 Anna Picca1,2†
Anna Picca1,2† Flora Guerra3†
Flora Guerra3† Riccardo Calvani1,2*
Riccardo Calvani1,2* Hélio José Coelho-Júnior4
Hélio José Coelho-Júnior4 Francesco Landi1,4Roberto Bernabei1,4*
Francesco Landi1,4Roberto Bernabei1,4* Roberta Romano3
Roberta Romano3 Cecilia Bucci3
Cecilia Bucci3 Emanuele Marzetti1,4
Emanuele Marzetti1,4- 1Fondazione Policlinico Universitario “Agostino Gemelli” IRCCS, Rome, Italy
- 2Aging Research Center, Department of Neurobiology, Care Sciences and Society, Karolinska Institutet and Stockholm University, Stockholm, Sweden
- 3Department of Biological and Environmental Sciences and Technologies, University of Salento, Lecce, Italy
- 4Università Cattolica del Sacro Cuore, Rome, Italy
Sterile inflammation develops as part of an innate immunity response to molecules released upon tissue injury and collectively indicated as damage-associated molecular patterns (DAMPs). While coordinating the clearance of potential harmful stimuli, promotion of tissue repair, and restoration of tissue homeostasis, a hyper-activation of such an inflammatory response may be detrimental. The complex regulatory pathways modulating DAMPs generation and trafficking are actively investigated for their potential to provide relevant insights into physiological and pathological conditions. Abnormal circulating extracellular vesicles (EVs) stemming from altered endosomal-lysosomal system have also been reported in several age-related conditions, including cancer and neurodegeneration, and indicated as a promising route for therapeutic purposes. Along this pathway, mitochondria may dispose altered components to preserve organelle homeostasis. However, whether a common thread exists between DAMPs and EVs generation is yet to be clarified. A deeper understanding of the highly complex, dynamic, and variable intracellular and extracellular trafficking of DAMPs and EVs, including those of mitochondrial origin, is needed to unveil relevant pathogenic pathways and novel targets for drug development. Herein, we describe the mechanisms of generation of EVs and mitochondrial-derived vesicles along the endocytic pathway and discuss the involvement of the endosomal-lysosomal in cancer and neurodegeneration (i.e., Alzheimer’s and Parkinson’s disease).
Introduction
Inflammation is part of the innate immunity response to pathogens or molecules released upon tissue injury, collectively indicated as damage-associated molecular patterns (DAMPs) (1). This non-specific first line of organismal defense is mounted upon binding of DAMPs to a set of pattern recognition receptors (PRRs), including Toll-like receptors (TLRs) and inflammasomes that sense DAMPs and elaborate an immune response (2, 3). Albeit DAMPs-triggered inflammation is protective towards harmful stimuli via the coordination of their clearance, promotion of tissue repair, and restoration of tissue homeostasis, an excessive inflammatory response in the setting of persistent stimuli may be detrimental. Indeed, if dysregulated or not timely resolved, inflammation contributes to the development of several disease conditions (e.g., autoimmune diseases, cardiovascular disease, neurodegeneration, and cancer) (4, 5). Hence, a hyper-resolution response aimed at limiting hyper-inflammation and triggered by DAMPs-activated/initialized innate immune cells is in place (6). This pro-resolving pathway is possibly mediated by suppressing/inhibiting inducible DAMPs (SAMPs) (6).
A large deal of research has been devoted to understanding the complex regulatory pathways involved in DAMPs production and trafficking. The endo-lysosomal system that includes a set of dynamic and inter-convertible intracellular compartments such as early-, recycling-, and late endosomes, and lysosomes is a major component of such response. Along with this, autophagosomes are autophagy executors that deliver intracellular contents to lysosomes (7). The fusion of endosomes and/or autophagosomes with lysosomes installs an acidic environment and enables cargo degradation for recycling unnecessary components into re-usable biological building blocks (e.g., carbohydrates, proteins, lipids, and nucleotides) within the cell (7). These events are accomplished via vesicle trafficking, protein sorting, and selective cargo degradation. In particular, two opposite sorting systems are in place: the endosomal sorting complex required for transport (ESCRT) that supports cargoes degradation and the retromer complex that allows specific retrograde cargo retrieval (7).
Mitochondria are highly interconnected organelles that form a dynamic network by contacting the endoplasmic reticulum (ER), lysosomes, and the actin cytoskeleton (8, 9). While inter-mitochondrial junctions allow mitochondrial membrane cristae remodeling between adjacent mitochondria (10), mitochondrial fusion enables the mixing of matrix and intermembrane space contents (11). Recently, an additional mechanism of mitochondrial interconnection based on tube-like protrusions (mitochondrial nanotunnels) has been described (9). Mitochondrial nanotunnels may be especially relevant in establishing connections between organelles immobilized within post-mitotic tissues (e.g., skeletal muscle, myocardium), in which fusion events are limited (9). Finally, Golgi-derived vesicles contribute to the maintenance of mitochondrial homeostasis through participating in mitochondrial dynamics (12).
Altered regulation of the endosomal-lysosomal system has been implicated in several age-related conditions, including cancer and neurodegeneration, and might therefore be targeted for therapeutic purposes (13). Remarkably, small extracellular vesicles (sEVs) isolated from primary fibroblasts of young humans have shown to ameliorate senescence biomarkers in cells obtained from old donors (14). A major task of the endosomal-lysosomal system is the disposal of dysfunctional, but not severely damaged mitochondria via a housekeeping process of mitochondrial quality control (MQC) (15). Herein, we provide an overview on vesicle trafficking along the endocytic pathway, the generation of exosome and mitochondrial-derived vesicles (MDVs), and discuss the involvement of the endosomal-lysosomal system in physiological and pathological conditions, including cancer and neurodegeneration [i.e., Alzheimer’s (AD) and Parkinson’s disease (PD)].
Genesis of Endo-Lysosomal Vesicles
Exosomes are EVs of endosomal origin with a diameter of 50-150 nm. The biogenesis of exosomes is associated with the generation and fate of multivesicular bodies (MVBs) (16). These organelles owe their name to the accumulation of intraluminal vesicles (ILVs) after inward budding of plasma membrane microdomains, fission, and release (16). ILVs have a small diameter (50-150 nm) and are identified as exosome precursors. As part of the endocytic trafficking, endosomal organelles undergo maturation and MVBs, moving from cell’s periphery to the center along microtubules, mature in late endosomes. For this reason, MVBs are considered to be newborn late endosomes derived from the maturation of early endosomes. However, according to an alternative model, MVBs are identified as intermediate transporters between early and late endosomes (17). Realistically, MVBs can follow two alternative directions: 1) toward fusion with other MVBs or late endosomes to undergo maturation and acidification, thus becoming lysosomes for cargo degradation or 2) toward the plasma membrane to fuse and release into the extracellular space ILVs, such as exosomes (16) (Figure 1).
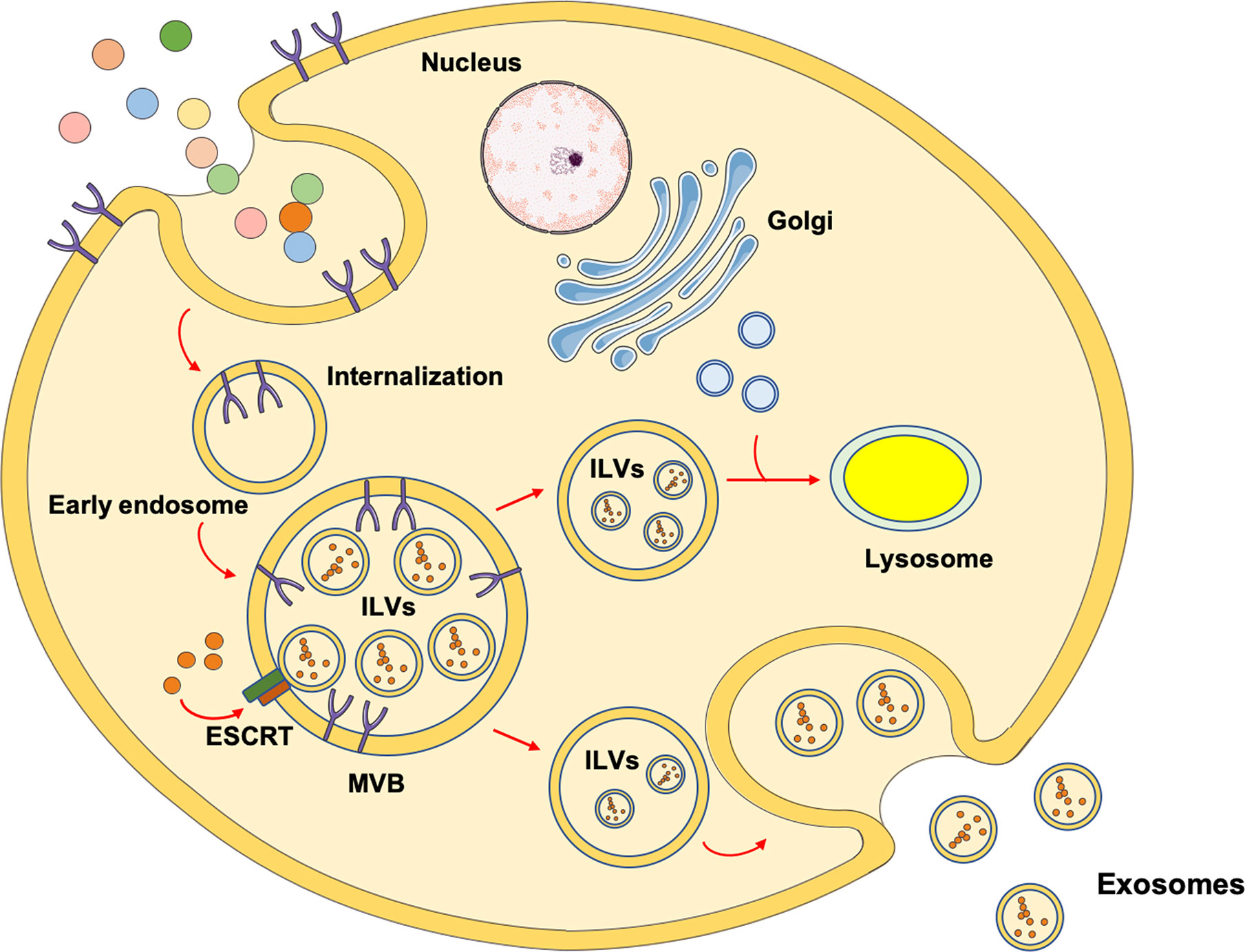
Figure 1 Schematic representation of the mechanisms involved in exosomes biogenesis. The most investigated mechanism through which exosomes are generated involves endocytosis after receptor/ligand binding at the cell’s membrane. After the ligand dissociates from its receptor, it is located into an early endosome. The receptor can either be recycled and relocated on the membrane surface or degraded into lysosomes. Through the activity of the endosomal sorting complex request for transport (ESCRT), the early endosome maturates into multivesicular bodies (MVBs) containing intraluminal vesicles (ILVs). Eventually, MVBs migrate toward the plasma membrane and fuse to release ILVs as exosomes. As an alternative route, MVBs can fuse with other MVBs or late endosomes and receive vesicles containing lysosomal enzymes from trans Golgi, evolving into lysosomes for degradative purposes.
Hence, the biogenesis of MVBs and exosomes is closely related. There are two different mechanisms that guide the origin of MVBs. In fact, they can originate via the sequential action of ESCRT or from endosomes containing lipid rafts (16, 18). The ESCRT system consists of five cytosolic complexes [i.e., ESCRT 0, I, II, III, and vacuolar protein sorting (VPS) 24] (19) and its role in exosome biogenesis has been proven by the identification of several ESCRT proteins in exosomes purified from different cell culture types or biological fluids. For this reason, ESCRT proteins are now used as exosomal markers (20).
ESCRT 0 recognizes a specific group of ubiquinated proteins on early endosomes referred to as phosphatidyl inositol monophosphate (PI3P) enriched domains. The recognition of ubiquitin and PI3P areas occurs through the interaction with the two subunits of the ESCRT 0 complex: HRS (hepatocyte growth factor regulated tyrosine kinase substrate) and STAM1/2 (signal transducing adaptor molecule 1/2). This is the first transition step from early endosomes to MVBs followed by the HRS-mediated recruitment of ESCTR I to endosomes (17, 21–23). ESCRT I in mammalian cells is a heterodimeric complex composed by tumor susceptibility gene 101 (TSG101), VPS28, VPS37A-D, and the ortholog of the yeast Mvb12 (22). In this step, the formation of stable vacuolar domains starts through TSG101 action carrying on the maturation of early endosome into MVBs (24). ESCRT I takes the place of ESCRT 0 and recruits ESCRT II that, in mammalian cells, is composed of ELL-associated protein of 30 kDa (EAP) 30, EAP20, and EAP45 (22). ESCRT III is a heterotetrameric complex composed by VPS20-CHromatin-Modifying Protein (CHMP) 6, Sucrose Non-Fermenting protein (SNF) 7-CHMP4, VPS24-CHMP3, and VPS2-CHMP2 subunits. ESCRT II recruits ESCRT III through the interaction between EAP20 and CHMP6, while CHMP-6 has been shown to regulate cargo sorting (25). ESCRT III has the role of recruiting deubiquitinating enzymes to remove ubiquitin residues from the protein with consequent complete invagination of the membrane and generation of ILVs. This is the last crucial step for the entry of cargoes into ILVs (22, 26). ESCRT III recruits accessory subunits, such as BRO1/ALIX (BCK1-like resistance to osmotic shock protein-1/apoptosis linked gene 2 interacting protein X) for cargo deubiquitination (27), and could also play a role in the fusion of MVBs with late endosomes (26, 28, 29). Finally, the interaction between ESCRT III and VPS4 allows the VPS4 ATPase activity to determine the final membrane budding, scission, and detachment of ESCRT subunit for recycling and cargo delivery (22). Thus, the whole process of ILV budding, cargo selection, membrane remodeling, and the incorporation of ILVs into MVBs is regulated by the ESCRT complex. However, only a few ESCRT components are necessary in this process, including HRS, TSG101, and STAM1 (ESCRT 0/I) (30). Indeed, the silencing of these proteins induces a decrease in exosome secretion, while an increase of exosome release is observed by inhibiting CHMP4C, VPS4B, VTA1 and ALIX (ESCRT III complex) (30). ALIX also interacts with several ESCRT proteins (e.g., TSG101 and CHMP4) and is involved in regulating protein composition/cargo loading, budding of ILVs, and MVB incorporation (31). Recent studies have also indicated that ALIX is crucial for the connection between syndecans and the ESCRT machinery through the binding of syntenins. Syntenins are soluble proteins acting as intracellular adaptors, via their PDZ domains that recruit syndecans. These latter are membrane proteins carrying heparan sulfate chains (HS) that are necessary to bind adhesion molecules and growth factors allowing them to interact with their receptors and assist in the endocytic process. This heterotrimeric complex is involved in endosomal budding and exosomes biogenesis (31, 32).
The exosome biogenesis can also follow an ESCRT-independent pathway. Indeed, even in the setting of simultaneous depletion of core ESCRT proteins, MVB and exosome biogenesis can still ensue via specific membrane lipid composition. Endosomes, which have domains enriched in cholesterol and sphingolipids, named lipid rafts, are able to curve inward and determine the formation of MVBs with the support of the pH gradient across the membrane (33). In this case, phospholipases mediate the synthesis of ceramides from sphingolipids and assure endosome membrane invaginations without ESCRT assistance. In fact, cone-shaped structures of ceramides, alone or associated with cholesterol, generate areas suitable for membrane deformation and ILV budding (34). The conversion of sphingomyelin in ceramide is catalyzed by neutral sphingomyelinases (SMases) which are enzymes located in the Golgi but also in the plasma membrane favoring exosomal biogenesis. Indeed, the inhibition of SMases reduces exosome secretion in specific cell types (35).
Originally identified in B lymphocytes and implicated in several cellular processes like cell fusion, cell migration and cell adhesion, the three tetraspanins CD9, CD81, and C63 are acknowledged as exosomal markers for their abundance in exosomes (36). These proteins generate the TEM domain (tetraspanin-enriched domain) and are composed by four transmembrane domains that interact with several other proteins, cholesterol, and gangliosides. Cargo sorting and formation of ILVs are mediated by the tetraspanins. Indeed, CD9 cooperates in the fusion of plasma membrane, while CD63 interacts with the PDZ syntenin domain (37).
The mechanisms through which MVBs move towards the plasma membrane for the release of exosomes instead of their fusing with lysosomes are presently unclear. Nevertheless, during the fusion of MVBs with the plasma membrane, the interaction between specific proteins and lipids determines exosome secretion, a process involving SNARE (soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptors) proteins and small GTPases (38). Indeed, exosomes secretion is inhibited by overexpression of R-SNARE VAMP7 (vesicle-associated membrane protein 7), which induces enlargement of MVBs and their clustering at the cell’s periphery (39). The transport of MVBs towards the plasma membrane is regulated by microtubules and microfilaments such that the modulation of the expression of cortactin induces changes in the release of exosomes (40). Moreover, members of the Ras-related in brain (RAB) protein family, known for their role in endosomal trafficking, are also involved in exosome biogenesis and release. In this regard, several studies have shown a pivotal role for RAB27 and RAB35 in the docking of MVBs at the plasma membrane (41–43), while the silencing of RAB7A, the master regulator of late endocytic pathway, decreases syntenin-mediated exosome secretion (31, 44) or increases the release of CD9- and CD81-positive exosomes in cisplatin resistant cancer cells (45, 46).
DAMPs of different nature can be shuttled via EVs. Of note, mitochondria can also exploit this pathway for preserving organelle homeostasis. The mechanisms assisting in the generation of EVs from mitochondria are discussed in the next paragraph.
Mitochondrial-Derived Vesicles
MDVs are generated by the selective incorporation of protein cargoes, including outer and inner membrane constituents, and matrix content. These vesicles have a uniform size (from 70 to 150 nm) and can follow two distinct fates: 1) they can fuse with MVBs and/or late endosomes for degradation (47) or extracellular secretion (13, 48); 2) they can be delivered to a subpopulation of peroxisomes (49).
Upon mitochondrial stress and isolation of mitochondria in vitro, it is possible to observe the formation of MDVs enriched in oxidized protein (50), revealing a mitochondrial stress-dependent selective cargo incorporation. An elegant work by Soubannier et al. (50) showed that MDVs carrying the outer membrane pore protein voltage-dependent anion channel (VDAC) are generated after the production of xanthine oxidase/xanthine-induced reactive oxygen species (ROS), while generation of ROS upon treatment with the complex III inhibitor antimycin A determines MDV formation without enrichment in VDAC, thus suggesting that MDVs can transport any oxidized cargo.
The protein kinase phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1) and the cytosolic ubiquitin E3 ligase Parkin are required for the generation of MDVs targeted to the endocytic pathway and, finally, to the lysosomes (51). Both mutated in familial forms of Parkinson’s disease (52, 53), PINK1 and Parkin are known relevant factors in MQC and inducers of the mitophagic pathway. PINK1 is targeted to mitochondria but is normally degraded very rapidly (54–56). Indeed, during the import process at the site of mitochondria, a set of matrix processing peptidases and presenilins-associated rhomboid-like protein (PARL) cleave PINK1, thereby allowing its release from the mitochondrial import channel and subsequent cytosolic proteolytic degradation (56). However, in the setting of damaged mitochondria, the import machinery is inactivated thus determining the trapping of PINK1 within or near the import channel at the mitochondrial outer membrane (55). Here, PINK1, by exposing its kinase domain to the cytosol, induces Parkin phosphorylation. As a consequence, a stable recruitment of Parkin at the mitochondria and a Parkin-dependent ubiquitination of several proteins at the mitochondrial surface occur (57). Finally, a set of autophagic adaptor proteins recognize mitochondrial Parkin-ubiquitinated proteins and deliver damaged organelles to the autophagosome for subsequent disposal (57).
Sugiura et al. (58) proposed a model in which they predicted a similar mechanism in PINK1- and Parkin-mediated MDV transport. The authors hypothesized that a local mitochondrial oxidative damage or complex assembly defects may induce protein aggregation at the mitochondrial import site that may clog the import process into the organelle. Along with this, the oxidation of phosphatidic acid and cardiolipin alters the membrane curvature which may support an early outward bending of the mitochondrial membrane, thus forming MDVs (59). Hence, a dual role for MDVs generation can be envisioned. On the one hand, MDVs can be considered as the first step of MQC, accomplished through the extrusion of damaged proteins as an attempt to avoid complete mitochondrial dysfunction. This would occur in the setting of mildly damaged organelles in which the autophagic pathway is not triggered (47, 51). On the other hand, severe mitochondrial dysfunction and uncoupling could induce a switch from local displacement of mitochondrial content to a complete arrest of PINK1 in all import channels, followed by the recruitment of autophagic mediators and degradation of the whole organelle (Figure 2).
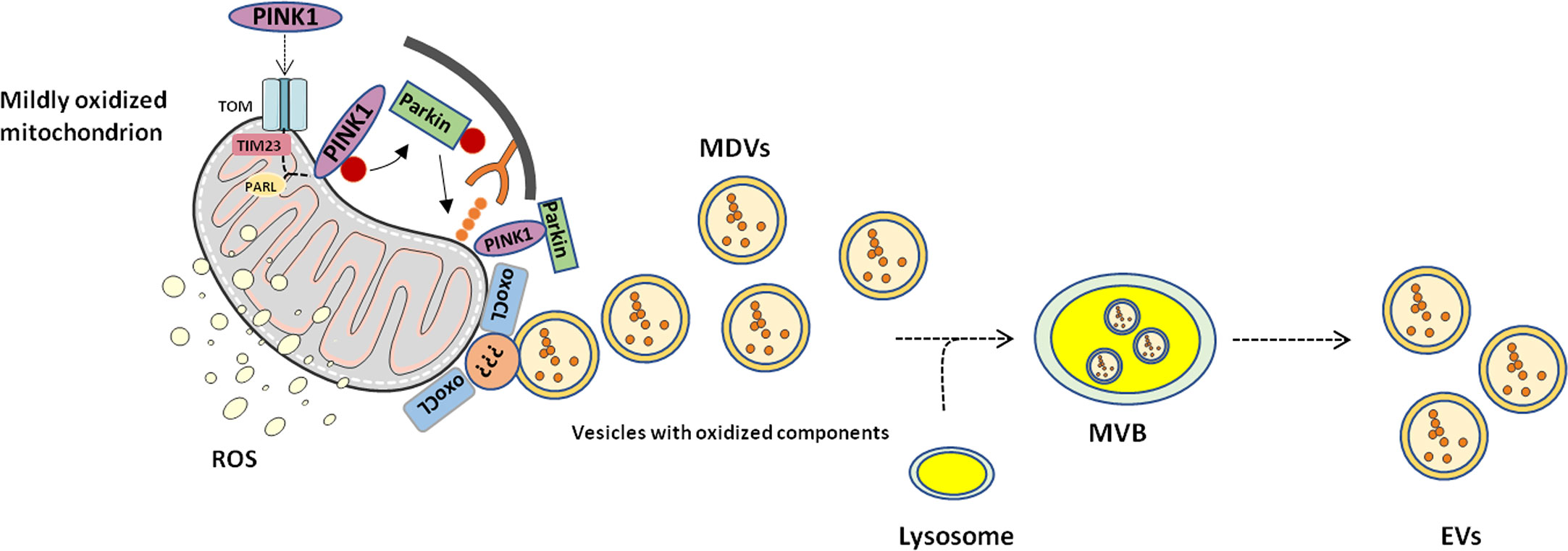
Figure 2 Proposed mechanism of mitochondrial-derived vesicle generation and release. Mitochondrial-derived vesicles (MDVs) may represent an additional level of mitochondrial quality control through which mildly damaged mitochondria are targeted and displaced. Phosphatase and tensin homolog-induced kinase 1 (PINK1) and Parkin prime damaged mitochondria for disposal. Membrane curvatures generated by oxidized cardiolipin (oxoCL) and other unknown proteins allow generation of MDVs that form multivesicular bodies (MVBs) within the endolysosomal system. Eventually, MVBs are extruded from the cell as extracellular vesicles (EVs). PARL, presenilin-associated rhomboid-like; ROS, reactive oxygen species; TIM23, translocase of inner mitochondrial membrane 23; TOM, translocase of the outer mitochondrial membrane.
Such a view supports the hypothesis of including the delivery of MDVs to lysosomes among MQC mechanisms. Indeed, cells perform MQCs via four different mechanisms: 1) degradation of unfolded and oxidized proteins within the mitochondrial matrix or intermembrane space by mitochondrial protease (60); 2) ubiquitination and delivery of mitochondrial outer membrane proteins to the cytosolic proteasome (61, 62); 3) activation of mitophagy to remove severely damaged mitochondria, whether linked to global protein misfolding or depolarization (63), and 4) generation and delivery of MDVs to lysosomes to protect the cell from premature mitophagy by removal of PINK1 and Parkin from each failing import channel.
Damage-Associated Molecular Patterns and Sterile Inflammation
Chronic sterile inflammation ensues in several pathological conditions for which a common thread may reside into dysregulated EV trafficking. Therefore, a deeper understanding of the pathways generating EVs and triggering innate immunity may help clarify the events linking cellular dyshomeostasis with peripheral changes. The generation of MDVs orchestrated by mitochondrial-lysosomal crosstalk (64) is a strong candidate mechanism linking the two processes. Indeed, while operating as an housekeeping system in healthy mitochondria (16), in the setting of failing mitochondrial fidelity pathways, the clearance of dysfunctional organelles via MDVs may release noxious material with the potential of triggering inflammation (64). This response, mediated by the release of interferons (IFNs), pro-inflammatory cytokines, and chemokines, is part of innate immunity and starts with the recognition of an infectious agent (either viral or bacterial) that binds and activates membrane or cytoplasmic immune sentinel molecules termed PRRs [reviewed in (65)]. Of these, membrane-bound TLRs and the cytosolic retinoic-acid-inducible gene I (RIG-I)-like receptors (RLRs, RIG-I, and MDA5) are the best characterized in the setting of viral infections (66). Upon detection of double-stranded RNA produced during viral genome replication (67), TLR3 located in the endolysosomal compartment signals the binding via Toll-interleukin-1 receptor domain-containing adaptor inducing IFN-β (TRIF) and activates the IκB kinase (IKK) complex and the IKK-related kinases TRAF family member-associated NF-κB activator (TANK)-binding kinase 1 (TBK1) and IKKε. As a result of this activation, the translocation of nuclear factor-kappa B (NF-κB) and IFN-regulatory factors (IRFs) to the nucleus and their activation occur, thus inducing the production of type I and III IFNs together with a set of inflammatory chemokines including the regulated on activation normal T cell expressed and secreted (RANTES), and IFN-γ-inducible protein 10 (IP-10) (68–70). Viral RNAs can also be sensed in the cytoplasm by the RLRs, which signal via the mitochondrial antiviral signaling protein (MAVS) adaptor located at the mitochondrial outer membrane. Following the RLR-MAVS pathway, the activation of IKK and IKK-related kinases and, subsequently, NF-κB and IRFs occurs (71–73). Once induced, IFNs upregulate the expression of hundreds of IFN-stimulated genes (ISGs), ultimately installing an antiviral response that halts viral replication and spread (74). Along shared pathways, mitochondrial DAMPs can also trigger inflammation. In particular, mitochondrial DNA (mtDNA), due to its bacterial ancestry and its hypomethylated CpG motifs, is a potent trigger of innate immunity response involving the release of pro-inflammatory mediators installing an inflammatory milieu (75, 76). Indeed, mtDNA can interact with PRRs including TLRs, but also NOD-like receptors (NLRPs), and the cyclic GMP-AMP synthase–stimulator of interferon genes (cGAS–STING) systems (77, 78). The TLR pathway is engaged by mtDNA via its binding to TLR9 at the endolysosomal level, followed by the recruitment of the innate immune signal transduction adaptor myeloid differentiation primary response 88 (MyD88). The latter, by activating the mitogen-activated protein kinase, triggers inflammation via NF-κB signaling (79–81). Alternatively, mtDNA can ignite inflammation as part of the innate immunity response either via inflammasome or cGAS–STING system activation at the cytosolic level (82–86). The cGAS–STING DNA-sensing pathway operates via the TBK1/IRFs/IFNs pathways described above as part of inflammation mounted in the presence of viral infections (84–86). The activation of the STING pathway is also triggered as part of neutrophil activation and neutrophil extracellular trap (NET) formation, a specific cell death route characterized by the extrusion of chromatin-bound cytosolic content (87). NETs have been implicated in the pathogenesis of autoimmune disorders. In particular, NETs enriched in oxidized mtDNA stimulate a type I IFN response and have been implicated in lupus-like diseases (88). In systemic lupus erythematosus, mtDNA binding to the histone-like protein mitochondrial transcription factor A (TFAM) has shown to assist in rerouting oxidized mtDNA of neutrophils to lysosomes for degradation (89). Once extruded, TFAM-oxidized mtDNA complexes are powerful immune system activators (89). Similarly, the release of activated platelet-derived microparticles enriched with high-mobility group box 1 (HMGB1) protein has been described in systemic sclerosis (90). This DAMP molecule might contribute to vasculopathy and tissue fibrosis possibly via the presentation of HMGB1 to neutrophils to induce their activation and consequent endothelial damage (90).
Finally, the engagement of NLRP3, the best studied multi-subunit inflammasome system, elicits caspase-1 signaling and promotes caspase-1-dependent cleavage and activation of interleukin (IL) 1 and 18 via binding to adaptor molecules (91). This route of inflammation is particularly relevant to mitochondrial dysfunction since the synergistic activation of redox-sensitive inflammation and inflammasome reinforce inflammation (92). The molecular triggers of the inflammatory response via inflammasome are unclear. However, bacterial-like motifs of mtDNA are sensed by NLRs (93). Furthermore, NLRP3 is involved in facilitating the organization of the mitochondrial transition pore and assist in mtDNA release (94). A self-sustaining circle involving mitochondrial damage, ROS production, and consequent mtDNA damage/DAMPs release triggered by NLRP3 activators has been hypothesized (83). In particular, damaged/oxidized mtDNA/DAMPs are preferentially sensed and bound by NLRP3 (83).
Following the view of MQC failure as a source of MDVs/DAMPs, we will discuss in the next section the main literature supporting the involvement of mitophagy impairment and DAMPs release in the setting of cancer and two common neurodegenerative diseases (AD and PD).
Implication of Extracellular Vesicles and Damage-Associated Molecular Patterns in Disease
Cancer
Although the involvement of DAMPs in cancer pathogenesis is debated, the installment of an inflammatory milieu is recognized as a factor favoring tumor progression (95, 96). In particular, increasing levels of pro-inflammatory mediators, including IFN-γ, IL1, IL6, lymphotoxin (LT)-β, tumor necrosis factor alpha (TNF-α), and transforming growth factor β, have been implicated in the promotion of carcinogenesis (95–97), for their potential role in modulating DAMPs expression and release (95, 98). Intracellular and extracellular DAMPs are, indeed, hallmarks of cancer that have been implicated in the early stages of carcinogenesis (95). While oxidative stress triggers the release of DAMPs in the extracellular space thus stimulating hyper-inflammation and immune injury, the loss of intracellular DAMPs, [i.e., HMGB1, histones, ATP, and DNA] induces genomic instability, epigenetic alterations, telomere attrition, reprogrammed metabolism, and impaired degradation (98). In the setting of such DAMPs-mediated pathogenic changes, cancer initiation and development are favored. Along with this, the release of ATP, IL1α, adenosine, and uric acid have also been implicated in carcinogenesis via induction of inflammation, immunosuppression, angiogenesis, and tumor cell proliferation (95) (Figure 3).
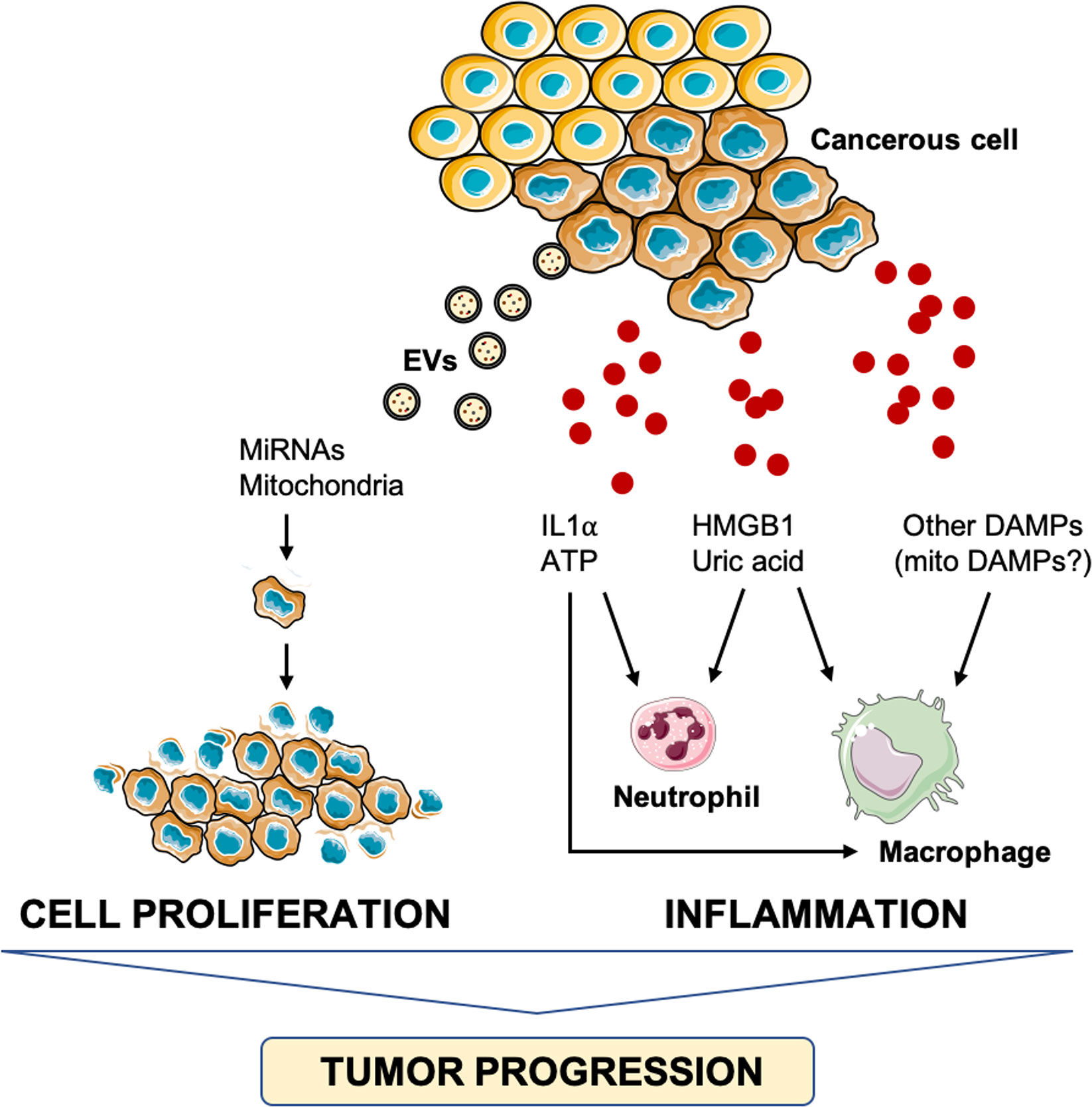
Figure 3 Schematic representation of the main pathways triggered by damage-associated molecular patterns and involved in tumor progression. ATP, adenosine triphosphate; DAMPs, damage-associated molecular patterns; HMGB1, high-mobility group box 1, IL1α, interleukin 1α; miRNA, micro RNA.
Strikingly, inflammatory pathways may also be activated by damaged mitochondrial constituents displaced within MDVs (65) that may trigger caspase-1 activation and secretion of pro-inflammatory cytokines (99). Interestingly, adaptive immunity responses are suppressed by PINK1 and Parkin that redirect MDVs toward lysosomal degradation to prevent endosomal loading with mitochondrial cargoes on major histocompatibility complex (MHC) class I molecules for antigen presentation purposes (48). Furthermore, the possibility that MDVs are used by cells as a homeostatic mechanism by horizontal mitochondrial transfer cannot be disregarded (100). Bone marrow mesenchymal stromal cells (BM-MSCs) eliminate damaged depolarized mitochondria through EVs and export them to neighbouring macrophages (101). Macrophages, in turn, recycle these MDVs to secrete exosomes which contain microRNAs (miRNAs) that inhibit TLR stimulation and induce macrophage tolerance to transferred damaged mitochondria (101). Moreover, cells with impaired mitochondria are able to transfer and take up fully-functional mitochondria displaced within MVDs to rescue aerobic respiration (102–105). A mitochondrial transfer was also shown between A549 mtDNA depleted (p0) lung cancer cell and BM-MSCs to rescue respiration in lung cancer cells lacking mtDNA-encoded subunits of the electron transport chain (ETC) (104). However, vesicles enriched in whole mitochondria or mitochondria void of envelops can also be released and serve as DAMPs in pathological conditions, including tissue injury and cancer (106). In particular, the release of mitochondria by damaged mesenchymal stem cells has been found to function as a danger signal to activate their rescue properties (107). The uptake of whole mitochondria by epidermal growth factor-activated human osteosarcoma cells via macropinocytosis has also been described (108).
Recent findings indicate that cancer cells can reprogram their energy metabolism to adapt and survive in unfavorable microenvironments via EVs (109). Indeed, an efficient mitochondrial respiration is required by cancer cells to maintain their tumorigenicity (110). Upon acquisition of mtDNA through EVs, estrogen receptor (ER)-positive breast cancer can evolve from hormonal therapy sensitive (HTS) to dormant (HTD) or resistant (HTR) with poorer outcome. EVs from patients with HTR disease contain full mitochondrial genome that might have been transferred to HTS/HTD cells to sustain oxidative phosphorylation, an exit from dormancy and the development of HTR disease (111). Additional findings show that EVs from melanoma, ovarian and breast cancer tissues contain mitochondrial membrane proteins and active mitochondrial enzymes that are not detected in healthy controls (112), thus corroborating the hypothesis that energy metabolism reprogramming in cancer cells may occur also via EVs.
Similar to HMGB1 and histones, miRNAs can also be releases in the extracellular space as DAMPs in cancer (113, 114). Recent work has shown an exosomes-dependent pathway to secrete miRNA in cancer cells (115). For instance, in pancreatic cancer cells, exosomes containing miR-212-3p are secreted and lead to decreased expression of MHC II in dendritic cells (DCs), thereby inducing immune tolerance (116). Another system used by cancer cells to escape their recognition by the immune system is based on PD-L1. This factor binds to the PD1 receptor on immune cells thereby inhibiting proliferation and survival of CD8+ cytotoxic T lymphocyte (117). A recent study has shown that exosomes derived from lung cancer express PD-L1 and this is implicated in immune escape and promotion of cancer growth (118). These mechanisms enable cancer cell survival, proliferation, and undisturbed dissemination into other bodily districts, even located at long distance from the primary neoplastic mass. Thus, exosomes are useful shuttles for cancer cells to elude the immune system’s response and achieve undisturbed survival and proliferation.
Moreover, cancer cells can also transfer miRNAs via exosomes to favor angiogenesis. These miRNAs of exosomal origin are ultimately DAMPs promoting cancer proliferation. Indeed, their secretion is induced under oxidative stress (119). An elegant work by Deng et al. (120) showed that gastric cancer cells released exosomes containing miR-155 to increase the expression of vascular endothelial growth factor (VEGF) and promote proliferation and tube formation of vascular cells. In further support to the role of exosomal miRNAs in promoting angiogenesis are findings showing a strong enhancement of angiogenesis and tumor growth in mice under the infusion of exosomes containing miR-155 (120).
Finally, DAMPs may also act as a suppressor of tumor progression by promoting immunogenic cell death. Under physiologic conditions, cell death linked to normal turnover is not immunogenic and does not activate PRRs, such as TLRs and NLRPs (121). In contrast, immunogenic cell death is essential for tumor suppression after chemotherapeutic treatments (122). Immunogenic and non-immunogenic cell death are characterized by different biochemical and metabolic events. In particular, during immunogenic cell death, antigens from dying cells are incorporated by DCs and presented bound to MHC to mount a T cell immune response. In this context, co-stimulatory signals and cytokines are required for differentiation of specific T cells (123). The preapoptotic exposure of calreticulin on the plasma membrane of dying cells promotes their uptake by DCs (124). Interestingly, the release of HMGB1 in the surroundings of dying cells (125) induces an increase in tumor antigen presentation and regulates the TLR4-dependent immune response (126). The role of the NLRP3 inflammasome is crucial for the immune response against dying tumor cells as it interacts with the adaptor molecule apoptosis-associated speck-like protein to induce caspase-1 activation (127). The caspase-1 pathway is involved in the production of proinflammatory cytokines (i.e., IL1β and IL18) which are essential to induce an immunogenic response (127). Notably, ATP released from dying tumor cells mediates immunogenic cell death via the activation of the NLRP3 inflammasome (128). Therefore, understanding the fine-tuning of DAMPs release may be crucial for unveiling new pathways that modulate tumor cell’s death vs. survival.
Neurodegeneration
As a first line of defense against microbes, microglial cells of the central nervous system (CNS) preserve tissue homeostasis by clearing out damaged neurons and limiting the spread of infections. This macrophage population accomplishes these housekeeping activities by triggering inflammation via the release of cytokines and by instigating ROS production (129). However, upon prolonged stressors, a persistent microglia activation installs a pro-inflammatory and pro-oxidant environment that impinges on tissue homeostasis. A state of chronic, low-grade inflammation is observed during aging (i.e., inflamm-aging) which has been associated also with metabolic changes in microglia (130, 131). The age-related microglial and metabolic reshaping plays relevant roles in the context of AD and PD (132, 133). Indeed, neuroinflammation may represent a common thread in a large set of neurological disorders for which DAMPs of different origins, including mitochondrial, may support disease progression (134) (Figure 4).
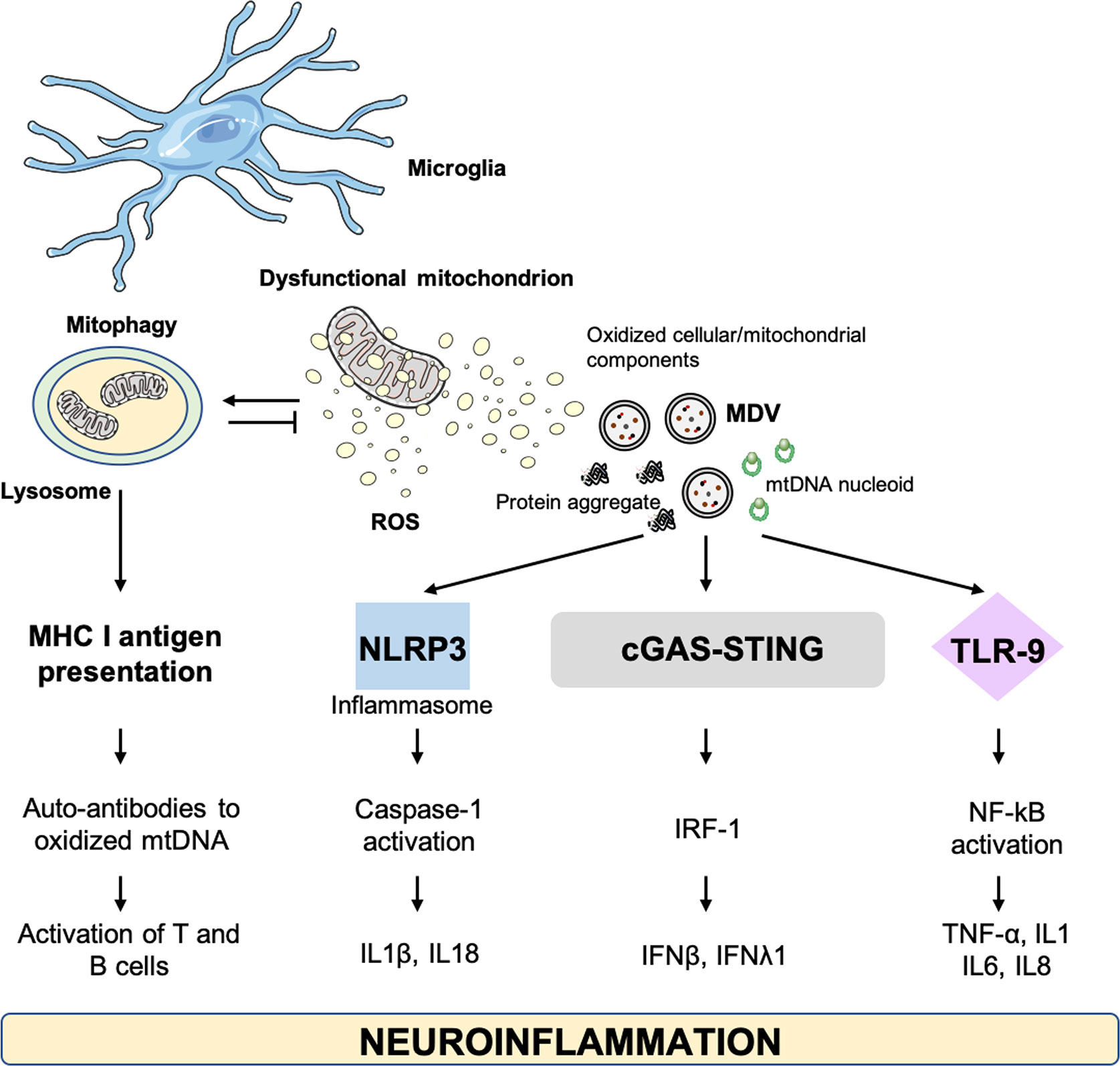
Figure 4 Cellular alterations and damage-associated molecular patterns involved in neuroinflammation. cGAS–STING, GMP-AMP synthase–stimulator of interferon genes; IFN, interferon; IL, interleukin; IRF-1, IFN-regulatory factor 1; MDV, mitochondrial derived vesicle; MHC, major histocompatibility complex; mtDNA, mitochondrial DNA; NLRP3, NOD-like receptor 3; NF-kB, nuclear factor kappa B; TLR, toll-like receptor; TNF-α, tumor necrosis factor alpha.
Alzheimer’s Disease
AD is the most common age-associated dementia and is characterized by neuronal degeneration mainly in the neocortex and the hippocampus (12). The extracellular deposition of amyloid beta (Aβ) aggregates and intracellular neurofibrillary tangles are distinctive histopathological traits of AD (12). Amyloid plaque deposition instigates microglia activation which, in turn, promotes the development of a pro-inflammatory environment through the release of inflammatory cytokines, including IL1β, IL6 and TNF-α (135). This neuroinflammatory response may represent an inter- and intracellular signaling system between microglia and astrocytes aimed at clearing damaged neuronal components (136). Indeed, in the setting of inefficient intracellular quality control (137), the persistence of damaged components and hyper-inflammation may favor the generation and spread of Aβ peptides, thereby triggering neurotoxicity (135).
Dysregulation of the endo-lysosomal system contributes to the generation of amyloid plaques and AD pathogenesis. Indeed, Aβ42 aggregates, the most pathogenic Aβ peptides, have been detected in the soma of neurons at the level of lysosomes or lysosome-derived components (138). Furthermore, neurons from AD transgenic mice show enlarged and dysfunctional MVBs in the presence of Aβ42 accrual (139). As a consequence of MVB dysfunction, higher levels of the amyloid precursor protein (APP) are secreted extracellularly in this murine model (139). In the endosomal compartment is also located the activity of the β-site APP-cleaving-enzyme (BACE1), a hub for the intracellular trafficking of APP and a relevant contributor to amyloid plaque generation (140). Conversely, a retrograde transport of APP from endosomes to the trans Golgi network is in place to reduce Aβ production (141). Notably, an impairment in the retromer complex activity has been involved in AD pathogenesis (141).
Circulating levels of HMGB1 and the soluble form of the receptor for advanced glycation end products (RAGE) have been detected in the serum of AD patients. The concentration of these DAMPs correlate with the extent of Aβ deposition (142). Moreover, HMGB1 and thrombin proteins have been identified as pro-inflammatory mediators contributing to dysfunction of the blood-brain barrier (BBB) (142). Similarly, serum levels of the brain-derived protein S100B have been associated with the severity of the disease (143). The administration of the S100B inhibitor pentamidine was able to reduce the levels of S100B and RAGE and blunt Aβ-induced gliosis and neuroinflammation in a mouse model of AD (144).
Mitochondrial dysfunction and the ensuing oxidative stress have also been involved in the pathogenesis of AD. Indeed, a lower copy number and a higher levels of mtDNA heteroplasmy have been found post-mortem in brains of people with AD (145–147). In addition, oxidative damage to mitochondrial components has been described as an early event in AD, which suggests a role for oxidative stress in disease pathogenesis (148, 149). Interestingly, Aβ peptide aggregates and neurofibrillary tangles can impact mitochondrial function by binding to proteins of the mitochondrial import machinery (150). As a result, increased ROS production occurs (151). The mitochondrial localization of fragments of the E4 variant of apolipoprotein E, the main susceptibility gene for sporadic AD, has also been reported and associated with mitochondrial dysfunction and oxidative stress in hippocampal neurons (152, 153).
While primary mitochondrial deficits have been observed in AD, aberrant mitochondria can also result from defective quality control mechanisms, especially mitophagy. In particular, a vicious circle between defective mitophagy and mitochondrial dysfunction may be triggered Aβ and phosphorylated Tau (p-Tau), ultimately leading to neuronal disruption (154–156). Altered expression of the mitophagy receptor disrupted-in-schizophrenia 1 (DISC1) has been reported in AD patients, transgenic AD mice, and cultured cells treated with Aβ (157). DISC1 is a promoter of mitophagy that binds to microtubule-associated proteins 1A/1B light chain 3 (LC3) and protects synaptic plasticity from the toxicity of Aβ accrual (157). The positive effect exerted by the pharmacological restoration of mitophagy on cognitive dysfunction and Aβ proteinopathy in APP/PS1 mice highlights the central role of defective mitophagy in AD pathogenesis (158). Following pro-mitophagy pharmacological treatments, reduced levels of Tau phosphorylation and mitigation of inflammation induced by microglia activation have also been observed (154). As such, a link between neuronal bioenergetic failure resulting from defective MQC, inflammation, and neuronal loss can also be hypothesized in AD (92). Following mitophagy impairment, cGAS–STING-DNA-mediated inflammation has been described in neurodegeneration (159) and NLRP3-induced inflammation has been observed in AD [reviewed in (160)].
A defective mitophagy and the resulting accrual of dysfunctional mitochondria in AD may instigate the extrusion of damaged organellar components with consequent stimulation of innate immunity (77, 78). Mitochondrial DAMPs have been retrieved within circulating EVs in several age-related conditions, including neurodegeneration (161, 162). Whether this mechanism is relevant to AD is worth being explored.
Parkinson’s Disease
PD is the second most common age-related neurodegenerative disorder (163) and is characterized by a progressive degeneration of dopaminergic neurons of the substantia nigra pars compacta and dopamine depletion in the striatum (164). These histopathological and biochemical abnormalities underlie a set of motor (i.e., bradykinesia, postural inability, rigidity, and tremor) and non-motor signs and symptoms (e.g., constipation, depression, sleep disorders, cognitive dysfunction) (164).
Neuroinflammation is a noticeable feature of PD (165). In particular, the HMGB1-TLR4 axis seems to plays an important role. Higher serum levels of HMGB1 and TLR4 protein have been detected in PD patients and correlated with disease stage (166). Moreover, the administration of anti-HMGB1 monoclonal antibody in a rat model of PD was able to reduce inflammation by preserving the BBB and lowering IL1β and IL6 secretion (167). The chemokine fractalkine (CX3CL1), which is mainly expressed by neurons and serves as a modulator of microglial-neuronal communication, has been indicated as a possible biomarker for PD (168). Increased levels of the S100B protein were also detected in the substantia nigra and cerebrospinal fluid of persons with PD and in the ventral midbrain of a murine PD model treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (169). Notably, the ablation of S100B in the murine model was neuroprotective by reducing microgliosis and the expression of both RAGE and TNF-α (169). Noticeably, a systemic inflammatory signature, involving IL8, IL9, and macrophage inflammatory protein 1α and 1β, has been identified in older adults with PD (170).
A defective cellular quality control, manifested by deposition of aberrant α-synuclein in dopaminergic neurons, is acknowledged as an important mechanism underlying neurodegeneration in PD (171). The accumulation of α-synuclein at the mitochondrial complex I has been shown to impair its activity (171). Such an inhibitory function, together with mutations in genes encoding for the mitochondrial regulators Parkin, PINK1, and protein deglycase DJ-1 have been linked with enhanced ROS generation in PD (172, 173) and α-synuclein aggregation (174–177). Derangements in mtDNA homeostasis, including large deletions, have been also detected in neuronal cells of the substantia nigra of persons with PD (178–180). These observations indicate that mitochondrial dysfunction plays a major role in the pathogenesis of familial PD (181). On the other hand, PD in its sporadic form recapitulates all major hallmarks of aging (182). Indeed, MQC derangements and the generation of DAMPs have been indicated as a major contributors to the co-occurrence of mitochondrial dysfunction and neuroinflammation in PD (159, 183, 184). An innate immune response triggered by defective autophagy and impaired disposal of damaged mitochondria has been described in mice lacking PINK1 or parkin gene (PARK2) (159). Moreover, the activity of the mitophagy mediator Parkin mediates a mitophagic control over inflammation (48). In particular, Parkin regulates adaptive immunity via the presentation of mitochondrial antigens to endosomes for loading onto MHC class I molecules (48). Similarly, the intracellular trafficking regulator RAB7A exerts also a mitochondrial antigen presentation role by controlling the fusion of MDVs with late endosome for their subsequent degradation (48). The function of RAB7A as a mitochondrial antigen-presenting system in immune cells via MDV trafficking ensures that the process can be finalized in the absence of PINK1 or Parkin (48). Indeed, alterations in PINK1/Parkin expression and activity in PD result in MQC dysregulation and possibly neuroinflammation via mitochondrial antigen presentation by MDVs (48). Recent work by our group described the presence of mitochondrial DAMPs among circulating EVs in older adults with PD along with a specific inflammatory signature (162). In particular, higher serum concentrations of small EVs including exosomes of endosomal origin were identified in older adults with PD (162). However, lower levels of MDVs were retrieved in people with PD relative to non-PD controls (162). A lower secretion of MDVs in older adults with PD is in keeping with the hypothesis of intracellular accrual of dysfunctional mitochondrial secondary to engulfed MQC system (162). According to this view, MDV generation may serve as a housekeeping mechanism that complements MQC to preserve cell homeostasis (15). A link between mitochondrial damage and inflammatory and metabolic disarrangements in PD has also been proposed (184, 185); however, the molecular mechanisms linking these processes are missing. An involvement of the cGAS–STING-DNA driven inflammation in neurodegeneration following mitophagy impairment has been reported (159). Indeed, higher circulating levels of the pro-inflammatory cytokines IL6 and IFNβ have been detected in Pink and Parkin knockout mice challenged with exhaustive exercise (159). Notably, the deletion of STING or the administration of IFNα/β receptor-blocking antibody was able to blunt this response, thus suggesting that the accrual of dysfunctional mitochondria may trigger inflammation in people with PD (159). Among the ever-growing list of molecules linking mitochondrial dysfunction to systemic inflammation in PD, the fibroblast growth factor 21 (FGF21) has emerged as a relevant mediator (162). Indeed, FGF21 has been indicated as a “mitokine” for its association with impaired MQC in neurons of murine models of tauopathy and prion disease (186). Taken as a whole, these findings suggest that a deeper understanding on the intracellular and extracellular trafficking of DAMPs and vesicles, including those of mitochondrial origin, may be key to unveiling relevant pathogenetic pathways of PD and, hence, novel targets for drug development.
Conclusion
Cells bearing DAMPs receptors sense and bind extracellular DAMPs as triggers of inflammation and fibrotic responses. Higher levels of circulating DAMPs have been identified during aging and related to inflamm-aging (3, 98, 187). A multicomponent senescence-associated secretory phenotype consisting of cytokines, chemokines (CXCLs), growth factors, and proteases has also been reported (188–191). While these secreted molecules contribute to preserving cell homeostasis in healthy tissues (192), the installment of an age-associated chronic secretory phenotype is a candidate pathway for the deployment of pathological hallmarks of aging, (e.g., inflamm-aging, tumorigenesis, loss of cell stemness). A core of circulating factors has been identified among plasma biomarkers of aging; however, their relationship with DAMPs is still unclear. The identification of circulating EVs stemming from altered regulation of the endosomal-lysosomal system in several age-related conditions, including cancer and neurodegeneration, holds hope for targeting this route for therapeutic purposes (13). Therefore, a deeper understanding of the complex, dynamic, intracellular and extracellular trafficking of DAMPs and vesicles, including those of mitochondrial origin, may be key to unveiling relevant pathogenic pathways and novel targets for drug development.
Author Contributions
Conceptualization: AP, CB, EM, and FG. Writing (original draft preparation): AP, CB, EM, and FG. Writing (review and editing): RC, HC-J, and RR. Supervision: FL and RB. Funding acquisition: CB and RB. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Innovative Medicine Initiative-Joint Undertaking (IMI-JU #115621), AIRC (Associazione Italiana per la Ricerca sul Cancro) Investigator grant 2016 #19068 to CB, Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR) to Consorzio Interuniversitario Biotecnologie (DM 1049, 29/12/2018; CIB N. 112/19 to CB), intramural research grants from the Università Cattolica del Sacro Cuore (D3.2 2020), and the nonprofit research foundation “Centro Studi Achille e Linda Lorenzon”.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The figures were drawn using the freely available Servier Medical Art resource (http://www.servier.com/Powerpoint-image-bank).
References
1. Albiger B, Dahlberg S, Henriques-Normark B, Normark S. Role of the innate immune system in host defence against bacterial infections: Focus on the Toll-like receptors. J Intern Med (2007) 261:511–28. doi: 10.1111/j.1365-2796.2007.01821.x
2. Lee MS, Kim Y-J. Pattern-recognition Receptor Signaling Initiated From Extracellular, Membrane, and Cytoplasmic Space. Mol Cells (2007) 23:1–10.
3. Feldman N, Rotter-Maskowitz A, Okun E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res Rev (2015) 24:29–39. doi: 10.1016/j.arr.2015.01.003
4. Medzhitov R. Inflammation 2010: New Adventures of an Old Flame. Cell (2010) 140:771–6. doi: 10.1016/j.cell.2010.03.006
5. Scrivo R, Vasile M, Bartosiewicz I, Valesini G. Inflammation as “common soil” of the multifactorial diseases. Autoimmun Rev (2011) 10:369–74. doi: 10.1016/j.autrev.2010.12.006
6. Relja B, Land WG. Damage-associated molecular patterns in trauma. Eur J Trauma Emerg Surg (2020) 46:751–75. doi: 10.1007/s00068-019-01235-w
7. Klumperman J, Raposo G. The complex ultrastructure of the endolysosomal system. Cold Spring Harb Perspect Biol (2014) 6:a016857. doi: 10.1101/cshperspect.a016857
8. Picca A, Calvani R, Coelho-Junior HJ, Landi F, Bernabei R, Marzetti E. Inter-Organelle Membrane Contact Sites and Mitochondrial Quality Control during Aging: A Geroscience View. Cells (2020) 9:598. doi: 10.3390/cells9030598
9. Vincent AE, Turnbull DM, Eisner V, Hajnóczky G, Picard M. Mitochondrial Nanotunnels. Trends Cell Biol (2017) 27:787–99. doi: 10.1016/j.tcb.2017.08.009
10. Picard M, McManus MJ, Csordás G, Várnai P, Dorn GW, Williams D, et al. Trans-mitochondrial coordination of cristae at regulated membrane junctions. Nat Commun (2015) 6:6259. doi: 10.1038/ncomms7259
11. Twig G, Elorza A, Molina AJA, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J (2008) 27:433–46. doi: 10.1038/sj.emboj.7601963
12. Rasmussen ML, Robertson GL, Gama V. Break on Through: Golgi-Derived Vesicles Aid in Mitochondrial Fission. Cell Metab (2020) 31:1047–9. doi: 10.1016/j.cmet.2020.05.010
13. Picca A, Guerra F, Calvani R, Coelho-Junior HJ, Bossola M, Landi F, et al. Generation and Release of Mitochondrial-Derived Vesicles in Health, Aging and Disease. J Clin Med (2020) 9:1440. doi: 10.3390/jcm9051440
14. Fafián-Labora JA, Rodríguez-Navarro JA, O’Loghlen A. Small Extracellular Vesicles Have GST Activity and Ameliorate Senescence-Related Tissue Damage. Cell Metab (2020) 32:71–86.e5. doi: 10.1016/j.cmet.2020.06.004
15. Todkar K, Chikhi L, Germain M. Mitochondrial interaction with the endosomal compartment in endocytosis and mitochondrial transfer. Mitochondrion (2019) 49:284–8. doi: 10.1016/j.mito.2019.05.003
16. Meldolesi J. Exosomes and Ectosomes in Intercellular Communication. Curr Biol (2018) 28:R435–44. doi: 10.1016/j.cub.2018.01.059
17. Gruenberg J, Stenmark H. The biogenesis of multivesicular endosomes. Nat Rev Mol Cell Biol (2004) 5:317–23. doi: 10.1038/nrm1360
18. Skotland T, Sandvig K, Llorente A. Lipids in exosomes: Current knowledge and the way forward. Prog Lipid Res (2017) 66:30–41. doi: 10.1016/j.plipres.2017.03.001
19. Henne WM, Buchkovich NJ, Emr SD. The ESCRT Pathway. Dev Cell (2011) 21:77–91. doi: 10.1016/j.devcel.2011.05.015
20. Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles (2018) 7:1535750. doi: 10.1080/20013078.2018.1535750
21. Falguières T, Luyet PP, Gruenberg J. Molecular assemblies and membrane domains in multivesicular endosome dynamics. Exp Cell Res (2009) 315:1567–73. doi: 10.1016/j.yexcr.2008.12.006
22. Williams RL, Urbé S. The emerging shape of the ESCRT machinery. Nat Rev Mol Cell Biol (2007) 8:355–68. doi: 10.1038/nrm2162
23. Bache KG, Brech A, Mehlum A, Stenmark H. Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J Cell Biol (2003) 162:435–42. doi: 10.1083/jcb.200302131
24. Razi M, Futter CE. Distinct roles for Tsg101 and Hrs in multivesicular body formation and inward vesiculation. Mol Biol Cell (2006) 17:3469–83. doi: 10.1091/mbc.e05-11-1054
25. Yorikawa C, Shibata H, Waguri S, Hatta K, Horii M, Katoh K, et al. Human CHMP6, a myristoylated ESCRT-III protein, interacts directly with an ESCRT-II component EAP20 and regulates endosomal cargo sorting. Biochem J (2005) 387:17–26. doi: 10.1042/BJ20041227
26. Stuffers S, Brech A, Stenmark H. ESCRT proteins in physiology and disease. Exp Cell Res (2009) 315:1619–26. doi: 10.1016/j.yexcr.2008.10.013
27. Luhtala N, Odorizzi G. Bro1 coordinates deubiquitination in the multivesicular body pathway by recruiting Doa4 to endosomes. J Cell Biol (2004) 166:717–29. doi: 10.1083/jcb.200403139
28. Babst M, Katzmann DJ, Estepa-Sabal EJ, Meerloo T, Emr SD. ESCRT-III: An endosome-associated heterooligomeric protein complex required for MVB sorting. Dev Cell (2002) 3:271–82. doi: 10.1016/S1534-5807(02)00220-4
29. Adell MAY, Vogel GF, Pakdel M, Müller M, Lindner H, Hess MW, et al. Coordinated binding of Vps4 to ESCRT-III drives membrane neck constriction during MVB vesicle formation. J Cell Biol (2014) 205:33–49. doi: 10.1083/jcb.201310114
30. Colombo M, Moita C, van Niel G, Kowal J, Vigneron J, Benaroch P, et al. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J Cell Sci (2013) 126:5553–65. doi: 10.1242/jcs.128868
31. Baietti MF, Zhang Z, Mortier E, Melchior A, Degeest G, Geeraerts A, et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat Cell Biol (2012) 14:677–85. doi: 10.1038/ncb2502
32. Friand V, David G, Zimmermann P. Syntenin and syndecan in the biogenesis of exosomes. Biol Cell (2015) 107:331–41. doi: 10.1111/boc.201500010
33. Hullin-Matsuda F, Taguchi T, Greimel P, Kobayashi T. Lipid compartmentalization in the endosome system. Semin Cell Dev Biol (2014) 31:48–56. doi: 10.1016/j.semcdb.2014.04.010
34. Conigliaro A, Corrado C, Fontana S, Alessandro R. “Exosome basic mechanisms”. In: Edelstein L, Smythies J, Quesenberry P, Noble D, editors. Exosomes: A Clinical Compendium. Cambridge, MA: Academic Press (2020). p. 1–21.
35. Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science (2008) 319:1244–7. doi: 10.1126/science.1153124
36. Charrin S, Jouannet S, Boucheix C, Rubinstein E. Tetraspanins at a glance. J Cell Sci (2014) 127:3641–8. doi: 10.1242/jcs.154906
37. Latysheva N, Muratov G, Rajesh S, Padgett M, Hotchin NA, Overduin M, et al. Syntenin-1 Is a New Component of Tetraspanin-Enriched Microdomains: Mechanisms and Consequences of the Interaction of Syntenin-1 with CD63. Mol Cell Biol (2006) 26:7707–18. doi: 10.1128/MCB.00849-06
38. Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci (2018) 75:193–208. doi: 10.1007/s00018-017-2595-9
39. Fader CM, Sánchez DG, Mestre MB, Colombo MI. TI-VAMP/VAMP7 and VAMP3/cellubrevin: two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim Biophys Acta (2009) 1793:1901–16. doi: 10.1016/j.bbamcr.2009.09.011
40. Sinha S, Hoshino D, Hong NH, Kirkbride KC, Grega-Larson NE, Seiki M, et al. Cortactin promotes exosome secretion by controlling branched actin dynamics. J Cell Biol (2016) 214:197–13. doi: 10.1083/jcb.201601025
41. Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol (2010) 12:19–30. doi: 10.1038/ncb2000
42. Bobrie A, Krumeich S, Reyal F, Recchi C, Moita LF, Seabra MC, et al. Rab27a supports exosome-dependent and -independent mechanisms that modify the tumor microenvironment and can promote tumor progression. Cancer Res (2012) 72:4920–30. doi: 10.1158/0008-5472.CAN-12-0925
43. Hsu C, Morohashi Y, Yoshimura SI, Manrique-Hoyos N, Jung SY, Lauterbach MA, et al. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A-C. J Cell Biol (2010) 189:223–32. doi: 10.1083/jcb.200911018
44. Wegener CS, Malerød L, Pedersen NM, Prodiga C, Bakke O, Stenmark H, et al. Ultrastructural characterization of giant endosomes induced by GTPase-deficient Rab5. Histochem Cell Biol (2010) 133:41–55. doi: 10.1007/s00418-009-0643-8
45. Guerra F, Paiano A, Migoni D, Girolimetti G, Perrone AM, De Iaco P, et al. Modulation of RAB7A Protein Expression Determines Resistance to Cisplatin through Late Endocytic Pathway Impairment and Extracellular Vesicular Secretion. Cancers (Basel) (2019) 11:52. doi: 10.3390/cancers11010052
46. Guerra F, Bucci C. Role of the RAB7 protein in tumor progression and cisplatin chemoresistance. Cancers (Basel) (2019) 11:1096. doi: 10.3390/cancers11081096
47. Soubannier V, McLelland G-L, Zunino R, Braschi E, Rippstein P, Fon EA, et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol (2012) 22:135–41. doi: 10.1016/j.cub.2011.11.057
48. Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M, et al. Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell (2016) 166:314–27. doi: 10.1016/j.cell.2016.05.039
49. Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, et al. Cargo-Selected Transport from the Mitochondria to Peroxisomes Is Mediated by Vesicular Carriers. Curr Biol (2008) 18:102–8. doi: 10.1016/j.cub.2007.12.038
50. Soubannier V, Rippstein P, Kaufman BA, Shoubridge EA, McBride HM. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PloS One (2012) 7:e52830. doi: 10.1371/journal.pone.0052830
51. McLelland G-L, Soubannier V, Chen CX, McBride HM, Fon EA. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J (2014) 33:282–95. doi: 10.1002/embj.201385902
52. Trinh J, Farrer M. Advances in the genetics of Parkinson disease. Nat Rev Neurol (2013) 9:445–54. doi: 10.1038/nrneurol.2013.132
53. Guerra F, Girolimetti G, Beli R, Mitruccio M, Pacelli C, Ferretta A, et al. Synergistic Effect of Mitochondrial and Lysosomal Dysfunction in Parkinson’s Disease. Cells (2019) 8:452. doi: 10.3390/cells8050452
54. Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol (2012) 2:120080. doi: 10.1098/rsob.120080
55. Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 Binding to the TOM Complex and Alternate Intracellular Membranes in Recruitment and Activation of the E3 Ligase Parkin. Dev Cell (2012) 22:320–33. doi: 10.1016/j.devcel.2011.12.014
56. Yamano K, Youle RJ. PINK1 is degraded through the N-end rule pathway. Autophagy (2013) 9:1758–69. doi: 10.4161/auto.24633
57. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol (2008) 183:795–803. doi: 10.1083/jcb.200809125
58. Sugiura A, McLelland G-L, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J (2014) 33:2142–56. doi: 10.15252/embj.201488104
59. Yurkova IL, Stuckert F, Kisel MA, Shadyro OI, Arnhold J, Huster D. Formation of phosphatidic acid in stressed mitochondria. Arch Biochem Biophys (2008) 480:17–26. doi: 10.1016/j.abb.2008.09.007
60. Tatsuta T, Langer T. AAA proteases in mitochondria: diverse functions of membrane-bound proteolytic machines. Res Microbiol (2009) 160:711–17. doi: 10.1016/j.resmic.2009.09.005
61. Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol (2010) 191:1367–80. doi: 10.1083/jcb.201007013
62. Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RLJ, et al. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet (2011) 20:1726–37. doi: 10.1093/hmg/ddr048
63. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol (2011) 12:9–14. doi: 10.1038/nrm3028
64. Picca A, Guerra F, Calvani R, Bucci C, Lo Monaco MR, Bentivoglio AR, et al. Mitochondrial Dysfunction and Aging: Insights from the Analysis of Extracellular Vesicles. Int J Mol Sci (2019) 20:805. doi: 10.3390/ijms20040805
65. Picca A, Lezza AMS, Leeuwenburgh C, Pesce V, Calvani R, Bossola M, et al. Circulating Mitochondrial DNA at the Crossroads of Mitochondrial Dysfunction and Inflammation During Aging and Muscle Wasting Disorders. Rejuvenation Res (2018) 21:350–9. doi: 10.1089/rej.2017.1989
66. Koyama S, Ishii KJ, Coban C, Akira S. Innate immune response to viral infection. Cytokine (2008) 43:336–41. doi: 10.1016/j.cyto.2008.07.009
67. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature (2001) 413:732–8. doi: 10.1038/35099560
68. Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, et al. IKKE and TBKI are essential components of the IRF3 signalling pathway. Nat Immunol (2003) 4:491–6. doi: 10.1038/ni921
69. Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, et al. The roles of two IκB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med (2004) 199:1641–50. doi: 10.1084/jem.20040520
70. Sharma S, TenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science (2003) 300:1148–51. doi: 10.1126/science.1081315
71. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol (2005) 6:981–8. doi: 10.1038/ni1243
72. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell (2005) 122:669–82. doi: 10.1016/j.cell.2005.08.012
73. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol (2004) 5:730–7. doi: 10.1038/ni1087
74. Crosse KM, Monson EA, Beard MR, Helbig KJ. Interferon-Stimulated Genes as Enhancers of Antiviral Innate Immune Signaling. J Innate Immun (2018) 10:85–93. doi: 10.1159/000484258
75. Cardon LR, Burge C, Clayton DA, Karlin S. Pervasive CpG suppression in animal mitochondrial genomes. Proc Natl Acad Sci USA (1994) 91:3799–803. doi: 10.1073/pnas.91.9.3799
76. Pollack Y, Kasir J, Shemer R, Metzger S, Szyf M. Methylation Pattern of Mouse Mitochondrial DNA. Nucleic Acids Res (1984) 12:4811–24. doi: 10.1093/nar/12.12.4811
77. Collins LV, Hajizadeh S, Holme E, Jonsson I-M, Tarkowski A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol (2004) 75:995–1000. doi: 10.1189/jlb.0703328
78. Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell (2014) 54:289–96. doi: 10.1016/j.molcel.2014.03.040
79. Takeuchi O, Akira S. Pattern Recognition Receptors and Inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
80. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464:104–7. doi: 10.1038/nature08780
81. Zhang Q, Itagaki K, Hauser CJ. Mitochondrial DNA is released by shock and activates neutrophils via P38 map kinase. Shock (2010) 34:55–9. doi: 10.1097/SHK.0b013e3181cd8c08
82. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469:221–5. doi: 10.1038/nature09663
83. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity (2012) 36:401–14. doi: 10.1016/j.immuni.2012.01.009
84. White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type i IFN production. Cell (2014) 159:1549–62. doi: 10.1016/j.cell.2014.11.036
85. Rongvaux A, Jackson R, Harman CCD, Li T, West AP, De Zoete MR, et al. Apoptotic caspases prevent the induction of type i interferons by mitochondrial DNA. Cell (2014) 159:1563–77. doi: 10.1016/j.cell.2014.11.037
86. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature (2015) 520:553–7. doi: 10.1038/nature14156
87. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
88. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med (2016) 22:146–53. doi: 10.1038/nm.4027
89. Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med (2016) 213:697–713. doi: 10.1084/jem.20151876
90. Maugeri N, Capobianco A, Rovere-Querini P, Ramirez GA, Tombetti E, Della Valle P, et al. Platelet microparticles sustain autophagy-associated activation of neutrophils in systemic sclerosis. Sci Transl Med (2018) 10:eaao3089. doi: 10.1126/scitranslmed.aao3089
91. Martinon F, Burns K, Tschopp J. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
92. Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature (2012) 481:278–86. doi: 10.1038/nature10759
93. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature (2018) 560:198–203. doi: 10.1038/s41586-018-0372-z
94. Nakahira K, Haspel JA, Rathinam VAK, Lee S-J, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12:222–30. doi: 10.1038/ni.1980
95. Hernandez C, Huebener P, Schwabe RF. Damage-associated molecular patterns in cancer: A double-edged sword. Oncogene (2016) 35:5931–41. doi: 10.1038/onc.2016.104
96. Grivennikov SI, Greten FR, Karin M. Immunity, Inflammation, and Cancer. Cell (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
97. Haybaeck J, Zeller N, Wolf MJ, Weber A, Wagner U, Kurrer MO, et al. A Lymphotoxin-Driven Pathway to Hepatocellular Carcinoma. Cancer Cell (2009) 16:295–308. doi: 10.1016/j.ccr.2009.08.021
98. Huang J, Xie Y, Sun X, Zeh HJ, Kang R, Lotze MT, et al. DAMPs, ageing, and cancer: The “DAMP Hypothesis.” Ageing Res Rev (2015) 24:3–16. doi: 10.1016/j.arr.2014.10.004
99. Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, et al. Emerging role DAMPs derived from mitochondria in inflammation. Trends Immunol (2011) 32:157–64. doi: 10.1016/j.it.2011.01.005
100. Al Amir Dache Z, Otandault A, Tanos R, Pastor B, Meddeb R, Sanchez C, et al. Blood contains circulating cell-free respiratory competent mitochondria. FASEB J (2020) 34:3616–30. doi: 10.1096/fj.201901917RR
101. Phinney DG, Di Giuseppe M, Njah J, Sala E, Shiva S, St Croix CM, et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat Commun (2015) 6:8472. doi: 10.1038/ncomms9472
102. Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, et al. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med (2012) 18:759–65. doi: 10.1038/nm.2736
103. Dong LF, Kovarova J, Bajzikova M, Bezawork-Geleta A, Svec D, Endaya B, et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. Elife (2017) 6:e22187. doi: 10.7554/eLife.22187
104. Spees JL, Olson SD, Whitney MJ, Prockop DJ. Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci USA (2006) 103:1283–8. doi: 10.1073/pnas.0510511103
105. Griessinger E, Moschoi R, Biondani G, Peyron J-F. Mitochondrial Transfer in the Leukemia Microenvironment. Trends Cancer (2017) 3:828–39. doi: 10.1016/j.trecan.2017.10.003
106. Rodriguez AM, Nakhle J, Griessinger E, Vignais ML. Intercellular mitochondria trafficking highlighting the dual role of mesenchymal stem cells as both sensors and rescuers of tissue injury. Cell Cycle (2018) 17:712–21. doi: 10.1080/15384101.2018.1445906
107. Mahrouf-Yorgov M, Augeul L, Da Silva CC, Jourdan M, Rigolet M, Manin S, et al. Mesenchymal stem cells sense mitochondria released from damaged cells as danger signals to activate their rescue properties. Cell Death Differ (2017) 24:1224–38. doi: 10.1038/cdd.2017.51
108. Patel D, Rorbach J, Downes K, Szukszto MJ, Pekalski ML, Minczuk M. Macropinocytic entry of isolated mitochondria in epidermal growth factor-activated human osteosarcoma cells. Sci Rep (2017) 7:12886. doi: 10.1038/s41598-017-13227-0
109. Payen VL, Porporato PE, Baselet B, Sonveaux P. Metabolic changes associated with tumor metastasis, part 1: Tumor pH, glycolysis and the pentose phosphate pathway. Cell Mol Life Sci (2016) 73:1333–48. doi: 10.1007/s00018-015-2098-5
110. Martinez-Outschoorn UE, Pavlides S, Sotgia F, Lisanti MP. Mitochondrial biogenesis drives tumor cell proliferation. Am J Pathol (2011) 178:1949–52. doi: 10.1016/j.ajpath.2011.03.002
111. Sansone P, Savini C, Kurelac I, Chang Q, Amato LB, Strillacci A, et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc Natl Acad Sci U.S.A. (2017) 114:E9066–75. doi: 10.1073/pnas.1704862114
112. Jang SC, Crescitelli R, Cvjetkovic A, Belgrano V, Olofsson Bagge R, Sundfeldt K, et al. Mitochondrial protein enriched extracellular vesicles discovered in human melanoma tissues can be detected in patient plasma. J Extracell Vesicles (2019) 8:1635420. doi: 10.1080/20013078.2019.1635420
113. Turchinovich A, Weiz L, Langheinz A, Burwinkel B. Characterization of extracellular circulating microRNA. Nucleic Acids Res (2011) 39:7223–33. doi: 10.1093/nar/gkr254
114. Allam R, Kumar SVR, Darisipudi MN, Anders H-J. Extracellular histones in tissue injury and inflammation. J Mol Med (2014) 92:465–72. doi: 10.1007/s00109-014-1148-z
115. Wang W, Lotze MT. Good things come in small packages: Exosomes, immunity and cancer. Cancer Gene Ther (2014) 21:139–41. doi: 10.1038/cgt.2014.14
116. Ding G, Zhou L, Qian Y, Fu M, Chen J, Chen J, et al. Pancreatic cancer-derived exosomes transfer miRNAs to dendritic cells and inhibit RFXAP expression via miR-212-3p. Oncotarget (2015) 6:29877–88. doi: 10.18632/oncotarget.4924
117. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature (2006) 439:682–7. doi: 10.1038/nature04444
118. Kim DH, Kim HR, Choi YJ, Kim SY, Lee JE, Sung KJ, et al. Exosomal PD-L1 promotes tumor growth through immune escape in non-small cell lung cancer. Exp Mol Med (2019) 51:1–13. doi: 10.1038/s12276-019-0295-2
119. Patel GK, Khan MA, Bhardwaj A, Srivastava SK, Zubair H, Patton MC, et al. Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK. Br J Cancer (2017) 116:609–19. doi: 10.1038/bjc.2017.18
120. Deng T, Zhang H, Yang H, Wang H, Bai M, Sun W, et al. Exosome miR-155 Derived from Gastric Carcinoma Promotes Angiogenesis by Targeting the c-MYB/VEGF Axis of Endothelial Cells. Mol Ther Nucleic Acids (2020) 19:1449–59. doi: 10.1016/j.omtn.2020.01.024
121. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124:783–801. doi: 10.1016/j.cell.2006.02.015
122. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol (2008) 8:59–73. doi: 10.1038/nri2216
123. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392:245–52. doi: 10.1038/32588
124. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med (2007) 13:54–61. doi: 10.1038/nm1523
125. Lotze MT, Zeh HJ, Rubartelli A, Sparvero LJ, Amoscato AA, Washburn NR, et al. The grateful dead: Damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev (2007) 220:60–81. doi: 10.1111/j.1600-065X.2007.00579.x
126. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β-dependent adaptive immunity against tumors. Nat Med (2009) 15:1170–8. doi: 10.1038/nm.2028
127. Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, et al. Critical role for NALP3/CIAS1/cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity (2006) 24:317–27. doi: 10.1016/j.immuni.2006.02.004
128. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med (2007) 13:1050–9. doi: 10.1038/nm1622
129. Yin J, Valin KL, Dixon ML, Leavenworth JW. The Role of Microglia and Macrophages in CNS Homeostasis, Autoimmunity, and Cancer. J Immunol Res (2017) 2017:5150678. doi: 10.1155/2017/5150678
130. von Bernhardi R, Eugenín-von Bernhardi L, Eugenín J. Microglial cell dysregulation in brain aging and neurodegeneration. Front Aging Neurosci (2015) 7:124. doi: 10.3389/fnagi.2015.00124
131. Koellhoffer E, McCullough L, Ritzel R. Old Maids: Aging and Its Impact on Microglia Function. Int J Mol Sci (2017) 18:769. doi: 10.3390/ijms18040769
132. Femminella GD, Dani M, Wood M, Fan Z, Calsolaro V, Atkinson R, et al. Microglial activation in early Alzheimer trajectory is associated with higher gray matter volume. Neurology (2019) 92:E1331–343. doi: 10.1212/WNL.0000000000007133
133. George S, Rey NL, Tyson T, Esquibel C, Meyerdirk L, Schulz E, et al. Microglia affect α-synuclein cell-to-cell transfer in a mouse model of Parkinson’s disease. Mol Neurodegener (2019) 14:34. doi: 10.1186/s13024-019-0335-3
134. Venegas C, Heneka MT. Danger-associated molecular patterns in Alzheimer’s disease. J Leukoc Biol (2017) 101:87–98. doi: 10.1189/jlb.3MR0416-204R
135. Wang WY, Tan MS, Yu JT, Tan L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med (2015) 3:136. doi: 10.3978/j.issn.2305-5839.2015.03.49
136. Kim S-M, Song J, Kim S, Han C, Park MH, Koh Y, et al. Identification of peripheral inflammatory markers between normal control and Alzheimer’s disease. BMC Neurol (2011) 11:51. doi: 10.1186/1471-2377-11-51
137. Neumann H, Kotter MR, Franklin RJM. Debris Clearance by Microglia: An Essential Link Between Degeneration and Regeneration. Brain (2009) 132:288–95. doi: 10.1093/brain/awn109
138. D’Andrea MR, Nagele RG, Wang HY, Peterson PA, Lee DHS. Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer’s disease. Histopathology (2001) 38:120–34. doi: 10.1046/j.1365-2559.2001.01082.x
139. Willén K, Edgar JR, Hasegawa T, Tanaka N, Futter CE, Gouras GK. Aβ accumulation causes MVB enlargement and is modelled by dominant negative VPS4A. Mol Neurodegener (2017) 12:61. doi: 10.1186/s13024-017-0203-y
140. Tang BL. Neuronal protein trafficking associated with Alzheimer disease: From APP and BACE1 to glutamate receptors. Cell Adhes Migr (2009) 3:118–28. doi: 10.4161/cam.3.1.7254
141. Zhang QY, Tan MS, Yu JT, Tan L. The Role of Retromer in Alzheimer’s Disease. Mol Neurobiol (2016) 53:4201–9. doi: 10.1007/s12035-015-9366-0
142. Festoff BW, Sajja RK, van Dreden P, Cucullo L. HMGB1 and thrombin mediate the blood-brain barrier dysfunction acting as biomarkers of neuroinflammation and progression to neurodegeneration in Alzheimer’s disease. J Neuroinflamm (2016) 13:194. doi: 10.1186/s12974-016-0670-z
143. Chaves ML, Camozzato AL, Ferreira ED, Piazenski I, Kochhann R, Dall’Igna O, et al. Serum levels of S100B and NSE proteins in Alzheimer’s disease patients. J Neuroinflamm (2010) 7:6. doi: 10.1186/1742-2094-7-6
144. Cirillo C, Capoccia E, Iuvone T, Cuomo R, Sarnelli G, Steardo L, et al. S100B Inhibitor Pentamidine Attenuates Reactive Gliosis and Reduces Neuronal Loss in a Mouse Model of Alzheimer’s Disease. BioMed Res Int (2015) 2015:508342. doi: 10.1155/2015/508342
145. Coskun PE, Beal MF, Wallace DC. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci USA (2004) 101:10726–31. doi: 10.1073/pnas.0403649101
146. Rodríguez-Santiago B, Casademont J, Nunes V. Is mitochondrial DNA depletion involved in Alzheimer’s disease? Eur J Hum Genet (2001) 9:279–85. doi: 10.1038/sj.ejhg.5200629
147. Wei W, Keogh MJ, Wilson I, Coxhead J, Ryan S, Rollinson S, et al. Mitochondrial DNA point mutations and relative copy number in 1363 disease and control human brains. Acta Neuropathol Commun (2017) 5:13. doi: 10.1186/s40478-016-0404-6
148. De La Monte SM, Wands JR. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J Alzheimer Dis (2006) 9:167–81. doi: 10.3233/JAD-2006-9209
149. Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, et al. Oxidative Damage Is the Earliest Event in Alzheimer Disease. J Neuropathol Exp Neurol (2001) 60:759–67. doi: 10.1093/jnen/60.8.759
150. Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci (2006) 26:9057–68. doi: 10.1523/JNEUROSCI.1469-06.2006
151. Eckert A, Schulz KL, Rhein V, Götz J. Convergence of amyloid-β and tau pathologies on mitochondria in vivo. Mol Neurobiol (2010) 4:107–14. doi: 10.1007/s12035-010-8109-5
152. Montagne A, Nation DA, Sagare AP, Barisano G, Sweeney MD, Chakhoyan A, et al. APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature (2020) 581:71–6. doi: 10.1038/s41586-020-2247-3
153. Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci USA (2006) 103:5644–51. doi: 10.1073/pnas.0600549103
154. Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci (2019) 22:401–12. doi: 10.1038/s41593-018-0332-9
155. Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, et al. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci (2017) 40:151–66. doi: 10.1016/j.tins.2017.01.002
156. Du F, Yu Q, Yan S, Hu G, Lue LF, Walker DG, et al. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain (2017) 140:3233–51. doi: 10.1093/brain/awx258
157. Wang ZT, Lu MH, Zhang Y, Ji WL, Lei L, Wang W, et al. Disrupted-in-schizophrenia-1 protects synaptic plasticity in a transgenic mouse model of Alzheimer’s disease as a mitophagy receptor. Aging Cell (2019) 18:e12860. doi: 10.1111/acel.12860
158. Fang EF. Mitophagy and NAD + inhibit Alzheimer disease. Autophagy (2019) 15:1112–4. doi: 10.1080/15548627.2019.1596497
159. Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature (2018) 561:258–62. doi: 10.1038/s41586-018-0448-9
160. Liu Y, Dai Y, Li Q, Chen C, Chen H, Song Y, et al. Beta-amyloid activates NLRP3 inflammasome via TLR4 in mouse microglia. Neurosci Lett (2020) 736:135279. doi: 10.1016/j.neulet.2020.135279
161. Picca A, Guerra F, Calvani R, Bucci C, Lo Monaco MR, Bentivoglio AR, et al. Mitochondrial-Derived Vesicles as Candidate Biomarkers in Parkinson’s Disease: Rationale, Design and Methods of the EXosomes in PArkiNson Disease (EXPAND) Study. Int J Mol Sci (2019) 20:2373. doi: 10.3390/ijms20102373
162. Picca A, Guerra F, Calvani R, Marini F, Biancolillo A, Landi G, et al. Mitochondrial Signatures in Circulating Extracellular Vesicles of Older Adults with Parkinson’s Disease: Results from the EXosomes in PArkiNson’s Disease (EXPAND) Study. J Clin Med (2020) 9:504. doi: 10.3390/jcm9020504
163. Ray Dorsey E, Elbaz A, Nichols E, Abd-Allah F, Abdelalim A, Adsuar JC, et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol (2018) 17:939–53. doi: 10.1016/S1474-4422(18)30295-3
164. Alexander GE. Biology of Parkinson’s disease: Pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialog Clin Neurosci (2004) 6:259–80.
165. Herrero MT, Estrada C, Maatouk L, Vyas S. Inflammation in Parkinson’s disease: Role of glucocorticoids. Front Neuroanat (2015) 9:32. doi: 10.3389/fnana.2015.00032
166. Yang Y, Han C, Guo L, Guan Q. High expression of the HMGB1–TLR4 axis and its downstream signaling factors in patients with Parkinson’s disease and the relationship of pathological staging. Brain Behav (2018) 8:e00948. doi: 10.1002/brb3.948
167. Sasaki T, Liu K, Agari T, Yasuhara T, Morimoto J, Okazaki M, et al. Anti-high mobility group box 1 antibody exerts neuroprotection in a rat model of Parkinson’s disease. Exp Neurol (2016) 275:220–31. doi: 10.1016/j.expneurol.2015.11.003
168. Angelopoulou E, Paudel YN, Shaikh MF, Piperi C. Fractalkine (CX3CL1) signaling and neuroinflammation in Parkinson’s disease: Potential clinical and therapeutic implications. Pharmacol Res (2020) 158:104930. doi: 10.1016/j.phrs.2020.104930
169. Sathe K, Maetzler W, Lang JD, Mounsey RB, Fleckenstein C, Martin HL, et al. S100B is increased in Parkinson’s disease and ablation protects against MPTP-induced toxicity through the RAGE and TNF-α pathway. Brain (2012) 135:3336–47. doi: 10.1093/brain/aws250
170. Calvani R, Picca A, Landi G, Marini F, Biancolillo A, Coelho-Junior HJ, et al. A novel multi-marker discovery approach identifies new serum biomarkers for Parkinson’s disease in older people: an EXosomes in PArkiNson Disease (EXPAND) ancillary study. GeroScience (2020) 42:1323–34. doi: 10.1007/s11357-020-00192-2
171. Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem (2008) 283:9089–100. doi: 10.1074/jbc.M710012200
172. Dodson MW, Guo M. Pink1, Parkin, DJ-1 and mitochondrial dysfunction in Parkinson’s disease. Curr Opin Neurobiol (2007) 17:331–7. doi: 10.1016/j.conb.2007.04.010
173. Ostrerova-Golts N, Petrucelli L, Hardy J, Lee JM, Farer M, Wolozin B. The A53T α-synuclein mutation increases iron-dependent aggregation and toxicity. J Neurosci (2000) 20:6048–54. doi: 10.1523/JNEUROSCI.20-16-06048.2000
174. Orth M, Tabrizi SJ, Tomlinson C, Messmer K, Korlipara LVP, Schapira AHV, et al. G209A mutant alpha synuclein expression specifically enhances dopamine induced oxidative damage. Neurochem Int (2004) 45:669–76. doi: 10.1016/j.neuint.2004.03.029
175. Junn E, Mouradian MM. Human α-Synuclein over-expression increases intracellular reactive oxygen species levels and susceptibility to dopamine. Neurosci Lett (2002) 320:146–50. doi: 10.1016/S0304-3940(02)00016-2
176. Paxinou E, Chen Q, Weisse M, Giasson BI, Norris EH, Rueter SM, et al. Induction of α-synuclein aggregation by intracellular nitrative insult. J Neurosci (2001) 21:8053–61. doi: 10.1523/JNEUROSCI.21-20-08053.2001
177. Ahn TB, Kim SY, Kim JY, Park SS, Lee DS, Min HJ, et al. α-Synuclein gene duplication is present in sporadic Parkinson disease. Neurology (2008) 70:43–9. doi: 10.1212/01.wnl.0000271080.53272.c7
178. Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet (2006) 38:515–7. doi: 10.1038/ng1769
179. Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet (2006) 38:518–20. doi: 10.1038/ng1778
180. Dölle C, Flønes I, Nido GS, Miletic H, Osuagwu N, Kristoffersen S, et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat Commun (2016) 7:13548. doi: 10.1038/ncomms13548
181. Park JS, Davis RL, Sue CM. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr Neurol Neurosci Rep (2018) 18:21. doi: 10.1007/s11910-018-0829-3
182. Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol (2019) 15:565–81. doi: 10.1038/s41582-019-0244-7
183. Cho B, Kim T, Huh YJ, Lee J, Lee Y II. Amelioration of mitochondrial quality control and proteostasis by natural compounds in Parkinson’s disease models. Int J Mol Sci (2019) 20:5208. doi: 10.3390/ijms20205208
184. White AJ, Wijeyekoon RS, Scott KM, Gunawardana NP, Hayat S, Solim IH, et al. The Peripheral Inflammatory Response to Alpha-Synuclein and Endotoxin in Parkinson’s Disease. Front Neurol (2018) 9:946. doi: 10.3389/fneur.2018.00946
185. Picca A, Calvani R, Landi G, Marini F, Biancolillo A, Gervasoni J, et al. Circulating amino acid signature in older people with Parkinson’s disease: A metabolic complement to the EXosomes in PArkiNson Disease (EXPAND) study. Exp Gerontol (2019) 128:110766. doi: 10.1016/j.exger.2019.110766
186. Restelli LM, Oettinghaus B, Halliday M, Agca C, Licci M, Sironi L, et al. Neuronal Mitochondrial Dysfunction Activates the Integrated Stress Response to Induce Fibroblast Growth Factor 21. Cell Rep (2018) 24:1407–14. doi: 10.1016/j.celrep.2018.07.023
187. Franceschi C, Campisi J. Chronic inflammation (Inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci (2014) 69:S4–9. doi: 10.1093/gerona/glu057
188. Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell (2008) 133:1006–18. doi: 10.1016/j.cell.2008.03.038
189. Coppé J-P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PloS Biol (2008) 6:2853–68. doi: 10.1371/journal.pbio.0060301
190. Coppé JP, Patil CK, Rodier F, Krtolica A, Beauséjour CM, Parrinello S, et al. Campisi J. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PloS One (2010) 5:e9188. doi: 10.1371/journal.pone.0009188
191. Kuilman T, Michaloglou C, Vredeveld LCW, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell (2008) 133:1019–31. doi: 10.1016/j.cell.2008.03.039
Keywords: Alzheimer’s disease, damage-associated molecular patterns, endo-lysosomal system, inflammation, innate immunity, mitochondrial-derived vesicles, Parkinson’s disease, quality control system
Citation: Picca A, Guerra F, Calvani R, Coelho-Júnior HJ, Landi F, Bernabei R, Romano R, Bucci C and Marzetti E (2020) Extracellular Vesicles and Damage-Associated Molecular Patterns: A Pandora’s Box in Health and Disease. Front. Immunol. 11:601740. doi: 10.3389/fimmu.2020.601740
Received: 01 September 2020; Accepted: 21 October 2020;
Published: 16 November 2020.
Edited by:
Sy Seong, Seoul National University, South KoreaReviewed by:
Angelo A. Manfredi, Vita-Salute San Raffaele University, ItalyRaymond B. Birge, Rutgers, The State University of New Jersey, United States
Copyright © 2020 Picca, Guerra, Calvani, Coelho-Júnior, Landi, Bernabei, Romano, Bucci and Marzetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Riccardo Calvani, cmljY2FyZG8uY2FsdmFuaUBndWVzdC5wb2xpY2xpbmljb2dlbWVsbGkuaXQ=; Roberto Bernabei, cm9iZXJ0by5iZXJuYWJlaUB1bmljYXR0Lml0
†These authors have contributed equally to this work