 Preethi Bala Balakrishnan
Preethi Bala Balakrishnan Elizabeth E. Sweeney
Elizabeth E. Sweeney- The George Washington University Cancer Center, School of Medicine and Health Sciences, George Washington University, Washington, DC, United States
Adoptive T cell therapy has emerged as a revolutionary immunotherapy for treating cancer. Despite immense promise and clinical success in some hematologic malignancies, limitations remain that thwart its efficacy in solid tumors. Particularly in tumors of the central nervous system (CNS), T cell therapy is often restricted by the difficulty in intratumoral delivery across anatomical niches, suboptimal T cell specificity or activation, and intratumoral T cell dysfunction due to immunosuppressive tumor microenvironments (TMEs). Nanoparticles may offer several advantages to overcome these limitations of T cell therapy, as they can be designed to robustly and specifically activate T cells ex vivo prior to adoptive transfer, to encapsulate T cell stimulating agents for co-localized stimulation, and to be conjugated onto T cells for added functionality. This perspective highlights recent preclinical advances in using nanoparticles to enhance T cell therapy, and discusses the potential applicability and constraints of nanoparticle-enhanced T cells as a new platform for treating CNS tumors.
Introduction
Adoptive T cell therapy, including antigen-specific T cells and chimeric antigen receptor (CAR) T cells, represents a promising avenue for treating cancer, as evidenced by numerous clinical trials in hematologic and solid tumors in both the adult and pediatric populations (1–7). Indeed, three T cell-based therapeutics, Kymriah, Yescarta, and Breyanzi have been approved by the US FDA for treating B cell leukemia and lymphoma (8–10). Despite immense potential in hematologic malignancies, T cell therapy inadequately controls tumor growth in many solid tumor contexts due to intrinsic limitations (11–14).
Firstly, effective T cell therapy can be hindered by the heterogeneous tumor antigen milieu, such that T cells engineered to target only one antigen may be ineffective in clearing the tumor. Abnormal blood vessels, typical of solid tumors (15), hinder T cell infiltration into the tumor microenvironment (TME), thus limiting their local bioavailability and function. Additionally, the TME of solid tumors is typically immunosuppressive. Thus, even when T cells are able to infiltrate and target the heterogeneous tumor-associated antigens, the TME signals suppression of T cell function, thereby blocking their therapeutic effect (16). Tumors of the central nervous system (CNS) are especially difficult to treat with T cell therapy, due to the increased difficulty of T cells to traffic to and infiltrate CNS anatomical regions, thereby dampening their effect (17, 18).
Nanotechnology offers advantages to overcome limitations of T cell therapy for solid tumors. Because of their size, surface area to volume ratio, and ability to encapsulate a variety of agents for controlled release, nanoparticles have long been studied for their advantages in drug delivery in cancer (19, 20). Nanoparticles offer a means to cross various anatomical niches, such as the blood brain barrier (BBB), to deliver therapeutic cargo, enabling both imaging and therapy of solid tumors. Over the past several decades, many nanoparticles have been explored and validated in preclinical brain tumor models (21–33). There is emerging research into how nanotechnology can be used to improve immunotherapies, including T cell therapy (34–38), but very few studies, if any, investigate the role to nanoparticles to improve T cell therapy for CNS tumors. Here, we aim to summarize the recent developments in nanoparticle-enhanced T cell therapy, and comment on hypothesized applications for treating CNS tumors. We highlight nanoparticle-mediated strategies to prepare T cell products for adoptive therapy, nanoparticles conjugated to T cells to overcome TME immunosuppression, and nanoparticles to enhance T cell tumor infiltration, activate T cells in situ, and/or add functionality to T cell therapy. Finally, we discuss how to apply the described principles and strategies of nanoparticle-enhanced T cells to treat tumors of the CNS.
Nanoparticles to Improve the Ex Vivo Generation of Therapeutic T Cell Products
Nanoparticles for Improving T Cell Manufacture
To improve ex vivo expansion of antigen-specific T cells, one group generated a nanostructured polyethylene glycol (PEG) hydrogel platform to stimulate T cells prior to adoptive transfer (39). The presented platform used gold nanoparticles conjugated with anti-CD3 antibodies to activate T cells, and integrin-activating peptides to initiate integrin-mediated cell adhesion of the hydrogel to T cells. Overall, the nanostructured hydrogel enabled T cell activation, proliferation, and memory (39).
In an effort to arm T cells to counter the immunosuppressive microenvironment typical of solid tumors, immunoliposomes were generated to encapsulate a small molecule TGF-β inhibitor (thereby avoiding toxic systemic administration of TGF-β blockade) and T cell targeting receptors CD45 or CD90 (40). The choice of the two receptors was to compare the effects of targeting a receptor likely to internalize the immunoliposomes (CD90) versus a receptor that would likely bind the immunoliposomes on the cell surface (CD45). These immunoliposomes enabled enhanced T cell activation and granzyme expression when incubated with T cells prior to adoptive transfer. Upon adoptive transfer, the T cells incubated with immunoliposomes targeting CD90 caused the most T cell-mediated anti-tumor activity and reduction in tumor growth in vivo in mice. These studies uncovered the importance of using an internalizing receptor (CD90) to target drug-loaded liposomes to T cells during manufacture (40).
Another strategy used small interfering RNA (siRNA) to downregulate immunosuppressive signaling pathways in the cytotoxic T cells before adoptive transfer, so as to improve their anti-tumor efficacy in vivo (41). Because the viability of primary T cells is sometimes affected by conventional or viral transduction or electroporation, the group attached gold nanoparticles to the T cells and used photoporation to gently heat the T cells and allow the membrane to transiently accept siRNA. They found successful gene silencing of those targeted by the siRNA, while maintaining the viability of the T cells (41). Significantly less T cell death occurred in response to transfection via the photoporation technique in comparison to traditionally employed nucleofection.
Polymeric nanocarriers that encapsulate mRNA have also been studied to transiently deliver mRNA to antigen-specific T cells prior to adoptive transfer (42). Researchers showed the ability of the nanocarriers to deliver mRNA to knock down immunosuppressive molecules in antigen-specific T cells, and induce transcriptional activity typical of a memory T cell phenotype. They illustrated the simple design of the strategy of mixing therapeutic T cells with the nanocarriers, and highlighted the broad applicability to overcoming various limitations of T cell therapy by targeting specific genes of interest (42).
Ionizable lipid nanoparticles can also mediate ex vivo mRNA delivery into T cells for transient CAR expression, thereby mitigating the toxic side effects seen in permanent CAR expression on T cells and circumventing classical mRNA delivery into cells (e.g. electroporation) which often impacts T cell viability (43). Indeed, a study showed effective CD19-specific CAR expression on T cells after lipid nanoparticle delivery of CD19 CAR mRNA, with decreased cell death as compared to electroporation-delivered CAR mRNA. Additionally, CAR T cells manufactured by this method via ionizable lipid nanoparticles induced functional protein expression and enabled anti-tumor efficacy in a leukemia model in vitro at equivalent levels to electroporated CAR T cells (43).
Nanoparticles as Artificial Antigen-Presenting Cells (aAPCs)
Ex vivo expanded antigen-specific T cells generated using antigen-presenting cells (APCs) can specifically target tumor cells and exert effector functions (44, 45). Artificial antigen-presenting cells (aAPCs) represent a new class of nanoparticles to mimic T cell recognition and stimulation ex vivo. Several nanoparticle platforms are being investigated for improving the manufacture of T cells prior to adoptive transfer. An emerging area of interest is using nanoparticle-based aAPCs to increase the activation, specificity, and functionality of T cell products in various applications (46). Carbon nanotubes have been a focus of research in T cell manufacture, as the high surface area to volume ratio of single-walled carbon nanotubes allows high density of immobilized antigens and/or T cell activation molecules (47). Researchers showed the ability of carbon nanotubes to present high concentrations of anti-CD3 antibody to T cells and stimulate T cells more than free anti-CD3 or anti-CD3 bound to beads, illustrating their potential to generate activated T cells prior to therapy (48). Additionally, they found that when the single-walled carbon nanotubes were functionalized with anti-CD3 and other antibodies to co-stimulate T cells (anti-CD28), the high potency of T cell activation was driven by the concentration of bound antigens on the nanotubes (48). This strategy enabled rapid expansion of T cells for the purpose of adoptive therapy. A carbon nanotube-polymer composite with conjugated T cell stimuli (MHC-I and anti-CD28) has been used as an aAPC in the presence of IL-2. This nano-composite allowed effective T cell expansion with 1000x less IL-2 than standard T cell manufacture, and enabled delayed melanoma growth in mice (49).
Another group generated aAPCs by immobilizing a peptide from a model antigen, ovalbumin (OVA), loaded onto MHC-I dimers, along with anti-CD28, on biocompatible polymer-based biodegradable microparticles containing IL-2 (50). Upon co-culture of T cells with these aAPCs, T cells were able to more efficiently expand and activate compared to treatment with exogenous IL-2, aAPCs, or aAPCs in combination with exogenous IL-2, allowing for significant generation of OVA-specific cytotoxic T cells (50). Paramagnetic nanoparticles have also been utilized as aAPCs (nano-aAPCs) for improved T cell manufacture for adoptive transfer (51). Nano-aAPCs enriched tumor-specific T cells (targeting melanoma-associated antigens such as TRP2 and GP100), and enabled their rapid expansion both in vitro and in vivo. T cells generated via nano-aAPCs were also effectively stimulated upon antigen recognition, ultimately illustrating the potential of the platform to rapidly generate high amounts of tumor-specific T cells (51).
In addition to particles functioning as aAPCs, a group also investigated the use of a biomimetic scaffold to mimic APCs for increased T cell product output (52). A lipid bilayer comprising T cell-activating and co-stimulatory molecules (i.e. anti-CD3, anti-CD28) was combined with silica microrods for sustained release of cytokines (i.e. IL-2) to ensure sustained T cell function. They posited that the T cell product expansion can be tuned based on the structure and pattern of the scaffold and its encapsulated contents, and demonstrated the improved efficacy of the aAPC scaffold to efficiently expand polyclonal and antigen-specific (i.e. specific for peptides from Epstein-Barr virus (EBV)-associated proteins) cytotoxic T cells for use in adoptive therapy, as compared to traditional methods for T cell product generation. They illustrate the efficacy of the platform to produce CD19-specific CAR T cells as well, by illustrating effective lymphoma regression after adoptive therapy in vivo (52).
Another group investigated the use of biomimetic magnetosomes as aAPCs (53). The biomimetic magnetosomes comprised magnetic nanoclusters coated with immune cell membranes, anti-CD28 to stimulate T cells, and peptide-loaded MHC-I complexes, which allowed for robust ex vivo T cell expansion prior to adoptive transfer, as well as a means to physically guide the activated T cells by a magnetic field. Ultimately, the combination of the biomimetic nanoclusters and T cells enabled delayed lymphoma growth in mice in vivo, thus representing a platform to both improve therapeutic T cell generation while adding functionality (magnetically controlled tumor accumulation) in vivo.
Nanoparticles to Overcome T Cell Immunosuppression in the TME
In addition to nanoparticle-mediated T cell engineering prior to adoptive transfer, other groups investigate the use of nanoparticles within the TME. Intratumoral T cell function is often dysregulated or inhibited in solid tumors, thereby preventing efficacy of adoptive T cell therapy (54–58). To overcome this, several groups have utilized nanoparticles loaded with various cargo aimed at maintaining T cell activation or blocking immunosuppressive pathway molecules. One potential solution is administering liposomes encapsulating T cell-activating antibodies or cytokines to target adopted T cells in vivo (59). Here, researchers used Thy1.1- and IL-2-conjugated liposomes to specifically target T cells and enable T cell proliferation, respectively (59). These studies illuminated the feasibility of effectively targeting T cells in vivo to deliver payloads (e.g. imaging agents, antibodies, cytokines, adjuvants, drugs) loaded within liposomes via surface targeting. Similarly, coating therapeutic T cells with nanoparticles loaded with adjuvant (i.e. IL-15 superagonist (IL-15SA) and IL-21) enabled adopted antigen-specific T cells to proliferate in vivo and effectively eliminate melanoma in a mouse model (60).
Another study illustrated a novel platform comprising T cells conjugated with nanogels loaded with proteins to enhance T cell function (61). Importantly, the protein cargo was designed to be released only upon antigen binding to T cell receptor, such that the likely activation site would be intratumoral. They found that when IL-15SA was encapsulated in the nanogel, transferred T cells had significantly higher expansion in tumors, and allowed effective CAR T cell-mediated tumor clearance in vivo (61). Alternatively, CAR T cells have been engineered to deliver liposomal nanoparticles encapsulating a small molecular adenosine antagonist in order to block the immunosuppressive function of adenosine within the TME of solid tumors that impairs T cell function (62). Because of the difficulty in delivering the antagonist specifically to T cells in the TME, the physical conjugation of the nanoparticles to the adoptive CAR T cells enables effective local delivery and provides a potential solution to overcoming an immunosuppressive pathway in solid tumors (62).
Other recent work has presented a novel nanoparticle platform for pre-treating the TME prior to CAR T cell therapy (63). There, lipid nanoparticles carrying a PI3K inhibitor were coated with a tumor-targeting peptide (iRGD). The nanoparticles also encapsulated α-GalCer, a component to activate NKT cells. The combination comprised in the nanoparticle platform was able to overcome the immunosuppressive TME that typically inhibits the success of CAR T cell therapy. Thus, CAR T cell therapy following nanoparticle treatment appeared successful in terms of T cell expansion and tumor regression (63). Other nanoparticles designed to remodel the TME could also play a role in improving the infiltration, activation, or functionality of adoptive T cell therapy (64).
These studies critically demonstrate the enhancement of T cell function through the use of conjugated nanoparticles. As nanoparticles can be designed to encapsulate innumerable disease- or cell-specific cargo, there is promise for vast applicability of platforms co-localizing nanoparticles and T cells.
Nanoparticles to Aid T Cell Tumor Infiltration and In Situ T Cell Activation
To overcome some of the physical barriers of T cell infiltration in addition to the local immunosuppression in solid tumors, one group employed photothermally activated nanoparticles (PLGA nanoparticles encapsulating ICG) to enable mild hyperthermia in the TME (65). They showed that pre-treating melanoma tumors with nanoparticle-mediated photothermal therapy enabled significantly more CAR T cell accumulation and better anti-tumor efficacy in vivo (65). The photothermal therapy allowed physical disruption of the TME, increased blood flow, and decreased pressure, but also released tumor-specific antigens that could importantly stimulate the CAR T cells upon infiltration. Nanoparticle-mediated photothermal therapy is not a clinically approved treatment strategy, but there have been clinical trials that completed testing its efficacy in other applications (66), thus suggesting its future clinical potential.
Other researchers employed magnetic nanoclusters that were synthesized with anti-PD-1 antibodies in order to bind to PD-1 expressing T cells prior to adoptive transfer. Then, a commercial neodymium magnet was used to magnetically recruit the nanocluster-T cells to the site of tumor, overcoming barriers of tumor infiltration, where the structures disassembled, allowing the simultaneous anti-tumor effects of T cells and anti-PD-1 antibodies (67). Although anti-PD-1 antibodies are FDA-approved for several applications, magnetically-responsive nanoparticles are still currently under investigation in clinical trials (68).
Another interesting strategy to combat transferred T cell infiltration issues uses DNA nanocarriers to transfect CAR genes into endogenous T cells, thereby circumventing the limitations involved in T cell manufacture. A group demonstrated feasibility of this approach using polymer-based nanoparticles carrying leukemia-specific CARs, showing effective generation of CAR T cells in vivo and anti-tumor efficacy at equivalent levels to traditionally infused 194-1BBz-specific CAR T cells (69). Another group investigated the use of circular bispecific aptamers to bypass the ex vivo generation of antigen-specific T cells (70). The aptamer was designed to bind both naïve T cells and tumor cells, thereby activating the T cells in situ. This strategy allowed T cell accumulation in the TME and a local activation of aptamer-bound T cells. A variety of antigen-specific aptamers could be used in order to treat a broad range of tumors (70). These designs may eliminate the need for cell engineering prior to adoptive transfer, although their clinical application has not yet been reported.
Nanoparticles to Add Functionality as Combination T Cell Therapy
Recently, researchers showed the potential of attaching Prussian blue nanoparticles enabling photothermal therapy to antigen-specific T cells for a combined nanoimmunotherapy (71). By combining disparate elements into a single platform, they demonstrated the improved efficacy of EBV-associated antigen-specific T cell function and nanoparticle-mediated photothermal therapy in vitro by increased target cell cytotoxicity (71). A different approach sought to combine the advantages of CAR T cells and nanoparticle-mediated photothermal therapy for hepatocellular carcinoma (72). IR780 nanoparticles were loaded on mesoporous silica, and subsequently coated with the membrane of CAR T cells specific to GPC3+ hepatocellular carcinoma. The CAR T cell membrane enabled the nanoparticles to target GPC3 on the surface of hepatocellular carcinoma cells in vitro and in vivo (72). This study represents a different approach to CAR T cell therapy, by isolating the functional component of CAR T cell targeting and adding it to a nanoparticle component. This work, however, relies on the photothermal capability of the nanoparticle to mediate cytotoxicity against tumor cells, rather than the cytotoxic functionality of CAR T cells, which could also introduce limitations, as the absence of T cells may reduce the generation of tumor-specific T cell expansion or T cell memory.
Discussion
Thus far, we have summarized the state of the field of nanoparticle-enhanced therapeutic T cells for treating cancer. The unique attributes of nanoparticles, for example, their ability to penetrate anatomical barriers, encapsulate or immobilize therapeutic cargo, and specifically target tumor cells, add to their potential for enhancing T cell therapy (Table 1) (19, 20).

Table 1 Types of nanoparticles and their application in enhancing T cell therapy for solid tumors.
Although there have been clinical studies demonstrating safety and feasibility of T cell therapy for glioblastoma (2, 73–75), tumors of the CNS remain difficult to treat with adoptive T cell therapy due to limitations typical of solid tumors in addition to the restrictive anatomical barriers that prevent adequate delivery (76, 77). Despite the benefits of using nanoparticles to enhance T cell therapy illustrated in the work described herein, no studies show efficacy in the CNS tumor setting. We hypothesize that the principles of nanoparticles enhancing T cell therapy for solid tumors can be applied successfully to treat brain and CNS tumors. We have summarized our hypothesized applications of nanoparticle-enhanced T cell therapy for CNS tumors in Figure 1.
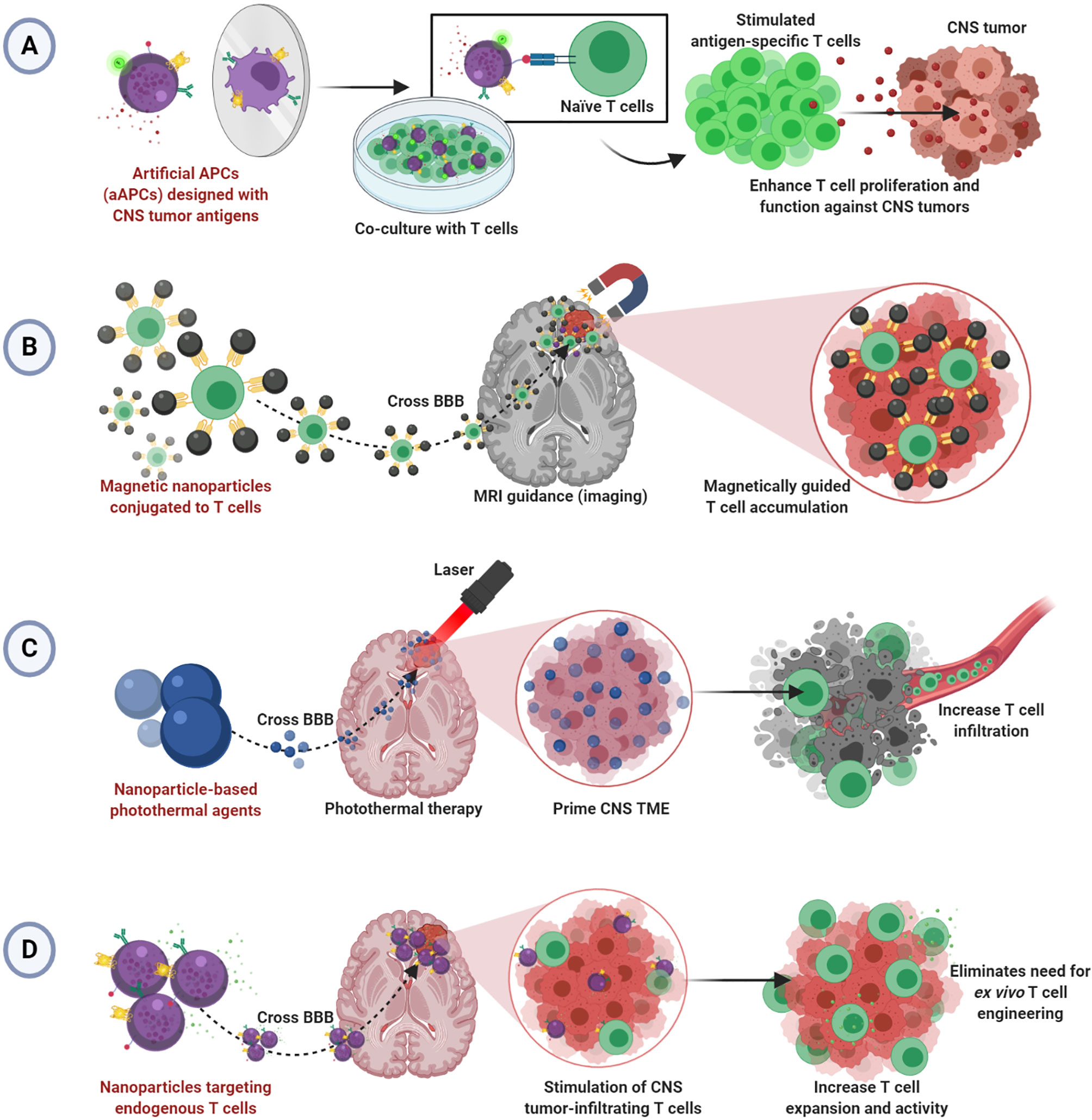
Figure 1 Hypothesized applications of nanoparticle-enhanced T cell therapy for CNS tumors. (A) Artificial aAPCs presenting multiple tumor-specific antigens may enable improved ex vivo generation of effective CNS tumor-specific T cells. (B) Magnetic nanoparticles conjugated to T cells may allow magnetically guided T cell delivery into CNS tumors. (C) Nanoparticle-mediated photothermal therapy may allow infiltration of T cells into primed CNS tumors. (D) Nanoparticles targeting T cells endogenous to CNS TME may bypass the BBB to enable enhanced T cell function without ex vivo T cell engineering.
Studies demonstrating improved T cell activation and expansion via aAPCs or nanoparticle-based T cell scaffolds prior to adoptive transfer can be refined for application in CNS tumors using CNS tumor-specific antigens. Since aAPCs can be engineered to express CNS tumor antigens, they could seamlessly change disease contexts. Indeed, there are antigen-specific and CAR T cell products to target CNS tumors (78, 79), and several have been investigated clinically (2, 73–75). We propose that aAPC-stimulated T cells may offer a more effective approach to development. Further, aAPCs using multiple antigens, as seen in neoantigen vaccines for glioblastoma (80, 81), could help to target the heterogeneous TME and induce a more complete T cell response (Figure 1A). However, because of the anatomical barriers hindering the infiltration of transferred T cells into the brain and spinal cord, we speculate that therapeutic T cells will need to be administered intratumorally in the absence of other TME manipulations or additional functionalities.
Several described studies offer insight into a hypothesized strategy for driving adopted T cell infiltration into CNS tumors. The research using MRI guidance and external magnets to drive magnetosome- or magnetic nanocluster-conjugated T cells into tumors could be applicable (53, 67). There, perhaps a magnetic field could magnetically attract CNS tumor-specific T cells armed with the magnetic nanoparticles as well, thereby overcoming the limitation of poor T cell penetration into tumors (Figure 1B). Alternatively, several noted studies critically illuminated a possible strategy for T cell delivery in CNS tumors by photothermal therapy (65, 71, 72). Photothermal therapy could be used to prime the tumor and/or BBB to allow increased vasodilation and T cell infiltration into CNS tumors. Thus, poor T cell infiltration would be mitigated (Figure 1C).
Lastly, endogenous T cells in CNS tumors can be exploited by employing methods to target T cells in situ as described (69, 70). These strategies rely on re-programming T cells already in the tumors to overcome immunosuppression and/or add functionality. Since nanoparticles could feasibility bypass the BBB to enter the brain and spinal cord, this design is particularly suited to treating CNS tumors (Figure 1D). However, efficacy may be limited depending on the number of endogenous T cells in the tumor. Recent work demonstrates the feasibility of BBB-permeable nanoparticles to drive infiltration of T cells into the brain TME, illustrating an interesting potential solution to this issue (82).
We have highlighted the state of the field of nanoparticle-enhanced T cell therapy and commented on the applicability of the principles described herein to treat CNS tumors. The ability of nanoparticles to efficiently activate therapeutic T cells prior to adoptive transfer, create an immunopermissive TME, and add specific targeting capabilities or functionalities, enables the efficacy of adoptive T cell therapy and CAR T cells against cancer. There are several promising avenues for T cell therapy to succeed in CNS tumors, but the anatomical barriers will narrow the applicable options. The research illustrated here suggests that nanotechnology can be key in solving many issues associated with treating CNS tumors with T cell therapy.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author Contributions
PBB and ES researched and wrote the article. Both authors contributed to the article and approved the submitted version.
Funding
Funding was provided by the GW Cancer Center, George Washington University, Washington, DC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Figure 1 was created with BioRender.com.
References
1. Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med (2019) 25(9):1408–14. doi: 10.1038/s41591-019-0549-5
2. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med (2016) 375(26):2561–9. doi: 10.1056/NEJMoa1610497
3. Curran KJ, Margossian SP, Kernan NA, Silverman LB, Williams DA, Shukla N, et al. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood (2019) 134(26):2361–8. doi: 10.1182/blood.2019001641
4. Pan J, Niu Q, Deng B, Liu S, Wu T, Gao Z, et al. CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia (2019) 33(12):2854–66. doi: 10.1038/s41375-019-0488-7
5. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N Engl J Med (2020) 382(14):1331–42. doi: 10.1056/NEJMoa1914347
6. Hont AB, Cruz CR, Ulrey R, O’Brien B, Stanojevic M, Datar A, et al. Immunotherapy of Relapsed and Refractory Solid Tumors With Ex Vivo Expanded Multi-Tumor Associated Antigen Specific Cytotoxic T Lymphocytes: A Phase I Study. J Clin Oncol (2019) 37(26):2349–59. doi: 10.1200/JCO.19.00177
7. Cruz CR, Micklethwaite KP, Savoldo B, Ramos CA, Lam S, Ku S, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood (2013) 122(17):2965–73. doi: 10.1182/blood-2013-06-506741
11. Zhang H, Ye ZL, Yuan ZG, Luo ZQ, Jin HJ, Qian QJ. New Strategies for the Treatment of Solid Tumors with CAR-T Cells. Int J Biol Sci (2016) 12(6):718–29. doi: 10.7150/ijbs.14405
12. Ma S, Li X, Wang X, Cheng L, Li Z, Zhang C, et al. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int J Biol Sci (2019) 15(12):2548–60. doi: 10.7150/ijbs.34213
13. Martinez M, Moon EK. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front Immunol (2019) 10:128. doi: 10.3389/fimmu.2019.00128
14. Knochelmann HM, Smith AS, Dwyer CJ, Wyatt MM, Mehrotra S, Paulos CM. CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies. Front Immunol (2018) 9:1740. doi: 10.3389/fimmu.2018.01740
15. Siemann DW. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by Tumor-Vascular Disrupting Agents. Cancer Treat Rev (2011) 37(1):63–74. doi: 10.1016/j.ctrv.2010.05.001
16. Akhavan D, Alizadeh D, Wang D, Weist MR, Shepphird JK, Brown CE. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol Rev (2019) 290(1):60–84. doi: 10.1111/imr.12773
17. Bechmann I, Galea I, Perry VH. What is the blood-brain barrier (not)? Trends Immunol (2007) 28(1):5–11. doi: 10.1016/j.it.2006.11.007
18. Owens T, Bechmann I, Engelhardt B. Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol (2008) 67(12):1113–21. doi: 10.1097/NEN.0b013e31818f9ca8
19. Brigger I, Dubernet C, Couvreur P. Nanoparticles in cancer therapy and diagnosis. Adv Drug Deliv Rev (2002) 54(5):631–51. doi: 10.1016/s0169-409x(02)00044-3
20. Begines B, Ortiz T, Perez-Aranda M, Martinez G, Merinero M, Arguelles-Arias F, et al. Polymeric Nanoparticles for Drug Delivery: Recent Developments and Future Prospects. Nanomaterials (Basel) (2020) 10(7):1403–41. doi: 10.3390/nano10071403
21. Pardridge WM. Vector-mediated drug delivery to the brain. Adv Drug Deliv Rev (1999) 36(2-3):299–321. doi: 10.1016/s0169-409x(98)00087-8
22. Pardridge WM. CNS drug design based on principles of blood-brain barrier transport. J Neurochem (1998) 70(5):1781–92. doi: 10.1046/j.1471-4159.1998.70051781.x
23. Kumar M, Medarova Z, Pantazopoulos P, Dai G, Moore A. Novel membrane-permeable contrast agent for brain tumor detection by MRI. Magn Reson Med (2010) 63(3):617–24. doi: 10.1002/mrm.22216
24. Kircher MF, de la Zerda A, Jokerst JV, Zavaleta CL, Kempen PJ, Mittra E, et al. A brain tumor molecular imaging strategy using a new triple-modality MRI-photoacoustic-Raman nanoparticle. Nat Med (2012) 18(5):829–34. doi: 10.1038/nm.2721
25. Nie G, Hah HJ, Kim G, Lee YE, Qin M, Ratani TS, et al. Hydrogel nanoparticles with covalently linked coomassie blue for brain tumor delineation visible to the surgeon. Small (2012) 8(6):884–91. doi: 10.1002/smll.201101607
26. He H, Li Y, Jia XR, Du J, Ying X, Lu WL, et al. PEGylated Poly(amidoamine) dendrimer-based dual-targeting carrier for treating brain tumors. Biomaterials (2011) 32(2):478–87. doi: 10.1016/j.biomaterials.2010.09.002
27. Sousa F, Mandal S, Garrovo C, Astolfo A, Bonifacio A, Latawiec D, et al. Functionalized gold nanoparticles: a detailed in vivo multimodal microscopic brain distribution study. Nanoscale (2010) 2(12):2826–34. doi: 10.1039/c0nr00345j
28. Dumont MF, Hoffman HA, Yoon PR, Conklin LS, Saha SR, Paglione J, et al. Biofunctionalized gadolinium-containing prussian blue nanoparticles as multimodal molecular imaging agents. Bioconjug Chem (2014) 25(1):129–37. doi: 10.1021/bc4004266
29. Dumont MF, Yadavilli S, Sze RW, Nazarian J, Fernandes R. Manganese-containing Prussian blue nanoparticles for imaging of pediatric brain tumors. Int J Nanomedicine (2014) 9:2581–95. doi: 10.2147/IJN.S63472
30. Kreuter J, Alyautdin RN, Kharkevich DA, Ivanov AA. Passage of peptides through the blood-brain barrier with colloidal polymer particles (nanoparticles). Brain Res (1995) 674(1):171–4. doi: 10.1016/0006-8993(95)00023-j
31. Orringer DA, Koo YE, Chen T, Kopelman R, Sagher O, Philbert MA. Small solutions for big problems: the application of nanoparticles to brain tumor diagnosis and therapy. Clin Pharmacol Ther (2009) 85(5):531–4. doi: 10.1038/clpt.2008.296
32. Sarin H, Kanevsky AS, Wu H, Brimacombe KR, Fung SH, Sousa AA, et al. Effective transvascular delivery of nanoparticles across the blood-brain tumor barrier into malignant glioma cells. J Transl Med (2008) 6:80. doi: 10.1186/1479-5876-6-80
33. Kaur IP, Bhandari R, Bhandari S, Kakkar V. Potential of solid lipid nanoparticles in brain targeting. J Control Release (2008) 127(2):97–109. doi: 10.1016/j.jconrel.2007.12.018
34. Nawaz W, Xu S, Li Y, Huang B, Wu X, Wu Z. Nanotechnology and immunoengineering: How nanotechnology can boost CAR-T therapy. Acta Biomater (2020) 109:21–36. doi: 10.1016/j.actbio.2020.04.015
35. Goldberg MS. Immunoengineering: how nanotechnology can enhance cancer immunotherapy. Cell (2015) 161(2):201–4. doi: 10.1016/j.cell.2015.03.037
36. Mi Y, Hagan CT4, BG V, Wang AZ. Emerging Nano-/Microapproaches for Cancer Immunotherapy. Adv Sci (Weinh) (2019) 6(6):1801847. doi: 10.1002/advs.201801847
37. Abdou P, Wang Z, Chen Q, Chan A, Zhou DR, Gunadhi V, et al. Advances in engineering local drug delivery systems for cancer immunotherapy. Wiley Interdiscip Rev Nanomed Nanobiotechnol (2020) 12(5):e1632. doi: 10.1002/wnan.1632
38. Cheung AS, Mooney DJ. Engineered Materials for Cancer Immunotherapy. Nano Today (2015) 10(4):511–31. doi: 10.1016/j.nantod.2015.06.007
39. Guasch J, Muth CA, Diemer J, Riahinezhad H, Spatz JP. Integrin-Assisted T-Cell Activation on Nanostructured Hydrogels. Nano Lett (2017) 17(10):6110–6. doi: 10.1021/acs.nanolett.7b02636
40. Zheng Y, Tang L, Mabardi L, Kumari S, Irvine DJ. Enhancing Adoptive Cell Therapy of Cancer through Targeted Delivery of Small-Molecule Immunomodulators to Internalizing or Noninternalizing Receptors. ACS Nano (2017) 11(3):3089–100. doi: 10.1021/acsnano.7b00078
41. Wayteck L, Xiong R, Braeckmans K, De Smedt SC, Raemdonck K. Comparing photoporation and nucleofection for delivery of small interfering RNA to cytotoxic T cells. J Control Release (2017) 267:154–62. doi: 10.1016/j.jconrel.2017.08.002
42. Moffett HF, Coon ME, Radtke S, Stephan SB, McKnight L, Lambert A, et al. Hit-and-run programming of therapeutic cytoreagents using mRNA nanocarriers. Nat Commun (2017) 8(1):389. doi: 10.1038/s41467-017-00505-8
43. Billingsley MM, Singh N, Ravikumar P, Zhang R, June CH, Mitchell MJ. Ionizable Lipid Nanoparticle-Mediated mRNA Delivery for Human CAR T Cell Engineering. Nano Lett (2020) 20(3):1578–89. doi: 10.1021/acs.nanolett.9b04246
44. Wolfl M, Greenberg PD. Antigen-specific activation and cytokine-facilitated expansion of naive, human CD8+ T cells. Nat Protoc (2014) 9(4):950–66. doi: 10.1038/nprot.2014.064
45. Castillo P, Wright KE, Kontoyiannis DP, Walsh T, Patel S, Chorvinsky E, et al. A New Method for Reactivating and Expanding T Cells Specific for Rhizopus oryzae. Mol Ther Methods Clin Dev (2018) 9:305–12. doi: 10.1016/j.omtm.2018.03.003
46. Neal LR, Bailey SR, Wyatt MM, Bowers JS, Majchrzak K, Nelson MH, et al. The Basics of Artificial Antigen Presenting Cells in T Cell-Based Cancer Immunotherapies. J Immunol Res Ther (2017) 2(1):68–79.
47. Fadel TR, Steenblock ER, Stern E, Li N, Wang X, Haller GL, et al. Enhanced cellular activation with single walled carbon nanotube bundles presenting antibody stimuli. Nano Lett (2008) 8(7):2070–6. doi: 10.1021/nl080332i
48. Fadel TR, Look M, Staffier PA, Haller GL, Pfefferle LD, Fahmy TM. Clustering of stimuli on single-walled carbon nanotube bundles enhances cellular activation. Langmuir (2010) 26(8):5645–54. doi: 10.1021/la902068z
49. Fadel TR, Sharp FA, Vudattu N, Ragheb R, Garyu J, Kim D, et al. A carbon nanotube-polymer composite for T-cell therapy. Nat Nanotechnol (2014) 9(8):639–47. doi: 10.1038/nnano.2014.154
50. Steenblock ER, Fahmy TM. A comprehensive platform for ex vivo T-cell expansion based on biodegradable polymeric artificial antigen-presenting cells. Mol Ther J Am Soc Gene Ther (2008) 16(4):765–72. doi: 10.1038/mt.2008.11
51. Perica K, Bieler JG, Schutz C, Varela JC, Douglass J, Skora A, et al. Enrichment and Expansion with Nanoscale Artificial Antigen Presenting Cells for Adoptive Immunotherapy. ACS Nano (2015) 9(7):6861–71. doi: 10.1021/acsnano.5b02829
52. Cheung AS, Zhang DKY, Koshy ST, Mooney DJ. Scaffolds that mimic antigen-presenting cells enable ex vivo expansion of primary T cells. Nat Biotechnol (2018) 36(2):160–9. doi: 10.1038/nbt.4047
53. Zhang Q, Wei W, Wang P, Zuo L, Li F, Xu J, et al. Biomimetic Magnetosomes as Versatile Artificial Antigen-Presenting Cells to Potentiate T-Cell-Based Anticancer Therapy. ACS Nano (2017) 11(11):10724–32. doi: 10.1021/acsnano.7b04955
54. Xia A, Zhang Y, Xu J, Yin T, Lu XJ. T Cell Dysfunction in Cancer Immunity and Immunotherapy. Front Immunol (2019) 10:1719. doi: 10.3389/fimmu.2019.01719
55. Shiao SL, Ganesan AP, Rugo HS, Coussens LM. Immune microenvironments in solid tumors: new targets for therapy. Genes Dev (2011) 25(24):2559–72. doi: 10.1101/gad.169029.111
56. Wang D, DuBois RN. Immunosuppression associated with chronic inflammation in the tumor microenvironment. Carcinogenesis (2015) 36(10):1085–93. doi: 10.1093/carcin/bgv123
57. Wu AA, Drake V, Huang HS, Chiu S, Zheng L. Reprogramming the tumor microenvironment: tumor-induced immunosuppressive factors paralyze T cells. Oncoimmunology (2015) 4(7):e1016700. doi: 10.1080/2162402X.2015.1016700
58. Croci DO, Zacarias Fluck MF, Rico MJ, Matar P, Rabinovich GA, Scharovsky OG. Dynamic cross-talk between tumor and immune cells in orchestrating the immunosuppressive network at the tumor microenvironment. Cancer Immunol Immunother (2007) 56(11):1687–700. doi: 10.1007/s00262-007-0343-y
59. Zheng Y, Stephan MT, Gai SA, Abraham W, Shearer A, Irvine DJ. In vivo targeting of adoptively transferred T-cells with antibody- and cytokine-conjugated liposomes. J Control Release (2013) 172(2):426–35. doi: 10.1016/j.jconrel.2013.05.037
60. Stephan MT, Moon JJ, Um SH, Bershteyn A, Irvine DJ. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat Med (2010) 16(9):1035–41. doi: 10.1038/nm.2198
61. Tang L, Zheng Y, Melo MB, Mabardi L, Castano AP, Xie YQ, et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat Biotechnol (2018) 36(8):707–16. doi: 10.1038/nbt.4181
62. Siriwon N, Kim YJ, Siegler E, Chen X, Rohrs JA, Liu Y, et al. CAR-T Cells Surface-Engineered with Drug-Encapsulated Nanoparticles Can Ameliorate Intratumoral T-cell Hypofunction. Cancer Immunol Res (2018) 6(7):812–24. doi: 10.1158/2326-6066.CIR-17-0502
63. Zhang F, Stephan SB, Ene CI, Smith TT, Holland EC, Stephan MT. Nanoparticles That Reshape the Tumor Milieu Create a Therapeutic Window for Effective T-cell Therapy in Solid Malignancies. Cancer Res (2018) 78(13):3718–30. doi: 10.1158/0008-5472.CAN-18-0306
64. Bai Y, Wang Y, Zhang X, Fu J, Xing X, Wang C, et al. Potential applications of nanoparticles for tumor microenvironment remodeling to ameliorate cancer immunotherapy. Int J Pharm (2019) 570:118636. doi: 10.1016/j.ijpharm.2019.118636
65. Chen Q, Hu Q, Dukhovlinova E, Chen G, Ahn S, Wang C, et al. Photothermal Therapy Promotes Tumor Infiltration and Antitumor Activity of CAR T Cells. Adv Mater (2019) 31(23):e1900192. doi: 10.1002/adma.201900192
66. Rastinehad AR, Anastos H, Wajswol E, Winoker JS, Sfakianos JP, Doppalapudi SK, et al. Gold nanoshell-localized photothermal ablation of prostate tumors in a clinical pilot device study. Proc Natl Acad Sci U S A (2019) 116(37):18590–6. doi: 10.1073/pnas.1906929116
67. Nie W, Wei W, Zuo L, Lv C, Zhang F, Lu GH, et al. Magnetic Nanoclusters Armed with Responsive PD-1 Antibody Synergistically Improved Adoptive T-Cell Therapy for Solid Tumors. ACS Nano (2019) 13(2):1469–78. doi: 10.1021/acsnano.8b07141
68. Taruno K, Kurita T, Kuwahata A, Yanagihara K, Enokido K, Katayose Y, et al. Multicenter clinical trial on sentinel lymph node biopsy using superparamagnetic iron oxide nanoparticles and a novel handheld magnetic probe. J Surg Oncol (2019) 120(8):1391–6. doi: 10.1002/jso.25747
69. Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji W, Reiman D, et al. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat Nanotechnol (2017) 12(8):813–20. doi: 10.1038/nnano.2017.57
70. Yang Y, Sun X, Xu J, Cui C, Safari Yazd H, Pan X, et al. Circular Bispecific Aptamer-Mediated Artificial Intercellular Recognition for Targeted T Cell Immunotherapy. ACS Nano (2020) 14(8):9562–71. doi: 10.1021/acsnano.9b09884
71. Burga RA, Patel S, Bollard CM, Cruz CRY, Fernandes R. Conjugating Prussian blue nanoparticles onto antigen-specific T cells as a combined nanoimmunotherapy. Nanomedicine (Lond) (2016) 11(14):1759–67. doi: 10.2217/nnm-2016-0160
72. Ma W, Zhu D, Li J, Chen X, Xie W, Jiang X, et al. Coating biomimetic nanoparticles with chimeric antigen receptor T cell-membrane provides high specificity for hepatocellular carcinoma photothermal therapy treatment. Theranostics (2020) 10(3):1281–95. doi: 10.7150/thno.40291
73. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Trans Med (2017) 9(399):eaaa0984. doi: 10.1126/scitranslmed.aaa0984
74. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol (2017) 3(8):1094–101. doi: 10.1001/jamaoncol.2017.0184
75. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin Cancer Res (2015) 21(18):4062–72. doi: 10.1158/1078-0432.CCR-15-0428
76. Long KB, Young RM, Boesteanu AC, Davis MM, Melenhorst JJ, Lacey SF, et al. CAR T Cell Therapy of Non-hematopoietic Malignancies: Detours on the Road to Clinical Success. Front Immunol (2018) 9:2740. doi: 10.3389/fimmu.2018.02740
77. Sampson JH, Gunn MD, Fecci PE, Ashley DM. Brain immunology and immunotherapy in brain tumours. Nat Rev Cancer (2020) 20(1):12–25. doi: 10.1038/s41568-019-0224-7
78. Ravanpay AC, Gust J, Johnson AJ, Rolczynski LS, Cecchini M, Chang CA, et al. EGFR806-CAR T cells selectively target a tumor-restricted EGFR epitope in glioblastoma. Oncotarget (2019) 10(66):7080–95. doi: 10.18632/oncotarget.27389
79. Masson F, Calzascia T, Di Berardino-Besson W, de Tribolet N, Dietrich PY, Walker PR. Brain microenvironment promotes the final functional maturation of tumor-specific effector CD8+ T cells. J Immunol (2007) 179(2):845–53. doi: 10.4049/jimmunol.179.2.845
80. Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature (2019) 565(7738):234–9. doi: 10.1038/s41586-018-0792-9
81. Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanovic S, Gouttefangeas C, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature (2019) 565(7738):240–5. doi: 10.1038/s41586-018-0810-y
Keywords: nanoparticles, nanotechnology, T cell therapy, immunoengineering, CNS tumors, solid tumors, artificial antigen-presenting cells, adoptive cell therapy
Citation: Balakrishnan PB and Sweeney EE (2021) Nanoparticles for Enhanced Adoptive T Cell Therapies and Future Perspectives for CNS Tumors. Front. Immunol. 12:600659. doi: 10.3389/fimmu.2021.600659
Received: 30 August 2020; Accepted: 08 March 2021;
Published: 23 March 2021.
Edited by:
Sujith K. Joseph, Baylor College of Medicine, United StatesReviewed by:
Amanda N. Rosewell Shaw, Baylor College of Medicine, United StatesSai Arun Batra, The Scripps Research Institute, United States
Copyright © 2021 Balakrishnan and Sweeney. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elizabeth E. Sweeney, bGl6aWVAZ3d1LmVkdQ==