 Alba Cortés1,2
Alba Cortés1,2 Simon Clare3
Simon Clare3 Alice Costain4
Alice Costain4 Alexandre Almeida3,5Catherine McCarthy3Katherine Harcourt3Cordelia Brandt3
Alexandre Almeida3,5Catherine McCarthy3Katherine Harcourt3Cordelia Brandt3 Charlotte Tolley3
Charlotte Tolley3 James Rooney1Matthew Berriman3
James Rooney1Matthew Berriman3 Trevor Lawley3
Trevor Lawley3 Andrew S. MacDonald4
Andrew S. MacDonald4 Gabriel Rinaldi3*
Gabriel Rinaldi3* Cinzia Cantacessi1*
Cinzia Cantacessi1*- 1Department of Veterinary Medicine, University of Cambridge, Cambridge, United Kingdom
- 2Departament de Farmàcia i Tecnologia Farmacèutica i Parasitologia, Facultat de Farmàcia, Universitat de València, València, Spain
- 3Wellcome Sanger Institute, Wellcome Genome Campus, Hinxton, United Kingdom
- 4Manchester Collaborative Centre for Inflammation Research, University of Manchester, Manchester, United Kingdom
- 5European Bioinformatics Institute (EMBL-EBI), Wellcome Genome Campus, Hinxton, United Kingdom
In spite of growing evidence supporting the occurrence of complex interactions between Schistosoma and gut bacteria in mice and humans, no data is yet available on whether worm-mediated changes in microbiota composition are dependent on the baseline gut microbial profile of the vertebrate host. In addition, the impact of such changes on the susceptibility to, and pathophysiology of, schistosomiasis remains largely unexplored. In this study, mice colonized with gut microbial populations from a human donor (HMA mice), as well as microbiota-wild type (WT) animals, were infected with Schistosoma mansoni, and alterations of their gut microbial profiles at 50 days post-infection were compared to those occurring in uninfected HMA and WT rodents, respectively. Significantly higher worm and egg burdens, together with increased specific antibody responses to parasite antigens, were observed in HMA compared to WT mice. These differences were associated to extensive dissimilarities between the gut microbial profiles of each HMA and WT groups of mice at baseline; in particular, the gut microbiota of HMA animals was characterized by low microbial alpha diversity and expanded Proteobacteria, as well as by the absence of putative immunomodulatory bacteria (e.g. Lactobacillus). Furthermore, differences in infection-associated changes in gut microbiota composition were observed between HMA and WT mice. Altogether, our findings support the hypothesis that susceptibility to S. mansoni infection in mice is partially dependent on the composition of the host baseline microbiota. Moreover, this study highlights the applicability of HMA mouse models to address key biological questions on host-parasite-microbiota relationships in human helminthiases.
Introduction
The study of the interactions between the resident microbiota of the vertebrate gut and both enteric and non-enteric parasitic worms is emerging as a key area of research (1–3). Indeed, over the last decade, several studies performed under both experimental and natural conditions of parasite infections, have provided evidence of quantitative and qualitative changes in populations of gut and/or fecal bacterial communities associated with worm establishment (4, 5). Some of these studies have shown that infection-associated alterations of the host gut microbiota impact on the pathogenesis and pathophysiology of helminthiases, e.g. via changes to worm burdens (6) and effects on parasite immune-modulatory properties (6–9). Other investigations suggest that the host gut flora contributes to the gastrointestinal pathology associated with helminth infection (10, 11). Nonetheless, disentangling the immune-molecular mechanisms that underpin these interactions is essential for establishing causal relationships between helminths and the gut microbiota. In addition, such knowledge will assist the development of intervention strategies for parasite control, and/or of novel therapeutics for the prevention and treatment of chronic inflammatory diseases, based on the rational manipulation of the host gut microbiota (12).
The causative agents of the major neglected tropical disease schistosomiasis are blood flukes (i.e. trematodes) of the genus Schistosoma, that includes S. mansoni, S. haematobium and S. japonicum among other, less prevalent, species (13). More than 250 million people are estimated to be infected with these parasites worldwide, mainly in tropical impoverished areas (13). In addition to the hepato-intestinal disease associated with S. mansoni and S. japonicum, and the urogenital pathology caused by S. haematobium, which includes bladder cancer, all forms of schistosomiasis are associated with systemic morbidities that include malnutrition, anemia, physical and/or cognitive impairment and stunted development in children (13). Over the last few years, a number of studies have demonstrated that infections by Schistosoma spp. are associated with quantitative and qualitative alterations of the gut microbial profiles of both humans (14–18) and experimentally-infected mice (10, 19–21). In particular, experiments conducted in murine models of hepato-intestinal schistosomiasis (i.e. by S. mansoni and S. japonicum, whose eggs penetrate the intestinal wall and are released into the environment via the feces) have suggested that, while both egg-related and -unrelated mechanisms contribute to the interactions between Schistosoma parasites and the host gut microbiota, the former exert a greater impact on intestinal microbial communities (19–21). In support of this hypothesis, we have previously reported dramatic changes in the gut microbiota profile of mice experimentally infected with S. mansoni cercariae that, while detectable during the pre-patent period, were most evident following the onset of egg-laying (19). In particular, features of microbial dysbiosis, including reduced alpha diversity and expansion of proinflammatory bacteria were observed during the patent phase of infection (19). In addition, Floudas et al. (20) have shown that perturbations in gut microbiota composition were more extensive in mice infected with mixed-sex S. mansoni than in mice colonized by single-sex (male) worms.
While the causality of Schistosoma-microbiome interactions remains unclear, it seems plausible that quantitative and qualitative changes in the host gut flora upon parasite colonization may contribute to the pathophysiology of schistosomiasis (10, 19, 21). However, experimental data on the impact of Schistosoma parasites on the host gut microbiota originate from studies conducted in wild type mouse models of infection, and no information is available on possible variations that changes in baseline microbiota communities might introduce into these systems. Determining whether the baseline gut microbiota composition of the vertebrate host affects the magnitude of changes observed upon Schistosoma infection is nonetheless pivotal, as it underpins the translatability of findings from mice to humans. In this study, we assessed quantitative and qualitative changes in gut microbiota composition of mice pre-colonized with a microbiome of human origin (i.e. human microbiota-associated [HMA] mice) following experimental infections with S. mansoni cercariae, and compared the findings with those observed in wild type (WT) animals. Strikingly, higher worm and egg burdens were observed in HMA mice compared to WT animals, and these findings were associated with specific changes in the microbiota composition of each rodent line.
Materials and Methods
Ethics Statement
The life cycle of S. mansoni (NMRI strain) is maintained at the Wellcome Sanger Institute (WSI) by breeding and infecting susceptible snails (Biomphalaria grabrata) and mice. All mouse infections and regulated procedures described in this study were presented to and approved by the Animal Welfare and Ethical Review Body (AWERB) of the WSI. All experiments were conducted under the Home Office Project Licenses (Procedure Project License - PPL) No. P77E8A062 held by GR, and No. P6D3B94CC held by TL. The AWERB is constituted as required by the UK Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012.
Generation of Human Microbiota-Associated Mice and Experimental Infections
The HMA mouse line was generated at the WSI. Briefly, fresh feces from a healthy human donor, processed within 1 h from transportation to the laboratory, were homogenized at 100 mg/ml in 1x D-PBS in an anaerobic cabinet (80% CO2, 10% H2, 10% N2). Five male and five female C57BL/6 germ-free mice, bred at the WSI, were inoculated by oral gavage with 200 μl of donor homogenates once a week, for 3 weeks. One week after the final gavage, mice were set up as breeding pairs in a decontaminated positive pressure isolator. Mice used in this study belonged to the 4th generation of breeding animals. All consumables that entered the isolator for maintenance of the colony were autoclaved at 121°C for 15 min. Feces from parental HMA mice (i.e. germ-free transplanted with human feces) were analyzed by 16S rRNA gene amplicon sequencing, in order to confirm engraftment of the human flora (unpublished data). Animals used for experiments were removed from the isolator in sealed ISOcages and maintained on a positive pressure ISOrack (Tecniplast).
A total of 10 HMA and 10 WT (C57BL/6, bred at the WSI) mice were exposed to 80 S. mansoni cercariae (Sm-exp), as described previously (22), and maintained for 50 days with access to food and water ad libitum. Additionally, 6 WT and HMA mice were kept uninfected and used as negative controls (Sm-). Experimental infections, with matched uninfected controls, were performed in two independent batches (B1 and B2), as indicated in Table 1.
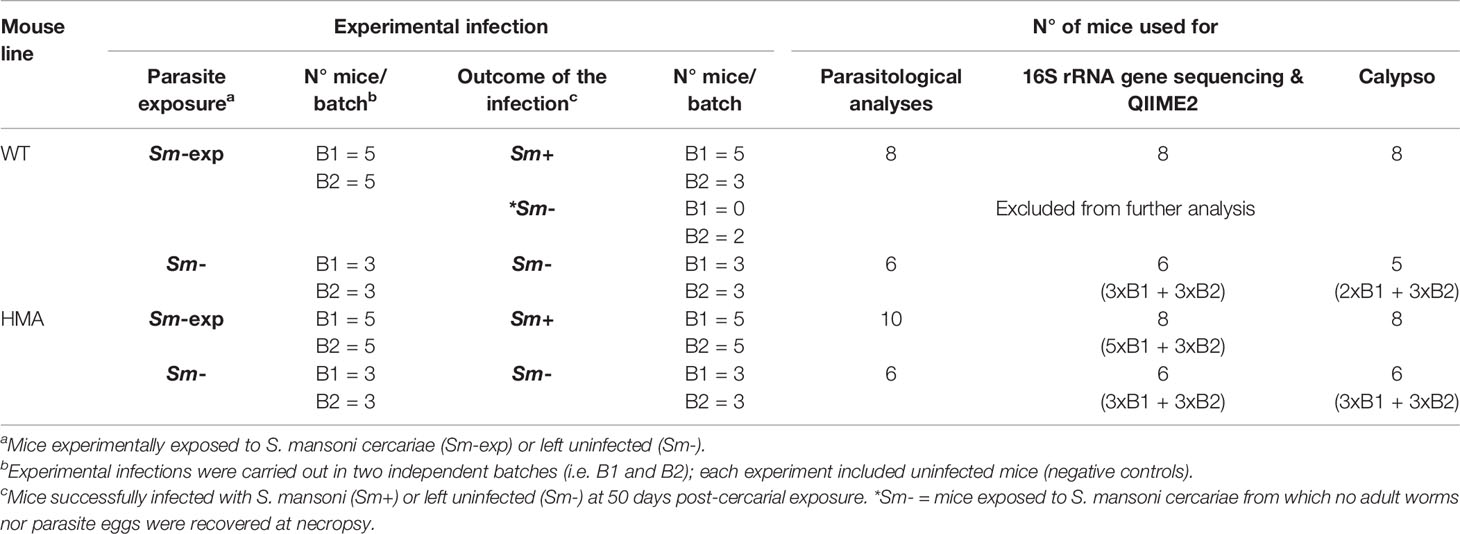
Table 1 Numbers of wild type (WT) and human microbiota-associated mice (HMA) successfully infected by Schistosoma mansoni and left uninfected, and number of samples from each rodent line processed for downstream parasitological and microbiota analyses.
Sample Collection and Parasitological Analyses
Serum samples were obtained from blood collected by puncture of the tail vein at days 28, 42, and 49 post-cercarial exposure (p.i.). at days 28, 42, and 49 post-infection. At day 50 p.i., Sm-exp mice were euthanized, S. mansoni adult worms were recovered by portal perfusion, and livers removed for parasite egg counts (22). Briefly, liver sections from the right lobe were weighed and digested overnight in 3 ml of freshly prepared 4% KOH in 1x PBS, at 37°C under gentle agitation. Eggs were counted in 20 aliquots of 10 µl each, and the number of eggs per gram (EPG) of liver was calculated by extrapolating the mean number of eggs per aliquot to the total volume, and dividing by the weight of liver tissue employed (Supplementary Table 1). Female fecundity, i.e. estimation of the number of eggs produced by individual female worms, was assessed by dividing the EPG of liver by the total number of female worms collected from each mouse. Statistically significant differences in worm burdens and EPG of liver between infected (Sm+) WT and HMA mice were assessed by unpaired two-tailed t-test, following the application of the Kolmogorov-Smirnov normality test for assessment of normal distribution of worm and egg counts datasets; differences in female fecundity were evaluated by Mann-Whitney test. Control Sm- mice were perfused under the same conditions as Sm-exp animals and the perfusates were discarded. For microbiota profiling, fecal pellets were collected directly from the colons of Sm-exp and Sm- mice of each line (i.e. WT and HMA) at necropsy. Samples were transferred to sterile tubes, snap frozen in dry ice, and stored at −80°C until DNA isolation, which was performed within 3 weeks from sample collection.
Specific Antibody Detection
A subset of mice, including six Sm+ and three Sm- animals from each HMA and WT groups were randomly selected to investigate the kinetics of S. mansoni-specific antibodies. Endotoxin-free soluble egg antigen (SEA) was generated from S. mansoni eggs isolated from trypsinized livers of infected mice, as previously described (23). The presence of IgG (total, IgG1, IgG2b, IgG2c and IgG3), and IgM reactive against SEA in mouse serum was determined by ELISA (24). Briefly, 96 well plates were coated with 5 μg/ml SEA overnight, before blocking for 1h at room temperature with 1x PBS containing 1% bovine serum albumin (BSA, Sigma). After blocking, and between each incubation step, plates were washed 5 times with PBS containing 0.05% Tween-20 (Sigma). Fifty microliters of diluted serum (1:1,000; PBS+1% BSA) were added to antigen coated wells and incubated for 1h at room temperature. SEA-specific isotypes were detected using alkaline phosphatase-conjugated goat anti-mouse IgG1, IgG2b, IgG2c, IgG3, or IgM antibodies (Southern Biotech). Bound IgG or IgM were detected by adding liquid p-nitrophenyl phosphate substrate (Southern Biotech), with absorbance read at 405 nm. Additionally, total serum IgE was measured using paired capture and detection antibodies (BD-Biosciences), and quantity was assessed via standard curve. Statistically significant differences in serum antibody levels between Sm+ and Sm- mice of each WT and HMA were determined at each p.i. time point by Mann-Whitney test.
DNA Isolation and Microbial 16S rRNA Gene Sequencing
Genomic DNA was isolated from fecal samples and (no-DNA template) negative controls, using the PowerSoil DNA Isolation Kit (Qiagen) according to manufacturers’ instructions. A total of 28 samples including eight WT and eight HMA Sm+ (selected based on highest DNA concentration and quality), six WT and six HMA Sm- mice, as well as four negative controls were processed for high-throughput sequencing of the bacterial 16S rRNA gene (Table 1). Sequencing libraries were prepared following the Illumina recommendations for bacterial 16S rRNA library preparation, with minor modifications. Briefly, the V3-V4 region of the bacterial 16S rRNA gene was amplified by conventional PCR using universal primers (25) containing the Illumina adapter overhang nucleotide sequences (Forward: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG; Reverse: 5’ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC), Q5® NEBNext Hot Start High-Fidelity DNA Polymerase (New England Biolabs), 5 ng/μl of template DNA and the following thermocycling protocol: 98°C for 2 min, 20 cycles of 98°C/15 s - 63°C/30 s - 72°C/30 s, and a final elongation step at 72°C for 5 min. Amplicons were purified using AMPure XP beads (Beckman Coulter) and set up for the index PCR using Q5® NEBNext Hot Start High-Fidelity DNA Polymerase (New England Biolabs) and Nextera XT index primers (Illumina) with thermocycling conditions as follows: 3 min at 95°C, eight cycles of 30 s at 95°C – 30 s at 55°C – 30 s at 72°C, and 5 min at 72°C. The indexed samples were purified using AMPure XP beads, quantified using Qubit™ dsDNA High Sensitivity Kit (Life Technologies) and equal DNA amounts from each sample were pooled. The resulting pooled library was quantified using the NEBNext® Library Quantification Kit for Illumina® (New England Biolabs), and sequenced using the v3 chemistry (2x300 bp paired-end reads, Illumina). DNA isolation and sequencing were performed separately for samples collected from each B1 and B2 mice (Table 1). Raw sequence data are available from the European Nucleotide Archive (ENA) database under accession number PRJEB40559, whilst curated data can be accessed via MICHELINdb (26).
Bioinformatics and Statistical Analyses
Paired-end demultiplexed Illumina sequencing reads were imported into the Quantitative Insights Into Microbial Ecology 2 (QIIME 2; 2019.4 distribution, https://qiime2.org) software suite for downstream analysis (27). Sequences were then quality filtered, dereplicated, chimeras identified, and paired-end reads merged in QIIME2 using DADA2 (28); quality filtering was performed using default settings, trimming was set at position 10 (forward and reverse), and truncation lengths were set at 250 and 240 for forward and reverse, respectively. A phylogenetic tree was generated using the align-to-tree-mafft-fasttree pipeline in the q2-phylogeny plugin. Bray-Curtis dissimilarity between samples was calculated using core-metrics-phylogenetic method from the q2-diversity plugin. Classification of Amplicon Sequence Variants (ASVs) was performed using a naïve Bayes algorithm trained using sequences representing the bacterial V3-V4 rRNA region available from the SILVA database (https://www.arb-silva.de/download/archive/qiime; Silva_132-99) (29), and the corresponding taxonomic classifications were obtained using the q2-feature-classifier plugin in QIIME2. The classifier was then employed to assign taxonomic information to representative sequences of each ASV.
Statistical analyses were performed using the Calypso software (cgenome.net/calypso/) (30). Relative abundances of individual microbial taxa were calculated by total sum normalization (TSS). Furthermore, for comparative analyses of taxa abundances between experimental groups, cumulative-sum scaling (CSS) was applied to the ASV table, followed by log2 transformation (CSS+log) to account for the non-normal distribution of taxonomic counts data. Statistical comparisons were performed between samples collected from uninfected WT and HMA mice, as well as between Sm+ and Sm- samples of each WT and HMA, as follows. Unsupervised Principal Coordinates Analysis (PCoA) on Bray-Curtis dissimilarities and supervised Canonical Correlation Analysis (CCA) were performed using ‘mouse line’ (i.e. WT and HMA) and ‘infection status’ (i.e. Sm- and Sm+) as explanatory variables, respectively. Beta diversity was calculated using Bray-Curtis dissimilarity and differences between groups were assessed using Analysis of Similarity (ANOSIM) (31). Differences in gut microbiota composition between experimental groups were assessed by ANOVA and by using the Linear discriminant analysis Effect Size (LEfSe) workflow (32). Correlations between differentially abundant bacterial genera and worm burdens or EPG of liver were examined via multiple linear regressions by incorporating worm burdens (or EPG of liver) as the dependent variable and bacterial genera as explanatory variables. Fecal microbial alpha diversity (Shannon index), richness and evenness were calculated using the ‘diversity’ and ‘specnumber’ functions of the vegan R package and differences between groups were evaluated by Mann-Whitney test.
Clusters of co-occurring bacteria and their association with Sm+ or Sm- mice in each line (i.e. WT and HMA) were identified by prediction of correlation networks, with microbial genera represented as nodes, genera abundances as node size, and edges representing positive associations among them. Networks were generated by computing associations among microbial taxa using Pearson’s correlation. The resulting pairwise correlations were converted into dissimilarities and used to ordinate the nodes in a two-dimensional plot by PCoA; thus, correlating nodes were located in close proximity, while anti-correlating nodes were placed at distant locations in the network. Taxa abundances were associated with the type of infection using Pearson’s correlation and the nodes were colored based on the strength of this association.
Results
Worm and Egg Burdens in HMA vs. WT Mice
All HMA mice of both experimental batches were successfully infected with S. mansoni, whereas no adult worms or eggs were recovered from two Sm-exp WT animals included in the second experiment at post mortem; the latter were thus removed from subsequent analyses (Table 1). Significantly higher worm burdens (p=0.017, t=2.67, df=16) and EPG of liver (p=0.008, t=3.02, df=16) were observed in HMA compared to WT mice (cf. Figures 1A, B; cf. Supplementary Table 1). Since differences in EPG of liver could be explained by higher worm burdens observed in HMA mice (Figure 1C), changes to parasite fecundity are unlikely. No significant differences were observed in the number of EPG of liver per female between the two groups of mice (p=0.897, U=38.00; Supplementary Table 1). Furthermore, the higher infection burdens observed in HMA compared to WT mice were accompanied by significantly higher levels of Schistosoma-specific antibodies in sera from HMA animals (Supplementary Figures 1 and 2).
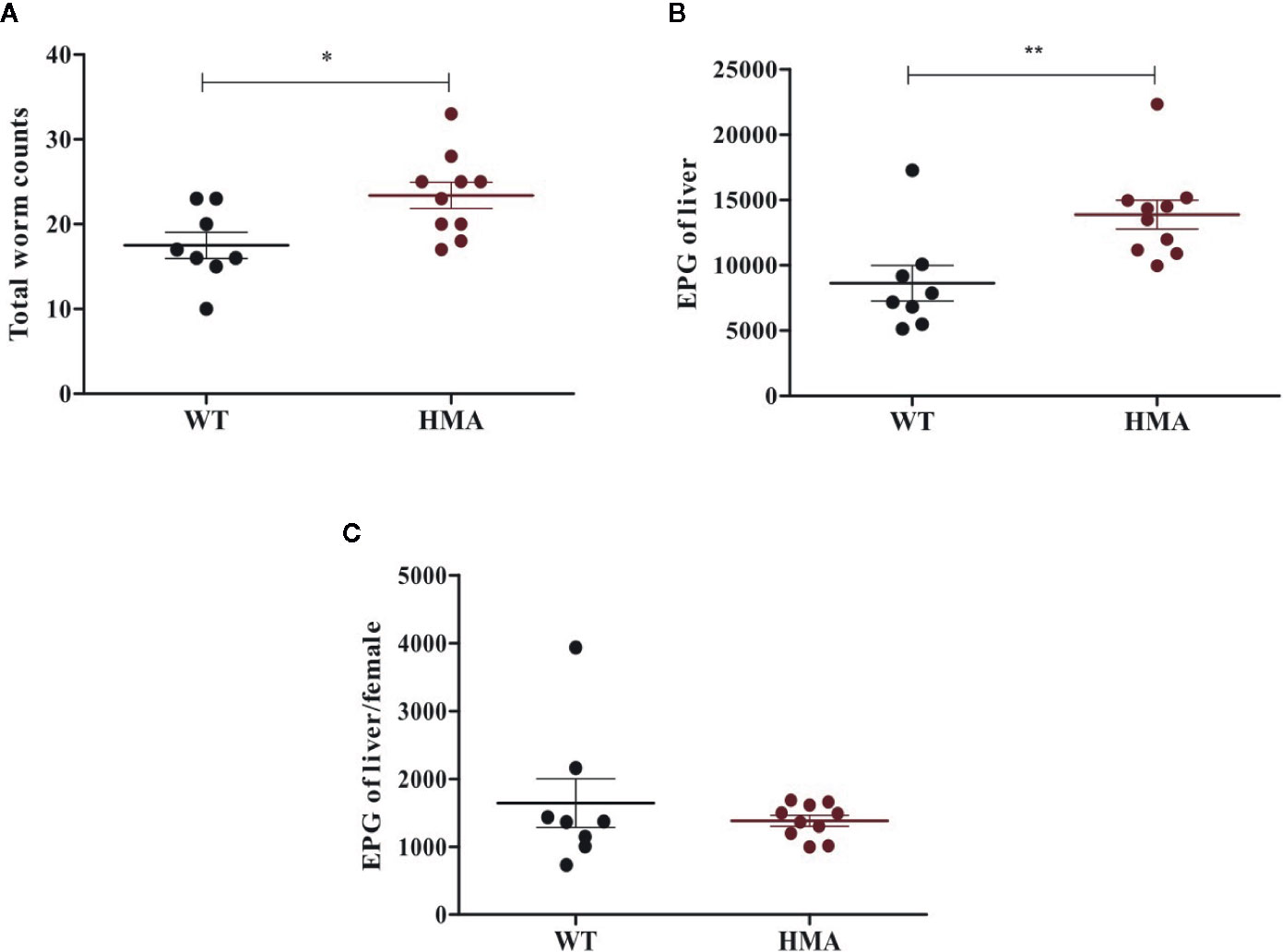
Figure 1 Host microbiome affects susceptibility to Schistosoma mansoni infection. Mean number of (± standard error) male and female worms (A), eggs per gram (EPG) of liver (B), and eggs per female worm (C) recovered from infected wild type (WT) and human microbiota-associated (HMA) mice at 50 days post cercarial exposure. Horizontal lines represent significant differences between mouse lines: *p < 0.05; **p < 0.01.
Diversity and Compositional Traits Differentiate the Gut Microbial Profiles of WT and HMA Mice at Baseline
The two rodent lines employed in this study were characterized by marked differences in (i) the origin of the microbiome (wildtype vs. heterologous from a human donor) and (ii) exposure to environmental microbes (maintained under specific-pathogen free [SPF] conditions vs. in isolation). We anticipated that these profound dissimilarities might significantly impact the composition of the gut microbiota of each mouse line; therefore, we first investigated structural and compositional differences between the gut microbial communities of these rodent lines at baseline. Fecal microbial profiles obtained from samples from Sm- WT and HMA were clustered separately by PCoA, which led to the identification of a putative outlier sample in the former group (Supplementary Figure 3). Dixon’s Q test for outliers, calculated on the Shannon diversity of all Sm- WT mice, supported this hypothesis (p=0.031; Q=0.66), and thus sequence data obtained from this sample were removed prior to further analyses (Table 1). PCoA analysis of gut microbial profiles of uninfected WT and HMA mice revealed profound differences between the gut microbiota compositions of these rodent lines (cf. Figure 2A). Notably, the gut microbiota composition of HMA mice was highly homogenous, unlike that of WT mice (Figure 2A). This observation was confirmed by ANOSIM, which revealed a significantly higher Bray-Curtis dissimilarity among WT compared to HMA mice (p=0.004, R=1; Supplementary Figure 4). Significant differences in fecal microbial alpha diversity were also detected between uninfected WT and HMA (Figure 2B), with significantly reduced microbial richness (p=0.004, U=0.0; Figure 2B) and evenness (p=0.004, F=0.0; Figure 3B) observed in the fecal microbiota of HMA compared to WT mice (Figure 2B).
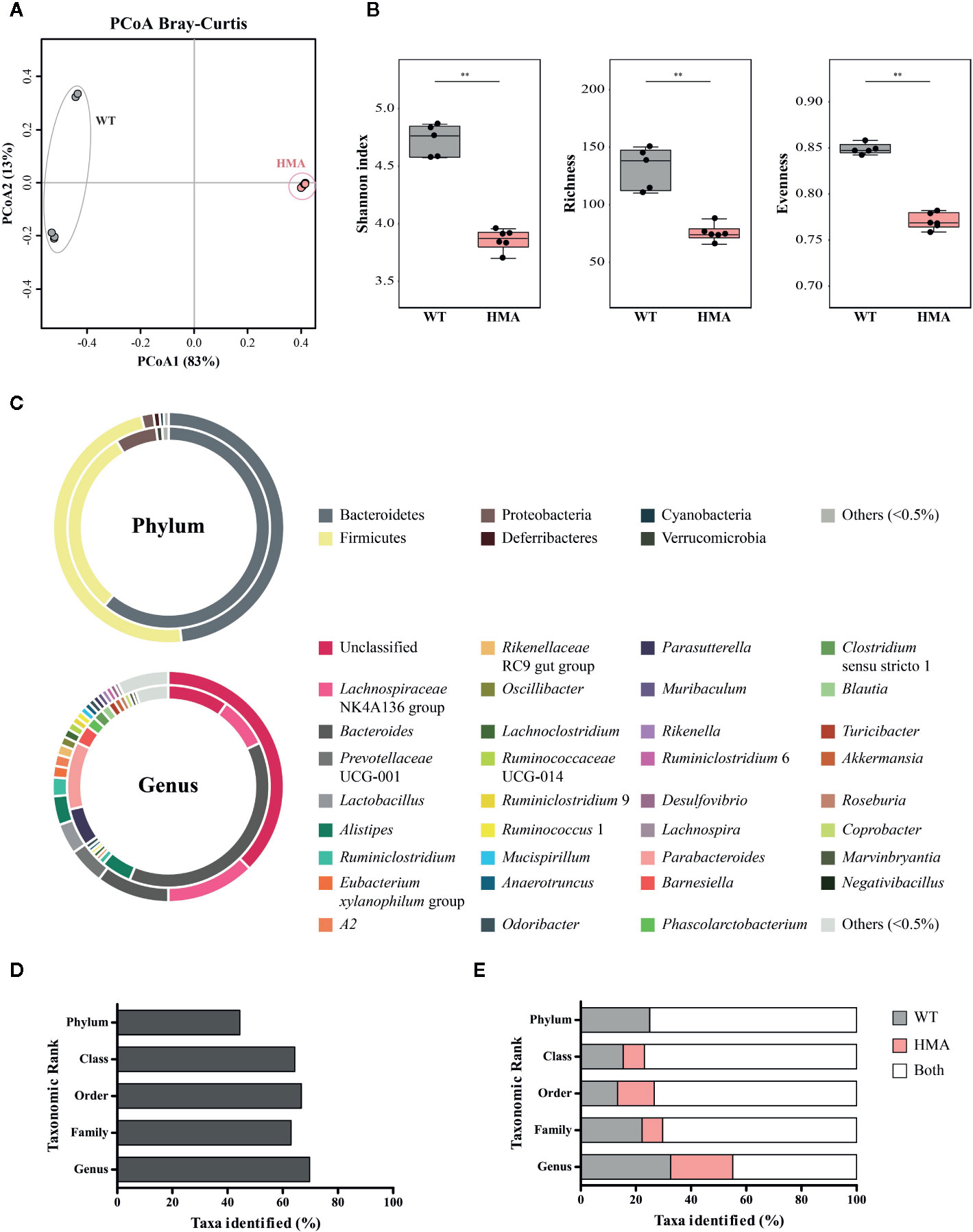
Figure 2 The composition of the baseline gut microbiota differs extensively between naïve wild type (WT) and human microbiota-associated (HMA) mice. (A) Principal Coordinate Analysis (PCoA) performed at amplicon sequence variant (ASV) level. (B) Differences in microbial alpha diversity; horizontal lines represent significant differences between mouse lines: **p < 0.01. (C) Doughnut charts representing the mean relative abundances (TSS-transformed data) of gut microbial phyla and genera identified in feces of naïve WT (outer ring) and HMA (inner ring) mice. Others = sum of all taxa individually representing >0.5% of the overall microbial community. Unclassified = sum of all unclassified genera. (D) Percentage of bacterial taxa (from phylum to genus) for which significant differences were detected between the gut of naïve WT and HMA mice, according to both LEfSe and ANOVA (see differentially abundant taxa in Supplementary Table 2). (E) Percentage of taxa identified (from phylum to genus) solely detected in the gut microbiota of either WT or HMA mice, and in both rodent lines.
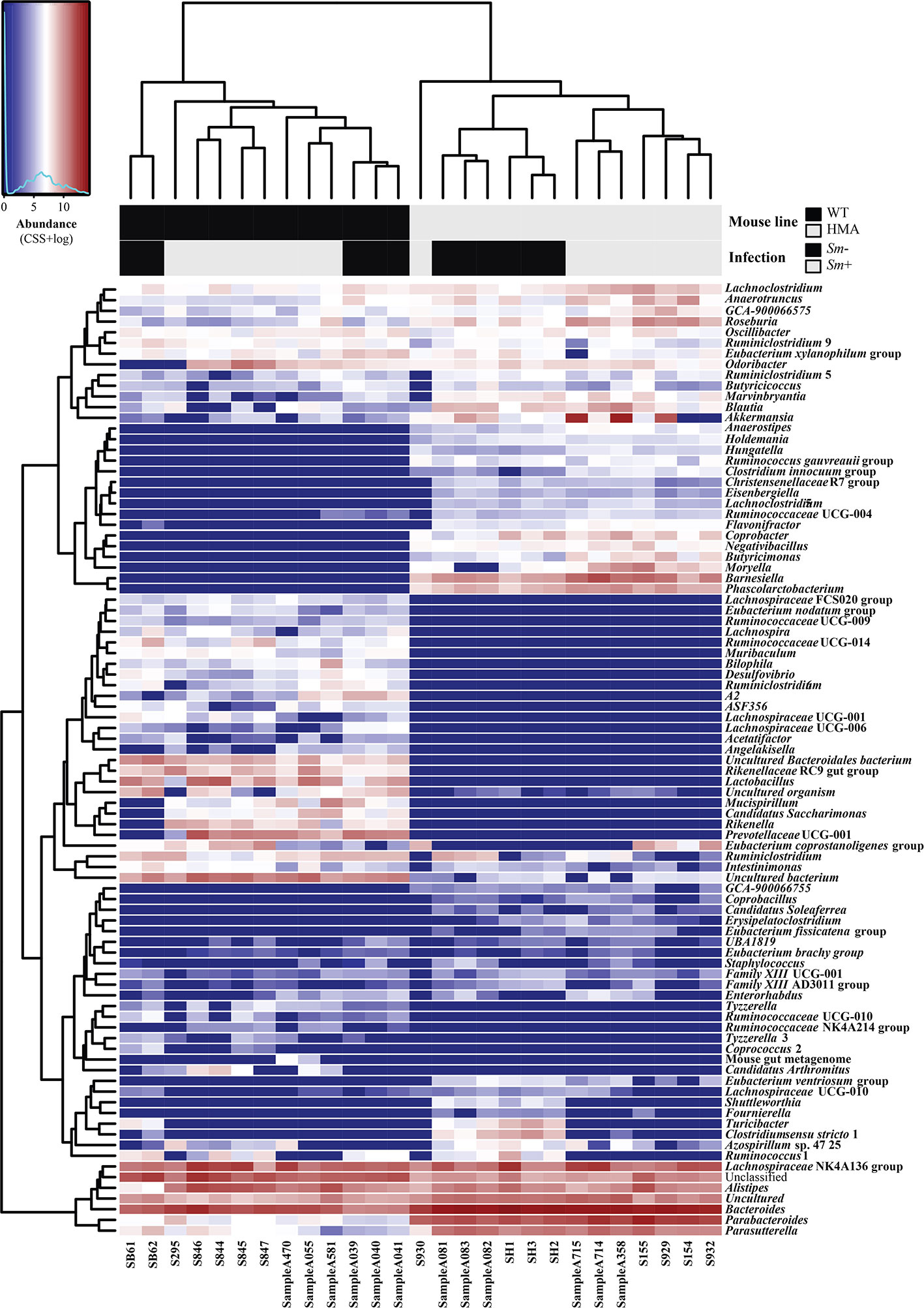
Figure 3 Microbial community composition of all fecal samples from Schistosoma mansoni-infected (Sm+) and uninfected (Sm-) wild type (WT) and human microbiota associated (HMA) mice analyzed in this study, at genus level. In the heatmap, columns represent samples and rows represent genera abundance, both ordered by hierarchical clustering. Explanatory variables (i.e. mouse line and infection status) are presented as a separate heatmap at the top of the figure.
In addition to differences in microbial community structure, significant dissimilarities were observed between the taxonomic composition of the gut microbiota of WT and HMA mice prior to parasite infection (Figures 2C–E and Supplementary Table 2). The phyla Bacteroidetes and Firmicutes were most abundant in the gut of both mouse lines (Figure 2C). However, in the gut microbiota of WT animals, similar proportions of these two phyla were observed (measured as mean TSS-transformed relative abundance ± standard deviation), i.e. 48.21 ± 3.61% Bacteroidetes compared with 47.94 ± 3.84% Firmicutes. In contrast, the phylum Bacteroidetes predominated in HMA mice, with 61.52 ± 3.52% compared with 30.73 ± 3.79% Firmicutes. Proteobacteria was the third most represented bacterial phylum in both rodent lines (1.72 ± 0.98% in WT and 6.70 ± 1.72% in HMA mice), followed by Deferribacteres and Cyanobacteria in WT (0.86 ± 1.23% and 0.58 ± 0.64%, respectively), and Verrucomicrobia in HMA (0.95 ± 0.93%; Figure 2C). No Deferribacteres or Patescibacteria were detected in the gut microbiota of HMA mice (Figure 2C). At genus level, the Lachnospiraceae NK4A136 group was most abundant in the gut of WT animals (12.41 ± 2.55%), followed by the genera Bacteroides (10.23 ± 3.62%), Prevotella UCG-001 (5.11 ± 4.84%) and Lactobacillus (4.28 ± 3.50%). In contrast, the genus Bacteroides was predominant in the gut of HMA mice (38.31 ± 3.69%), followed by Parabacteroides (10.82 ± 0.94%), Lachnospiraceae NK4A136 group (8.19 ± 4.89%) and Parasutterella (6.12 ± 1.75%) (Figure 2C). Furthermore, large differences in the proportions of unclassified microbial genera were observed between mouse lines, together representing 37.56% of the fecal microbial communities in WT animals vs. the 9.73% in HMA mice (Figure 2C).
Differences in the abundances of individual microbial taxa in the gut of uninfected WT and HMA mice were assessed by LEfSe and ANOVA on CSS+log transformed 16S rRNA sequence data (Figures 2D, E and Supplementary Table 2A, B). In particular, significant differences (LEfSe: LDA score (log10)>3; ANOVA p<0.05) were detected in ~45% of the phyla, and >60% of the microbial classes, orders, families and genera composing the gut microbiota of the two mouse lines (Figure 2D; Supplementary Table 2). Among these, the genera Eubacterium coprostanoligenes group and Oscillibacter, both belonging to the family Ruminoccocaceae, were significantly more abundant in the gut of WT mice, while several genera, including Blautia, Roseburia, Clostridium sensu stricto 1, Bacteroides and Parabacteroides, among others, showed greater abundances in HMA animals (Supplementary Table 2).
Additionally, a large number of bacterial taxa were solely detected in either WT or HMA (Figure 3; Supplementary Table 2). For instance, at genus level, >50% of the taxa identified in this study were exclusively detected in either WT or HMA mice (Figure 2E). Of these, Prevotellaceae UCG-001 (5.11% ± 4.84%) and Lactobacillus (4.28% ± 3.50%) were exclusively detected in WT, while Barnesiella (3.00 ± 1.10%) in HMA mice (Figures 2C and 3). However, the majority of these taxa were low-abundant bacteria, each representing <1% of the overall gut microbial population. In particular, numerous ‘line-exclusive’ genera belonged to the family Lachnospiraceae (Supplementary Table 2), the second most abundant family in the gut of both mouse lines. Furthermore, the orders Deferribacterales and Saccharimonadales, along with the families Lactobacillaceae and Muribaculaceae were detected exclusively in the gut of WT animals, whereas the orders Slenomonadales and DTU014 were solely found in the gut of the HMA hosts (Supplementary Table 2).
Schistosoma mansoni Infection Is Associated With Differences in Gut Microbiota Alterations Between HMA and WT Mice
The extensive differences observed between the gut microbiota composition of HMA and WT mice at baseline prevented us from conducting direct comparisons between infected and uninfected animals of these rodent lines; therefore, we next investigated the impact of S. mansoni infection on the gut microbiota profile of each host. Colonization by S. mansoni was associated with overall changes in the gut microbiota composition of both WT and HMA mice, as determined by CCA (WT: p=0.002, F=2.68; HMA: p=0.001, F=4.65; Figure 4A). In both mouse lines, these changes included a significant decrease in microbial alpha diversity post-infection, as well as variations in the abundances of selected populations of gut bacteria (Figure 4, Supplementary Figure 5 and Supplementary Table 3). Notably, the reduction in microbial alpha diversity observed in Sm+ WT mice (Shannon index [ASV], p=0.006, U=2.0) was associated with a significant decrease in evenness (p=0.002, U=0.0), whereas that observed in Sm+ HMA mice (Shannon index [ASV], p=0.008, U=4.0) was linked to a significant decrease in ASV richness (p=0.005, U=3.0) (Figure 4B).
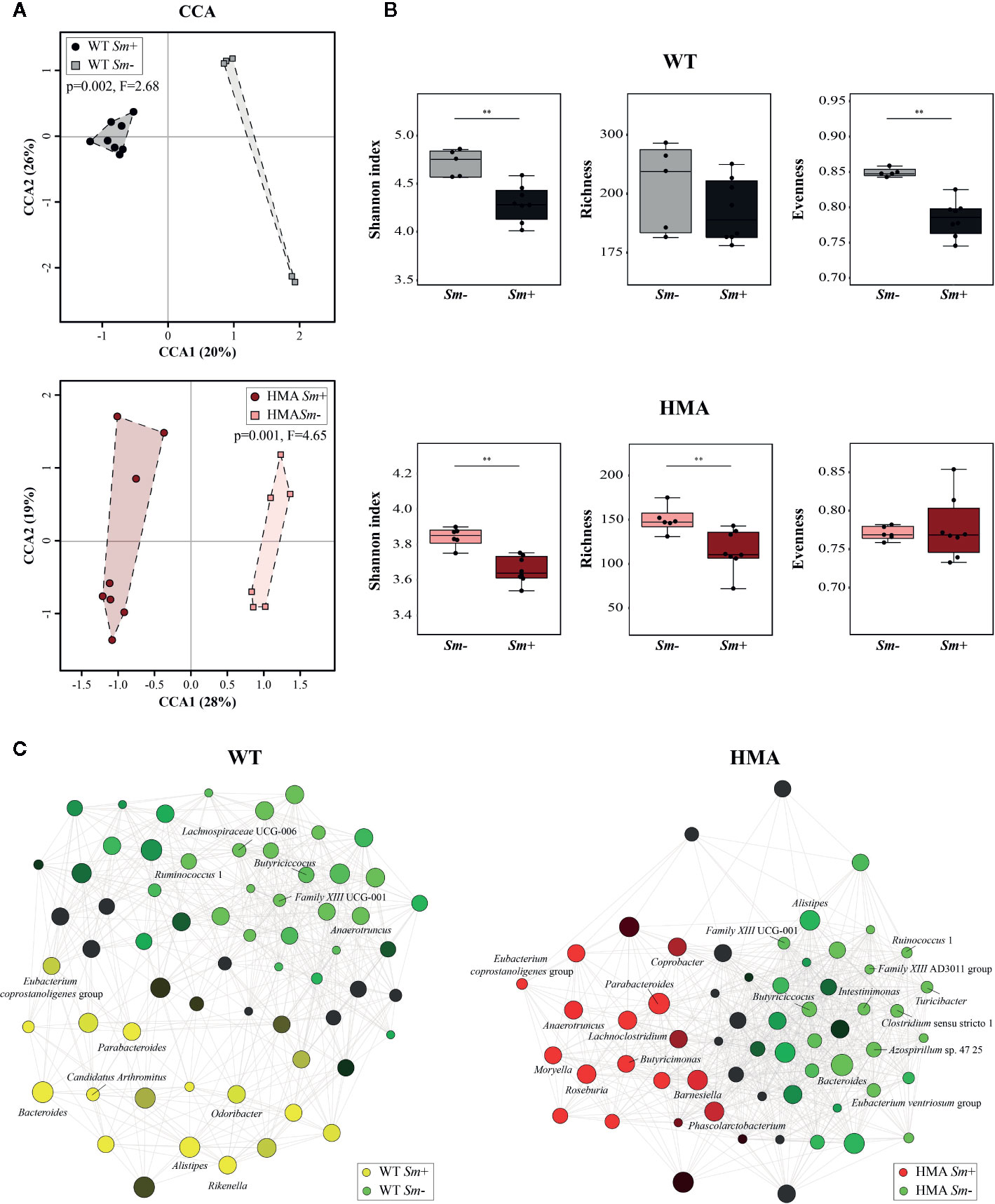
Figure 4 Infection by Schistosoma mansoni is associated with significant alterations of the fecal microbiota composition of wild type (WT) and human microbiota-associated (HMA) mice. (A) Canonical Correlation Analysis (CCA) of microbial profiles of WT and HMA mice, performed at amplicon sequence variant (ASV) level and clustered according to infection status [i.e. infected (Sm+) vs. uninfected (Sm-)]. (B) Differences in microbial alpha diversity between the fecal microbial profiles of infected and uninfected animals in each rodent line; horizontal lines represent significant differences between rodent lines: **p < 0.01. (C) Clustering of microbial genera co-occurring in the fecal microbiota of infected and uninfected WT and HMA mice (the full list of differentially abundant taxa is available from Supplementary Table 3).
Variations in the abundances of individual microbial taxa in the gut of WT and HMA mice following infection by S. mansoni were assessed by LEfSe and ANOVA on CSS+log transformed 16S rRNA sequence data (Supplementary Table 3A–C). Furthermore, network analyses led to the identification of populations of gut bacteria associated with infection by S. mansoni in each mouse line (Figure 4C), while information on the abundances of individual microbial genera across all samples is shown in Figure 3. At day 50 p.i. significant alterations in the abundances of several families and genera belonging mainly to the orders Clostridiales and Bacteroidales were observed in Sm+ WT and HMA mice, compared to Sm- (Supplementary Table 3). Within the Clostridiales, members of the family Lachnospiraceae, such as Lachnospiraceae UCG-006 and UCG-10, were significantly reduced in the gut microbiota of WT mice following infection, while Lachnospiraceae FCS020 group was more abundant in the gut of these animals (Figure 4C and Supplementary Table 3). In the HMA line, selected Lachnospiraceae, i.e. Shuttleworthia and Eubacterium ventriosum group, were significantly reduced in the gut microbiota of infected animals, whereas other genera, including Moryella, Roseburia or Lachnoclostridium, were significantly expanded (Figure 4C and Supplementary Table 3). Similarly, several members of the family Ruminococcaceae were either reduced or expanded in the gut microbiota of each mouse line post-infection (Supplementary Table 3). For instance, the genus Anaerotruncus was negatively associated with Sm+ WT mice based on both ANOVA and network analyses; in contrast, a positive correlation between the abundance of this bacterial genus and helminth infection was observed in HMA rodents by both LEfSe and network analyses (Figure 4C and Supplementary Table 3). The family Clostridiaceae 1 was significantly associated with infected and uninfected WT and HMA mice, respectively. In WT, this finding was linked to a significant expansion of the genus Candidatus Arthromitus following S. mansoni infection, while a significant contraction in populations of Clostridium sensu stricto 1 was detected in Sm- HMA mice (Figure 4C and Supplementary Table 3).
Among members of Bacteroidales whose abundances were altered following S. mansoni infection, the genus Parabacteroides was associated with helminth colonization in both mouse lines by network analysis (Figure 4C); however, LEfSe and ANOVA only detected significant infection-associated increases in populations of Parabacteroides in HMA animals (Supplementary Table 3). Furthermore, while significant expansions of populations of Bacteroides and Alistipes were observed in the gut of infected WT mice (Figure 4C and Supplementary Table 3), network analysis revealed a negative association between these genera and infection by S. mansoni in HMA animals (Figure 4C). Nevertheless, significant differences in intestinal populations of Bacteroides or Alistipes were not detected between Sm+ and Sm- HMA mice by either LEfSe or ANOVA (Supplementary Table 3). In addition, within the Bacteroidales, LEfSe and network analyses detected positive associations between the genera Odoribacter and Rikenella (the latter also supported by ANOVA) and S. mansoni infection in WT mice (Figure 4C and Supplementary Table 3). In HMA mice, however, infection was associated with significant expansions of bacteria within the family Barnesiellaceae, and the related genera Barnesiella and Coprobacter (detected by LEfSe and network analysis), as well as the genus Butyricimonas (detected by LEfSe, ANOVA and network analysis) (Figure 4C and Supplementary Table 3); notably, these taxa were solely detected in the gut of HMA mice (Supplementary Table 2).
Besides changes in the abundances of members of the Clostridiales and Bacteroidales, S. mansoni infection was associated with significant alterations in populations of Alphaproteobacteria, and of the genus Azospirillum sp. 47 25 in particular, which was positively and negatively linked to parasite colonization in WT and HMA mice, respectively (Figure 4C and Supplementary Table 3). Furthermore, in HMA animals, helminth infection was linked to significant reductions in populations of the class Bacilli (and order Bacillales), and expansions of the class Negativicutes (order Selenomonadales, family Acidaminococcaceae and genus Phascolarctobacterium); the latter bacterial class (and related taxa) was solely detected in the gut microbiota of HMA mice (Supplementary Table 3).
For each rodent line, correlations between the abundances of individual gut bacteria and infection burdens (i.e. worm counts and EPG of liver) were investigated by multiple regression. While the genera Lachnospiraceae UCG-006 and Family XIII UCG-001 were negatively correlated with worm counts and EPG of liver in WT mice, the abundances of Bacteroides and Rikenella were positively correlated with infection burdens (worm counts and/or EPG of liver) in Sm+ WT hosts (Supplementary Table 4). In contrast, in HMA mice, parasite burdens were negatively correlated with fecal populations of Turicibacter, Shuttleworthia, Ruminococcus 1 and Clostridium sensu stricto 1, and positively correlated with those of Moryella and Lachnoclostridium, among other genera. The complete list of bacterial genera whose abundances correlated with infection burdens in either line is available from Supplementary Table 4. These findings were consistent with data obtained by LEfSe (Supplementary Table 3A), ANOVA (Supplementary Table 3B, C) and/or taxa correlation network analyses (Figure 4C).
Fecal Bacteria Correlated With Infection Burdens in Both WT and HMA Mice
While the study of the alterations of the gut microbial composition associated with S. mansoni infection in HMA and WT mice revealed substantial differences between rodent lines, some common traits were identified. In order to identify fecal microbial taxa consistently associated with infection in both mouse lines, the LEfSe workflow was applied to all metagenomics datasets, while controlling for both infection status and rodent line. In accordance with the findings obtained from individual analyses of each rodent line, the genus Eubacterium coprostanoligenes group was positively associated to S. mansoni infection when the two variables (i.e. ‘infection status’ and ‘mouse line’) were considered, whereas the genera Ruminococcus 1, Butyricicoccus and Family XIII UCG-001, among others, were associated to Sm- samples (Supplementary Table 5). Correlations between the fecal abundances of key bacterial genera identified by LEfSe with worm counts and EPG of liver were investigated by multiple regression analysis. Significant negative associations were detected between infection burden and fecal abundances of Ruminococcus 1, Butyricicoccus, Family XIII UCG-001 and Family XIII AD3011 in both rodent lines, while a positive correlation was observed between the abundance of Eubacterium coprostanoligenes group and total worm counts (Supplementary Table 5B). Linear regression data of worm burdens/EPG of liver vs. taxa abundance in the two rodent lines showed similar trends to those observed in each WT and HMA (Supplementary Figure 6).
Discussion
Animal models of infection are used extensively to address causal relationships between helminth colonization, alterations in gut microbial composition, and changes in the immune and/or metabolic functions of the host (12). Nevertheless, helminth-gut microbiota interactions are highly heterogeneous depending on the host-parasite pair under consideration (4). Thus, comparing data between different host-parasite systems, even when these involve the same helminth species, may lead to misinterpretations. On the other hand, studies on naturally infected humans may provide data on associations between gut microbial composition and infection with one or more parasites (33); nevertheless, such studies often lack proof of causality between gut microbial signatures and susceptibility to, or pathophysiology of, infection (34, 35). Therefore, complementary studies involving naturally-infected human populations in endemic areas and experimental animal models are pivotal to advance our knowledge of the contribution of the gut microbiota in human parasitic diseases. Towards this aim, novel animal models of infection involving mice colonized by gut microbes of human origin (i.e. HMA mice) may offer unique research opportunities. Thus far, these experimental systems have been applied to investigations of the contribution of human dysbiotic microbiomes to immune- and metabolic-mediated diseases such as diabetes, obesity, inflammatory bowel disease and cancer (34). In this study, HMA mice were used to explore the links between S. mansoni infections and host gut microbiota composition. To the best of our knowledge, this is the first study investigating the relationships between helminth infections and the gut microbiota of the definitive host via the use of HMA mice.
Furthermore, prior to our study, no data was available on the associations between the baseline composition of the host gut microbiota and host susceptibility to Schistosoma colonization and/or pathogenicity. Signatures of gut microbial dysbiosis (i.e. expanded Proteobacteria) have been associated with S. haematobium infection in adolescents from an endemic region (17), as well as to overt clinical signs of schistosomiasis mansoni (i.e. blood in stool and/or splenomegaly) and adverse effects to praziquantel treatment in children (16). However, it remains unclear whether dysbiosis represents an outcome of infection or a risk factor leading to the development of severe disease in young susceptible individuals (17).
In this study, associations between the outcome of S. mansoni infections and the gut microbiota composition at the time of infection were detected in WT and HMA mice. The baseline gut microbiota profile of HMA mice is influenced by multiple evolutionary and ecological factors that impact on the colonization, establishment and growth of selected groups of bacteria (among other microbes) at the expense of others, generally resulting in intestinal dysbiosis (34). Interestingly, in our study, HMA mice were colonized by higher worm burdens – resulting in higher egg loads in the liver – and displayed an enhanced production of anti-S. mansoni SEA antibodies than WT animals, suggesting the occurrence of an association between baseline gut microbiota composition and susceptibility to parasite infection. Of note, gut microbial dysbiosis in naïve HMA mice was characterized by enlarged proportions of Proteobacteria and low microbial richness and overall alpha diversity (compared to WT animals), which is partially consistent with data from studies performed in humans from endemic areas, where no alterations in alpha diversity had been reported (16, 17).
While the mechanisms underpinning the higher worm and egg burdens observed in HMA hosts are yet to be discovered, one hypothesis might be linked to the modified immune system that characterizes these mice. Indeed, the gut microbiome of vertebrate animals is essential for the maturation and proper functioning of their immune system (36, 37). Germ-free mice are known for their deficient innate and adaptive immunity (38) that, in turn, had been linked to enhanced susceptibility to S. mansoni colonization (39). Similarly, HMA mice have been reported to display altered immune responses and susceptibility to intestinal bacterial infections (38, 40, 41). In humans, gut microbial dysbiosis has been associated with a number of pathological conditions (42); importantly, in resource-limited settings, such as those where schistosomiasis is endemic (13), young children are often affected by environmental enteric dysfunction, a multifactorial disorder associated with early childhood malnutrition, growth retardation and impaired immune function (43, 44). Intestinal dysbiosis is key to the pathogenesis of environmental enteric dysfunction (43, 44); therefore, causal relationships may occur between gut microbial imbalances in children from these areas and susceptibility to schistosomiasis that deserve to be thoroughly investigated.
The large number of bacterial taxa that were solely detected in either HMA or WT mice is noteworthy and, together with the substantial differences in the relative proportions of several other bacteria detected between the two rodent lines, might be linked to the different burdens of S. mansoni observed in these experiments. Indeed, altered populations of immune-modulatory bacteria (i.e. lactobacilli) in the mouse gut have been linked to differences in immune responses and susceptibility to infection by the gastrointestinal helminth Heligmosomoides polygyrus (6). In our study, Lactobacillaceae (including the genus Lactobacillus) were only detected in the gut of WT mice, although populations of these bacteria were not altered by infection. Previously, transient expansions of Lactobacillus were reported in the gut of Swiss-Webster mice exposed to large numbers of S. mansoni cercariae (19). The role of lactobacilli in regulating S. mansoni infection remains, however, unknown, and further studies are necessary in order to ascertain whether the absence of these bacteria in the gut of HMA mice might be linked to the higher infection burdens observed in these animals.
The genus Barnesiella was solely identified in the feces of HMA mice, and was significantly expanded in the gut of infected animals. While members of this genus have been regarded as immunomodulatory, current evidence is inconsistent. Expanded populations of Barnesiella spp. have been linked to Th1 environments in diverse murine models (45–47). In contrast, in humans, negative correlations between fecal abundances of Barnesiella and the ability of peripheral blood mononuclear cells to produce IFN-γ following stimulation with either lipopolysaccharide or Bacteroides fragilis have been described (48). IFN-γ has been reported to mediate protection against S. mansoni infection in vaccine studies (49–51). Moreover, Th1 responses are dominant during pre-patent schistosomiasis (52). Therefore, given the known links between Barnesiella and Th1-mediated immunity in mice and humans, the potential role(s) of this genus of bacteria in modulating the response against S. mansoni in HMA mice merit(s) further investigations.
Structural and compositional differences between the gut microbiota of WT and HMA mice at baseline might explain the large dissimilarities observed in infection-associated changes in microbial profiles. However, some common traits were noted. For instance, the genus Ruminococcus 1 (the abundance of which was negatively correlated with infection burdens in both WT and HMA mice) includes mucin-degrading bacteria (53, 54) whose growth might be affected by variations in mucosal carbohydrate availability (55). Alterations in intestinal mucus production and glycosylation are known to occur during gastrointestinal helminth infections (56–58) and, given the essential role of the IL-4/IL-13 pathway in goblet cell hyperplasia, mucus secretion and glycosylation (59), are likely to arise during intestinal schistosomiasis. Therefore, it is plausible that reduced populations of Ruminococcus 1 in the gut of Sm+ mice might be linked to changes in mucin glycan composition; this, in turn, might be associated to egg-induced immunity and disruption of the intestinal barrier. On the other hand, selected Ruminococcus spp. play roles in the breakdown of dietary polysaccharides and production of immunomodulatory short-chain fatty acids (SCFAs) (60, 61). Other populations of SCFAs-producing bacteria, such as Butyricicoccus (62), and the genera Shuttleworthia (63) and Intestinimonas (64, 65), were also negatively correlated with S. mansoni infection burdens (the latter two in HMA mice only); thus, reductions in the abundances of these populations of gut bacteria might lead to an impaired regulation of intestinal immunity and barrier function in infected animals (66).
While these findings suggest the occurrence of associations between changes in gut microbiota composition and alterations of host immune and intestinal barrier function over the course of schistosomiasis, the profound differences between the gut microbial profiles of HMA and WT mice call for caution when selecting bacterial candidates for follow-up experiments aimed at ascertaining the role of the host gut microbiota in modulating immunity to infection. For instance, despite the abovementioned reduction in several SCFA-producing bacteria in the intestines of both Sm+ WT and HMA mice, butyrate-producing Roseburia (67) were significantly more abundant in the gut of HMA hosts at baseline, and were further expanded post schistosome infection. Hence, similar changes in gut microbial composition of both lines might not result in comparable immune responses in each host. Nevertheless, laboratory rodent lines characterized by different susceptibility to infection, and with predefined profiles of immunomodulatory gut bacteria might be useful to investigate the contribution of the host gut flora to resistance to Schistosoma infection via, for instance, fecal transplants or gut recolonization with selected bacteria (68). In particular, HMA mice might represent invaluable models for mechanistic studies aimed to reveal causal relationships between alpha diversity and/or selected populations of immunomodulatory bacteria (and other microscopic inhabitants of the gut including prokaryotes, eukaryotes and viruses) and immunity to infection.
Furthermore, given that HMA mice are derived from germ-free animals and maintained under strict conditions of isolation, it is tempting to speculate that, besides the gut microbiota, microbial communities colonizing other organs such as the skin or the respiratory tract might also be dysbiotic. The skin microflora plays important roles in shaping cutaneous immunity and preventing pathogen colonization (69), while interactions between the gut and lung microbiome (i.e. the so-called gut-lung axis) have been linked to alterations of the lung immune function (70). The contributing roles of skin and lung microbiomes in regulating immunity to and survival of schistosomulae are yet to be investigated; this, together with the current lack of data on the composition of the dermal and respiratory microbiota of HMA mice, prevents us from further speculating about the potential role(s) of skin and/or lung dysbiosis in enhanced susceptibility to S. mansoni infection. Nevertheless, our results suggest that HMA mouse models could be useful to test the contribution of such microbial communities to the defense against the juvenile stages of Schistosoma spp.
Conclusions and Future Perspectives
Overall, our results suggest that the baseline composition of the host gut microbiome might impact host susceptibility to S. mansoni colonization, as well as infection-associated changes in gut microbial profiles. However, further studies are necessary to ascertain the impact that gut microbial dysbiosis (i.e. reduced alpha diversity) and/or selected human microbes exerts on S. mansoni infection rates. In particular, such studies should include (i) experimental infections of additional HMA mouse lines, (ii) groups of control rodents derived from germ-free mice transplanted with the fecal microbiota of WT mice, and (iii) antibiotic-treated WT mice. Moreover, predictive models linking infection burdens to the abundances of key gut microbes and/or alpha diversity prior to infection could be useful to identify gut microbial features that might facilitate parasite colonization. This knowledge will underpin future mechanistic studies to untangle the complex interactions between the gut microbiome, the host immune response and the outcome of schistosome infection.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by Animal Welfare and Ethical Review Body (AWERB).
Author Contributions
ACor, GR and CC designed the research. ACor, SC, ACos, CM, KH, CB, CT, and GR carried out the research. ACor, ACos, AA and JR performed data processing and analyses. ACor, GR, and CC wrote the main manuscript text with input from AM, TL, and MB. The figures were prepared by ACor and ACos. All authors reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
ACor is supported by Red de Investigación Colaborativa en Enfermedades Tropicales—RICET (RD16/0027/0023—Ministerio de Ciencia, Innovación y Universidades, Madrid, Spain). Wellcome provided core-funding support to the Wellcome Sanger Institute, award number 206194. JR is the grateful recipient of a PhD scholarship by the UK Biotechnology and Biological Sciences Research Council (BBSRC). Research in the CC laboratory is funded by grants by the Isaac Newton Trust and the University of Cambridge.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.593838/full#supplementary-material
Supplementary Figure 1 | IgG responses to Schistosoma mansoni soluble egg antigen (SEA) in the sera of wild type (WT) and human microbiota-associated (HMA) mice. Lines connect samples collected from individual mice from two independent batches of infection (i.e., B1 = dashed, and B2 = solid). OD = Optical density. Asterisks indicate statistically significant differences between the sera of infected (Sm+) and uninfected (Sm-) mice (*p<0.05).
Supplementary Figure 2 | IgM responses to Schistosoma mansoni soluble egg antigen (SEA) and total IgE in the sera of wild type (WT) and human microbiota-associated (HMA) mice. Lines connect samples collected from individual mice from two independent batches of infection (i.e., B1 = dashed, and B2 = solid). OD = Optical density. Asterisks indicate statistically significant differences between the sera of infected (Sm+) and uninfected (Sm-) mice (*p<0.05).
Supplementary Figure 3 | Principal Coordinate Analysis (PCoA) performed at amplicon sequence variant (ASV) level (A) and estimated microbial alpha diversity (B) of microbial profiles of uninfected wild type mice (WT); red circles indicate the outlier sample identified in this experimental group, which was removed prior to further downstream processing. (C) PCoA (ASV) depicting the microbial profiles of uninfected human microbiota-associated mice (HMA).
Supplementary Figure 4 | Microbial beta diversity of uninfected wild type (WT) and human microbiota-associated (HMA) mice. Differences between mouse lines were assessed by ANOSIM on Bray-Curtis dissimilarities (**p<0.01). Between = beta diversity between groups.
Supplementary Figure 5 | Gut microbial profiles of wild type (WT) and human microbiota-associated (HMA) mice following infection by Schistosoma mansoni. Doughnut charts representing the mean relative abundances (TSS-transformed data) of gut microbial genera detected in the feces of uninfected (outer ring) and S. mansoni-infected (inner ring) WT and HMA mice. Others = sum of all genera individually representing less than 0.5% of the overall microbial community. Unclassified = sum of all unclassified genera.
Supplementary Figure 6 | Correlations between Schistosoma mansoni worm burdens and eggs per gram (EPG) of liver, and abundances of selected bacterial genera in feces of wild type (WT) and human microbiota associated (HMA) mice.
Abbreviations
ASV, Amplicon Sequence Variant; EPG, Eggs per gram; HMA, Human microbiota-associated; SCFA, Short Chain Fatty Acid; SEA, Soluble egg antigen; Sm-exp, Exposed to Schistosoma mansoni cercariae; Sm-, Uninfected; Sm+, Infected with Schistosoma mansoni; WT, Wildtype.
References
1. Leung JM, Graham AL, Knowles SCL. Parasite-Microbiota Interactions With the Vertebrate Gut: Synthesis Through an Ecological Lens. Front Microbiol (2018) 9:843. doi: 10.3389/fmicb.2018.00843
2. Peachey LE, Jenkins TP, Cantacessi C. This Gut Ain’t Big Enough for Both of Us. Or Is It? Helminth-Microbiota Interactions in Veterinary Species. Trends Parasitol (2017) 33:619–32. doi: 10.1016/j.pt.2017.04.004
3. Saltykova IV, Petrov VA, Brindley PJ. Opisthorchiasis and the Microbiome. Adv Parasitol (2018) 102:1–23. doi: 10.1016/bs.apar.2018.07.001
4. Cortés A, Peachey L, Scotti R, Jenkins TP, Cantacessi C. Helminth-microbiota cross-talk - A journey through the vertebrate digestive system. Mol Biochem Parasitol (2019) 233:111222. doi: 10.1016/j.molbiopara.2019.111222
5. Rapin A, Harris NL. Helminth-Bacterial Interactions: Cause and Consequence. Trends Immunol (2018) 39:724–33. doi: 10.1016/j.it.2018.06.002
6. Reynolds LA, Smith KA, Filbey KJ, Harcus Y, Hewitson JP, Redpath SA, et al. Commensal-pathogen interactions in the intestinal tract: lactobacilli promote infection with, and are promoted by, helminth parasites. Gut Microbes (2014) 5:522–32. doi: 10.4161/gmic.32155
7. Fricke WF, Song Y, Wang A-J, Smith A, Grinchuk V, Mongodin E, et al. Type 2 immunity-dependent reduction of segmented filamentous bacteria in mice infected with the helminthic parasite Nippostrongylus brasiliensis. Microbiome (2015) 3:40. doi: 10.1186/s40168-015-0103-8
8. Su C, Su L, Li Y, Long SR, Chang J, Zhang W, et al. Helminth-induced alterations of the gut microbiota exacerbate bacterial colitis. Mucosal Immunol (2018) 11:144–57. doi: 10.1038/mi.2017.20
9. Zaiss MM, Rapin A, Lebon L, Dubey LK, Mosconi I, Sarter K, et al. The Intestinal Microbiota Contributes to the Ability of Helminths to Modulate Allergic Inflammation. Immunity (2015) 43:998–1010. doi: 10.1016/j.immuni.2015.09.012
10. Holzscheiter M, Layland LE, Loffredo-Verde E, Mair K, Vogelmann R, Langer R, et al. Lack of host gut microbiota alters immune responses and intestinal granuloma formation during schistosomiasis. Clin Exp Immunol (2014) 175:246–57. doi: 10.1111/cei.12230
11. Cortés A, Wills J, Su X, Hewitt RE, Robertson J, Scotti R, et al. Infection with the sheep gastrointestinal nematode Teladorsagia circumcincta increases luminal pathobionts. Microbiome (2020) 8:60. doi: 10.1186/s40168-020-00818-9
12. Cortés A, Toledo R, Cantacessi C. Classic Models for New Perspectives: Delving into Helminth-Microbiota-Immune System Interactions. Trends Parasitol (2018) 34:640–54. doi: 10.1016/j.pt.2018.05.009
13. Colley DG, Bustinduy AL, Secor WE, King CH. Human schistosomiasis. Lancet Lond Engl (2014) 383:2253–64. doi: 10.1016/S0140-6736(13)61949-2
14. Kay GL, Millard A, Sergeant MJ, Midzi N, Gwisai R, Mduluza T, et al. Differences in the Faecal Microbiome in Schistosoma haematobium Infected Children vs. Uninfected Children. PLoS Negl Trop Dis (2015) 9:e0003861. doi: 10.1371/journal.pntd.0003861
15. Adebayo AS, Suryavanshi MV, Bhute S, Agunloye AM, Isokpehi RD, Anumudu CI, et al. The microbiome in urogenital schistosomiasis and induced bladder pathologies. PLoS Negl Trop Dis (2017) 11:e0005826. doi: 10.1371/journal.pntd.0005826
16. Schneeberger PHH, Coulibaly JT, Panic G, Daubenberger C, Gueuning M, Frey JE, et al. Investigations on the interplays between Schistosoma mansoni, praziquantel and the gut microbiome. Parasit Vectors (2018) 11:168. doi: 10.1186/s13071-018-2739-2
17. Ajibola O, Rowan AD, Ogedengbe CO, Mshelia MB, Cabral DJ, Eze AA, et al. Urogenital schistosomiasis is associated with signatures of microbiome dysbiosis in Nigerian adolescents. Sci Rep (2019) 9:829. doi: 10.1038/s41598-018-36709-1
18. Osakunor DNM, Munk P, Mduluza T, Petersen TN, Brinch C, Ivens A, et al. The gut microbiome but not the resistome is associated with urogenital schistosomiasis in preschool-aged children. Commun Biol (2020) 3:155. doi: 10.1038/s42003-020-0859-7
19. Jenkins TP, Peachey LE, Ajami NJ, MacDonald AS, Hsieh MH, Brindley PJ, et al. Schistosoma mansoni infection is associated with quantitative and qualitative modifications of the mammalian intestinal microbiota. Sci Rep (2018) 8:12072. doi: 10.1038/s41598-018-30412-x
20. Floudas A, Aviello G, Schwartz C, Jeffery IB, O’Toole PW, Fallon PG. Schistosoma mansoni Worm Infection Regulates the Intestinal Microbiota and Susceptibility to Colitis. Infect Immun (2019) 87:e00275. doi: 10.1128/IAI.00275-19
21. Zhao Y, Yang S, Li B, Li W, Wang J, Chen Z, et al. Alterations of the Mice Gut Microbiome via Schistosoma japonicum Ova-Induced Granuloma. Front Microbiol (2019) 10:352:352. doi: 10.3389/fmicb.2019.00352
22. Crosnier C, Brandt C, Rinaldi G, McCarthy C, Barker C, Clare S, et al. Systematic screening of 96 Schistosoma mansoni cell-surface and secreted antigens does not identify any strongly protective vaccine candidates in a mouse model of infection. Wellcome Open Res (2019) 4:159. doi: 10.12688/wellcomeopenres.15487.1
23. MacDonald AS, Straw AD, Bauman B, Pearce EJ. CD8- dendritic cell activation status plays an integral role in influencing Th2 response development. J Immunol (2001) 167:1982–8. doi: 10.4049/jimmunol.167.4.1982
24. Phythian-Adams AT, Cook PC, Lundie RJ, Jones LH, Smith KA, Barr TA, et al. CD11c depletion severely disrupts Th2 induction and development in vivo. J Exp Med (2010) 207:2089–96. doi: 10.1084/jem.20100734
25. Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res (2013) 41:e1. doi: 10.1093/nar/gks808
26. Scotti R, Southern S, Boinett C, Jenkins TP, Cortés A, Cantacessi C. MICHELINdb: a web-based tool for mining of helminth-microbiota interaction datasets, and a meta-analysis of current research. Microbiome (2020) 8:10. doi: 10.1186/s40168-019-0782-7
27. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods (2010) 7:335–6. doi: 10.1038/nmeth.f.303
28. Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods (2016) 13:581–3. doi: 10.1038/nmeth.3869
29. Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res (2013) 41:D590–6. doi: 10.1093/nar/gks1219
30. Zakrzewski M, Proietti C, Ellis JJ, Hasan S, Brion M-J, Berger B, et al. Calypso: a user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics (2017) 33:782–3. doi: 10.1093/bioinformatics/btw725
31. Clarke KR. Non-parametric multivariate analyses of changes in community structure. Austral Ecol (1993) 18:117–43. doi: 10.1111/j.1442-9993.1993.tb00438.x
32. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol (2011) 12:R60. doi: 10.1186/gb-2011-12-6-r60
33. Cortés A, Peachey LE, Jenkins TP, Scotti R, Cantacessi C. Helminths and microbes within the vertebrate gut - not all studies are created equal. Parasitology (2019) 146:1371–8. doi: 10.1017/S003118201900088X
34. Arrieta M-C, Walter J, Finlay BB. Human Microbiota-Associated Mice: A Model with Challenges. Cell Host Microbe (2016) 19:575–8. doi: 10.1016/j.chom.2016.04.014
35. Nguyen TLA, Vieira-Silva S, Liston A, Raes J. How informative is the mouse for human gut microbiota research? Dis Model Mech (2015) 8:1–16. doi: 10.1242/dmm.017400
36. Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E. Dysbiosis and the immune system. Nat Rev Immunol (2017) 17:219–32. doi: 10.1038/nri.2017.7
37. Makki K, Deehan EC, Walter J, Bäckhed F. The Impact of Dietary Fiber on Gut Microbiota in Host Health and Disease. Cell Host Microbe (2018) 23:705–15. doi: 10.1016/j.chom.2018.05.012
38. Kennedy EA, King KY, Baldridge MT. Mouse Microbiota Models: Comparing Germ-Free Mice and Antibiotics Treatment as Tools for Modifying Gut Bacteria. Front Physiol (2018) 9:1534. doi: 10.3389/fphys.2018.01534
39. Viera LQ, Moraes-Santos T. Schistosomiasis mansoni: evidence for a milder response in germfree mice. Rev Inst Med Trop Sao Paulo (1987) 29:37–42. doi: 10.1590/s0036-46651987000100006
40. Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell (2012) 149:1578–93. doi: 10.1016/j.cell.2012.04.037
41. Heimesaat MM, Mrazek K, Bereswill S. Murine Fecal Microbiota Transplantation Alleviates Intestinal and Systemic Immune Responses in Campylobacter jejuni Infected Mice Harboring a Human Gut Microbiota. Front Immunol (2019) 10:2272. doi: 10.3389/fimmu.2019.02272
42. Weiss GA, Hennet T. Mechanisms and consequences of intestinal dysbiosis. Cell Mol Life Sci (2017) 74:2959–77. doi: 10.1007/s00018-017-2509-x
43. Bartelt LA, Bolick DT, Guerrant RL. Disentangling Microbial Mediators of Malnutrition: Modeling Environmental Enteric Dysfunction. Cell Mol Gastroenterol Hepatol (2019) 7:692–707. doi: 10.1016/j.jcmgh.2018.12.006
44. Tickell KD, Walson JL. Nutritional Enteric Failure: Neglected Tropical Diseases and Childhood Stunting. PLoS Negl Trop Dis (2016) 10:e00045237. doi: 10.1371/journal.pntd.0004523
45. Daillère R, Vétizou M, Waldschmitt N, Yamazaki T, Isnard C, Poirier-Colame V, et al. Enterococcus hirae and Barnesiella intestinihominis Facilitate Cyclophosphamide-Induced Therapeutic Immunomodulatory Effects. Immunity (2016) 45:931–43. doi: 10.1016/j.immuni.2016.09.009
46. Faas MM, Liu Y, Borghuis T, van Loo-Bouwman CA, Harmsen H, de Vos P. Microbiota Induced Changes in the Immune Response in Pregnant Mice. Front Immunol (2019) 10:2976. doi: 10.3389/fimmu.2019.02976
47. Qi Q, Liu Y-N, Jin X-M, Zhang L-S, Wang C, Bao C-H, et al. Moxibustion treatment modulates the gut microbiota and immune function in a dextran sulphate sodium-induced colitis rat model. World J Gastroenterol (2018) 24:3130–44. doi: 10.3748/wjg.v24.i28.3130
48. Schirmer M, Smeekens SP, Vlamakis H, Jaeger M, Oosting M, Franzosa EA, et al. Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity. Cell (2016) 167:1125–1136.e8. doi: 10.1016/j.cell.2016.10.020
49. Mountford AP, Coulson PS, Pemberton RM, Smythies LE, Wilson RA. The generation of interferon-gamma-producing T lymphocytes in skin-draining lymph nodes, and their recruitment to the lungs, is associated with protective immunity to Schistosoma mansoni. Immunology (1992) 75:250–6. doi: 10.1016/j.cell.2012.04.037
50. Smythies LE, Pemberton RM, Coulson PS, Mountford AP. Wilson RA. T cell-derived cytokines associated with pulmonary immune mechanisms in mice vaccinated with irradiated cercariae of Schistosoma mansoni. J Immunol (1992) 148:1512–8.
51. Smythies LE, Coulson PS, Wilson RA. Monoclonal antibody to IFN-gamma modifies pulmonary inflammatory responses and abrogates immunity to Schistosoma mansoni in mice vaccinated with attenuated cercariae. J Immunol (1992) 149:3654–8.
52. Pearce EJ, MacDonald AS. The immunobiology of schistosomiasis. Nat Rev Immunol (2002) 2:499–511. doi: 10.1038/nri843
53. Crost EH, Tailford LE, Monestier M, Swarbreck D, Henrissat B, Crossman LC, et al. The mucin-degradation strategy of Ruminococcus gnavus: The importance of intramolecular trans-sialidases. Gut Microbes (2016) 7:302–12. doi: 10.1080/19490976.2016.1186334
54. Owen CD, Tailford LE, Monaco S, Šuligoj T, Vaux L, Lallement R, et al. Unravelling the specificity and mechanism of sialic acid recognition by the gut symbiont Ruminococcus gnavus. Nat Commun (2017) 8:2196. doi: 10.1038/s41467-017-02109-8
55. Wu H, Rebello O, Crost EH, Owen CD, Walpole S, Bennati-Granier C, et al. Fucosidases from the human gut symbiont Ruminococcus gnavus. Cell Mol Life Sci (2020). doi: 10.1007/s00018-020-03514-x
56. Hasnain SZ, Evans CM, Roy M, Gallagher AL, Kindrachuk KN, Barron L, et al. Muc5ac: a critical component mediating the rejection of enteric nematodes. J Exp Med (2011) 208:893–900. doi: 10.1084/jem.20102057
57. Hasnain SZ, Thornton DJ, Grencis RK. Changes in the mucosal barrier during acute and chronic Trichuris muris infection. Parasite Immunol (2011) 33:45–55. doi: 10.1111/j.1365-3024.2010.01258.x
58. Cortés A, Muñoz-Antoli C, Sotillo J, Fried B, Esteban JG, Toledo R. Echinostoma caproni (Trematoda): differential in vivo mucin expression and glycosylation in high- and low-compatible hosts. Parasite Immunol (2015) 37:32–42. doi: 10.1111/pim.12159
59. Sharpe C, Thornton DJ, Grencis RK. A sticky end for gastrointestinal helminths; the role of the mucus barrier. Parasite Immunol (2018) 40:e12517. doi: 10.1111/pim.12517
60. Ze X, Duncan SH, Louis P, Flint HJ. Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J (2012) 6:1535–43. doi: 10.1038/ismej.2012.4
61. Crost EH, Le Gall G, Laverde-Gomez JA, Mukhopadhya I, Flint HJ, Juge N. Mechanistic insights into the cross-feeding of Ruminococcus gnavus and Ruminococcus bromii on host and dietary carbohydrates. Front Microbiol (2018) 9:2558. doi: 10.3389/fmicb.2018.02558
62. Devriese S, Eeckhaut V, Geirnaert A, Van den Bossche L, Hindryckx P, Van de Wiele T, et al. Reduced mucosa-associated Butyricicoccus activity in patients with ulcerative colitis correlates with aberrant claudin-1 expression. J Crohns Colitis (2017) 11:229–36. doi: 10.1093/ecco-jcc/jjw142
63. Downes J, Munson MA, Radford DR, Spratt DA, Wade WG. Shuttleworthia satelles gen. nov., sp. nov., isolated from the human oral cavity. Int J Syst Evol Microbiol (2002) 52:1469–75. doi: 10.1099/00207713-52-5-1469
64. Bui TPN, Ritari J, Boeren S, de Waard P, Plugge CM, de Vos WM. Production of butyrate from lysine and the Amadori product fructoselysine by a human gut commensal. Nat Commun (2015) 6:10062. doi: 10.1038/ncomms10062
65. Bui TPN, Shetty SA, Lagkouvardos I, Ritari J, Chamlagain B, Douillard FP, et al. Comparative genomics and physiology of the butyrate-producing bacterium Intestinimonas butyriciproducens. Environ Microbiol Rep (2016) 8:1024–37. doi: 10.1111/1758-2229.12483
66. Parada Venegas D, De la Fuente MK, Landskron G, González MJ, Quera R, Dijkstra G, et al. Short Chain Fatty Acids (SCFAs)-Mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol (2019) 10:277. doi: 10.3389/fimmu.2019.00277
67. Duncan SH, Aminov RI, Scott KP, Louis P, Stanton TB, Flint HJ. Proposal of Roseburia faecis sp. nov., Roseburia hominis sp. nov. and Roseburia inulinivorans sp. nov., based on isolates from human faeces. Int J Syst Evol Microbiol (2006) 56:2437–41. doi: 10.1099/ijs.0.64098-0
68. Ericsson AC, Franklin CL. Manipulating the Gut Microbiota: Methods and Challenges. ILAR J (2015) 56:205–17. doi: 10.1093/ilar/ilv021
69. Byrd AL, Belkaid Y, Segre JA. The human skin microbiome. Nat Rev Microbiol (2018) 16:143–55. doi: 10.1038/nrmicro.2017.157
Keywords: helminth-gut microbiota interactions, Schistosoma mansoni, human-microbiota associated mouse models, gut microbial diversity, dysbiosis, immune-modulation
Citation: Cortés A, Clare S, Costain A, Almeida A, McCarthy C, Harcourt K, Brandt C, Tolley C, Rooney J, Berriman M, Lawley T, MacDonald AS, Rinaldi G and Cantacessi C (2020) Baseline Gut Microbiota Composition Is Associated With Schistosoma mansoni Infection Burden in Rodent Models. Front. Immunol. 11:593838. doi: 10.3389/fimmu.2020.593838
Received: 11 August 2020; Accepted: 20 October 2020;
Published: 18 November 2020.
Edited by:
Marina Moraes Mourão, Oswaldo Cruz Foundation (Fiocruz), BrazilReviewed by:
Gabriel Rocha Fernandes, Oswaldo Cruz Foundation (Fiocruz), BrazilGuillaume Sarrabayrouse, Vall d’Hebron Research Institute (VHIR), Spain
Copyright © 2020 Cortés, Clare, Costain, Almeida, McCarthy, Harcourt, Brandt, Tolley, Rooney, Berriman, Lawley, MacDonald, Rinaldi and Cantacessi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriel Rinaldi, Z3IxMEBzYW5nZXIuYWMudWs=; Cinzia Cantacessi, Y2M3NzlAY2FtLmFjLnVr