 Geoffrey P. Dobson
Geoffrey P. Dobson Erik Biros
Erik Biros Hayley L. Letson
Hayley L. Letson Jodie L. Morris
Jodie L. Morris- Heart, Trauma and Sepsis Research Laboratory, College of Medicine and Dentistry, James Cook University, Townsville, QLD, Australia
We present a brief history of the immune response and show that Metchnikoff’s theory of inflammation and phagocytotic defense was largely ignored in the 20th century. For decades, the immune response was believed to be triggered centrally, until Lafferty and Cunningham proposed the initiating signal came from the tissues. This shift opened the way for Janeway’s pattern recognition receptor theory, and Matzinger’s danger model. All models failed to appreciate that without inflammation, there can be no immune response. The situation changed in the 1990s when cytokine biology was rapidly advancing, and the immune system’s role expanded from host defense, to the maintenance of host health. An inflammatory environment, produced by immune cells themselves, was now recognized as mandatory for their attack, removal and repair functions after an infection or injury. We explore the cellular programs of the immune response, and the role played by cytokines and other mediators to tailor the right response, at the right time. Normally, the immune response is robust, self-limiting and restorative. However, when the antigen load or trauma exceeds the body’s internal tolerances, as witnessed in some COVID-19 patients, excessive inflammation can lead to increased sympathetic outflows, cardiac dysfunction, coagulopathy, endothelial and metabolic dysfunction, multiple organ failure and death. Currently, there are few drug therapies to reduce excessive inflammation and immune dysfunction. We have been developing an intravenous (IV) fluid therapy comprising adenosine, lidocaine and Mg2+ (ALM) that confers a survival advantage by preventing excessive inflammation initiated by sepsis, endotoxemia and sterile trauma. The multi-pronged protection appears to be unique and may provide a tool to examine the intersection points in the immune response to infection or injury, and possible ways to prevent secondary tissue damage, such as that reported in patients with COVID-19.
Living Systems: The Steady-State and Design Tolerances
A superficial consideration of the totality of the processes in the living being immediately shows that in the strictest sense a dynamic equilibrium is never present.
E. Pfluger (1877) (1) p57
Living systems are not equilibrium states; they are steady-states requiring a continual flow of matter, energy and exchange with the environment (2, 3). The concept of a steady-state began in 1877 with Pfluger’s “natural adjustments”, Bernard’s concept of “milieu intérieur” (1878), and Richet’s “living beings were stable but modifiable” (1900) (3, 4). It was not until the early 1920s that Walter Cannon combined these ideas into a unified scheme of homeostasis (5). Cannon argued that the living organism was in a dynamic state of constancy, with its constituent parts and processes being actively maintained in balance despite external fluctuations. He believed homeostasis was a systems phenomenon: “peculiar to living beings - involving, as they may, the brain and nerves, the heart, lungs, kidneys and spleen, all working cooperatively” (5). The addition of negative and positive feedback circuits to support homeostasis was not Cannon’s idea, but entered in the mid-1930s from Russian physiologist Pyotr Anokhin’s theory of functional systems (6). The steady-state was now viewed as comprising the sum of negative and positive feedback mechanisms that maintains a living system within a range of operational limits or tolerances. A stress, injury or infection was a challenge to the body’s steady-state, and the major goal of any new drug therapy, device or intervention is to restore homeostatic balance.
Exceeding The Body’s Defense Capability After A Barrier Breach
Except on few occasions, the patient appears to die from the body’s response to infection rather than from it.
Sir William Osler (1904)
During the life of an organism, the immune system continually senses and responds to barrier breaches and threats. If a breach occurs, the number of blood-borne and tissue-resident immune cells can change dramatically in seconds (7, 8). “Tissue residency” refers to immune cells that already reside in the tissue parenchyma or stroma, where they can roam freely without moving from tissue to tissue (9). A breach is defined as a break in epithelial continuity, external or internal, that may arise from a pathogen (e.g. viruses, bacteria, fungi, protozoa, or helminth) or sterile injury. Sterile injury is defined as a trauma in the absence of pathogen. However, trauma is rarely sterile and can be colonized by opportunistic pathogens, soon after a penetrating injury, which may lead to secondary infection. From our hunter-gatherer, protohuman origins, the body has developed, through natural selection, a defense system against infection or injury that is normally robust, self-limiting and restorative (4, 10, 11). The immune response normally neutralizes a pathogen or promotes wound healing. However, if the threat overwhelms the body’s internal tolerances, such as witnessed in some patients during the current COVD-19 pandemic or after major trauma, excessive sympathetic outflows, inflammation, coagulopathy, endothelial and metabolic dysfunction can occur leading to multiple organ failure and death (11, 12). Currently, it is not known, for example, why COVID-19 triggers such an explosive inflammatory response in some patients, and not in others (13–16). Understanding the mechanisms responsible for these different responses resides in the control of the immune system (17).
In this review we will: 1) present a brief history of the host’s immune response to infection or trauma, 2) discuss the importance of inflammation and underlying molecular defense mechanisms in the context of an infectious diseases like COVID-19, and 3) discuss how a new therapeutic approach using adenosine, lidocaine and magnesium (ALM) may alter the host’s phenotype to prevent or resolve hyperinflammation, and help return the system to “normal” operating conditions. We address the following questions:
• How does the host mount an immune defence against a pathogen?
• Is the 20th century self/nonself discrimination still a useful concept?
• How does the host discriminate infection from sterile injury?
• What role does local and systemic inflammation play in host defence?
• Are there common intersection or checkpoint points that could lead to new drug development to bolster the hosts’ immune defence against infection and sterile injury?
Brief History of Immune Defense: From Macrophages to Clonal Theory
When I first put forward the biological theory of inflammation eight years ago, I expressed the idea that this reaction is effected by the intermediation of a physiological continuity between “the cells of the connective tissue, those of the endothelial wall and the leucocytes, which form a complete chain and play the principal part in the inflammation of vertebrates.” The connective tissue cells which are first attacked, would, I thought, transmit the action to the vascular wall, the cells of which would contract to facilitate the passage of the white corpuscles.
E. Metchnikoff (1893) (18) p191
Origins of Cell and Humoral Theories of Immunity
Around 130 years ago, Russian zoologist and pathologist Elie Metchnikoff (1845–1916) was among the first to develop a cellular theory of inflammation and phagocytic defense against pathogens (18). The theory was built on Rudolf Virchow’s cellular basis of disease (19), and Pasteur’s pioneering research on vaccine development (20). For his model, Metchnikoff embraced Darwin’s laws of natural selection, and although he was not the first to observe phagocytosis and inflammation, he does appear to be the first to highlight the importance of blood-borne and tissue-resident macrophages (21, 22). Metchnikoff viewed inflammation as a highly integrative and restorative process (23). He also intuitively drew parallels between phagocytes devouring the tadpole’s tail, which was “eaten” at the appropriate time of metamorphosis, to wound repair and bacterial killing from amoeba to humans (23). As a sideline, but related, Metchnikoff anticipated the importance of the gut “microbiome” to immune daily health by eating yogurt, or other types of sour milk, to cultivate beneficial bacteria for host health (24).
Opponents of Metchnikoff’s cellular scheme advocated the antibody (or “antitoxin”) theory of immunity, which became more popular. The new movement was largely driven by Emil von Behring and Japanese bacteriologist Shibasaburo Kitasato, who argued that antibodies provide greater specificity to ward off foreign invaders than a freely mobile phagocyte in an inflammatory environment (25, 26). They showed “serum therapies” extracted from the blood of naturally or artificially immunized animals induced immunity in sick patients suffering diphtheria or tetanus, a practice used today in some COVID pandemic patients (27). The partial success of serum therapies, and later experiments in mice, led German immunochemist Paul Ehrlich to develop his “side-chain theory” of immunity. Ehrlich believed that living cells were covered with chemical side-chains that formed links with foreign toxins, and when under threat, the cell would produce more side-chains to bind the toxin (or antigen) (26). For every antigen in nature, there is at least one side-chain that will bind it, and once detected, more cells can be made with the same side-chain, which are released into the blood as “antibodies”, in a feed-forward manner. Over time, Ehrlich believed that the host builds a “memory” immunity “ready-made” in their blood to protect against subsequent exposures to the same infection. Despite its ingenious specificity and refinement, Ehrlich’s humoral hypothesis had a number of shortcomings, as it did not explain why some cells possessed the ability to make side-chains, and others did not (28). Metchnikoff and Ehrlich shared the Nobel prize in 1908 for their different theories, and the modern era of immunology was born (29).
Clonal Selection Theory of Immunity: One Cell Makes One Antibody
Ehrlich’s idea that antibodies were already present in blood was also rejected in 1930 by Fritz Breinl and Felix Haurowitz (28, 30). In its place, they developed the “template instructive hypothesis” of immunity, which was later adopted by Linus Pauling (28, 30, 31). The theory proposed that foreign antigens served as “templates” of antibody globulin production, and helped to explain how Karl Landsteiner could stimulate the formation of antibodies from artificially generated substances, known as haptens (28). Although popular, the “template” theory did not explain a number of critical experiments that showed humans at birth already had a “pre-immune” antibody repertoire (in absence of antigen), or why antibody production during one’s life was enhanced after a second inoculation (adjuvant) with faster, stronger and long-lasting immunity (30). In 1955, Danish immunologist Niels Jerne proposed an alternative natural selection theory of antibody formation, where he argued that antibody diversity was part of the host’s “in-built” memory, which anticipated antigenic interactions rather than being a consequence of antigen exposure (31–33). This theory was a game-changer because it moved the focus of immunity from the antigen-antibody response to the host’s antibody-producing cells themselves, which were known to display memory-like functions (23).
In the late 1950s, Jerne’s work stimulated immunologists David Talmage and Frank Macfarlane Burnet to propose two cell selection theories (32). According to both theories, a foreign “antigen” binds to the host’s antigen-specific lymphocyte, generates a signal, and activates the lymphocyte to rapidly divide and make exact copies (26, 32) (Figure 1). While sharing some similarities, Burnet’s clonal theory explained in great detail: 1) why there was such a high probability of warding off a foreign attack from a lymphocyte recognizing one epitope and dividing, and 2) how “self” was protected from attack because it was developed early during embryogenesis in an environment where there was no external threats (26, 32). According to Burnet, it was only after birth, beginning with a clean slate, that an individual’s immune response discriminated self from non-self (32, 34, 35). During lymphocyte development, the embryo removed or inactivated any autoreactive clones, and the remaining lymphocytes only become responsive after birth. Burnet’s idea for germline selection, and the specificity that one lymphocyte makes one antibody, was later supported by the studies of Peter Medawar and Gus Nossal (26, 32). Burnet’s separation of the germline selection theory (innate) and adaptive (during life) immunity meant that the latter can respond to millions of different foreign antigens in a highly specific way, without causing harm to the host. Today, the adaptive immune system comprises humoral immune responses, orchestrated by B-lymphocytes (B cells), and cell-mediated immune responses, orchestrated by cellular T-lymphocytes (T cells), that are developed successively over a lifetime (Figure 1). The adaptive immune responses are distinct from innate immunity, since they involve specificity and immunological memory (36).
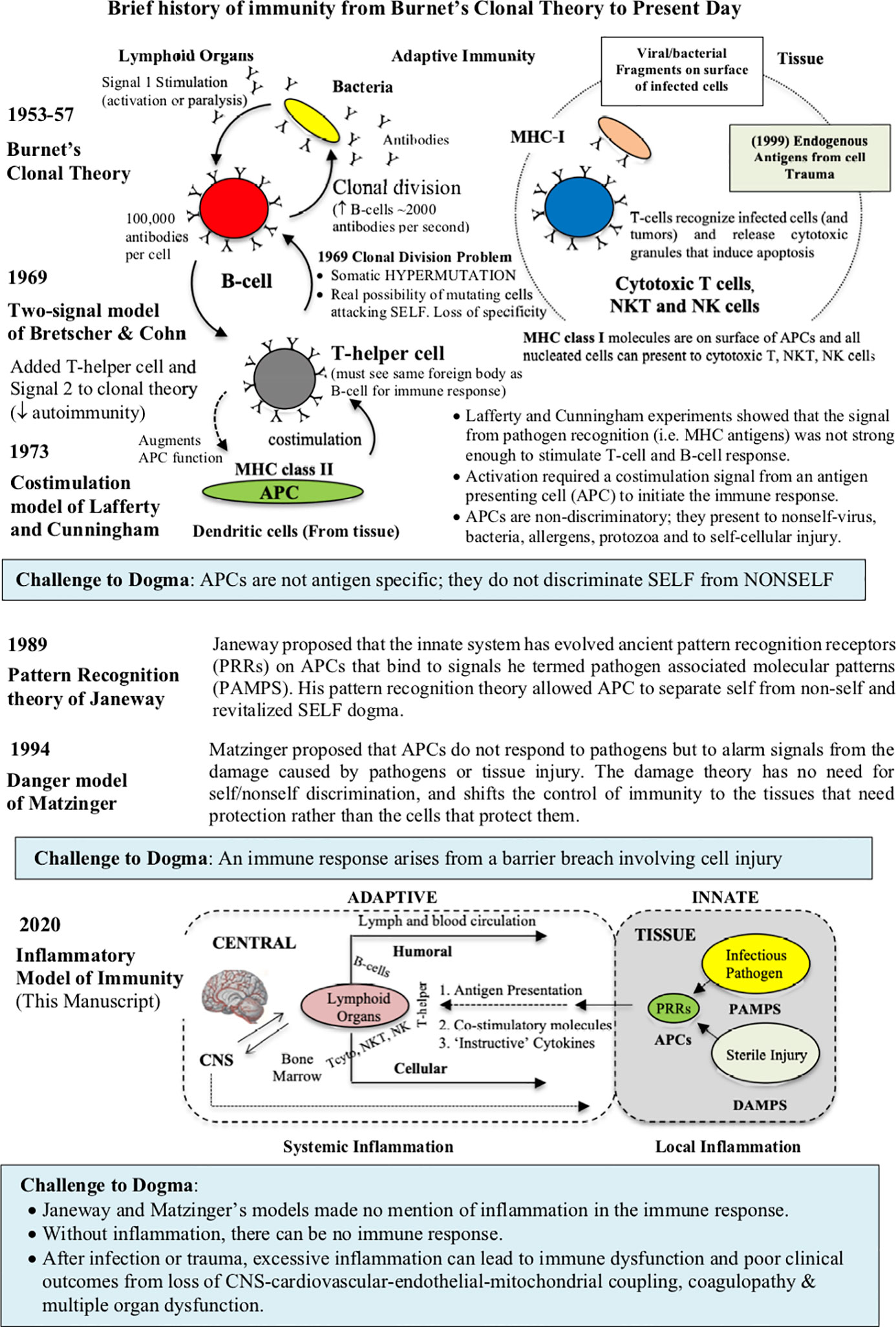
Figure 1 Brief history of the working models of immunity from Burnet’s clonal theory, Bretscher and Cohn two-signal model, Janeway’s pattern recognition receptor (PRR) theory, and Matzinger’s danger theory. In 1989, Janeway’s model, building on the earlier work of Lafferty and Cunningham, ingeniously relegated the innate function of antigen presenting cells (APCs), that roam the tissues, to recognize highly conserved microbial components of ancient origin. He called these microbial components pathogen-associated molecular patterns (PAMPS), which serve as ligands for a broad class of PRRs located on innate cells. While Matzinger’s danger theory recognizes Janeway’s contribution, APCs are more responsive to cellular damage, irrespective of the presence of pathogen or sterile injury (DAMPS). Despite their different individual contributions, no model is complete as they fail to appreciate the major contribution of inflammation. An inflammatory model has been added to the current theories of immunity (see text for more information). Depending upon the type and location of antigen, DCs can activate two arms of adaptive immunity; humoral activation (B cell via MHC class II molecules) from extracellular pathogens and toxins, and cell-mediated immunity (cytotoxic T cells, NKT, NK and macrophages via MHC class 1 molecules) from intracellular pathogens or tumors.
Post-Clonal Theory Era: Signaling was a Problem
Despite its simplicity and elegance, Burnet’s clonal model had a number of limitations. In the 1960s, the first big unknown was what kind of signal was generated to initiate an immune response, and how was it presented to the host, and subsequently processed (37). On the question of antigen presentation, Bretscher and Cohn argued on theoretical grounds that Burnet’s one signal-one response lacked specificity [38, 39 Bretscher, 2019 #5959]. They argued, given the tens of billions of circulating cells in blood, one signal increased the likelihood of a lymphocyte hypermutation that could attack “self” (37–39). In its place, they suggested two signals; one by the receptor antibody of the B cell (Signal 1), and a second ‘partner’ signal by T-helper lymphocytes, that recognized the same antigen (Signal 2) (Figure 1) (37, 40). The idea that lymphocyte activation required lymphocyte cooperation provided a check-point over mistaken identity and untoward antigenic activation (37). Bretscher and Cohn further suggested that the second “partner” signal may involve a membrane/membrane interaction between the two interacting cells (37). Today, this two-signal hypothesis has been experimentally verified; B-cell activation requires interaction with a specific ligand (CD40), which is expressed on the surface of an activated helper T cell (CD4+ T cells) (37, 41, 42).
The next major advance to antigenic signaling came from studies of Lafferty and Cunningham who showed that the two-signal model suffered from submaximal activation, and required some form of booster ‘signal’ (43, 44). The antigen to antibody signal (even with T-helpers) was too weak to stimulate an immune response. They proposed a costimulation signal, which they believed must come from another cell or cells located in the peripheral tissues, which they termed antigen presenting cells (APCs) (44). In 1973, the dendritic cell (DC) was the first APC discovered by Canadian physician Steinman and Cohn in lymph nodes and spleen (45) (Figure 1). Mature macrophages, and other tissue-resident immune cells, also have this antigen presenting capacity, though not to the extent of DCs (46–49). The “costimulatory” APC signal of Lafferty and Cunningham was an on/off signal, and spatially separated from the “priming” or “helper” signal of Bretscher and Cohn. In addition, shifting the initiating signal from the central lymphoid tissues to the periphery directly challenged the concept of self/nonself discrimination, since APCs cannot differentiate between the two (23, 50). These were exciting times for the field of immunology.
Janeway’s Pattern Recognition Receptor Theory: Relegating Innate Immunity to Ancient Times
I will argue that the solution to both problems (diversity and specificity) existed prior to the development of the adaptive immune response and persists in contemporary mammalian immune systems.
C. Janeway (1992) (51) p11
The apparent irrelevance of self/nonself discrimination by APCs captivated immunologist Charles Janeway in 1989 and challenged him to rethink the problem. Janeway began by exposing the “immunologist’s dirty little secret”, namely, that foreign antigen alone was insufficient to elicit the adaptive immune response (52–54). He further questioned why T helper cells were required to activate B and T cells, and yet foreign antigen could either activate or inactivate the same cells in the absence of T helper cells. Janeway’s basic assumptions for his scheme are listed in Table 1. After exposing the weaknesses of prevailing models, Janeway ingeniously postulated that costimulation was only switched ‘on’ if the host’s APC’s possessed specific pattern recognition receptors (PRRs) that recognized some common pathogen‐associated molecular pattern (PAMP) from an invading pathogen (54) (Figure 1).

Table 1 Major assumptions that led Charles Janeway to develop his pattern recognition receptor (PRR) theory of innate and adaptive immunity.
Janeway’s concept of PRRs has been experimentally supported with a long list of PAMPS, which led him, and colleague Ruslan Medzhitov, to clone the first human Toll-like receptor (TLR). Together, they showed that TLR stimulation activated signaling pathways required for the development of adaptive immunity (55, 56). This demonstration provided strong support on the significance of TLRs, and their ligands, to initiate an immune response (57). Janeway and Medzhitov further proposed that host innate recognition had ancient roots, and if PAMPS were not present or detected, no immune response would occur. Key to Janeway’s thinking was that the host’s innate system was part of the germline selection process, that APCs had evolved receptors that recognize infectious bacteria or their components, and that they were evolutionary distinct from clonal receptors derived from adaptive immunity after birth (52, 55). Here, the remnants of Burnett’s clonal theory are apparent, with modifications driven by intuition and experiment. Moreover, since PAMPS are foreign, and not produced by the host, the immune system can efficiently discriminate self from nonself, and instruct the adaptive immune system to respond accordingly (51). Janeway and Medzhitov believed they had solved this fundamental problem posed by Lafferty and Cunningham having with regard to APC surveillance in the periphery. Much debate exists as to why Janeway and Medzhitov were not awarded the 2011 Nobel Prize for their unification theory, and TLR signaling pathway demonstrations (57). The prize was shared between Jules Hoffman (described Toll genes in fruit flies, 1996), Bruce Beutler (showed LPS activated TLR-4 receptor, 1998) and Ralph Steinman (discovered DCs, 1973) for their discoveries on innate immunity (57).
Matzinger’s Danger Model: Moving Beyond the Immune Self/Nonself Dichotomy
Although this (self/nonself) paradigm has often served us well, years of detailed examination have revealed a number of inherent problems. This viewpoint outlines a model of immunity based on the idea that the immune system is more concerned with entities that do damage than with those that are foreign.
P. Matzinger (2002) (58) p301
In 1994, Polly Matzinger challenged Janeway’s theory and his PAMPS self/nonself dichotomy. She proposed the primary function of the immune system “is the need to detect and protect against danger” (59). Notwithstanding the special case of foreign antigen recognition, Matzinger argued the host’s innate immune cells must also recognize molecular signals from cellular damage (59). Matzinger termed these danger or alarm signals, damage-associated molecular patterns (DAMPs) (35, 39, 59, 60). Danger or alarm signals arise from cellular damage caused by pathogens or tissue trauma. Up until now tissue injury had received very little attention, and Matzinger’s views were intuitive and transforming. She proposed that DAMPS and PAMPS share the common property of signaling cellular damage, irrespective of its origin. The danger theory also challenged Janeway’s concept of “foreign”, meaning the host’s infectious “nonself” (Figure 1). Vertebrates have evolved harmoniously with many billions of friendly “foreign” microbes living in their intestines, mouth and epithelial surfaces without causing harm. Indeed, animals and humans have evolved two genomes; their own and the gut microbiome (61, 62). Today, we know there are more bacteria residing in our gut (1015) than cells in our body (1014), which are not infectious unless there is a barrier breach (39, 60, 61). Every day we inhale hundreds of thousands of bacteria per cubic meter of air, and many of these inhabit our nasal-pharyngeal passages (63). These friendly “foreigners” contribute to our immune health as part of the gut–brain–immune axis (64), and have the potential to become pathogenic when a breach occurs from ischemia, trauma or disease (62).
Conceptually and operationally, Matzinger’s theory shifted the molecular specificity of innate activation to damage in the tissues. While sharing many features of Janeway’s PRR theory, Matzinger concluded there was no need for a self/nonself binary distinction to explain immunity. She wrote: “to say that specificity is important is different from saying that a discrimination between self and nonself is necessary” (39). The immune response was now viewed as a response to tissue damage or alarm “signals” that don’t discriminate (60). Over the decades, Matzinger’s danger model has received a great deal of experimental support, with a long list of DAMPs from damaged or dying cells (35, 58, 60, 65). Despite their differences, Matzinger and Janeway’s theories largely focused on the early molecular specificity of the innate immune response, and not the regulation of adaptive immunity (66).
Inflammation: A Missing Piece of the Puzzle
Our understanding of inflammation started with research in the field of leukocyte migration, meticulously observed under the microscope by Metchnikoff and colleagues. … He can be considered the father of innate immunity.
B. Imhof (2016) (22) p655
From an historical perspective, it is remarkable that the models of immunity rarely included Metchnikoff’s theory of inflammation and phagocytotic defense (67). There was no mention of ‘inflammation’ in any of the key papers of Janeway and Matzinger (51, 59), or in later reviews on the “danger theory; 20 years later” (35). To be fair, cytokine biology was in its infancy and the first drugs targeting the inflammatory system were not developed until the 1990s (68). However, given that cytokines were known to be produced by lymphocytes and macrophages in the 1970s (69), and Lafferty and Cunningham had shifted the initiating immune signal from lymphoid tissues to the periphery (1975), it is still curious why there was no eureka moment, at least in their writings, linking the innate immune response to inflammation, and recognizing that immune cells were inflammatory cells. It appears that Janeway and Matzinger were more interested in molecular aspects of the initiating signals and the concept of self/nonself discrimination, rather than immune cell interactions and the changing milieu underpinning this response.
Like most paradigm shifts in science, it is difficult to pin-point when inflammation entered the different models of immunity. It appears to have occurred in the 1990s when cytokine biology and technologies were rapidly advancing. For example, in 1993 Ferrara reported an explosive inflammatory response with profound immune dysregulation in patients receiving allogenic tissue graft transplantation (70, 71). Ferrara coined the term “cytokine storm” to describe this response, whereby donor T cells produce excessive quantities of proinflammatory cytokines that induced damage and pathology in the recipient’s tissues (70). A few years earlier, Beutler and colleagues showed that cytokines, such as IL-1, TNF-α, and IL-6, played a direct role in the pathogenesis of endotoxic shock (72), and Cavaillon showed experimentally that individual immune cells possessed the ability to produce their own source of regulatory cytokines that modulated their effector functions (69, 73). There are many other examples showing that cytokines, inflammation, immune function and disease were increasingly being recognized as part of the same intertwined process. Another aspect not widely reported until the 1990s, is that the main role of the immune system expanded from host defense to maintenance of host health (Figure 2). This new role involved a “low-level” innate inflammatory response with housekeeping functions such as removal of stressed or aged cells and replacing them with new ones, maintaining symbiotic exchanges with the gut microbiome, maintaining CNS-cardiac health via multiple feedback networks, integrating inter-organ exchange, endothelial health, control of the stress response via shared receptors and hormones, restoring the steady-state after trauma, wound healing, and preventing chronic diseases, including autoimmune diseases and cancer (Figure 2).
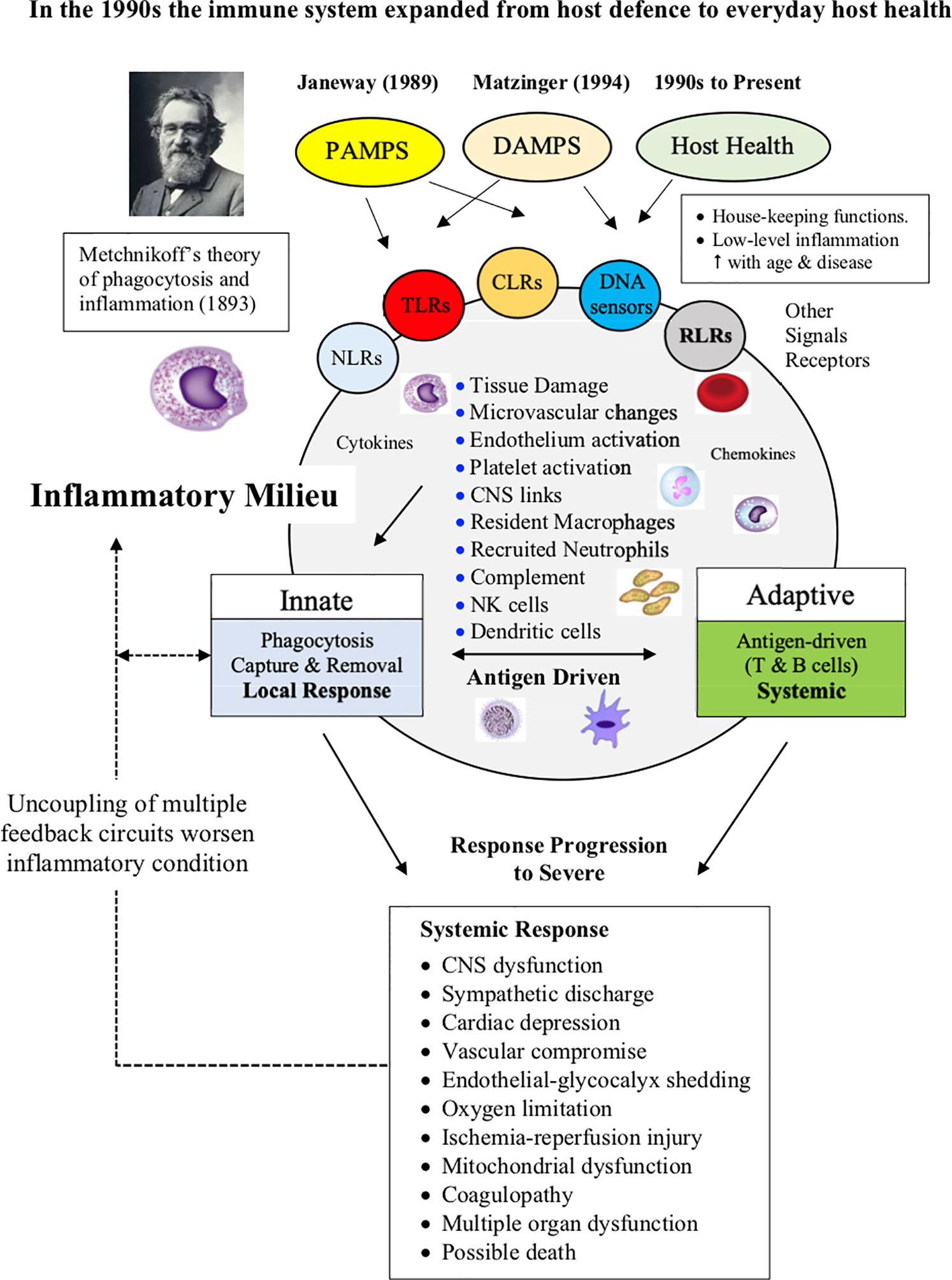
Figure 2 Without inflammation there is no immune response. Changes in the inflammatory milieu and low oxygen drives the type and extent of the immune response. Metchnikoff’s theory of inflammation and phagocytotic defense was largely ignored throughout the 20th century. In the 1990s, the role of the immune system expanded from defense to maintaining host health, which includes a low-level of inflammation associated with general housekeeping functions and maintenance of whole body steady-state. The adaptive response proceeds only if there is sufficient antigen to drive the process. In severe cases, inflammation can spread to become a systemic response and activate a CNS-sympathetically-driven “fight-or-flight” stress response, and if not controlled, can escalate into widespread immune, cardiovascular and metabolic dysfunction, multiple organ failure and death.
Control of inflammation is key for immune defence and everyday host health. Without inflammation there is no immune response.
Inflammation: A Brief History
The Roman Celsus is credited as first documenting (1st century AD) the four cardinal signs of inflammation: rubor et tumor cum calore et dolore (redness and swelling with heat and pain).
Scott and colleagues (2004) (74)
The term inflammation is derived from Latin inflammare (to set on fire) (74, 75). Before the 18th century, acute inflammation was regarded more as a disease, involving Celsus’ redness, pain, heat and swelling, and a surcharge of blood in the tissues (74). In the third century AD, Galen also believed that inflammation was the body’s reaction to an injury, an idea taken up in the mid-1700s by surgeon and anatomist John Hunter (1728–1793). “This inflammation”, Hunter wrote, “will generally be in proportion to the degree of injury done, the nature of the parts injured, and the state of the constitution at the time” (76) p243. Hunter also appreciated that inflammation was universal, beneficial and restorative: “But from whatever cause inflammation arises, it appears to nearly the same in all, for it is an effect intended to bring about a reinstatement of the parts nearly to their natural functions” (76) p286. In the mid-19th century, Virchow viewed inflammation as inherently pathological, and in the second half of the century, as changing cell populations in blood and the tissues, which provided a backdrop to Metchnikoff’s phagocytosis theory (74).
Today, a typical inflammatory response consists of at least five components (77–81) (Figure 3):
1. Initiating signals: DAMPS and PAMPS
2. Innate pattern recognition receptors: initial sensing of DAMPS and PAMPS
3. Cell signaling transduction pathways
4. Effector responses to remove the threat (local and systemic)
5. Resolution or containment strategies to promote healing

Figure 3 Overview of the immune response. The time-course depends upon the precise nature and severity of the initial threat. The process comprises five main components; 1) initiator signals (DAMPS and PAMPS), 2) tissue-resident “innate” cells, and their receptors (PRRs), 3) cell signal transduction pathways, 4) effector responses to neutralize the threat (innate and adaptive), and 5) active resolution to promote healing. Inflammation is key to the selection, recruitment, and phenotypic expression of the various immune cells during the immune response. How damage control is communicated among immune cells, and their subsequent go/no-go “decision-making criteria”, to start healing with epithelial closure, are not well understood.
Below, we discuss the five components of inflammation and the linkages and possible intersection points that activate the process, and its timely resolution for optimal healing.
1) Initiating Signals: DAMPS and PAMPS
The innate immune response resides with the tissue resident macrophages (82), DCs (47), a subset of B memory cells (83), and mast cells detecting a barrier breach (84). Some T cells of ancient origins are also resident in tissues and include memory (TRM) cells, intraepithelial lymphocyte (IELs), invariant natural killer (iNKT) cells, and gamma-delta T-cell subsets (γδT cells) (85–87). These cells detect a myriad of molecular stimuli, the DAMPS and PAMPS, which comprise a complex mixture of proteins, lipoproteins, nucleic acids and saccharides (65, 88, 89). As mentioned above, DAMPS are released from damaged, stressed or dying cells, including extracellular and cell membrane, cytosolic, cytoskeleton, nuclear mitochondrial, vascular endothelial, and blood components (65, 90).
Some DAMPS include fibrinogen, annexins, platelet components, fibronectin, S100 proteins, syndecan-1, F-actin, ATP, histones, DNA, TFAM, mitoROS, cytochrome C, IL-1α, HMGB1, heparan sulfate, tenascin C, defensins, amyloid-β, and many others (90). PAMPS, on the other hand, are molecular signals from pathogens, or their components, that may directly or indirectly breach barriers such as skin, lung epithelium, or the lining of the gut, or from tissue injury that becomes infected. PAMPs can be derived from viruses, bacteria, fungi, and protozoa and helminths. Examples include double-stranded RNA from viruses, LPS from gram-negative bacteria, flagellin products from bacteria, DNA from pathogens, surface glycoproteins, lipoproteins, and other membrane components (53, 55, 65). Importantly, DAMPS and PAMPS are not mutually exclusive and may share co-receptors and accessory molecules, and form “partnerships” to coordinate a response (91). Antigens, key drivers of adaptive immune responses, are not normally considered PAMPs, and largely comprise proteins or polysaccharides, although small molecules coupled to carrier proteins can also be antigenic (e.g. haptens).
2) Innate Pattern Recognition Receptors: Initial Sensing of DAMPS and PAMPS
The LPS sensing role of TLR4 was a huge surprise.
Bruce Beutler (Nobel Laureate) Quoted from Ravindran (2013) (92)
DAMPS and PAMPs are detected by Janeway’s PRRs located on immune cells and non-immune cell types (e.g. astrocytes, neurones, cardiomyocytes, hepatocytes, gut and muscle cells) (93, 94). PRRs serve as “sensors” to communicate the nature and severity of the breach (94). Most, if not all, cells express at least five families of PRRs, including toll-like receptors (TLRs), RIG-I-like receptors (RLRs), NOD-like receptors (NLRs), C-type lectin-like receptors (CLRs), and cytosolic DNA sensors (95–97). These different PRRs are located either on the cell surface or intracellularly. TLRs are the best studied, and in humans, ten different motifs have been characterized (96–99). Structurally, TLRs are integral glycoproteins characterized by an extracellular ligand-binding domain containing leucine-rich repeat motifs, and a cytoplasmic signaling Toll/IL-1 receptor homology (TIR) domain (100). Ligand binding to TLRs through PAMP or DAMPS induces receptor oligomerization, which subsequently triggers intracellular signal transduction (90, 101). For example, TLRs can mediate cellular responses to bacterial LPS (TLR4), lipopeptides (TLRs 1, 2, and 6), flagellum (TLR5), and microbial RNA and DNA nucleotide sequences (TLRs 3, 7, 8, and 9) (102). As mentioned, TLRs are also expressed in most tissue cells. The most important immune cell types expressing TLRs are macrophages, DCs, mast cells and B cells (100). TLR9, for example, can be modulated on cardiomyocytes to reduce myocardial ischemia/reperfusion injury (101), and are believed to be involved in normal cardiovascular function and disease (103). Another important PRR that senses a wide range of PAMPS and DAMPS is the NLRP3 protein that activates a cytoplasmic multiprotein platform assembly known as the inflammasome (104). The inflammasome amplifies the inflammatory response via caspase-1 activation and IL-1β and IL-18 maturation, and is critical for host defenses against bacterial, fungal, viral infections and trauma (105, 106).
3) Cell Signaling Transduction Pathways: Linking Receptor Activation to Gene Expression
Once PRRs are activated, the intracellular pathways that modulate the inflammatory response involve a network of cascades and interconnections. There are at least five major signaling transduction pathways; NF-κB, MAPK, JAK/STAT3, TGF-β/Smad and PI3K/AKT (Figure 4) (88, 94, 107–111). If TLRs are activated, different signals may be directed into two distinct pathways with different cytokine products: 1) the myeloid differentiation primary response protein 88 (MyD88)-dependent pathways, which is activated by most TLRs and IFN-γ, or 2) the TIR domain-containing adaptor-inducing IFN-β (TRIF)-dependent pathway, which responds to only a few TLRs, such as TLR3 and TLR4 (111, 112). If a robust innate immune response is required, both MyD88 and TRIF pathways can be activated (113), which leads to activation of three transcriptional factors, NF-κB, activating protein-1 (AP-1) and IFN regulatory factor 3 (IRF3) (114–116) (Figure 4). Similarly, “booster” coactivation of NF-κB and AP-1 can occur via the MAPK pathway (117), and additionally FOXO and cAMP Response Element Binding (CREB), if the phosphatidylinositol-3-kinase (PI3K/AKT) pathway is triggered (118) (Figure 4). Other transcriptional factors STAT3, p53, estrogen receptor, ATF3 (CREB family), Smad3 and 4, and NFATs all have the ability to intersect and regulate NF-κB (115, 119). Possible reasons for why NF-κB was selected as a principal regulator of inflammation, and check-point for multiple pathway and crosstalk interactions, may be to expand gene expression in a more robust, controlled manner to deal with threats of diverse origins. From an evolutionary perspective, having multiple networks to finely tune the inflammatory response in a context- and time-specific manner may have conferred the host a major survival advantage to a pathogen or injury. Single pathway activation may not have been sufficient to induce an adequate effector response (113).
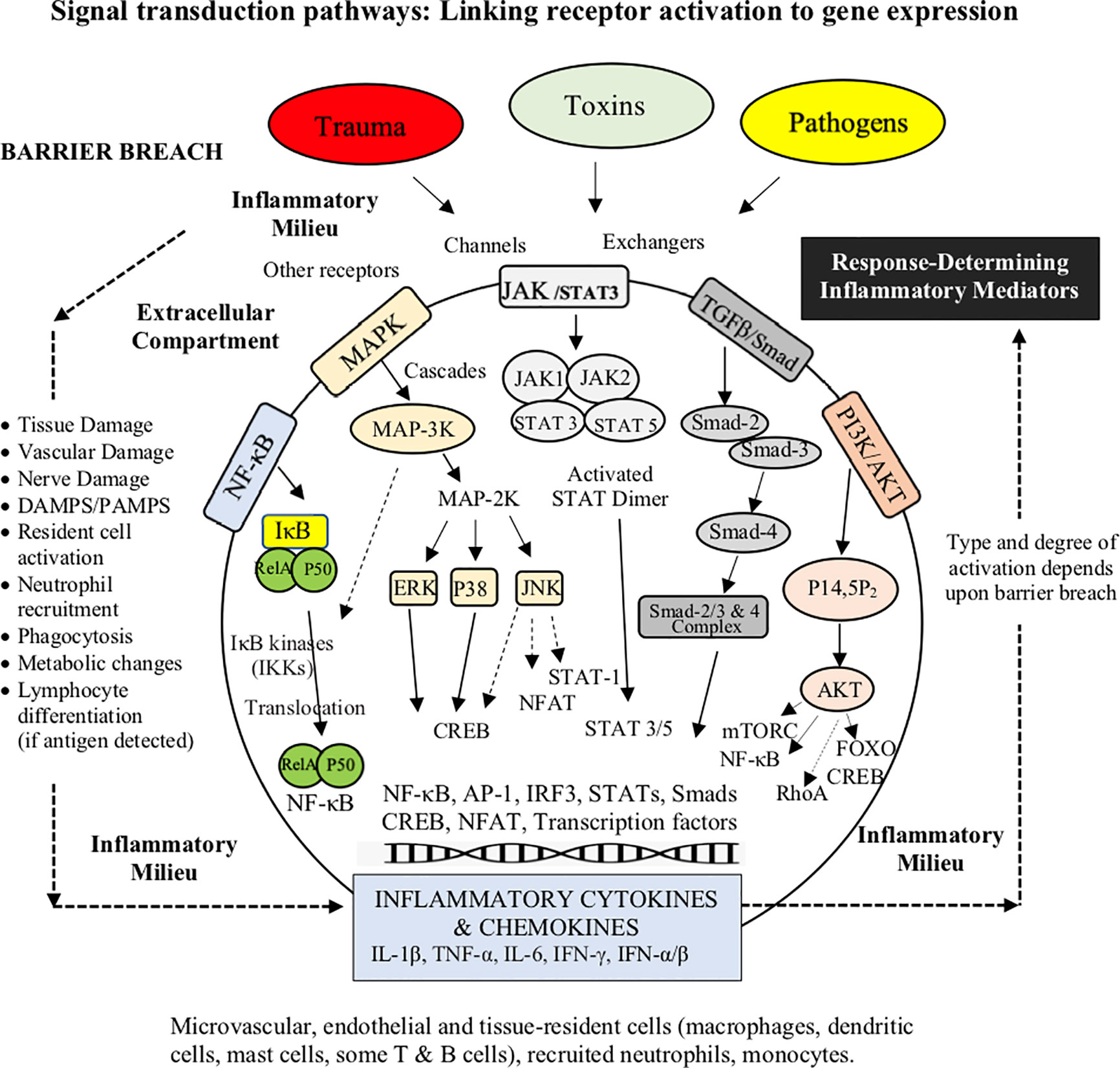
Figure 4 Broad schematic of the main signaling transduction pathways and the changing role of the inflammatory environment. The link between receptor activation and control of inflammatory cytokine and chemokine production involves at least five signaling transduction pathways; NF-κB, MAPK, JAK/STAT3, TGF-β/Smad and the PI3K. The cytokine TGFβ1 can also regulate NFκB and JNK pathways via TRAF6 convergence. Although the five major pathways are regulated by different mechanisms, they share many PRRs, stressor signals (e.g. adrenergic, cholinergic, receptors/ion channels) and accessory mediators (e.g. growth factors, hormones), and converge at common intersections points, such as NF-κB, AP-1 and the CREB family. NFκB signaling (not shown) also participates in the regulation of the NLRP3 inflammasome, which is involved in the rapid amplification of inflammation and its resolution.
The ultimate goal of PRR signaling is to change immune cell activation states, and production of inflammatory mediators (107). For example, TLR stimulation via both MyD88 and TRIF signaling induces the maturation of DCs, specifically the upregulation of costimulatory molecules (e.g. CD40, CD80 and CD86) and the production of proinflammatory cytokines (e.g. IL-6, and TNF-α) (120). The main function of cytokines is intercellular communication among and between immune and non-immune cells as autocrine, paracrine, or endocrine messengers to modulate the host’s effector response (121, 122). Other inflammatory mediators include chemokines, vasoactive amines (histamine and serotonin), adhesion molecules, neurogenic receptors (e.g. dopamine, adrenergic, glutamate acetylcholine and serotonin), vasoactive peptides (Substance P), complement components (C3a, C4a and C5a), lipid mediators (leukotriene B4, prostaglandins and platelet-activating factors), regulators of the extracellular matrix, pro-resolving mediators (lipoxins, resolvins, protectins, maresins, adenosine), and proteolytic enzymes (elastin, cathepsins and matrix metalloproteinases) (77, 78, 121–126). Upregulation or downregulation of inflammatory gene expression is also influenced by changes in the intracellular environment such as calcium handling, pH, redox coupling, nitric oxide, hypoxia status, endosomal and lysosomal activity, glycolytic and mitochondrial energy metabolism, and the generation of reactive oxygen species (ROS) (115, 119, 127–131). Calcium handling and oxidative stress, for example, if not tightly regulated, can lead to pathway dysregulation, mitochondrial dysfunction and genomic instability (131). This often occurs after major trauma or infection and can lead to immunodeficiency, septic shock, or induction of autoimmunity (see below) (94). Orchestration and timing of production of these inflammatory mediators shapes the type and extent of acute immune response to a threat (107).
4) Effector Inflammatory Responses to Remove the Threat (Local and Systemic)
In acute inflammation, we find, as a general rule, vascular dilatation accompanied by an active condition of the endothelium of the vessel-walls and an exudation with diapedesis, that is to say, three events which concur in producing a considerable afflux of leucocytes towards the injured spot.
E. Metchnikoff (1893) (18) p171
The innate effector response can range from a local Metchnikoffian phagocytotic engulfing, digesting, debris clearance and tissue repair, with no further action, to a full-blown, centrally-coordinated, clonal expansion of lymphocytes, if the host becomes overwhelmed with antigen (Figures 1 and 2) (41). This innate strategy may apply to small wounds which are open to opportunistic bacterial or viral intrusion. Thus, if antigen has not reached threshold, it appears a pathogen threat can be dealt with locally without activating the adaptive immune response (132–134). While trauma per se does not activate adaptive immunity in a conventional manner, an increasing number of studies involving CNS and musculoskeletal injury, have shown that T cells and B cells can be activated by endogenous “antigens”, independent of a pathogen (135, 136). The endogenous “antigens” are believed to belong to a class of DAMPS that stimulate autoantibodies, that may assist in cellular defense and repair (136). However, like inflammation, where there is overactivation of T or B cells, there is potential for collateral tissue damage and detrimental autoimmunity (94). In the case of major trauma, the immune cell effector response can quickly become a CNS-sympathetically-driven “fight-or-flight” stress response, and if not controlled, can escalate into widespread cardiovascular and metabolic dysfunction, systemic inflammation, immunosuppression, multiple organ failure and death (11, 12, 135, 137–139).
At the breach site, irrespective of the type of threat, there is an increase in blood supply and a myriad of local signals that prime and instruct innate immune cells to respond. These signals are derived from activated vascular endothelial and tissue cells, leakage of serum components (component), damaged nerve cells, platelets, neutrophils, macrophages, mast cells, monocytes, natural killer (NK) cells and DCs (140–145). These different immune and non-immune cells, through their cytokine networks, play pivotal roles both as producer cells and target effector cells to produce the right response. Recruited neutrophils produce ROS to help them neutralize rogue/damaged cells via phagocytosis, degranulation and extracellular traps (109, 146). In this proinflammatory environment, macrophages also switch to a M1 killer phenotype (induced by IFN-γ or TNF-α) and with the help from mast cells, recruit more neutrophils from the circulation to swarm into the site (7, 146–149). M1 macrophages are also activated by complement receptors (C3a, C5a, and C5b) (independent of antibody), which can induce the activation of the NLRP3 inflammasome to amplify the inflammatory response (147, 150). Resident innate NK cells also secrete cytokines, such as IFN-γ and TNF-α, and interact with macrophages, and other immune cells, to enhance the response (151). As the attack progresses, more neutrophils swarm in and blood monocytes are recruited to resupply tissue macrophages (126, 152).
In the case of infection, the bridge between innate and adaptive immunity, and induction of classical immune memory in lymphocytes, begins at the tissues with the activation of resident DCs (via complement and cytokines) (36, 46, 47, 153). In peripheral tissues, DCs: 1) capture and process antigens, 2) express lymphocyte co-stimulatory molecules, 3) migrate to secondary lymphoid organs and 3) secrete “instructive” cytokines to initiate and drive lymphocyte differentiation and clonal expansion (Figure 1) (128). Depending upon the type and location of antigen, DCs can activate two arms of adaptive immunity; 1) humoral activation from extracellular pathogens and toxins, and 2) cell-mediated immunity from intracellular pathogens or tumors (Figure 1). Humoral immunity is initiated by T-cell-dependent and -independent antigens, and leads to B cell activation, clonal expansion and antibody secretion (Figure 1). Cell-mediated responses, on the other hand, involve activation of cytotoxic T cells, NK, NKT and macrophages, for the purpose of destroying abnormal or infected cells.
DCs do not present antigens directly to T-helper cells of both arms but first internalize and process them as antigenic peptides that are presented by major histocompatibility complexes (MHCs) located on their surface. DCs can present antigen on both MHC class I and class II complexes depending upon the source (128). Antigens arising from an intracellular source and presented on MHC class I will activate CD8 (cytotoxic) T cells (facilitated by IL-12 and type 1 IFN); while antigenic peptides from an extracellular (or foreign) source are presented on MHC class II molecules and activate CD4 (helper) T cells (128). In contrast to MHC class II molecules, MHC class I are more ubiquitous and found on all nucleated cells, presumably, conferring an evolutionary advantage for wider interorgan communications and defense (53, 58, 142, 153). Importantly, DCs are generally classified as the master regulators of adaptive immunity because they have greater capacity to transfer the three signals (see above) to initiate the response compared to any other APC (128). It is also noteworthy, that mitochondrial metabolism is key for the regulation of the effector response. In pro-inflammatory cells, such as M1 macrophages and activated T and B cells, the metabolic energy in the form of ATP is generated largely by glycolysis, while in regulatory cells, such as M2 macrophages or regulatory T cells, energy is generated by increased reliance on mitochondrial function and beta-oxidation (131).
5) Resolution Strategies to Halt the Inflammatory Process and Promote Healing
Studies in recent years have unequivocally shown that resolution of inflammation is an actively controlled processes rather than a passive procedure in which the proinflammatory immune cascade in inflammation simply fizzles.
Markus F. Neurath (2019) (154) p627
After the threat has been neutralized, immune cells begin to switch effector status and metabolism to their healing phenotypes or they undergo programmed apoptosis. Macrophages switch to an anti-inflammatory M2 phenotype (155–159), neutrophils undergo self-destruct (160), and T cells change their population from killer Th1 to a Th2 healing phenotype (via IL-10 and IL-4 produced by DCs) (141, 161). Th2 cytokines also help to maintain the presence of the M2 phenotype (155–159). In addition, DCs induce the production of T regulatory cells that suppress the destructive pro-inflammatory activities of Th1 cells by secreting cytokines, such as TGF-β1, IL-10 and IL-35, and enhance M2 responses to promote cell clearance (47, 160, 162, 163). Collectively, these changes limit inflammation and create a permissive healing environment (Figure 3).
As part of the healing processes, a further distinction is often made between anti-inflammatory and pro-resolving responses (126, 164–166). The term “pro-resolving” generally refers to the active suppression of the inflammatory processes, such as recruitment of immune cells, apoptosis and clearance of cell debris, whereas the anti-inflammatory targets refer more to specific inhibitory or blocking actions of one or more pathways, which may involve inhibiting PRRs, signal transduction, transcriptional/translational shuttling, and/or gene expression (Figure 3) (77, 165, 167). Molecules that fulfill the criteria of pro-resolving mediators include specialized lipid mediators (lipoxins, resolvins, protectins, and maresins) (126, 166), proteins and peptides (annexin A1, adrenocorticotropic hormone), gaseous mediators (H2S and CO), purines (adenosine), as well as neuromodulators (acetylcholine and other neuropeptides) released under the control of the vagus nerve (126, 165, 167–169).
Inflammatory inhibitors, on the other hand, include inhibitors or antagonists of TLR, NF-κB, TNF-α, Type I interferons (IFN-α and β), Type II IFN-γ, IL-1β, TGF-β1, and suppressor of cytokine signaling (SOCS) proteins, which have attracted much interest because of their ability to inhibit cytokine signaling pathways, and many others (111, 170). Notwithstanding the separation of process and pathway, much overlap exists between anti-inflammatory mediators and pro-resolving mechanisms. The successful development of new anti-inflammatory drugs with pro-resolving properties will likely involve inhibition of a “systems” process rather than targeting a single pro-inflammatory cytokine or pathway (see below).
CNS Control of Inflammation: Crosstalk Between the Nervous and the Immune System
The defense of the organism against deleterious agencies, which is at first confined to the phagocytic mechanisms and the somatic system of nerves, by and by spreads to and is undertaken by the psychical nervous apparatus … One function of these psychical cells has been to develop a complete science for the defense of the organism against hostile influences.
E. Metchnikoff (1893) (18) p195
A topic that is often overlooked in discussions of immune response is the regulatory role of the CNS. Over a hundred years ago Metchnikoff recognized the importance of the “psychical nervous apparatus” in the host’s defense (above quote) (18). Today, the evidence suggests that the CNS interacts with immune cells in a bidirectional manner through shared receptors and neurotransmitters (126, 171–173). Macrophages, neutrophils, mast cells, DCs, blood monocytes, B cells and T lymphocytes all express many neurogenic receptors, including dopamine, adrenergic (β -2, α-1 and 2), glutamate acetylcholine and serotonin receptors (146, 148, 171, 173, 174). At the site of infection or injury, this would allow short-range communication between immune cells and recruitment of local neuronal signals to fine tune the immune response (172, 173). Dopamine (DA) bidirectional pathways are particularly interesting because dopamine has emerged as a fundamental regulator of inflammation (175). To this end, the primary and secondary lymphoid organs are highly innervated by sympathetic nerve cell terminals that store DA (176). Uncontrolled activation of dopamine pathways is believed to contribute to excessive inflammation and longer-term autoimmune pathologies (176). Recent studies have shown that the CNS-immune crosstalk is linked to increases in circulating neurohormones and cytokines, such as IL-1β and IL-6, which in turn are known to activate the HPA axis (173).
We have argued elsewhere that acute changes in the HPA axis-sympathetic-parasympathetic outflows are associated with immune dysfunction following different trauma states and sepsis (11, 12), and responsible for “low-level” persistent inflammation in most chronic inflammatory diseases, such as osteoarthritis and cardiovascular diseases (61, 64). Recent studies further demonstrate that the vagus nerve can modulate the host immune response after an infectious or sterile barrier breach (126, 168, 173), which may also have feedback inputs from changes to the CNS-gut-microbiome-immune axis (61, 64, 177). Increased vagal outflow to the spleen leads to reduced activation of circulating neutrophils by modulating the expression of CD11b (146). This indicates that the CNS can control neutrophil recruitment, and may be vitally important for timely resolution of inflammation after a barrier breach. Some catecholaminergic/cholinergic neurotransmitters can also modulate centrally-driven T cell-mediated immunity, although the underlying mechanisms are not well understood. To sum up, peripheral nerve cell interactions with immune cells is emerging as a major player in controlling the type and extent of inflammation and the immune response (146, 173). From a clinical perspective, modulating the different neuro-immune circuits, and their interactions, can potentially modify inflammatory processes and pathways, that may lead to better outcomes after major trauma, infection or disease.
Developing New Therapies to Reduce Excessive Inflammation: Choosing the Right Animal and the Conditions for Safety and Translation to Humans
Achieving FDA approval for only one-in-ten drug indications that enter the clinic is a concerning statistic for drug developers, regulators, investors and patients.
Hay and colleagues (2014) (178)
Currently, there are few safe and effective drug therapies targeting excessive inflammation and immune dysfunction after trauma or infection. The widely used non-steroidal anti-inflammatory drugs (NSAIDs), COX-2 inhibitors and TNF-α inhibitors do not appear to be pro-resolving, and may in fact exacerbate the resolution process (166). The reasons for lack of progress in drug development in this area is complex. The 90% failure rate of new drugs reaching FDA approval is one of the major impediments because of the high risks of failure and high costs (178). Furthermore, if a drug does reach approval, around 30% have been shown to have a postmarket safety event (139, 179). This adverse event statistic is startling given the high level of scrutiny from independent institutional review boards and pre-market safety and regulatory oversight guidelines imposed by the FDA. We propose at least five reasons for “translational failure”:
1. Choice of animal model and gut microbiome status
2. Male animal species and human study bias
3. Heterogeneity of the human response to infection or injury
4. Poor clinical trial design in a real-world environment
5. Ignoring the systems approach
In our view, choosing the right animal model is one of the single determinants for failure of new drugs to translate (61, 62, 177, 180, 181). An overlooked variable is the composition of gut microbiome. In the 1960s, disease was an unwanted variable in small animal experiments, and potentially harmful bacteria were selectively bred out of animal colonies (61, 62, 177). These animals are called specific pathogen-free (SPF) and the gut composition varies from institution to institution, and different animal suppliers. The problem with modifying gut microbiomes is that the microflora ‘mix’ can lead to host immune alterations and responses that are often not representative of the human condition (61). In a landmark study, Beura and colleagues demonstrated that SPF adult mice have “immature” immune systems that were more prone to inflammation and infection than conventionally-bred mice (182). Their SPF mice lacked effector-differentiated and mucosally-distributed memory T cells, and when co-housed and bred with pet store mice, their immune system shifted closer to adult humans (182). Similarly, we have reported that SPF animals are not suitable for studying traumatic injury or hemorrhagic shock (62, 177).
Male bias in animal and human studies is another important variable affecting drug translation. In the past, the exclusive use of male animals assumes that male and female animals are biologically identical (183, 184). This assumption is false (137, 183, 185). Women have twofold higher mortality after equivalent burn injury than men, and have lower incidence of sepsis and mortality after major surgery or blunt force trauma (186). Women also have different responses to many FDA-approved drugs than men, including anti-inflammatory drugs, and in some cases with adverse outcomes (184, 187–189). Problems with translation also depend on the heterogeneity of the population including socio-economic, age and health status, that can impact clinical trials (179, 181, 190). While randomized control trials are considered the “gold standard”, they may not always mimic real-life treatment situations because of their strict inclusion/exclusion criteria, and highly controlled settings (191). In addition, many trials suffer from poor design and low statistical power to support primary and secondary endpoints (184, 191).
Another major contributor to “translational failure”, that receives little attention, is the flawed practice of drugs targeting single nodal steps (61, 62, 180). Targeting individual pro-inflammatory cytokines, for example, or any single step along a signaling pathway, ignores the importance of the system. Single-nodal thinking rarely solves a medical problem unless the site is believed to be a central hub or intersection point. The IL-1 receptor has been proposed to be such a site, and while anakinra (IL-1 antagonist) has an excellent safety record, further trials are required to demonstrate its clinical efficacy after an infection and trauma (192, 193). Reductionism in scientific discovery is important in breaking a system into its constituent parts, and studying them, however, it does not do away with the system (61, 62, 177). This flawed way of thinking, we believe, is responsible for a high failure rate of translating promising new drugs, and why there are so many failed clinical trials (194). A systems approach is much more likely to increase animal to human success of translation.
Toward a System-Based Drug to Protect Against Excessive Inflammation After Infection And Sterile Injury
What we anticipate seldom occurs; what we least expect generally happens.
Benjamin Disraeli (1804–81), Henrietta Temple (3)
Brief History: Teaching Old Drugs New Tricks
Twenty years ago, the first author (GPD) asked if it was possible to pharmacologically manipulate the human heart to operate more like the heart of a natural hibernator for improved protection during adult and pediatric cardiopulmonary bypass or valvular surgery (2, 3, 195). Within 10 years, high dose ALM cardioplegia was translated from isolated rat heart experiments into human cardiac surgery. We chose adenosine (A) to inhibit the sinoatrial node and reduce the atrial and ventricular action potential (AP) duration (A1 receptor subtype and A1 linked opening of KATP channels), lidocaine (L) to reduce AP amplitude by arresting Na+ fast channels, and magnesium (M) to stabilize the membrane and protect against reperfusion arrhythmias (196). We theorized this strategy would arrest the heart at its resting membrane potential and avoid the use of commonly used high potassium, which depolarizes the membrane and promotes ‘ischemic’ injury currents (197, 198). Two prospective, randomized, clinical trials have shown the ALM cardioplegia to be superior to high potassium cardioplegia with less days in hospital (199, 200). After surgery, the heart is reanimated in sinus rhythm with 10-times lower concentrations of ALM in a heart. This resuscitation strategy led to a second idea; namely, the application of ALM to resuscitate and protect the heart after major trauma such as hemorrhagic shock, cardiac arrest, heart attack and stroke and trauma of surgery itself (3).
Adenosine, Lidocaine, and Mg2+ Immune-Modulatory Functions
We will now briefly review our efforts to develop an intravenous (IV) fluid therapy comprising adenosine, lidocaine and Mg2+ (ALM) that uniquely protects against trauma and infection, and may be useful as an immune-modulatory agent for coronavirus 2 (SARS-CoV-2), and other viral challenges. Our early experiments focused on traumatic injury (139, 201–205) and subsequently we showed the same therapy protects against sepsis and endotoxemia in different animal models (3, 206, 207). The individual effects of A, L or M also have been shown to confer some protection, but not to the same extent as ALM combined (12, 208). Some of these effects on immune cells, and key regulatory sites, are listed in Tables 2 and 3. Each drug exerts anti-inflammatory and immunoregulatory effects from molecular to cellular and tissue-level responses (209–214), including 1) differential PRRs expression (e.g. TLRs) and activity of cell surface receptors, 2) the activation of signal transduction pathways and 3) the transcription factors responsible for regulating inflammatory cytokine production, and preservation of cellular energy metabolism (3, 211, 215–238) (Tables 2 and 3). In addition, A, L or M individually have been shown to support endothelial-mediated inflammatory/immune functions (3, 239), however, they do not do so to the same extent as ALM combined (12, 208). The precise mechanisms of how ALM confers a survival benefit at the level of the immune system, and the intersection between trauma and infection, are not known (3, 139). What follows is a characterization of the ALM survival phenotype and why it may be applicable to reduce inflammation from a viral attack.
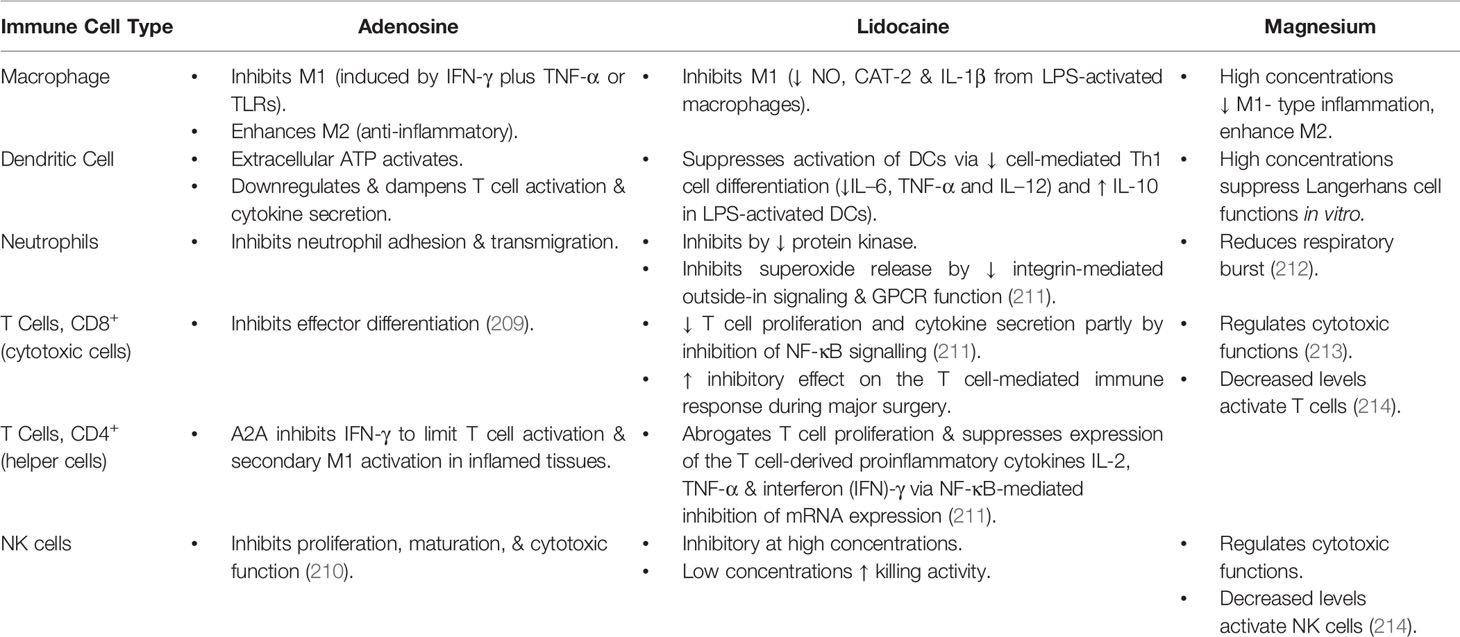
Table 2 Summary of the effects of adenosine, lidocaine and magnesium alone on immune cells.
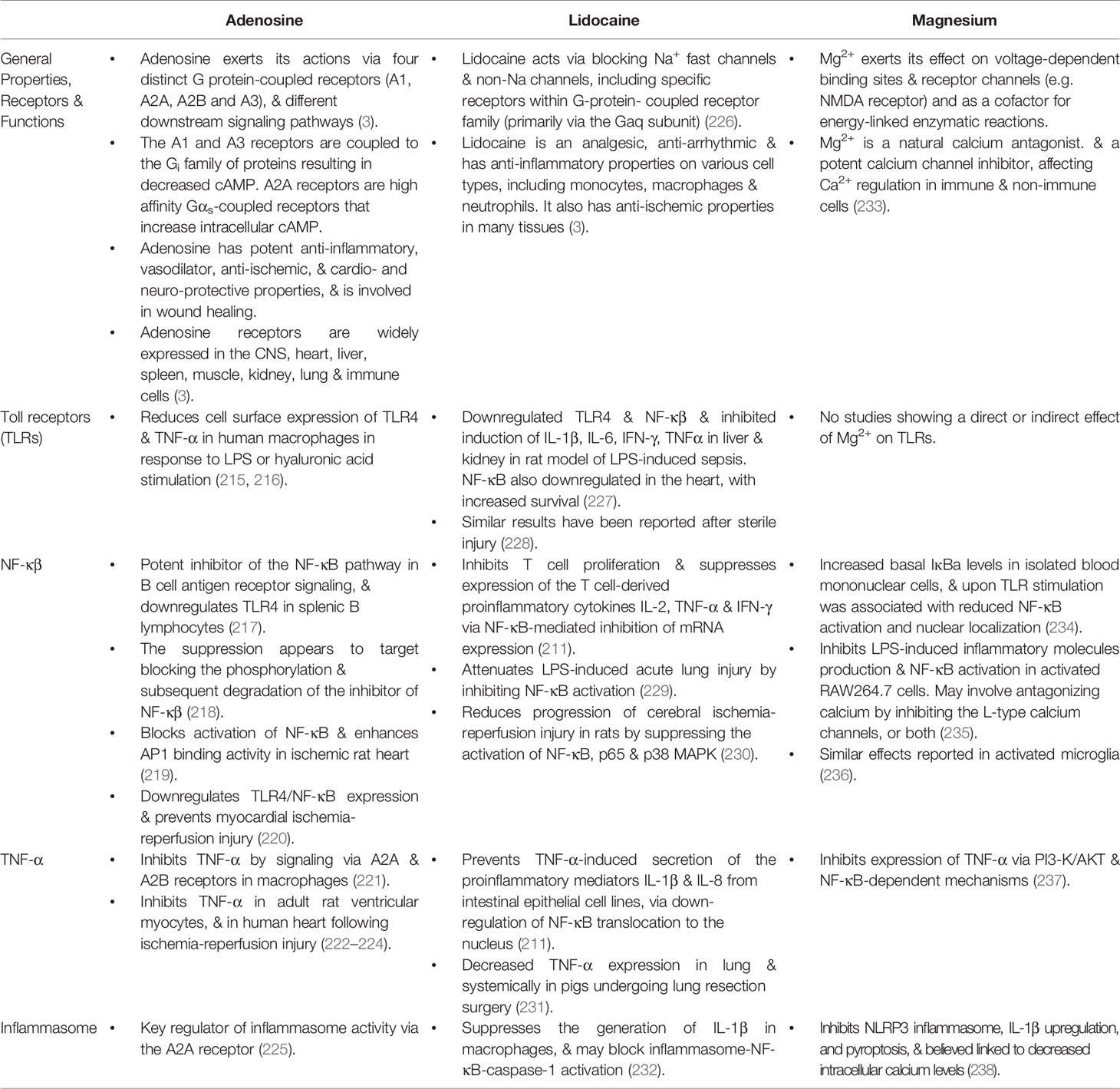
Table 3 Effects of Adenosine, Lidocaine and Magnesium alone on Toll-like receptors, NF-κβ, TNF-α and inflammasome activity.
Adenosine, Lidocaine, and Mg2+ Defends Against Infection and Trauma
A 4-hour infusion of ALM after polymicrobial sepsis led to 6-day survival (without antibiotics), while controls died from different levels of immune dysfunction on days 2 and 4 (3, 207). ALM survival was associated with suppression of the systemic inflammatory response. Interestingly, blood IL-1β levels remained at baseline levels, which we argued was associated with inhibition of the inflammasome (207). As discussed, IL-1β is a potent inducer of inflammation via the inflammasome and activation of NF-κB (112, 240). In another porcine model of LPS endotoxemia, ALM infusion blunted inflammation and bolstered the cardiovascular system to produce a high flow, hypotensive, vasodilatory state with improved O2 delivery over a 4-hour period (241).
With respect to trauma, the ALM therapy suppressed inflammation after hemorrhagic shock, major surgery or traumatic brain injury (139, 201–205, 242). After hemorrhagic shock, ALM extended survival to 3 days, which was associated with improved cardiac function, correction of coagulopathy, preservation of platelets, and differential expression of the master genes of metabolism (ampk, sirt-1, PGC-1α and mtCO3). Metabolic genes were upregulated in heart and brain and downregulated in the periphery (liver, gut) at 3 days. Increased expression of sirt-1 in heart is clinically relevant because sirt-1 activation has been shown to inhibit NF-κB signaling, and reduce inflammation (243). Similarly, upregulation of heart mtCO3 indicates improved mitochondrial metabolism by improving cytochrome c oxidase to drive ATP synthesis (204). TFAM (transcription factor A, mitochondrial), a gene involved in mitochondrial biogenesis, was also significantly increased in heart and brain at 3 days. From a systems perspective, the ALM survival phenotype appears to be associated with changes to the CNS, with downregulation of sympathetic activation and upregulation of parasympathetic outflow during recovery (3). As discussed above, an increased reliance of parasympathetic outflows is consistent an anti-inflammatory survival phenotype (146).
Future Studies and Outstanding Questions
Currently, we don’t know how and when the host “switches” from an injury phenotype to a survival one, or if protection can be extended from 3 to 7 days, or longer. Neither do we know if the therapy confers the same whole-body protection if administered at later times after insult? Other questions include what are the underlying mechanisms responsible for ALM’s ability to reduces the “cytokine storm”? What roles do different immune cells, such as macrophages and neutrophils, and platelets play in inflammatory suppression? What is the role of adenosine receptors, lidocaine Na+ fast channels and magnesium ions to PRR signal activation and transduction of inflammatory pathways? What role does ALM play in regulating intracellular Ca2+ and the metabolic pathways involved in maturation of the immune response? Are there common intersection points at NF-κB in the regulation of the immune response? How is ALM’s dual protection capability linked to altering CNS-cardiac-endothelial-mitochondrial coupling and improved survival after trauma and infection?
Adenosine, Lidocaine, and Mg2+ as a System-Based Therapy
On the basis of the available evidence, ALM appears to confer a “systems’ effect” of dual protection, which has led us to propose a Systems Hypothesis of Trauma (SHOT) (11, 12). SHOT is discussed in detail elsewhere and begins with improved CNS control of cardiovascular function, support of endothelial-glycocalyx integrity, and improved O2 delivery and mitochondrial metabolism, that is not replicated with the individual actives A, L or M (3, 11, 12). ALM’s dual protection capability may offer a unique opportunity to control excessive inflammation and secondary injury in patients suffering infectious diseases, such as COVID-19 presenting with cardiac, metabolic, pulmonary and immune dysfunction (193, 244, 245) (Figure 5).
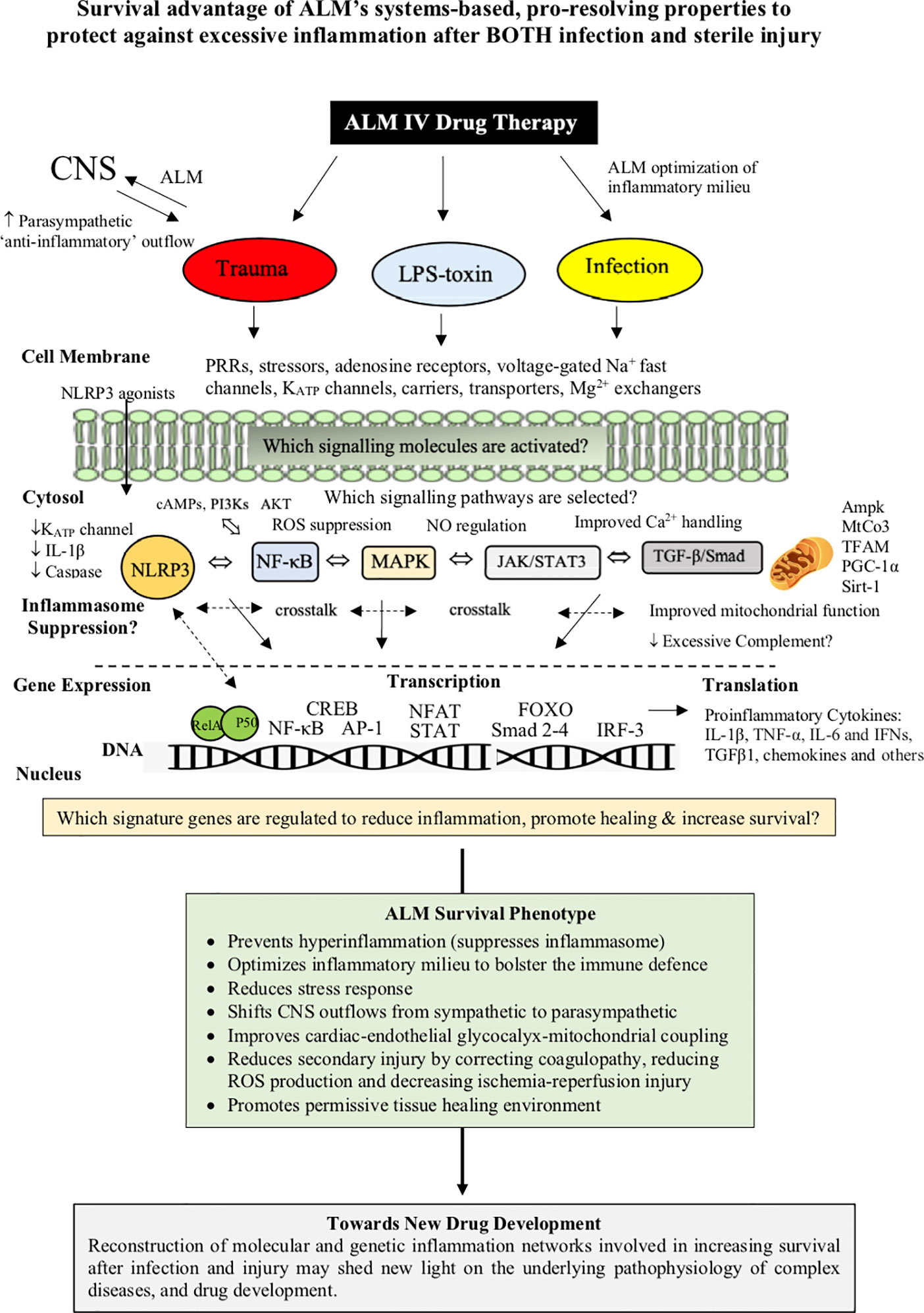
Figure 5 ALM therapy drug development has revealed a duality of protection against infection/toxin and major trauma. The ALM survival phenotype involves an early genetic switch and reprograming of the whole body to provide CNS-cardiac-endothelial and mitochondrial support and early bolstering of the immune system with suppression of hyperinflammation and secondary injury processes. The underlying mechanisms have been shown to involve the differential expression of the master genes of metabolism, however, it is not known what transcriptional signaling mechanisms are involved in ALM suppressing of inflammation after infection or trauma. Genomic and proteomic analysis arrays may help unmask the changes in patterns of gene expression, and the nature of the ALM survival switch that protects against an infection/toxin or major trauma.
Conclusions
For much of the 20th century, the different models of immunity rarely included Metchnikoff’s theory of inflammation and phagocytotic defense. Today, inflammation is viewed as a key component of innate and adaptive immunity that creates a milieu that removes and resolves infectious and non-infectious threats. The type and extent of immune response is determined by the mix of cytokines and other neural and inflammatory mediators that determine the selection, activation, recruitment and fate of immune effector cells. Although inflammation is essential for threat neutralization and healing, if left unresolved, it can lead to immune dysfunction and further tissue damage with coagulopathy, endothelial dysfunction, mitochondrial dysfunction, organ failure and death. We have been developing an intravenous (IV) fluid therapy comprising adenosine, lidocaine and Mg2+ (ALM) that confers a survival advantage by preventing excessive inflammation associated with sepsis, endotoxemia and sterile trauma. ALM may provide a therapeutic option for treating COVID‐19.
Author Contributions
All authors contributed to the article and approved the submitted version.
Funding
This work was supported by USSOCOM, USAMRMC proposal SO150053 under Award No. W81XWH-USSOCOM-BAA-15-1. The opinions, interpretations, and conclusions are those of the authors and are not necessarily endorsed by the US Department of Defense.
Conflict of Interest
GPD is the sole inventor of the ALM concept for cardioplegia and trauma.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would particularly like to thank the US Department of Defense for continued support of our work, and also the College of Medicine and Dentistry at James Cook University.
References
1. Pfluger E. The teleogical mechanism of living nature. Pflugers Arch (1877) 15:57–103. doi: 10.1007/BF01628340
2. Dobson GP. Organ arrest, protection and preservation: natural hibernation to cardiac surgery: a review. Comp Biochem Physiol Part B (2004) 139:469–85. doi: 10.1016/j.cbpc.2004.06.002
3. Dobson GP, Letson HL. Adenosine, Lidocaine and Mg2+ (ALM): From Cardiac Surgery to Combat Casualty Care: Teaching Old Drugs New Tricks. J Trauma Acute Care Surg (2016) 80(1):135–45. doi: 10.1097/TA.0000000000000881
4. Dobson GP. Addressing the global burden of trauma in major surgery. Front Surg (2015) 2(Sept):43. doi: 10.3389/fsurg.2015.00043
5. Cannon WB. Organisation for physiological homeostasis. Physiol Rev (1929) 9(3):399–431. doi: 10.1152/physrev.1929.9.3.399
6. Egiazaryan GG, Sudakov KV. Theory of functional systems in the scientific school of P.K. Anokhin. J Hist Neurosci (2007) 16(1-2):194–205. doi: 10.1080/09647040600602805
7. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol (2013) 14(10):986–96. doi: 10.1038/ni.2705
8. Di Benedetto P, Ruscitti P, Vadasz Z, Toubi E, Giacomelli R. Macrophages with regulatory functions, a possible new therapeutic perspective in autoimmune diseases. Autoimmun Rev (2019) 18(10):102369. doi: 10.1016/j.autrev.2019.102369
9. Huang Q, Belz GT. Parallel worlds of the adaptive and innate immune cell networks. Curr Opin Immunol (2019) 58:53–9. doi: 10.1016/j.coi.2019.04.008
10. Dragoş D, Tănăsescu MD. The effect of stress on the defense systems. J Med Life (2010) 3(1):10–8.
11. Dobson GP. Trauma of Major Surgery: A Global Problem that is not going away. Int J Surg (2020) 81:47–54. doi: 10.1016/j.ijsu.2020.07.017
12. Dobson GP, Morris JL, Davenport LM, Letson HL. Traumatic-Induced Coagulopathy as a Systems Failure: A New Window into Hemostasis. Semin Thromb Hemost (2020) 46(2):199–214. doi: 10.1055/s-0039-1701018
13. Leyfman Y, Erick TK, Reddy SS, Galwankar S, Nanayakkara PWB, Di Somma S, et al. Potential Immunotherapeutic Targets For Hypoxia Due to COVI-FLU. Shock (2020) 54(4):438–50. doi: 10.1097/SHK.0000000000001627
14. Sanders JM, Monogue ML, Jodlowski TZ, Cutrell JB. Pharmacologic Treatments for Coronavirus Disease 2019 (COVID-19): A Review. JAMA (2020) 323(18):1824–36. doi: 10.1001/jama.2020.6019
15. Shi Y, Wang Y, Shao C, Huang J, Gan J, Huang X, et al. COVID-19 infection: the perspectives on immune responses. Cell Death Differ (2020) 27:1451–4. doi: 10.1038/s41418-020-0530-3
16. Garcia LF. Immune Response, Inflammation, and the Clinical Spectrum of COVID-19. Front Immunol (2020) 11:1441. doi: 10.3389/fimmu.2020.01441
17. Dobson GP. Science and the war on truth and coronavirus. Front Med (2020) 7:563. doi: 10.3389/fmed.2020.00563
18. Metchnikoff E. Lectures on the Comparative Pathology of Inflammation, Delivered at the Pasteur Institute in 1891. In: Metchnikoff E, editor. Lectures on the Comparative Pathology of Inflammation. (Translated by F. A. Starling and E. H. Starling.) 1891. New York: Dover Publications (1968).
20. Allen P. Pasteur’s life and pioneering work. Lancet (2002) 360(July 6):93. doi: 10.1016/S0140-6736(02)09363-7
21. Gordon S. Elie Metchnikoff, the Man and the Myth. J Innate Immun (2016) 8:223–7. doi: 10.1159/000443331
22. Underhill DM, Gordon S, Imhof BA, Nunez G, Bousso P. Élie Metchnikoff (1845–1916): celebrating 100 years of cellular immunology and beyond. Nat Rev Immunol (2016) 16:651–5. doi: 10.1038/nri.2016.89
23. Tauber AI. Metchnikoff and the phagocytosis theory. Nat Rev (Mol Cell Biol) (2003) 4(Nov):897–901. doi: 10.1038/nrm1244
24. Mantovani A. From phagocyte diversity and activation to probiotics: back to Metchnikoff. Eur J Immunol (2008) 38(12):3269–73. doi: 10.1002/eji.200838918
25. Winau F, Winau R. Emil von Behring and serum therapy. Microbes Infect (2002) 4(2):185–8. doi: 10.1016/S1286-4579(01)01526-X
26. Ada G. The enunciation and impact of Macfarlane Burnet’s clonal selection theory of acquired immunity. Immun Cell Biol (2008) 86:116–8. doi: 10.1038/sj.icb.7100156
27. Casadevall A, Pirofski LA. The convalescent sera option for containing COVID-19. J Clin Invest (2020) 130(4):1545–8. doi: 10.1172/JCI138003
29. Silverstein AM. Cellular versus humoral immunology: a century-long dispute. Nat Immunol (2003) 4(5):425–8. doi: 10.1038/ni0503-425
31. Hodgkin PD, Heath WR, Baxter AG. The clonal selection theory: 50 years since the revolution. Nat Immunol (2007) 8:1019–26. doi: 10.1038/ni1007-1019
32. Burnet FM. A modification of Jerne’s theory of antibody production using the concept of clonal selection. Aust J Sci (1957) 20:67–9.
34. Rosin DL, Okusa MD. Dangers Within: DAMP Responses to Damage and Cell Death in Kidney Disease. J Am Soc Nephrol (2011) 22:416–25. doi: 10.1681/ASN.2010040430
35. Pradeu T, Cooper EL. The danger theory: 20 years later. Front Immunol (2012) 3:1–8. doi: 10.3389/fimmu.2012.00287
36. Netea MG, Schlitzer A, Placek K, Joosten LAB, Schultze JL. Innate and Adaptive Immune Memory: an Evolutionary Continuum in the Host’s Response to Pathogens. Cell Host Microbe (2019) 25(1):13–26. doi: 10.1016/j.chom.2018.12.006
37. Bretscher P. The history of the two-signal model of lymphocyte activation: A personal perspective. Scand J Immunol (2019) 89(6):e12762. doi: 10.1111/sji.12762
38. Cunningham AJ. Evolution in microcosm: The rapid somatic diversification of lymphocytes. Cold Spring Harbor Symp Quant Biol (1977) 41:761–70. doi: 10.1101/SQB.1977.041.01.087
39. Matzinger P. Essay 1: The Danger Model in Its Historical Context. Scand J Immunol (2001) 54:4–9. doi: 10.1046/j.1365-3083.2001.00974.x
40. Bretscher P, Cohn M. A theory of self-nonself discrimination. Science (1970) 169(3950):1042–9. doi: 10.1126/science.169.3950.1042
41. Crotty S. A brief history of T cell help to B cells. Nat Rev Immunol (2015) 15(3):185–9. doi: 10.1038/nri3803
42. Hua Z, Hou B. The role of B cell antigen presentation in the initiation of CD4+ T cell response. Immunol Rev (2020) 296:1–12. doi: 10.1111/imr.12859
43. Lafferty KJ, Cunningham AJ. A new analysis of allogeneic interactions. Aust J Exp Biol Med Sci (1975) 53:27–42. doi: 10.1038/icb.1975.3
44. Lafferty KJ, Warren HS, Woolnough JA, Talmage DW. Immunological induction of T lymphocytes: role of antigen and the lymphocyte costimulator. Blood Cells (1978) 4:395–406.
45. Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med (1973) 137:1142–62. doi: 10.1084/jem.137.5.1142
46. Adema GJ. Dendritic cells from bench to bedside and back. Immunol Lett (2009) 122(2):128–30. doi: 10.1016/j.imlet.2008.11.017
47. Dai H, Thomson AW, Rogers NM. Dendritic Cells as Sensors, Mediators, and Regulators of Ischemic Injury. Front Immunol (2019) 10:1–11. doi: 10.3389/fimmu.2019.02418
48. Geginat J, Nizzoli G, Paroni M, Maglie S, Larghi P, Pascolo S, et al. Immunity to Pathogens Taught by Specialized Human Dendritic Cell Subsets. Front Immunol (2015) 6:527. doi: 10.3389/fimmu.2015.00527
49. Schülke S. Induction of Interleukin-10 Producing Dendritic Cells As a Tool to Suppress Allergen-Specific T Helper 2 Responses. Front Immunol (2018) 9:455. doi: 10.3389/fimmu.2018.00455
50. Pradeu T, Carosella ED. The self model and the conception of biological identity in immunology. Biol Philosophy (2006) 21:235–52. doi: 10.1007/s10539-005-8621-6
51. Janeway CA. The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol Today (1992) 13(1):11–6. doi: 10.1016/0167-5699(92)90198-G
52. Janeway CAJ. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harbor Symp Quant Biol (1989) 54:1–13. doi: 10.1101/SQB.1989.054.01.003
53. Janeway CAJ, Medzhitov R. Innate immune recognition. Annu Rev Immunol (2002) 20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359
54. Gayed PM. Toward a Modern Synthesis of Immunity: Charles A. Janeway Jr. and the Immunologist’s Dirty Little Secret. Yale J Biol Med (2011) 84(2):131–8.
55. Medzhitov R, Janeway CA. Innate immunity: impact on the adaptive immune response. Curr Opin Immunol (1997) 9:4–9. doi: 10.1016/S0952-7915(97)80152-5
56. Medzhitov R. Inflammation 2010: New Adventures of an Old Flame. Cell (2010) 140(6):771–6. doi: 10.1016/j.cell.2010.03.006
57. Allison JP, Benoist C, Chervonsky AV. Nobels: Toll pioneers deserve recognition. Nature (2011) 479:178. doi: 10.1038/479178a
58. Matzinger P. The Danger Model: A Renewed Sense of Self. Science (2002) 296:301–5. doi: 10.1126/science.1071059
59. Matzinger P. Tolerance, Danger, and the Extended Family. Ann Rev Immunol (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
60. Matzinger P. The evolution of the danger theory. Expert Rev Clin Immunol (2012) 8(4):311–7. doi: 10.1586/eci.12.21
61. Dobson GP, Letson HL, Biros E, Morris J. Specific pathogen-free (SPF) animal status as a variable in biomedical research: Have we come full circle? EBioMed (Lancet) (2019) 41(March):42–3. doi: 10.1016/j.ebiom.2019.02.038
62. Dobson GP, Morris J, Biros E, Letson HL. Specific Pathogen-Free Animals for Civilian and Military Trauma: A Cautionary Note in the Translation of New Drug Therapies. Shock (2020) 54(2):232–6. doi: 10.1097/SHK.0000000000001495
63. Kumpitsch C, Koskinen K, Schopf V, Moissl-Eichinger C. The microbiome of the upper respiratory tract in health and disease. BMC Biol (2019) 17(1):87. doi: 10.1186/s12915-019-0703-z
64. Morris JL, Letson HL, Gillman R, Hazratwala K, Wilkinson M, McEwen P, et al. The CNS theory of osteoarthritis: Opportunities beyond the joint. Semin Arthritis Rheumatol (2019) 49:331–6. doi: 10.1016/j.semarthrit.2019.03.008
65. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol (2007) 81(1):1–5. doi: 10.1189/jlb.0306164
66. Pacheco-Tena C, Gonzalez-Chavez SA. The danger model approach to the pathogenesis of the rheumatic diseases. J Immunol Res (2015) 2015:506089. doi: 10.1155/2015/506089
67. Merien F. A Journey with Elie Metchnikoff: From Innate Cell Mechanisms in Infectious Diseases to Quantum Biology. Front Public Health (2016) 4:125. doi: 10.3389/fpubh.2016.00125
68. Dinarello CA. Historical insights into cytokines. Eur J Immunol (2007) 37(Suppl 1):S34–45. doi: 10.1002/eji.200737772
69. Cavaillon JM. Cytokines and macrophages. BioMed Pharmacother (1994) 48:445–53. doi: 10.1016/0753-3322(94)90005-1
70. Ferrara JL. Cytokine dysregulation as a mechanism of graft versus host disease. Curr Opin Immunol (1993) 5(Oct):794–79. doi: 10.1016/0952-7915(93)90139-J
71. D’Elia RV, Harrison K, Oyston PC, Lukaszewski RA, Clark GC. Targeting the “cytokine storm” for therapeutic benefit. Clin Vaccine Immunol (2013) 20(3):319–27. doi: 10.1128/CVI.00636-12
72. Beutler B, Milsark IW, Cerami AC. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science (1985) 229:869–71. doi: 10.1126/science.3895437
73. Cavaillon JM, Fitting C, Haeffner-Cavaillon N, Kirsch SJ, Warren HS. Cytokine Response by Monocytes and Macrophages to Free and Lipoprotein-Bound Lipopolysaccharide. Infect Immun (1990) 58(July):2735–82. doi: 10.1128/IAI.58.7.2375-2382.1990
74. Scott A, Khan KM, Cook JL, Duronio V. What is “inflammation”? Are we ready to move beyond Celsus? Br J Sports Med (2004) 38(3):248–9. doi: 10.1136/bjsm.2003.011221
75. Rocha E Silva M. A brief survey of the history of inflammation. Agents Actions (1978) 8(1-2):45–8. doi: 10.1007/BF01972401
76. Hunter J. A Treatise on the Blood, Inflammation, and Gunshot Wounds. Philadelphia: Haswell, Barrington and Haswell (1840). p. 611.
77. Nathan C. Points of control in inflammation. Nature (2002) 420(6917):846–52. doi: 10.1038/nature01320
78. Medzhitov R. Origin and physiological roles of inflammation. Nature (2008) 454(July):428–35. doi: 10.1038/nature07201
79. Nunez G. Intracellular sensors of microbesand danger. Immunol Rev (2011) 243:5–8. doi: 10.1111/j.1600-065X.2011.01058.x
80. Shen H, Kreisel D, Goldstein DR. Processes of Sterile Inflammation. J Immunol (2013) 191:2857–63. doi: 10.4049/jimmunol.1301539
81. Hagemann JH, Haegele H, Muller S, Anders H-J. Danger control programs cause tissue injury and remodeling. Int J Mol Sci (2013) 14(6):11319–46. doi: 10.3390/ijms140611319
82. Dorrington MG, Fraser IDC. NF-kappaB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front Immunol (2019) 10:705. doi: 10.3389/fimmu.2019.00705
83. Allie SR, Randall TD. Resident Memory B Cells. Viral Immunol (2020) 33(4):282–93. doi: 10.1089/vim.2019.0141
84. Galli SJ, Gaudenzio N. Human mast cells as antigen-presenting cells: When is this role important in vivo? J Allergy Clin Immunol (2018) 141(1):92–3. doi: 10.1016/j.jaci.2017.05.029
85. Fan X, Rudensky AY. Hallmarks of Tissue-Resident Lymphocytes. Cell (2016) 164(6):1198–211. doi: 10.1016/j.cell.2016.02.048
86. Roehr B. Tissue resident memory cells emerging as key player in health and disease. Proc Natl Acad Sci USA (2017) 114(46):12092–3. doi: 10.1073/pnas.1715754114
87. Chou C, Li MO. Tissue-Resident Lymphocytes Across Innate and Adaptive Lineages. Front Immunol (2018) 9(Sep 21):2104. doi: 10.3389/fimmu.2018.02104
88. Venereau E, Ceriotti C, Bianchi ME. DAMPs from Cell Death to New Life. Front Immunol (2015) 6:422. doi: 10.3389/fimmu.2015.00422
89. Hauser CJ, Otterbein LE. Danger signals from mitochondrial DAMPS in trauma and post-injury sepsis. Eur J Trauma Emerg Surg (2018) 44(3):317–24. doi: 10.1007/s00068-018-0963-2
90. Roh JS, Sohn DH. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw (2018) 18(4):e27. doi: 10.4110/in.2018.18.e27
91. Piccinini AM, Midwood KS. DAMPening Inflammation by Modulating TLR Signalling. Mediators Inflammation (2010) 2010:1–21. doi: 10.1155/2010/672395
92. Ravindran S. Profile of Bruce A. Beutler. Proc Natl Acad Sci U S A (2013) 110(32):12857–8. doi: 10.1073/pnas.1311624110
93. Kigerl KA, de Rivero Vaccari JP, Dietrich WD, Popovich PG, Keane RW. Pattern recognition receptors and central nervous system repair. Exp Neurol (2014) 258:5–16. doi: 10.1016/j.expneurol.2014.01.001
94. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2020) 140(6):805–20. doi: 10.1016/j.cell.2010.01.022
95. Kawai T, Akira S. Pathogen recognition with Toll-like receptors. Curr Opin Immunol (2005) 17(4):338–44. doi: 10.1016/j.coi.2005.02.007
96. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124:783–801. doi: 10.1016/j.cell.2006.02.015
97. Vijay K. Toll-like receptors in immunity and infl ammatory diseases: Past, present, and future. Int Immunopharmacol (2018) 59(June):391–412. doi: 10.1016/j.intimp.2018.03.002
98. O’Neill LAJ, Golenbock D, Bowie AG. The history of Toll-like receptors — redefining innate immunity. Nat Rev Immun (2013) 13:453–60. doi: 10.1038/nri3446
99. Gao W, Xiong Y, Li Q, Yang H. Inhibition of Toll-Like Receptor Signaling as a Promising Therapy for Inflammatory Diseases: A Journey from Molecular to Nano Therapeutics. Front Physiol (2017) 8:508. doi: 10.3389/fphys.2017.00508
100. Mogensen TH. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin Microbiol Rev (2009) 22(2):240–73. doi: 10.1128/CMR.00046-08
101. Kitazume-Taneike R, Taneike M, Omiya S, Misaka T, Nishida K, Yamaguchi O, et al. Ablation of Toll-like receptor 9 attenuates myocardial ischemia/reperfusion injury in mice. Biochem Biophys Res Commun (2019) 515(3):442–7. doi: 10.1016/j.bbrc.2019.05.150
102. Wen AY, Sakamoto KM, Miller LS. The Role of the Transcription Factor CREB in Immune Function. J Immunol (2010) 185(11):6413–9. doi: 10.4049/jimmunol.1001829
103. Goulopoulou S, McCarthy CG, Webb RC. Toll-like Receptors in the Vascular System: Sensing the Dangers Within. Pharmacol Rev (2016) 68(1):142–67. doi: 10.1124/pr.114.010090
104. Kanneganti T-D. Central roles of NLRs and inflammasomes in viral infection. Nat Rev Immunol (2010) 10(10):688–98. doi: 10.1038/nri2851
105. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140(6):821–32. doi: 10.1016/j.cell.2010.01.040
106. Vecchie A, Bonaventura A, Toldo S, Dagna L, Dinarello CA, Abbate A. IL-18 and infections: Is there a role for targeted therapies? J Cell Physiol (2020) 1–20. doi: 10.1002/jcp.30008
107. Newton K, Dixit VM. Signaling in Innate Immunity and Inflammation. Cold Spring Harb Perspect Biol (2012) 4:a006049. doi: 10.1101/cshperspect.a006049
108. Zhou G-N, Jiang D-S, Li H. Interferon regulatory factors: at the crossroads of immunity, metabolism,and disease. Biochim Biophys Acta (2015) 1852:365–78. doi: 10.1016/j.bbadis.2014.04.030
109. Vono M, Lin A, Norrby-Teglund A, Koup RA, Liang F, Loré K. Neutrophils acquire the capacity for antigen presentation to memory CD4+ T cells in vitro and ex vivo. Blood (2017) 129(14):1991–2001. doi: 10.1182/blood-2016-10-744441
110. Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal Transduct Target Ther (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
111. Adelaja A, Hoffmann A. Signaling Crosstalk Mechanisms That May Fine-Tune Pathogen-Responsive NFkappaB. Front Immunol (2019) 10:433. doi: 10.3389/fimmu.2019.00433
112. McGuire VA, Arthur JMC. Subverting Toll-Like Receptor Signaling by Bacterial Pathogens. Front Immunol (2015) 6:607. doi: 10.3389/fimmu.2015.00607
113. Lin B, Dutta B, Fraser IDC. Systematic Investigation of Multi-TLR Sensing Identifies Regulators of Sustained Gene Activation in Macrophages. Cell Syst (2017) 5(1):25–37 e3. doi: 10.1016/j.cels.2017.06.014
114. Stetson DB, Medzhitov R. Type I Interferons in Host Defense. Immunity (2006) 25:373–81. doi: 10.1016/j.immuni.2006.08.007
115. Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer (2013) 12:86. doi: 10.1186/1476-4598-12-86
116. Heissig B, Salama Y, Takahashi S, Osada T, Hattori K. The multifaceted role of plasminogen in inflammation. Cell Signal (2020) 75:109761. doi: 10.1016/j.cellsig.2020.109761
117. Fujioka S, Niu J, Schmidt C, Sclabas GM, Peng B, Uwagawa T, et al. NF-kappaB and AP-1 connection: mechanism of NF-kappaB-dependent regulation of AP-1 activity. Mol Cell Biol (2004) 24(17):7806–19. doi: 10.1128/MCB.24.17.7806-7819.2004
118. Hawkins PT, Stephens LR. PI3K signalling in inflammation. Biochim Biophys Acta (2015) 1851(6):882–97. doi: 10.1016/j.bbalip.2014.12.006
119. Koga Y, Tsurumaki H, Aoki-Saito H, Sato M, Yatomi M, Takehara K, et al. Roles of Cyclic AMP Response Element Binding Activation in the ERK1/2 and p38 MAPK Signalling Pathway in Central Nervous System, Cardiovascular System, Osteoclast Differentiation and Mucin and Cytokine Production. Int J Mol Sci (2019) 20(6):1346. doi: 10.3390/ijms20061346
120. Shen H, Tesar BM, Walker WE, Goldstein DR. Dual Signaling of MyD88 and TRIF are Critical for Maximal TLR4-Induced Dendritic Cell Maturation. J Immunol (2008) 181(3):1849–58. doi: 10.4049/jimmunol.181.3.1849
121. Wrona D. Neural-immune interactions: an integrative view of the bidirectional relationship between the brain and immune systems. J Neuroimmunol (2006) 172:38–58. doi: 10.1016/j.jneuroim.2005.10.017
122. Bennett JM, Reeves G, Billman GE, Sturmberg JP. Inflammation-Nature’s Way to Efficiently Respond to All Types of Challenges: Implications for Understanding and Managing “the Epidemic” of Chronic Diseases. Front Med (Lausanne) (2018) 5:316. doi: 10.3389/fmed.2018.00316
123. Neher MD, Weckbach S, Flierl MA, Huber-Lang MS, Stahel PF. Molecular mechanisms of inflammation and tissue injury after major trauma-is complement the “bad guy”? J BioMed Sci (2011) 18:90. doi: 10.1186/1423-0127-18-90
124. Xu L, Botchway BOA, Zhang S, Zhou J, Liu X. Inhibition of NF-kappaB Signaling Pathway by Resveratrol Improves Spinal Cord Injury. Front Neurosci (2018) 12:690. doi: 10.3389/fnins.2018.00690
125. Nakayama H, Otsu K. Mitochondrial DNA as an inflammatory mediator in cardiovascular diseases. Biochem J (2018) 475:839–52. doi: 10.1042/BCJ20170714
126. Serhan CN, de la Rosa X, Jouvene C. Novel mediators and mechanisms in the resolution of infectious inflammation: evidence for vagus regulation. J Intern Med (2019) 286(3):240–58. doi: 10.1111/joim.12871
127. Haack KKV, Mitra AK, Zucker IH. NF-κB and CREB Are Required for Angiotensin II Type 1 Receptor Upregulation in Neurons. PLoS One (2013) 8(11):e78695. doi: 10.1371/journal.pone.0078695
128. Gleeson PA. The role of endosomes in innate and adaptive immunity. Semin Cell Dev Biol (2014) 31:64–72. doi: 10.1016/j.semcdb.2014.03.002
129. Vilborg A, Passarelli MC, Steitz JA. Calcium signaling and transcription: elongation, DoGs, and eRNAs. Receptors Clin Invest (2016) 3(1):e1169. doi: 10.14800/rci.1169
130. Bodgdan C. Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol (2015) 36(3):161–78. doi: 10.1016/j.it.2015.01.003
131. Faas MM, de Vos P. Mitochondrial function in immune cells in health and disease. Biochim Biophys Acta Mol Basis Dis (2020) 1866(10):165845. doi: 10.1016/j.bbadis.2020.165845
132. Karst SM, Wobus CE, Lay M, Davidson J, Virgin HW 4th. STAT1-dependent innate immunity to a Norwalk-like virus. Science (2003) 299:1575–8. doi: 10.1126/science.1077905
133. Renn CN, Sanchez DJ, Ochoa MT, Legaspi AJ, Oh C-K, Liu PT, et al. TLR activation of langerhans cell-like dendritic cells triggers an antiviral immune response. J Immunol (2006) 177:298–305. doi: 10.4049/jimmunol.177.1.298
134. Stavitsky AB. The innate immune response to infection, toxins and trauma evolved into networks of interactive, defensive, reparative, regulatory, injurious and pathogenic pathways. Mol Immunol (2007) 44:2787–99. doi: 10.1016/j.molimm.2007.01.011
135. Stahel PF, Smith WR, Moore EE. Role of biological modifiers regulating the immune response after trauma. Injury (2007) 38:1409–22. doi: 10.1016/j.injury.2007.09.023
136. Raad M, Nohra E, Chams N, Itani M, Talih F, Mondello S, et al. Autoantibodies in traumatic brain injury and central nervous system trauma. Neuroscience (2014) 281:16–23. doi: 10.1016/j.neuroscience.2014.08.045
137. Choudhry MA, Bland KI, Chaudry IH. Trauma and immune response–effect of gender differences. Injury (2007) 38(12):1382–91. doi: 10.1016/j.injury.2007.09.027
138. Tschoeke SK, Ertel W. Immunoparalysis after multiple trauma. Injury (2007) 38(12):1346–57. doi: 10.1016/j.injury.2007.08.041
139. Dobson GP, Letson HL. Far Forward Gaps in Hemorrhagic Shock and Prolonged Field Care: An Update of ALM Fluid Therapy for Field Use. J Spec Oper Med (2020) 20(3):78–84.
140. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8(12):958–69. doi: 10.1038/nri2448
141. Muraille E, Leo O, Moser M. Th1/Th2 Paradigm Extended: Macrophage Polarization as an Unappreciated Pathogen-Driven Escape Mechanism? Front Immunol (2014) 5:603. doi: 10.3389/fimmu.2014.00603
142. Mills CD. Anatomy of a Discovery: M1 and M2 Macrophages. Front Immunol (2015) 6(May 5):212. doi: 10.3389/fimmu.2015.00212
143. Gordon S, Plüddemann A. Tissue macrophages: heterogeneity and functions. BMC Biol (2017) 15:53. doi: 10.1186/s12915-017-0392-4
144. Atri C, Guerfali FZ, Laouini D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int J Mol Sci (2018) 19(6):1801. doi: 10.3390/ijms19061801
145. Mu X, Li Y, Fan G-C. Tissue-Resident Macrophages in the Control of Infection and Resolution of Inflammation. Shock (2020). doi: 10.1097/SHK.0000000000001601
146. Kanashiro A, Hiroki CH, Morais da Fonseca M, Birbrair A, Gomes Ferreira R, Shimizu Bassi G, et al. The role of neutrophils in neuro-immune modulation. Pharmacol Res (2020) 151:104580. doi: 10.1016/j.phrs.2019.104580
147. Bohlson SS, O’Conner SD, Hulsebus HJ, Ho M-M, Fraser DA. Complement, c1q, and c1q-related molecules regulate macrophage polarization. Front Immunol (2014) 5:402. doi: 10.3389/fimmu.2014.00402
148. Green DP, Limjunyawong N, Gour N, Pundir P, Dong X. A Mast-Cell-Specific Receptor Mediates Neurogenic Inflammation and Pain. Neuron (2019) 101(3):412–20.e3. doi: 10.1016/j.neuron.2019.01.012
149. Abraham SN, St John AL. Mast cell-orchestrated immunity to pathogens. Nat Rev Immunol (2010) 10(6):440–52. doi: 10.1038/nri2782
150. Li Y, Wang W, Yang F, Xu Y, Feng C, Zhao Y. The regulatory roles of neutrophils in adaptive immunity. Cell Commun Signal (2019) 17(1):147. doi: 10.1186/s12964-019-0471-y
151. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science (2011) 331(6013):44–9. doi: 10.1126/science.1198687
152. Linke B, Schreiber Y, Picard-Willems B, Slattery P, Nusing RM, Harder S, et al. Activated Platelets Induce an Anti-Inflammatory Response of Monocytes/Macrophages through Cross-Regulation of PGE2 and Cytokines. Mediators Inflammation (2017) 2017:1463216. doi: 10.1155/2017/1463216
153. Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol (2006) 311:17–58. doi: 10.1007/3-540-32636-7_2
154. Neurath MF. Resolution of inflammation: from basic concepts to clinical application. Semin Immunopath (2019) 41:627–31. doi: 10.1007/s00281-019-00771-2
155. Tugal D, Liao X, Jain MK. Transcriptional Control of Macrophage Polarization. Arterioscler Thromb Vasc Biol (2013) 33:1135–44. doi: 10.1161/ATVBAHA.113.301453
156. Rőszer T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediators Inflammation (2015) 2015:816460. doi: 10.1155/2015/816460
157. Ley K. M1 Means Kill; M2 Means Heal. J Immunol (2017) 199:2191–3. doi: 10.4049/jimmunol.1701135
158. Bahia D, Satoskar AR, Dussurget O. Editorial: Cell Signaling in Host-Pathogen Interactions: The Host Point of View. Front Immunol (2018) 9:221. doi: 10.3389/fimmu.2018.00221
159. Shrivastava R, Shukla N. Attributes of Alternatively Activated (M2) Macrophages. Life Sci (2019) 224(May 1):222–31. doi: 10.1016/j.lfs.2019.03.062
160. Li JJ, Tan J, Martino MM, Lui KO. Regulatory T-Cells: Potential Regulator of Tissue Repair and Regeneration. Front Immunol (2018) 9:585. doi: 10.3389/fimmu.2018.00585
161. Corthay A. How do Regulatory T Cells Work? Scand J Immunol (2009) 70:326–36. doi: 10.1111/j.1365-3083.2009.02308.x
162. Wan YY, Flavell RA. ‘Yin-Yang’ functions of TGF-β and Tregs in immune regulation. Immunol Rev (2007) 220(Dec):199–213. doi: 10.1111/j.1600-065X.2007.00565.x
163. Okeke EB, Uzonna JE. The Pivotal Role of Regulatory T Cells in the Regulation of Innate Immune Cells. Front Immunol (2019) 10:680. doi: 10.3389/fimmu.2019.00680
164. Nathan C, Ding A. Nonresolving inflammation. Cell (2010) 140(6):871–82. doi: 10.1016/j.cell.2010.02.029
165. Headland SE, Norling LV. The resolution of inflammation: Principles and challenges. Semin Immunol (2015) 27(3):149–60. doi: 10.1016/j.smim.2015.03.014
166. Panigrahy D, Gilligan MM, Huang S, Gartung A, Cortes-Puch I, Sime PJ, et al. Inflammation resolution: a dual-pronged approach to averting cytokine storms in COVID-19? Cancer Metastasis Rev (2020) 39(2):337–40. doi: 10.1007/s10555-020-09889-4
167. Sugimoto MA, Sousa LP, Pinho V, Perretti M, Teixeira MM. Resolution of Inflammation: What Controls Its Onset? Front Immunol (2016) 7:160. doi: 10.3389/fimmu.2016.00160
168. Dalli J, Serhan CN. Immunoresolvents signaling molecules at intersection between the brain and immune system. Curr Opin Immunol (2018) 50:48–54. doi: 10.1016/j.coi.2017.10.007
169. Spite M, Serhan CN. Novel Lipid Mediators Promote Resolution of Acute Inflammation: Impact of Aspirin and Statins. Circ Res (2010) 107:1170–84. doi: 10.1161/CIRCRESAHA.110.223883
170. Duncan SA, Baganizi DR, Sahu R, Singh SS, Dennis VA. SOCS Proteins as Regulators of Inflammatory Responses Induced by Bacterial Infections: A Review. Front Microbiol (2017) 8:2431. doi: 10.3389/fmicb.2017.02431
171. Pacheco R, Riquelme E, Kalergis EM. Emerging Evidence for the Role of Neurotransmitters in the Modulation of T Cell Responses to Cognate Ligands. Cent Nerv Syst Agents Med Chem (2010) 10(1):65–83. doi: 10.2174/187152410790780154
172. Procaccini C, Pucino V, De Rosa V, Marone G, Matarese G. Neuro-endocrine networks controlling immune system in health and disease. Front Immunol (2014) 5:143. doi: 10.3389/fimmu.2014.00143
173. Dantzer R. Neuroimmune Interactions: From the Brain to the Immune System and Vice Versa. Physiol Rev (2018) 98(1):477–504. doi: 10.1152/physrev.00039.2016
174. Besedovsky H, Sorkin E. Network of immune-neuroendocrine interactions. Clin Exp Immunol (1977) 27(1):1–12.
175. Pinoli M, Marino F, Cosentino M. Dopaminergic Regulation of Innate Immunity: A Review. J Neuroimmune Pharmacol (2017) 12(4):602–23. doi: 10.1007/s11481-017-9749-2
176. Prado C, Contreras F, Gonzalez H, Diaz P, Elgueta D, Barrientos M, et al. Stimulation of dopamine receptor D5 expressed on dendritic cells potentiates Th17-mediated immunity. J Immunol (2012) 188(7):3062–70. doi: 10.4049/jimmunol.1103096
177. Letson HL, Morris J, Biros E, Dobson GP. Conventional and Specific-Pathogen Free Rats Respond Differently to Anesthesia and Surgical Trauma. Sci Rep (2019) 9(1):9399. doi: 10.1038/s41598-019-45871-z
178. Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol (2014) 32(1):40–51. doi: 10.1038/nbt.2786
179. Downing NS, Shah ND, Aminawung JA, Pease AM, Zeitoun J-D, Krumholz HM, et al. Postmarket Safety Events Among Novel Therapeutics Approved by the US Food and Drug Administration Between 2001 and 2010. JAMA (2017) 317(18):1854–63. doi: 10.1001/jama.2017.5150
180. Dobson GP. Addressing the global burden of sepsis: Importance of a systems-based approach. Crit Care Med (2014) 42(Dec 12):e797–8. doi: 10.1097/CCM.0000000000000595
181. Pound P, Ritskes-Hoitinga M. Is it possible to overcome issues of external validity in preclinical animal research? Why most animal models are bound to fail. J Transl Med (2018) 16(1):304. doi: 10.1186/s12967-018-1678-1
182. Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature (2016) 532:512–6. doi: 10.1038/nature17655
183. Lee SK. Sex as an important biological variable in biomedical research. BMB Rep (2018) 51(4):167–73. doi: 10.5483/BMBRep.2018.51.4.034
184. Franconi F, Campesi I, Colombo D, Antonini P. Sex-Gender Variable: Methodological Recommendations for Increasing Scientific Value of Clinical Studies. Cells (2019) 8(5):476. doi: 10.3390/cells8050476
185. Mauvais-Jarvis F, Bairey Merz N, Barnes PJ, Brinton RD, Carrero J-J, DeMeo DL, et al. Sex and gender: modifiers of health, disease, and medicine. Lancet (2020) 396(10250):565–82. doi: 10.1016/S0140-6736(20)31561-0
186. Valvis SM, Waithman J, Wood FM, Fear MW, Fear VS. The Immune Response to Skin Trauma Is Dependent on the Etiology of Injury in a Mouse Model of Burn and Excision. J Invest Dermatol (2015) 135(8):2119–28. doi: 10.1038/jid.2015.123
187. Demyanets S, Wojta J. Sex Differences in Effects and Use of Anti-inflammatory Drugs., in Sex and Gender Differences in Pharmacology. Handbook of Experimental Pharmacology. V R-Z, editor. Berlin: Springer (2012) p. 443–72.
188. Gölz C, Kirchhoff FP, Westerhorstmann J, Schmidt M, Hirnet T, Rune GM, et al. Sex hormones modulate pathogenic processes in experimental traumatic brain injury. J Neurochem (2019) 150(2):173–87. doi: 10.1111/jnc.14678
189. Doran SJ, Ritzel RM, Glaser EP, Henry RJ, Faden AI, Loane DJ. Sex Differences in Acute Neuroinflammation after Experimental Traumatic Brain Injury Are Mediated by Infiltrating Myeloid Cells. J Neurotrauma (2019) 36(7):1040–53. doi: 10.1089/neu.2018.6019
190. Van Norman GA. Phase II Trials in Drug Development and Adaptive Trial Design. JACC: Basic Trans Sci (2019) 4(3):428–37. doi: 10.1016/j.jacbts.2019.02.005
191. Lewis SC, Warlow CP. How to spot bias and other potential problems in randomised controlled trials. J Neurol Neurosurg Psychiatry (2004) 75:181–7. doi: 10.1136/jnnp.2003.025833
192. Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev (2018) 281(1):8–27. doi: 10.1111/imr.12621
193. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet (2020) 395(10229):1033–4. doi: 10.1016/S0140-6736(20)30628-0
194. Moore TJ, Zhang H, Anderson G, Alexander GC. Estimated Costs of Pivotal Trials for Novel Therapeutic Agents Approved by the US Food and Drug Administration, 2015-2016. JAMA Intern Med (2018) 178(11):1451–7. doi: 10.1001/jamainternmed.2018.3931
195. Dobson GP. On Being the Right Size: Heart Design, mitochondrial efficiency, and lifespan potential. Clin Exp Pharm Physiol (2003) 30(8):590–7. doi: 10.1046/j.1440-1681.2003.03876.x
196. Dobson GP, Faggian G, Onorati F, Vinten-Johansen J. Hyperkalemic cardioplegia in adult and pediatric cardiac surgery: end of an Era? Front Clin Trans Physiol (2013) 4(Aug 28):1–28. doi: 10.3389/fphys.2013.00228
197. Dobson GP, Jones MW. Adenosine and Lignocaine: a new concept in non-depolarising surgical arrest, protection and preservation. J Thoracic Cardiovas Surg (2004) 127(3):794–805. doi: 10.1016/S0022-5223(03)01192-9
198. Dobson GP. Membrane Polarity: A target for myocardial protection and reduced inflammation in adult and pediatric cardiothoracic surgery (Editorial - Free Standing). J Thorac Cardiovasc Surg (2010) 140(December):1213–7. doi: 10.1016/j.jtcvs.2010.05.040
199. Onorati F, Santini F, Dandale R, Ucci G, Pechlivandis K, Menon T, et al. “Polarizing” microplegia improves cardiac cycle efficiency after CABG for unstable angina. Int J Cardiol (2013) 167(6):2739–46. doi: 10.1016/j.ijcard.2012.06.099
200. Onorati F, Dobson GP, San Biagio L, Abbasciano R, Fanti D, Covajes C, et al. Superior Myocardial Protection using ‘Polarizing’ Adenosine, Lidocaine, and Mg2+ (ALM) Cardioplegia in Humans. J Am Coll Cardiol (2016) 67(14):1751–3. doi: 10.1016/j.jacc.2015.12.071
201. Letson HL, Dobson GP. 3.0% NaCl Adenosine, Lidocaine, Mg2+ (ALM) bolus and 4 hours ‘drip’ infusion reduces non-compressible hemorrhage by 60% in a rat model. J Trauma Acute Care Surg (2017) 82(6):1063–72. doi: 10.1097/TA.0000000000001454
202. Letson HL, Dobson GP. Adenosine, Lidocaine and Mg2+ (ALM) fluid therapy attenuates systemic inflammation, platelet dysfunction and coagulopathy after non-compressible truncal hemorrhage. PloS One (2017) 12(11):e0188144. doi: 10.1371/journal.pone.0188144
203. Letson HL, Dobson GP. Adenosine, Lidocaine and Mg2+ (ALM) resuscitation fluid protects against experimental traumatic brain injury. J Trauma Acute Care Surg (2018) 84(6):908–16. doi: 10.1097/TA.0000000000001874
204. Letson HL, Morris JL, Biros E, Dobson GP. ALM fluid therapy leads to 72 hr survival after hemorrhagic shock: a model for studying differential gene expression and extending biological time. J Trauma Acute Care Surg (2019) 87(3):606–13. doi: 10.1097/TA.0000000000002397
205. Letson HL, Granfeldt A, Jensen TH, Mattson TH, Dobson GP, et al. ALM supports a high flow, hypotensive, vasodilatory state with improved O2 delivery and cerebral protection in a pig model of non-compressible hemorrhage. J Surg Res (2020) 253(Sept):127–38. doi: 10.1016/j.jss.2020.03.048
206. Griffin MJ, Letson HL, Dobson GP. Adenosine, Lidocaine and Mg2+ (ALM) induces a reversible hypotensive state, reduces lung edema and prevents coagulopathy in the rat model of polymicrobial sepsis. J Trauma Acute Care Surg (2014) 77(3):471–8. doi: 10.1097/TA.0000000000000361
207. Griffin MJ, Letson HL, Dobson GP. Small-volume Adenosine, lidocaine and Mg2+ (ALM) 4 hour infusion leads to 88% survival after 6 days of experimental sepsis in the rat without antibiotics. Clin Vaccine Immunol (2016) 23(11):863–72. doi: 10.1128/CVI.00390-16
208. Torres Filho IP, Torres LN, Salgado C, Dubick MA, et al. Novel Adjunct Drugs Reverse Endothelial Glycocalyx Damage After Hemorrhagic Shock in Rats. Shock (2017) 48(5):583–9. doi: 10.1097/SHK.0000000000000895
209. Linnemann C, Schildberg FA, Schurich A, Diehl L, Hegenbarth SI, Endl E, et al. Adenosine regulates CD8 T-cell priming by inhibition of membrane-proximal T-cell receptor signalling. Immunology (2009) 128(1 Suppl):e728–37. doi: 10.1111/j.1365-2567.2009.03075.x
210. Chambers AM, Wang J, Lupo KB, Yu H, Atallah Lanman NM, Matosevic S. Adenosinergic Signaling Alters Natural Killer Cell Functional Responses. Front Immunol (2018) 9:2533. doi: 10.3389/fimmu.2018.02533
211. Lahat A, Ben-Horin S, Lang A, Fudim E, Picard O, Chowers Y. Lidocaine down-regulates nuclear factor-kB signalling and inhibits cytokine production and T cell proliferation. Clin Exp Immunol (2008) 152:320–7. doi: 10.1111/j.1365-2249.2008.03636.x
212. Cairns CB, Krafi M. Magnesium attenuates the neutrophil respiratory burst in adult asthmatic patients. Acad Emerg Med (1996) 3(12):1093–7. doi: 10.1111/j.1553-2712.1996.tb03366.x
213. Chaigne-Delalande B, Li F-Y, O’Connor GM, Lukacs MJ, Jiang P, Zheng L, et al. Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D. Science (2013) 341(6142):186–91. doi: 10.1126/science.1240094
214. Diao B, Huang X, Guo S, Yang C, Liu G, Chen Y, et al. MAGT1-mediated disturbance of Mg(2+) homeostasis lead to exhausted of HBV-infected NK and CD8(+) T cells. Sci Rep (2017) 7(1):13594. doi: 10.1038/s41598-017-11522-4
215. Haas B, Leonard F, Ernens I, Rodius S, Vausort M, Rolland-Turner M, et al. Adenosine reduces cell surface expression of toll-like receptor 4 and inflammation in response to lipopolysaccharide and matrix products. J Cardiovasc Transl Res (2011) 4(6):790–800. doi: 10.1007/s12265-011-9279-x
216. Yang Y, Lv J, Jiang S, Ma Z, Wang D, Hu W, et al. The emerging role of Toll-like receptor 4 in myocardial inflammation. Cell Death Dis (2016) 7:e2234. doi: 10.1038/cddis.2016.140
217. Minguet S, Huber M, Rosenkranz L, Schamel WWA, Reth M, Brummer T. Adenosine and cAMP are potent inhibitors of the NF-kappa B pathway downstream of immunoreceptors. Eur J Immunol (2005) 35(1):31–41. doi: 10.1002/eji.200425524
218. Gessi S, Varani K, Merighi S, Fogli E, Sacchetto V, Benini A, et al. Adenosine and lymphocyte regulation. Purinerg Signal (2007) 3(1-2):109–16. doi: 10.1007/s11302-006-9042-y
219. Li C, Ha T, Liu L, Browder W, Kao RL. Adenosine prevents activation of transcription factor NF-kappa B and enhances activator protein-1 binding activity in ischemic rat heart. Surgery (2000) 127(2):161–9. doi: 10.1067/msy.2000.101582
220. Fuentes-Antras J, Loan AM, Tunon J, Egido J, Lorenzo O. Activation of toll-like receptors and inflammasome complexes in the diabetic cardiomyopathy-associated inflammation. Int J Endocrinol (2014) 2014:847827. doi: 10.1155/2014/847827
221. Hamidzadeh K, Mosser DM. Purinergic Signaling to Terminate TLR Responses in Macrophages. Front Immunol (2016) 7(2):1–6. doi: 10.3389/fimmu.2016.00074
222. Wagner DR, Combes A, McTiernan C, Sanders VJ, Lemster B, Feldman AM. Adenosine inhibits lipopolysaccharide-induced cardiac expression of tumor necrosis factor-alpha. Circ Res (1998) 82(1):47–56. doi: 10.1161/01.RES.82.1.47
223. Haskó G, Cronstein B. Regulation of inflammation by adenosine. Front Immunol (2013) 4(Apr 8):85. doi: 10.3389/fimmu.2013.00085
224. Cain BS, Harken AH, Meldrum DR. Therapeutic strategies to reduce TNF-alpha mediated cardiac contractile depression following ischemia and reperfusion. J Mol Cell Cardiol (1999) 31(5):931–47. doi: 10.1006/jmcc.1999.0924
225. Ghosh G, Ya-Fan Wang V, Huang D-B, Fusco A. NF-kappaB regulation: lessons from structures. Immunol Rev (2012) 246(1):36–58. doi: 10.1111/j.1600-065X.2012.01097.x
226. Mussbacher M, Salzmann M, Brostjan C, Hoesel B, Schoergenhofer C, Datler H, et al. Cell Type-Specific Roles of NF-kappaB Linking Inflammation and Thrombosis. Front Immunol (2019) 10:85. doi: 10.3389/fimmu.2019.00085
227. Liu J, Zhang H, Qi Z, Zheng X. Lidocaine protects against renal and hepatic dysfunction in septic rats via downregulation of Toll−like receptor 4. Mol Med Rep (2014) 9(1):118–24. doi: 10.3892/mmr.2013.1799
228. Sirait RH, Hatta M, Ramli M, Siagian C, Suprayogi B, Simanjuntak TP. Lidocaine suppressed hyperinflammation in BALB/c mice model sterile injury via downregulation of toll-like receptor 4. Egyptian J Anaesthesia (2019) 34(4):135–7. doi: 10.1016/j.egja.2018.07.002
229. Feng G, Liu S, Wang G-L, Liu G-J. Lidocaine Attenuates Lipopolysaccharide-Induced Acute Lung Injury through Inhibiting NF-κB Activation. Pharmacology (2008) 81:32–40. doi: 10.1159/000107792
230. Jiang R, Liao J, Yang M-C, Deng J, Hu Y-X, Li P, et al. Lidocaine mediates the progression of cerebral ischemia/reperfusion injury in rats via inhibiting the activation of NF-kappaB p65 and p38 MAPK. Ann Transl Med (2020) 8(8):548. doi: 10.21037/atm-20-3066
231. Garutti I, Rancan L, Simon C, Cusati G, Sanchez-Pedrosa G, Moraga F, et al. Intravenous lidocaine decreases tumor necrosis factor alpha expression both locally and systemically in pigs undergoing lung resection surgery. Anesth Analg (2014) 119(4):815–28. doi: 10.1213/ANE.0000000000000360
232. Wen F, Liu Y, Wang H, Tang W, Hou Y-D, Wang H-L. Lidocaine inhibits the production of IL-1β from macrophages RAW264.7 induced with lipopolysaccharide. Int J Clin Exp Pathol (2017) 10(6):6582–8.
233. Brandao K, Deason-Towne F, Perraud A-L, Schmitz C. The role of Mg2+ in immune cells. Immunol Res (2013) 55:261–9. doi: 10.1007/s12026-012-8371-x
234. Sugimoto J, Romani AM, Valentin-Torres AM, Luciano AA, Ramirez Kitchen CM, Funderburg N, et al. Magnesium decreases inflammatory cytokine production: a novel innate immunomodulatory mechanism. J Immunol (2012) 188(12):6338–46. doi: 10.4049/jimmunol.1101765
235. Lin CY, Tsai PS, Hung YC, Huang CJ. L-type calcium channels are involved in mediating the anti-inflammatory effects of magnesium sulphate. Br J Anaesth (2010) 104(1):44–51. doi: 10.1093/bja/aep336
236. Gao F, Ding B, Zhou L, Gao X, Guo H, Xu H. Magnesium sulfate provides neuroprotection in lipopolysaccharide-activated primary microglia by inhibiting NF-kappaB pathway. J Surg Res (2013) 184(2):944–50. doi: 10.1016/j.jss.2013.03.034
237. Yu X, Guan P-P, Zhu D, Liang Y-Y, Wang T, Wang Z-Y, et al. Magnesium Ions Inhibit the Expression of Tumor Necrosis Factor alpha and the Activity of gamma-Secretase in a beta-Amyloid Protein-Dependent Mechanism in APP/PS1 Transgenic Mice. Front Mol Neurosci (2018) 11:172. doi: 10.3389/fnmol.2018.00172
238. Chang YY, Kao M-C, Lin J-A, Chen T-Y, Cheng C-F, Wong C-S, et al. Effects of MgSO4 on inhibiting Nod-like receptor protein 3 inflammasome involve decreasing intracellular calcium. J Surg Res (2018) 221:257–65. doi: 10.1016/j.jss.2017.09.005
239. Piegeler T, Votta-Velis EG, Bakhshi FR, Mao M, Carnegie G, Bonini MG, et al. Endothelial Barrier Protection by Local Anesthetics. Anesthesiology (2014) 120(6):1414–28. doi: 10.1097/ALN.0000000000000174
240. Kaminska B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy—from molecular mechanisms to therapeutic benefits. Biochim Biophys Acta (2005) 1754:253–62. doi: 10.1016/j.bbapap.2005.08.017
241. Granfeldt A, Letson HL, Dobson GP, Shi W, Vinten-Hohansen J, Tonnesen E. Adenosine, lidocaine and Mg2+ improves cardiac and pulmonary function, induces reversible hypotension and exerts anti-inflammatory effects in an endotoxemic porcine model. Crit Care (2015) 18(6):682. doi: 10.1186/s13054-014-0682-y
242. Davenport L, Letson HL, Dobson GP. Immune-inflammatory activation after a single laparotomy in a rat model: effect of adenosine, lidocaine and Mg2+ infusion to dampen the stress response. Innate Immun (2017) 23(5):482–94. doi: 10.1177/1753425917718921
243. Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal (2013) 25(10):1939–48. doi: 10.1016/j.cellsig.2013.06.007
244. Pearce L, Davidson SM, Yellon DM. The cytokine storm of COVID-19: a spotlight on prevention and protection. Expert Opin Ther Targ (2020) 27:1–8. doi: 10.1080/14728222.2020.1783243
Keywords: coronavirus, inflammation, trauma, infection, immune system, evolution
Citation: Dobson GP, Biros E, Letson HL and Morris JL (2021) Living in a Hostile World: Inflammation, New Drug Development, and Coronavirus. Front. Immunol. 11:610131. doi: 10.3389/fimmu.2020.610131
Received: 30 September 2020; Accepted: 25 November 2020;
Published: 22 January 2021.
Edited by:
Jean-Marc Cavaillon, Institut Pasteur, FranceReviewed by:
Didier Payen, Université Sorbonne Paris Cité, FranceMatthijs Kox, Radboud University Nijmegen Medical Centre, Netherlands
Copyright © 2021 Dobson, Biros, Letson and Morris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Geoffrey P. Dobson, Z2VvZmZyZXkuZG9ic29uQGpjdS5lZHUuYXU=