 Jaime Díaz-Zúñiga1
Jaime Díaz-Zúñiga1 Jamileth More2
Jamileth More2 Samanta Melgar-Rodríguez1
Samanta Melgar-Rodríguez1 Matías Jiménez-Unión1
Matías Jiménez-Unión1 Francisca Villalobos-Orchard1
Francisca Villalobos-Orchard1 Constanza Muñoz-Manríquez1
Constanza Muñoz-Manríquez1 Gustavo Monasterio1
Gustavo Monasterio1 José Luis Valdés3,4
José Luis Valdés3,4 Rolando Vernal1
Rolando Vernal1 Andrea Paula-Lima3,4,5*
Andrea Paula-Lima3,4,5*- 1Periodontal Biology Laboratory, Faculty of Dentistry, Universidad de Chile, Santiago, Chile
- 2Centro de Investigación Clínica Avanzada (CICA), Hospital Clínico Universidad de Chile, Santiago, Chile
- 3Biomedical Neuroscience Institute, Faculty of Medicine, Universidad de Chile, Santiago, Chile
- 4Department of Neuroscience, Faculty of Medicine, Universidad de Chile, Santiago, Chile
- 5Faculty of Dentistry, Institute for Research in Dental Sciences, Universidad de Chile, Santiago, Chile
Periodontal disease is a disease of tooth-supporting tissues. It is a chronic disease with inflammatory nature and infectious etiology produced by a dysbiotic subgingival microbiota that colonizes the gingivodental sulcus. Among several periodontal bacteria, Porphyromonas gingivalis (P. gingivalis) highlights as a keystone pathogen. Previous reports have implied that chronic inflammatory response and measurable bone resorption are observed in young mice, even after a short period of periodontal infection with P. gingivalis, which has been considered as a suitable model of experimental periodontitis. Also, encapsulated P. gingivalis strains are more virulent than capsular-defective mutants, causing an increased immune response, augmented osteoclastic activity, and accrued alveolar bone resorption in these rodent experimental models of periodontitis. Recently, P. gingivalis has been associated with Alzheimer’s disease (AD) pathogenesis, either by worsening brain pathology in AD-transgenic mice or by inducing memory impairment and age-dependent neuroinflammation middle-aged wild type animals. We hypothesized here that the more virulent encapsulated P. gingivalis strains could trigger the appearance of brain AD-markers, neuroinflammation, and cognitive decline even in young rats subjected to a short periodontal infection exposure, due to their higher capacity of activating brain inflammatory responses. To test this hypothesis, we periodontally inoculated 4-week-old male Sprague-Dawley rats with K1, K2, or K4 P. gingivalis serotypes and the K1-isogenic non-encapsulated mutant (GPA), used as a control. 45-days after periodontal inoculations with P. gingivalis serotypes, rat´s spatial memory was evaluated for six consecutive days in the Oasis maze task. Following functional testing, the animals were sacrificed, and various tissues were removed to analyze alveolar bone resorption, cytokine production, and detect AD-specific biomarkers. Strikingly, only K1 or K2 P. gingivalis-infected rats displayed memory deficits, increased alveolar bone resorption, pro-inflammatory cytokine production, changes in astrocytic morphology, increased Aβ1-42 levels, and Tau hyperphosphorylation in the hippocampus. None of these effects were observed in rats infected with the non-encapsulated bacterial strains. Based on these results, we propose that the bacterial virulence factors constituted by capsular polysaccharides play a central role in activating innate immunity and inflammation in the AD-like pathology triggered by P. gingivalis in young rats subjected to an acute experimental infection episode.
Introduction
Periodontitis is a chronic non-communicable disease caused by a dysbiotic subgingival microbiota (1). Porphyromonas gingivalis (P. gingivalis) has been identified as a keystone pathogen among several bacteria present in this dysbiotic microbiota, invoking increased secretion of pro-inflammatory cytokines, matrix metalloproteinases, and pro-bone resorptive mediators that promote periodontal tissue breakdown (2, 3). P. gingivalis displays a wide variety of virulence factors, which have major roles in bacterial adhesion, colonization and invasion. Among the virulence factors described for P. gingivalis, the most important are the gingipains, lipopolysaccharide (LPS) and capsular polysaccharides, but its antigenicity is mainly related to its extracellular capsule’s polysaccharide components (4, 5).
Several studies have evaluated the virulence of the K-antigen of P. gingivalis for its capacity to cause an immune response in various immune cells. Encapsulated serotypes K1 and K2 are known to induce increased production of interleukin (IL)-1β, IL-6, IL-17, interferon (IFN)-γ, and tumor necrosis factor (TNF)-α in macrophages and dendritic cells, as compared with the other serotypes and the non-encapsulated strains (6, 7). Similarly, serotypes K1 and K2 induce higher differentiation of CD4+ T helper type 1 (Th1) and type 17 (Th17) lymphocytes compared with the others (8, 9). Besides, the K1 serotype induces bone-resorption and activates Th1 and Th17 immune response, while mice infected with the GPA strain and sham controls do not exhibit these effects (10). Furthermore, in subjects affected by periodontitis, IgG forms reactive to the K antigen are detected, and the presence of serotypes K1 or K2 is observed more frequently in periodontal sites (11, 12).
Conversely, non-encapsulated and K4 strains—associated with periodontal health—induce an immune regulatory response predominantly (8, 9). While non-encapsulated and K4 strains generate Treg response and localized abscesses, encapsulated serotypes are resistant to phagocytosis, generate distant abscesses and sepsis (8, 9, 13). The generation of the W50 strain (serotype K1)-based isogenic capsular-deficient mutants was possible after the identification and characterization of the capsular polysaccharide (K-antigen) locus of P. gingivalis, which contributed to the current understanding of the importance of capsule in the induction of the host immune response (14). The analysis of these mutants indicated that the presence of capsule associates with the deregulated production of pro-inflammatory cytokines by fibroblasts and macrophages, thus further supporting the idea that encapsulation is a hallmark of the virulence heterogeneity of P. gingivalis (15).
P. gingivalis can destroy the periodontal tissues and distribute in different organs and tissues, where it can produce coronary and aortic atherogenesis, favor preterm birth, and deregulate the adequate metabolic control in diabetes mellitus-affected patients (16–20). Also, recent studies have used wild-type and transgenic mice to establish the association between P. gingivalis and Alzheimer’s disease (AD)-related cognitive decline, by assessing the amyloid β (Aβ) peptide accumulation and pro-inflammatory cytokines production, as well as the performance in spatial memory-dependent tasks in rodents orally infected with P. gingivalis (21–25).
AD is a neurodegenerative disorder that affects behavioral and cognitive functions, with an early, prominent, and progressive dysfunction in the hippocampus (26, 27). The amyloid cascade hypothesis begins with the accumulation of Aβ peptide, which induces a microglial M1 inflammatory response that activates astrocytes, triggering energy, metabolic, and oxidative imbalance that leads to hyperphosphorylation of the microtubule-associated protein Tau in neurons (28, 29). A new hypothesis proposes sporadic AD as the result of independent, yet intersecting age-related pathologies that affect the aging human brain, many of which are related to neuroinflammation (30). It is believed that over time such newly generated, divergent activated microglia could sustain a chronic type of neuroinflammation in AD. This inflammatory environment, where activated microglia and reactive-astrocytes are actively producing inflammatory mediators, enhances Aβ production; its accumulation causes Tau hyperphosphorylation, which constitutes the histopathological hallmarks of AD (31). This finding led to the “neuroinflammation hypothesis” that emphasizes chronic systemic inflammatory diseases as major risk factors for AD (27).
Among chronic systemic diseases, periodontitis is the inflammatory disease more frequent in humans. Recently, brains of patients who died due to AD revealed frequent detection of P. gingivalis, particularly in the fourth ventricle, hippocampus, and cerebrospinal fluid (CSF) (32–34). Interestingly, brain expression levels of the pro-inflammatory cytokines and memory of mice after 6 weeks of P. gingivalis oral infection in young versus middle-aged (12 months old) mice. After receiving oral gavage with a P. gingivalis strain of reduced virulence, only middle-aged P. gingivalis infected mice exhibited memory impairment and increased expression levels of the pro-inflammatory cytokines TNF-α, IL-6, and IL-1β in the brain tissues, suggesting that P. gingivalis effects in the brain are dependent on age (22, 23).
Interestingly, the possibility that the immune response induced in the brain might differ depending on the infecting capsular P. gingivalis serotype has never been exploited. Microglia are known to discriminate against different bacterial antigens, triggering the activation of divergent pathways according to the antigens recognized (35–38). Thus, it is possible that some P. gingivalis strains might be more virulent than others in causing Alzheimer´s like-disease. Accordingly, this study aimed to evaluate whether the capsulated strains of P. gingivalis display divergent capacities of inducing cognitive impairment, neuroinflammation and AD-like neuropathology in young rats subjected to a short period of infection with different serotypes of this periodontal-pathogen.
Materials and Methods
Animals
Sprague-Dawley young male rats (4 weeks old) were obtained from the Animal Care Facility of the Faculty of Medicine at the Universidad de Chile. Rats were housed individually in a controlled environment with a 12 h light/dark cycle at 22 ± 0.5°C, 40%–70% relative humidity, with food and water ad libitum except when indicated otherwise; all animals were handled daily for 2 weeks before bacterial inoculation. During the 4 weeks previous to bacterial injection, all rats in each group inhabited the same cage and were fed and hydrated from the same sources. After injecting the bacteria, they were isolated in separate filter-containing cages. The experimental protocols complied and were approved by the Bioethics Committees on Animal Research of the Faculty of Medicine (protocol #CBA 0755 FMUCH) and the Faculty of Dentistry (protocol #17085-ODO-UCH), Universidad de Chile.
Experimental Periodontitis
Each experimental group consisted of six rats housed in separate boxes conditioned with air filters maintained under standard conditions, as described above. Experimental periodontal infections were performed under general anesthesia with 2% isoflurane. Animals received an injection into the palatal mucosa of 1010 CFU/ml of the P. gingivalis strains W50 (serotype K1), HG184 (K2), or ATCC® 49417™ (K4). As controls, animals were inoculated with the non-encapsulated W50 ΔPG0116-PG0120 mutant strain (GPA) of P. gingivalis. A final volume of 100 μl was inoculated into the palatal gingival tissues, between the first and second molar, repeating this procedure after 7 days (i.e., each rat received two palatal injections). The volume and the CFU concentration used in the palatal injections were determined based on a previous pilot study reported by our group (10). Sham-rats that received vehicle without bacteria were used for comparisons. After 45 days, animals were subjected to the Oasis Maze task to evaluate spatial memory.
OASIS Maze Spatial Memory Task
The Oasis maze—a dry version of the classical Morris water maze memory task—was used to evaluate the hippocampal-dependent spatial memory of rats, as previously described (39–41). Animal behavior was recorded with a video camera installed in the zenithal position, and video recordings were analyzed using the Virtual Dub software (Microsoft Windows, Microsoft Corporation, Redmond, WA). The position of animals was tracked, and navigation was reconstructed and analyzed with the Matlab® software (MathWorks, Cleve Moler, MA, USA). This model was chosen to replace the classical Morris Water Maze (42) to avoid oral dysbiosis by colonizing bacteria resident in the water used in the maze.
Euthanasia and Tissue Sampling
After concluding the Oasis maze task, rats were euthanized by cervical dislocation following the American Veterinary Medical Association recommendations. Samples of maxillae, CSF, and hippocampus were immediately obtained. Extraction of CSF and blood (by cardiac puncture with 5 ml hypodermic syringe) was performed according to standard protocols (43). Blood was maintained at 37°C/2 h and at 4°C/30 min for serum isolation. Brains were removed and homogenized. Samples were centrifuged at 10.000 x g for 5 min at 4°C; the supernatant was recovered, its protein concentration was measured in a spectrophotometer (Bio-Tek, Winooski, VT, USA), and aliquots of 200 μl were stored at -80°C until further analysis. For histochemical analysis, brains were fixed after finishing the Oasis maze task according to published protocols (39, 40).
Bone Destruction Quantification
To determine the pathogenicity of P. gingivalis, we evaluated morphological changes in the alveolar bone. The maxillary alveolar bone was scanned by µCT (Bruker microCT, SkyScan 1278; Bruker, Kontich, Belgium), and 3D image reconstructions were used to quantify bone resorption in the maxillae of rats as previously described (10, 44). The Nrecon software (Bruker, Kontich, Belgium) was used to perform the morphometric analysis, executing a linear measurement from the cementum-enamel junction (CEJ) to the alveolar bone-crest (ABC) on the 1st, 2nd, and 3rd left and right upper molars.
Immunofluorescence Analysis
For immunofluorescence analysis, 1 h after the last test session, adult rats were transcardially perfused with 300 ml of saline flush and 300 ml of 4% paraformaldehyde in 0.1 M PBS, pH 7.4. After perfusion, brains were removed, postfixed in 4% paraformaldehyde for 2 h at room temperature, and incubated for 72 h at 4°C in a solution containing 30% sucrose, 0.002% sodium azide for cryopreservation, as previously described (39). Afterward, brains were cut in the coronal plane with a sliding frozen microtome at -30°C. Free-floating 40 µm thickness sections were immersed in a blocking solution (PBS containing 0.25% Triton X-100 plus 3% donkey) serum for 2 h at room temperature. Sections were incubated overnight at 4°C with blocking solution containing primary antibodies to visualize neurons (anti-TubIII, Abcam, Ab18207), astrocytes (anti-GFAP, Abcam, ab10062), and the cytoskeleton-associated protein Tau, in its phosphorylated (anti-phosphoTau, Phospho S404, Abcam, ab92676) and total (anti-Tau, Abcam, ab64193) forms. Sections were washed three times for 5 min with PBS and incubated for 2 h with secondary fluorescent anti-mouse, goat or rabbit antibodies (Alexa Fluor® 488, AlexaFluor® 594 or Alexa Fluor® 647, Abcam US). Nuclei were stained with Hoechst (1:10,000, Sigma, St Louis, MI, USA). Z-stack of 1.5 μm sections were captured from the hippocampus in a confocal microscope (Nikon C2+, Melville, NY.). Fluorescence intensity was measured with the confocal microscope NIS-Elements software viewer 4.0 (Nikon, Melville, NY) and with ImageJ free viewer software (National Institutes of Health, MD, USA; https://imagej.nih.gov/ij/). Sholl analysis was performed to evaluate changes in neuronal morphology. To this aim, the neuronal nucleus is located in the center of the measurement, and the software establishes concentric circles at a distance of 10 μm and calculates the number of projections that intersect for each radius and the maximum distance it reaches the most extensive projection of the cell. For the quantification of different molecules, the brains of sham rats were used as control.
Cytokines and Aβ1-42 Peptide Measurements
The levels of IL-1β, IL-4, IL-6, IL-10, TNF-α, IFN-γ, in hippocampal homogenates, serum and CSF samples, and the levels of Aβ1-42 peptide in hippocampal homogenates and CSF were quantified using ELISA assays (R&D Systems, Minneapolis, USA or Invitrogen, Thermo Fisher Scientific, MA, USA), by evaluating the absorbance at 450 and 560 nm in an automated spectrophotometer (Bio-Tek, Winooski, VT, USA), as previously described (37). The Aβ1-42 peptide ELISA assay detects and quantifies mouse and rat Aβ1-42, both natural and synthetic. For the quantification of the different molecules, each animal was considered individually. No sample was held as a pool.
Malondialdehyde Quantification
We used the thiobarbituric acid reactive substances (TBARS) method to measure lipid peroxidation in the hippocampus of the rats, following the manufacturer’s recommendations (Invitrogen, Thermo Fisher Scientific, MA, USA). In brief, 300 μl of hippocampus homogenates were treated with 0.6 N trichloroacetic acid and clarified by precipitating the interfering proteins. Thus, the absorbance of all samples was measured at 450 nm and 560 nm using a plate spectrophotometer (Bio-Tek, Winooski, VT, USA), as indicated by the manufacturer. This assay recognizes oxidized lipids by a reaction between malondialdehyde (MDA) and thiobarbituric acid in the presence of heat. The reaction produces TBARS, which are detected and quantified by a spectrophotometer and compared with a standard curve.
Porphyromonas gingivalis Detection
The presence of P. gingivalis was determined by quantifying by qPCR the contents of the 16S rRNA subunit and the RgpA and Kgp genes. Total DNA was purified following the manufacturer’s instructions (FavorPrep™ Tissue Genomic DNA extraction Mini Kit, Favorgen Biotech Corp.). Briefly, samples were incubated in 200 μl FATG1 buffer and 20 μl of proteinase K and incubated at 60°C. After tissue elution, samples were incubated with 4 μl of RNAase A for 2 min, followed by incubation with 200 μl of FATG1 buffer at 70°C for 1 h, and then 200 μl of 100% ethanol were added. Each sample was inserted in a collection tube and centrifuged for 1 min at 18,000 x g and then washed with 400 μl of FATG buffer. Next, 2 washes were performed: 400 μl of W1 buffer and 1 min of centrifugation at 18,000 x g and 750 μl of wash buffer followed by 1 min of centrifugation at 18,000 x g. Finally, the column was incubated with 200 μl of elution buffer (pH 7.5-9.0) for 3 min and centrifuged at maximum speed for 2 min to elute the total DNA. The samples were quantified using a spectrophotometer (Bio-Tek). Genes were quantified from 50 ng of DNA by qPCR, using specific primers (Table 1), a KAPA SYBR® FAST qPCR kit (Kapa Biosystems, MA, USA) according to the manufacturer’s instructions and using equipment from StepOne Plus. As negative controls, no-template control (NTC) and a no-amplification control omitting DNA polymerase were performed, and samples were analyzed in triplicates in each experiment.

Table 1 Primers used in the detection of P. gingivalis by qPCR.
To determine the presence of P. gingivalis in the hippocampus, we performed an immunofluorescence assay to visualize neurons and the bacteria. Fixing and pre-treatments were performed as described above, but here, samples were incubated overnight at 4°C with PBS-TX containing primary antibodies anti-tubIII (1:300, Abcam, ab18207) and anti-RgpA1 (1:200, RgpA R1, Biorbyt, UK). Sections were washed 4-times for 5 min in PBS and incubated for 2 h with secondary fluorescent antibodies (Alexa Fluor® 488 1:300 or Alexa Fluor® 633 1:300). The Hoechst reagent (1:10,000, Sigma, St Louis, MI, USA) was used for nuclear staining. Endogenous tissue background immunofluorescence control was performed by the direct observation of the hippocampal tissue under the confocal microscope. Also, a primary antibody control staining was performed to discard any background that could interfere with the analysis of RgpA1 staining. A z-stack of 1.5 μm sections was captured from the CA1 hippocampal region in a confocal microscope (Nikon C2+, Melville, NY). Fluorescence intensity was measured with the confocal microscope NIS-Elements software viewer 4.0 and with ImageJ free viewer software.
Data Analysis
The OASIS maze data were expressed as the mean ± SD of distance (cm) or time (s) units. Cytokine production was expressed as the mean ± SD of concentration (pg/ml). Alveolar bone resorption was represented in terms of distance units (nm) and expressed as the mean ± SD. The Shapiro-Wilk test was used to determine the normal distribution of the data, which were analyzed using the ANOVA-Tukey or Kruskal Wallis-Dunn tests using the SPSS v.15.0 software (IBM, NY, USA). Differences were considered as statistically significant when p-value <0.05.
Results
Bone Resorption Quantification
In order to determine whether the inoculation of P. gingivalis induces bone resorption, each maxilla was reconstructed in 3D images (Figures 1A–E). Three-dimensional quantification of alveolar bone loss was performed by determining the CEJ-ABC distances (Figure 1F). P. gingivalis-infected rats with capsular serotypes K1 or K2 displayed significantly increased alveolar bone loss compared to both P. gingivalis-infected rats with K4, GPA, and sham rats, as indicated by an increase in CEJ-bone crest distance of each root in the palatal root of 1st molar (Figure 1G). These results were also consistent when 2nd and 3rd molars (Supplementary Figure 1) were analyzed.
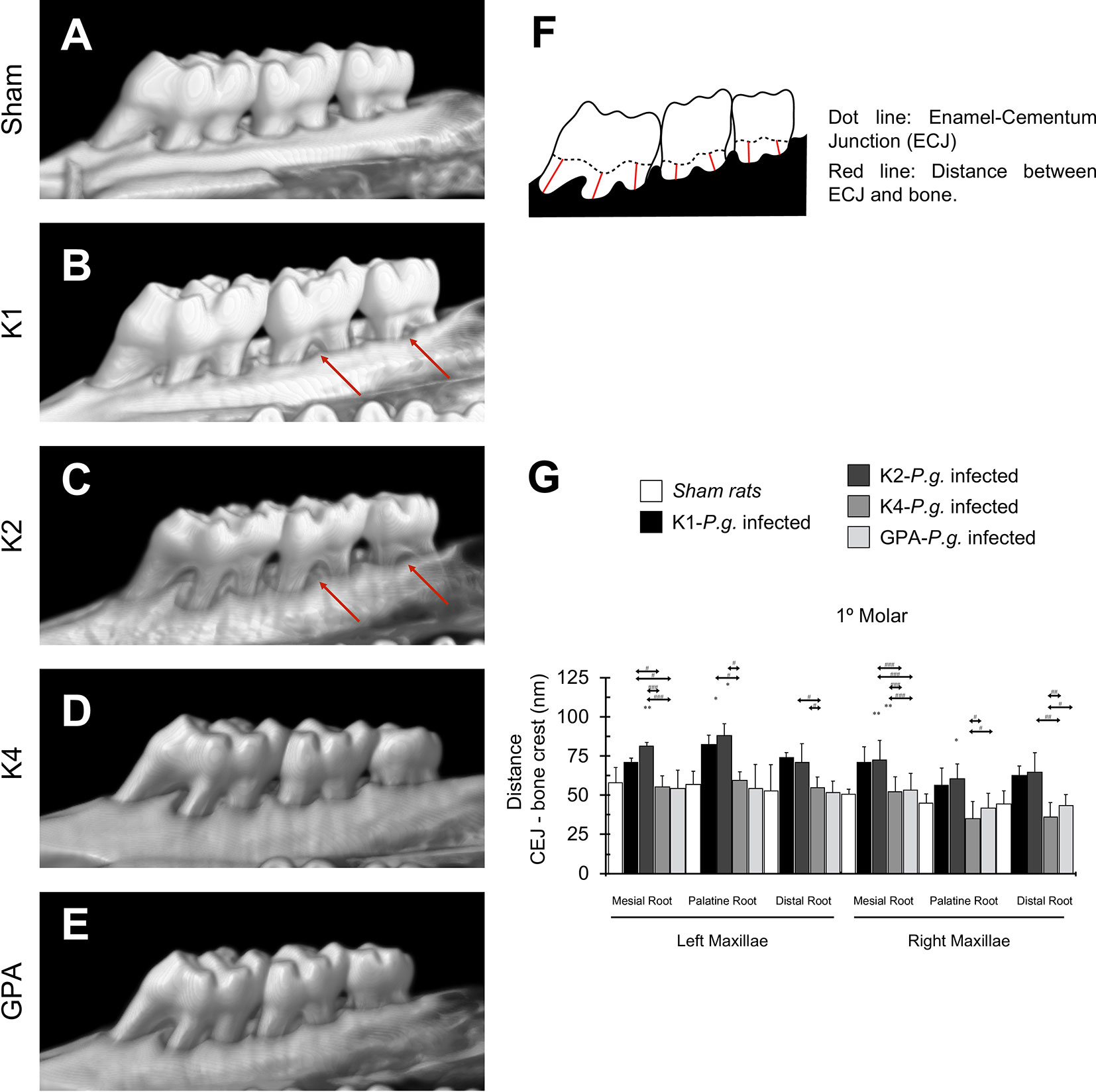
Figure 1 Bone resorption in the 1st molar. (A) micro-CT reconstruction of the left and right maxillae of sham rats. (B–E) micro-CT reconstruction of maxillae of rats inoculated with strains K1, K2, K4, or GPA of Porphyromonas gingivalis. Red arrows indicate macroscopic changes in the alveolar bone, highlighting the presence of furcation, which exists when periodontal disease has caused resorption of bone. (F) Scheme representing the measures design for bone resorption quantification. (G) Bone resorption quantification of 1st molar. Data, mean ± SD, are represented as distance (nm) for six independent experiments. *p < 0.05, **p < 0.01, compared to the control group; #p < 0.05, ##p < 0.01, ###p < 0.001, compared to the group indicated.
Spatial Memory and Learning
Along the daily training sessions composed of 15 trials, since the 5th day of training rats infected with capsular-serotypes K1 or K2 exhibited worse spatial memory than animals infected with K4, GPA, or sham (Supplementary Figure 2). On the 6th day (last day of the task), sham rats and those infected with K4 or GPA displayed significantly higher hit rates, which are defined as the number of times the rat found the reward over the 15 trials, compared to rats infected with K1 or K2 capsular serotypes (Figure 2A). Likewise, the average latency to find the reward was significantly lower in the control groups and K4 or GPA infected rats, when compared to K1 or K2 infected-rats (Figure 2B). Finally, the ratio of the observed over the expected (shortest) distance covered by the animals while searching for the reward was higher in the P. gingivalis K1 or K2 injected group (Figure 2C) than that displayed by the sham group or by rats injected with K4 or GPA.
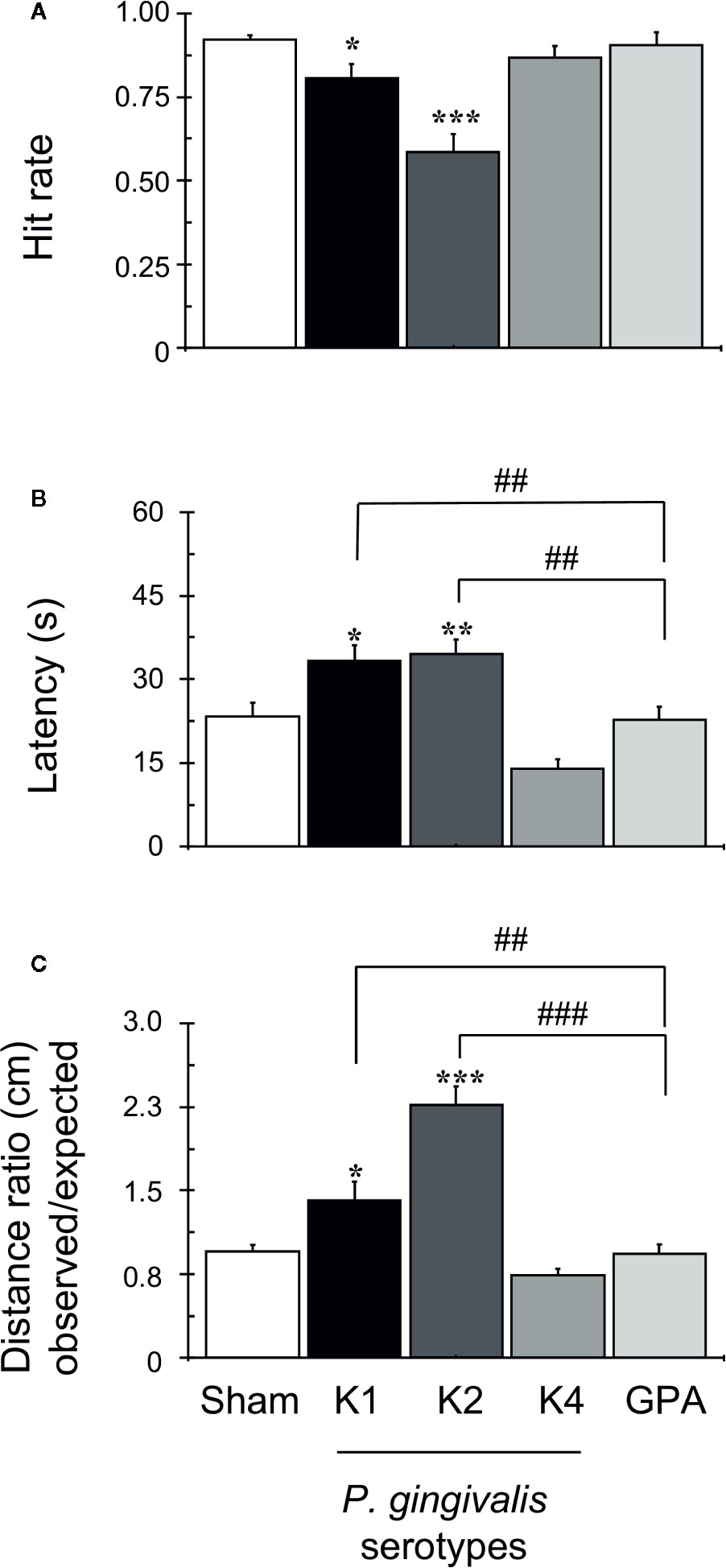
Figure 2 OASIS Maze task. The graph corresponds to the performance of all groups on day 6 of the task, as indicated. Each bar represents the mean ± SD of 90 videos: 15 videos were collected for each animal, out of a total of six animals per group. (A) Hit rate achieved during training in OASIS Maze by rats infected with P. gingivalis, which represents the percent of the times that the animal obtained the reward over the total trials per day. (B) Latency, defined as the total time (s) rats take to find the reward and complete the task, represented as mean ± SD. (C) The distance ratio represents the ratio between the observed/expected distances walked by rats during the task, represented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, compared to the control group; ##p < 0.01, ###p < 0.001, compared to the group indicated.
Cytokine Production
We quantified the secretion levels of the cytokines IL-1β, IL-4, IL-6, IL-10, TNF-α, and IFN-γ production in the hippocampus, CSF and serum by ELISA (Figure 3A–C, respectively), the levels of extracellular Aβ42 peptide on the hippocampus and CSF (Figures 3C, D) and calculated the Aβ42 hippocampus/CSF ratio (Figure 3E). Statistically significant increased production of IL-1β was detected in the hippocampus, CSF, and serum of rats infected with the capsular-serotypes K1 or K2 compared to sham, which exhibited IL-1β concentration values similar to those observed in rats infected with the capsular-serotypes K4 or GPA. IL-6 levels were also significantly increased in the hippocampus and serum of rats infected with the capsular-serotypes K1 or K2 compared to sham, which exhibited no differences in samples from rats infected with the capsular-serotypes K4 or GPA. However, in CSF, only in samples from rats infected with the capsular-serotype K1 a significant increase in IL-6 levels in comparison to all the other conditions was observed. IFN-γ levels were significantly increased in the hippocampus and serum of rats infected with the capsular-serotypes K1 or K2 compared to sham, which exhibited IL-1β concentration values similar to those observed in rats infected with the capsular-serotypes K4 or GPA. Of note, the effect observed in serum from rats infected with the capsular-serotype K2 was significantly higher than in serum obtained from rats infected with the capsular-serotype K1. However, no differences in the IFN-γ levels were detected in the CSF for all these experimental conditions. Also, increased production of TNF-α was detected in serum from K1 or K2 infected-rats compared with sham, K4 or GPA injected rats. In the CSF, the increase in the concentration of TNF-α was only observed in samples from rats injected with the capsular-serotype K2.
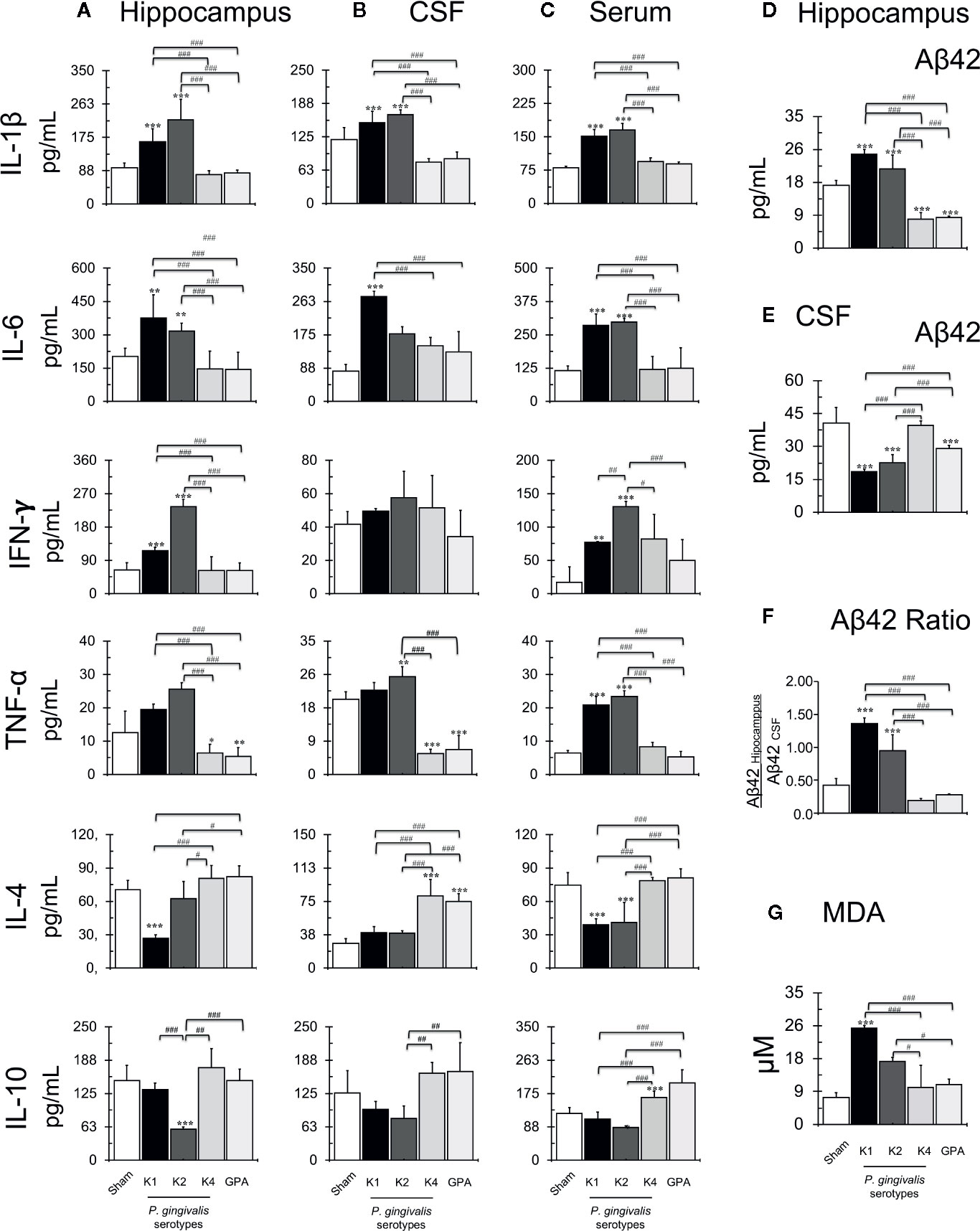
Figure 3 Quantification of secretion levels of pro-inflammatory molecules. Secretion levels of IL-1β, IL-4, IL-6, IL-10, TNF-α, and IFN-γ determined in the hippocampus (A), serum (B) or CSF (C) from sham and P. gingivalis infected-rats. Data are presented as cytokine concentration (pg/ml); mean ± SD of 4 independent experiments. (B) Secretion levels of Aβ1-42 peptide on the hippocampus (D), CSF (E) and ratio (F) from sham or P. gingivalis infected-rats. Data are presented as molecule concentration (pg/ml); mean ± SD for 4 independent experiments. (D) Secretion levels of Aβ1-42 peptide on CSF from sham or P. gingivalis infected-rats. Data are presented as molecule concentration (pg/ml); mean ± SD for 4 independent experiments. (E) The ratio of the secretion levels of Aβ1-42 on the hippocampus and CSF from sham rats or P. gingivalis infected-rats. Production levels of MDA (G) on the hippocampus from sham or P. gingivalis infected-rats. Data are presented as molecule concentration (pg/ml); mean ± SD for four independent experiments. Each experiment was performed in duplicate. **p < 0.01, ***p < 0.001, compared to the control group; #p < 0.05, ##p < 0.01, ###p < 0.001, compared to the group indicated. IL, interleukin, TNF, tumor necrosis factor, IFN, interferon, CSF, cerebrospinal fluid.
Interestingly, the injection of the K1-isogenic non-encapsulated mutant GPA and the K4 serotype P. gingivalis strain resulted in a statistically significant decrease in the levels of TNF-α. Also, significantly increased IL-4 and IL-10 secretion levels were detected in the hippocampus and CSF from rats infected with K4 or GPA, compared with those infected with K1 or K2. Also, in rats inoculated with K1 or K2 serotypes, a statistically significant increase in Aβ42 hippocampus/CSF ratio was detected, compared with the other conditions. The K1-isogenic non-encapsulated mutant and the K4 serotype P. gingivalis strain injections resulted in a significant decrease in Aβ42 levels in the hippocampus. Still, this difference was not significant when the Aβ42 hippocampus/CSF ratio was calculated in comparison to the sham infected control. Finally, to determine if all these stressful changes induce lipid peroxidation, we quantified MDA production in the hippocampus of periodontitis rat models (Figure 3F, G). An increase in MDA levels was detected in the hippocampus from K1 and K2 infected rats compared with sham, K4, or GPA infected rats.
Glial Activation and Tau Protein Phosphorylation
After the Oasis maze task, rat hippocampal samples were evaluated for the occurrence of astrogliosis and phosphorylation of Tau protein by immunohistochemistry (Figures 4, 5). For this purpose, we visualized neurons and astrocytes in the CA1, CA3, and DG regions of the hippocampus (Figures 4A–C) and quantified the total number of cells in the CA1 region by counting DAPI-stained nuclei, the number of TubIII-positive and GFAP-positive cells for all experimental groups (Figures 4D–G). We observed no differences in the total number of astrocytes and neurons when comparing all the conditions tested. However, we noted that astrocytes of rats infected with the K1 or K2 serotype exhibited an unusual, distinctive morphology in the CA1, CA3, and DG regions. We thus performed a Sholl analysis to compare the morphology of these cells in the CA1 region, in the hippocampus of sham, K1 or GPA infected rats and observed that K1 infection promoted a slight increase in the size and the complexity of astrocytic processes (Figure 4H and Supplementary Figure 3).
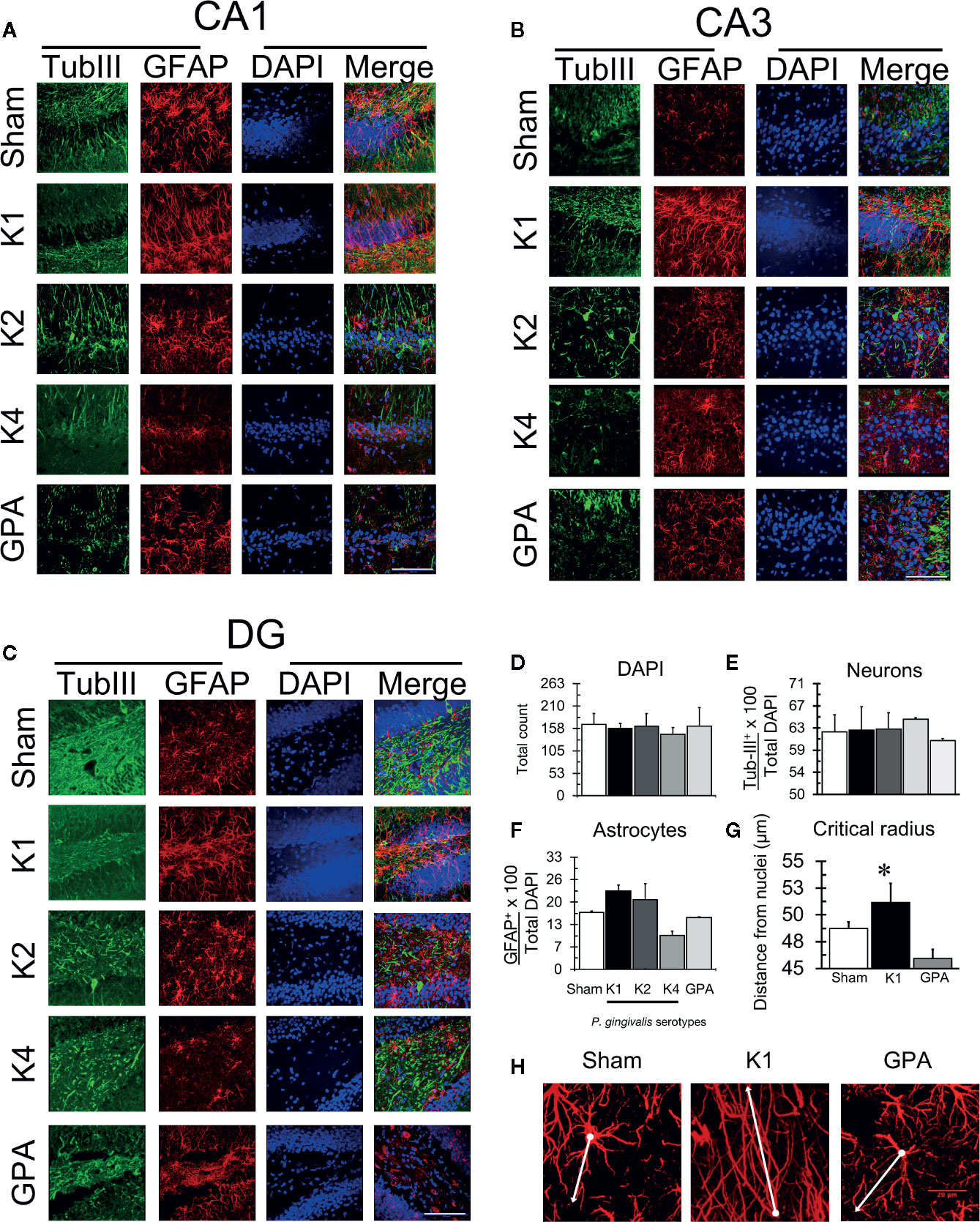
Figure 4 Astrogliosis detection by immunofluorescence. Representative confocal microscopy images of a 36-slices 3D projection of (A) CA1, (B) CA3, and (C) DG hippocampal regions from sham or P. gingivalis infected-rats, fixed after the sixth training session and labeled with glial fibrillary acidic protein (GFAP)- antibody (green), neuronal-specific cytoskeleton protein tubulin III (Tub-III)—antibody (red) and DAPI for the nucleus (blue). A merged image of the triple-staining is also shown. Scale Bar: 100 µm. (D) Total DAPI quantification. (E) Total neuron quantification. (F) Total astrocyte quantification. (G) Sholl analysis of the critical radius of astrocytes. (H) Representative confocal images show the determination of the critical radius of astrocytes from K1 P. gingivalis-infected rats versus sham or GPA infected rats. The white dot identifies the nuclei and the dash the most extensive prolongation. A white line joins dot and dash. Each image corresponds to a 36-slice 3D projection. Scale Bar: 20 µm. The primary monoclonal antibodies used were: anti-tubIII (1:300, ab18207), and anti-GFAP (1:300, ab10062). The secondary monoclonal antibodies were conjugated with Alexa Fluor®488 1:300 or Alexa Fluor®633 1:300. *p < 0.05.
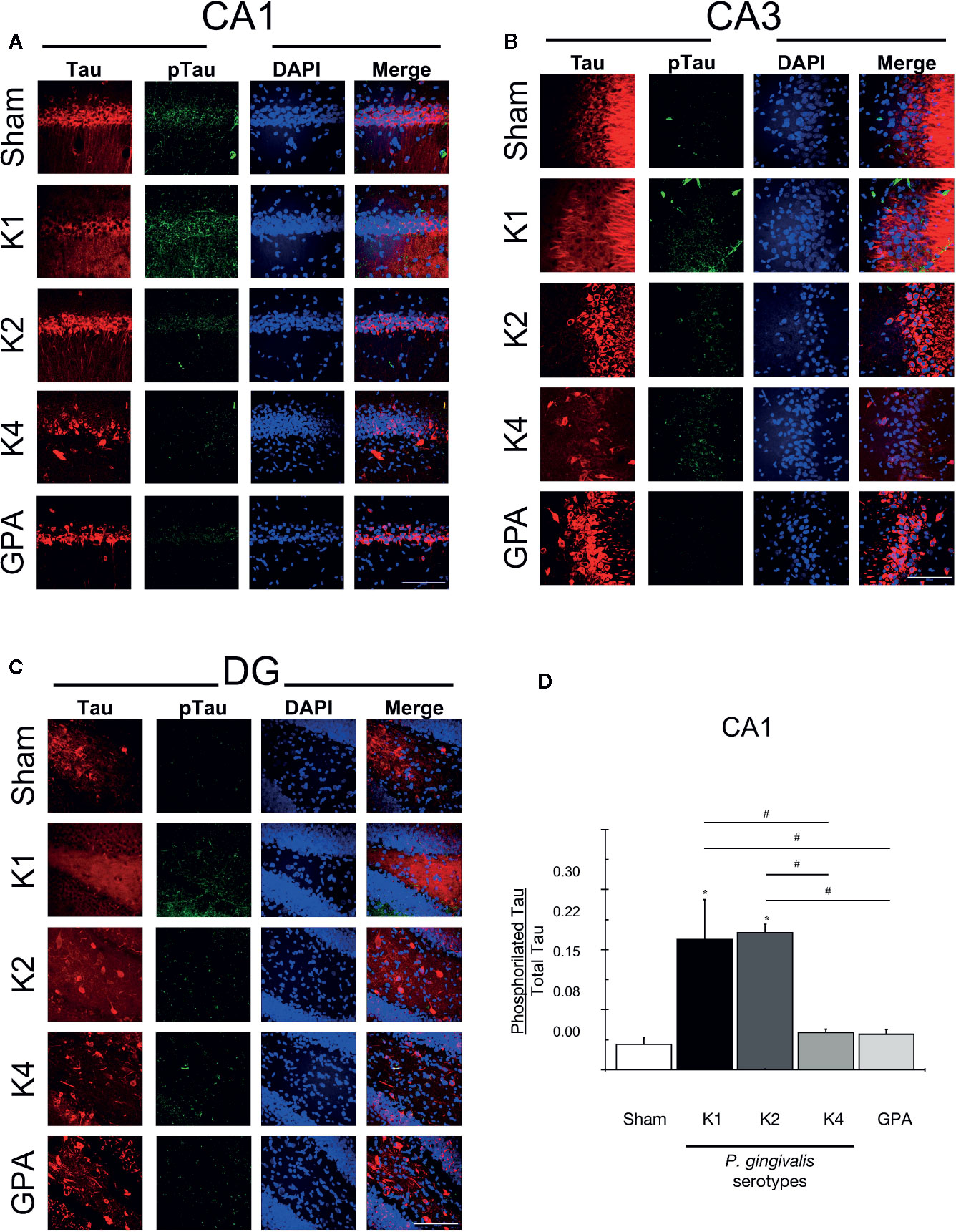
Figure 5 Tau and phosphorylated Tau detection by immunofluorescence. Representative confocal microscopy 36-sliceS 3D projection images of (A) CA1, (B) CA3, and (C) DG hippocampal regions from sham and P. gingivalis infected-rats, fixed after the sixth training session and labeled with anti-Tau (red), anti-phosphorylated (phospho Tau) antibody (green) and DAPI for the nucleus (blue). A merged image of the triple-staining is also shown. Scale Bar: 100 µm. (D) pTau/Tau ratio. An increase in pTau stain was observed in rats infected with encapsulated serotypes K1 or K2 compared with the other conditions in the CA1 region. *p < 0.05, compared to control group; #p < 0.05, compared to the group indicated. The primary monoclonal antibodies used were: anti-Tau (1:300, ab64193), and anti-phospho-Tau (1:300, Phospho S404, ab64193). The secondary monoclonal antibodies were conjugated with Alexa Fluor®488, 1:300, or Alexa Fluor®633 1:300.
Finally, to further search for AD-markers in the brains of orally-infected rats, we performed immunohistochemistry to stain Tau protein and its phosphorylated-form (pTau) and quantified the pTau/Tau ratio, which increase is considered a characteristic feature of the disease (Figures 5A-C). An increase in pTau staining was observed in the CA1 hippocampal region from rats infected with the serotypes K1 or K2 compared with sham rats and with K4 or GPA infected rats (Figure 5D).
Detection of P. gingivalis
Further, to investigate the potential spreading capacity of P. gingivalis, we set to detect the presence of the bacteria in serum, cerebrospinal fluid (CSF), and hippocampus. To this aim, we measured the bacterial load by quantifying the 16S rRNA subunit of P. gingivalis by qPCR (Figure 6A). We found that after 55 days of the first inoculation of 1 x 1010 CFU/ml in palatal mucosa, the different serotypes were detectable in serum, CSF and Hippocampus. In serum, we found 1 x 103 CFU/ml, in CSF 1 x 104 CFU/ml and in hippocampus, 1 x 105 CFU/ml for all bacterial serotypes, but no significant differences were found among the different conditions in hippocampus and serum. However, in CSF, K1 was increased compared to K4 serotype. We quantified also RgpA and Kgp gingipain genes in hippocampal tissue by qPCR (Figure 6B). We found that both genes were detected in hippocampus, but no differences were found among the different serotypes. Finally, to visualize the localization of the bacteria in hippocampal tissue, the gingipain RgpA component of P. gingivalis was immunodetected in the CA1 region of the hippocampi collected from rats exposed to all the experimental and control conditions (Figure 6C). Immunodetection of RgpA in the CA1 region indicated its co-localization with different cell types, including neuronal cells, for all the experimental conditions.
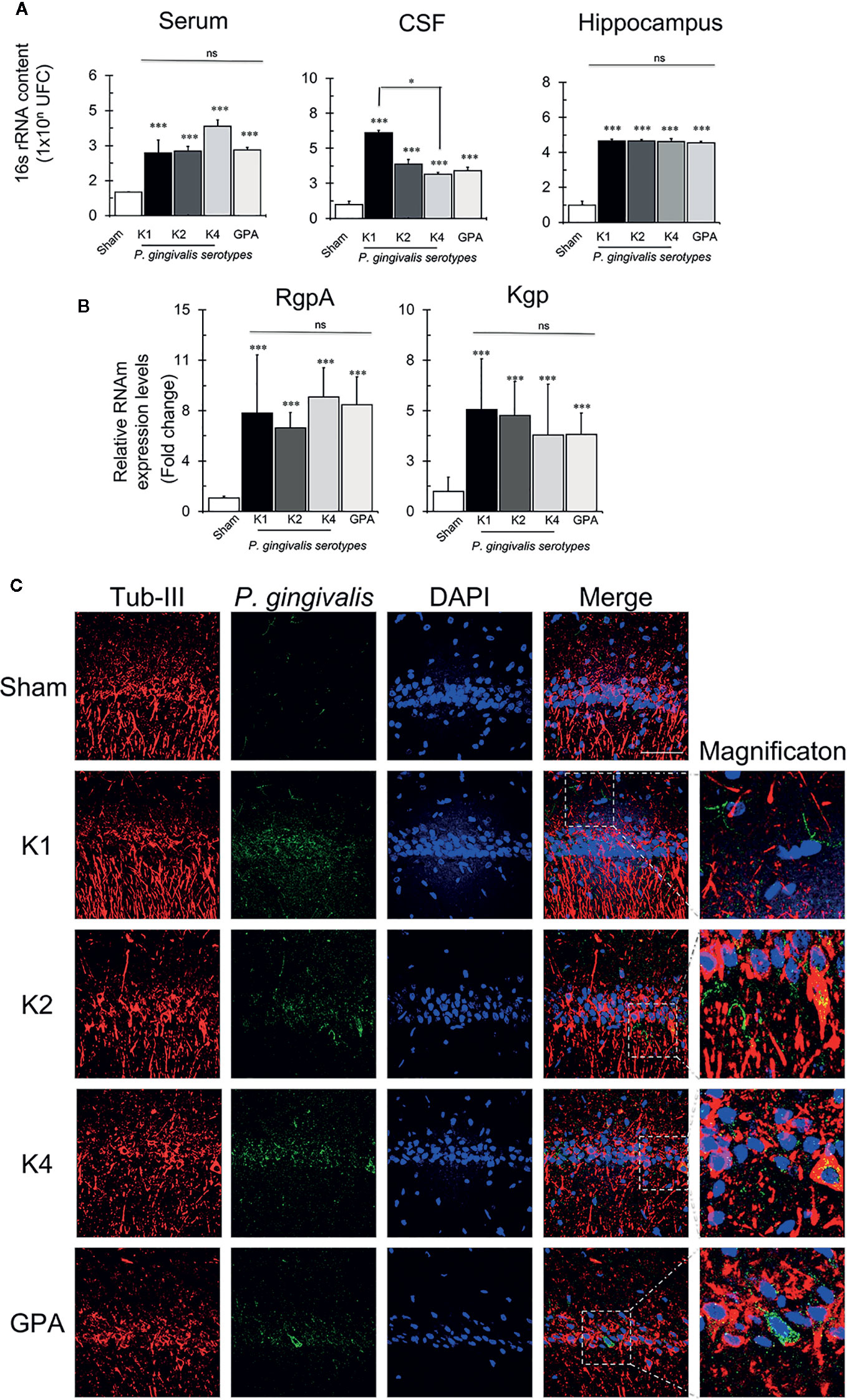
Figure 6 P. gingivalis detection by qPCR and immunofluorescence. (A) Absolute quantification of the 16S subunit of P. gingivalis in serum, CSF and hippocampus of sham- or rats infected with the serotype K1, K2, K4, or GPA. Results expresses as mean± SEM of four independent rats in each condition. CFU, colony-forming units, CSF, cerebrospinal fluid. *p < 0.05. (B) Detection of RgpA and Kgp genes of P. gingivalis in the hippocampus. Relative expression levels of RgpA and Kgp of P. gingivalis in hippocampi of sham rats infected with the serotype K1, K2, K4, or GPA ***p < 0.001. Results expresses as mean± SEM of four independent rats in each condition (C) Immunofluorescence detection of P. gingivalis. Immunofluorescence of the CA1 region of the hippocampus of Sprague-Dawley rats for the detection of gingipain R1 (RgpA). In red, neurons labeled with Tubulin III), in green P. gingivalis labeled with RgpA, in blue the nuclei (DAPI) and in merge. The superposition of the three channels. 2D images obtained from the 35-slice 3D projection taken from the two hemispheres from two independent rats in each condition. Scale Bar: 20 μm. ns, non significative.
Discussion
Different studies have reported that the presence of chronic inflammatory disease, such as periodontitis, would increase inflammatory mediators, anaerobic bacteria, or virulence factors in the systemic circulation. Chronic oral infection of mice with P. gingivalis results in a specific immune response, significant increases in oral bone resorption, aortic inflammation, and viable bacteria in oral epithelium and aorta, allowing constant exposure of periodontal bacteria to systemic circulation (45). According to the infectious origin of neuroinflammation, bacteria, virulence factors, and systemic pro-inflammatory mediators could spread to the central nervous system through the systemic circulation or brain nerves (27). Also, it is proposed that different microorganisms would use the Aβ peptide as a substrate or source of adhesion (46). Once in the brain, these agents can induce microglial pro-inflammatory cytokine secretion, characterized by significant increases in the levels of IL-1β, IL-6, TNF-α, and IFN-γ, which induce a reactive-astrocyte phenotype (29). Reactive astrocytes express increased levels of the three components necessary for Aβ production: APP, β-secretase, and γ-secretase, the enzymes that jointly cleave APP to produce Aβ. Reactive astrocytes secrete 7-fold more Aβ42 than neurons, and the Aβ42 is 60% truncated by non-enzymatic modifications, which induce its hydrophobicity and synaptotoxicity (47). This chronic inflammatory response in the brain would thus increase Aβ secretion and/or decrease its clearance causing its accumulation in the brain (48, 49). An increase in the concentration or accumulation of Aβ42 facilitates peptide aggregation in oligomeric and fibrillar forms linked to synaptotoxicity. As a consequence, the intracellular cytoskeleton-associated protein suffers abnormal Tau hyperphosphorylation, which in turn induces loss of neuronal function (50), and these sequential events would lead to neuronal death and spatial memory and learning deficits (26, 51).
Several studies have analyzed the relationship between periodontitis and AD and established that P. gingivalis cause cognitive impairment and induce brain pro-inflammatory cytokine release (21–25, 32, 34, 45, 52). From them, some studies used transgenic mice (21, 23, 25, 32, 45) and other studies used wild-type animal models (22–24, 32, 52). Only one of these studies reported a direct association between periodontitis-related alveolar bone resorption and deficits in learning and memory (21). For periodontitis induction, some studies used oral bacterial irrigation or oral gavage, (21, 23, 32, 52) two studies intraperitoneal injections of LPS, (22, 24), and two studies direct inoculation of LPS or lysed bacteria to the brain (25, 34). This technical variability to induce periodontitis makes it challenging to compare the reported results. Although all these studies demonstrate, to a greater or lesser extent, that cognitive deterioration is associated with periodontitis, it remains to be elucidated whether these effects are a consequence of some periodontitis molecular mediator, the direct activity of the bacteria, general sepsis, or alteration of the intestinal microbiota, based on direct interaction between periodontal and gut microbiota. Accordingly, to identify the role of a bacterium or virulence factor in periodontitis, the model we chose was the palatal inoculation, which has been previously used by our research group in mice and known by causing an increased Th1/Th17 immune response, osteoclast activity, and alveolar bone resorption (10, 44).
LPS constitutes an important virulence factor for P. gingivalis, being composed of lipid A, a central core of oligosaccharides and O-polysaccharide or O-antigen (O-Ag) (53). For lipid A, five isoforms have been described: 2 tetra-acylated and 3 penta-acylated, which induce different immune responses (54, 55). Although lipid A has potential immunogenicity, these effects have not been observed in humans, suggesting that human immune cells are not able to recognize the structural differences of lipid A, in the same way as animal immune cells do (55–57). One reason for this might reside in the fact that P. gingivalis-secreted gingipains degrade the CD14 receptor, the primary lipid A receptor in macrophages (58–61). In addition to lipid A, structural variability has also been described in the O-antigen, but its role in the pathogenicity remains unclear (62). All P. gingivalis strains possess LPS. Unfortunately, the studies that induced periodontal infection using LPS to evaluate the consequences in the brain, do not specify the strain of origin of the LPS used (22, 24, 25, 63). As above mentioned, it is relevant to know the origin of the LPS, since the variability of Lipid A of the LPS of P. gingivalis is important when analyzing the immunogenicity of this antigen, which is not necessarily associated with the pathogenicity of P. gingivalis. Lipid A can be tetra or penta-acylated. If it is tetra-acylated, it is immunogenic and is capable of inducing a CD14-mediated response in immune cells, but penta-acylated LPS does not induce an immune response (54, 55, 57, 62, 64). In general, clinical isolates of P. gingivalis from healthy subjects have a modified LPS with substitutions and lack O-antigen (65, 66) and also, their main receptor, the CD14 molecule, is a substrate of gingipains (58–60).
Besides LPS, gingipains and capsular polysaccharides are central in the mechanisms of pathogenesis of P. gingivalis infection. P. gingivalis is the only bacterium that produces gingipains, which are the endopeptidases responsible for 85% of its proteolytic activity (4, 5). They can be detected and isolated in aggregates with hemagglutinin, hemoglobin or other gingipains, or attached to the outer membrane of P. gingivalis (67, 68). In general terms, gingipains are essential for P. gingivalis growth, attachment and migration, and also for fimbria maturation (5, 69, 70). The gingipain knock-out is not compatible with bacterial life (71). Interestingly, gingipains degrade the receptors that recognize LPS in the host cells, and this degradation is considered a mechanism of evasion of the immune response (58–60). It was recently reported that gingipain inhibition reduces the bacterial load of an established P. gingivalis brain infection, decreases neuroinflammation, and rescues neurons in the hippocampus, revealing that this endopeptidase has a pivotal role in the mechanism of brain damage induced by P. gingivalis (34). Gingipains are likely to participate in the process of neuronal tissue invasion (38).
Without detriment to other virulence factors that P. gingivalis possesses, the capsular polysaccharides or K-antigens, which constitute the main macromolecule of its surface, are responsible for its serotypification, defining the taxonomic classification and contributing to the virulence (72, 73). The immunogenic role of P. gingivalis capsule has been previously demonstrated, and the structural variability of its polysaccharide component has been directly associated with its virulence potential (6–10, 74). Several encapsulated and non-encapsulated (K1-K7) serotypes of P. gingivalis have been described; among them, the encapsulated serotypes display more virulence in experimental infections (10, 72, 75, 76). Also, a P. gingivalis K1-isogenic non-encapsulated knock-out mutant ΔPG0116-PG0120 (GPA) has been developed, allowing to test the effects of the absence of capsule (14).
Given that there is differential microbiological susceptibility to periodontitis, which is determined by the presence of strains belonging to different serotypes in the oral cavity, it is reasonable to postulate that this susceptibility could also be observable in brain cells. Therefore, we decided to investigate the potential deleterious effects of each serotype of P. gingivalis in the brain. In the present study we investigated, for the first time, the effects of palatal injections of different serotypes of P. gingivalis on rat spatial memory and hippocampal histopathology. To this aim, animals received two injections separated by an interval of 1 week, into the palatal mucosa of 1010 CFU/ml of the strains W50 (serotype K1), HG184 (K2), or ATCC® 49417™ (K4) of P. gingivalis. Control animals were inoculated with the non-encapsulated W50 ΔPG0116-PG0120 mutant strain (GPA) of P. gingivalis. Making a parallel with the normal daily bacterial load from chronic periodontal disease, these values in the humans depends on the patient’s diagnosis: in healthy subjects the burden of P. gingivalis ranges from 6 x 102 to 8.6 x 104 CFU/ml. In patients with stage III periodontitis, it can vary from 4 x 106 to 9 x 105, or even 1 x 1012CFU/ml. However, as the severity of the disease increases, the burden of P. gingivalis decreases (77, 78).
Our study ratified the higher capacity of the serotypes K1 and K2 of P. gingivalis to induce alveolar bone resorption during experimental periodontitis. Besides, we report for the first time that periodontal infections induced with strains K1, K2, K4, or GPA of P. gingivalis differentially affected the rat brain. The presence of P. gingivalis was determined by quantifying the total bacterial load in serum and hippocampus. Although the identification of the bacteria in the serum has been previously reported (10), the presence of all P. gingivalis serotypes in the brain is the most intriguing and novel finding of our work. After 55 days, all P. gingivalis strains were detected in both samples, demonstrating that it is possible to detect P. gingivalis in the brain, regardless of the capsule and its virulence. The explanation to the fact that even the GPA strain entered the brain may reside on fimbria. Fimbria is the most extensive portion of the virulence factors of P. gingivalis, which allows it to adhere to any surface, its maturation is dependent on gingipains and is one of the migration mechanisms (5, 69, 79, 80). Indeed, fimbria allows P. gingivalis to invade the cells or tissues of the host and even the W50 (K1) strain, in the absence of the long fimbria, maintains its adherent capacity (81–83). In mutant strains of gingipains, P. gingivalis loses its ability to adhere and migrate, but in mutant capsule strains, LPS, gingipains and fimbria are not affected, evidenced in the maintenance of the adherence and migration capacity of these strains (10, 14, 84, 85).
Our results indicate that despite all the strains can achieve the brain, only the most virulent strains of P. gingivalis, K1 and K2, effectively induced pro-inflammatory cytokine production, astrogliosis, Aβ42 secretion, Tau hyperphosphorylation, and cognitive decline in young rats, after a short period of infection exposure. Since our experimental model consists of wild type rats injected with P. gingivalis and examined for the presence of brain Aβ amyloid 55 days after bacterial injection, we did not expect yet to detect the presence of accumulated Aβ in plaques. Nevertheless, our protocol did detect monomeric forms as well as soluble oligomeric conformations of Aβ, which are pivotal to trigger Tau hyperphosphorylation, and neuronal dysfunction, leading to cognitive deficits in AD pathology (39, 86–91). We show that periodontal infections with the encapsulated serotypes K1 or K2 of P. gingivalis induced neuroinflammation, astrogliosis, cognitive decline, and histopathological signs of AD in the hippocampus, as compared with the less virulent K4 strain and GPA. It worth noting that, instead, K4 and GPA strains promoted the increase in the secretion levels of immunomodulatory cytokines, such as IL-4 and IL-10, and that astrogliosis was considerably reduced in these rats. Unlike in several of the previous studies, (21, 22, 24, 34, 52) we carried out periodontal infections using inoculation of complete bacteria, with their full antigenic potential, which is considered the standard protocol that allows determining the local and systemic effect of a single bacterium (92). We show that, in contrast to the deleterious effects triggered by the serotypes K1 and K2 on the brain, periodontal infections induced with strains K4 or GPA provoked no significant changes in comparison to sham-controls. The present work is the first study to demonstrate the role of the encapsulated and non-encapsulated strains of P. gingivalis in the pathogenesis of AD and the differential capacity to induce neuroinflammation in the hippocampus, with the concomitant spatial memory loss. This study corroborates and adds to recent findings showing that P. gingivalis infection impairs cognition in wild-type or transgenic mice for amyloid precursor protein (APP), Cathepsin-β, or ApoE-/- (21, 22, 24, 25, 32, 34, 52). However, we further show that only the pathogenic serotypes of the bacteria selectively activate the inflammatory response in the brain.
Compared to previous reports utilizing the entire P. gingivalis, our study present novel findings, since all of these works used ATCC®33277™ or FDC 38 P. gingivalis strains, which are less virulent strains. Some of them have been unable to capture all AD hallmark features like Tau hyperphosphorylation as we did. One of these studies compared the effects of ATCC®33277™ strain in young and middle-age rats, and showed that P. gingivalis oral infection induced memory impairment and neuroinflammation only in the middle-age mice, suggesting that the effect of the bacteria is age-dependent. In contrast, using a short-term infection in young rats, we were able to find all the above-mentioned AD-features simultaneously in wild type rats, when they were orally infected with the most virulent, encapasulated K1 and K2 P. gingivalis serotypes. These results indicate that despite the fact that all serotypes were capable of invading the brain, their capsular K antigens were the fundamental determinants of the host response and vulnerability. Of note, our results may also shed light on previous studies in postmortem brains, in which the presence of the P. gingivalis 16S rRNA gene was detected not only in the AD brains but also in five of the six nondemented control brains examined. The PCR analysis of P. gingivalis in the postmortem brains and CSF reported by these authors did not differentiate between P. gingivalis strains (34). Therefore, the possibility that some strains might be more virulent than others in causing Alzheimer´s disease and that healthy brains exhibit less virulent strains should be evaluated in the future.
One of the limitations of our study is that our experimental model, which can be considered a short-term inflammatory condition, is rather like an infectious state in humans, equivalent to a condition such as sepsis. The provision of anti-inflammatory medication would be really useful to demonstrate whether the cognitive function is a short-term effect of the pro-inflammatory state or a long-term degenerative change. Also, considering that periodontitis is a multibacterial disease, it would be interesting to contemplate the virulence effects of the other periodontal and gastrointestinal bacteria to which the human body is exposed to on a daily basis. In this regard, Aggregatibacter actinomycetemcomitans is recognized to be highly virulent; studies are in progress in our research group to determine whether this bacterium can also generate neuroinflammation in vivo, since we previously reported that serotype b of Aggregatibacter actinomycetemcomitans triggers pro-inflammatory responses and amyloid beta secretion in hippocampal cell cultures (37).
Conclusions
Periodontitis is a major risk factor for Alzheimer´s disease, and P. gingivalis is a keystone pathogen linking these diseases. Although different P. gingivalis serotypes can access the brain, its particular capsular types play a central role in the chronic inflammatory assault and cognitive impairment induced by short-term oral infection. The more virulent encapsulated serotypes are likely more prompt to lead to AD-like-pathology, probably speeding up the pathogenic process compared to less virulent serotypes.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The experimental protocols were approved by the Bioethics Committees on Animal Research of the Faculty of Medicine (protocol #CBA 0755 FMUCH) and the Faculty of Dentistry (protocol #17085-ODO-UCH), Universidad de Chile.
Author Contributions
JD-Z and AP-L substantially contributed to conception and design of the experiments, acquisition, analysis, and interpretation of data and they drafted and critically revised the manuscript. JM, JV and RV contributed to conception and design of the experiments, acquisition of data and they also critically revised the manuscript. SM-R, MJ-U, FV-O, CM-M, and GM contributed to acquisition of data and critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was financed by grants (FONDECYT 1150736, FONDECYT 1181780) from the Chilean Governmental Agencia Nacional de Investigación y Desarrollo (ANID), by ICM P-09-015, by a grant (FIOUCh 17/019) from the Faculty of Dentistry and by the Regional Development Program of the International Association for Dental Research (RDP-IADR).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Darna Venegas and Andrea Birkner for technical assistance, Dr. Cecilia Hidalgo, for her critical contribution to this article, and the Plataforma Experimental Bio-CT, Faculty of Dentistry from Universidad de Chile (FONDEQUIP EQM150010) for performing the micro-CT analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.588036/full#supplementary-material
References
1. Hajishengallis G. Immuno-microbial pathogenesis of periodontitis: Keystones, pathobionts, and the host response. Trends Immunol (2014) 35:3–11. doi: 10.1016/j.it.2013.09.001
2. Hajishengallis G. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol (2015) 15:30–44. doi: 10.1038/nri3785
3. Meyle J, Chapple I. Molecular aspects of the pathogenesis of periodontitis. J Periodontol (2015) 69:7–17. doi: 10.1111/prd.12104
4. Potempa J, Pike R, Travis J. The multiple forms of trypsin-like activity present in various strains of Porphyromonas gingivalis are due to the presence of either Arg-gingipain or Lys-gingipain. Infect Immun (1995) 63:1176–82. doi: 10.1128/IAI.63.4.1176-1182.1995
5. Li N, Collyer CA. Gingipains from Porphyromonas gingivalis - Complex domain structures confer diverse functions. Eur J Microbiol Immunol (2011) 1:41–58. doi: 10.1556/EuJMI.1.2011.1.7
6. d’Empaire G, Baer MT, Gibson F. The K1 serotype capsular polysaccharide of Porphyromonas gingivalis elicits chemokine production from murine macrophages that facilitates cell migration. Infect Immun (2006) 74:6236–43. doi: 10.1128/IAI.00519-06
7. Vernal R, León R, Silva A, van Winkelhoff AJ, García-Sanz J, Sanz M. Differential cytokine expression by human dendritic cells in response to different Porphyromonas gingivalis capsular serotypes. J Clin Periodontol (2009) 36:823–9. doi: 10.1111/j.1600-051X.2009.01462.x
8. Vernal R, Díaz-Guerra E, Silva A, Sanz M, García-Sanz J. Distinct human T-lymphocyte responses triggered by Porphyromonas gingivalis capsular serotypes. J Clin Periodontol (2014) 41:19–30. doi: 10.1111/jcpe.12176
9. Vernal R, Díaz-Zúñiga J, Melgar-Rodríguez S, Pujol M, Díaz-Guerra E, Silva A, et al. Activation of RANKL-induced osteoclasts and memory T lymphocytes by Porphyromonas gingivalis is serotype dependant. J Clin Periodontol (2014) 41:451–9. doi: 10.1111/jcpe.12236
10. Monasterio G, Fernández B, Castillo F, Rojas C, Cafferata EA, Rojas L, et al. Capsular-defective Porphyromonas gingivalis mutant strains induce less alveolar bone resorption the W50 wild-type strain due to a decreased Th1/Th17 immune response and less osteoclast activity. J Periodontol (2018) 90:522–34. doi: 10.1002/JPER.18-0079
11. Califano JV, Schifferle RE, Gunsolley JC, Best AM, Schenkein HA, Tew JG. Antibody reactive with Porphyromonas gingivalis serotypes K1-6 in adult and generalized early-onset periodontitis. J Periodontol (1999) 70:730–5. doi: 10.1902/jop.1999.70.7.730
12. Sims TJ, Schifferle RE, Ali RW, Skaug N, Page RC. Immunoglobulin G response of periodontitis patients to Porphyromonas gingivalis capsular carbohydrate and lipopolysaccharide antigens. Oral Microbiol Immunol (2001) 16:193–201. doi: 10.1034/j.1399-302X.2001.160401.x
13. Laine ML, van Winkelhoff AJ. Virulence of six capsular serotypes of Porphyromonas gingivalis in a mouse model. Oral Microbiol Immunol (1998) 13:322–5. doi: 10.1111/j.1399-302X.1998.tb00714.x
14. Aduse-Opoku J, Slaney JM, Hashim A, Gallagher A, Gallagher RP, Rangarajan M, et al. Identification and characterization of the capsular polysaccharide (K-antigen) locus ofPophyromonas gingivalis. Infect Immun (2006) 74:449–60. doi: 10.1128/IAI.74.1.449-460.2006
15. Singh A, Wyant T, Anaya-Bergman C, Aduse-Opoku J, Brunner J, Laine ML, et al. The capsule of Porphyromonas gingivalis leads to a reduction in the host inflammatory response, evasion of phagocytosis, and increase in virulence. Infect Immun (2011) 79:4533–42. doi: 10.1128/IAI.05016-11
16. Demmer RT, Papapanou PN, Jacobs DR Jr, Dezvarieux M. Evaluating clinical periodontal measures as surrogates for bacterial exposure: the Oral Infections and Vascular Disease Epidemiology Study (INVEST). BMC Med Res Method (2010) 10:2. doi: 10.1186/1471-2288-10-2
17. Nakajima M, Arimatsu K, Kato T, Matsuda Y, Minagawa T, Takahashi N, et al. Oral Administration of P. gingivalis Induces Dysbiosis of Gut Microbiota and Impaired Barrier Function Leading to Dissemination of Enterobacteria to the Liver. PloS One (2015) 10:e0134234. doi: 10.1371/journal.pone.0134234
18. Bale BF, Doneen AL, Vigerust DJ. High-risk periodontal pathogens contribute to the pathogenesis of atherosclerosis. Postgraduate Med J (2017) 93:215–20. doi: 10.1136/postgradmedj-2016-134279
19. Kramer CD, Simas AM, He X, Ingalls RR, Weinberg EO, Genco CA. Distinct roles for dietary lipids and Porphyromonas gingivalis infection on atherosclerosis progression and the gut microbiota. Anaerobe (2017) 45:19–30. doi: 10.1016/j.anaerobe.2017.04.011
20. Udagawa S, Katagiri S, Maekawa S, Takeuchi Y, Komazaki R, Ohtsu A, et al. Effect of Porphyromonas gingivalis infection in the placenta and umbilical cord in pregnant mice with low birth weight. Acta Odontol Scand (2018) 76:433–41. doi: 10.1080/00016357.2018.1426876
21. Ishida N, Ishihara Y, Ishida K, Tada H, Funaki-Kato Y, Hagiwara M, et al. Periodontitis induced by bacterial infection exacerbates features of Alzheimer’s disease in transgenic mice. NPJ Aging Mech Dis (2017) 3:15. doi: 10.1038/s41514-017-0015-x
22. Wu Z, Ni J, Liu Y, Teeling JL, Takayama F, Collcutt A, et al. Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav Immun (2017) 65:350–61. doi: 10.1016/j.bbi.2017.06.002
23. Ding Y, Ren J, Yu H, Yu W, Zhou Y. Porphyromonas gingivalis, a periodontitis causing bacterium, induces memory impairment and age-dependent neuroinflammation in mice. Immun Ageing (2018) 15:6. doi: 10.1186/s12979-017-0110-7
24. Zhang J, Yu C, Zhang X, Chen H, Dong J, Lu W, et al. Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signaling pathway in C57BL/6 mice. J Neuroinflamm (2018) 15:37–52. doi: 10.1186/s12974-017-1052-x
25. Hayashi K, Hasegawa Y, Takemoto Y, Cao C, Takeya H, Komohara Y, et al. Continous intracerebroventricular injection of Porphyromonas gingivalis lipopolysaccharide induces systemic organ dysfunction in a mouse model of Alzheimer’s disease. Exp Gerontol (2019) 120:1–5. doi: 10.1016/j.exger.2019.02.007
26. Paula-Lima AC, Brito-Moreira J, Ferreira ST. Deregulation of excitatory neurotransmission underlying synapse failure in Alzheimer’s disease. J Neurochem (2013) 126:191–202. doi: 10.1111/jnc.12304
27. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol (2015) 14:388–405. doi: 10.1016/S1474-4422(15)70016-5
28. Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol (2005) 76:77–98. doi: 10.1016/j.pneurobio.2005.06.004
29. Frost GR, Li YM. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol (2017) 7:170228. doi: 10.1098/rsob.170228
30. Tse KH, Herrup K. Re-imagining Alzheimer’s disease - the diminishing importance of amyloid and a glimpse of what lies ahead. J Neurochem (2017) 143:432–44. doi: 10.1111/jnc.14079
31. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med (2016) 8:595–608. doi: 10.15252/emmm.201606210
32. Poole S, Singhrao SK, Chukkapalli S, Rivera M, Velsko I, Kesavalu L, et al. Active invasion of Porphyromonas gingivalis and infection-induced complement activation in ApoE-/- mice brains. J Alzheimers Dis (2015) 43:67–80. doi: 10.3233/JAD-140315
33. Carvajal P, Gómez M, Gomes S, Costa R, Toledo A, Solanes F, et al. Prevalence, severity, and risk indicators of gingival inflammation in a multi-center study on South American adults: a cross sectional study. J Appl Oral Sci (2016) 24:524–34. doi: 10.1590/1678-775720160178
34. Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromons gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv (2019) 5:eaau3333. doi: 10.1126/sciadv.aau3333
35. Rodríguez AM, Delpino MV, Miraglia MC, Costa Franco MM, Barrionuevo P, Dennis VA, et al. Brucella abortus-activated microglia induce neuronal death through primary phagocytosis. Glia (2017) 65:1137–51. doi: 10.1002/glia.23149
36. Zhang Y, Feng S, Nie K, Li Y, Gao Y, Gan R, et al. TREM2 modulates microglia phenotypes in the neuroinflammation of Parkinson’s disease. Biochem Biophys Res Commun (2018) 499:797–802. doi: 10.1016/j.bbrc.2018.03.226
37. Díaz-Zúñiga J, Muñoz Y, Melgar-Rodríguez S, More J, Bruna B, Lobos P, et al. Serotype b ofAggregatibacter actinomycetemcomitanstriggers pro-inflammatory responses and amyloid beta secretion in hippocampal cells: a novel link between periodontitis and Alzheimer’s disease? J Oral Microbiol (2019) 11:1586423. doi: 10.1080/20002297.2019.1586423
38. Haditsch U, Roth T, Rodriguez L, Hancock S, Cecere T, Nguyen M, et al. Alzhiemer’s Disease-like neurodegeneration in Porphyromonas gingivalis infected neurons with persistent expression of active gingipains. J Alzheimers Dis (2020) 75:1361–76. doi: 10.3233/JAD-200393
39. More J, Galusso N, Veloso P, Montecinos L, Finkelstein JP, Sanchez G, et al. N-Acetylcysteine Prevents the Spatial Memory Deficits and the Redox-Dependent RyR2 Decrease Displayed by an Alzheimer’s Disease Rat Model. Front Aging Neurosci (2018) 10:399. doi: 10.3389/fnagi.2018.00399
40. More JY, Bruna BA, Lobos PE, Galaz JL, Figueroa PL, Namias S, et al. Calcium Release Mediated by Redox-Sensitive RyR2 Channels Has a Central Role in Hippocampal Structural Plasticity and Spatial Memory. Antioxid Redox Signaling (2018) 29:1125–46. doi: 10.1089/ars.2017.7277
41. Concha-Miranda M, More J, Grispun N, Sánchez C, Paula-Lima AC, Valdés JL. Differential navigational strategies during spatial learning in a new modified version of the Oasis Maze. Behav Brain Res (2020) 385:112555. doi: 10.1016/j.bbr.2020.112555
42. Morris R. Deveptoments of a Water-Maze procedure for studying spatial learning in the rat. J Neurosci Methods (1984) 11:47–60. doi: 10.1016/0165-0270(84)90007-4
43. Mahat MYA, Ahamed FA, Chandrasekaran S, Rajagopal S, Narayanan S, Surendran N. An improved method of transcutaneous cisterna magna puncture for cerebrospinal fluid sampling in rats. J Neurosci Methods (2012) 211:272–9. doi: 10.1016/j.jneumeth.2012.09.013
44. Monasterio G, Castillo F, Ibarra JP, Guevara J, Rojas L, Alvarez C, et al. Alveolar bone resorption and Th1/Th17-associated immune response triggered during Aggregatibacter actinomycetemcomitans-induced experimental periodontitis are serotype-dependent. J Periodontol (2018) 89:1249–61. doi: 10.1002/JPER.17-0563
45. Velsko IM, Chukkapalli SS, Rivera-Kweh MF, Zheng D, Aukhil I, Lucas AR, et al. Periodontal pathogens invade gingiva and aortic adventitia and elicit inflammasome activation in avB6 integrin-deficient mice. Infect Immun (2015) 83:4582–93. doi: 10.1128/IAI.01077-15
46. Ezzak K, Pernemalm M, Pålsson S, Roberts TC, Järver P, Dondalska A, et al. The viral protein corona directs viral pathogenesis and amyloid aggregation. Nat Commun (2019) 10:2331. doi: 10.1038/s41467-019-10192-2
47. Oberstein TJ, Spitzer P, Klafki HW, Linning P, Neff F, NKnölker HJ, et al. Astrocytes and microglia but not neurons preferentially generate N-terminally truncated AB peptides. Neurobiol Dis (2015) 73:24–35. doi: 10.1016/j.nbd.2014.08.031
48. Fulop T, Witkowski JM, Bourgade K, Khalil A, Zerif E, Larbi A, et al. Can an infection hypothesis explain the Beta amyloid hypothesis of Alzheimer’s disease? Front Aging Neurosci (2018) 10:224. doi: 10.3389/fnagi.2018.00224
49. Block J. Alzheimer’s disease might depend on enabling pathogens which do not necessarily cross the blood-brain barrier. Med Hypotheses (2019) 125:129–36. doi: 10.1016/j.mehy.2019.02.044
50. Forny-Germano L, Silva NML, Batista AF, Brito-Moreira J, Gralle M, Boehnke SE, et al. Alzheimer’s disease-like pathology induced by amyloid-B oligomers in nonhuman primates. J Neurosci (2014) 34:13629–43. doi: 10.1523/JNEUROSCI.1353-14.2014
51. Jones DT, Graff-Radford J, Lowe VJ, Wiste HJ, Gunter JL, Senjem ML, et al. Tau, amyloid, and cascading network failure across the Alzheimer’s disease spectrum. Cortex (2017) 97:143–59. doi: 10.1016/j.cortex.2017.09.018
52. Ilievski V, Zuchowska PK, Green SJ, Toth PT, Ragozzino ME, Le K, et al. Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PloS One (2018) 13:e0204941. doi: 10.1371/journal.pone.0204941
53. Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem (2002) 71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414
54. Darveau RP, Pham TT, Lemley K, Reife RA, Bainbridge BW, Coats SR, et al. Pophyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both toll-like receptors 2 and 4. Infect Immun (2004) 72:5051–1. doi: 10.1128/IAI.72.9.5041-5051.2004
55. Reife RA, Coats SR, Al-Qutub M, Dixon DR, Braham PA, Billharz RJ, et al. Porphyromonas gingivalis lipopolysaccharide lipid A heterogeneity: differential activities of tetra- and penta-acylated lipid A structures on E-selectin expression and TLR4 recognition. Cell Microbiol (2006) 8:857–68. doi: 10.1111/j.1462-5822.2005.00672.x
56. Blufstein A, Behm C, Nguyen PQ, Rausch-Fan X, Andrukhov O. Human periodontal ligament cells exhibit no endotoxin tolerance upon stimulation with Porphyromonas gingivalis polysaccharide. J Periodontal Res (2018) 53:589–97. doi: 10.1111/jre.12549
57. Olsen I, Singhrao SK. Importance of heterogeneity in Porphyromonas gingivalis lipopolysaccharide lipid A in tissue specific inflammatory signalling. J Oral Microbiol (2018) 10:1440128. doi: 10.1080/20002297.2018.1440128
58. Sugawara S, Nemoto E, Tada H, Miyake K, Imamura T, Takada H. Proteolysis of human monocyte CD14 by cysteine proteinases (gingipains) from Porphyromonas gingivalis leading to lipopolysaccharide hyporesponsiveness. J Immunol (2000) 165:411–8. doi: 10.4049/jimmunol.165.1.411
59. Tada H, Sugawara S, Nemoto E, Takahashi N, Imamura T, Potempa J, et al. Proteolysis of CD14 on human gingival fibroblasts by arginine-specific cysteine proteinases from Porphyromonas gingivalis leading to down-regulation of lipopolysaccharide-induced interleukin-8 production. Infect Immun (2002) 70:3304–7. doi: 10.1128/IAI.70.6.3304-3307.2002
60. Wang PL, Ohura K. Porphyromonas gingivalis lipopolysaccharide signaling in gingival fibroblasts-CD14 and Toll-like receptors. Crit Rev Oral Biol Med (2002) 13:132–42. doi: 10.1177/154411130201300204
61. Bao K, Belibasakis GN, Thurnheer T, Aduse-Opoku J, Curtis MA. Role of Porphyromonas gingivalis gingipains in multi-species biofilm formation. BMC Microbiol (2014) 14:258–66. doi: 10.1186/s12866-014-0258-7
62. Rangarajan M, Aduse-Opoku J, Hashim A, McPhail G, Luklinska Z, Haurat MF, et al. LptO (PG0027) is required for lipid A 1-phosphatase activity in Porphyromonas gingivalis W50. J Bacteriol (2017) 199:e00751–16. doi: 10.1128/JB.00751-16
63. Hu Y, Li H, Zhang J, Zhang X, Xia X, Qiu C, et al. Periodontitis induced by P. gingivalis-LPS is associated with neuroinflammation and learning and memory impairment in Sprague-Dawley Rats. Front Neurosci (2020) 14:1–15. doi: 10.3389/fnins.2020.00658
64. Jain S, Chang AM, Singh M, McLean AS, Coats SR, Kramer RW, et al. Identification of PGN_1123 as the gene encoding Lipid A deacylase, an enzyme required for Toll-like receptor 4 evasion, in Prophyromonas gingivalis. J Bacteriol (2019) 201:e00683–18. doi: 10.1128/JB.00683-18
65. Martinic M, Hoare A, Contreras I, Alvarez SA. Contribution of the lipopolysaccharide to resistance of Shigella flexneri 2a to extreme acidity. PloS One (2011) 6:e25557. doi: 10.1371/journal.pone.0025557
66. Díaz L, Hoare A, Soto C, Bugueño I, Silva N, Dutzan N, et al. Changes in lipopolysaccharide profile of Porphyromonas gingivalis clinical isolates correlate with changes in colony morphology and polymyxin B resistance. Anaerobe (2015) 33:25–32. doi: 10.1016/j.anaerobe.2015.01.009
67. Pike R, McGraw J, Potempa J, Travis J. Lysine- and argininespecific proteinases from Porphyromonas gingivalis. J Biol Chem (1994) 270:1007–10.
68. Slakeski NS, Cleal M, Bhogal PS, Reynolds EC. Characterization of a Porphyromonas gingivalis gene prtK that encodes a lysinespecific cysteine proteinase and three sequence-related adhesins. Oral Microbiol Immunol (1999) 14:92–7. doi: 10.1034/j.1399-302X.1999.140203.x
69. Kadowaki T, Nakayama F, Yoshimura F, Okamoto NA, Yamamoto K. Arg-gingipain acts as a major processing enzyme for various cell surface proteins in Porphyromonas gingivalis. J Biol Chem (1998) 273:29072–6. doi: 10.1074/jbc.273.44.29072
70. Yasuhara R, Miyamoto Y, Takami M, Imamura T, Potempa J, Yoshimura F, et al. Lysine-specific gingipain promotes lipopolysaccharide- and active-vitamin D3-induced osteoclast differentiation by degrading osteoprotegerin. Biochem J (2009) 419:159–66. doi: 10.1042/BJ20081469
71. Shi Y, Ratnayake DB, Okamoto K, Abe N, Yamamoto K, Nakayama K. Genetic analyses of proteolisis, hemoglobin binding, and hemagglutination of Prophyromonas gingivalis. J Biol Chem (1999) 274:17955–60. doi: 10.1074/jbc.274.25.17955
72. van Winkelhoff AJ, Applemelk BJ, Kippuw N, de Graaff J. K-antigens in Porphyromonas gingivalis are associated with virulence. Oral Microbiol Immunol (1993) 8:259–65. doi: 10.1111/j.1399-302X.1993.tb00571.x
73. Whitfield C, Roberts IS. Structure, assembly and regulation of expression of capsules in Escherichia coli. Mol Microbiol (1999) 31:1307–19. doi: 10.1046/j.1365-2958.1999.01276.x
74. Vernal R, León R, Herrera D, García-Sanz J, Silva A, Sanz M. Variability in the response of human dendritic cells stimulated with Porphyromonas gingivalis or Aggregatibacter actinomycetemcomitans. J Periodontal Res (2008) 43:689–97. doi: 10.1111/j.1600-0765.2007.01073.x
75. Laine ML, Applemelk BJ, van Winkelhoff AJ. Novel polysaccharide capsular serotypes in Porphyromonas gingivalis. J Periodontal Res (1996) 31:278–84. doi: 10.1111/j.1600-0765.1996.tb00494.x
76. Laine ML, Applemelk BJ, van Winkelhoff AJ. Prevalence and distribution of six capsular serotypes of Porphyromonas gingivalis in periodontitis patients. J Dental Res (1997) 76:1840–4. doi: 10.1177/00220345970760120601
77. Lau L, Sanz M, Herrera D, Morillo JM, Martin C, Silva A. Quantitative real-time polymerase chain reaction versus culture: a comparison between two methods for the detection and quantification of Actinobacillus actinomycetemcomitans, Porphyromonas gingivalis and Tannerella forsythensis in subgingival plaque samples. J Clinicasl Periodontol (2004) 31:1061–9. doi: 10.1111/j.1600-051X.2004.00616.x
78. Padmalatha GV, Bavle RM, Satyakiran GV, Paremala K, Sudhakara M, Makarla S. Quantification of Porphyromonas gingivalis in chronic periodontitis patients associated with diabetes mellitus using real-time polymeraase chain reaction. J Oral Maxillofacial Pathol (2016) 20:413–8. doi: 10.4103/0973-029X.190933
79. Enersen M, Nakano K, Amano A. Porphyromonas gingivalis fimbriae. J Oral Microbiol (2013) 5. doi: 10.3402/jom.v5i0.20265
80. Hall M, Hasegawa Y, Yoshimura F, Persson K. Structural and functional characterization of shaft, anchor, and tip proteins of the Mfa1 fimbria from the periodontal pathogen Porphyromonas gingivalis. Sci Rep (2018) 8:1793. doi: 10.1038/s41598-018-20067-z
81. Potempa J, Mikolajczyk-Pawlinska J, Brassell D, Nelson S, Thogersen IB, Enghild JJ, et al. Comparative properties of two cysteine proteinases )gingipains R), the products of two related but individual genes of Porphyromonas. J Biol Chem (1998) 273:21648–57. doi: 10.1074/jbc.273.34.21648
82. Umemoto T, Hamada N. Characterization of biologically active cell surface components of a periodontal pathogen. The roles of major and minor fimbriae of Porphyromonas gingivalis. J Periodontol (2003) 74:119–22. doi: 10.1902/jop.2003.74.1.119
83. Bostanci N, Belibasakis GN. Porphyromonas gingivalis: an invasive and evasive opportunistic oral pathogen. FEMS Microbiol Letter (2012) 333:1–9. doi: 10.1111/j.1574-6968.2012.02579.x
84. Kato T, Kawai S, Nakano K, Inaba H, Kubinowa M, Nakagawa I. Virulence of Porphyromonas gingivalis is altered by substitution of fimbria gene with different genotype. Cell Microbiol (2007) 9:753–65. doi: 10.1111/j.1462-5822.2006.00825.x
85. Kato T, Tsuda T, Omori H, Kato T, Yoshimori T, Amano A. Maturation of fimbria precursor protein by exogenous gingipains in Porphyromonas gingivalis gingipain-null mutant. FEMS Microbiol Letter (2007) 273:96–102. doi: 10.1111/j.1574-6968.2007.00779.x
86. Brito-Moreira J, Paula-Lima AC, Bomfim TR, Oliveira FB, Sepúlveda FJ, De Mello FG, et al. AB oligomers induce glutamate release from hipocampal neurons. Curr Alzheimer Res (2011) 8:552–62. doi: 10.2174/156720511796391917
87. Paula-Lima AC, Adasme T, San Martín C, Sebollela A, Hetz C, Carrasco MA, et al. Amyloid β-peptide oligomers stimulate RyR-mediated Ca2+ release inducing mitochondrial fragmentation in hippocampal neurons and prevent RyR-mediated dendritic spine remodeling produced by BDNF. Antioxid Redox Signaling (2011) 14:1209–23. doi: 10.1089/ars.2010.3287
88. San Martín CD, Adasme T, Hidalgo C, Paula-Lima AC. The antioxidant N-acetylcysteine prevents the mitochondrial fragmentation induced by soluble amyloid-B peptide oligomers. Neurodegenerative Dis (2012) 10:34–7. doi: 10.1159/000334901
89. Lobos P, Bruna B, Cordova A, Barattini P, Galaz JL, Adasme T, et al. Astaxathin protects primary hipocampal neurons against noxious effects of AB-oligomers. Neural Plasticity (2016) 2016:3456783. doi: 10.1155/2016/3456783
90. San Martín CD, Veloso P, Adasme T, Lobos P, Bruna B, Galaz J, et al. RyR2-Mediated Ca2+ Release and mitochondrial ros generation partake in the synaptic dysfunction caused by amyloid β peptide oligomers. Front Mol Neurosci (2017) 10:115. doi: 10.3389/fnmol.2017.00115
91. Muñoz Y, Paula-Lima AC, Núñez M-T. Reactive oxygen species released from astrocytes treated with amyloid beta oligomers elicit neuronal calcium signals that decrease phospho-Ser727-STAT3 nuclear content. Free Radical Biol Med (2018) 117:132–44. doi: 10.1016/j.freeradbiomed.2018.01.006
92. Trombone AP, Ferreira SBJ, Raimundo FM, de Moura KC, Avila-Campos MJ, Silva JS, et al. Experimental periodontitis in mice selected for maximal or minimal inflammatory reactions: increased inflammatory immune responsiveness drives increased alveolar bone loss without enhancing the control of periodontal infection. J Periodontal Res (2009) 44:443–51. doi: 10.1111/j.1600-0765.2008.01133.x
Keywords: periodontitis, Alzheimer’s disease, memory, glia, neuroinflammation
Citation: Díaz-Zúñiga J, More J, Melgar-Rodríguez S, Jiménez-Unión M, Villalobos-Orchard F, Muñoz-Manríquez C, Monasterio G, Valdés JL, Vernal R and Paula-Lima A (2020) Alzheimer’s Disease-Like Pathology Triggered by Porphyromonas gingivalis in Wild Type Rats Is Serotype Dependent. Front. Immunol. 11:588036. doi: 10.3389/fimmu.2020.588036
Received: 28 July 2020; Accepted: 14 October 2020;
Published: 09 November 2020.
Edited by:
Teun J. De Vries, VU University Amsterdam, NetherlandsReviewed by:
Jan Potempa, University of Louisville, United StatesStJohn Crean, University of Central Lancashire, United Kingdom
Copyright © 2020 Díaz-Zúñiga, More, Melgar-Rodríguez, Jiménez-Unión, Villalobos-Orchard, Muñoz-Manríquez, Monasterio, Valdés, Vernal and Paula-Lima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Paula-Lima, YWNwYXVsYWxpbWFAdS51Y2hpbGUuY2w=