 Erik M. Matson
Erik M. Matson Miranda L. Abyazi
Miranda L. Abyazi Kayla A. Bell
Kayla A. Bell Kevin M. Hayes
Kevin M. Hayes Paul J. Maglione
Paul J. Maglione- Pulmonary Center, Section of Pulmonary, Allergy, Sleep & Critical Care Medicine, Department of Medicine, Boston University School of Medicine, Boston Medical Center, Boston, MA, United States
Common variable immunodeficiency (CVID) is the most frequently diagnosed primary antibody deficiency. About half of CVID patients develop chronic non-infectious complications thought to be due to intrinsic immune dysregulation, including autoimmunity, gastrointestinal disease, and interstitial lung disease (ILD). Multiple studies have found ILD to be a significant cause of morbidity and mortality in CVID. Yet, the precise mechanisms underlying this complication in CVID are poorly understood. CVID ILD is marked by profound pulmonary infiltration of both T and B cells as well as granulomatous inflammation in many cases. B cell depletive therapy, whether done as a monotherapy or in combination with another immunosuppressive agent, has become a standard of therapy for CVID ILD. However, CVID is a heterogeneous disorder, as is its lung pathology, and the precise patients that would benefit from B cell depletive therapy, when it should administered, and how long it should be repeated all remain gaps in our knowledge. Moreover, some have ILD recurrence after B cell depletive therapy and the relative importance of B cell biology remains incompletely defined. Developmental and functional abnormalities of B cell compartments observed in CVID ILD and related conditions suggest that imbalance of B cell signaling networks may promote lung disease. Included within these potential mechanisms of disease is B cell activating factor (BAFF), a cytokine that is upregulated by the interferon gamma (IFN-γ):STAT1 signaling axis to potently influence B cell activation and survival. B cell responses to BAFF are shaped by the divergent effects and expression patterns of its three receptors: BAFF receptor (BAFF-R), transmembrane activator and CAML interactor (TACI), and B cell maturation antigen (BCMA). Moreover, soluble forms of BAFF-R, TACI, and BCMA exist and may further influence the pathogenesis of ILD. Continued efforts to understand how dysregulated B cell biology promotes ILD development and progression will help close the gap in our understanding of how to best diagnose, define, and manage ILD in CVID.
Introduction
Primary antibody deficiencies (PADs) are the most prevalent form of immunodeficiency and are defined by disruption of a patient’s ability to generate functional antibodies. They are further classified by the mechanism of disruption and type of antibody affected. For example, X-linked agammaglobulinemia is an antibody deficiency defined by a reduction in all antibody classes due to a severe block in B cell differentiation, and hyper IgM syndrome is a deficiency characterized by defective B cell isotype class switching that results in lower levels of IgG and IgA, and higher IgM (1–3). The lack of a complete antibody arsenal typically predisposes PAD patients to recurrent bacterial and viral infections; however, the severity and prevalence of symptoms varies with type of PAD as well as individual manifestations of those with the same PAD.
The most prevalent symptomatic PAD is common variable immune deficiency (CVID) which is classified by profound reduction in IgG as well as IgA or IgM due to impaired B cell differentiation (4). Affecting 1:25,000 individuals, patients are typically diagnosed between the ages of 20 and 40 (5). Immunoglobulin replacement therapies can be used to limit infections, however about half of CVID patients develop non-infectious complications such as autoimmunity, lung and/or gastrointestinal disease, and malignancy despite this therapy (6). Moreover, these non-infectious complications occur in CVID more frequently than other forms of PAD for reasons that are poorly understood (7, 8). This suggests the presence of genetic, immunological, and/or environmental factors, and not simply antibody deficiency alone, drive the development of inflammatory complications in PAD. Yet, these complex etiologies remain poorly understood. Consequently, non-infectious complications are the leading cause of morbidity and mortality in CVID (9, 10).
The lung, as a mucosal surface regularly exposed to exogenous pathogens, is one of the organs most affected by the infectious and non-infectious complications of CVID. Upper respiratory tract infections by encapsulated bacteria are common in patients, leading to airway inflammation, impaired host defense, permanent tissue damage, and frequently bronchiectasis - an irreversible dilation of the bronchial airways (11). While bronchiectasis is likely the most common pulmonary complication of CVID, interstitial lung disease (ILD) also occurs in about 1 out of 3 CVID patients and accounts for a larger percentage of mortality (9, 10, 12). Radiological findings that distinguish CVID ILD typically include pulmonary nodules, ground glass opacities, and mediastinal lymphadenopathy (13). Additionally, biopsies typically reveal benign lymphoproliferation and granulomatous inflammation leading this form of interstitial lung disease to be labeled granulomatous-lymphocytic interstitial lung disease (GLILD) (1, 13). The exact cause of ILD in CVID remains unclear and does not require the presence of bronchiectasis or history of pneumonia, suggesting that infection is not an underlying cause in many cases (14). Immunoglobulin replacement therapy typically does not ameliorate the development of ILD in CVID, and current therapeutic approaches rely on immunomodulatory drugs (15). While treating ILD, these immunomodulatory drugs may also increase the risk of infection or malignancy in these patients already vulnerable for these complications, particularly because a therapeutic endpoint is often unclear (16). Greater understanding of ILD pathogenesis in CVID is needed to develop safer and more effective therapeutic approaches.
Perhaps a key to understanding ILD pathogenesis in CVID is the fact that it frequently occurs together with other non-infectious complications, like autoimmune cytopenia and splenomegaly, which are driven by mechanisms of immune dysregulation (17). Additionally, there are a number of monogenic antibody deficiency syndromes that present with ILD of a similar pathology to that seen in CVID patients (18). These include patients with gain-of-function mutations of PI3KD that develop the CVID-like activated PI3Kδ syndrome defined by lymphoid hyperplasia, which can affect the airways. Activated PI3Kδ syndrome can be ameliorated by rapamycin, which reduces resultant hyperactive mTOR signaling in lymphocytes, or targeted inhibition with the PI3Kδ inhibitor leniolisib (19, 20). Similarly, patients with genetic deficiency of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) or a protein vital for its vesicular trafficking, lipopolysaccharide (LPS)-responsive and beige-like anchor protein (LRBA), develop inflammatory complications that are responsive to CTLA-4-Ig, known as abatacept (21, 22). These examples highlight the potential of precision immunomodulatory treatments for ILD as well as other non-infectious complications of CVID based upon identification of an underlying genetic lesion.
Despite CVID being defined by impaired antibody production, B cells appear to play an important role in ILD pathogenesis. Pulmonary B cell hyperplasia is a defining feature of CVID ILD, particularly in patients with biopsy proven follicular bronchiolitis, lymphocytic interstitial pneumonia, and nodular lymphoid hyperplasia of the lungs (23). Notably, ILD occurs far less commonly in X-linked agammaglobulinemia, a form of PAD where B cells are absent (7). Numerous studies have found B cell-depletive therapy with rituximab to be efficacious for CVID ILD (23–27). We conducted the largest study of rituximab monotherapy for CVID ILD, finding clear efficacy of this intervention over supportive care (28). ILD recurred after rituximab in about 1/3rd of subjects, but this recurrence could be limited by additional immunosuppression with azathioprine or mycophenolate. ILD recurrence was associated with increased levels of B cell activating factor (BAFF) in the blood and lungs, a key cytokine for B cell activation and survival (28). While these results do not prove that B cells are pathogenic in CVID ILD, they provide justification for deeper consideration and further research efforts to understand how these lymphocytes may contribute to disease. In the effort to summarize our understanding of how B cells may contribute to CVID ILD, we will review mechanisms of B cell dysfunction described in CVID and non-CVID lung diseases alike. We apply particular focus upon BAFF-related B cell biology given the considerable research in CVID and other lung diseases that has been recently conducted.
It is important to note that not all ILD found in CVID may be the same. It has been suggested that there are diverse forms of ILD afflicting CVID patients (29). We have found evidence of B cell hyperplasia and heightened BAFF responses in CVID, specifically with biopsy-proven forms of benign lymphoproliferative interstitial lung disease. This is a spectrum of pulmonary pathology that starts with follicular bronchiolitis, when disease is limited to peribronchial areas, and progresses to lymphocytic interstitial pneumonia and nodular lymphoid hyperplasia, when inflammation becomes more diffuse within the lung parenchyma (30). CVID ILD can also manifest as other types of pathology, such as non-specific interstitial pneumonia, prominent granulomatous inflammation, or organizing pneumonia (12, 14). It may be important to confirm ILD by performing lymphocyte phenotyping of biopsies to gain a specific pathology diagnosis, like lymphocytic interstitial pneumonia, rather than label all forms of presumed ILD on CT scan as GLILD and treat them the same. It is likely that CVID ILD pathology with prominent B cell follicles, such as follicular bronchiolitis and lymphocytic interstitial pneumonia, may be more responsive to B cell-targeted therapy. Variability among CVID ILD pathology may mean that some cases are more responsive to BAFF or B cell-targeted therapy than others.
Biology of BAFF and Its Receptors
BAFF and a proliferation-inducing ligand (APRIL), are members of the tumor necrosis factor family of ligands that share receptors to promote activation and survival of B cells. BAFF and APRIL are elevated in the blood of CVID patients (31, 32). BAFF may contribute to lung disease in CVID as its levels were found to be highest in CVID patients with progressive ILD (28). APRIL levels were not found to be also elevated in this study. A variety of cell types are capable of producing BAFF in response to type I and type II interferons as well as pattern recognition receptor engagement, including dendritic cells, monocytes, and neutrophils (33). BAFF is expressed as a type II transmembrane protein that is processed at a furin cleavage site to release soluble BAFF (33, 34). Upon release from the cell membrane, BAFF can assemble into homotrimers or oligomeric, capsid-like 60-mers (35). Alternative splicing of BAFF generates a shorter isoform (ΔBAFF) that is co-expressed and associates with BAFF but interferes with proteolytic cleavage at the membrane (36). Thus, soluble BAFF can have distinct functional impact upon B cells depending on its abundance, multimeric state, and isoform.
The effects of BAFF are influenced by the specific receptor it binds. BAFF can signal via three receptors, BAFF receptor (BAFF-R), transmembrane activator and CAML interactor (TACI), and B cell maturation antigen (BCMA), while APRIL signals through TACI and BCMA only (Table 1) (37). BAFF receptors are differentially expressed across developmental subsets of B cells to regulate intracellular signaling pathways related to B cell activation, survival, and maturation (37–39). Expression of BAFF-R is absent on pre-B cells in the bone marrow until development into immature B cells, coinciding with establishment of BAFF-R as the predominant BAFF receptor in naive and transitional B cells (39). TACI expression increases with development into marginal zone and memory B cells as well antibody producing cells (38, 40). Expression of BCMA is mainly restricted to plasma cells (38, 41–43).

Table 1 Important characteristics of the receptors for BAFF.
Along with differences in expression during B cell maturation, there are distinguishing features regarding BAFF-R signaling compared to other receptors for BAFF (Figure 1). In addition to activating the canonical NF-κB and phosphoinositide 3-kinase pathways, BAFF-R engagement of trimeric or oligomeric BAFF activates the non-canonical NF-κB pathway and upregulates expression of proteins in the Bcl-2 family that enhance B cell survival (44–46). Non-canonical NF-κB signaling requires activation of NF-κB-inducing kinase (NIK), a kinase that is targeted for constitutive degradation while in complex with TNF receptor-associated factor 3 (TRAF3) in unstimulated B cells (47, 48). Ligation of BAFF to BAFF-R induces the targeted degradation of TRAF3, allowing NIK to accumulate and induce IκB kinase (IKKα)-dependent cleavage of p100 into p52 which associates with RelB to alter transcriptional activity (49–54). TRAF molecules such as TRAF2, TRAF3, TRAF5, and TRAF6, are recruited to the intracellular domain of BAFF receptors to mediate downstream signaling pathways in B cells through the canonical and non-canonical NF-κB pathways, AP-1 signaling, and MyD88-dependent class switch recombination in cooperation with TLRs (51, 55–58). B cell survival is also enhanced through the cooperation of BAFF-R with CD19 in regulating the activity of phosphoinositide 3-kinase (59). BAFF-R has greater affinity for BAFF compared to TACI and BCMA (60, 61). Due to its ability to promote survival through the non-canonical NF-κB and phosphoinositide 3-kinase pathways, expression from early stages of B cell maturation, and high affinity for BAFF, BAFF-R is positioned as a chief mediator of BAFF activity. Further efforts are needed to determine how significant the role of BAFF-R is in the pathogenesis of CVID-related complications.
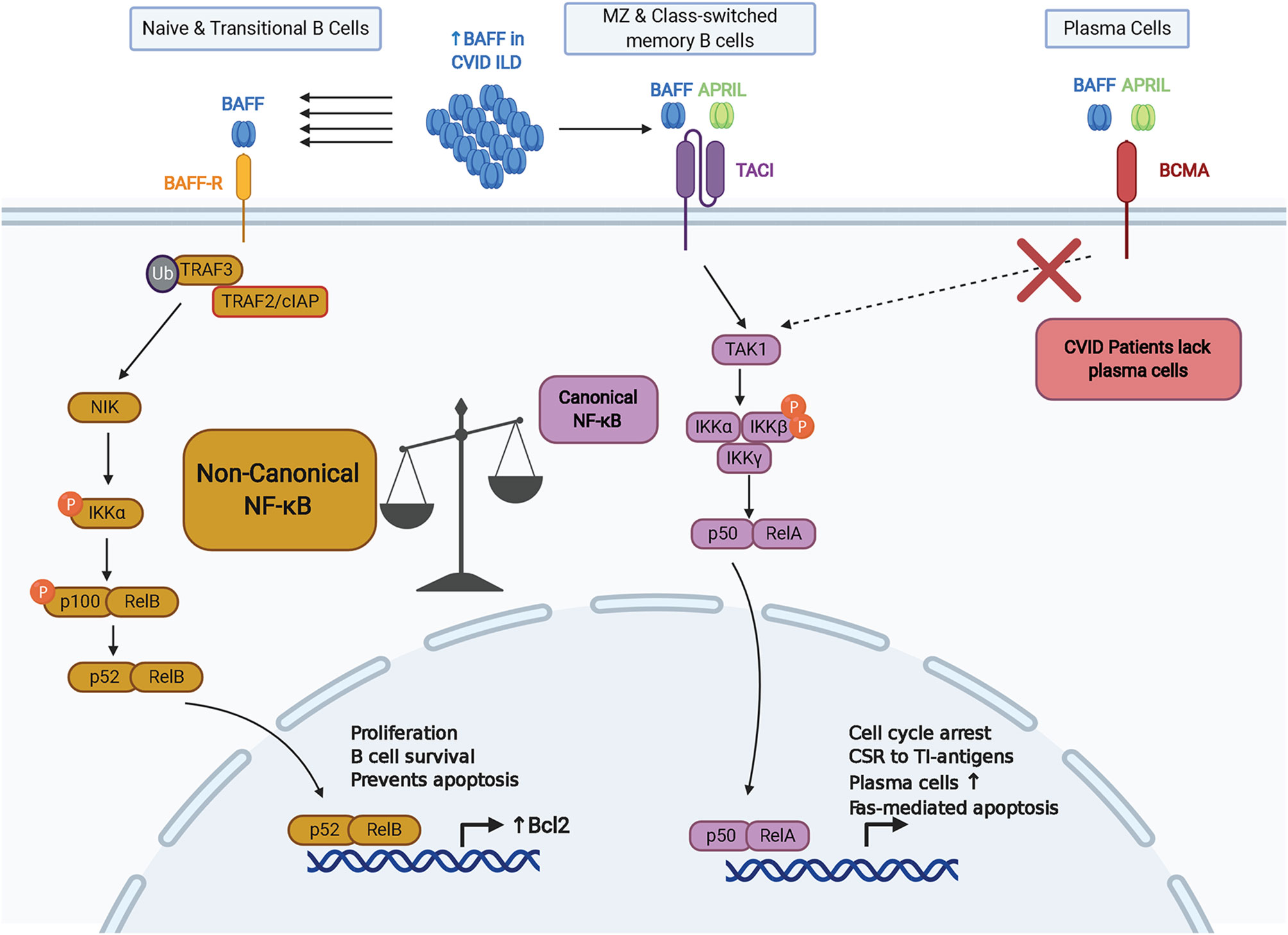
Figure 1 Key aspects of BAFF-R, TACI, and BCMA signaling within the context of CVID. BAFF-R is distinguished by its ability to signal via the non-canonical NF-kB pathway to induce Bcl-2 and other pro-survival factors. Lack of memory B cells and plasma cells expressing TACI and BCMA in CVID may increase signaling via BAFF-R. CSR, class-switch recombination; TI, T-independent.
Unlike BAFF-R, TACI signal activation requires binding to a higher-order oligomeric BAFF complex, such as the BAFF 60mer (62). BAFF signaling through TACI activates the canonical NF-κB pathway and upregulates expression of genes involved with cell cycle arrest, cell death, and class switch recombination (CSR) in response to T cell-independent (TI) antigens (45, 63, 64). In line with the role of TACI in TI responses, BAFF and APRIL induce IgG and IgA CSR via TACI through MyD88 (64). TACI interacts with mechanistic target of rapamycin (mTOR) via MyD88 to contribute to TACI-mediated NF-κB activation, association with TLRs, and IgG class switching in response to TI antigens (65). TACI also appears to have a regulatory role in antibody production from B cells stimulated with BAFF and CD40, which indicates a homeostatic role in regulating T cell-independent versus T cell-dependent antibody production (66). TACI can also signal through the nuclear factor of activated T cells (NFAT) pathway (67). Alternative splicing of TACI transcripts can generate a short isoform that induces strong activation of the NF-κB pathway and has distinct localization within B cells compared to the full-length isoform (68, 69). Importantly, TACI signaling promotes expression of BLIMP-1, a transcription factor that induces cell cycle arrest and plasma cell differentiation by inhibiting expression of Bcl-6 and Pax5 (63, 70). Interestingly, Pax5 has been characterized as a lineage biomarker for a subset of rituximab-treated B cell lymphoma patients who relapse with CD20-negative B cells (71–74). However, the role of Pax5 in the development and progression of non-infectious complications in CVID remains to be characterized.
BAFF and Its Receptors in CVID
Germline mutations in TNFRSF13B, the gene that encodes TACI, are observed in 5-10% of CVID patients (75, 76). TACI-deficient patients are known to have an increased rate of autoimmunity and lymphoproliferative disease in CVID in association with increased autoreactive B cell selection and survival (77, 78). There may a greater risk of progressive ILD in CVID patients with certain TACI mutations compared to other CVID patients (28). The C104R and A181E variants are the most common variants in TACI that are considered likely pathogenic (Figure 2). The C104R mutation disrupts a disulfide bond in the extracellular cysteine rich domain 2 (CRD2) to diminish TACI ligand binding capacity and TACI-mediated activation of canonical NF-κB signaling (79). The A181E TACI variant affects the CAML binding site located in the transmembrane domain does not interfere with ligand binding or surface expression but fails to activate NF-κB signaling (79). Several other CVID-associated genetic variants of TACI have been identified in clinical settings and further characterization of these variants may provide insight into TACI’s role in regulation of the BAFF/APRIL signaling axis in CVID and other diseases (75–77, 79–82). A global cohort analysis revealed that although mutations in TNFRSF13B are prevalent in CVID and healthy populations, there is an excess of rare derived alleles of TNFRSF13B in CVID cohorts compared to healthy individuals of the same population, indicating that defects in TACI are contributory toward manifestations of CVID (80). However, given the prevalence of the same variants in healthy populations, TNFRSF13B mutations are likely disease-modifying rather than disease-causing.
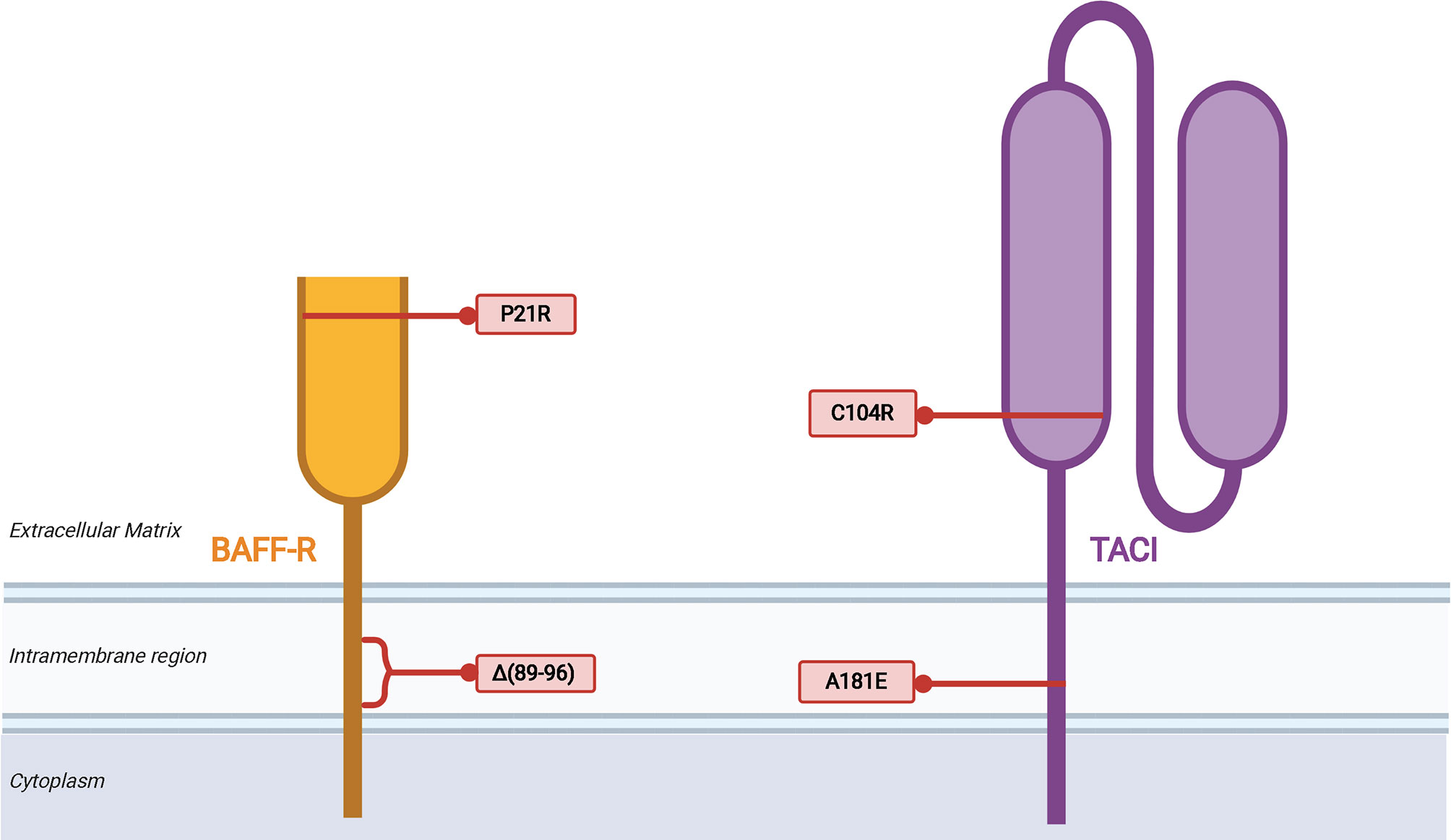
Figure 2 Mutations of TNFRSF13B (TACI) associated with CVID. The variants listed are limited to the two most common, C104R and A181E, which are discussed in the text, as well as two other illustrative examples of how disruption of TACI can impair B cell function. Proposed mechanisms of biochemical disruption of certain variants included.
Regarding BAFF-R, a homozygous in-frame deletion that results in the loss of eight amino acids within the transmembrane region was identified in siblings with hypogammaglobulinemia (83). The two siblings had reduced serum IgG and IgM, but normal level of IgA. Class-switched memory B cells were lacking in these patients, and they did not have a medical history of autoimmune or lymphoproliferative complications. Also, a P21R variant of BAFF-R has been identified that interferes with BAFF-R complex formation, has reduced capacity to bind BAFF, and impairs BAFF-mediated NF-κB2 activation (84). B cells from patients with the BAFF-R P21R mutation lacked an increase in cell number and IgM secretion in response after stimulation with CpG DNA, anti-IgM, and BAFF. The BAFF-R P21R allele is found in 10.2% of CVID patients and 6.7% of healthy controls. Three additional heterozygous BAFF-R variants have been identified in a CVID cohort, all of which are present in healthy controls as well and their role in CVID remains to be defined (85).
Soluble BAFF Receptors
Each of the three BAFF receptors can be proteolytically processed to generate soluble molecules that function as decoy receptors in circulation (Figure 3). These soluble BAFF receptors add another layer to regulation of BAFF and APRIL-mediated homeostasis in B cells, prompting investigations into their utility in pharmacologic and diagnostic applications (86). Upon binding to BAFF, the extracellular domain of BAFF-R is processed by a metalloprotease (ADAM10) only in cells that also express TACI (87). This regulated processing is different from that of TACI and BCMA, receptors that undergo constitutive processing to release soluble fragments (88, 89). The BAFF trimer induces processing of BAFF-R by ADAM10, whereas TACI processing is unaffected by BAFF trimer stimulation (87). BAFF 60-mers are capable of stimulating processing of BAFF-R and TACI by both ADAM10 and ADAM17 (87). In the same study, the two metalloproteases, ADAM10 and ADAM17, demonstrated differential activity with respect to the activity state of B cells with increased ADAM10 activity on resting and TLR9-activated B cells, and ADAM17 processes BAFF-R on dark zone and germinal center B cells. Inhibition of ADAM10, responsible for processing of BAFF-R and TACI, was then shown to increase BAFF-dependent survival and secretion of IgM from B cells.
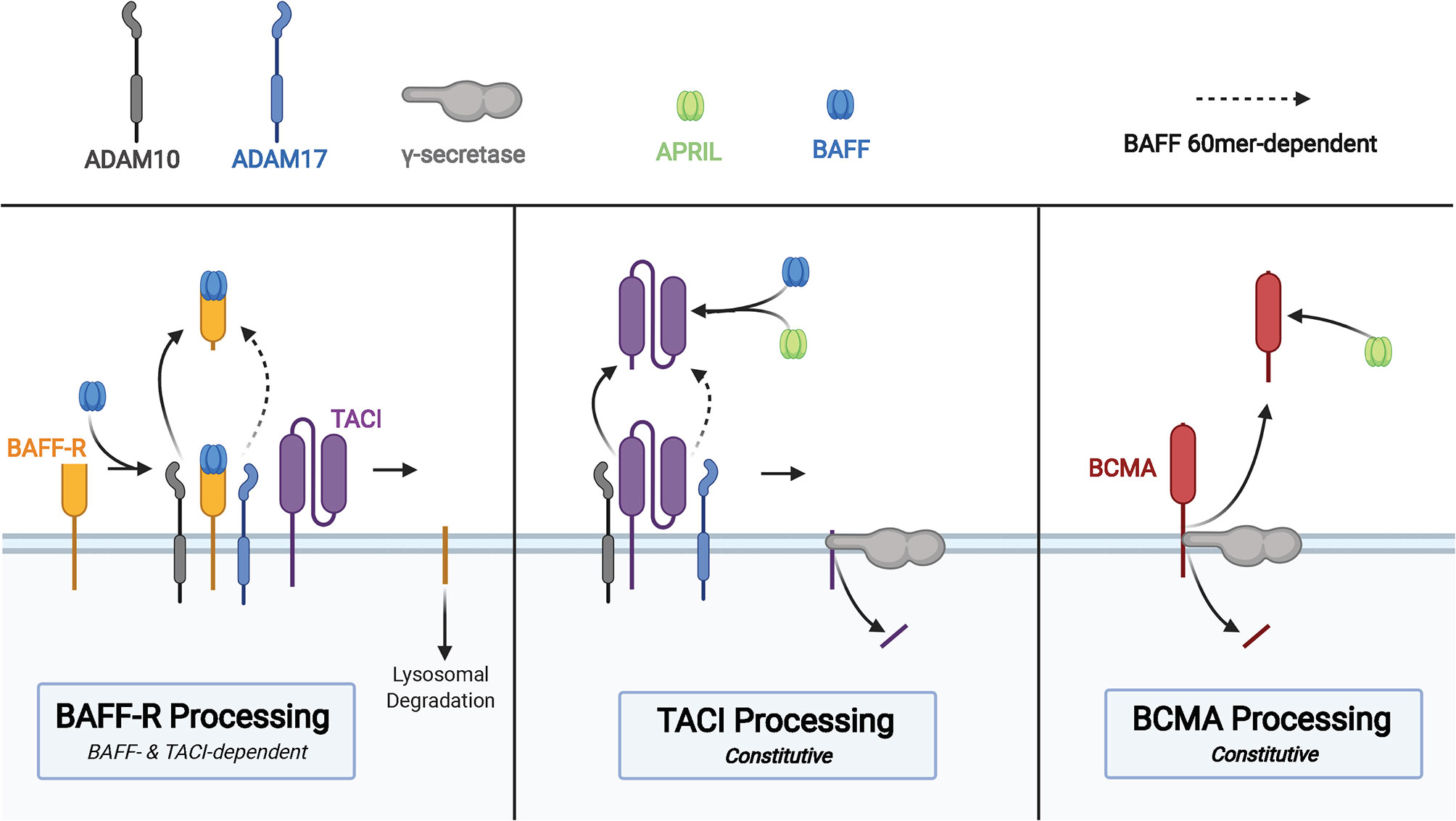
Figure 3 Membrane processing of human BAFF receptors. Cleavage of the BAFF-R ectodomain is induced by BAFF binding in cells that co-express TACI. Processing of BAFF-R by ADAM10 is induced by binding to BAFF trimers and binding of BAFF 60mer to BAFF-R induces ADAM17 processing of BAFF-R. The membrane-bound C-terminal fragment of BAFF-R is degraded in lysosomes after cleavage of the ectodomain. TACI is cleaved in a constitutive manner by ADAM10, followed by cleavage of the membrane-bound C-terminal fragment by γ-secretase. sTACI exhibits homotypic assembly and binds to BAFF and APRIL to reduce NF-κB activation and B cell survival, with TACI-Fc demonstrating similar capabilities. BCMA is constitutively cleaved by γ-secretase to release sBCMA consisting of the ectodomain and a portion of the transmembrane domain of BCMA. sBCMA is a decoy for APRIL-induced NF-κB activation but does not block BAFF-mediated NF-κB activation. However, BCMA-Fc is capable of binding both APRIL and BAFF to block NF-κB activation.
TACI is constitutively processed by ADAM10 on the surface of B cells to release the soluble extracellular domain of TACI capable of binding to BAFF and APRIL (88). Then γ-secretase, an intramembranous protease, cleaves the remaining membrane-proximal TACI fragment to prevent receptor-dependent activation of canonical NF-κB signaling (88). There is conflicting evidence supporting the capacity of the extracellular domain of TACI fused to an immunoglobulin Fc domain (TACI-Fc) to induce reverse signaling in macrophages through membrane bound BAFF and APRIL (90, 91). Studies that interrogate the role of soluble TACI must take into consideration differences in amino acid composition of endogenous sTACI compared to that of TACI-Fc due to demonstrated differences in BAFF/APRIL binding between sBCMA and BCMA-Fc (89). Subtle differences in the amino acid composition may have drastic effects on ligand binding capacity of the extracellular domain, as point mutations in TACI are capable of diminishing affinity for ligand, processing of TACI, and even processing of BAFF-R (87). Thus, the biological impact of soluble TACI remains incompletely understood.
BCMA is constitutively processed by γ-secretase, a process that acts to reduce surface BCMA and consequently regulate the number of plasma cells in the bone marrow, given the importance of this receptor for plasma cell survival (89). Although BCMA is able to bind BAFF and APRIL to induce canonical NF-κB signaling, soluble BCMA (sBCMA) is able to bind APRIL but does not block BAFF-mediated activation of NF-κB in HEK cells transfected with BCMA (89). The same study also found recombinant BCMA-Fc to bind BAFF and APRIL, leading to inhibition of BAFF and APRIL-mediated NF-κB signaling through BCMA. Quantification of serum BCMA revealed markedly reduced levels among patients with severe PAD, such as CVID and XLA (92). Evaluation of immunoglobulin deficiencies in CVID and other PADs often requires repeated vaccine challenges and discontinuation of immunoglobulin replacement therapy, which increase patient susceptibility to infection and may take several weeks (93–95). Methods of diagnosing PAD requiring immunoglobulin replacement that reduces diagnostic delay and does not require treatment discontinuation, such as is the case with sBCMA measurement, could significantly improve clinical care and quality of life in those with PAD.
The Potential Contribution of BAFF to CVID ILD
CVID patients can have a significant increase in serum IgM corresponding to progression of ILD as determined by pulmonary function decline (96). This serum IgM increase is associated with hyperplasia of ectopic pulmonary B cells expressing IgM (28). B cell depletion with rituximab ameliorates CVID ILD, corresponding with improved pulmonary function and reduction of serum IgM, compared to those receiving supportive care (28). Moreover, the ILD recurrence that occurred in 1/3rd of study subjects within 2 years of receiving rituximab was also associated with serum IgM elevation (28). Thus, the presence and reemergence of B cells, corresponding with rising levels of serum IgM, may be quite fundamental to CVID ILD pathogenesis.
CVID patients who experienced ILD progression after rituximab had significantly elevated levels of BAFF in blood and lung tissue compared to CVID patients with stable ILD, no ILD, and healthy controls (28). IFN-γ upregulates signal transducer and activator of transcription 1 (STAT1) expression to act as a potent stimulus of BAFF production (97). Numerous reports that have found elevation of IL-12, IFN-γ, and related T helper type 1 cytokines in CVID patients with inflammatory complications (28, 98–105). Furthermore, plasma IFN-γ levels and STAT1 expression were elevated in CVID patients with progressive ILD and correlated with BAFF expression, and CD14+ monocytes were identified as a prominent source of IFN-y-induced BAFF production and STAT1 expression in CVID patients with progressive ILD (28). Together, these results implicate an IFN-γ:STAT1:BAFF axis in pathogenesis of ILD in CVID. Efforts to unravel fundamental biology and clinical importance of this IFN-γ and BAFF relationship in CVID are underway.
Heterozygous mutations of TACI found in CVID appear to be key for the persistence of autoreactive B cells through interaction with toll-like receptor (TLR) 7 and TLR9 (106). Moreover, when BAFF is elevated in non-CVID patients it has been shown that autoantigen-engaged B cells demonstrate enhanced survival and migration to follicular zone and marginal zone niches where they would normally be excluded (107, 108). While the relationship between B cell autoreactivity and ILD is unclear in CVID, it is possible that enhanced BAFF-R signaling in the absence of counterbalancing signals from TACI promotes pathogenic pulmonary B cell hyperplasia. Indeed, 3 patients with TACI mutations in our study of CVID all had progressive ILD that recurred after rituximab (28). Thus, in addition to the greater prevalence of progressive ILD in CVID patients with TACI mutations there was apparently greater resistance to B cell depletive therapy, possibly due to elevated signaling through BAFF-R.
BAFF-R is the predominant BAFF receptor expressed by the IgD+ B cells that make up the ectopic pulmonary follicles observed in CVID ILD, while TACI is expressed in the extrafollicular areas of the lung harboring plasmablasts expressing IgM and the proliferation marker Ki67 (28). BAFF-R is the principal BAFF receptor on B cells in CVID patients with autoimmune and lymphoid hyperplasia due to the lack of marginal zone, memory, and plasma cells in these patients that would otherwise express TACI and/or BCMA (109, 110). Elevated levels of BAFF enhance BAFF-R-mediated activation of the non-canonical NF-κB pathway to upregulate Bcl-2 survival signals and impair B cell apoptosis (45, 48). The expanded subset of naïve B cells in CVID ILD were observed to induce expression of Bcl-2 and RelB to a level that is significantly greater in CVID patients with progressive ILD compared to healthy controls (28). Enhanced activity of BAFF-R signaling in response to elevated BAFF not only drives proliferation and resistance to apoptosis in naïve B cells, but may concurrently impair B cell maturation by drowning out BAFF-mediated maturation signals from TACI (45, 63). Excessive BAFF inhibits autophagy in B cells and reduces autophagosome marker LC3-II through mechanisms that depend on active Akt/mTOR signaling, suggesting that elevated BAFF can drive B cell survival through multiple mechanisms (111).
The extent of B cell contributions to pathogenesis of CVID ILD remains to be sufficiently defined. Like B cells, T cells are a prominent feature of CVID ILD pathology, and treatment with azathioprine or mycophenolate mofetil in combination with rituximab improved clinical chest radiography scores and components of pulmonary function testing in patients with CVID ILD (15). A considerable portion of patients in this study relapsed after receiving this immunosuppressive combination in association with elevated B cells and activated CD4+ T cells. Variations in the extent of immune cell compartment imbalance in CVID may enhance the progression of ILD due to mechanisms that remain unclear. T cells from CVID patients demonstrate increased frequencies of activated, memory, and effector populations with a lack of naïve and regulatory T cell subsets (112). The enhanced state of T cell activation and effector function in CVID may further contribute to the B cell hyperplasia observed in CVID ILD due to a lack of T cell-mediated regulation of B cell activity in addition to upregulation of non-canonical NF-κB signaling in B cells as a result of more widespread stimulation of CD40 through CD40L expressed on activated T cells (113, 114). The notable variability of clinical manifestations and aberrant immune cell compartments in CVID suggests that multiple aspects of immune system dysregulation may contribute to CVID ILD. Furthermore, efficacy of therapeutic depletion of B cells may stem from indirect effects upon leukocytes, such as T cells, that closely interact with B cells in CVID ILD.
Studies of lung disease with pathologic similarities to that observed in CVID ILD may also prove to be informative. For example, lymphocytic interstitial pneumonia makes up 15% of interstitial lung disease affecting Sjogren’s syndrome patients (115). Similar to CVID, B cells appear to play a central role in the development of ILD in Sjogren’s syndrome. Specifically, elevated levels of BAFF can be found in the serum, saliva, and salivary glands of Sjogren’s syndrome patients in comparison to healthy controls (116–118). BAFF levels in these patients are also positively associated with the presence of autoantibodies, including anti-SSA and anti-SSB (119). Also, like we found in CVID ILD, elevated levels of BAFF seen in Sjogren’s is associated with heightened interferon signaling through the JAK/STAT pathway in monocytes (28, 120). Elevated levels of BAFF in Sjogren’s syndrome ultimately enables prolonged survival of B cells, which have been shown to aggregate into inducible bronchus-associated lymphoid tissue structures with pulmonary B cell follicles as in CVID ILD (121). A double-blind, randomized, placebo-controlled, multi-center, multi-national clinical trial (NCT02631538) that investigated the effects of rituximab and belimumab administration in 86 pSS patients was recently completed in June 2020. This trial contained four groups, including a placebo group, a group that received only belimumab, a group that received only rituximab, and a group that received both belimumab and rituximab. Results from this study have not been published yet, but they will put the implication of BAFF and aberrant B cell survival and signaling found in Sjogren’s syndrome patients to the test.
Another chronic lung disease where there is increasing evidence for a role of B cells and BAFF is chronic obstructive pulmonary disease (COPD). Although COPD is commonly associated with smoking, anywhere between 25 and 45% of COPD patients have never smoked, suggesting that other factors contribute to the pathogenesis of this lung disease (122). The implication of the adaptive immune system in the development and progression of COPD becomes evident when considering the fact that there is a significantly greater number of B cells and CD4+ and CD8+ T cells in the airways and parenchyma of the lungs of COPD patients (123, 124). These excess B and T cells arise from induced bronchus-associated lymphoid tissue (iBALT) and form pulmonary follicles containing germinal center B cells and follicular T cells (123). Moreover, significantly more lymphoid follicles were found in the lungs of those who were diagnosed with COPD in comparison to smokers without COPD (124). Also, when categorizing COPD patients on the Global Initiative for Chronic Obstructive Lung Disease (GOLD) scale, a significant increase in the number and size of lymphoid follicles was seen in later-stage COPD patients in comparison to those in the earlier stages (124). The same study also performed immunofluorescence on lung samples and found the size of the lymphoid follicles identified in each of the aforementioned groups to be directly correlated to the percentage of BAFF-positive B cells, which co-localized with BAFF-R (124, 125). BAFF expression was also found to be elevated in the blood of COPD patients in comparison to non-smoking and smoking control subjects (124). Healthy, smoking controls and the early-stage COPD subjects, on the other hand, had a higher proportion of caspase-3-positive B cells, indicating apoptosis, in their pulmonary follicles in comparison to later-stage COPD subjects. These findings implicate dysregulation of the BAFF : BAFF-R axis in the progression of COPD, with the anti-apoptotic signals of BAFF-R promoting the B cell follicles that are a major component of pulmonary pathology, similar to what was found in CVID ILD.
Conclusion
There is increasing evidence that dysregulated B cell responses, such as those exacerbated by BAFF, promote the progression of ILD in CVID. This is supported by the adoption of B cell depletive therapy, either alone or in combination with other immunosuppression, as a fundamental component of CVID ILD treatment. Continued suppression of B cell activation through administration of immunosuppressive antimetabolite agents such as azathioprine or mycophenate, or potentially through inhibition of BAFF may help maintain CVID ILD in remission. B cell hyperplasia is a defining aspect of CVID ILD and is perpetuated via survival signals mediated by BAFF through BAFF-R. In addition to B cells, CVID ILD consists of prominent T cell infiltration which appears to also improve with B cell depletive therapy (23, 28). The link between B cells and T cells in the CVID lungs remains undefined, and whether depletion of B cells removes a vital antigen-presenting cell, lymphoid structure, source of chemokines, and/or another component required for T cell recruitment and persistence in the lungs is unknown. Further research is necessary to prove whether B cells fundamentally contribute to pathogenesis of CVID ILD, define the best way to achieve safe long-lasting suppression of dysregulated B cell responses, and accurately identify the individual CVID patients who would most benefit from B cell-targeted therapy. Moreover, we must elucidate mechanisms by which the IFN-γ/STAT1/BAFF axis is elevated in CVID and other disorders. Further efforts to unravel the mechanisms by which BAFF and B cells become dysregulated in CVID offer potential to address these knowledge gaps in CVID and other forms of autoimmune and inflammatory disease.
Author Contributions
EM, MA, KB, and KH drafted the manuscript. PM provided guidance and revisions. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Institutes of Health grants AI137183 and AI151486, a faculty development grant from the American Association of Allergy, Asthma and Immunology Foundation, and a Boston University Career Investment Award.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Figures were created with BioRender.com. This work is dedicated to the memory of co-author KB.
Abbreviations
APRIL, a proliferation-inducing ligand; BAFF, B cell activating factor; BAFF-R, BAFF receptor; BCMA, B cell maturation antigen; COPD, chronic obstructive pulmonary disease; CSR, class-switch recombination; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; CVID, common variable immunodeficiency; GLILD, granulomatous lymphocytic interstitial lung disease; GOLD, Global Initiative for Chronic Obstructive Lung Disease; IKKα, IκB kinase; iBALT, induced bronchus-associated lymphoid tissue; ILD, interstitial lung disease; LRBA, lipopolysaccharide (LPS)-responsive and beige-like anchor protein; NIK, NF-κB-inducing kinase; PAD, primary antibody deficiency; STAT1, signal transducer and activator of transcription 1; TACI, transmembrane activator and CAML interactor; TI, T cell-independent; TRAF3, TNF receptor-associated factor 3.
References
1. Schussler E, Beasley MB, Maglione PJ. Lung Disease in Primary Antibody Deficiencies. J Allergy Clin Immunol Pract (2016) 4:1039–52. doi: 10.1016/j.jaip.2016.08.005
2. Conley ME, Dobbs AK, Farmer DM, Kilic S, Paris K, Grigoriadou S, et al. Primary B cell immunodeficiencies: comparisons and contrasts. Annu Rev Immunol (2009) 27:199–227. doi: 10.1146/annurev.immunol.021908.132649
3. Davies EG, Thrasher AJ. Update on the hyper immunoglobulin M syndromes. Br J Haematol (2010) 149:167–80. doi: 10.1111/j.1365-2141.2010.08077.x
4. Cunningham-Rundles C, Maglione PJ. Common variable immunodeficiency. J Allergy Clin Immunol (2012) 129:1425–6.e3. doi: 10.1016/j.jaci.2012.03.025
5. Cunningham-Rundles C. Common variable immune deficiency: case studies. Blood (2019) 134:1787–95. doi: 10.1182/blood.2019002062
6. Cunningham-Rundles C. Common variable immune deficiency: Dissection of the variable. Immunol Rev (2019) 287:145–61. doi: 10.1111/imr.12728
7. Weinberger T, Fuleihan R, Cunningham-Rundles C, Maglione PJ. Factors Beyond Lack of Antibody Govern Pulmonary Complications in Primary Antibody Deficiency. J Clin Immunol (2019) 39:440–7. doi: 10.1007/s10875-019-00640-5
8. Swain S, Selmi C, Gershwin ME, Teuber SS. The clinical implications of selective IgA deficiency. J Transl Autoimmun (2019) 2:100025. doi: 10.1016/j.jtauto.2019.100025
9. Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood (2008) 112:277–86. doi: 10.1182/blood-2007-11-124545
10. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood (2012) 119:1650–7. doi: 10.1182/blood-2011-09-377945
11. Baumann U, Routes JM, Soler-Palacín P, Jolles S. The Lung in Primary Immunodeficiencies: New Concepts in Infection and Inflammation. Front Immunol (2018) 9:1837. doi: 10.3389/fimmu.2018.01837
12. Bates CA, Ellison MC, Lynch DA, Cool CD, Brown KK, Routes JM. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol (2004) 114:415–21. doi: 10.1016/j.jaci.2004.05.057
13. Maglione PJ. Chronic Lung Disease in Primary Antibody Deficiency: Diagnosis and Management. Immunol Allergy Clin North Am (2020) 40:437–59. doi: 10.1016/j.iac.2020.03.003
14. Maglione PJ, Overbey JR, Radigan L, Bagiella E, Cunningham-Rundles C. Pulmonary radiologic findings in common variable immunodeficiency: clinical and immunological correlations. Ann Allergy Asthma Immunol (2014) 113:452–9. doi: 10.1016/j.anai.2014.04.024
15. Verbsky JW, Hintermeyer MK, Simpson PM, Feng M, Barbeau J, Rao N, et al. Rituximab and antimetabolite treatment of granulomatous and lymphocytic interstitial lung disease in common variable immunodeficiency. J Allergy Clin Immunol (2020) 1:S0091-6749(20)31069-1. doi: 10.1016/j.jaci.2020.07.021
16. Chien SH, Liu CJ, Hong YC, Teng CJ, Hu YW, Shen CC, et al. Use of azathioprine for graft-vs-host disease is the major risk for development of secondary malignancies after haematopoietic stem cell transplantation: a nationwide population-based study. Br J Cancer (2015) 112:177–84. doi: 10.1038/bjc.2014.523
17. Maglione PJ. Autoimmune and Lymphoproliferative Complications of Common Variable Immunodeficiency. Curr Allergy Asthma Rep (2016) 16:19. doi: 10.1007/s11882-016-0597-6
18. Gereige JD, Maglione PJ. Current Understanding and Recent Developments in Common Variable Immunodeficiency Associated Autoimmunity. Front Immunol (2019) 10:2753. doi: 10.3389/fimmu.2019.02753
19. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol (2014) 15:88–97. doi: 10.1038/ni.2771
20. Rao VK, Webster S, Dalm V, Šedivá A, van Hagen PM, Holland S, et al. Effective “activated PI3Kδ syndrome”-targeted therapy with the PI3Kδ inhibitor leniolisib. Blood (2017) 130:2307–16. doi: 10.1182/blood-2017-08-801191
21. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science (2015) 349:436–40. doi: 10.1126/science.aaa1663
22. Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol (2018) 142:1932–46. doi: 10.1016/j.jaci.2018.02.055
23. Maglione PJ, Ko HM, Beasley MB, Strauchen JA, Cunningham-Rundles C. Tertiary lymphoid neogenesis is a component of pulmonary lymphoid hyperplasia in patients with common variable immunodeficiency. J Allergy Clin Immunol (2014) 133:535–42. doi: 10.1016/j.jaci.2013.08.022
24. Boursiquot JN, Gerard L, Malphettes M, Fieschi C, Galicier L, Boutboul D, et al. Granulomatous disease in CVID: retrospective analysis of clinical characteristics and treatment efficacy in a cohort of 59 patients. J Clin Immunol (2013) 33:84–95. doi: 10.1007/s10875-012-9778-9
25. Chase NM, Verbsky JW, Hintermeyer MK, Waukau JK, Tomita-Mitchell A, Casper JT, et al. Use of combination chemotherapy for treatment of granulomatous and lymphocytic interstitial lung disease (GLILD) in patients with common variable immunodeficiency (CVID). J Clin Immunol (2013) 33:30–9. doi: 10.1007/s10875-012-9755-3
26. Jolles S, Carne E, Brouns M, El-Shanawany T, Williams P, Marshall C, et al. FDG PET-CT imaging of therapeutic response in granulomatous lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID). Clin Exp Immunol (2017) 187:138–45. doi: 10.1111/cei.12856
27. Zdziarski P, Gamian A. Lymphoid Interstitial Pneumonia in Common Variable Immune Deficiency - Case Report With Disease Monitoring in Various Therapeutic Options: Pleiotropic Effects of Rituximab Regimens. Front Pharmacol (2018) 9:1559. doi: 10.3389/fphar.2018.01559
28. Maglione PJ, Gyimesi G, Cols M, Radigan L, Ko HM, Weinberger T, et al. BAFF-driven B cell hyperplasia underlies lung disease in common variable immunodeficiency. JCI Insight (2019) 4(5):e122728. doi: 10.1172/jci.insight.122728
29. Patel S, Anzilotti C, Lucas M, Moore N, Chapel H. Interstitial lung disease in patients with common variable immunodeficiency disorders: several different pathologies? Clin Exp Immunol (2019) 198:212–23. doi: 10.1111/cei.13343
30. Carrillo J, Restrepo CS, Rosado de Christenson M, Ojeda Leon P, Lucia Rivera A, Koss MN. Lymphoproliferative lung disorders: a radiologic-pathologic overview. Part I: Reactive disorders. Semin Ultrasound CT MR (2013) 34:525–34. doi: 10.1053/j.sult.2013.05.002
31. Knight AK, Radigan L, Marron T, Langs A, Zhang L, Cunningham-Rundles C. High serum levels of BAFF, APRIL, and TACI in common variable immunodeficiency. Clin Immunol (2007) 124:182–9. doi: 10.1016/j.clim.2007.04.012
32. Jin R, Kaneko H, Suzuki H, Arai T, Teramoto T, Fukao T, et al. Age-related changes in BAFF and APRIL profiles and upregulation of BAFF and APRIL expression in patients with primary antibody deficiency. Int J Mol Med (2008) 21:233–8. doi: 10.3892/ijmm.21.2.233
33. Nardelli B, Belvedere O, Roschke V, Moore PA, Olsen HS, Migone TS, et al. Synthesis and release of B-lymphocyte stimulator from myeloid cells. Blood (2001) 97:198–204. doi: 10.1182/blood.V97.1.198
34. Schneider P, MacKay F, Steiner V, Hofmann K, Bodmer JL, Holler N, et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med (1999) 189:1747–56. doi: 10.1084/jem.189.11.1747
35. Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol (2009) 9:491–502. doi: 10.1038/nri2572
36. Gavin AL, Aït-Azzouzene D, Ware CF, Nemazee D. DeltaBAFF, an alternate splice isoform that regulates receptor binding and biopresentation of the B cell survival cytokine, BAFF. J Biol Chem (2003) 278:38220–8. doi: 10.1074/jbc.M306852200
37. Vincent FB, Saulep-Easton D, Figgett WA, Fairfax KA, Mackay F. The BAFF/APRIL system: emerging functions beyond B cell biology and autoimmunity. Cytokine Growth Factor Rev (2013) 24:203–15. doi: 10.1016/j.cytogfr.2013.04.003
38. Darce JR, Arendt BK, Wu X, Jelinek DF. Regulated expression of BAFF-binding receptors during human B cell differentiation. J Immunol (2007) 179:7276–86. doi: 10.4049/jimmunol.179.11.7276
39. Sakai J, Akkoyunlu M. The Role of BAFF System Molecules in Host Response to Pathogens. Clin Microbiol Rev (2017) 30:991–1014. doi: 10.1128/CMR.00046-17
40. Garcia-Carmona Y, Cols M, Ting AT, Radigan L, Yuk FJ, Zhang L, et al. Differential induction of plasma cells by isoforms of human TACI. Blood (2015) 125(11):1749–58. doi: 10.1182/blood-2014-05-575845
41. Pieper K, Grimbacher B, Eibel H. B-cell biology and development. J Allergy Clin Immunol (2013) 131:959–71. doi: 10.1016/j.jaci.2013.01.046
42. Naradikian MS, Perate AR, Cancro MP. BAFF receptors and ligands create independent homeostatic niches for B cell subsets. Curr Opin Immunol (2015) 34:126–9. doi: 10.1016/j.coi.2015.03.005
43. Coquery CM, Erickson LD. Regulatory roles of the tumor necrosis factor receptor BCMA. Crit Rev Immunol (2012) 32:287–305. doi: 10.1615/CritRevImmunol.v32.i4.10
44. Gardam S, Brink R. Non-Canonical NF-κB Signaling Initiated by BAFF Influences B Cell Biology at Multiple Junctures. Front Immunol (2014) 4:509. doi: 10.3389/fimmu.2013.00509
45. Yang S, Li JY, Xu W. Role of BAFF/BAFF-R axis in B-cell non-Hodgkin lymphoma. Crit Rev Oncol Hematol (2014) 91:113–22. doi: 10.1016/j.critrevonc.2014.02.004
46. Endo T, Nishio M, Enzler T, Cottam HB, Fukuda T, James DF, et al. BAFF and APRIL support chronic lymphocytic leukemia B-cell survival through activation of the canonical NF-kappaB pathway. Blood (2007) 109:703–10. doi: 10.1182/blood-2006-06-027755
47. Sevdali E, Katsantoni E, Smulski CR, Moschovi M, Palassopoulou M, Kolokotsa EN, et al. BAFF/APRIL System Is Functional in B-Cell Acute Lymphoblastic Leukemia in a Disease Subtype Manner. Front Oncol (2019) 9:594. doi: 10.3389/fonc.2019.00594
48. Gasparini C, Celeghini C, Monasta L, Zauli G. NF-κB pathways in hematological malignancies. Cell Mol Life Sci (2014) 71:2083–102. doi: 10.1007/s00018-013-1545-4
49. Senftleben U, Cao Y, Xiao G, Greten FR, Krähn G, Bonizzi G, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science (2001) 293:1495–9. doi: 10.1126/science.1062677
50. Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell (2001) 7:401–9. doi: 10.1016/S1097-2765(01)00187-3
51. Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng PH, Keats JJ, Wang H, et al. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat Immunol (2008) 9:1364–70. doi: 10.1038/ni.1678
52. Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol (2008) 9:1371–8. doi: 10.1038/ni.1676
53. Chan TD, Gardam S, Gatto D, Turner VM, Silke J, Brink R. In vivo control of B-cell survival and antigen-specific B-cell responses. Immunol Rev (2010) 237:90–103. doi: 10.1111/j.1600-065X.2010.00942.x
54. Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat Immunol (2002) 3:958–65. doi: 10.1038/ni842
55. Lin WW, Hildebrand JM, Bishop GA. A Complex Relationship between TRAF3 and Non-Canonical NF-κB2 Activation in B Lymphocytes. Front Immunol (2013) 4:477. doi: 10.3389/fimmu.2013.00477
56. Gardam S, Sierro F, Basten A, Mackay F, Brink R. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity (2008) 28:391–401. doi: 10.1016/j.immuni.2008.01.009
57. Hildebrand JM, Luo Z, Manske MK, Price-Troska T, Ziesmer SC, Lin W, et al. and reveals new insights into BAFF-R signaling. J Exp Med (2010) 207:2569–79. doi: 10.1084/jem.20100857
58. Xie P. TRAF molecules in cell signaling and in human diseases. J Mol Signal (2013) 8:7. doi: 10.1186/1750-2187-8-7
59. Jellusova J, Miletic AV, Cato MH, Lin WW, Hu Y, Bishop GA, et al. Context-specific BAFF-R signaling by the NF-κB and PI3K pathways. Cell Rep (2013) 5:1022–35. doi: 10.1016/j.celrep.2013.10.022
60. Day ES, Cachero TG, Qian F, Sun Y, Wen D, Pelletier M, et al. Selectivity of BAFF/BLyS and APRIL for binding to the TNF family receptors BAFFR/BR3 and BCMA. Biochemistry (2005) 44:1919–31. doi: 10.1021/bi048227k
61. Hymowitz SG, Patel DR, Wallweber HJ, Runyon S, Yan M, Yin J, et al. Structures of APRIL-receptor complexes: like BCMA, TACI employs only a single cysteine-rich domain for high affinity ligand binding. J Biol Chem (2005) 280:7218–27. doi: 10.1074/jbc.M411714200
62. Bossen C, Cachero TG, Tardivel A, Ingold K, Willen L, Dobles M, et al. TACI, unlike BAFF-R, is solely activated by oligomeric BAFF and APRIL to support survival of activated B cells and plasmablasts. Blood (2008) 111:1004–12. doi: 10.1182/blood-2007-09-110874
63. Hase H, Kanno Y, Kojima M, Hasegawa K, Sakurai D, Kojima H, et al. BAFF/BLyS can potentiate B-cell selection with the B-cell coreceptor complex. Blood (2004) 103:2257–65. doi: 10.1182/blood-2003-08-2694
64. He B, Santamaria R, Xu W, Cols M, Chen K, Puga I, et al. The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nat Immunol (2010) 11:836–45. doi: 10.1038/ni.1914
65. Sintes J, Gentile M, Zhang S, Garcia-Carmona Y, Magri G, Cassis L, et al. mTOR intersects antibody-inducing signals from TACI in marginal zone B cells. Nat Commun (2017) 8:1462. doi: 10.1038/s41467-017-01602-4
66. Sakurai D, Kanno Y, Hase H, Kojima H, Okumura K, Kobata T. TACI attenuates antibody production costimulated by BAFF-R and CD40. Eur J Immunol (2007) 37:110–8. doi: 10.1002/eji.200636623
67. von Bülow GU, Bram RJ. NF-AT activation induced by a CAML-interacting member of the tumor necrosis factor receptor superfamily. Science (1997) 278:138–41. doi: 10.1126/science.278.5335.138
68. Garcia-Carmona Y, Cols M, Ting AT, Radigan L, Yuk FJ, Zhang L, et al. Differential induction of plasma cells by isoforms of human TACI. Blood (2015) 125:1749–58. doi: 10.1182/blood-2014-05-575845
69. Garcia-Carmona Y, Ting AT, Radigan L, Athuluri Divakar SK, Chavez J, Meffre E, et al. TACI Isoforms Regulate Ligand Binding and Receptor Function. Front Immunol (2018) 9:2125. doi: 10.3389/fimmu.2018.02125
70. Schebesta A, McManus S, Salvagiotto G, Delogu A, Busslinger GA, Busslinger M. Transcription factor Pax5 activates the chromatin of key genes involved in B cell signaling, adhesion, migration, and immune function. Immunity (2007) 27:49–63. doi: 10.1016/j.immuni.2007.05.019
71. Chu PG, Loera S, Huang Q, Weiss LM. Lineage determination of CD20- B-Cell neoplasms: an immunohistochemical study. Am J Clin Pathol (2006) 126:534–44. doi: 10.1309/3WG32YRAMQ7RB9D4
72. Rasheed AA, Samad A, Raheem A, Hirani SI, Shabbir- Moosajee M. Cd20 Expression and Effects on Outcome of Relapsed/ Refractory Diffuse Large B Cell Lymphoma after Treatment with Rituximab. Asian Pac J Cancer Prev (2018) 19:331–5. doi: 10.22034/APJCP.2018.19.2.331
73. Cioc AM, Vanderwerf SM, Peterson BA, Robu VG, Forster CL, Pambuccian SE. Rituximab-induced changes in hematolymphoid tissues found at autopsy. Am J Clin Pathol (2008) 130:604–12. doi: 10.1309/UXLE9RHL968TER7B
74. Desouki MM, Post GR, Cherry D, Lazarchick J. PAX-5: a valuable immunohistochemical marker in the differential diagnosis of lymphoid neoplasms. Clin Med Res (2010) 8:84–8. doi: 10.3121/cmr.2010.891
75. Salzer U, Chapel HM, Webster AD, Pan-Hammarström Q, Schmitt-Graeff A, Schlesier M, et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet (2005) 37:820–8. doi: 10.1038/ng1600
76. Castigli E, Wilson SA, Garibyan L, Rachid R, Bonilla F, Schneider L, et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet (2005) 37:829–34. doi: 10.1038/ng1601
77. Zhang L, Radigan L, Salzer U, Behrens TW, Grimbacher B, Diaz G, et al. Transmembrane activator and calcium-modulating cyclophilin ligand interactor mutations in common variable immunodeficiency: clinical and immunologic outcomes in heterozygotes. J Allergy Clin Immunol (2007) 120:1178–85. doi: 10.1016/j.jaci.2007.10.001
78. Jacobs HM, Thouvenel CD, Leach S, Arkatkar T, Metzler G, Scharping NE, et al. Cutting Edge: BAFF Promotes Autoantibody Production via TACI-Dependent Activation of Transitional B Cells. J Immunol (2016) 196:3525–31. doi: 10.4049/jimmunol.1600017
79. Fried AJ, Rauter I, Dillon SR, Jabara HH, Geha RS. Functional analysis of transmembrane activator and calcium-modulating cyclophilin ligand interactor (TACI) mutations associated with common variable immunodeficiency. J Allergy Clin Immunol (2011) 128:226–8.e1. doi: 10.1016/j.jaci.2011.01.048
80. Sazzini M, Zuntini R, Farjadian S, Quinti I, Ricci G, Romeo G, et al. An evolutionary approach to the medical implications of the tumor necrosis factor receptor superfamily member 13B (TNFRSF13B) gene. Genes Immun (2009) 10:566–78. doi: 10.1038/gene.2009.43
81. Pulvirenti F, Zuntini R, Milito C, Specchia F, Spadaro G, Danieli MG, et al. Clinical Associations of Biallelic and Monoallelic TNFRSF13B Variants in Italian Primary Antibody Deficiency Syndromes. J Immunol Res (2016) 2016:8390356. doi: 10.1155/2016/8390356
82. Almejun MB, Cols M, Zelazko M, Oleastro M, Cerutti A, Oppezzo P, et al. Naturally occurring mutation affecting the MyD88-binding site of TNFRSF13B impairs triggering of class switch recombination. Eur J Immunol (2013) 43:805–14. doi: 10.1002/eji.201242945
83. Warnatz K, Salzer U, Rizzi M, Fischer B, Gutenberger S, Böhm J, et al. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc Natl Acad Sci USA (2009) 106:13945–50. doi: 10.1073/pnas.0903543106
84. Pieper K, Rizzi M, Speletas M, Smulski CR, Sic H, Kraus H, et al. A common single nucleotide polymorphism impairs B-cell activating factor receptor’s multimerization, contributing to common variable immunodeficiency. J Allergy Clin Immunol (2014) 133:1222–5. doi: 10.1016/j.jaci.2013.11.021
85. Losi CG, Silini A, Fiorini C, Soresina A, Meini A, Ferrari S, et al. Mutational analysis of human BAFF receptor TNFRSF13C (BAFF-R) in patients with common variable immunodeficiency. J Clin Immunol (2005) 25:496–502. doi: 10.1007/s10875-005-5637-2
86. Meinl E, Thaler FS, Lichtenthaler SF. Shedding of BAFF/APRIL Receptors Controls B Cells. Trends Immunol (2018) 39:673–6. doi: 10.1016/j.it.2018.07.002
87. Smulski CR, Kury P, Seidel LM, Staiger HS, Edinger AK, Willen L, et al. BAFF- and TACI-Dependent Processing of BAFFR by ADAM Proteases Regulates the Survival of B Cells. Cell Rep (2017) 18:2189–202. doi: 10.1016/j.celrep.2017.02.005
88. Hoffmann FS, Kuhn PH, Laurent SA, Hauck SM, Berer K, Wendlinger SA, et al. The immunoregulator soluble TACI is released by ADAM10 and reflects B cell activation in autoimmunity. J Immunol (2015) 194:542–52. doi: 10.4049/jimmunol.1402070
89. Laurent SA, Hoffmann FS, Kuhn PH, Cheng Q, Chu Y, Schmidt-Supprian M, et al. γ-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat Commun (2015) 6:7333. doi: 10.1038/ncomms8333
90. Jeon ST, Kim WJ, Lee SM, Lee MY, Park SB, Lee SH, et al. Reverse signaling through BAFF differentially regulates the expression of inflammatory mediators and cytoskeletal movements in THP-1 cells. Immunol Cell Biol (2010) 88:148–56. doi: 10.1038/icb.2009.75
91. Nys J, Smulski CR, Tardivel A, Willen L, Kowalczyk C, Donzé O, et al. No evidence that soluble TACI induces signalling via membrane-expressed BAFF and APRIL in myeloid cells. PloS One (2013) 8:e61350. doi: 10.1371/journal.pone.0061350
92. Maglione PJ, Ko HM, Tokuyama M, Gyimesi G, Soof C, Li M, et al. Serum B-Cell Maturation Antigen (BCMA) Levels Differentiate Primary Antibody Deficiencies. J Allergy Clin Immunol Pract (2020) 8:283–91.e1. doi: 10.1016/j.jaip.2019.08.012
93. Jolles S, Chapel H, Litzman J. When to initiate immunoglobulin replacement therapy (IGRT) in antibody deficiency: a practical approach. Clin Exp Immunol (2017) 188:333–41. doi: 10.1111/cei.12915
94. Jackson LA, Benson P, Sneller VP, Butler JC, Thompson RS, Chen RT, et al. Safety of revaccination with pneumococcal polysaccharide vaccine. Jama (1999) 281:243–8. doi: 10.1001/jama.281.3.243
95. Wijetilleka S, Jayne DR, Mukhtyar C, Ala A, Bright PD, Chinoy H, et al. Recommendations for the management of secondary hypogammaglobulinaemia due to B cell targeted therapies in autoimmune rheumatic diseases. Rheumatol (Oxford) (2019) 58:889–96. doi: 10.1093/rheumatology/key394
96. Maglione PJ, Overbey JR, Cunningham-Rundles C. Progression of Common Variable Immunodeficiency Interstitial Lung Disease Accompanies Distinct Pulmonary and Laboratory Findings. J Allergy Clin Immunol Pract (2015) 3:941–50. doi: 10.1016/j.jaip.2015.07.004
97. Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, et al. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol (2002) 3:822–9. doi: 10.1038/ni829
98. Park J, Munagala I, Xu H, Blankenship D, Maffucci P, Chaussabel D, et al. Interferon signature in the blood in inflammatory common variable immune deficiency. PloS One (2013) 8:e74893. doi: 10.1371/journal.pone.0074893
99. Cambronero R, Sewell WA, North ME, Webster AD, Farrant J. Up-regulation of IL-12 in monocytes: a fundamental defect in common variable immunodeficiency. J Immunol (2000) 164:488–94. doi: 10.4049/jimmunol.164.1.488
100. Martinez-Pomar N, Raga S, Ferrer J, Pons J, Munoz-Saa I, Julia MR, et al. Elevated serum interleukin (IL)-12p40 levels in common variable immunodeficiency disease and decreased peripheral blood dendritic cells: analysis of IL-12p40 and interferon-gamma gene. Clin Exp Immunol (2006) 144:233–8. doi: 10.1111/j.1365-2249.2006.03063.x
101. Cols M, Rahman A, Maglione PJ, Garcia-Carmona Y, Simchoni N, Ko HM, et al. Expansion of inflammatory innate lymphoid cells in patients with common variable immune deficiency. J Allergy Clin Immunol (2016) 137:1206–1215.e6. doi: 10.1016/j.jaci.2015.09.013
102. Cunill V, Clemente A, Lanio N, Barceló C, Andreu V, Pons J, et al. Follicular T Cells from smB(-) Common Variable Immunodeficiency Patients Are Skewed Toward a Th1 Phenotype. Front Immunol (2017) 8:174. doi: 10.3389/fimmu.2017.00174
103. Unger S, Seidl M, van Schouwenburg P, Rakhmanov M, Bulashevska A, Frede N, et al. The T(H)1 phenotype of follicular helper T cells indicates an IFN-γ-associated immune dysregulation in patients with CD21low common variable immunodeficiency. J Allergy Clin Immunol (2018) 141:730–40. doi: 10.1016/j.jaci.2017.04.041
104. Turpin D, Furudoi A, Parrens M, Blanco P, Viallard JF, Duluc D. Increase of follicular helper T cells skewed toward a Th1 profile in CVID patients with non-infectious clinical complications. Clin Immunol (2018) 197:130–8. doi: 10.1016/j.clim.2018.09.006
105. Hultberg J, Ernerudh J, Larsson M, Nilsdotter-Augustinsson Å, Nyström S. Plasma protein profiling reflects T(H)1-driven immune dysregulation in common variable immunodeficiency. J Allergy Clin Immunol (2020) 146:417–28. doi: 10.1016/j.jaci.2020.01.046
106. Romberg N, Chamberlain N, Saadoun D, Gentile M, Kinnunen T, Ng YS, et al. CVID-associated TACI mutations affect autoreactive B cell selection and activation. J Clin Invest (2013) 123:4283–93. doi: 10.1172/JCI69854
107. Thien M, Phan TG, Gardam S, Amesbury M, Basten A, Mackay F, et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity (2004) 20:785–98. doi: 10.1016/j.immuni.2004.05.010
108. Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity (2004) 20:441–53. doi: 10.1016/S1074-7613(04)00079-2
109. Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood (2008) 111:77–85. doi: 10.1182/blood-2007-06-091744
110. Carsetti R, Rosado MM, Donnanno S, Guazzi V, Soresina A, Meini A, et al. The loss of IgM memory B cells correlates with clinical disease in common variable immunodeficiency. J Allergy Clin Immunol (2005) 115:412–7. doi: 10.1016/j.jaci.2004.10.048
111. Dong X, Qin J, Ma J, Zeng Q, Zhang H, Zhang R, et al. BAFF inhibits autophagy promoting cell proliferation and survival by activating Ca(2+)-CaMKII-dependent Akt/mTOR signaling pathway in normal and neoplastic B-lymphoid cells. Cell Signal (2019) 53:68–79. doi: 10.1016/j.cellsig.2018.09.012
112. Caldirola MS, Martínez MP, Bezrodnik L, Zwirner NW, Gaillard MI. Immune Monitoring of Patients With Primary Immune Regulation Disorders Unravels Higher Frequencies of Follicular T Cells With Different Profiles That Associate With Alterations in B Cell Subsets. Front Immunol (2020) 11:576724. doi: 10.3389/fimmu.2020.576724
113. Hömig-Hölzel C, Hojer C, Rastelli J, Casola S, Strobl LJ, Müller W, et al. Constitutive CD40 signaling in B cells selectively activates the noncanonical NF-kappaB pathway and promotes lymphomagenesis. J Exp Med (2008) 205:1317–29. doi: 10.1084/jem.20080238
114. Hostager BS, Bishop GA. CD40-Mediated Activation of the NF-κB2 Pathway. Front Immunol (2013) 4:376. doi: 10.3389/fimmu.2013.00376
115. Flament T, Bigot A, Chaigne B, Henique H, Diot E, Marchand-Adam S. Pulmonary manifestations of Sjögren’s syndrome. Eur Respir Rev (2016) 25:110–23. doi: 10.1183/16000617.0011-2016
116. Daridon C, Devauchelle V, Hutin P, Le Berre R, Martins-Carvalho C, Bendaoud B, et al. Aberrant expression of BAFF by B lymphocytes infiltrating the salivary glands of patients with primary Sjögren’s syndrome. Arthritis Rheum (2007) 56:1134–44. doi: 10.1002/art.22458
117. Pers JO, d’Arbonneau F, Devauchelle-Pensec V, Saraux A, Pennec YL, Youinou P. Is periodontal disease mediated by salivary BAFF in Sjögren’s syndrome? Arthritis Rheum (2005) 52:2411–4. doi: 10.1002/art.21205
118. Groom J, Kalled SL, Cutler AH, Olson C, Woodcock SA, Schneider P, et al. Association of BAFF/BLyS overexpression and altered B cell differentiation with Sjögren’s syndrome. J Clin Invest (2002) 109:59–68. doi: 10.1172/JCI0214121
119. Mariette X, Roux S, Zhang J, Bengoufa D, Lavie F, Zhou T, et al. The level of BLyS (BAFF) correlates with the titre of autoantibodies in human Sjögren’s syndrome. Ann Rheum Dis (2003) 62:168–71. doi: 10.1136/ard.62.2.168
120. Brkic Z, Maria NI, van Helden-Meeuwsen CG, van de Merwe JP, van Daele PL, Dalm VA, et al. Prevalence of interferon type I signature in CD14 monocytes of patients with Sjogren’s syndrome and association with disease activity and BAFF gene expression. Ann Rheum Dis (2013) 72:728–35. doi: 10.1136/annrheumdis-2012-201381
121. Peng M, Wang W, Qin L, Liu H, Qin M, Zheng W, et al. Association between nonspecific interstitial pneumonia and presence of CD20+ B lymphocytes within pulmonary lymphoid follicles. Sci Rep (2017) 7:16912. doi: 10.1038/s41598-017-17208-1
122. Salvi SS, Barnes PJ. Chronic obstructive pulmonary disease in non-smokers. Lancet (2009) 374:733–43. doi: 10.1016/S0140-6736(09)61303-9
123. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med (2004) 350:2645–53. doi: 10.1056/NEJMoa032158
124. Polverino F, Cosio BG, Pons J, Laucho-Contreras M, Tejera P, Iglesias A, et al. An Orchestrator of Lymphoid Follicles in Severe Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med (2015) 192:695–705. doi: 10.1164/rccm.201501-0107OC
Keywords: common variable immune deficiency, CVID, GLILD, interstitial lung disease, TACI, BAFF-R, rituximab, B cell activating factor
Citation: Matson EM, Abyazi ML, Bell KA, Hayes KM and Maglione PJ (2021) B Cell Dysregulation in Common Variable Immunodeficiency Interstitial Lung Disease. Front. Immunol. 11:622114. doi: 10.3389/fimmu.2020.622114
Received: 27 October 2020; Accepted: 23 December 2020;
Published: 05 February 2021.
Edited by:
John R. Hurst, University College London, United KingdomReviewed by:
David Andrew Fulcher, Australian National University, AustraliaJavier Chinen, Baylor College of Medicine, United States
Copyright © 2021 Matson, Abyazi, Bell, Hayes and Maglione. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul J. Maglione, cG1hZ2xpb25AYnUuZWR1