 Jin Wo
Jin Wo Feng Zhang2†
Feng Zhang2† Chenghong Sun
Chenghong Sun Guodong Sun
Guodong Sun- 1Department of Orthopedics, First Affiliated Hospital, Jinan University, Guangzhou, China
- 2Intensive Care Unit, First Affiliated Hospital, Jinan University, Guangzhou, China
- 3State Key Laboratory of Generic Manufacture Technology of Chinese Traditional Medicine, Linyi, China
Gamma-delta (γδ) T cells are a subset of T cells that promote the inflammatory responses of lymphoid and myeloid lineages, and are especially vital to the initial inflammatory and immune responses. Given the capability to connect crux inflammations of adaptive and innate immunity, γδ T cells are responsive to multiple molecular cues and can acquire the capacity to induce various cytokines, such as GM-CSF, IL-4, IL-17, IL-21, IL-22, and IFN-γ. Nevertheless, the exact mechanisms responsible for γδ T cell proinflammatory functions remain poorly understood, particularly in the context of the central nervous system (CNS) diseases. CNS disease, usually leading to irreversible cognitive and physical disability, is becoming a worldwide public health problem. Here, we offer a review of the neuro-inflammatory and immune functions of γδ T cells, intending to understand their roles in CNS diseases, which may be crucial for the development of novel clinical applications.
Introduction
Together, gamma-delta (γδ) and alpha-beta (αβ) T cells represent two different T cell lineages that have been defined by their expression of αβ or γδ T cell receptors (TCRs) (1). Although γδ T cells share many effector capabilities with αβ T cells (for example, cytotoxicity and cytokine production), the lineages exhibit different biological properties, such as thymic-dependent or -independent development, major histocompatibility complex (MHC) restriction, and recognition of soluble protein and non-protein antigens of endogenous origin (2–5).
Unlike αβ T cells, γδ T cells are a relatively minor subset of T lymphocytes in the peripheral blood (PB), comprising only 1–5% of lymphocytes circulating (6). However, γδ T cells are abundant at barrier sites such as the skin, gut, lung, and reproductive tract; up to 20% of intraepithelial lymphocytes in the human colon express the γδ TCRs (7).
γδ T cells are divided according to the type of Vγ and Vδ chain they express at the TCRs. Concerning the Vγ chains, a unique feature of murine γδ T cells is the preferential expression of different Vγ segments in different tissues. For example, Vγ5+ γδ T cells are present in the skin, Vγ7+ γδ T cells lie in the intestinal, Vγ6+ γδ T cells localize to the reproductive mucosa, and Vγ1+ or Vγ4+ γδ T cells are found in secondary lymphoid organs (8, 9). The previous studies related to human γδ T cells have identified Vγ9 as the most frequently used Vγ chain in the PB (10). Vγ9 chain associates with Vδ2 in most cases, defining a Vγ9Vδ2 T cell population (account for 50–95% of γδ T cells in the PB) that is unique to humans and other primates (11, 12).
Vγ9Vδ2 T cells are known to identify microbe-derived [HMBPP, (E)-4-hydroxy-3methyl-but-2-enyl pyrophosphate] and host-derived (IPP, isopentenyl pyrophosphate) phosphorylated metabolites originating from the isoprenoid metabolic mevalonate and non-mevalonate pathways, through association with butyrophilin 3A1 (BTN3A1) and BTN3A2 (13–16). Moreover, Vδ1+ γδ T cells frequently coexpress functional receptors of innate immune cells, such as activating natural killer (NK) receptors such as NKG2D (17–20). It includes MHC class I polypeptide-related chains (MIC) A and B, and UL16 binding proteins (ULBP) (21–24). Although first described for Vδ1+ γδ T cells, interactions of the ULBP and MIC-A/B molecules with NKG2D are now recognized to stimulate Vδ2+ γδ T cells (21, 22). Besides, Vδ1+ γδ T cells recognize lipids and glycolipids presented by CD1 molecules (25, 26). Furthermore, both Vδ1+ and Vδ2+ γδ T cells are activated by heat shock proteins (HSP) (27–29).
Recently, some discrete population of T cells that coexpressed αβ-γδ TCRs and Vγ-Cβ TCRs have been identified (30, 31). Among them, the αβ-γδ T cells protected against infection by licensing encephalitogenic Th17 cells, triggered inflammatory and immune in the central nervous system (CNS). Moreover, our research group found that, in addition to diseases of the CNS, such as multiple sclerosis (MS) and stroke, immune responses induced by γδ T cells are also critically implicated in neuroinflammation associated with spinal cord injury (SCI) (32–34). These findings raise significant questions concerning the inflammatory and immune functions of γδ T cells in CNS disease that have yet to be addressed (35–37). CNS disease, which can result in irreversible sensory, motor, and autonomic impairments, is a severe health problem worldwide. As a central pathological process in CNS diseases, the inflammatory response is vital to clinical prognosis. Here, we provide a review of recent advances in the understanding of γδ T cells with relevance to their inflammatory and immune roles in CNS disease, which suggest potential approaches for future treatment of CNS diseases (Table 1).
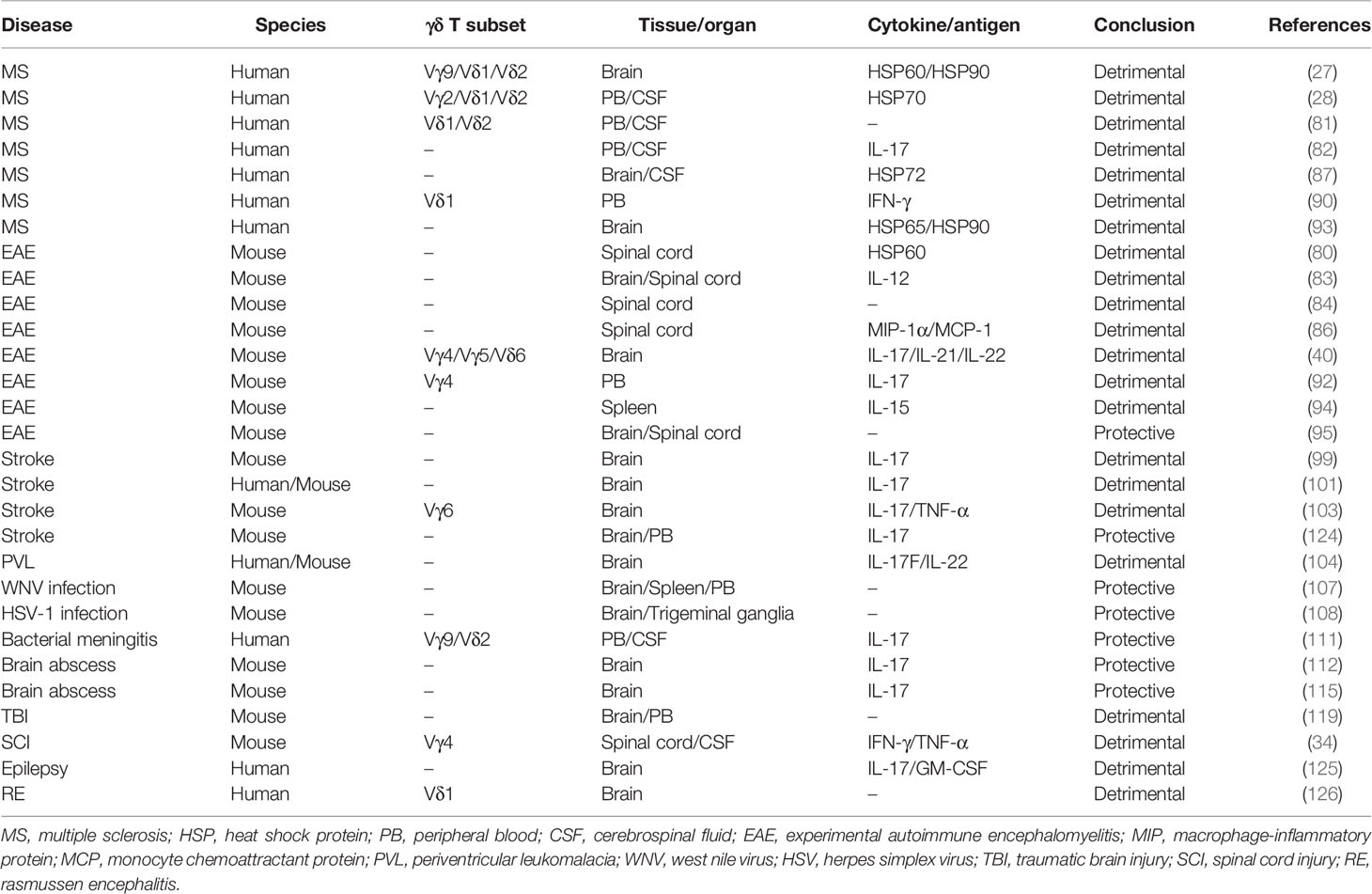
Table 1 The role of γδ T cells in CNS diseases.
Proinflammatory Cytokines Induced by γδ T Cells in the CNS
Activation and development of γδ T cells promoting CNS inflammation are chiefly mediated by dendritic cells (DCs). The immunostimulatory component induces IL-1β, IL-6, IL-18, and IL-23 by DCs via caspase-1 and inflammasome complex. γδ T cells secrete IL-17 in response to IL-1β, IL-18, and IL-23 in the absence of TCR (38–40). During this process, the retinoid-related orphan receptor (ROR) -γt and IL-7 coordinate the B and T lymphocyte attenuator (BTLA) expression, thus regulating γδ T cell inflammatory responses (41–44). Moreover, Shibata et al. demonstrated that signal transducer and activator of transcription 3 (STAT3) is dispensable for the development of IL-17-producing γδ T (γδT17) cells (45). Also, IL-23-activated γδ T cells suppress the factor forkhead box P3+ (Foxp3) -expressing Treg cells conversion, as well as promoting effector T (Te) cells response (46, 47). The capacity of γδ T cells to produce a burst of IL-17 in the absence of activated αβ T cells is crucial for the initiation of CNS inflammation (48).
Activated DCs also promotes the induction of other proinflammatory cytokines from γδ T cells, such as granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-21, and IL-22 (30, 40) (Figure 1). While IL-17A, IL-17F, and IL-22 are prominently expressed in CNS inflammation, they may only marginally contribute to disease development (49–51); however, McGinley et al. recently demonstrated that IL-17 might recruit IL-1β-secreting myeloid cells that prime pathogenic γδ T cells in CNS inflammation (52).
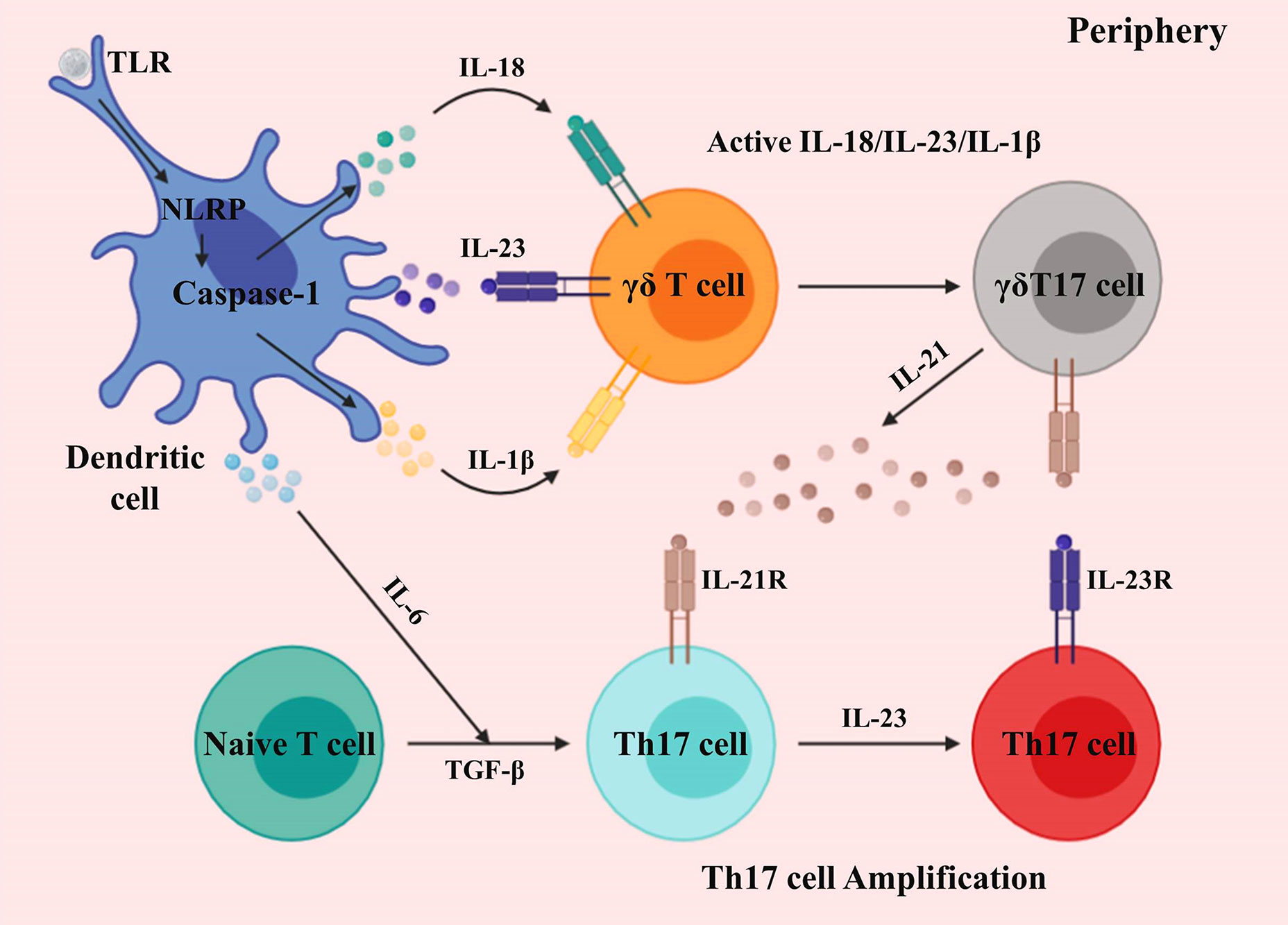
Figure 1 Activation and development of γδ T cells in the periphery. Differentiated dendritic cells and macrophages generate proinflammatory cytokines via toll-like and NOD-like receptors. γδ T cells sense IL-1β, IL-18, and IL-23, producing an initial burst of IL-17. The differentiation of Th17 cells is induced by IL-6 and TGF-β. γδT17 cells secrete IL-21, which further amplifies their proliferation, and also that of Th17 cells.
Different from γδ T cells, which can produce IL-17 in response to cytokine (IL-1β, IL-18, and IL-23) signals alone, in the absence of primary (TCR) and secondary (costimulation) signals, IL-17-producing T helper (Th17) cells require primary, secondary, and cytokine (IL-6 and TGF-β) signals to generate IL-17 (40) (Figure 1). Seminal studies demonstrated that IL-6 and TGF-β induce Th17 cell differentiation, in which TGF-β is critical for T cells to differentiate into Foxp3+ Treg or Th17 cells (53–58). Moreover, TGF-β is also critical to γδT17 cells (59). Besides, IL-21 is induced by IL-6 in Th17 cells, which establishes a feed-forward loop to support Th17 cell amplification, in which STAT3 and ROR-γt mediate lineage specification (54, 55, 60–63).
During this process, IL-23 acts as a maturation factor for Th17 cells, and both IL-23 and IL-21 can induce IL-17 expression independently of IL-6 (55, 64–66). Therefore, mice lacking IL-23 are resistant to Th17-mediated CNS inflammation (46). To demonstrate the role of IL-23, Awasthi et al. substituted the green fluorescent protein for the intracellular domain of IL-23R, to generate a “knock-in” mouse, which demonstrated that IL-23 is crucial for Th17 cell function (67). IL-23 created a positive feedback loop, whereby GM-CSF secreted by Th17 cells induced the generation of IL-23 (68, 69).
Alongside IL-17, GM-CSF is also essential for CNS inflammation. Further, the activation of the microglial cell, but not macrophage in the periphery, is a GM-CSF-dependent process (70). El-Behi et al. demonstrated that GM-CSF neutralization attenuated CNS inflammation (68). Although both IL-12 and IL-23 can induce Te cells to generate GM-CSF, IL-23 is crucially required for GM-SCF generation (69, 71). In addition to DCs and Th17 cells, γδ T cells generate large amounts of GM-CSF, resulting in neuroinflammation (72).
γδ T Cells in CNS Diseases
Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis
MS is a chronic inflammatory demyelinating CNS disease, resulting in progressive cognitive, sensory, and motor disorders. Experimental autoimmune encephalomyelitis (EAE), a murine MS model, is used to research the proinflammatory mechanism underlying CNS (73). Before the discovery of Th17 cells, IFN-γ-producing Th1 cells were considered the primary pathogenic cell inducing MS and EAE, which puzzled immunologists for many years, since both IFN-γ−/− and IFN-γR−/− mice enhanced EAE development (74–77). Besides, deficiencies of IL-12 and IL-12R, which are critical to the development of Th1 cells, also exhibited exacerbated EAE (64). Together, findings to date indicate that Th1 cells are not the initial T cell involved in EAE. IL-12 and IFN-γ (Th1-associated molecules) negatively regulate tissue inflammation in EAE (78). Nonetheless, Th1 cells are vital to EAE, as they are detected in active EAE.
Subsequently, the identification of IL-23 and Th17 cells partly worked out this issue (61, 65) (Figure 2). IL-23p40−/− and IL-23p19−/− mouse strains are both resistant to EAE (64). The depletion of Th17 cells or IL-17 resulted in reduced EAE severity (78). Although Th17 cells are thought to be the major mediators of EAE, γδT cells can also be a potent producer of IL-17, and are dominant over Th17 cells in CNS inflammation (79). Several researchers demonstrated that γδ T cells are frequently present in the peripheral blood (PB) and cerebrospinal fluid (CSF) of MS patients, as well as in the brains of mice with EAE (27, 28, 80–82). During the chronic and acute phases of EAE, the absence of γδ T cells notably reduces the CNS inflammation, suggesting that γδ T cells are significant in EAE, and their inflammatory mobilization is related to the pathogenesis of CNS autoimmunity (83–87). Indeed, an enormous population of CD4+ T cells (IL-17 and IFN-γ double-positive) is observed at the peak of EAE (88). Using a fate-tracking system, 5–10% of γδT17 cells were shown to express IFN-γ in the CNS, indicating that IL-17-IFN-γ-γδ T cells might be consequential intermediates in EAE pathogenesis (89).
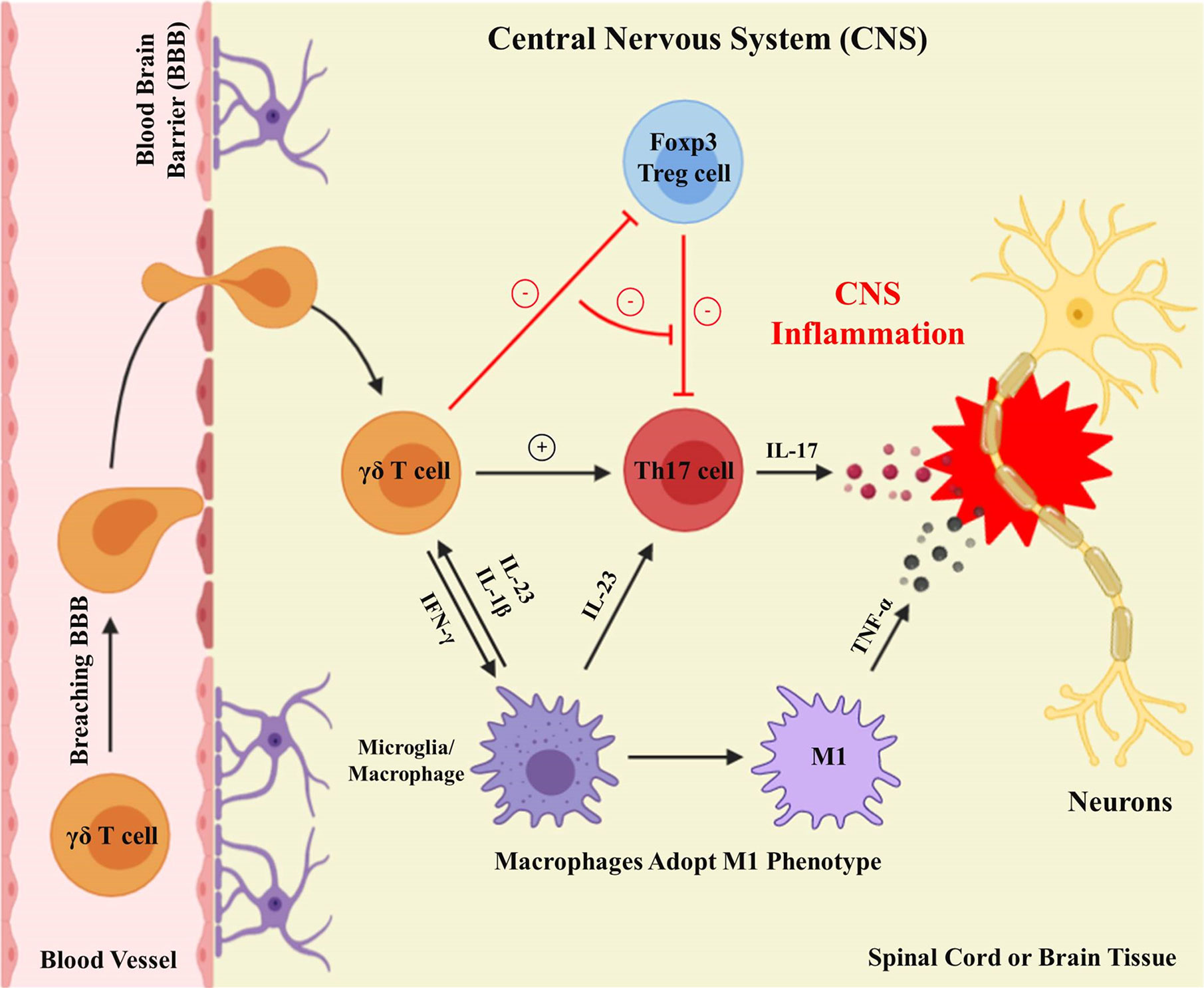
Figure 2 γδ T cells execute proinflammatory functions in the CNS. Activated γδ T cells breach the blood-brain barrier to carry out proinflammatory functions in the CNS. Differentiated microglia/macrophages secrete IL-23 within the CNS to facilitate the production of γδ T cells and Th17 cells. γδ T cells result in CNS inflammation by improving Th17 cell effector functions, restraining Tregs cell suppressive functions, and generating IFN-γ, to induce M1 phenotype macrophages secreting TNF-α.
Moreover, Vγ4+ γδ T cells were identified as the major γδT17 cells in EAE, while Vγ5+ and Vδ6+ γδ T cells were present (40). Vδ1+, Vδ2+, and Vγ9+ γδ T cells were also observed in acute demyelinating plaques of MS patients (27, 90). Besides, the biological drugs designed to suppress the activity of γδ T cells, such as fingolimod (FTY720) and Natalizumab, partly contribute to the clinical therapeutic effects of MS. (90–92). Further, HSP60 and HSP90 compared with normal CNS tissues are overexpressed in MS plaques, while Selmaj et al. showed the colocalization of HSP65 and γδ T cells in immature oligodendrocytes in MS lesions (27, 80, 93). The multitude of γδ T cells collected from MS patients proliferated in response to HSP70, but not to HSP65, revealing that HSPs may be the antigens responsible for promoting the γδ T cells proliferation (28). CNS inflammation is associated with altered expression of HSPs, which may function as targets in the development of the chronic disease. Interestingly, IL-15-producing γδ T (γδT15) cells, another subset of γδ T cell, were recently discovered; however, whether these cells produce other proinflammatory cytokines in EAE is not well elucidated (94).
In contrast to the above observations, γδ T cells are also reported have a protective function in EAE. Ponomarev et al. reported that γδ T cells of wild-type (WT) reconstitute γδ T cell−/− mice, but not FasL dysfunctional γδ T cells, diminishing inflammation in EAE (95). These findings suggest that the γδ T cell-mediated Fas/FasL-induced T cells apoptosis regulates CNS inflammation. Indeed, the mechanism by which γδ T cells regulate proinflammatory chemokine and cytokine expression in CNS, as well as infiltrating cell heterogeneity, warrant detailed investigation.
Ischemic Brain Injury
The main consequence of ischemic brain injury is manifested as the CNS tissue necrosis, due to the loss of nutrition. The tissue necrosis leads to a secondary inflammation, involving the accumulation of specific immune cells, especially neutrophils, macrophages, and T cells, which is a critical factor to the entire pathophysiology (96, 97).
IL-17 has a specific role in the delayed phase of the ischemic brain injury inflammatory cascade (98). Shichita et al. demonstrated that γδT17 cells play a significant role during late-stage ischemic brain injury, and that they, rather Th17 cells, are (surprisingly) the major origin of IL-17 (99). Moreover, increased IL-17 levels are present in the PB of patients who have suffered a stroke, relative to healthy individuals (100). IL-23, generated by macrophages or monocytes from stroke initiation, is an essential contributor for inducing IL-17 by γδ T cells during the delayed phase of encephalic ischemia. Thereby, IL-23p19−/− mice illustrated a diminishment in infarct extent only 1 day after the ischemic injury, whereas IL-17 deficiency led to reduced infarct size after 4 days. Long-term, deficiencies of IL-17 and IL23 demonstrated obviously diminished CNS injury, relative to WT, or even IFN-γ−/−, mice (99). Gelderblom et al. demonstrated that injection of IL-17-neutralizing antibody to mice within the post-stroke 3h could reduce infarct volume and improve disease prognosis after 3 days (101).
Overall, the available evidence demonstrates that γδ T cells are the main source of IL-17. The mechanism of antigen-independent T cell activation post-stroke remains unclear; however, it has been owed chiefly to γδ T cells. Nevertheless, Kleinschnitz et al. demonstrated that γδ T cell-deficient mice remain susceptible to ischemic insult, indicating an extra function for other immune cells in ischemic brain injury. Furthermore, the fact that transgenic-TCR mice are susceptible to stroke implies that, besides γδ T cells, Th17 cells may also have a prominent role in stroke, whereas the precise function of Th17 cells in inducing stroke is not exact (102).
In addition, astrocytes can respond to IL-17 and promote stroke induction and development (101). For instance, IL-17 produced by γδ T cells and TNF-α secreted by macrophages act synergistically on astrocytes, by inducing the expression of CXCL1, a neutrophil chemoattractant (101, 103). Recently, periventricular leukomalacia (PVL), a distinctive form of brain injury in premature infants, was demonstrated to be caused by developmental immaturity of the cerebral vasculature in mid to late gestational age, and large numbers of γδ T cells were observed in postmortem brains from preterm infants (104). Although there were increased IL-17 and IL-22 in mouse brains after injury, neither cytokine contributes to preterm brain injury (104).
In summary, γδ T cells and IL-17 have essential roles in ischemic brain injury. Hence, γδ T cells and IL-17 should be considered potential therapeutic targets to decrease secondary inflammation after ischemic brain injury (105, 106).
Central Nervous System Infection
The CNS infections commonly lead to the disruption of the blood-brain barrier (BBB) protectiveness and subsequent tissue inflammation; however, inflammation is also crucial to CNS immunity, as reduced γδ T cell expansion leads to increased host vulnerability to viral infection (107, 108). For example, MS patients treated with Natalizumab, a monoclonal antibody against α4-integrin, undergo fatal viral infections, due to the immune cells fail to infiltrate the CNS and eliminate the infection (109).
In contrast to viral infection, there are some (although limited) researches involving the function of γδ T cells in models of CNS bacterial infection (110). For example, children with bacterial meningitis exhibit high γδ T cell fractions in the CSF (111). Nichols et al. suggested that the γδ T cell was an alternative pathway available to respond to Grampositive bacteria CNS infection. They found that TLR2−/− brain abscess mice (TLR2 is a critical receptor for eliciting responses to Grampositive bacteria) were detected elevated IL-17, and γδ T cells were the source of IL-17 (112–114). Similarly, IL-17R signaling regulates γδ T cell infiltration, as well as bacterial clearance, during S. aureus-induced brain abscess formation (115). Also, IL-17 expression is augmented in the CNS of mice infected with Toxoplasma gondii (116). An increased percentage of γδT17 cells was observed in the PB and lesion in children with bacterial meningitis, and the condition was reversed after antibacterial therapy (111). High levels of IL-17 can also be detected in the abscess formation of humans; however, such researches are only associated, since no direct evidence can be demonstrated (117). Nevertheless, evidence for the involvement of γδ T cells in any CNS infection is sparse, and more studies are needed to establish a relation between γδ T cells and CNS infections.
Central Nervous System Traumatic Diseases
Immune responses and neuroinflammation involving γδ T cells are also critically involved in CNS traumatic diseases. Diseases resulting from CNS trauma usually involve irreversible sensory, motor, and autonomic impairments (118). Peripheral immune mechanisms establishment is related to the pathological processes of traumatic brain injury (TBI). Richard et al. found that CD4+ and CD8+ T cells, Tregs, and γδ T cells, increased in number within 24 h after TBI (119).
Further, recent results from our laboratory found that γδ T cells, particularly Vγ4+ γδ T cells, exert a detrimental role in SCI, probably by providing an important origin of IFN-γ, which induces macrophages to adopt the M1 phenotype, with increased secretion of inflammatory cytokines, such as TNF-α (34) (Figure 2). Moreover, one significant discovery from our studies was that bone marrow-derived macrophages (BMDMs) respond to IFN-γ. This was supported by two sets of findings. First, IFN-γR−/− mice, chimeras with IFN-γR−/− bone marrow, and mice receiving adoptively transferred IFN-γR−/− peritoneal macrophages, all showed similar recovery following SCI. Second, numbers of M1 macrophages and proinflammatory cytokines are reduced in IFN-γR−/− compared with WT controls (34). Besides, the treatment of SCI with anti-Vγ4 antibodies has a beneficial effect, similar to that obtained with anti-TNF-α (34). In conclusion, manipulation of γδ T cell functions may be a potential treatment approach for future CNS traumatic diseases.
Other Central Nervous System Diseases
The pathological and clinical outcome of CNS diseases can also be affected by the intestinal microflora in the context of autoimmunity (120–124). This relationship has been particularly well established for the response to bacteria, including pathogens and commensals, within the intestinal compartment and its effects on the CNS, a connection that was recently termed the gut-brain axis. The gut environment has been found to significantly influence CNS diseases such as MS, EAE, and ischemic brain injury; however, immune cell mechanisms are unclear. In addition, the pathogenesis of intractable epilepsy is related to γδ T cells, where proinflammatory γδ T cells were concentrated in epileptogenic lesions, and their numbers positively associated with disease severity (125–127).
Conclusion
Since the identification of γδ T cells, there has been a boom in related studies and discoveries. Equipped with functions of both innate and adaptive immune cells, γδ T cells can provide consequential functions in the development of CNS diseases, such as recognizing a diverse array of antigens, rapid production of inflammatory mediators, and influencing the differentiation of their αβ counterparts. Recently, understanding the inflammatory and immune roles of γδ T cells has resulted in the development of many prospective therapies for CNS diseases. However, the exact mechanisms behind their contributions are yet to be fully elucidated.
The pandemic of 2019 coronavirus disease COVID-19, caused by the SARS-CoV-2 virus infection, has caused worldwide mortality (128). Past pandemics have demonstrated that COVID-19 is accompanied by diverse neuropsychiatric symptoms, such as encephalopathy, neuromuscular dysfunction, or demyelinating processes (129). Whether recovered SARS-CoV-2 patients will exhibit an increased incidence of MS symptomatology or other delayed neurologic sequelae, is an important, yet unanswered. Nevertheless, more substantial shreds of evidence are required on different subtypes of γδ T cells for defining their opposing roles in CNS inflammation and explaining the confounding findings on their pathogenic or protective role in CNS diseases. In summary, this review discusses recent notable studies of the neuro-inflammatory and immune functions of γδ T cells, intending to understand their roles in CNS disease, which may be crucial for the effective immunotherapies.
Author Contributions
JW and FZ contributed to editing the manuscript. ZL and CS provided administrative support. GS and WZ helped the manuscript editing and discussions. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (No. 31970862), the Natural Science Foundation of Guangdong Province (Nos. 2018A030313576, 2019A1515011335), and the Science and Technology Program of Guangzhou (No. 201803010001).
Conflict of Interest
CS was employed by Lunan Pharmaceutical Group Co. Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to thank Kui Shen for critical comments, and Samuel Gill for revising and editing the manuscript.
References
1. Nanno M, Shiohara T, Yamamoto H, Kawakami K, Ishikawa H. gamma delta T cells: firefighters or fire boosters in the front lines of inflammatory responses. Immunol Rev (2007) 215:103–13. doi: 10.1111/j.1600-065X.2006.00474.x
2. Correa I, Bix M, Liao NS, Zijlstra M, Jaenisch R, Raulet D. Most gamma delta T cells develop normally in beta 2-microglobulin-deficient mice. Proc Natl Acad Sci U S A (1992) 89:653–7. doi: 10.1073/pnas.89.2.653
3. Bigby M, Markowitz JS, Bleicher PA, Grusby MJ, Simha S, Siebrecht M, et al. Most gamma delta T cells develop normally in the absence of MHC class II molecules. J Immunol (1993) 151:4465–75.
4. Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity (2009) 31:321–30. doi: 10.1016/j.immuni.2009.06.020
5. Dieli F, Poccia F, Lipp M, Sireci G, Caccamo N, Di Sano C, et al. Differentiation of effector/memory Vdelta2 T cells and migratory routes in lymph nodes or inflammatory sites. J Exp Med (2003) 198:391–7. doi: 10.1084/jem.20030235
6. Carding SR, Egan PJ. gamma delta T cells: Functional plasticity and heterogeneity. Nat Rev Immunol (2002) 2:336–45. doi: 10.1038/nri797
7. Mayassi T, Jabri B. Human intraepithelial lymphocytes. Mucosal Immunol (2018) 11:1281–9. doi: 10.1038/s41385-018-0016-5
8. Heilig JS, Tonegawa S. Diversity of murine gamma genes and expression in fetal and adult T lymphocytes. Nature (1986) 322:836–40. doi: 10.1038/322836a0
9. Melandri D, Zlatareva I, Chaleil RAG, Dart RJ, Chancellor A, Nussbaumer O, et al. The gamma delta TCR combines innate immunity with adaptive immunity by utilizing spatially distinct regions for agonist selection and antigen responsiveness. Nat Immunol (2018) 19:1352–65. doi: 10.1038/s41590-018-0253-5
10. Andreu-Ballester JC, Garcia-Ballesteros C, Benet-Campos C, Amigo V, Almela-Quilis A, Mayans J, et al. Values for alphabeta and gammadelta T-lymphocytes and CD4+, CD8+, and CD56+ subsets in healthy adult subjects: assessment by age and gender. Cytometry B Clin Cytom (2012) 82:238–44. doi: 10.1002/cyto.b.21020
11. Kalyan S, Kabelitz D. Defining the nature of human gamma delta T cells: a biographical sketch of the highly empathetic. Cell Mol Immunol (2013) 10:21–9. doi: 10.1038/cmi.2012.44
12. Poggi A, Zocchi MR. gamma delta T lymphocytes as a firs line of immune defense: old and new ways of antigen recognition and implications for cancer immunotherapy. Front Immunol (2014) 5:1–6. doi: 10.3389/fimmu.2014.00575
13. Gu SY, Nawrocka W, Adams EJ. Sensing of pyrophosphate metabolites by V gamma 9 delta 2T cells. Front Immunol (2015) 5:688. doi: 10.3389/fimmu.2014.00688
14. De Libero G, Lau SY, Mori L. Phosphoantigen Presentation to TCR gammadelta Cells, a Conundrum Getting Less Gray Zones. Front Immunol (2014) 5:679. doi: 10.3389/fimmu.2014.00679
15. Vantourout P, Laing A, Woodward MJ, Zlatareva I, Apolonia L, Jones AW, et al. Heteromeric interactions regulate butyrophilin (BTN) and BTN-like molecules governing gamma delta T cell biology. Proc Natl Acad Sci U S A (2018) 115:1039–44. doi: 10.1073/pnas.1701237115
16. Harly C, Guillaume Y, Nedellec S, Peigne CM, Monkkonen H, Monkkonen J, et al. Key implication of CD277/butyrophilin-3 (BTN3A) in cellular stress sensing by a major human gamma delta T-cell subset. Blood (2012) 120:2269–79. doi: 10.1182/blood-2012-05-430470
17. Hudspeth K, Silva-Santos B, Mavilio D. Natural cytotoxicity receptors: broader expression patterns and functions in innate and adaptive immune cells. Front Immunol (2013) 4:1–15. doi: 10.3389/fimmu.2013.00069
18. Correia DV, Lopes A, Silva-Santos B. Tumor cell recognition by gamma delta T lymphocytes T-cell receptor vs. NK-cell receptors. Oncoimmunology (2013) 2:e22892. doi: 10.4161/onci.22892
19. Simoes AE, Di Lorenzo B, Silva-Santos B. Molecular Determinants of Target Cell Recognition by Human gamma delta T Cells. Front Immunol (2018) 9:929. doi: 10.3389/fimmu.2018.00929
20. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science (1999) 285:727–9. doi: 10.1126/science.285.5428.727
21. Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol (2003) 3:781–90. doi: 10.1038/nri1199
22. Bahram S, Inoko H, Shiina T, Radosavljevic M. MIC and other NKG2D ligands: from none to too many. Curr Opin Immunol (2005) 17:505–9. doi: 10.1016/j.coi.2005.07.016
23. Crowley MP, Fahrer AM, Baumgarth N, Hampl J, Gutgemann I, Teyton L, et al. A population of murine gamma delta T cells that recognize an inducible MHC class lb molecule. Science (2000) 287:314–6. doi: 10.1126/science.287.5451.314
24. Bonneville M, O’Brien RL, Born WK. Gamma delta T cell effector functions: a blend of innate programming and acquired plasticity. Nat Rev Immunol (2010) 10:467–78. doi: 10.1038/nri2781
25. Luoma AM, Castro CD, Adams EJ. gamma delta T cell surveillance via CD1 molecules. Trends Immunol (2014) 35:613–21. doi: 10.1016/j.it.2014.09.003
26. de Jong A. Activation of human T cells by CD1 and self-lipids. Immunol Rev (2015) 267:16–29. doi: 10.1111/imr.12322
27. Wucherpfennig KW, Newcombe J, Li H, Keddy C, Cuzner ML, Hafler DA. Gamma delta T-cell receptor repertoire in acute multiple sclerosis lesions. Proc Natl Acad Sci U S A (1992) 89:4588–92. doi: 10.1073/pnas.89.10.4588
28. Stinissen P, Vandevyver C, Medaer R, Vandegaer L, Nies J, Tuyls L, et al. Increased frequency of gamma delta T cells in cerebrospinal fluid and peripheral blood of patients with multiple sclerosis. Reactivity, cytotoxicity, and T cell receptor V gene rearrangements. J Immunol (1995) 154:4883–94.
29. Hirsh MI, Junger WG. Roles of Heat Shock Proteins and gamma delta T Cells in Inflammation. Am J Resp Cell Mol (2008) 39:509–13. doi: 10.1165/rcmb.2008-0090TR
30. Edwards SC, Sutton CE, Ladell K, Grant EJ, McLaren JE, Roche F, et al. A population of proinflammatory T cells coexpresses alphabeta and gammadelta T cell receptors in mice and humans. J Exp Med (2020) 217:1–16. doi: 10.1084/jem.20190834
31. Bowen S, Sun P, Livak F, Sharrow S, Hodes RJ. A Novel T Cell Subset with Trans-Rearranged V gamma-C beta b TCRs Shows V beta Expression Is Dispensable for Lineage Choice and MHC Restriction. J Immunol (2014) 192:169–77. doi: 10.4049/jimmunol.1302398
32. Billiau A, Heremans H, Vandekerckhove F, Dijkmans R, Sobis H, Meulepas E, et al. Enhancement of experimental allergic encephalomyelitis in mice by antibodies against IFN-gamma. J Immunol (1988) 140:1506–10.
33. Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med (2008) 205:1535–41. doi: 10.1084/jem.20080159
34. Sun GD, Yang SX, Cao GC, Wang QH, Hao JL, Wen Q, et al. Gamma delta T cells provide the early source of IFN-gamma to aggravate lesions in spinal cord injury. J Exp Med (2018) 215:521–35. doi: 10.1084/jem.20170686
35. Jensen KDC, Su X, Shin S, Li L, Youssef S, Yarnasaki S, et al. Thymic selection determines gamma delta T cell effector fate: Antigen-naive cells make interleukin-17 and antigen-experienced cells make interferon gamma. Immunity (2008) 29:90–100. doi: 10.1016/j.immuni.2008.04.022
36. Muñoz-Ruiz M, Ribot JC, Grosso AR, Gonçalves-SoU S A N, Pamplona A, Pennington DJ, et al. TCR signal strength controls thymic differentiation of discrete proinflammatory γδ T cell subsets. Nat Immunol (2016) 17:721–7. doi: 10.1038/ni.3424
37. Prinz I, Sansoni A, Kissenpfennig A, Ardouin L, Malissen M, Malissen B. Visualization of the earliest steps of gammadelta T cell development in the adult thymus. Nat Immunol (2006) 7:995–1003. doi: 10.1038/ni1371
38. Lalor SJ, Dungan LS, Sutton CE, Basdeo SA, Fletcher JM, Mills KHG. Caspase-1-processed cytokines IL-1 beta and IL-18 promote IL-17 production by gamma delta and CD4 T cells that mediate autoimmunity. J Immunol (2011) 186:5738–48. doi: 10.4049/jimmunol.1003597
39. Ribot JC, deBarros A, Pang DJ, Neves JF, Peperzak V, Roberts SJ, et al. CD27 is a thymic determinant of the balance between interferon-gamma-and interleukin 17-producing gamma delta T cell subsets. Nat Immunol (2009) 10:427–36. doi: 10.1038/ni.1717
40. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity (2009) 31:331–41. doi: 10.1016/j.immuni.2009.08.001
41. Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol (2010) 10:479–89. doi: 10.1038/nri2800
42. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
43. Marks BR, Nowyhed HN, Choi JY, Poholek AC, Odegard JM, Flavell RA, et al. Thymic self-reactivity selects natural interleukin 17-producing T cells that can regulate peripheral inflammation. Nat Immunol (2009) 10:1125–U108. doi: 10.1038/ni.1783
44. Bekiaris V, Šedý JR, Macauley MG, Rhode-Kurnow A, Ware CF. The inhibitory receptor BTLA controls γδ T cell homeostasis and inflammatory responses. Immunity (2013) 39:1082–94. doi: 10.1016/j.immuni.2013.10.017
45. Shibata K, Yamada H, Sato T, Dejima T, Nakamura M, Ikawa T, et al. Notch-Hes1 pathway is required for the development of IL-17-producing gamma delta T cells. Blood (2011) 118:586–93. doi: 10.1182/blood-2011-02-334995
46. Petermann F, Rothhammer V, Claussen MC, Haas JD, Blanco LR, Heink S, et al. Gamma delta T cells enhance autoimmunity by restraining regulatory T cell responses via an interleukin-23-dependent mechanism. Immunity (2010) 33:351–63. doi: 10.1016/j.immuni.2010.08.013
47. Riol-Blanco L, Ordovas-Montanes J, Perro M, Naval E, Thiriot A, Alvarez D, et al. Nociceptive sensory neurons drive interleukin-23-mediated psoriasiform skin inflammation. Nature (2014) 510:157–61. doi: 10.1038/nature13199
48. Chien YH, Zeng X, Prinz I. The natural and the inducible interleukin (IL)-17-producing gamma delta T cells. Trends Immunol (2013) 34:151–4. doi: 10.1016/j.it.2012.11.004
49. Haak S, Croxford AL, Kreymborg K, Heppner FL, Pouly S, Becher B, et al. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest (2009) 119:61–9. doi: 10.1172/JCI35997
50. Kreymborg K, Etzensperger R, Dumoutier L, Haak S, Rebollo A, Buch T, et al. IL-22 is expressed by Th17 cells in an IL-23-dependent fashion, but not required for the development of autoimmune encephalomyelitis. J Immunol (2007) 179:8098–104. doi: 10.4049/jimmunol.179.12.8098
51. Lukens JR, Barr MJ, Chaplin DD, Chi H, Kanneganti TD. Inflammasome-derived IL-1beta regulates the production of GM-CSF by CD4(+) T cells and gammadelta T cells. J Immunol (2012) 188:3107–15. doi: 10.4049/jimmunol.1103308
52. McGinley AM, Sutton CE, Edwards SC, Leane CM, DeCourcey J, Teijeiro A, et al. Interleukin-17A serves a priming role in autoimmunity by recruiting IL-1β-producing myeloid cells that promote pathogenic T cells. Immunity (2020) 52:342–56. doi: 10.1016/j.immuni.2020.01.002
53. McGeachy MJ, Cua DJ. Th17 cell differentiation: The long and winding road. Immunity (2008) 28:445–53. doi: 10.1016/j.immuni.2008.03.001
54. Nurieva R, Yang XXO, Martinez G, Zhang YL, Panopoulos AD, Ma L, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature (2007) 448:480–U8. doi: 10.1038/nature05969
55. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol (2007) 8:967–74. doi: 10.1038/ni1488
56. Bettelli E, Carrier YJ, Gao WD, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector T(H)17 and regulatory T cells. Nature (2006) 441:235–8. doi: 10.1038/nature04753
57. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGF beta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity (2006) 24:179–89. doi: 10.1016/j.immuni.2006.01.001
58. Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature (2006) 441:231–4. doi: 10.1038/nature04754
59. Do JS, Fink PJ, Li L, Spolski R, Robinson J, Leonard WJ, et al. Cutting edge: spontaneous development of IL-17-producing gamma delta T cells in the thymus occurs via a TGF-beta 1-dependent mechanism. J Immunol (2010) 184:1675–9. doi: 10.4049/jimmunol.0903539
60. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4(+) effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol (2005) 6:1123–32. doi: 10.1038/ni1254
61. Park H, Li ZX, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol (2005) 6:1133–41. doi: 10.1038/ni1261
62. Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature (2007) 448:484–U9. doi: 10.1038/nature05970
63. Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3(+) regulatory T cells. Proc Natl Acad Sci U S A (2008) 105:18460–5. doi: 10.1073/pnas.0809850105
64. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature (2003) 421:744–8. doi: 10.1038/nature01355
65. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med (2005) 201:233–40. doi: 10.1084/jem.20041257
66. Sutton C, Brereton C, Keogh B, Mills KHG, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med (2006) 203:1685–91. doi: 10.1084/jem.20060285
67. Awasthi A, Riol-Blanco L, Jager A, Korn T, Pot C, Galileos G, et al. Cutting edge: IL-23 receptor gfp reporter mice reveal distinct populations of IL-17-producing cells. J Immunol (2009) 182:5904–8. doi: 10.4049/jimmunol.0900732
68. El-Behi M, Ciric B, Dai H, Yan YP, Cullimore M, Safavi F, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1-and IL-23-induced production of the cytokine GM-CSF. Nat Immunol (2011) 12:568–U241. doi: 10.1038/ni.2031
69. Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. ROR gamma t drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol (2011) 12:560–U248. doi: 10.1038/ni.2027
70. Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol (2007) 178:39–48. doi: 10.4049/jimmunol.178.1.39
71. Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, et al. The Cytokine GM-CSF Drives the Inflammatory Signature of CCR2(+) Monocytes and Licenses Autoimmunity. Immunity (2015) 43:502–14. doi: 10.1016/j.immuni.2015.08.010
72. Shi YF, Liu CH, Roberts AI, Das JT, Xu GW, Ren GW, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don’t know. Cell Res (2006) 16:126–33. doi: 10.1038/sj.cr.7310017
73. Mokhtarian F, McFarlin DE, Raine CS. Adoptive transfer of myelin basic protein-sensitized T cells produces chronic relapsing demyelinating disease in mice. Nature (1984) 309:356–8. doi: 10.1038/309356a0
74. Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, et al. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J Immunol (1996) 156:5–7.
75. Panitch HS, Hirsch RL, Haley AS, Johnson KP. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet (1987) 1:893–5. doi: 10.1016/s0140-6736(87)92863-7
76. Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med (2004) 200:79–87. doi: 10.1084/jem.20031819
77. Gao Y, Yang W, Pan M, Scully E, Girardi M, Augenlicht LH, et al. Gamma delta T cells provide an early source of interferon gamma in tumor immunity. J Exp Med (2003) 198:433–42. doi: 10.1084/jem.20030584
78. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol (2009) 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
79. Roark CL, Simonian PL, Fontenot AP, Born WK, O’Brien RL. gamma delta T cell: an important source of IL-17. Curr Opin Immunol (2008) 20:353–7. doi: 10.1016/j.coi.2008.03.006
80. Gao YL, Brosnan CF, Raine CS. Experimental autoimmune encephalomyelitis. Qualitative and semiquantitative differences in heat shock protein 60 expression in the central nervous system. J Immunol (1995) 154:3548–56.
81. Shimonkevitz R, Colburn C, Burnham JA, Murray RS, Kotzin BL. Clonal expansions of activated gamma/delta T cells in recent-onset multiple sclerosis. Proc Natl Acad Sci U S A (1993) 90:923–7. doi: 10.1073/pnas.90.3.923
82. Schirmer L, Rothhammer V, Hemmer B, Korn T. Enriched CD161high CCR6+ γδ T cells in the cerebrospinal fluid of patients with multiple sclerosis. JAMA Neurol (2013) 70:345–51. doi: 10.1001/2013.jamaneurol.409
83. Odyniec A, Szczepanik M, Mycko MP, Stasiolek M, Raine CS, Selmaj KW. gamma delta T cells enhance the expression of experimental autoimmune encephalomyelitis by promoting antigen presentation and IL-12 production. J Immunol (2004) 173:682–94. doi: 10.4049/jimmunol.173.1.682
84. Rajan J, Gao YL, Raine CS, Brosnan CF. A pathogenic role for gamma delta T cells in relapsing-remitting experimental allergic encephalomyelitis in the SJL mouse. J Immunol (1996) 157:941–9.
85. Rezende RM, Nakagaki BN, Moreira TG, Lopes JR, Kuhn C, Tatematsu BK, et al. γδ T Cell-Secreted XCL1 Mediates Anti-CD3-Induced Oral Tolerance. J Immunol (2019) 203:2621–9. doi: 10.4049/jimmunol.1900784
86. Rajan AJ, Asensio VC, Campbell IL, Brosnan CF. Experimental autoimmune encephalomyelitis on the SJL mouse: Effect of gamma delta T cell depletion on chemokine and chemokine receptor expression in the central nervous system. J Immunol (2000) 164:2120–30. doi: 10.4049/jimmunol.164.4.2120
87. Freedman MS, Ruijs TC, Selin LK, Antel JP. Peripheral blood gamma-delta T cells lyse fresh human brain-derived oligodendrocytes. Ann Neurol (1991) 30:794–800. doi: 10.1002/ana.410300608
88. Duhen R, Glatigny S, Arbelaez CA, Blair TC, Oukka M, Bettelli E. Cutting edge: The pathogenicity of IFN-gamma-producing Th17 cells is independent of T-bet. J Immunol (2013) 190:4478–82. doi: 10.4049/jimmunol.1203172
89. Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol (2011) 12:255–U95. doi: 10.1038/ni.1993
90. Singh AK, Novakova L, Axelsson M, Malmeström C, Zetterberg H, Lycke J, et al. High interferon-γ uniquely in Vδ1 T cells correlates with markers of inflammation and axonal damage in early multiple sclerosis. Front Immunol (2017) 8:260. doi: 10.3389/fimmu.2017.00260
91. Bloomgren G, Richman S, Hotermans C, Subramanyam M, Goelz S, Natarajan A, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med (2012) 366:1870–80. doi: 10.1056/nejmoa1107829
92. Maeda Y, Seki N, Kataoka H, Takemoto K, Utsumi H, Fukunari A, et al. IL-17-producing Vγ4+ γδ T cells require sphingosine 1-phosphate receptor 1 for their egress from the lymph nodes under homeostatic and inflammatory conditions. J Immunol (2015) 195:1408–16. doi: 10.4049/jimmunol.1500599
93. Selmaj K, Brosnan CF, Raine CS. Colocalization of lymphocytes bearing gamma delta T-cell receptor and heat shock protein hsp65+ oligodendrocytes in multiple sclerosis. Proc Natl Acad Sci U S A (1991) 88:6452–6. doi: 10.1073/pnas.88.15.6452
94. Wang XQ, Wei YX, Liu XL, Xing C, Han GC, Chen GJ, et al. IL-15-secreting gamma delta T cells induce memory T cells in experimental allergic encephalomyelitis (EAE) mice. Mol Immunol (2015) 66:402–8. doi: 10.1016/j.molimm.2015.04.021
95. Ponomarev ED, Dittel BN. gamma delta T cells regulate the extent and duration of inflammation in the central nervous system by a Fas ligand-dependent mechanism. J Immunol (2005) 174:4678–87. doi: 10.4049/jimmunol.174.8.4678
96. Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke (2009) 40:1849–57. doi: 10.1161/Strokeaha.108.534503
97. Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation (2006) 113:2105–12. doi: 10.1161/Circulationaha.105.593046
98. Gelderblom M, Gallizioli M, Ludewig P, Thom V, Arunachalam P, Rissiek B, et al. IL-23-producing conventional dendritic cells control the detrimental IL-17 response in stroke. Stroke (2018) 49:155–64. doi: 10.1161/strokeaha.117.019101
99. Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, et al. Pivotal role of cerebral interleukin-17-producing gammadelta T cells in the delayed phase of ischemic brain injury. Nat Med (2009) 15:946–50. doi: 10.1038/nm.1999
100. Kostulas N, Pelidou SH, Kivisakk P, Kostulas V, Link H. Increased IL-1beta, IL-8, and IL-17 mRNA expression in blood mononuclear cells observed in a prospective ischemic stroke study. Stroke (1999) 30:2174–9. doi: 10.1161/01.str.30.10.2174
101. Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, Steinbach K, et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood (2012) 120:3793–802. doi: 10.1182/blood-2012-02-412726
102. Kleinschnitz C, Schwab N, Kraft P, Hagedorn I, Dreykluft A, Schwarz T, et al. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood (2010) 115:3835–42. doi: 10.1182/blood-2009-10-249078
103. Arunachalam P, Ludewig P, Melich P, Arumugam TV, Gerloff C, Prinz I, et al. CCR6 is essential for the migration of detrimental natural interleukin-17-producing gammadelta T cells in stroke. Stroke (2017) 48:1957–65. doi: 10.1161/strokeaha.117.016753
104. Albertsson AM, Zhang X, Vontell R, Bi D, Bronson RT, Supramaniam V, et al. gammadelta T cells contribute to injury in the developing brain. Am J Pathol (2018) 188:757–67. doi: 10.1016/j.ajpath.2017.11.012
105. Kuo PC, Scofield BA, Yu IC, Chang FL, Ganea D, Yen JH. Interferon-beta Modulates Inflammatory Response in Cerebral Ischemia. J Am Heart Assoc (2016) 5:2–15. doi: 10.1161/jaha.115.002610
106. Xie L, Sun F, Wang J, Mao X, Xie L, Yang SH, et al. mTOR signaling inhibition modulates macrophage/microglia-mediated neuroinflammation and secondary injury via regulatory T cells after focal ischemia. J Immunol (2014) 192:6009–19. doi: 10.4049/jimmunol.1303492
107. Xie G, Luo H, Pang L, Peng BH, Winkelmann E, McGruder B, et al. Dysregulation of toll-like receptor 7 compromises innate and adaptive T cell responses and host resistance to an attenuated west nile virus infection in old mice. J Virol (2016) 90:1333–44. doi: 10.1128/jvi.02488-15
108. Sciammas R, Kodukula P, Tang Q, Hendricks RL, Bluestone JA. T cell receptor-gamma/delta cells protect mice from herpes simplex virus type 1-induced lethal encephalitis. J Exp Med (1997) 185:1969–75. doi: 10.1084/jem.185.11.1969
110. Deknuydt F, Roquilly A, Cinotti R, Altare F, Asehnoune K. An in vitro model of mycobacterial granuloma to investigate the immune response in brain-injured patients. Crit Care Med (2013) 41:245–54. doi: 10.1097/ccm.0b013e3182676052
111. Caccamo N, La Mendola C, Orlando V, Meraviglia S, Todaro M, Stassi G, et al. Differentiation, phenotype, and function of interleukin-17-producing human V gamma 9V delta 2 T cells. Blood (2011) 118:129–38. doi: 10.1182/blood-2011-01-331298
112. Nichols JR, Aldrich AL, Mariani MM, Vidlak D, Esen N, Kielian T. TLR2 deficiency leads to increased Th17 infiltrates in experimental brain abscesses. J Immunol (2009) 182:7119–30. doi: 10.4049/jimmunol.0802656
113. Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity (1999) 11:443–51. doi: 10.1016/s1074-7613(00)80119-3
114. Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol (2000) 165:5392–6. doi: 10.4049/jimmunol.165.10.5392
115. Vidlak D, Kielian T. Differential effects of interleukin-17 receptor signaling on innate and adaptive immunity during central nervous system bacterial infection. J Neuroinflamm (2012) 9:128. doi: 10.1186/1742-2094-9-128
116. Stumhofer JS, Laurence A, Wilson EH, Huang E, Tato CM, Johnson LM, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol (2006) 7:937–45. doi: 10.1038/ni1376
117. Bajpai A, Prasad KN, Mishra P, Singh AK, Gupta RK, Ojha BK. Distinct cytokine pattern in response to different bacterial pathogens in human brain abscess. J Neuroimmunol (2014) 273:96–102. doi: 10.1016/j.jneuroim.2014.05.009
118. Rubiano AM, Carney N, Chesnut R, Puyana JC. Global neurotrauma research challenges and opportunities. Nature (2015) 527:S193–7. doi: 10.1038/nature16035
119. Tobin RP, Mukherjee S, Kain JM, Rogers SK, Henderson SK, Motal HL, et al. Traumatic brain injury causes selective, CD74-dependent peripheral lymphocyte activation that exacerbates neurodegeneration. Acta Neuropathol Com (2014) 2:197. doi: 10.1186/s40478-014-0143-5
120. Berer K, Mues M, Koutrolos M, Al Rasbi Z, Boziki M, Johner C, et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature (2011) 479:538–U266. doi: 10.1038/nature10554
121. Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A (2011) 108:4615–22. doi: 10.1073/pnas.1000082107
122. Kadowaki A, Saga R, Lin Y, Sato W, Yamamura T. Gut microbiota-dependent CCR9+CD4+ T cells are altered in secondary progressive multiple sclerosis. Brain (2019) 142:916–31. doi: 10.1093/brain/awz012
123. Kadowaki A, Miyake S, Saga R, Chiba A, Mochizuki H, Yamamura T. Gut environment-induced intraepithelial autoreactive CD4(+) T cells suppress central nervous system autoimmunity via LAG-3. Nat Commun (2016) 7:116–39. doi: 10.1038/ncomms11639
124. Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M, et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal gamma delta T cells. Nat Med (2016) 22:516–23. doi: 10.1038/nm.4068
125. Xu D, Robinson AP, Ishii T, Duncan DS, Alden TD, Goings GE, et al. Peripherally derived T regulatory and γδ T cells have opposing roles in the pathogenesis of intractable pediatric epilepsy. J Exp Med (2018) 215:1169–86. doi: 10.1084/jem.20171285
126. Owens GC, Erickson KL, Malone CC, Pan C, Huynh MN, Chang JW, et al. Evidence for the involvement of gamma delta T cells in the immune response in Rasmussen encephalitis. J Neuroinflamm (2015) 12:134. doi: 10.1186/s12974-015-0352-2
127. Al Nimer F, Jelcic I, Kempf C, Pieper T, Budka H, Sospedra M, et al. Phenotypic and functional complexity of brain-infiltrating T cells in Rasmussen encephalitis. Neurol Neuroimmunol Neuroinflamm (2018) 5:e419. doi: 10.1212/nxi.0000000000000419
128. Guan W, Ni Z, Hu Y, Liang W, Ou C, He J, et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med (2020) 382:1708–20. doi: 10.1056/NEJMoa2002032
Keywords: γδ T cell, Th17 cell, cytokines, inflammation, central nervous system, IL-17
Citation: Wo J, Zhang F, Li Z, Sun C, Zhang W and Sun G (2020) The Role of Gamma-Delta T Cells in Diseases of the Central Nervous System. Front. Immunol. 11:580304. doi: 10.3389/fimmu.2020.580304
Received: 05 July 2020; Accepted: 30 September 2020;
Published: 23 October 2020.
Edited by:
Weifeng He, Army Medical University, ChinaReviewed by:
Kingston H. Mills, Trinity College Dublin, IrelandMichele Kay Anderson, University of Toronto, Canada
Song Zhang, Nankai University, China
Xue Yao, Tianjin Medical University, China
Copyright © 2020 Wo, Zhang, Li, Sun, Zhang and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guodong Sun, c2dkOTZAam51LmVkdS5jbg==; Wencai Zhang, emh3YzUwQHNvaHUuY29t
†These authors have contributed equally to this work