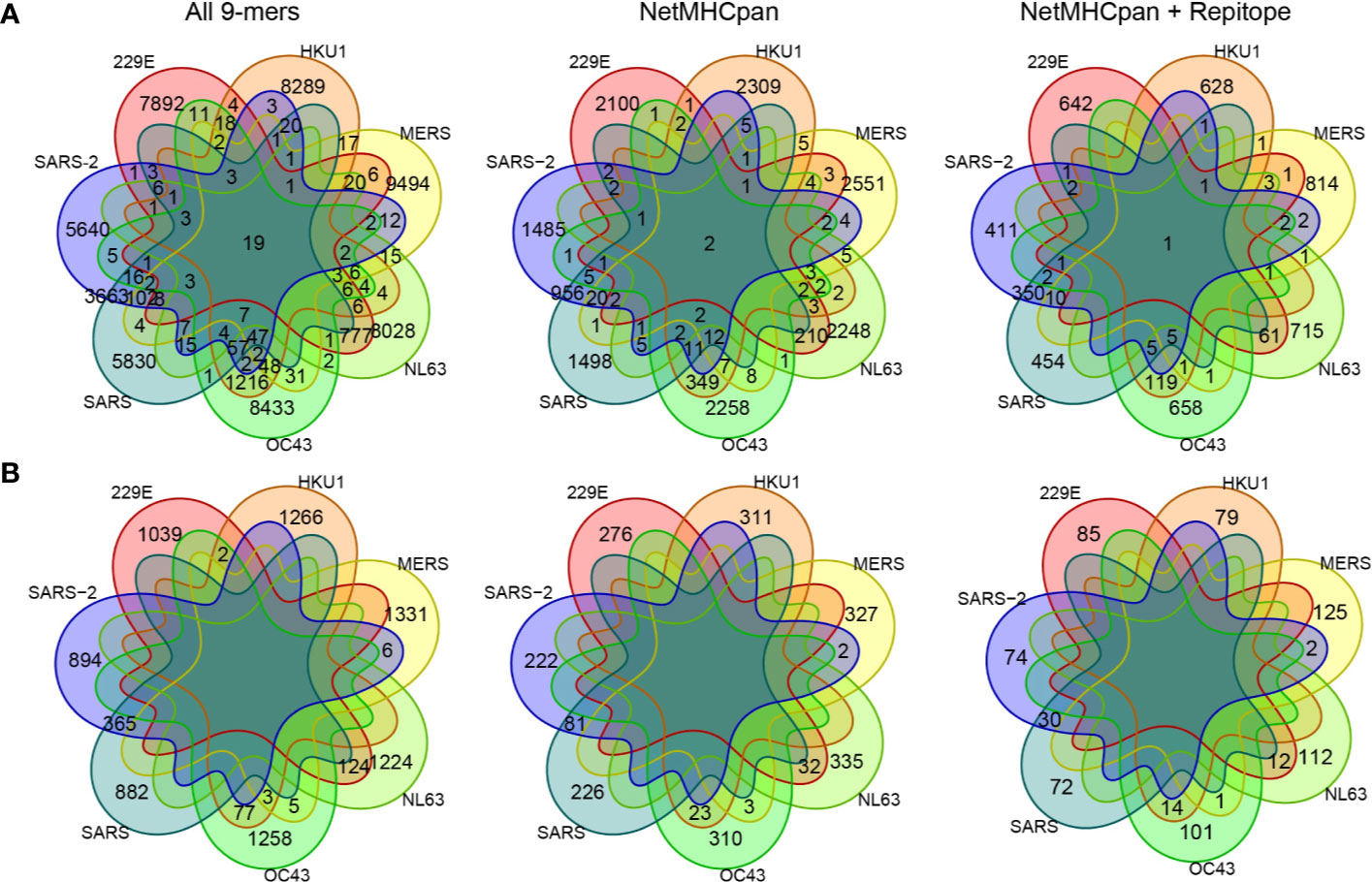
Figure 3 Comparing private and public 9-mer peptides from a complete set of peptides to after MHC presentation prediction by NetMHCpan and immunogenicity prediction by Repitope. Venn diagram is colored by strains. (A) Peptides derived from all encoded proteins. (B) Peptides derived from spike protein only.
Here, we conducted predictions of HLA binding and immunogenicity on 9-mer peptides as they were shown most prevalent for MHC binding compared to peptides of other lengths (16). However, it is now clear that peptides between lengths of 8–15aa can bind to HLA-I molecules and elicit immune responses (17, 18). Therefore, in parallel to the analysis of 9-mer peptides, we studied publicness of 10-mer predicted epitopes (Supplementary Figure 6A, Supplementary Table 2). In general given the longer sequences, there were less public peptides across coronavirus strains. Notably, as peptide length affects binding of HLA alleles differently (17), fewer peptides were predicted to bind HLA-B*08:01, HLA-C*04:01, HLA-C*07:01, HLA-C*07:02 (Supplementary Figure 6B). Peptides of other lengths can be readily investigated in the same manner.
Validating Predictions by Epitopes Deposited in IEDB
For a validation of predicted epitopes, we have compared the list of predicted epitopes with epitopes characterized by in vitro T cell assays and deposited in Immune Epitope Database and Analysis Resource (IEDB). We retrieved all 7,869 linear epitopes in IEDB (as of 05-05-2020), presented on MHC-I, reported positive in T cell assays and have human as the host organism then compared with the list of 4,894 unique predicted epitopes from SARS-CoV-2.
From the 7,869 IEBD immunogenic peptides, there were 34 unique peptides derived from coronavirusstrains, one from OC43, two from 229E, and the remaining from SARS-CoV. We identified 25 unique9-mer predicted epitopes from four coronavirus strains, 229E, NL63, SARS-CoV, and SARS-CoV-2 having matching pattern with 24 IEDB peptides (Table 1). The matching epitopes from NL63 and SARS-CoV-2 found in our analysis were shared with 229E and SARS-CoV respectively (Supplementary Table 3). Consequently, all 229E and NL63 predicted epitopes were matching with 229E IEBD peptides, whereas all SARS-CoV and SARS-CoV-2 predicted epitopes were matching with SARS-CoV IEBD peptides. None of our predicted epitopes matched with peptides derived from non-coronavirus microorganisms, suggesting a restricted potential for cross-reactivity between coronavirus and other virus strains.
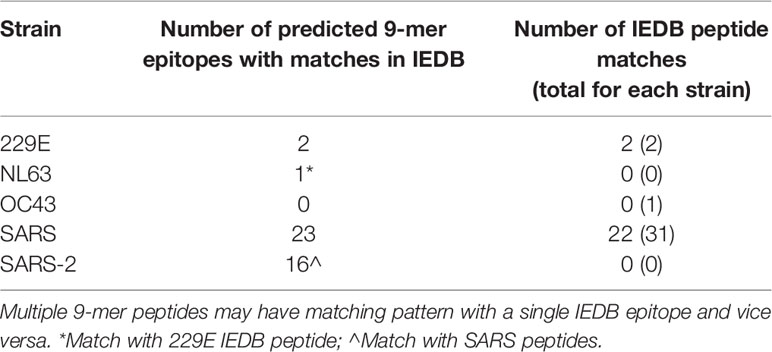
Table 1 Number of unique peptides by strain having matching pattern with immunogenic peptides deposited in IEDB.
Notably, 22 out of 31 (71%) SARS-derived immunogenic peptides in IEDB matched with our 23 SARS-derived 9-mer predicted epitopes (Table 1). Among the nine false negative SARS-derived IEDB peptides, seven were predicted MHC nonbinders and two were predicted non-immunogenic by our analysis. Despite the false negatives, 71% of true positives were retrieved from the shortlisted predicted epitopes, indicating a higher probability of identifying immunogenic peptides to facilitate targeted identification and validation of vaccine candidates.
Public Epitopes Between SARS-CoV-2 and Other Coronavirus Strains by High Sequence Similarity
In addition to public peptides by exact matches, predicted epitopes with a high sequence similarity may also trigger cross-reactivity across coronavirus strains. Here, we expanded the previous set of public predicted epitopes of SARS-CoV-2 to include those with up to two amino acid mismatches (Figure 4A, Supplementary Table 4). In addition to 383 predicted epitopes from SARS-CoV-2 shared with other coronavirus strains, there can be an increase of 173 and 159 unique public epitopes by allowing one or two amino acid mismatches respectively (Supplementary Figure 7). Given the long sequence of Orf1ab, the majority of SARS-CoV-2 predicted epitopes shared with 229E, HKU1, NL63, OC43, and MERS-CoV were derived from Orf1ab, while those derived from other proteins were predominantly shared with SARS-CoV (Supplementary Figure 7).
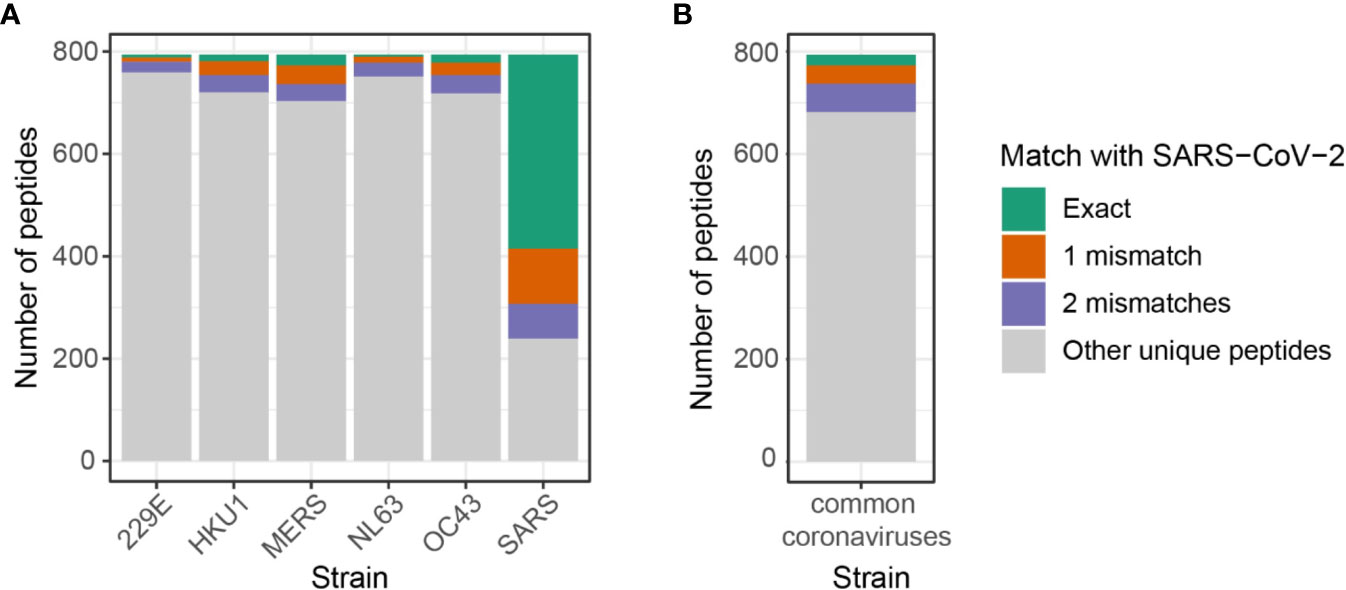
Figure 4 Number of shared predicted epitopes between SARS-CoV-2 and other coronavirus strains by allowing up to two mismatches. (A) Map of public peptides out of 794 SARS-CoV-2 predicted epitopes expanded by allowing up to two amino acids difference. Note that it includes duplicated peptides that may be shared across coronavirus strains, i.e. peptides shared across SARS-CoV-2, SARS-CoV, and MERS-CoV are counted in both SARS and MERS. Numbers can be retrieved from Supplementary Figure 7. (B) Unique predicted epitopes from four common coronaviruses, 229E, HKU1, NL63, and OC43, shared with 794 SARS-CoV-2 predicted epitopes. Numbers can be retrieved from Supplementary Figure 8.
While SARS-CoV and MERS-CoV shared the highest numbers of predicted epitopes with SARS-CoV-2, individuals have a greater exposure to common coronaviruses, 229E, HKU1, NL63, and OC43. To analyze the extent of cross-reactive responses that can be conferred by common coronaviruses, we investigated the number of unique SARS-CoV-2 predicted epitopes shared with either one of the four coronaviruses, namely 229E, HKU1, NL63, and OC43. There were 112 unique peptides out of 794 (14%) SARS-CoV-2 predicted epitopes shared with four common coronaviruses with a high sequence similarity (Figure 4B, Supplementary Figure 8). Among the 112 peptides, 21 (/794, 2.6%) had exact matches with SARS-CoV-2 peptides, while 36 (/794, 4.5%) and 55 (/794, 6.9%) peptides had one and two mismatches, respectively.
Potential Risk of Self-Reactivity by SARS-CoV-2 Derived Predicted Epitopes
Viral peptides identical or highly similar to those of the host organism might not give rise to high-affinity T cell responses because most of the T cells binding with high affinity to self peptides are eliminated during thymic negative selection. However, some of these potentially self-reactive T cells escape thymic selection, and when primed in the context of an infection they might cause autoimmune response (19, 20). Thus, when looking for potential candidates for vaccine design, it is important to assess the sequence similarity of predicted epitopes with their best matching counterparts in human proteome.
By comparing predicted epitopes from SARS-CoV-2 with the human proteome, none of the predicted epitopes had identical match but we detected 10 and 184 epitopes with one and two amino acid mismatches respectively with their best matching human proteome counterparts (Figure 5A). The predicted epitopes differing by only one amino acid are listed in Table 2.
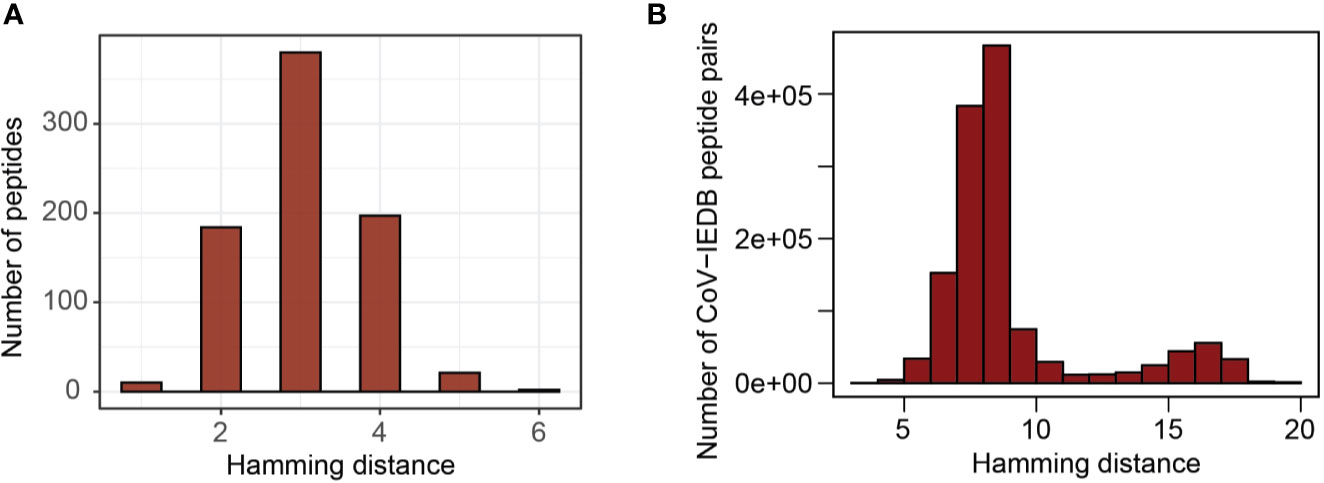
Figure 5 Sequence similarity with human proteome and influenza virus epitopes deposited in IEDB. (A) Distribution of hamming distance between SARS-CoV-2 derived peptides and human proteome counterparts (the region most similar to corresponding virus peptides). (B) Distribution of hamming distance between coronavirus derived peptides and all influenza virus epitopes deposited in IEDB.
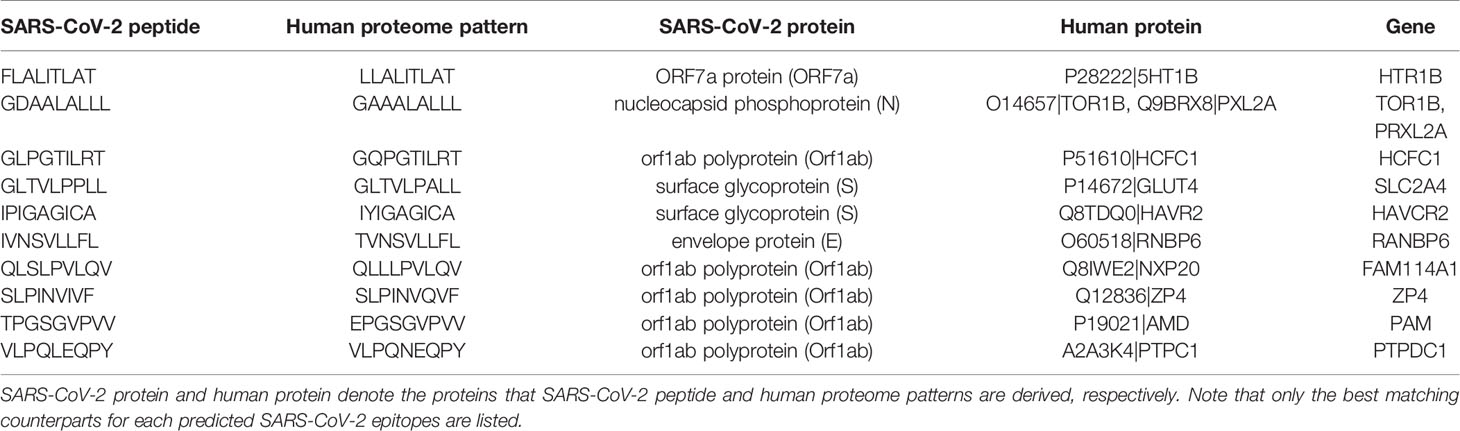
Table 2 Predicted epitopes from SARS-CoV-2 that are single amino acid variants of human proteome counterparts.
Cross-Reactivity Between Coronavirus and Influenza Virus
In order to compare theoretical cross-reactivity between different coronaviruses with the one from unrelated viruses, we compared the 4,894 unique predicted epitopes from all coronavirus strains with 1334 unique MHC-I influenza virus-derived epitopes (1,298 influenza A virus and 36 influenza B virus) deposited in IEDB.
Due to a relative sequence dissimilarity between influenza virus and coronavirus, there were no peptides with identical match between two strains and all peptides were distinct by at least three amino acids (Figure 5B). This indicates a minimal potential for cross-protection against coronavirus, especially SARS-CoV-2, conferred by influenza virus.
Of note, among those with three amino acid differences, there were public predicted epitopes shared across multiple coronavirus strains as exemplified in Table 3 (full list provided in Supplementary Figure 9). Despite a limited potential for cross-reactivity, these public peptides may cross-protect within coronavirus strains and given that they share a modest sequence similarity with epitopes derived from influenza virus, may pose a marginal potential to cross-react against influenza virus.
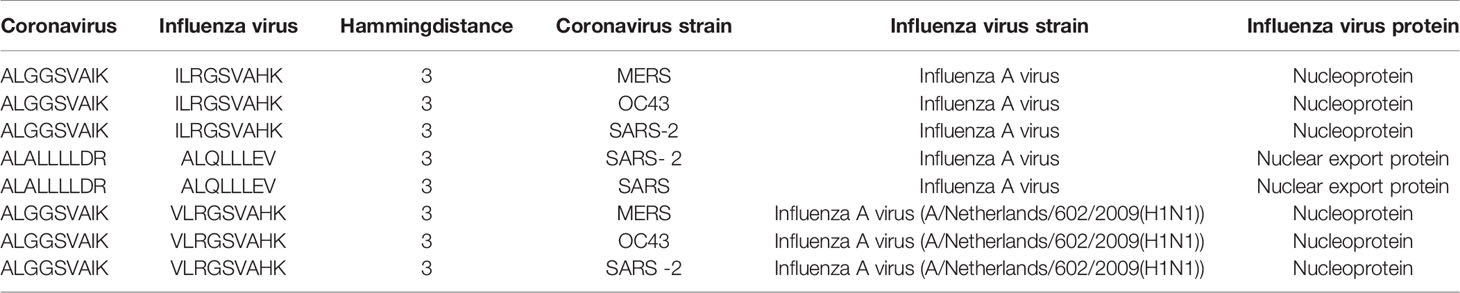
Table 3 Example of public predicted epitopes from coronavirus strains with modest sequence similarity with influenza virus epitopes.
Discussion
The severity and recurrence of coronavirus disease outbreaks pose ongoing global threat, and prompts the need for better understanding of potential cross-protection by prior infection of familial coronaviruses to mitigate the current spread and prevent future pandemics. Hereby, in continuation of our previous study to identify vaccine target for SARS-CoV-2 (21), we sought to determine the extent of antigen cross-reactivity amongst coronavirus strains.
Taking a step ahead of previous efforts to study potential immune recognition of SARS-CoV-2 by earlier infections of common coronaviruses based on MHC presentation predictions (22–25), we i) shortlisted predicted epitopes by taking immunogenicity potential of predicted binders into account, ii) validated the prediction by comparing with epitopes deposited in IEDB, iii) expanded the map of public predicted epitopes to accommodate up to two amino acid variants, and iv) analyzed sequence similarity against human proteome to eliminate self-peptides from vaccine targets. In comparison to Grifoni et al. (24, 26), the same algorithm has been adapted to predict HLA binding but moderate differences in HLA alleles and additional filtering strategies for immunogenicity led to differences in predicted epitope list. Detailed comparisons have been discussed in Supplementary Figure 10.
Due to the prevalence of disease caused by influenza virus and its tendency to gain mutations (antigenic drift) and reassortment among subtypes of virus (antigenic shift), there have been multiple attempts to develop generic vaccines protective against all influenza viruses to reduce the severity of infection and spread of disease (27, 28). Along with cross-reactivity among influenza virus strains, the cross-reactivity between coronavirus and influenza virus would benefit the community to combat recurrence of diseases caused by both strains. To aid design of vaccines targeting both coronaviruses and influenza viruses, we analyzed correlates of the predicted epitopes from coronaviruses with epitopes from influenza virus. However, due to a high sequence dissimilarity between these two virus families, there were no shared predicted epitopes, implying a limited potential of CD8 cross-protection against coronaviruses conferred by influenza virus. Nonetheless, we analyzed sequence similarity on 9-mers only and cannot neglect potential cross-reactivity conferred by shorter peptides or from CD4 + T cells.
Along with the in silico profile of predicted epitopes shared across coronaviruses, we evaluated the potential risk of self-reactivity imposed by high sequence similarity with the human proteome. Considering the reports of lung, heart, liver, intestine, genital and kidney failures by autoimmune disorders in COVID-19 patients (29), peptides from SARS-CoV-2 may carry high risk of immunopathology and should be carefully selected to proceed for vaccination.
In this study, we determined the potential immunogenicity by peptide sequence alone but other factors, such as turnover rate and expression level of parent proteins, may contribute to selection of immunodominant epitopes. For example, Bojkova et al. characterized changes in the protein level of SARS-CoV-2 infected Caco-2 cells, and showed 3–6 folds (in log2) increase in protein expressions after 20 h compared to 2 h post infection, with nucleocapsid and membrane proteins having the highest and the replicase polyprotein 1ab having the lowest fold change, respectively (30). Therefore, although fewer number of predicted epitopes were found from the membrane, nucleoprotein and spike proteins compared to the replicase polyprotein 1ab, their higher abundance might have a major impact in determining their immunodominance, and skew the immune response against their epitopes.
Comparing predicted epitopes with those characterized and deposited in IEDB, we could detect 64% of coronavirus IEDB epitopes from the prediction. In regards to the accuracy of predictive models utilized in this study, the algorithms to predict MHC presentation has matured significantly in the last decade by training with extensive datasets, especially for the most common HLA types. On the other hand, it is worth noting that predicting immunogenicity is challenging and not a fully solved problem. For example, these algorithms do not take into account other factors influencing the immunogenicity of a given epitope, such as abundance, expression pattern, and localization. Therefore, although the best performing models have been used for classifying immunogenicity, the predictions are suboptimal and should be taken with caution.
Along with the accuracy of predictive models, it is worth noting that classification thresholds of these algorithms have intrinsic tradeoff between sensitivity and specificity. In the case of SARS-CoV derived peptides, strong binding of HLA-A*02:01 has shown to correlate with stimulation of IFNγ secretion in the PBMC from HLA-A2 SARS-recovered donors ex vivo (31, 32), but we nevertheless cannot disregard potential immunogenicity from low-binding SARS-CoV-2 peptides. While we generated in silico map based on recommended thresholds of NetMHCpan and Repitope, stringent or relaxed thresholds can be applied based on the availability of resources for functional validations.
In this study, to validate our in silico identified epitopes, we compared our predicted epitopes with those characterized by in vitro T cell assays deposited in IEDB, and identified 22 out of 31 SARS-CoV IEDB epitopes matching our predictions. Please note that coronavirus-related peptides in IEDB are of varying lengths of up to 25aa, therefore multiple 9-mer epitopes could match with a single peptide in IEDB. In order to gauge the inevitable false negative rate, we compared peptides that didn’t pass our filters to IEDB peptides. We observed that 23 out of 43,732 predicted non-binders and 2 out of 11,202 predicted binder-non-immunogenic peptides matched with 10 coronavirus-derived epitopes in IEDB (9 from SARS-CoV and 1 from OC43). While the presence of multiple 9-mers matching with the same IEDB epitope is not surprising, exclusion of some matches by the MHC binding and immunogenicity filtering may facilitate determining the regions of peptide that are most likely to elicit the T cell response.
In a separate study, Nelde et al. (33) predicted 1,739 SARS-CoV-2 derived HLA-I peptides to be immunogenic based on integration of two algorithms, SYFPEITHI and NetMHCpan. They then selected 100 from 1,739 peptides to proceed for functional validation and reported 29/100 (29%) peptides as naturally occurring CD4+ or CD8+ T-cell epitopes. Of these 100 peptides, we compared only 9-mer peptides (59 peptides) with our predictions and found 39/59 (66%) peptides to agree with our predictions (Supplementary Figure 11). Out of these 39 common 9mers, we have identified 5 true positive and 34 true negative that leads to a predictive accuracy of 66%. However, one may notice that an additional filter also imposes a risk of increasing false negatives. Suitable filters should be decided based on the capacity of testing immunogenicity of peptides.
Notably, this study further demonstrated presence of T cell responses in unexposed individuals to 31% of their validated HLA class I epitopes, which hints to presence of CD8+ T cell cross-reactivity with common coronaviruses. In agreement with our prediction, Orf1ab was the protein most recognized by the class I epitopes they found in healthy donors, while membrane (M) and nucleocapsid (N) proteins were not recognized. This points to the potential cross-reactivity with other coronavirus strains and suggests the significance of conceptualizing the map of cross-reactive potential among coronavirus strains.
We believe that our comprehensive profile of private and public predicted epitopes across coronaviruses will assists biologists with targeted function validation, and facilitate design of vaccines capable of protecting against multiple prevalent virus strains. Further validations of the predicted profile would help estimate the extent of cross-protection and pave the way for better understanding of heterogeneity in the susceptibility and severity to the disease.
Data Availability Statement
All datasets presented in this study are included in the article/Supplementary Material.
Author Contributions
HK and GN conceived and designed the study. CL conceived and designed computational approaches, conducted data analysis, and generated figures and tables. PB conducted immunogenicity prediction. IW conducted MHC presentation prediction. CL MP, GO, AS, HK, and GN wrote the manuscript, and IW and PB contributed to methods. AS and HK supervised the project. All authors contributed to the article and approved the submitted version.
Conflict of Interest
GO has served on advisory boards or holds consultancies or equity with Eli Lilly, Novartis, Janssen, Sanofi, Orbit Discovery, and UCB Pharma, and has undertaken clinical trials with Atopix, Regeneron/Sanofi, and Roche.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work has been supported by Medical Research Council. HK and GN are funded by MRC Human Immunology Unit core funding. CL is funded by UK National Institute of Health Research (NIHR). AS is funded by a Wellcome Investigator Award (219523/Z/19/Z), the UK Medical Research Council, NIHR, awards from Bristol-Myers Squibb and UCB. MP and GN are funded by the department of Health and Social Care (project number:16/107/01) as part of the UK Vaccine Network. AS and GO are NIHR Senior Investigators and acknowledge support from the Oxford NIHR Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health, UK. This manuscript has been released as a pre-print at bioRxiv (34).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.579480/full#supplementary-material
Supplementary Table 1 | List of predicted scores for immunogenicity. Immunogenicity predicted by Repitope for all 9-mer peptides that have passed MHC presentation threshold.
Supplementary Table 2 | List of predicted scores for MHC presentation and immunogenicity of 10-mer peptides.
Supplementary Table 3 | List of predicted epitopes referenced in IEDB.
Supplementary Table 4 | List of public predicted epitopes up to two mismatches. Expansion of shared epitopes to include up to two amino acid mismatches.
References
1. Caramelo F, Ferreira N, Oliveiros B. Estimation of risk factors for COVID-19 mortality - preliminary results. medRxiv (2020), 2020.02.24.20027268. doi: 10.1101/2020.02.24.20027268
2. Dowd JB, Andriano L, Brazel DM, Rotondi V, Block P, Ding X, et al. Demographic science aids in understanding the spread and fatality rates of COVID-19. Proc Natl Acad Sci USA (2020) 117:9696. doi: 10.1073/pnas.2004911117
3. Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol (2020) 20:363–74. doi: 10.1038/s41577-020-0311-8
4. Verity R, Okell LC, Dorigatti I, Winskill P, Whittaker C, Imai N, et al. Estimates of the severity of coronavirus disease 2019: a model-based analysis. Lancet Infect Dis (2020) 20(6):669–77. doi: 10.1016/S1473-3099(20)30243-7
5. Epstein SL, Price GE. Cross-protective immunity to influenza A viruses. Expert Rev Vaccines (2010) 9:1325–41. doi: 10.1586/erv.10.123
6. Frank SA. Immunological Variability of Hosts. Immunology and Evolution of Infectious Disease. Princeton (NJ): Princeton University Press (2002).
7. Auladell M, Jia X, Hensen L, Chua B, Fox A, Nguyen THO, et al. Recalling the Future: Immunological Memory Toward Unpredictable Influenza Viruses. Front Immunol (2019) 10:1400. doi: 10.3389/fimmu.2019.01400
8. Nickbakhsh S, Ho A, Marques DFP, McMenamin J, Gunson RR, Murcia P. Epidemiology of Seasonal Coronaviruses: Establishing the Context for the Emergence of Coronavirus Disease 2019. J Infect Dis (2020) 222:17–25. doi: 10.1101/2020.03.18.20037101
9. Khan S, Nakajima R, Jain A, de Assis RR, Jasinskas A, Obiero JM, et al. Analysis of Serologic Cross-Reactivity Between Common Human Coronaviruses and SARS-CoV-2 Using Coronavirus Antigen Microarray. bioRxiv (2020) 2020.03.24.006544. doi: 10.1101/2020.03.24.006544
10. Cheng M, Chan CWL, Cheung RCF, Bikkavilli RK, Zhao Q, Au SWN, et al. Cross-reactivity of antibody against SARS-coronavirus nucleocapsid protein with IL-11. Biochem Biophys Res Commun (2005) 338:1654–60. doi: 10.1016/j.bbrc.2005.10.088
11. Lim YX, Ng YL, Tam JP, Liu DX. Human Coronaviruses: A Review of Virus–Host Interactions. Diseases (2016) 4(3):26. doi: 10.3390/diseases4030026
12. Common Human Coronaviruses | CDC. (2020). https://www.cdc.gov/coronavirus/general-information.html.
13. Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: Improved Peptide–MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J Immunol (2017) 199:3360–8. doi: 10.4049/jimmunol.1700893
14. Maiers M, Gragert L, Klitz W. High-resolution HLA alleles and haplotypes in the United States population. Hum Immunol (2007) 68:779–88. doi: 10.1016/j.humimm.2007.04.005
15. Ogishi M, Yotsuyanagi H. Quantitative Prediction of the Landscape of T Cell Epitope Immunogenicity in Sequence Space. Front Immunol (2019) 10:827. doi: 10.3389/fimmu.2019.00827
16. Trolle T, McMurtrey CP, Sidney J, Bardet W, Osborn SC, Kaever T, et al. The Length Distribution of Class I–Restricted T Cell Epitopes Is Determined by Both Peptide Supply and MHC Allele–Specific Binding Preference. J Immunol (2016) 196:1480–7. doi: 10.4049/jimmunol.1501721
17. Rist MJ, Theodossis A, Croft NP, Neller MA, Welland A, Chen Z, et al. HLA Peptide Length Preferences Control CD8+ T Cell Responses. J Immunol (2013) 191:561–71. doi: 10.4049/jimmunol.1300292
18. Hassan C, Chabrol E, Jahn L, Kester MGD, de Ru AH, Drijfhout JW, et al. Naturally Processed Non-canonical HLA-A*02:01 Presented Peptides. J Biol Chem (2015) 290:2593–603. doi: 10.1074/jbc.M114.607028
19. Frankild S, Boer RJ, Lund O, Nielsen M, Kesmir C.. Amino Acid Similarity Accounts for T Cell Cross-Reactivity and for “Holes” in the T Cell Repertoire. PloS One (2008) 3:e1831. doi: 10.1371/journal.pone.0001831
20. Petrova G, Ferrante A, Gorski J. Cross-Reactivity of T Cells and Its Role in the Immune System. Crit Rev Immunol (2012) 32:349–72. doi: 10.1615/CritRevImmunol.v32.i4.50
21. Lee CH, Koohy H. In silico identification of vaccine targets for 2019-nCoV. F1000Res (2020) 9:145. doi: 10.12688/f1000research.22507.2
22. Nguyen A, David JK, Maden SK, Wood MA, Weeder BR, Nellore A, et al. Human leukocyte antigen susceptibility map for SARS-CoV-2. J Virol (2020) JVI.00510-20:jvi;JVI.00510–20v1. doi: 10.1128/JVI.00510-20
23. Dijkstra JM, Hashimoto K. Expected immune recognition of COVID-19 virus by memory from earlier infections with common coronaviruses in a large part of the world population. F1000Res (2020) 9:285. doi: 10.12688/f1000research.23458.2
24. Grifoni A, Sidney J, Zhang Y, Scheuermann RH, Peters B, Sette A. A Sequence Homology and Bioinformatic Approach Can Predict Candidate Targets for Immune Responses to SARS-CoV-2. Cell Host Microbe (2020) 27:671–680.e2. doi: 10.1016/j.chom.2020.03.002
25. Peters B, Nielsen M, Sette A. T Cell Epitope Predictions. Annu Rev Immunol (2020) 38:123–45. doi: 10.1146/annurev-immunol-082119-124838
26. Grifoni A, Weiskopf D, Ramirez S II, Mateus J, Dan JM, Moderbacher CR, et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell (2020) 181:1489–1501.e15. doi: 10.1016/j.cell.2020.05.015
27. Eisenstein M. Towards a universal flu vaccine. Nature (2019) 573:S50–2. doi: 10.1038/d41586-019-02751-w
28. Koutsakos M, Illing PT, Nguyen THO, Mifsud NA, Crawford JC, Rizzetto S, et al. Human CD8 + T cell cross-reactivity across influenza A, B and C viruses. Nat Immunol (2019) 20:613–25. doi: 10.1038/s41590-019-0320-6
29. How COVID-19 kills: Explaining the mechanisms, symptoms, and diagnosis of the new coronavirus. ScienceDaily. Available at: https://www.sciencedaily.com/releases/2020/05/200513081810.htm.
30. Bojkova D, Klann K, Koch B, Widera M, Krause D, Ciesek S, et al. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature (2020) 583:469–72. doi: 10.1038/s41586-020-2332-7
31. Tsao Y-P, Lin J-Y, Jan J-T, Leng C-H, Chu C-C, Yang Y-C, et al. HLA-A∗0201 T-cell epitopes in severe acute respiratory syndrome (SARS) coronavirus nucleocapsid and spike proteins. Biochem Biophys Res Commun (2006) 344:63–71. doi: 10.1016/j.bbrc.2006.03.152
32. Zhou M, Xu D, Li X, Li H, Shan M, Tang J, et al. Screening and Identification of Severe Acute Respiratory Syndrome-Associated Coronavirus-Specific CTL Epitopes. J Immunol (2006) 177:2138–45. doi: 10.4049/jimmunol.177.4.2138
33. Nelde A, Bilich T, Heitmann JS, Maringer Y, Salih HR, Roerden M, et al. SARS-CoV-2-derived peptides define heterologous and COVID-19-induced T cell recognition. Nat Immunol (2020) 1–12. doi: 10.1038/s41590-020-00808-x
Keywords: cross-reactivity, antigen presentation, predict immunogenicity, epitopes, CD8+ T cell recognition, COVID-19, SARS-CoV-2
Citation: Lee CH, Pinho MP, Buckley PR, Woodhouse IB, Ogg G, Simmons A, Napolitani G and Koohy H (2020) Potential CD8+ T Cell Cross-Reactivity Against SARS-CoV-2 Conferred by Other Coronavirus Strains. Front. Immunol. 11:579480. doi: 10.3389/fimmu.2020.579480
Received: 02 July 2020; Accepted: 13 October 2020;
Published: 05 November 2020.
Edited by:
Francisco Sanchez-Madrid, Autonomous University of Madrid, SpainReviewed by:
Brian M. Baker, University of Notre Dame, United StatesJonathan E. Boyson, University of Vermont, United States
Balbino Alarcon, Consejo Superior de Investigaciones Científicas (CSIC), Spain
Copyright © 2020 Lee, Pinho, Buckley, Woodhouse, Ogg, Simmons, Napolitani and Koohy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hashem Koohy, aGFzaGVtLmtvb2h5QHJkbS5veC5hYy51aw==