 Sophie Liot1†
Sophie Liot1† Jonathan Balas1†
Jonathan Balas1† Alexandre Aubert1
Alexandre Aubert1 Laura Prigent1
Laura Prigent1 Perrine Mercier-Gouy1
Perrine Mercier-Gouy1 Bernard Verrier1
Bernard Verrier1 Philippe Bertolino2
Philippe Bertolino2 Ana Hennino2
Ana Hennino2 Ulrich Valcourt1
Ulrich Valcourt1 Elise Lambert1*
Elise Lambert1*- 1Laboratoire de Biologie Tissulaire et Ingénierie Thérapeutique (LBTI), UMR CNRS 5305, Université Lyon 1, Institut de Biologie et Chimie des Protéines, Lyon, France
- 2Cancer Research Center of Lyon, UMR INSERM 1052, CNRS 5286, Lyon, France
Pancreatic cancer is the seventh leading cause of cancer-related deaths worldwide and is predicted to become second in 2030 in industrialized countries if no therapeutic progress is made. Among the different types of pancreatic cancers, Pancreatic Ductal Adenocarcinoma (PDAC) is by far the most represented one with an occurrence of more than 90%. This specific cancer is a devastating malignancy with an extremely poor prognosis, as shown by the 5-years survival rate of 2–9%, ranking firmly last amongst all cancer sites in terms of prognostic outcomes for patients. Pancreatic tumors progress with few specific symptoms and are thus at an advanced stage at diagnosis in most patients. This malignancy is characterized by an extremely dense stroma deposition around lesions, accompanied by tissue hypovascularization and a profound immune suppression. Altogether, these combined features make access to cancer cells almost impossible for conventional chemotherapeutics and new immunotherapeutic agents, thus contributing to the fatal outcomes of the disease. Initially ignored, the Tumor MicroEnvironment (TME) is now the subject of intensive research related to PDAC treatment and could contain new therapeutic targets. In this review, we will summarize the current state of knowledge in the field by focusing on TME composition to understand how this specific compartment could influence tumor progression and resistance to therapies. Attention will be paid to Tenascin-C, a matrix glycoprotein commonly upregulated during cancer that participates to PDAC progression and thus contributes to poor prognosis.
Introduction
Pancreatic cancer is relatively rare and represents 2.5% of all cancers worldwide in 2018 (1). However, the fatal outcome of this disease is almost inevitable which consequently ranks this cancer site as the most devastating one. This poor survival is mainly inherent to the fact that this cancer evolves with few specific symptoms and is therefore mostly diagnosed at an advanced stage when the cancer presents a very aggressive behavior (4). Upon cancer detection, resection is possible in 10–20% of the cases, depending on tumor stage, and localization. Before or after surgery, or for unresectable tumors, various treatments including chemotherapeutic agents (gemcitabine, nab-paclitaxel, 5-fluorouacil, or FOLFIRINOX) and radiotherapy are generally used, but demonstrate little improvement of patient survival (4–6). Therefore, the discovery of new therapeutics and/or earlier detection of the disease before the onset of signs and symptoms is mandatory to improve patient survival rate.
Acinar cells are the predominant cell type in the pancreas and present an intrinsic plasticity enabling them to perform metaplasia to ductal-like cells. This metaplastic process called acinar-to-ductal metaplasia (ADM), is observed during acute and chronic pancreatitis and may represent the initial step toward the formation of pancreatic intraepithelial neoplasia (PanIN), which may then progress to PDAC. PanIN lesions are classified in different grades, from PanIN1A to PanIN3, characterized by the evolution of epithelial cell morphology (Figure 1).
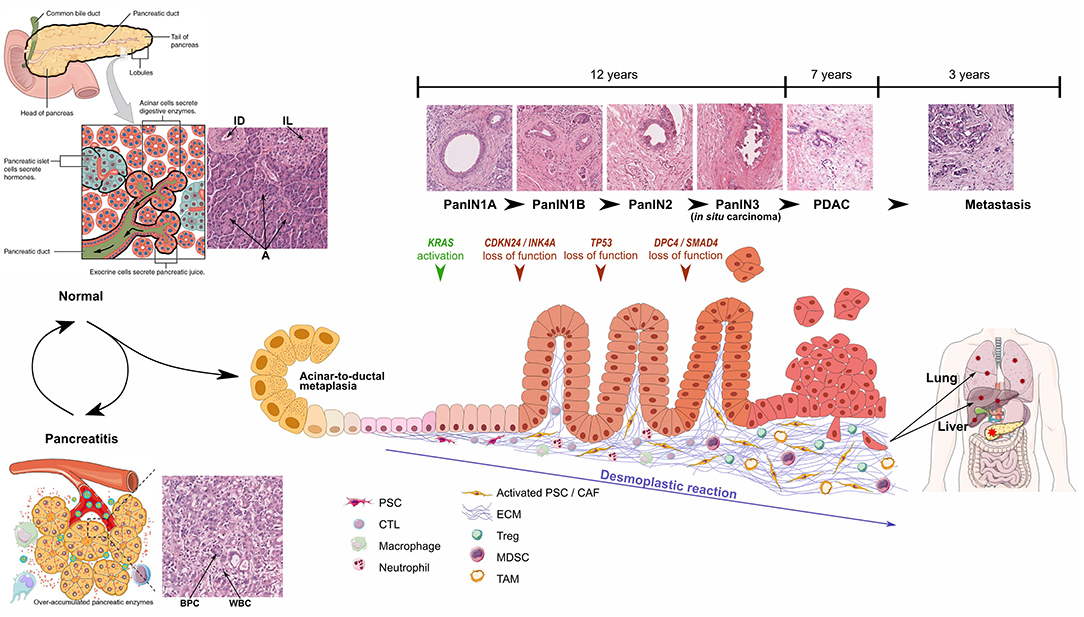
Figure 1. Schematic representations and optical microscopic images of normal pancreas, pancreatitis, and progression from PanIN1 to invasive PDAC. Particular focus is made on development of desmoplastic stroma. Pancreatitis schematic representation from Yao et al. (2), PanIN progression inspired from Morris et al. (3). ID, Interlobular Duct; IL, Islet of Langerhans; A, Acini; BPC, Benign Pancreatic Cells; WBC, White Blood Cells. Healthy pancreas scheme has been obtained from OpenStax College - Anatomy & Physiology. http://cnx.org/content/col11496/1.6/.
Pancreatic carcinogenesis is a multi-stage process resulting primarily from the accumulation of genetic alterations (average of 63 mutations per patient) in the somatic DNA of normal cells as well as inherited mutations (7). Among the numerous referenced alterations, KRAS, CDKN2A, TP53, and SMAD4 are the four most frequently mutated genes. KRAS proto-oncogene mutations have been detected in 92% of PDAC and are already detectable in precursor lesions, including early preinvasive intraepithelial neoplasia. Interestingly, SMAD4 mutations are associated with tumor size, lymphatic invasion, and metastasis and no survival at 5 years (8).
Besides the dramatic modifications in epithelial tissue morphology and genome, PDAC formation is also characterized by the desmoplastic reaction induced by tumor cells, which corresponds to a profound modification of the connective tissue through (1) recruitment and activation of specific fibroblasts and (4) intense ECM deposition. Initially corresponding to around 5% of pancreas mass, the connective tissue thus largely develops up to 90% of tumor area (60% on average) (9). Changes in stroma composition also lead to modifications in local immune system and vascularization, which dramatically influence prognosis (10, 11). However, the TME also contains anti-tumor components, which could explain why strategies depleting connective tissue cells have been so ineffective or even deleterious (12, 13).
The purpose of this review will thus be to describe the changes (1) in the cellular composition of the TME, as well as (4) in the ECM composition by attempting to identify which proteins have a potential pro- or anti-tumoral role with the ultimate aim of bringing out new therapeutic targets.
Cellular Composition of the Stroma
Cancer-Associated Fibroblasts
Among the various PDAC stromal cell types, the Cancer-Associated Fibroblasts (CAFs) are the most abundant. The CAF population presents a high heterogeneity and diversity of functions, presumably due to the multiple origins of these cells (14). Indeed, they can originate from tissue-resident fibroblasts that are activated under the control of growth factors such as TGFβ or following genetic mutations such as TP53 or PTEN (15). Another cellular origin of CAFs, and probably the most frequent one, is the Pancreatic Stellate Cells (PSCs) (16). PSC activation occurs following pancreatic injury, or upon PDGF or TGFβ stimulation, and leads to (1) morphological changes from a star-like shape into spindle-like cells, (4) loss of vitamin-A droplets and (5) increase in cell nucleus volume (17–19). CAFs may also derive from the recruitment and differentiation of bone marrow-derived mesenchymal stem cells or from the trans-differentiation of non-fibroblastic lineages such as adipocytes or epithelial cells (12, 20, 21). Various markers can be used to distinguish the multiple subsets of CAFs, such as PDGF-receptor α and β (PDGFRα/β), α-SMA, FAP, and S100A4 (Ca2+-binding protein), but none of them is exclusively expressed by CAFs, further highlighting CAF heterogeneity (21, 22). CAFs are responsible for the deposition of a dense tumor stroma, which subsequently can function as a physical barrier against immune infiltration or as a structural scaffold for cell interactions. In addition, CAFs secrete MMPs, which consequently ensure ECM degradation and the subsequent release of various factors leading to the recruitment of specific cells and/or cell dissemination. Finally, CAFs also produce many growth factors and proinflammatory cytokines such as TGFβ, vascular endothelial growth factor (VEGF), interleukin-6 (IL-6) and CXC-chemokine ligand 12 (CXCL12), thereby promoting tumor growth, angiogenesis and recruitment of immunosuppressive cells into the TME to assist in immune evasion (20, 23). In an effort to depict the fibroblast heterogeneity, 3 different CAF subpopulations have been identified according to their function or gene signature: the “inflammatory,” “myofibroblastic” and “antigen-presenting” CAFs (24, 25).
Endothelial Cells
Despite the obvious production of pro-angiogenic factors by CAFs and pancreatic tumor cells, PDAC is characterized by a low microvascular density compared to other types of cancers (11). Indeed, the dense fibrotic stroma forms a physical barrier that inhibits the formation and the proper functioning of vasculature, resulting in sparse constricted blood and lymphatic vessels that are only partially functional and physically separated from the cancer cells. This feature is deleterious for patient survival since low vascularity is associated with poor patient survival due to poor anti-cancer immune cell infiltration and chemotherapeutic drug delivery (26). Consequently, addition of anti-angiogenic drug (bevacizumab) to standard chemotherapy demonstrated no improvement in PDAC outcome (27). On contrary, vascular normalization aiming at improving drug delivery could be a good strategy for this type of carcinoma (27).
Infiltrating Immune Cells
Chronic pancreatitis is a risk factor for the development of PDAC as well as of systemic diseases characterized by chronic low-grade inflammation, such as metaflammation in patients with the metabolic syndrome or diabetes (28, 29). Interestingly, chronic pancreatitis and PDAC tissues show similarities in their desmoplasia and inflammatory infiltrates, indicating overlapping inflammatory responses.
The prevention and elimination of cancer cells are dependent on the immune system around the tumor. The PDAC immune microenvironment is characterized by (1) the exhaustion of anti-cancer immune cytotoxic T lymphocytes notably due to high mechanical constraints within the tumor and (4) the infiltration of multiple types of tumor-promoting immune cells, including myeloid-derived suppressor cells, tumor-associated macrophages and regulatory T cells (10, 30). Those tumor-promoting immune cells, in combination with CAFs and cancer cells secrete various pro-inflammatory cytokines such as TGFβ, TNFα, and different interleukins which subsequently favor immune evasion, PDAC development and metastasis formation (31). Various strategies are currently developed to treat PDAC by restoring proper immune system function: enzymatic digestion of TME, vascular normalization and neutralization of immune system modulators (21).
ECM Evolution During Pancreatic Carcinogenesis
PDAC is characterized by an intense desmoplastic reaction, defined as the fibrotic response of healthy tissue to invasive carcinoma and consisting of an abnormal accumulation of ECM components, mostly collagen fibers (32). This new TME acts as a physical barrier preventing (1) proper angiogenesis, and subsequent drug delivery, and (4) anti-cancer immune infiltration (33, 34). Consequently, TME has been considered as deleterious for patient prognosis and CAFs, which are responsible for dense ECM deposition, have been the target of clinical trials. However, CAF depletion resulted in an apparent paradoxical accelerated disease progression and encouraged a more detailed analysis of the most differentially regulated ECM components in the pancreatic tumors vs. healthy tissue, in order to identify new therapeutic targets within the TME (35, 36). We hereafter describe these matrix components according to the matrisome classification [Table 1; (37)].
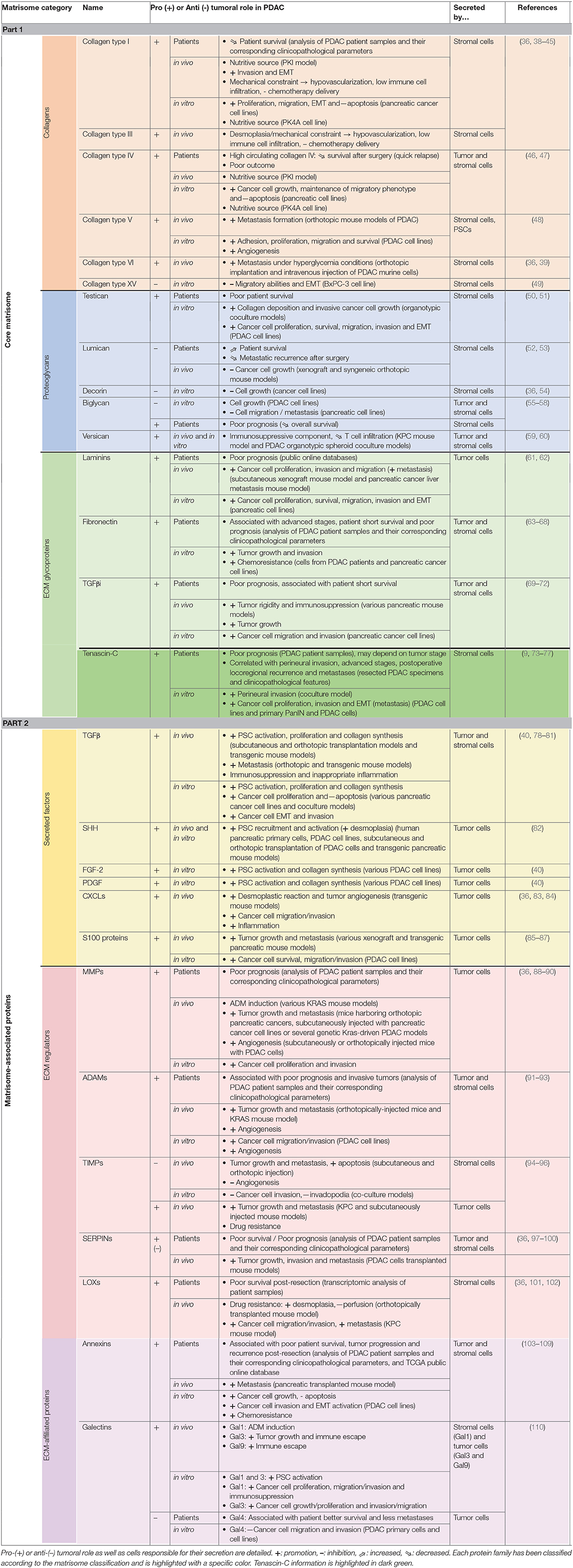
Table 1. ECM proteins involved in PDAC, presented according to the matrisome classification..
Core Matrisome
Collagens
Collagens are by far the most represented constituents of the connective tissue of normal and pathological pancreas (>90% of ECM proteins), with the type I and III fibrillar collagens accounting for >90% of all collagen mass (36, 38). Protein level for those collagens increases 2.6-fold during pancreatic tumor progression, which explains desmoplasia and justifies them as crucial targets. Additionally, the stroma undergoes intense rearrangement, leading to highly aligned collagen fibers, associated with bad prognosis for patients following pancreatic cancer resection (111). Despite their increased deposition, no ratio variation is observed for type I and III collagens between healthy and PDAC connective tissues, thus encouraging attention to other collagens differentially expressed during pancreatic carcinogenesis (37). Among them, type IV, V, VI, VII, XII, XIV, and XV collagens are key players in pancreatic tumorigenesis and act either as beneficial or detrimental molecules. For instance, collagen IV, which is an essential constituent of the basement membrane (BM), is produced by cancer cells, favors cancer cell growth, migration and protect them from apoptosis. Consequently, high serum level of collagen IV is associated with quick relapse after surgery and thus poor survival (46, 47). On contrary, another BM component, collagen XV, is lost during pancreatic tumorigenesis and its overexpression reduces the migratory abilities of PDAC cells in type I collagen-rich matrices (49). Interestingly, collagen VI is highly expressed during PDAC progression, induces metastatic colonization particularly in a hyperglycemic context and could therefore be targeted especially in diabetic patients (36, 39).
Besides their architectural and signaling role enabling tumor progression, collagens also serve as a nutritive source. Indeed, under PDAC specific conditions low in oxygen and nutrients, tumor cells metabolize collagen molecules, and thus collagen-derived proline enables PDAC cell proliferation (112). Therefore, this could explain the correlation between high serum collagen fragment levels in serum and significantly shorter overall survival, and prompts detailed analysis of collagen fragment role during PDAC progression (113).
Proteoglycans
Proteoglycans consist of one or more glycosaminoglycan (GAG) chain(s)—representing around 85% of the molecule mass—covalently attached to a core protein, and are categorized depending of their GAG chain nature and size (114). Among the small proteoglycans which are mainly expressed by TME, testican acts as a pro-tumoral molecule by affecting collagen deposition and thus favoring tumor cell growth and invasion, therefore leading to a poor patient survival, whereas lumican interferes with tumor progression and is associated with prolonged patient survival by limiting cancer cell growth and metastasis (50, 52, 53). Decorin is also considered as an anti-tumoral constituent by reducing tumor cell growth (36, 54) whereas the pro- or anti-tumoral status of biglycan in PDAC is still under debate. Indeed, its expression by stromal and epithelial cells (1) is induced by TGFβ, (4) has been described to inhibit pancreatic cancer cell growth and migration (55–57), but (5) is associated with poor prognosis (58). Finally, versican, which corresponds to a large proteoglycan expressed by both stromal and epithelial cells in PDAC, acts as an immunosuppressive component by reducing T cell infiltration and is thus considered as a deleterious molecule for patient survival (59).
Glycoproteins
Laminins are a family of ECM glycoproteins representing the major non-collagenous constituent in BM. Most of their subunits are over-expressed in PDAC and associated with poor outcome for patient survival (61). Fibronectin (FN1), which supports cell-ECM interactions, is essential for wound healing, development, and tissue homeostasis under physiological context. FN1 is also upregulated in PDAC which leads to tumor growth, invasion and metastasis formation and is consequently associated with poor prognosis in PDAC patients (63).
Transforming Growth Factor beta-induced (TGFβi) protein, also named βig-h3, is able to modulate cell adhesion through various integrins, including αvβ3, α1β1, and αvβ5.This glycoprotein is increased during pancreatic cancer and acts either directly on tumor CD8+ T cells by reducing their proliferation and activation or on tumor cells by promoting their migration and invasion (69, 115). TGFβi could thus be regarded for its double therapeutic potential to increase local anti-tumor immunity and subsequently induce cancer cell apoptosis or inhibit metastasis (69–72).
Among the four members of the Tenascin (TN) family, Tenascin-C (TNC) is by far the most well-characterized and is commonly described as being widely distributed in embryonic tissues, restricted in some adult tissues, such as stem cell niches and tendons (116), and de novo re-expressed during physio-pathological contexts such as wound healing and tumor progression (117). In PDAC, TNC protein is restrained to the tumor stroma and is not found in epithelial tumor cells or adjacent normal pancreatic tissue (9). High TNC expression, and downstream signaling through the Annexin II receptor, have been initially correlated with poor prognosis but this association is still controversial and could depend on the stage and grade of the pancreatic tumor or the specific location of TNC (9, 73, 118). So, high perineural TNC expression is associated with perineural invasion and poor prognosis with high loco-regional recurrence (74). In the same line, a recent study highlighted TNC as a prominent protein found in exosomal compartment and associated with local invasion and distant metastasis (75).
We recently demonstrated TNXB gene and TNX protein were significantly downregulated in the six cancers with the highest incidence and mortality worldwide (i.e., lung, breast, prostate, stomach, colorectal, and liver carcinomas) and low TNX levels were associated with poor prognosis in patients suffering from lung and breast carcinomas (119). In the same study, TNX protein expression was also decreased in tumor samples from PDAC patients (119).
Matrisome-Associated Proteins
Secreted Factors
In pancreatic cancer, Various Factors Are Mainly produced by cancer cells to favor tumor progression. Among them, TGFβ role is complex and mediates both pro- and anti-tumoral activities in cancer cells depending on their context, in space and time and their microenvironment. Indeed, in normal pancreatic cells and at early stages of pancreatic carcinogenesis, TGFβ exerts a tumor suppressive effect through SMAD4-regulated genes. However, in the late phase, SMAD4 is inactivated whereas TGFβ expression is upregulated leading to PI3K/Akt, Ras/ERK, p38MAPK, and Rho/GTPase pathway activation and subsequent tumor progression (120). Then, TGFβ invariably induces (1) proliferation and survival of PDAC cells, (4) EMT, invasion, and metastasis, (5) production of a dense fibrotic stroma and (6) deregulation of the immune microenvironment toward immunosuppression and inappropriate inflammation. Thus, various promising pre-clinical and clinical trials have already evaluated the potential of TGFβ-targeting therapies, through TGFβ regulator (losartan), TGFβ neutralizing antibodies or TGFβ receptor inhibitors (78–81, 121, 122). Other signaling factors are secreted by pancreatic cancer cells, enabling PSC recruitment and activation, and subsequent desmoplastic response inducing collagen synthesis. Thus, Sonic HedgeHog (SHH), Fibroblast Growth Factor-2 (FGF-2) and Platelet-derived Growth Factor (PDGF) are overexpressed during PDAC and interfering with their signaling corresponds to valuable strategies for PDAC treatment (40, 82). However, clinical trials using IPI-926, vismodegib and sonidegib that target the hedgehog pathway have so far been disappointing (123). PDAC cells also overexpress several CXC ligands, which are involved in desmoplastic reaction, immune modulation and tumor angiogenesis (36, 124). Thus, blocking the CXCLs-CXCR2 axis improves survival in a PDAC developing mouse model by reducing cell invasion and inflammation and could be a therapeutic approach against PDAC progression (83, 125). Finally, proteomic analyses of ECM during PDAC progression demonstrated that various members of the S100 Ca2+-binding protein family, notably S100P and S100A4, are upregulated in this disease and their high levels are associated with poor prognosis, thus shedding light on their receptor, i.e., the Receptor for Advanced Glycation End products (RAGE) and the RAGE/S100 ligand axis as a promising therapeutic approach (85). Therefore, various S100 monoclonal antibodies, S100 protein inhibitors or RAGE antagonist have already demonstrated a reduction of tumor growth and metastasis formation in mouse models (85).
ECM Regulators
Many proteins overexpressed during pancreatic tumorigenesis are responsible for ECM remodeling and are therefore potential targets for pancreatic cancer treatment. Matrix MetalloProteinases (MMPs), A Disintegrin And Metalloproteinases (ADAMs), and A Disintegrin And Metalloproteinase with ThromboSpondin motifs (ADAMTSs) are zinc-dependent endopeptidases that are able to degrade all ECM proteins. Their activities are tightly regulated by proteolytic activation and inhibition via their natural inhibitors, Tissue Inhibitors of MetalloProteinases (TIMPs) (126). The imbalance between the expression of metalloproteinases and TIMPs is thus of crucial interest in cancer development and metastasis (127). With some exceptions, those proteases are overexpressed during pancreatic cancer progression and are the targets of numerous pre-clinical and clinical trials, which for some of them were disappointing or less powerful than expected, probably due to (1) aspecific metalloproteinase targeting (use of broad-spectrum inhibitors) or (4) compensation mechanisms set up by tumor cells (88).
Several SERPIN family members are also importantly differentially regulated during PDAC development, mainly promoting tumor growth, invasion, and are associated with poor survival, but their activities have to be analyzed individually with particular attention paid to their original cells (36, 97–99). Finally, Lysyl Oxidases (LOX), a family of extracellular copper-dependent enzymes involved in ECM cross-linking, are also important matrix regulators over-represented during pancreatic tumorigenesis (36). Their inhibition in mouse model prolonged tumor-free survival by interfering with stroma stiffness (101, 102).
ECM-Affiliated Proteins
Among the ECM-affiliated proteins significantly deregulated during PDAC development, numerous members belong to the vertebrate “A subgroup” of the annexin superfamily coding a calcium- and membrane-binding protein (36). This subgroup consists of at least 12 members (A1-A11 and A13), all of which are suspected to be involved in tumor development (103). In PDAC, annexins are known to favor tumor cell growth, EMT, invasion and metastasis and to inhibit apoptosis. Additionally, they interact with various peri-cellular proteins such as S100 proteins and TNC, which are upregulated during PDAC progression. Therefore, annexin overexpression is associated with poor patient prognosis and could inspire new therapeutic strategies (128). Galectins, which are a family of carbohydrate-binding proteins, are also upregulated during PDAC progression (36, 110). Besides galectin-4, which has been described as a tumor suppressor by inhibiting tumor cell migration and invasion, the other galectins favor pancreatic tumor. Consequently, galectin inhibitors are considered as promising opportunities for pancreatic cancer therapeutic interventions, either alone or combined with current chemo- and/or immunotherapies (110).
Conclusions
During pancreatic tumorigenesis, important stromal modifications occur both at the cell landscape level and the matrix molecular composition in response to tumor signals. Herein, we have listed these major changes by focusing only on the proteins belonging to the matrisome. However, other extracellular components have not been underlined, but can drastically influence pancreatic tumor progression, as it is the case for hyaluronic acid (HA) (129–136). Indeed, in PDAC mouse model, HA deposition (1) was observed very early during tumor formation in an intralobular position in ADM regions and close to PanIN lesions and (4) preceded collagen deposition around lesions that will progress to PDAC (129). Besides making the ECM denser, HA deposition may be related to an inflammatory stage allowing the recruitment of immune cells in ADM areas, which further underlines the value of HA as a therapeutic target for PDAC treatment. Various drugs targeting HA have been developed such as pegylated hyaluronidase (PEGPH20) and Minnelide (132, 137). However, PEGPH20 in combination with conventional chemotherapies failed to demonstrate an improvement in median overall survival, leading to the recent discontinuation of PEGPH20 development after a phase 3 clinical trial. Minnelide, corresponding to an active substance extracted from thunder god vine is still under investigation and its mechanism of action seems multimodal (132, 138–140). Additionally, abnormal glycosylation of ECM components such as proteoglycans and glycoproteins can significantly influence tumor growth, neoplastic progression, metastasis and chemoresistance and thus should be considered for new drug design (141, 142). So far, despite promising results in preclinical models, no therapeutic strategy targeting the stroma compartment has brought conclusive results in clinical settings. This could be explained by differences in pharmacokinetics, pharmacodynamics and metabolism and the failure to accurately model the tumor microenvironment of patients using preclinical mouse models. However, a better understanding of the tumor stroma is expected to open up new possibilities for the development of new drugs.
Author Contributions
SL and JB: data curation, investigation and writing—original draft. AA, LP, and PM-G: writing—review and editing. BV: funding acquisition. PB and UV: writing—review and editing and funding acquisition. AH: investigation and writing—review and editing. EL: conceptualization, supervision, and writing—original draft. All authors: contributed to the article and approved the submitted version.
Funding
This work was supported by the Ligue Nationale contre le Cancer, Comité du Rhône, Comité de l'Allier, Comité de Savoie, and Comité de la Drôme, Comité Régional Auvergne-Rhône-Alpes et Saône-et-Loire (2019) as well as by the Fondation ARC pour la recherche sur le cancer (PJA 20141201790), Bristol Myers Squibb Foundation (2018–2019), and Inserm Transfert (2018–2020).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Piñeros M, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. (2019) 144:1941–53. doi: 10.1002/ijc.31937
2. Yao Q, Jiang X, Zhai Y-Y, Luo L-Z, Xu H-L, Xiao J, et al. Protective effects and mechanisms of bilirubin nanomedicine against acute pancreatitis. J ControlRelease. (2020) 322:312–25. doi: 10.1016/j.jconrel.2020.03.034
3. Morris JP, Wang SC, Hebrok M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer. (2010) 10:683–95. doi: 10.1038/nrc2899
4. Eibl ASG. Pancreatic Ductal Adenocarcinoma. Pancreapedia: The Exocrine Pancreas Knowledge Base. (2015). Available online at: /reviews/pancreatic-ductal-adenocarcinoma (accessed September 19, 2020).
5. Lambert A, Schwarz L, Borbath I, Henry A, Van Laethem J-L, Malka D, et al. An update on treatment options for pancreatic adenocarcinoma. Ther Adv Med Oncol. (2019) 11:1758835919875568. doi: 10.1177/1758835919875568
6. Oba A, Ho F, Bao QR, Al-Musawi MH, Schulick RD, Del Chiaro M. neoadjuvant treatment in pancreatic cancer. Front Oncol. (2020) 10:245. doi: 10.3389/fonc.2020.00245
7. Jones S, Zhang X, Parsons DW, Lin JC-H, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. (2008) 321:1801–6. doi: 10.1126/science.1164368
8. Oshima M, Okano K, Muraki S, Haba R, Maeba T, Suzuki Y, et al. Immunohistochemically detected expression of 3 major genes (CDKN2A/p16, TP53, and SMAD4/DPC4) strongly predicts survival in patients with resectable pancreatic cancer. Ann Surg. (2013) 258:336–46. doi: 10.1097/SLA.0b013e3182827a65
9. Leppänen J, Lindholm V, Isohookana J, Haapasaari K-M, Karihtala P, Lehenkari PP, et al. Tenascin C, Fibronectin, and tumor-stroma ratio in pancreatic ductal adenocarcinoma. Pancreas. (2019) 48:43–8. doi: 10.1097/MPA.0000000000001195
10. Li K-Y, Yuan J-L, Trafton D, Wang J-X, Niu N, Yuan C-H, et al. Pancreatic ductal adenocarcinoma immune microenvironment and immunotherapy prospects. Chronic Dis Transl Med. (2020) 6:6–17. doi: 10.1016/j.cdtm.2020.01.002
11. Longo V, Brunetti O, Gnoni A, Cascinu S, Gasparini G, Lorusso V, et al. Angiogenesis in pancreatic ductal adenocarcinoma: A controversial issue. Oncotarget. (2016) 7:58649–58. doi: 10.18632/oncotarget.10765
12. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. (2014) 25:735–47. doi: 10.1016/j.ccr.2014.04.021
13. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. (2014) 25:719–34. doi: 10.1016/j.ccr.2014.04.005
14. Öhlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med. (2014) 211:1503–23. doi: 10.1084/jem.20140692
15. Arina A, Idel C, Hyjek EM, Alegre M-L, Wang Y, Bindokas VP, et al. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proc Natl Acad Sci USA. (2016) 113:7551–6. doi: 10.1073/pnas.1600363113
16. Neuzillet C, Tijeras-Raballand A, Ragulan C, Cros J, Patil Y, Martinet M, et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J Pathol. (2019) 248:51–65. doi: 10.1002/path.5224
17. Xu Z, Pothula SP, Wilson JS, Apte MV. Pancreatic cancer and its stroma: a conspiracy theory. World J Gastroenterol. (2014) 20:11216–29. doi: 10.3748/wjg.v20.i32.11216
18. Moir JAG, Mann J, White SA. The role of pancreatic stellate cells in pancreatic cancer. Surgical Oncology. (2015) 24:232–8. doi: 10.1016/j.suronc.2015.05.002
19. Apte MV, Xu Z, Pothula S, Goldstein D, Pirola RC, Wilson JS. Pancreatic cancer: The microenvironment needs attention too!. Pancreatology. (2015). 15 (4. Suppl):S32–8. doi: 10.1016/j.pan.2015.02.013
20. Sun Q, Zhang B, Hu Q, Qin Y, Xu W, Liu W, et al. The impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer. Theranostics. (2018) 8:5072–87. doi: 10.7150/thno.26546
21. Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, et al. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol. (2019) 12:86. doi: 10.1186/s13045-019-0770-1
22. Norton J, Foster D, Chinta M, Titan A, Longaker M. Pancreatic cancer associated fibroblasts (caf): under-explored target for pancreatic cancer treatment. Cancers (Basel). (2020) 12:12051347. doi: 10.3390/cancers12051347
23. Huang T-X, Guan X-Y, Fu L. Therapeutic targeting of the crosstalk between cancer-associated fibroblasts and cancer stem cells. Am J Cancer Res. (2019) 9:1889–904.
24. Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. (2017) 214:579–96. doi: 10.1084/jem.20162024
25. Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. (2019) 9:1102–23. doi: 10.1158/2159-8290.CD-19-0094
26. Katsuta E, Qi Q, Peng X, Hochwald SN, Yan L, Takabe K. Pancreatic adenocarcinomas with mature blood vessels have better overall survival. Sci Rep. (2019) 9:1310. doi: 10.1038/s41598-018-37909-5
27. Li S, Xu H-X, Wu C-T, Wang W-Q, Jin W, Gao H-L, et al. Angiogenesis in pancreatic cancer: current research status and clinical implications. Angiogenesis. (2019) 22:15–36. doi: 10.1007/s10456-018-9645-2
28. Kirkegård J, Mortensen FV, Cronin-Fenton D. Chronic pancreatitis and pancreatic cancer risk: a systematic review and meta-analysis. Am J Gastroenterol. (2017) 112:1366–72. doi: 10.1038/ajg.2017.218
29. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. (2017) 542:177–85. doi: 10.1038/nature21363
30. Hosein AN, Brekken RA, Maitra A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol. (2020) 17:487–505. doi: 10.1038/s41575-020-0300-1
31. Padoan A, Plebani M, Basso D. Inflammation and pancreatic cancer: focus on metabolism, cytokines, and immunity. Int J Mol Sci. (2019) 20:20030676. doi: 10.3390/ijms20030676
32. Nielsen MFB, Mortensen MB, Detlefsen S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J Gastroenterol. (2016) 22:2678–700. doi: 10.3748/wjg.v22.i9.2678
33. Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. (2012) 18:4266–76. doi: 10.1158/1078-0432.CCR-11-3114
34. Whatcott CJ, Posner RG, Von Hoff DD, Han H. Desmoplasia and chemoresistance in pancreatic cancer. In: Grippo PJ, Munshi HG, editors. Pancreatic Cancer and Tumor Microenvironment [Internet]. Trivandrum (India): Transworld Research Network (2012) (accessws April 23, 2020). Available online at: http://www.ncbi.nlm.nih.gov/books/NBK98939/
35. Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Na Med. (2013) 19:1410–22. doi: 10.1038/nm.3389
36. Tian C, Clauser KR, Öhlund D, Rickelt S, Huang Y, Gupta M, et al. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc Natl Acad Sci USA. (2019) 116:19609–18. doi: 10.1073/pnas.1908626116
37. Naba A, Clauser KR, Ding H, Whittaker CA, Carr SA, Hynes RO. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. (2016) 49:10–24. doi: 10.1016/j.matbio.2015.06.003
38. Imamura T, Iguchi H, Manabe T, Ohshio G, Yoshimura T, Wang ZH, et al. Quantitative analysis of collagen and collagen subtypes I, III, and V in human pancreatic cancer, tumor-associated chronic pancreatitis, and alcoholic chronic pancreatitis. Pancreas. (1995) 11:357–64. doi: 10.1097/00006676-199511000-00007
39. Jian Z, Cheng T, Zhang Z, Raulefs S, Shi K, Steiger K, et al. Glycemic variability promotes both local invasion and metastatic colonization by pancreatic ductal adenocarcinoma. Cell Mol Gastroenterol Hepatol. (2018) 6:429–49. doi: 10.1016/j.jcmgh.2018.07.003
40. Bachem MG, Schünemann M, Ramadani M, Siech M, Beger H, Buck A, et al. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology. (2005) 128:907–21. doi: 10.1053/j.gastro.2004.12.036
41. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. (2012) 21:418–29. doi: 10.1016/j.ccr.2012.01.007
42. Whatcott CJ, Diep CH, Jiang P, Watanabe A, LoBello J, Sima C, et al. Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin Cancer Res. (2015) 21:3561–8. doi: 10.1158/1078-0432.CCR-14-1051
43. Weniger M, Honselmann KC, Liss AS. The extracellular matrix and pancreatic cancer: a complex relationship. Cancers. (2018) 10:316. doi: 10.3390/cancers10090316
44. Shintani Y, Hollingsworth MA, Wheelock MJ, Johnson KR. Collagen i promotes metastasis in pancreatic cancer by activating c-jun nh2-terminal kinase 1 and up-regulating n-cadherin expression. Cancer Res. (2006) 66:11745–53. doi: 10.1158/0008-5472.CAN-06-2322
45. Armstrong T, Packham G, Murphy LB, Bateman AC, Conti JA, Fine DR, et al. Type I collagen promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Clin Cancer Res. (2004) 10:7427–37. doi: 10.1158/1078-0432.CCR-03-0825
46. Ohlund D, Lundin C, Ardnor B, Oman M, Naredi P, Sund M. Type IV collagen is a tumour stroma-derived biomarker for pancreas cancer. Br J Cancer. (2009) 101:91–7. doi: 10.1038/sj.bjc.6605107
47. Öhlund D, Franklin O, Lundberg E, Lundin C, Sund M. Type IV collagen stimulates pancreatic cancer cell proliferation, migration, and inhibits apoptosis through an autocrine loop. BMC Cancer. (2013) 13:1–11. doi: 10.1186/1471-2407-13-154
48. Berchtold S, Grünwald B, Krüger A, Reithmeier A, Hähl T, Cheng T, et al. Collagen type V promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Cancer Lett. (2015) 356(2, Part B):721–32. doi: 10.1016/j.canlet.2014.10.020
49. Clementz AG, Mutolo MJ, Leir S-H, Morris KJ, Kucybala K, Harris H, et al. Collagen XV inhibits epithelial to mesenchymal transition in pancreatic adenocarcinoma cells. PLoS ONE. (2013) 8:e72250. doi: 10.1371/journal.pone.0072250
50. Veenstra VL, Damhofer H, Waasdorp C, Steins A, Kocher HM, Medema JP, et al. Stromal SPOCK1 supports invasive pancreatic cancer growth. Mol Oncol. (2017) 11:1050–64. doi: 10.1002/1878-0261.12073
51. Li J, Ke J, Fang J, Chen J-P. A potential prognostic marker and therapeutic target: SPOCK1 promotes the proliferation, metastasis, and apoptosis of pancreatic ductal adenocarcinoma cells. J Cell Biochem. (2020) 121:743–54. doi: 10.1002/jcb.29320
52. Li X, Truty MA, Kang Y, Chopin-Laly X, Zhang R, Roife D, et al. Extracellular lumican inhibits pancreatic cancer cell growth and is associated with prolonged survival after surgery. Clin Cancer Res. (2014) 20:6529–40. doi: 10.1158/1078-0432.CCR-14-0970
53. Li X, Kang Y, Roife D, Lee Y, Pratt M, Perez MR, et al. Prolonged exposure to extracellular lumican restrains pancreatic adenocarcinoma growth. Oncogene. (2017) 36:5432–8. doi: 10.1038/onc.2017.125
54. Köninger J, Giese NA, Mola di FF, Berberat P, Giese T, Esposito I, et al. Overexpressed decorin in pancreatic cancer: potential tumor growth inhibition and attenuation of chemotherapeutic action. Clin Cancer Res. (2004) 10:4776–83. doi: 10.1158/1078-0432.CCR-1190-03
55. Weber CK, Sommer G, Michl P, Fensterer H, Weimer M, Gansauge F, et al. Biglycan is overexpressed in pancreatic cancer and induces G1-arrest in pancreatic cancer cell lines. Gastroenterology. (2001) 121:657–67. doi: 10.1053/gast.2001.27222
56. Chen W-B, Lenschow W, Tiede K, Fischer JW, Kalthoff H, Ungefroren H. Smad4/DPC4-dependent regulation of biglycan gene expression by transforming growth factor-beta in pancreatic tumor cells. J Biol Chem. (2002) 277:36118–28. doi: 10.1074/jbc.M203709200
57. Otterbein H, Lehnert H, Ungefroren H. Negative control of cell migration by rac1b in highly metastatic pancreatic cancer cells is mediated by sequential induction of nonactivated smad3 and biglycan. Cancers (Basel). (2019) 11: 11121959. doi: 10.3390/cancers11121959
58. Aprile G, Avellini C, Reni M, Mazzer M, Foltran L, Rossi D, et al. Biglycan expression and clinical outcome in patients with pancreatic adenocarcinoma. Tumour Biol. (2013) 34:131–7. doi: 10.1007/s13277-012-0520-2
59. Rainiero HR, Emmerich PB, Sievers CK, Maloney CJ, Pitera RT, Payne SN, et al. Abstract 1904: Versican production is driven by both epithelial and stromal cells in pancreatic cancer. Cancer Res. (2019) 79(13 Supplement):1904–904. doi: 10.1158/1538-7445.AM2019-1904
60. Skandalis SS, Kletsas D, Kyriakopoulou D, Stavropoulos M, Theocharis DA. The greatly increased amounts of accumulated versican and decorin with specific post-translational modifications may be closely associated with the malignant phenotype of pancreatic cancer. Biochim Biophys Acta. (2006) 1760:1217–25. doi: 10.1016/j.bbagen.2006.03.021
61. Yang C, Liu Z, Zeng X, Wu Q, Liao X, Wang X, et al. Evaluation of the diagnostic ability of laminin gene family for pancreatic ductal adenocarcinoma. Aging (Albany NY). (2019) 11:3679–703. doi: 10.18632/aging.102007
62. Zhang H, Pan Y-Z, Cheung M, Cao M, Yu C, Chen L, et al. LAMB3 mediates apoptotic, proliferative, invasive, and metastatic behaviors in pancreatic cancer by regulating the PI3K/Akt signaling pathway. Cell Death Dis. (2019) 10:230. doi: 10.1038/s41419-019-1320-z
63. Hu D, Ansari D, Zhou Q, Sasor A, Said Hilmersson K, Andersson R. Stromal fibronectin expression in patients with resected pancreatic ductal adenocarcinoma. World J Surg Oncol. (2019) 17:29. doi: 10.1186/s12957-019-1574-z
64. Porter RL, Magnus NKC, Thapar V, Morris R, Szabolcs A, Neyaz A, et al. Epithelial to mesenchymal plasticity and differential response to therapies in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci USA. (2019) 16:26835–45. doi: 10.1073/pnas.1914915116
65. Hu D, Ansari D, Pawłowski K, Zhou Q, Sasor A, Welinder C, et al. Proteomic analyses identify prognostic biomarkers for pancreatic ductal adenocarcinoma. Oncotarget. (2018) 9:9789–807. doi: 10.18632/oncotarget.23929
66. Topalovski M, Brekken RA. Matrix control of pancreatic cancer: New insights into fibronectin signaling. Cancer Letters. (2016) 381:252–8. doi: 10.1016/j.canlet.2015.12.027
67. Hiroshima Y, Kasajima R, Kimura Y, Komura D, Ishikawa S, Ichikawa Y, et al. Novel targets identified by integrated cancer-stromal interactome analysis of pancreatic adenocarcinoma. Cancer Letters. (2020) 469:217–27. doi: 10.1016/j.canlet.2019.10.031
68. Amrutkar M, Aasrum M, Verbeke CS, Gladhaug IP. Secretion of fibronectin by human pancreatic stellate cells promotes chemoresistance to gemcitabine in pancreatic cancer cells. BMC Cancer. (2019) 19:596. doi: 10.1186/s12885-019-5803-1
69. Goehrig D, Nigri J, Samain R, Wu Z, Cappello P, Gabiane G, et al. Stromal protein βig-h3 reprogrammes tumour microenvironment in pancreatic cancer. Gut. (2019) 68:693–707. doi: 10.1136/gutjnl-2018-317570
70. Turtoi A, Musmeci D, Wang Y, Dumont B, Somja J, Bevilacqua G, et al. Identification of novel accessible proteins bearing diagnostic and therapeutic potential in human pancreatic ductal adenocarcinoma. J Proteome Res. (2011) 10:4302–13. doi: 10.1021/pr200527z
71. Sato T, Muramatsu T, Tanabe M, Inazawa J. Identification and characterization of transforming growth factor beta-induced in circulating tumor cell subline from pancreatic cancer cell line. Cancer Sci. (2018) 109:3623–33. doi: 10.1111/cas.13783
72. Costanza B, Rademaker G, Tiamiou A, De Tullio P, Leenders J, Blomme A, et al. Transforming growth factor beta-induced, an extracellular matrix interacting protein, enhances glycolysis and promotes pancreatic cancer cell migration. Int J Cancer. (2019) 145:1570–84. doi: 10.1002/ijc.32247
73. Yoneura N, Takano S, Yoshitomi H, Nakata Y, Shimazaki R, Kagawa S, et al. Expression of annexin II and stromal tenascin C promotes epithelial to mesenchymal transition and correlates with distant metastasis in pancreatic cancer. Int J Mol Med. (2018) 2:821–30. doi: 10.3892/ijmm.2018.3652
74. Furuhashi S, Sakaguchi T, Murakami T, Fukushima M, Morita Y, Ikegami K, et al. Tenascin C in the tumor-nerve microenvironment enhances perineural invasion and correlates with locoregional recurrence in pancreatic ductal adenocarcinoma. Pancreas. (2020) 49:442–54. doi: 10.1097/MPA.0000000000001506
75. Qian S, Tan X, Liu X, Liu P, Wu Y. Exosomal Tenascin-c induces proliferation and invasion of pancreatic cancer cells by WNT signaling. Onco Targets Ther. (2019) 12:3197–205. doi: 10.2147/OTT.S192218
76. Xu Y, Li Z, Jiang P, Wu G, Chen K, Zhang X, et al. The co-expression of MMP-9 and Tenascin-C is significantly associated with the progression and prognosis of pancreatic cancer. Diagn Pathol. (2015) 10:211. doi: 10.1186/s13000-015-0445-3
77. Cai J, Lu W, Du S, Guo Z, Wang H, Wei W, et al. Tenascin-C modulates cell cycle progression to enhance tumour cell proliferation through AKT/FOXO1 signalling in pancreatic cancer. Journal of Cancer. (2018) 9:4449–62. doi: 10.7150/jca.25926
78. Löhr M, Schmidt C, Ringel J, Kluth M, Müller P, Nizze H, et al. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. (2001) 61:550–5.
79. Neuzillet C, de Gramont A, Tijeras-Raballand A, de Mestier L, Cros J, Faivre S, et al. Perspectives of TGF-β inhibition in pancreatic and hepatocellular carcinomas. Oncotarget. (2013) 5:78–94. doi: 10.18632/oncotarget.1569
80. Shen W, Tao G, Zhang Y, Cai B, Sun J, Tian Z. TGF-β in pancreatic cancer initiation and progression: two sides of the same coin. Cell Biosci. (2017) 7:39. doi: 10.1186/s13578-017-0168-0
81. Principe DR, Park A, Dorman MJ, Kumar S, Viswakarma N, Rubin J, et al. TGFβ blockade augments pd-1 inhibition to promote t-cell-mediated regression of pancreatic cancer. Mol Cancer Ther. (2019) 18:613–20. doi: 10.1158/1535-7163.MCT-18-0850
82. Bailey JM, Swanson BJ, Hamada T, Eggers JP, Singh PK, Caffery T, et al. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res. (2008) 14:5995–6004. doi: 10.1158/1078-0432.CCR-08-0291
83. Sano M, Ijichi H, Takahashi R, Miyabayashi K, Fujiwara H, Yamada T, et al. Blocking CXCLs - CXCR2 axis in tumor-stromal interactions contributes to survival in a mouse model of pancreatic ductal adenocarcinoma through reduced cell invasion/migration and a shift of immune-inflammatory microenvironment. Oncogenesis. (2019) 8:1–12. doi: 10.1038/s41389-018-0117-8
84. Ijichi H, Chytil A, Gorska AE, Aakre ME, Bierie B, Tada M, et al. Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J Clin Invest. (2011) 121:4106–17. doi: 10.1172/JCI42754
85. Leclerc E, Vetter SW. The role of S100 proteins and their receptor RAGE in pancreatic cancer. Biochim Biophys Acta. (2015) 1852:2706–11. doi: 10.1016/j.bbadis.2015.09.022
86. Arumugam T, Ramachandran V, Sun D, Peng Z, Pal A, Maxwell DS, et al. Designing and developing S100P inhibitor 5-methyl cromolyn for pancreatic cancer therapy. Mol Cancer Ther. (2013) 12:654–62. doi: 10.1158/1535-7163.MCT-12-0771
87. Dakhel S, Padilla L, Adan J, Masa M, Martinez JM, Roque L, et al. S100P antibody-mediated therapy as a new promising strategy for the treatment of pancreatic cancer. Oncogenesis. (2014) 3:e92. doi: 10.1038/oncsis.2014.7
88. Slapak EJ, Duitman J, Tekin C, Bijlsma MF, Spek CA. Matrix metalloproteases in pancreatic ductal adenocarcinoma: key drivers of disease progression? Biology. (2020) 9:80. doi: 10.3390/biology9040080
89. Awasthi N, Mikels-Vigdal AJ, Stefanutti E, Schwarz MA, Monahan S, Smith V, et al. Therapeutic efficacy of anti-MMP9 antibody in combination with nab-paclitaxel-based chemotherapy in pre-clinical models of pancreatic cancer. J Cell Mol Med. (2019) 23:3878–87. doi: 10.1111/jcmm.14242
90. Rath N, Morton JP, Julian L, Helbig L, Kadir S, McGhee EJ, et al. ROCK signaling promotes collagen remodeling to facilitate invasive pancreatic ductal adenocarcinoma tumor cell growth. EMBO Mol Med. (2017) 9:198–218. doi: 10.15252/emmm.201606743
91. Oria VO, Lopatta P, Schmitz T, Preca B-T, Nyström A, Conrad C, et al. ADAM9 contributes to vascular invasion in pancreatic ductal adenocarcinoma. Mol Oncol. (2019) 13:456–79. doi: 10.1002/1878-0261.12426
92. Schlomann U, Koller G, Conrad C, Ferdous T, Golfi P, Garcia AM, et al. ADAM8 as a drug target in pancreatic cancer. Nat Commun. (2015) 6:6175. doi: 10.1038/ncomms7175
93. Veenstra VL, Damhofer H, Waasdorp C, van Rijssen LB, van de Vijver MJ, Dijk F, et al. ADAM12 is a circulating marker for stromal activation in pancreatic cancer and predicts response to chemotherapy. Oncogenesis. (2018) 7:87. doi: 10.1038/s41389-018-0096-9
94. Bloomston M, Shafii A, Zervos EE, Rosemurgy AS. TIMP-1 overexpression in pancreatic cancer attenuates tumor growth, decreases implantation and metastasis, and inhibits angiogenesis. J Surg Res. (2002) 102:39–44. doi: 10.1006/jsre.2001.6318
95. Benzing C, Lam H, Tsang CM, Rimmer A, Arroyo-Berdugo Y, Calle Y, et al. TIMP-2 secreted by monocyte-like cells is a potent suppressor of invadopodia formation in pancreatic cancer cells. BMC Cancer. (2019) 19:1214. doi: 10.1186/s12885-019-6429-z
96. D'Costa Z, Jones K, Azad A, van Stiphout R, Lim SY, Gomes AL, et al. Gemcitabine-Induced TIMP1 attenuates therapy response and promotes tumor growth and liver metastasis in pancreatic cancer. Cancer Res. (2017) 77:5952–62. doi: 10.1158/0008-5472.CAN-16-2833
97. Mardin WA, Ntalos D, Mees ST, Spieker T, Senninger N, Haier J, et al. SERPINB5 promoter hypomethylation differentiates pancreatic ductal adenocarcinoma from pancreatitis. Pancreas. (2016) 45:743–7. doi: 10.1097/MPA.0000000000000526
98. Harris NLE, Vennin C, Conway JRW, Vine KL, Pinese M, Cowley MJ, et al. SerpinB2 regulates stromal remodelling and local invasion in pancreatic cancer. Oncogene. (2017) 36:4288–98. doi: 10.1038/onc.2017.63
99. Tian C, Öhlund D, Rickelt S, Lidström T, Huang Y, Hao L, et al. Cancer cell-derived matrisome proteins promote metastasis in pancreatic ductal adenocarcinoma. Cancer Res. (2020) 80:1461–74. doi: 10.1158/0008-5472.CAN-19-2578
100. Bianconi D, Herac M, Spies D, Kieler M, Brettner R, Unseld M, et al. SERPINB7 expression predicts poor pancreatic cancer survival upon gemcitabine treatment. Transl Oncol. (2019) 12:15–23. doi: 10.1016/j.tranon.2018.08.019
101. Miller BW, Morton JP, Pinese M, Saturno G, Jamieson NB, McGhee E, et al. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol Med. (2015) 7:1063–76. doi: 10.15252/emmm.201404827
102. Le Calvé B, Griveau A, Vindrieux D, Maréchal R, Wiel C, Svrcek M, et al. Lysyl oxidase family activity promotes resistance of pancreatic ductal adenocarcinoma to chemotherapy by limiting the intratumoral anticancer drug distribution. Oncotarget. (2016) 7:32100–12. doi: 10.18632/oncotarget.8527
103. Mussunoor S, Murray GI. The role of annexins in tumour development and progression. J Pathol. (2008) 216:131–40. doi: 10.1002/path.2400
104. O'Sullivan D, Dowling P, Joyce H, McAuley E, McCann A, Henry M, et al. A novel inhibitory anti-invasive MAb isolated using phenotypic screening highlights AnxA6 as a functionally relevant target protein in pancreatic cancer. Br J Cancer. (2017) 117:1326–35. doi: 10.1038/bjc.2017.306
105. Karanjawala ZE, Illei PB, Ashfaq R, Infante JR, Murphy K, Pandey A, et al. New markers of pancreatic cancer identified through differential gene expression analyses: claudin 18 and annexin A8. Am J Surg Pathol. (2008) 32:188–96. doi: 10.1097/PAS.0b013e31815701f3
106. Zheng L, Foley K, Huang L, Leubner A, Mo G, Olino K, et al. Tyrosine 23 phosphorylation-dependent cell-surface localization of annexin A2 is required for invasion and metastases of pancreatic cancer. PLoS ONE. (2011) 6:e19390. doi: 10.1371/journal.pone.0019390
107. Takano S, Togawa A, Yoshitomi H, Shida T, Kimura F, Shimizu H, et al. Annexin II overexpression predicts rapid recurrence after surgery in pancreatic cancer patients undergoing gemcitabine-adjuvant chemotherapy. Ann Surg Oncol. (2008) 15:3157–68. doi: 10.1245/s10434-008-0061-5
108. Takahashi H, Katsuta E, Yan L, Dasgupta S, Takabe K. High expression of Annexin A2 is associated with DNA repair, metabolic alteration, and worse survival in pancreatic ductal adenocarcinoma. Surgery. (2019) 166:150–6. doi: 10.1016/j.surg.2019.04.011
109. Zhu J, Wu J, Pei X, Tan Z, Shi J, Lubman DM. Annexin A10 is a candidate marker associated with the progression of pancreatic precursor lesions to adenocarcinoma. PLoS ONE. (2017) 12:e0175039. doi: 10.1371/journal.pone.0175039
110. Manero-Rupérez N, Martínez-Bosch N, Barranco LE, Visa L, Navarro P. The galectin family as molecular targets: hopes for defeating pancreatic cancer. Cells. (2020) 9:689. doi: 10.3390/cells9030689
111. Drifka CR, Loeffler AG, Esquibel CR, Weber SM, Eliceiri KW, Kao WJ. Human pancreatic stellate cells modulate 3D collagen alignment to promote the migration of pancreatic ductal adenocarcinoma cells. Biomed Microdevices. (2016) 18:105. doi: 10.1007/s10544-016-0128-1
112. Olivares O, Mayers JR, Gouirand V, Torrence ME, Gicquel T, Borge L, et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat Commun. (2017) 8:16031. doi: 10.1038/ncomms16031
113. Willumsen N, Ali SM, Leitzel K, Drabick JJ, Yee N, Polimera HV, et al. Collagen fragments quantified in serum as measures of desmoplasia associate with survival outcome in patients with advanced pancreatic cancer. Sci Rep. (2019) 9:19761. doi: 10.1038/s41598-019-56268-3
114. Iozzo RV, Schaefer L. Proteoglycan form and function: a comprehensive nomenclature of proteoglycans. Matrix Biol. (2015) 42:11–55. doi: 10.1016/j.matbio.2015.02.003
115. Patry M, Teinturier R, Goehrig D, Zetu C, Ripoche D, Kim I-S, et al. βig-h3 Represses T-cell activation in type 1 diabetes. Diabetes. (2015). 64:4212–9. doi: 10.2337/db15-0638
116. Chiquet-Ehrismann R, Orend G, Chiquet M, Tucker RP, Midwood KS. Tenascins in stem cell niches. Matrix Biol. (2014) 37:112–23. doi: 10.1016/j.matbio.2014.01.007
117. Midwood KS, Chiquet M, Tucker RP, Orend G. Tenascin-C at a glance. J Cell Sci. (2016) 129:4321–7. doi: 10.1242/jcs.190546
118. Esposito I, Penzel R, Chaib-Harrireche M, Barcena U, Bergmann F, Riedl S, et al. Tenascin C and annexin II expression in the process of pancreatic carcinogenesis. J Pathol. (2006) 208:673–85. doi: 10.1002/path.1935
119. Liot S, Aubert A, Hervieu V, Kholti NE, Schalkwijk J, Verrier B, et al. Loss of Tenascin-X expression during tumor progression: A new pan-cancer marker. Matrix Biol Plus. (2020) 100021. doi: 10.1016/j.mbplus.2020.100021
120. Ahmed S, Bradshaw A-D, Gera S, Dewan MZ, Xu R. The TGF-β/smad4 signaling pathway in pancreatic carcinogenesis and its clinical significance. J Clin Med. (2017) 6:10005. doi: 10.3390/jcm6010005
121. Murphy JE, Wo JY, Ryan DP, Clark JW, Jiang W, Yeap BY, et al. Total Neoadjuvant therapy with folfirinox in combination with losartan followed by chemoradiotherapy for locally advanced pancreatic cancer. JAMA Oncol. (20190) 5:1020–7. doi: 10.1001/jamaoncol.2019.0892
122. Hauge A, Rofstad EK. Antifibrotic therapy to normalize the tumor microenvironment. J Transl Med. (2020) 18:207. doi: 10.1186/s12967-020-02376-y
123. Jiang B, Zhou L, Lu J, Wang Y, Liu C, You L, et al. Stroma-targeting therapy in pancreatic cancer: one coin with two sides? Front Oncol. (2020) 10:576399. doi: 10.3389/fonc.2020.576399
124. Susek KH, Karvouni M, Alici E, Lundqvist A. The role of CXC chemokine receptors 1-4 on immune cells in the tumor microenvironment. Front Immunol. (2018) 9:2159. doi: 10.3389/fimmu.2018.02159
125. Cheng Y, Ma X-L, Wei Y-Q, Wei X-W. Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochim Biophys Acta Rev Cancer. (2019) 1871:289–312. doi: 10.1016/j.bbcan.2019.01.005
126. Raeeszadeh-Sarmazdeh M, Coban M, Sankaran B, Radisky E. Engineering protein therapeutics for cancer based on the natural matrix metalloproteinase inhibitor TIMP-1. The FASEB Journal. (2020) 34:1. doi: 10.1096/fasebj.2020.34.s1.04889
127. Bramhall SR. The matrix metalloproteinases and their inhibitors in pancreatic cancer. From molecular science to a clinical application. Int J Pancreatol. (1997) 21:1–12.
128. Schloer S, Pajonczyk D, Rescher U. Annexins in translational research: hidden treasures to be found. Int J Mol Sci. (2018) 19:19061781. doi: 10.3390/ijms19061781
129. Johnson BL, Salazar M, d'Alincourt, Mackenzie-Dyck S, D'Apuzzo M, Shih HP, Manuel ER, et al. Desmoplasia and oncogene driven acinar-to-ductal metaplasia are concurrent events during acinar cell-derived pancreatic cancer initiation in young adult mice. PLoS ONE. (2019) 14:e0221810. doi: 10.1371/journal.pone.0221810
130. Sato N, Kohi S, Hirata K, Goggins M. Role of hyaluronan in pancreatic cancer biology and therapy: once again in the spotlight. Cancer Sci. (2016) 107:569–75. doi: 10.1111/cas.12913
131. Sato N, Cheng X-B, Kohi S, Koga A, Hirata K. Targeting hyaluronan for the treatment of pancreatic ductal adenocarcinoma. Acta Pharm Sin B. (2016) 6:101–5. doi: 10.1016/j.apsb.2016.01.002
132. Banerjee S, Modi S, McGinn O, Zhao X, Dudeja V, Ramakrishnan S, et al. Impaired synthesis of stromal components in response to minnelide improves vascular function, drug delivery, and survival in pancreatic cancer. Clin Cancer Res. (2016) 22:415–25. doi: 10.1158/1078-0432.CCR-15-1155
133. Nakazawa H, Yoshihara S, Kudo D, Morohashi H, Kakizaki I, Kon A, et al. 4-methylumbelliferone, a hyaluronan synthase suppressor, enhances the anticancer activity of gemcitabine in human pancreatic cancer cells. Cancer Chemother Pharmacol. (2006). 57:165–70. doi: 10.1007/s00280-005-0016-5
134. Cheng X-B, Sato N, Kohi S, Yamaguchi K. Prognostic impact of hyaluronan and its regulators in pancreatic ductal adenocarcinoma. PLoS ONE. (2013) 8:e80765. doi: 10.1371/journal.pone.0080765
135. van Mackelenbergh MG, Stroes CI, Spijker R, van Eijck CHJ, Wilmink JW, Bijlsma MF, et al. Clinical trials targeting the stroma in pancreatic cancer: a systematic review and meta-analysis. Cancers (Basel). (2019) 11:588. doi: 10.3390/cancers11050588
136. Ho WJ, Jaffee EM, Zheng L. The tumour microenvironment in pancreatic cancer - clinical challenges and opportunities. Nat Rev Clin Oncol. (2020) 17:527–40. doi: 10.1038/s41571-020-0363-5
137. Hingorani SR, Zheng L, Bullock AJ, Seery TE, Harris WP, Sigal DS, et al. HALO 202: randomized phase ii study of pegph20 plus nab-paclitaxel/gemcitabine versus nab-paclitaxel/gemcitabine in patients with untreated, metastatic pancreatic ductal adenocarcinoma. J Clin Oncol. (2018) 36:359–66. doi: 10.1200/JCO.2017.74.9564
138. Banerjee S, Saluja A. Minnelide, a novel drug for pancreatic and liver cancer. Pancreatology. (2015) 15(4 Suppl.):S39–43. doi: 10.1016/j.pan.2015.05.472
139. Dauer P, Nomura A, Saluja A, Banerjee S. Microenvironment in determining chemo-resistance in pancreatic cancer: Neighborhood matters. Pancreatology. (2017) 17:7–12. doi: 10.1016/j.pan.2016.12.010
140. Jentzsch V, Davis JAA, Djamgoz MBA. Pancreatic Cancer (PDAC): introduction of evidence-based complementary measures into integrative clinical management. Cancers (Basel). (2020) 12:3096. doi: 10.3390/cancers12113096
141. Pan S, Brentnall TA, Chen R. Proteome alterations in pancreatic ductal adenocarcinoma. Cancer Lett. (2020) 469:429–36. doi: 10.1016/j.canlet.2019.11.020
Keywords: pancreatic ductal adenocarcinoma, stroma, tumor microenvironment, extracellular matrix, tenascin
Citation: Liot S, Balas J, Aubert A, Prigent L, Mercier-Gouy P, Verrier B, Bertolino P, Hennino A, Valcourt U and Lambert E (2021) Stroma Involvement in Pancreatic Ductal Adenocarcinoma: An Overview Focusing on Extracellular Matrix Proteins. Front. Immunol. 12:612271. doi: 10.3389/fimmu.2021.612271
Received: 30 September 2020; Accepted: 23 February 2021;
Published: 06 April 2021.
Edited by:
Gertraud Orend, INSERM Immuno Rhumatologie Moléculaire (IRM), FranceReviewed by:
Rolf A. Brekken, University of Texas Southwestern Medical Center, United StatesI. Velázquez-Quesada, Temple University, United States
Copyright © 2021 Liot, Balas, Aubert, Prigent, Mercier-Gouy, Verrier, Bertolino, Hennino, Valcourt and Lambert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elise Lambert, ZWxpc2UubGFtYmVydEBpYmNwLmZy
†These authors have contributed equally to this work