 Amit Rawat1*†
Amit Rawat1*† Pandiarajan Vignesh1†
Pandiarajan Vignesh1† Murugan Sudhakar1‡
Murugan Sudhakar1‡ Madhubala Sharma1‡
Madhubala Sharma1‡ Deepti Suri1
Deepti Suri1 Ankur Jindal1
Ankur Jindal1 Anju Gupta1
Anju Gupta1 Jitendra Kumar Shandilya1
Jitendra Kumar Shandilya1 Sathish Kumar Loganathan1
Sathish Kumar Loganathan1 Gurjit Kaur1Sanchi Chawla1Pratap Kumar Patra1
Gurjit Kaur1Sanchi Chawla1Pratap Kumar Patra1 Alka Khadwal2
Alka Khadwal2 Biman Saikia3
Biman Saikia3 Ranjana Walker Minz3
Ranjana Walker Minz3 Vaishali Aggarwal1
Vaishali Aggarwal1 Prasad Taur4
Prasad Taur4 Ambreen Pandrowala4
Ambreen Pandrowala4 Vijaya Gowri4
Vijaya Gowri4 Mukesh Desai4
Mukesh Desai4 Manasi Kulkarni5
Manasi Kulkarni5 Gauri Hule5
Gauri Hule5 Umair Bargir5
Umair Bargir5 Priyanka Kambli5
Priyanka Kambli5 Manisha Madkaikar5
Manisha Madkaikar5 Sagar Bhattad6Chetan Ginigeri6Harish Kumar6Ananthvikas Jayaram7
Sagar Bhattad6Chetan Ginigeri6Harish Kumar6Ananthvikas Jayaram7 Deenadayalan Munirathnam8
Deenadayalan Munirathnam8 Meena Sivasankaran8
Meena Sivasankaran8 Revathi Raj9
Revathi Raj9 Ramya Uppuluri9Fouzia Na10
Ramya Uppuluri9Fouzia Na10 Biju George10Harsha Prasada Lashkari11
Biju George10Harsha Prasada Lashkari11 Manas Kalra12Anupam Sachdeva12Shishir Seth13Tapas Sabui14Aman Gupta15
Manas Kalra12Anupam Sachdeva12Shishir Seth13Tapas Sabui14Aman Gupta15 Karin van Leeuwen16
Karin van Leeuwen16 Martin de Boer16
Martin de Boer16 Koon Wing Chan17Kohsuke Imai18,19
Koon Wing Chan17Kohsuke Imai18,19 Osamu Ohara20
Osamu Ohara20 Shigeaki Nonoyama18
Shigeaki Nonoyama18 Yu Lung Lau17
Yu Lung Lau17 Surjit Singh1
Surjit Singh1- 1Allergy Immunology Unit, Advanced Pediatrics Centre, Department of Pediatrics, Post Graduate Institute of Medical Education and Research, Chandigarh, India
- 2Bone Marrow Transplantation Unit, Department of Internal Medicine, Post Graduate Institute of Medical Education and Research, Chandigarh, India
- 3Department of Immunopathology, Post Graduate Institute of Medical Education and Research, Chandigarh, India
- 4Department of Immunology, Bai Jerbai Wadia Hospital for Children, Mumbai, India
- 5ICMR—National Institute of Immunohaematology, Mumbai, India
- 6Department of Pediatrics, Aster CMI Hospital, Bengaluru, India
- 7Neuberg Anand Diagnostic and Research Centre, Bengaluru, India
- 8Department of Pediatric Hematology and Oncology, Kanchi Kamakoti Child Trust Hospital, Chennai, India
- 9Apollo Children’s Hospitals, Chennai, India
- 10Christian Medical College, Vellore, India
- 11Department of Pediatrics, Kasturba Medical College, Mangalore, India
- 12Sir Ganga Ram Hospital, Rajendra Nagar, New Delhi, India
- 13Apollo Cancer Institute, Indraprastha Apollo Hospitals, Savita Vihar, New Delhi, India
- 14R G Kar Medical College, Kolkata, India
- 15Department of Pediatric Rheumatology & Immunology, MEDENS Hospital, Panchkula, India
- 16Sanquin Research and Landsteiner Laboratory, Amsterdam Medical Center, University of Amsterdam, Amsterdam, Netherlands
- 17Department of Paediatrics and Adolescent Medicine, The University of Hong Kong, Hong Kong, China
- 18Department of Pediatrics, National Defense Medical College, Saitama, Japan
- 19Department of Community Pediatrics, Perinatal and Maternal Medicine, Tokyo Medical and Dental University, Tokyo, Japan
- 20Kazusa DNA Research Institute, Chiba, Japan
Background: Chronic granulomatous disease (CGD) is an inherited defect in phagocytic respiratory burst that results in severe and life-threatening infections in affected children. Single center studies from India have shown that proportion of autosomal recessive (AR) CGD is more than that reported from the West. Further, affected patients have high mortality rates due to late referrals and difficulties in accessing appropriate treatment. However, there is lack of multicentric collaborative data on CGD from India.
Objective: To describe infection patterns, immunological, and molecular features of CGD from multiple centers in India.
Methods: A detailed proforma that included clinical and laboratory details was prepared and sent to multiple centers in India that are involved in the care and management of patients with inborn errors of immunity. Twelve centers have provided data which were later pooled together and analyzed.
Results: Of the 236 patients analyzed in our study, X-linked and AR-CGD was seen in 77 and 97, respectively. Male female ratio was 172:64. Median age at onset of symptoms and diagnosis was 8 and 24 months, respectively. Common infections documented include pneumonia (71.6%), lymphadenitis (31.6%), skin and subcutaneous abscess (23.7%), blood-stream infection (13.6%), osteomyelitis (8.6%), liver abscess (7.2%), lung abscess (2.9%), meningoencephalitis (2.5%), splenic abscess (1.7%), and brain abscess (0.9%). Forty-four patients (18.6%) had evidence of mycobacterial infection. Results of molecular assay were available for 141 patients (59.7%)—CYBB (44.7%) gene defect was most common, followed by NCF1 (31.9%), NCF2 (14.9%), and CYBA (8.5%). While CYBA variants were documented only in Southern and Western parts of India, a common dinucleotide deletion in NCF2 (c.835_836delAC) was noted only in North Indian population. Of the 174 patients with available outcome data, 67 (38.5%) had expired. Hematopoietic stem cell transplantation was carried out in 23 patients, and 12 are doing well on follow-up.
Conclusions: In India, proportion of patients with AR-CGD is higher as compared to Western cohorts, though regional differences in types of AR-CGD exist. Clinical profile and mortality rates are similar in both X-linked and AR-CGD. However, this may be a reflection of the fact that milder forms of AR-CGD are probably being missed.
Introduction
Chronic granulomatous disease (CGD) is an inborn error of immunity (IEI) characterized by a defective respiratory burst in phagocytes, resulting in defective clearance of phagocytosed microorganisms (1). It is caused by mutations in genes encoding different protein subunits of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzyme complex (2). Clinical manifestations vary from an immunodeficient phenotype with repeated infections (3, 4) to those characterized by uncontrolled hyper-inflammation (5) and other autoimmune manifestations. Common patterns of infections in CGD include pneumonia, lymphadenitis, hepatosplenomegaly, and abscesses (6).
Laboratory diagnosis of CGD can be made by the nitroblue tetrazolium dye reduction test (NBT) (7, 8) or by flow-cytometry based dihydrorhodamine (DHR) assay (9, 10). The DHR assay is now considered to be the preferred screening test due to its higher reproducibility, sensitivity, rapidity, and ability to detect X linked carriers (11, 12). While several centers in India have published their individual experiences on CGD (13–16), countrywide data have never been collated before. Such collaborative efforts are the need of the hour and are especially important for uncommon conditions like CGD, for which there is paucity of data on disease burden in the country. The present work is the first multi-centric study in India to provide data on clinical, immunological, and molecular features of CGD.
Methods
We contacted all centers that are recognized as Foundation for Primary Immunodeficiency Diseases (FPID) regional centers for diagnosis or treatment for primary immunodeficiencies in India, and also other Indian institutions involved in care of patients with IEI. All centers were requested to provide clinical and laboratory details of patients with CGD on a pre-designed Excel sheet. Data collection was completed by July 2020. Details included demographic information, family history including consanguinity, clinical manifestations, immunological investigations, genetic diagnosis, treatment, and follow-up.
Participating institutes included—Postgraduate Institute of Medical Education and Research (PGIMER), Chandigarh, North India (80 patients); Bai Jerbai Wadia Hospital for Children, Mumbai, West India (57 patients); Aster CMI Hospital, Bengaluru, South India (22 patients); National Institute of Immunohematology (NIIH), Mumbai, West India (22 patients); Kanchi Kamakoti Childs Trust Hospital, Chennai, South India (21 patients); Apollo Hospitals, Chennai, South India (17 patients); Christian Medical College, Vellore, South India (five patients); Kasturba Medical College, Mangalore, South-West, India (five patients); Sir Ganga Ram Hospital (SGRH), New Delhi, North India (four patients) and one patient each from R G Kar Medical College, Kolkata, East India; Indraprastha Apollo Hospital, New Delhi, North India; and Medens Hospital, Haryana, North India. Data were collated and subsequently translated on to a database and analyzed (Figure 1). Before the analysis, all patient identity details were anonymized. Three (3) female patients had a probable skewed X-linked inactivation. These patients have been described separately.
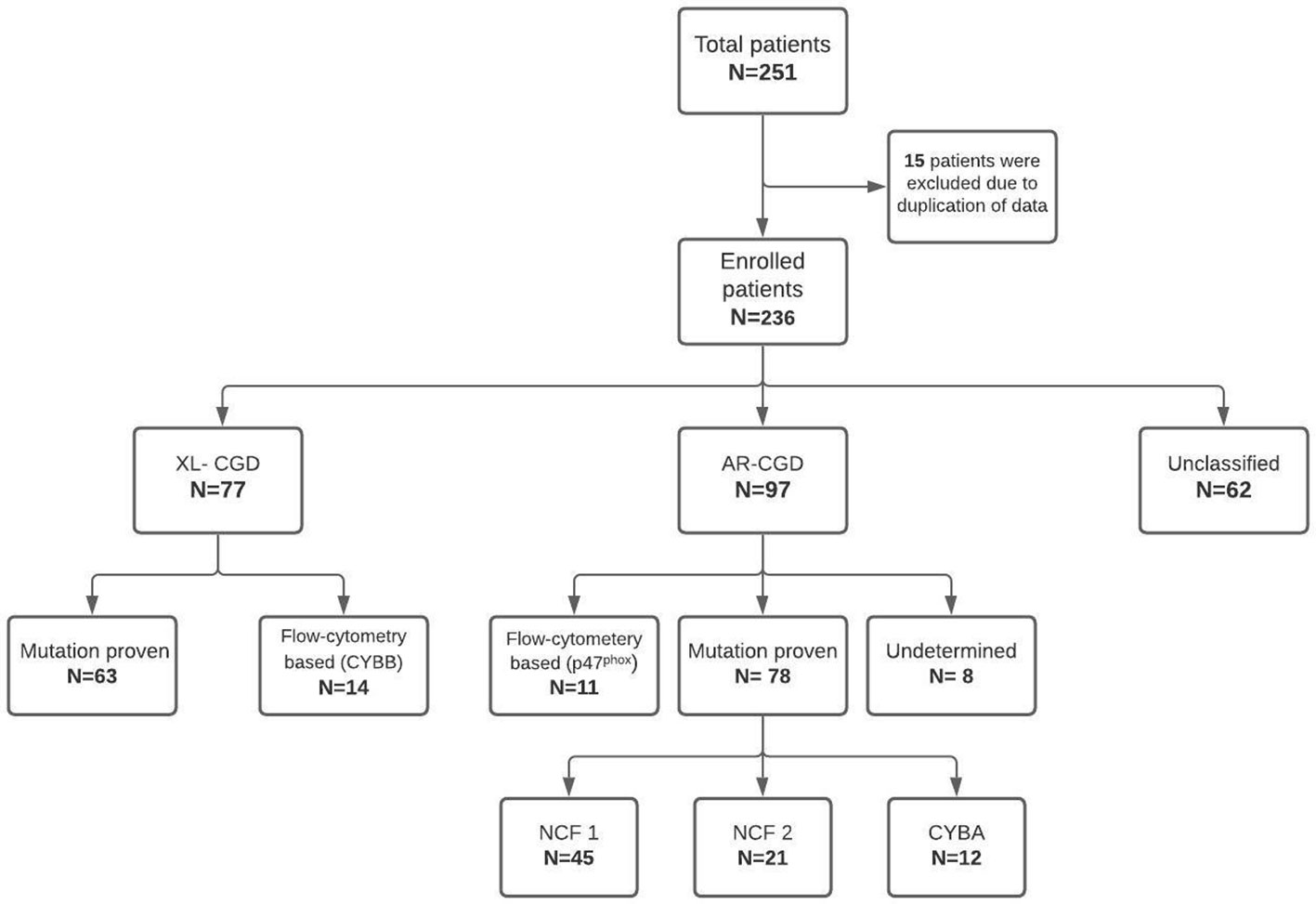
Figure 1 Flowchart showing the classification of patients analyzed in the current study.
Nitroblue Tetrazolium and Dihydro-Rhodamine Tests
Diagnosis of CGD was based on an abnormal granulocyte oxidative burst evaluated by either NBT or flow-cytometry based DHR assay or both. Nitroblue tetrazolium test was available in almost all the centers. However, flow cytometry based DHR was available only in six centers. Analysis of NADPH oxidase components by flow cytometry was carried out only in two centers: National Institute of Immunohematology (NIIH), Mumbai, and Post Graduate Institute of Medical Education and Research (PGIMER), Chandigarh. Details of flow cytometry methods for DHR and NADPH oxidase components at NIIH, Mumbai (13, 14) and PGIMER, Chandigarh (15–17) have been described previously.
Molecular Assays
Diagnosis was confirmed by molecular analysis in 60% of the patients. At most centers, molecular analysis was carried out at private laboratories (Medgenome Laboratories Pvt. Ltd., India; Strand Genomics Pvt. Ltd., India; Neuberg Anand Diagnostics Pvt. Ltd., India). Illumina platform was used for Next-Generation sequencing (NGS) in these private laboratories with coverage of >80×. Sanger sequencing was used to confirm variants obtained by Next-Generation Sequencing (NGS).
Two centers had in-house facilities for molecular analysis—NIIH, Mumbai and PGIMER, Chandigarh. Molecular assays carried out at NIIH, Mumbai have been detailed in a previous publication (18). Molecular analysis for some patients at PGIMER, Chandigarh (before 2016) was performed at centers in Hong Kong (Department of Pediatrics and Adolescent Medicine, The University of Hong Kong) and Japan (Kazusa DNA Research Institute, Chiba; Tokyo Medical and Dental University, Tokyo; National Defense Medical College, Saitama).
Sanger Sequencing and Next-Generation Sequencing at PGIMER, Chandigarh
Genomic DNA was extracted from peripheral blood using DNA extraction kits (QIAamp DNA Blood mini kit, Germany). Depending upon flow-cytometry evaluation for NADPH oxidase component expression, polymerase chain reaction (PCR) was performed for respective genes i.e. CYBB (13 exons) for X-linked CGD and NCF1 (11 Exons), NCF2 (16 Exons), CYBA (six Exons) for AR-CGD. PCR products from genomic DNA were sequenced on an auto fluorescent sequencer (ABI 3500, Applied Biosystems™; Thermo Fisher Scientific, USA) using BigDye™ Terminator (V3.1 Applied Biosystems™). Sequencing primers were the same as those used for PCR. Upon sequencing the results were obtained in.abi format and were analyzed using Codon-code aligner for DNA sequence assembly (4.2.5/2013). Patient sequence was compared with reference human sequence obtained from Ensemble database (https://asia.ensembl.org/index.html), and novel variants were further assessed for their disease-causing effect on protein. Prediction tools such as SIFT, PolyPhen-2, Mutation Taster, and CADD score were used to evaluate their pathogenicity. Human Splicing Finder tool was used for splice-site variants.
Since 2018, the center has started performing targeted NGS using a modest 44 gene panel for patients with primary immunodeficiency diseases. Genomic DNA was quantified using Qubit™ Fluorometer (ThermoFisher Scientific, USA) followed by target amplification using PID 2X 2-primer pool panel. Amplified products were partially digested followed by adapter ligation, barcoding, library purification, and amplification. Further, DNA fragments were immobilized on Ion sphere particles and clonally amplified using Ion One Touch™ 2 Instrument. This emulsion PCR results in beads containing amplified and cloned DNA fragment. Elimination of empty beads was carried out using a robotic enrichment system (Ion One Touch™ ES). Finally, the beads containing clonal population of DNA were loaded on to Ion530 Chip. Sequencing was done using Ion S5™ instrument and simultaneously processed on an Ion torrent server for further analysis. Variant calling and analysis were performed using Ion reporter software (ThermoFisher Scientific, USA). The identified variants were validated using Sanger sequencing.
Gene-Scan Analysis at PGIMER, Chandigarh
The most common defect in NCF1 is a dinucleotide deletion, c.75_76delGT at the beginning of exon 2. Conventional Sanger Sequencing or NGS can miss this defect as NCF1 has two flanking pseudogenes (ΨNCF1) with 99% sequence homology with functional gene. So, fluorochrome labeled primers were used to amplify NCF1 gene and ΨNCF1 using a method described earlier (Roos et al, 2001). The amplification yielded a mixture of labeled amplicons from the NCF1 gene and ΨNCF1 differing by 2 bp in length. This product mixture was analyzed on an Applied Biosystems 3500 Genetic Analyzer using GeneMapper software (Life Technologies, Carlsbad, CA, USA). The amplification product from the pseudogene, being shorter than that from the functional gene, had a shorter retention time and could thus be resolved as distinct peak from the one resulting from the amplicon from the functional gene. Ratio between the two peak heights denotes the relative number of genes and pseudogenes in a test sample.
Statistical Analysis
Descriptive tests such as medians and ranges were used for continuous variables. Counts and percentages were used for categorical variables. Mann–Whitney U test was used to compare continuous variables between groups. Categorical variables were compared by using the chi-square (x2) test or Fisher’s exact test wherever needed. Wilcoxon rank sum test was used to assess efficacy of intervention in a single group. Kaplan–Meier method was used to estimate the survival analysis. Death from any cause was considered as an event, and log-rank test was used to compare the groups. All p values were two-sided and considered significant when p <0.05.
Results
Patient Profile
Of the 236 patients, 172 (72.8%) were boys and 64 (27.1%) girls. Details about consanguinity were available for 91 patients, and 40 of them had a history of consanguineous marriage (43.9%). XL-CGD was seen in 77 patients, AR-CGD in 97 children, and the remaining 62 patients could not be categorized as X-linked or AR due to insufficient details of flow cytometry results or molecular analysis (Figure 1). AR-CGD was the commonest type in our cohort (XL: AR ratio, 0.8). Three female patients who had skewed X-linked inactivation have been described separately and were not included in the survival analysis.
Median age of onset of initial symptom was 8 months (interquartile range [IQR]: 3–24, range: 0.1 months–20 years), and median age of establishing the diagnosis was 24 months (IQR: 8–60 months, range: 2 weeks–35 years). Median delay in diagnosis was 8 months (IQR: 1.9 months–24 months, range: 0–33.2 years). Follow-up data were available for 98 patients, and median duration of follow-up was 1 year (IQR: 0.1–3 years, range: 1 month–24 years).
Comparison of Clinical and Laboratory Characteristics Between XL-CGD and AR-CGD
Median age at diagnosis was earlier in XL-CGD than AR-CGD (p = 0.002). However, there was no difference in median age of onset symptoms between these groups (p = 0.074) (Table 1). Similarly, median values of stimulation index (SI) derived from DHR assay were also comparable between both groups (p = 0.741) (Table 1).

Table 1 Comparison of clinical, and laboratory characteristics between XL-CGD, and AR-CGD.
Number of episodes of pneumonia was more in AR-CGD (n = 70) than XL-CGD (n = 45) (p = 0.003). Number of patients with >three episodes of pneumonia was also higher in AR-type as compared to XL-CGD (p 0.007). There was no statistical difference between AR-CGD and XL-CGD with regard to following parameters—episodes of superficial abscess (p = 0.462); deep abscess (p = 0.488); lymphadenitis (p = 0.272); and osteomyelitis (p = 0.510). Mortality rate was also comparable in both groups (p = 0.489) (Table 1).
On comparing the individual subtypes (CYBB, NCF1, NCF2, and CYBA defects), age of onset of symptoms and age at diagnosis were earlier in XL-CGD compared to other subtypes (Table 2). Mortality rates were low in NCF1 defect (13.3%) compared to other forms (p 0.040) (Table 2).
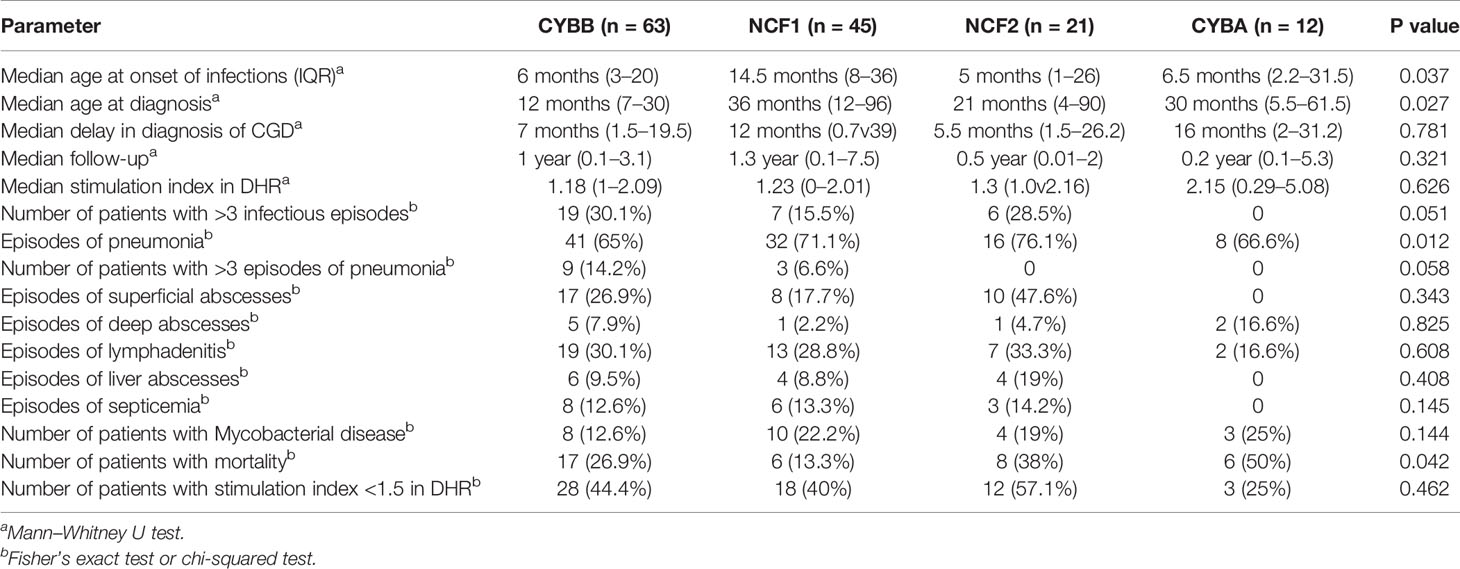
Table 2 Comparison of clinical, and laboratory characteristics between CYBB, NCF1, NCF2, and CYBA defects.
Clinical Characteristics of p47phox Defect in Comparison to p67phox- Defect
Median age at symptom onset was higher in p47phox defect (14 months) when compared to p67phox defect (5 months). The difference was, however, not statistically significant. Median age at diagnosis and median delay in diagnosis was comparable between two groups (Supplementary Table 1). There was also no statistical difference between p47phox and p67phox deficiency with regard to the following parameters: number of patients with >three infectious episodes (p 0.326); number of patients with more than three episodes of pneumonia (p = 0.545); and colitis (p 0.441) (Supplementary Table 1).
Infection Profile
We document 559 episodes of infections in 236 patients over 298.6 patient–years of follow-up (Table 1). Median number of episodes of infections was two (IQR 1–3 episodes).
Localization of Infection
Lung
Pneumonia was seen in 169 patients (71.6%), and the total number of episodes of pneumonia was 242. Due to indolent presentation of pneumonia in CGD, and India being an endemic country for tuberculosis, several patients with difficult to treat pneumonia were empirically started on antitubercular therapy (ATT) before the diagnosis of CGD could be ascertained. Fifty-three (54%) of 96 patients with persistent pneumonia had received empirical ATT in our cohort. Number of ATT courses ranged from one to three. Three patients with CGD had extensive bronchiectasis with pulmonary hypertension at time of diagnosis—all three had received multiple courses of empirical ATT for many months before the diagnosis of CGD was confirmed in them.
In our cohort, fungal pneumonia was commonest (26.8%), followed by bacterial (23.9%) and mycobacterial infections (11.5%) (Table 3). The commonest organisms were Aspergillus sp., Mycobacterium sp., Staphylococcus aureus, Pseudomonas sp., Klebsiella sp., and B. cepacia (Table 2). Confirmed fungal etiology either on lung histopathology or on microbiological cultures from affected tissues was documented in 18 of 62 episodes of suspected fungal infection. The remaining patients also probably had a fungal infection as they had positivity for biomarkers such as galactomannan and/or beta-D-glucan. Twenty patients (8.4%) required mechanical ventilation due to severe lung disease or associated septicemia. Other pulmonary complications include lung abscesses (n = 7; 2.9%), bronchiectasis (n = 5; 2.1%), and contiguous rib osteomyelitis (n = 5; 2.1%).
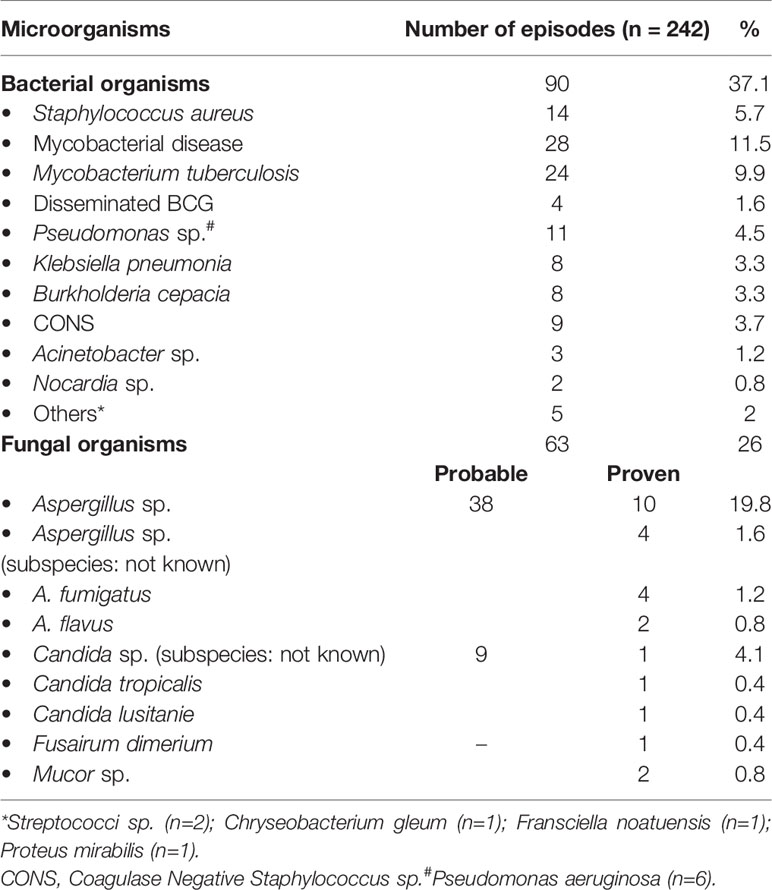
Table 3 Profile of microorganisms in patients with pneumonia.
Mycobacterial infections were documented in 44 patients (18.6%)—Mycobacterium tuberculosis in 25, Bacillus Calmette Guerin (BCG) related complications in 17 (localized disease in 13, and disseminated disease in four), and infection due to non-tuberculous mycobacteria in two (Table 4). Two patients with disseminated tuberculosis had a stormy course—one patient had anterior chest wall cold abscess, hepatosplenomegaly, and computed tomography (CT) guided lung biopsy showed granulation tissue with AFB positivity; one patient presented with pericardial and pleural effusions. Both of them also had lymphadenopathy and rib osteomyelitis at time of diagnosis. While one improved after initiation of conventional ATT, the other succumbed to the illness due to concomitant Mucorales infection in the lung.
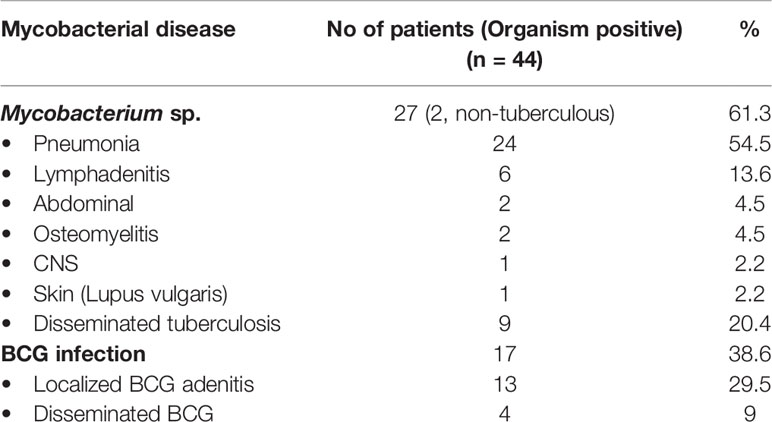
Table 4 Spectrum of mycobacterial disease in our cohort.
Blood-Stream Infections
Though patients with CGD usually do not have a proclivity for dissemination of infection, 21 patents (9%) in our cohort had septicemia. Blood-stream infections (BSI) were found in 75 of 551 infectious episodes (Table 4). Bacterial infections were commonest (70/75 episodes, 93.3%). Some of the signature organisms that were isolated included S. aureus (21.3%); Burkholderia cepacia (13.3%); Pseudomonas sp. (9.3%); Klebsiella sp. (8%); followed by other organisms (Table 5).
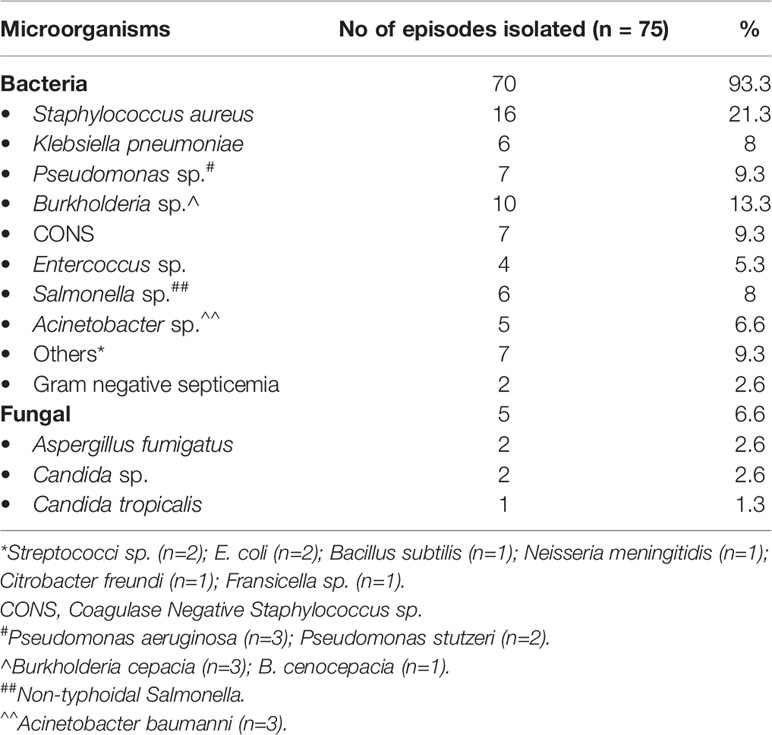
Table 5 Isolation of microorganisms in blood stream infections.
Lymphadenitis
Lymphadenitis was seen in 74/236 patients (31.6%), and 94 episodes were documented in our cohort. Microorganisms were isolated from lymph node aspirate in 25/74 patients (33.7%). The commonest organisms isolated were Staphylococcus sp. (40%), Mycobacterium sp. infections (36%) followed by Burkholderia sp., Klebsiella sp., and Pseudomonas sp. (Table 6). Two patients underwent surgical resection for recurrent lymphadenitis that was recalcitrant to medical therapy.
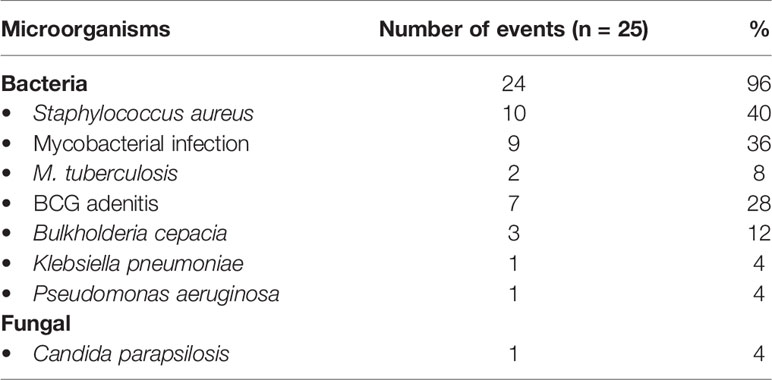
Table 6 Organism profile in lymphadenitis.
Skin and Subcutaneous Abscesses
Skin and subcutaneous abscesses were found in 56 patients (23.7%), and a microorganism could be isolated from pus culture in 17/56 patients (30.3%) (Table 7). Majority (88.2%) were due to bacterial infections and among these, S. aureus was isolated in eight patients (53.3%). Perianal abscesses were found in six patients (2.5%) with CGD.
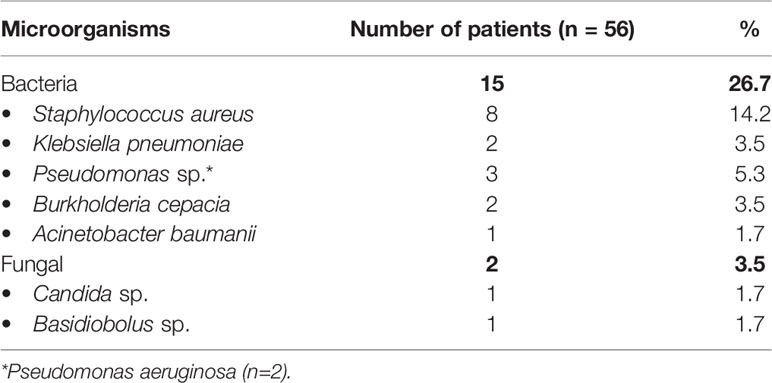
Table 7 Spectrum of microorganisms in patients with skin and subcutaneous abscesses.
Liver Abscess
Seventeen patients presented with liver abscesses (7.2%), and one among these had a recurrent liver abscess (19). Four patients with liver abscess had concurrent other site abscesses (staphylococcal skin pustules in one patient, Pseudomonas aeruginosa cervical abscess in one patient, and multiple deep-seated abscesses noted in two other patients).
S. aureus was isolated from pus in 5/17 patients (29.4%). Three patients with liver abscess were administered corticosteroids besides intravenous antibiotics. Rupture of liver abscess warranting surgical intervention was documented in one child who had not received corticosteroids.
Other Deep-Seated Abscesses
Seven patients had lung abscess—Nocardia sp. and Aspergillus sp. was isolated in one patient each. Splenic abscess was found in four patients. Two patients developed brain abscesses, and Aspergillus fumigatus was isolated from the pus in both. One child also had retropharyngeal and parapharyngeal abscess.
Bone
Infective osteomyelitis was found in 20 patients (8.6%). Site of involvement was variable: ribs—five; lower extremities [tibia (n = 2), fibula (n= 1)]; vertebrae—one; radius—one; skull—one; and small bone osteomyelitis of the hands in two patients. Aspergillus fumigatus was isolated from four patients; Enterobacter sp., Chromobacterium violaceum, A. terreus, and Serratia marcescens were isolated in one patient each (16, 19, 20). Mycobacterial infection was responsible for two cases of osteomyelitis (one with M. tuberculosis, and another with atypical mycobacterial disease). Surgical resection of ribs was required in two patients, while other patients were managed conservatively.
Other Sites
Meningoencephalitis was seen in six patients. Two children had complicated meningitis with hydrocephalus. Aspergillus nidulans was isolated from CSF from one of them. Ten children had otitis media (4.2%). Urinary tract infection (UTI) was seen in 12 patients (5.1%).
Non-Infectious Manifestations
Twelve patients (5.1%) had colitis. Twenty-three episodes of colitis were noted in the cohort, and one patient had had six relapses. One child had been diagnosed to have inflammatory bowel disease elsewhere. He had received infliximab for recalcitrant colitis before the diagnosis of CGD and had developed severe pneumonia following the therapy. Glucocorticoids, mesalamine, and azathioprine were used in seven, five, and three patients respectively. A child with XL-CGD had complete remission of colitis following hematopoietic stem cell transplantation (HSCT).
Lung granuloma was documented in 16 patients (6.8%), and liver granuloma in two patients. Secondary hemophagocytic lymphohistiocytosis (HLH) was documented in six patients (2.5%) (21). Common infective triggers identified were blood stream septicaemia (n = 3) [Francisella noatuensis (one), Burkholderia cenocepacia (one), Candida albicans (one)], pneumonia (n = 4) [Nocardia sp. (n = 1), probable Aspergillus sp. (n = 1)], and disseminated BCGosis (n = 1) (21). Other non-infective manifestations found in our cohort included chilblains (n = 2), HLA-B27 related arthritis (n = 1), Kawasaki disease with coronary artery aneurysm (n = 1), unexplained chronic kidney disease (n = 1), and intestinal obstruction in one patient.
Molecular Diagnosis
Results of genetic analysis were available for 141 patients (59.7%). Pathogenic variants in the CYBB were the most common (63/141; 44.7%), followed by NCF1 gene variants (45/141; 31.9%) whereas genetic variants in NCF2 and CYBA comprised 14.9% (21/142) and 8.5% (12/141) of all the variants, respectively (Supplementary Table 2) (22–30). Fifteen of the 141 genetic variants were novel; 10 in the CYBB gene, three in the NCF2 gene, and two in the NCF1 gene. Nonsense variants comprised majority of CYBB gene variants (21/63; 33.3%), followed by missense variants (11/63; 17.4%), splice-site variants (10/63; 15.8%), deletions (9/63; 14.3%), insertions (5/63; 7.9%), duplications (4/63; 6.3%), and promoter-site variant (n = 1) (Figure 2). Five patients had a previously reported synonymous variant c.252G>A; p.A84(=) which is located proximal to the splice donor site of intron 3 and results in skipping of exon 3 of CYBB. Most patients with autosomal recessive CGD due to gene variants in the NCF1 gene had the common dinucleotide deletion in exon 2 of the gene. However, variants were also detected in other exons of the gene including a large deletion involving exons 2–10 of the NCF1 gene and nonsense variants in exon 7 in two other patients (Figures 3, 4). NCF2 gene variants were more common in patients from North India (15/22; 68.1%) compared to patients from West or South India (7/22; 31.9%). Majority of the patients with NCF2 gene variants from North India (10/15; 66.7%) had a common dinucleotide deletion in exon 9 (c.835_836delAC; p.Thr279GlyTer16) (31) of the gene resulting in alteration of the reading frame and termination (Figures 3, 4). CYBA gene variants in contrast were exclusively seen in patients from South and West India. Most of the CYBA gene variants were located in exon 4 of the gene (6/12; 50%) and one patient had a large deletion involving exons 2–4 of the CYBA gene (Figure 3).
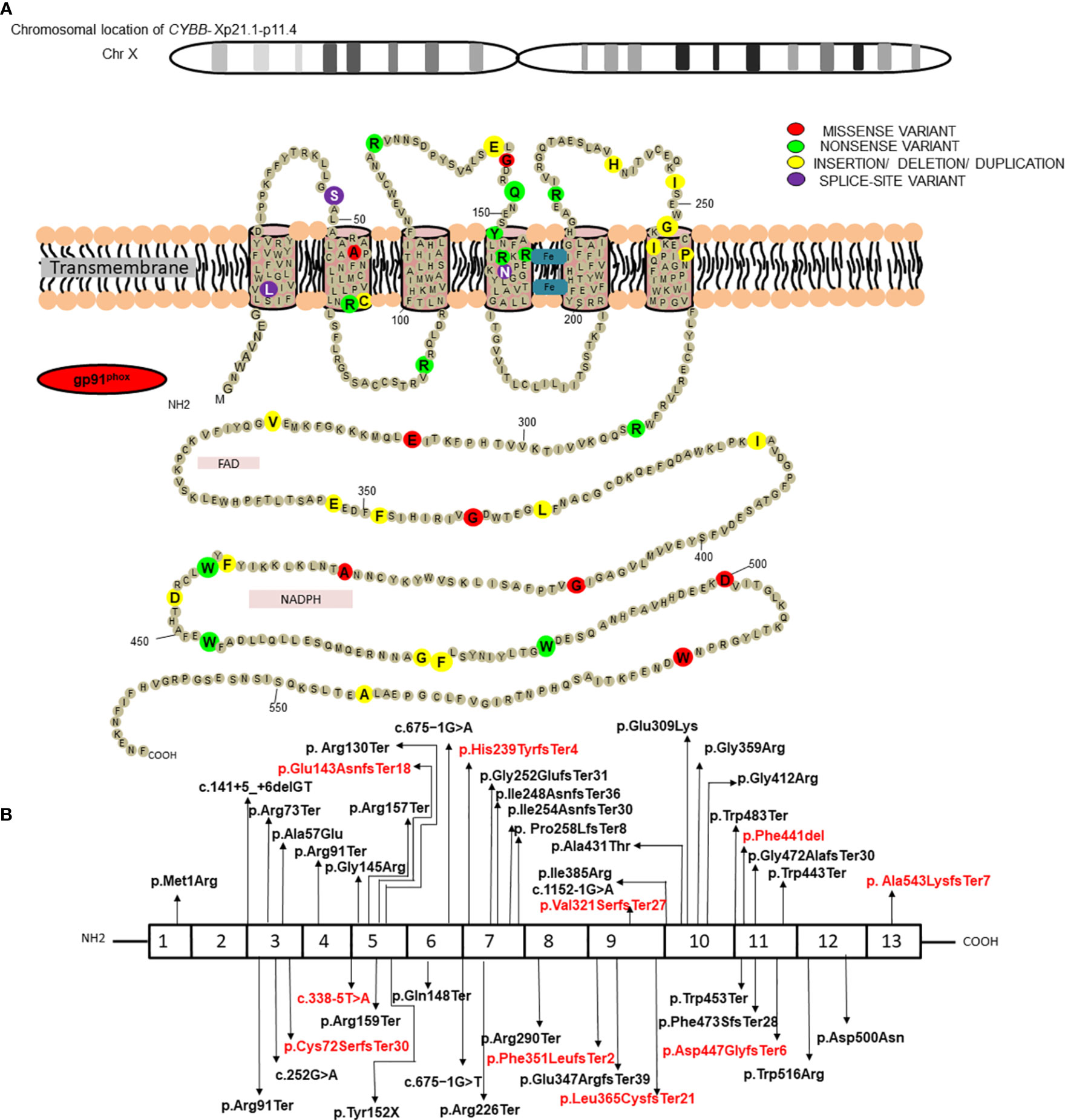
Figure 2 (A) Position of protein change in gp91phox for the molecular variants in CYBB identified in current study; (B) Molecular variants identified in different exons of CYBB and corresponding protein domains. Novel variants have been highlighted in red.
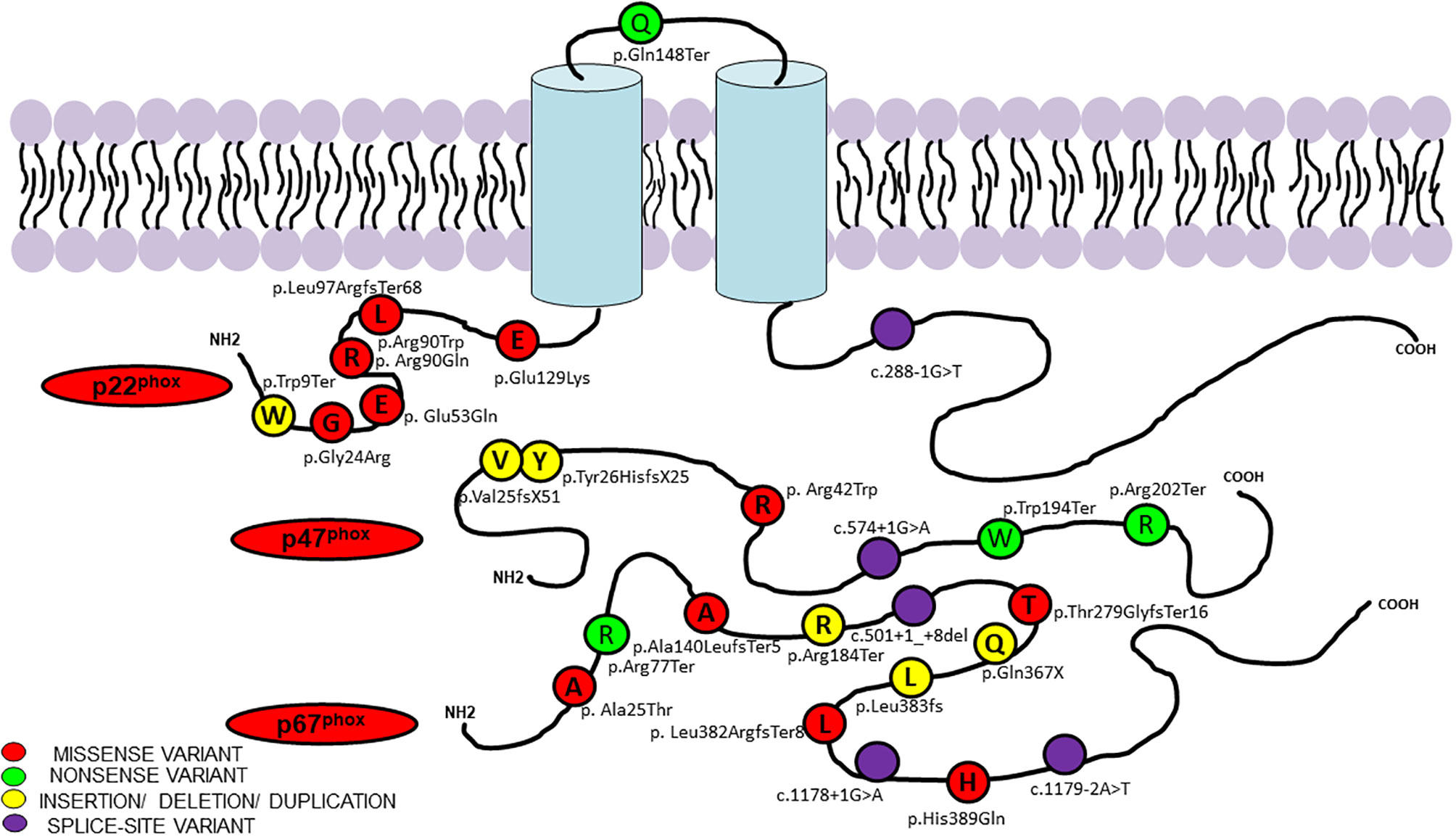
Figure 3 Molecular variants and position of protein change in p47phox, p67phox, and p22phox identified in current study.
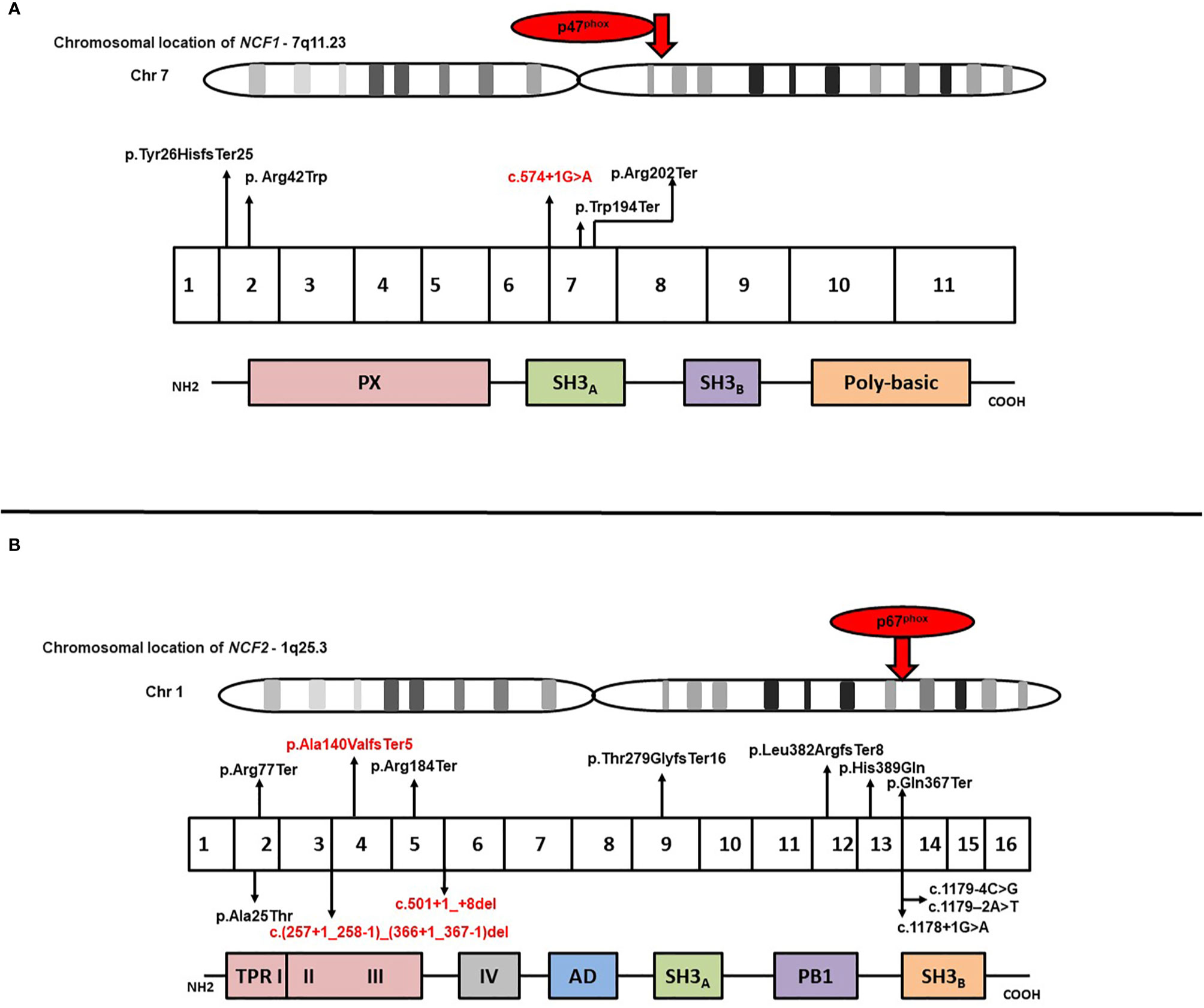
Figure 4 (A) Molecular variants identified in different exons of NCF1 and corresponding protein domains; (B) Molecular variants identified in different exons of NCF2 and corresponding protein domains. Novel variants have been highlighted in red.
Three X-linked carriers of CYBB defect had manifestations of CGD in form of severe infections probably as a result of skewed inactivation (32, 33). A carrier of X-linked CGD developed manifestations of severe lupus malar rash, arthritis, positivity for anti-nuclear and anti-double stranded DNA antibodies, seizures, and demyelinating lesions in the brain and spinal cord (21). She ultimately succumbed to her illness.
Treatment
All patients were started on prophylactic antimicrobials, usually a combination of cotrimoxazole and itraconazole. Data on breakthrough infections after initiation of antimicrobial prophylaxis were available for 78 children. Statistical analysis showed a significant reduction in episodes of pneumonia; lymphadenitis (p = 0.003); skin, and subcutaneous abscesses (p < 0.001); deep-seated abscesses (p = 0.007); osteomyelitis (p = 0.035); liver abscess (p = 0.02); and septicemia (p = 0.007) in those compliant to antimicrobials (Table 8). Corticosteroids were used in four children with liver abscess, two patients with pneumonia, and in two patients with HLH.
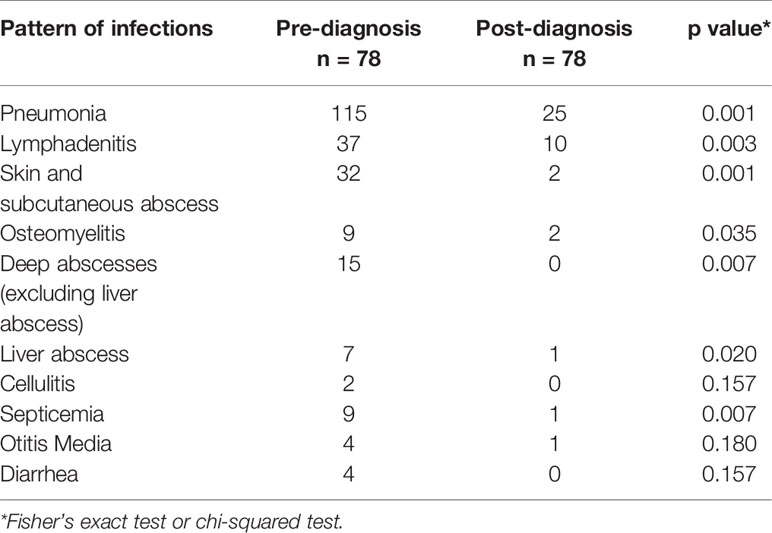
Table 8 Comparison of infectious episodes before and after prophylactic therapy in 78 patients.
Twenty-three patients (9.7%) in the present cohort underwent an HSCT [X-linked—12; AR—three (one each in NCF1, NCF2, and CYBA); unclassified—eight]. Eight patients underwent transplantation with matched related donors. Haploidentical transplantations were carried out in seven patients, while matched unrelated transplantations were performed in three patients, and umbilical cord blood transplantation in one patient. Acute graft versus host disease (GVHD) occurred in seven of 23 transplant recipients, and two patients succumbed to acute GVHD. Complete donor chimerism was attained in 16/23 recipients (68.1%). Six patients succumbed to transplant related complications (two patients had GVHD; one patient had primary graft failure; and one patient had CNS complications).
Survival Analysis
Outcome details were available for 174 patients (73.7%), and follow-up duration was available for 98 patients (292.6 patient-years of follow-up). Sixty-seven of 174 patients (38.5%) had expired at the time of analysis (XL-CGD: 27; AR-CGD: 28; and undetermined: 12) (Figure 5A). The cumulative survival of XL-CGD and AR-CGD was comparable on Kaplan–Meier analysis (p: 0.152) (Figures 5B, C). Cumulative survival comparisons between individual subtypes (CYBB, NCF1, NCF2, and CYBA) showed better survival in NCF1 defect compared to other subtypes (p 0.012) (Figures 5D, E).
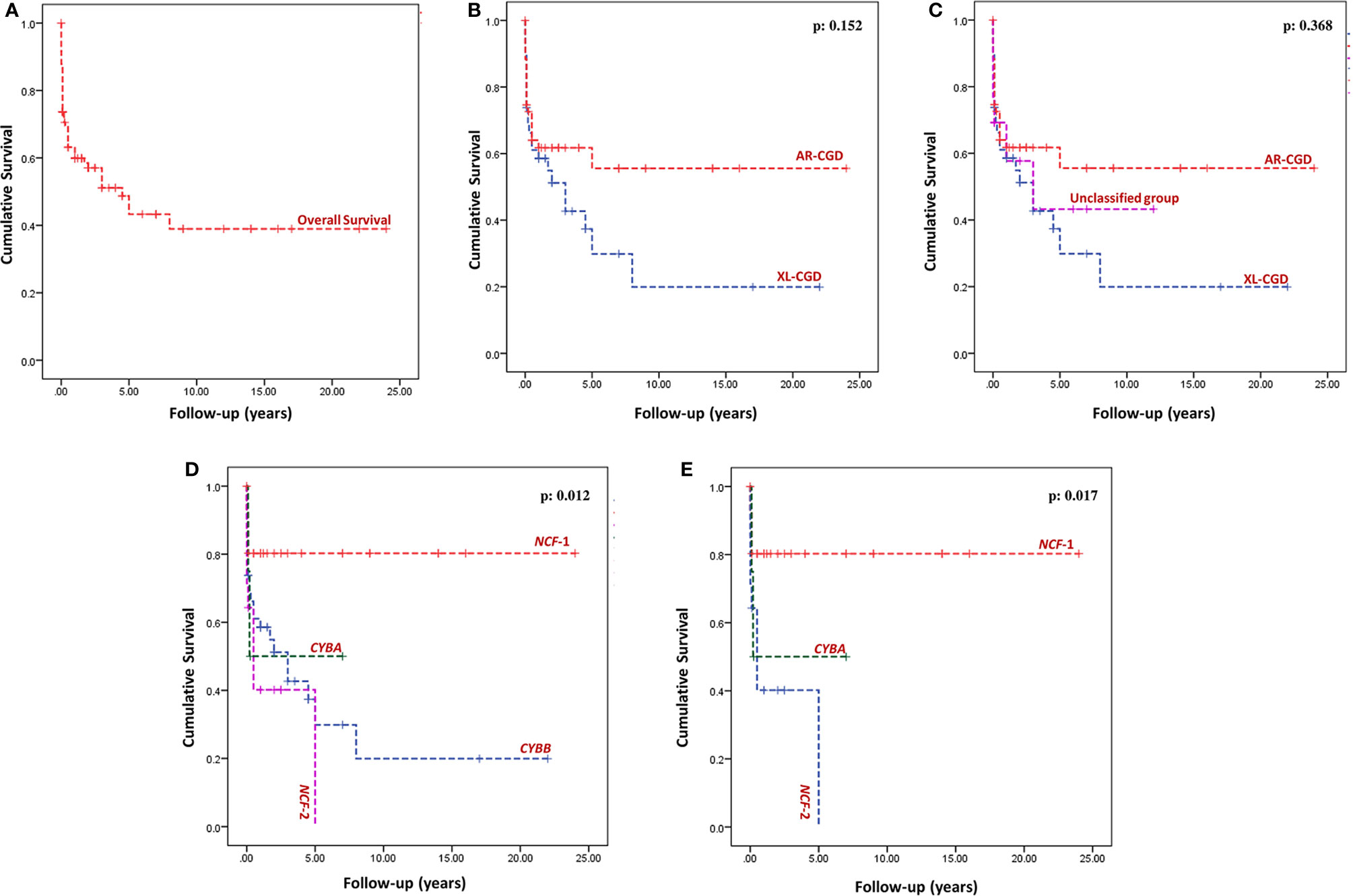
Figure 5 Kaplan–Meier survival curves—(A) Overall survival of the entire cohort; (B) Comparison of overall survival between AR-CGD and XL-CGD, p = 0.152 (Log Rank Mantel–Cox); (C) Comparison of overall survival between Unclassified group, AR-CGD and XL-CGD, p = 0.368 (Log Rank Mantel-Cox); (D) Comparison of overall survival between individual subtypes (CYBB, NCF1, NCF2, and CYBA), p = 0.012 (Log Rank Mantel–Cox); (E) Comparison of overall survival between NCF1, NCF2, and CYBA defects p = 0.017 (Log Rank Mantel–Cox).
Median age at diagnosis in individuals who succumbed to illness was 14 months (IQR 7–40 months), and the median age at time of death was 19 months (IQR 7.8–53.6 months). All patients were under antibiotic prophylaxis. Details of terminal events were available only for 37 out of the 61 patients who did not undergo HSCT. Terminal events that were observed include pneumonia in 30 patients (81%); septicemia (10 patients, 27%); brain abscess (two patients, 5.4); lung abscess, liver abscess, and meningitis in one patient each (2.7%). Three patients (8.1%) also had concomitant HLH at terminal illness.
Details of microbiological profile were available in 22 of 37 patients (59.4%). Aspergillus sp. was isolated in eight patients (26.6%) including A. fumigatus in two patients and A. flavus in one patient; M. tuberculosis, Mucorales, Nocardia sp. in two patients each (6.6%); Chryseobacterium gleum, Pseudomonas aeruginosa, Klebsiella pneumoniae, Candida lusitanea, and cytomegalovirus were isolated in one patient each (3.3%). Out of 10 patients with septicemia, microorganisms could be isolated in eight patients [S. aureus (two), Candida sp. (one), C. tropicalis (one), P. aeruginosa (one), P. stutzeri (one), Acinetobacter sp. (one), B. cepacia (one), B. cenocepacia (one), and K. pneumoniae (one)]. Aspergillus fumigatus was isolated in a patient with brain abscesses, and A. nidulans was isolated in cerebrospinal fluid in a patient who succumbed to meningitis.
Discussion
CGD was first described in four male children from Minnesota in the year 1954 (34) and was later referred as ‘fatal granulomatosus of childhood’ by Berendes et al. in 1957 (35). Subsequently, molecular mechanism of disease pathogenesis (36) and genetic defect (CYBB) was discovered in the years 1967 and 1986 (37, 38), respectively. CGD is now one of the commonly recognized IEI in children. Prognosis of affected children has improved significantly over the last few decades with the introduction of long-term antimicrobial prophylaxis and initiation of curative therapies such as HSCT. Incidence of CGD differs between populations, and it ranges from an estimated one per 200,000 lakh live births in the US and Europe (1, 6), to 1.5 per 100,000 live births among Arabs in Israel where consanguinity rates are high (39).
Initial reports of CGD from India date back to late 1990s (40). Subsequently, the two Indian Council of Medical Research Centres for Advanced Research in Primary Immunodeficiency Diseases (PGIMER, Chandigarh and NIIH, Mumbai) published large case series on CGD from their respective centres (13, 15, 16). Several case reports and case series from other centers have also emanated in the last few years. The present multi-centric study is an effort to collate the data of patients with CGD diagnosed at multiple centers across the country to understand epidemiology, profile of infections, and molecular spectrum of CGD in India. For this study, we obtained data on 236 patients with CGD diagnosed at 12 different centers in India.
We observed a higher proportion of AR forms of CGD as compared to X-linked forms. This is consistent with the previously published data on CGD from India (15). Higher proportion of AR-CGD may be a reflection of high rates of consanguinity and endogamous marriages in the country. Median age at diagnosis in our cohort was 24 months (IQR: 8–60 months), which is comparable with data from other large multi-centric studies from the USA, Europe, and China (1, 6, 41, 42) (Supplementary Table 3). Patients with X-linked CGD have an earlier age of presentation and are reported to have a more severe illness as compared to patients with AR-CGD. We also observed that age at onset of infections and diagnosis of AR-CGD was significantly higher than XL-CGD. However, unlike reports from several other countries (1, 43), there was no difference in survival and mortality rates between AR and XL types of CGD. We attribute this difference to three factors—delays in diagnosis of patients with AR-CGD, a probable missed diagnosis of milder forms of AR-CGD in many patients probably due to lack of awareness especially among the adult physicians, and lack of appropriate laboratory facilities for diagnosis of CGD at many hospitals in India. At present, NBT test can be performed at most centers involved in care of IEI and DHR is currently performed only at six centers. However, many tertiary care hospitals in the country still do not have the facility to perform either NBT or DHR. Median follow-up duration of our cohort is 1 year suggesting that majority of patients are only diagnosed only in the recent years. Increase in awareness of IEI and upscaling of laboratory facilities for carrying out basic immunological investigations are the needs of the hour.
Among the breakthrough infections, pneumonia was most common followed by abscesses (skin, subcutaneous, and deep abscesses, inclusive of liver abscesses), lymphadenitis, and BSI. India, being a country endemic for tuberculosis, it is not surprising that many children with persistent pneumonia (54%) in our cohort had received empirical ATT in our cohort. This has led to significant delays in diagnosis of CGD. Pediatricians and internists practising in developing countries such as India need to keep in mind that persistent pneumonia may be a presentation of CGD. Empirical ATT may not be warranted in such cases.
Aspergillus sp. is the most common etiology for pneumonia in our cohort followed by gram-negative bacteria, Mycobacterium sp., and Staphylococcus sp. Microbiological profile of pneumonia is similar to cohorts from China (44), Iran (45), and Turkey (43). Pneumonia spreading to contiguous tissues such as ribs and vertebra has been documented only with pneumonia due to fungal or mycobacterial etiology (Aspergillus sp.—four; Mucorales sp. —one; Mycobacterium sp. —two). Pulmonary infection due to Mucor sp. was documented in two patients who had not been exposed to corticosteroids. Delays in diagnosis, malnutrition, and prolonged antibiotic therapy for pneumonia could be the reasons for the development of Mucorales infection in our cohort. Nocardia sp. has been isolated in two patients—one with a lung abscess and the other with persistent pneumonia. Unusual bacteria documented with pneumonia include C. gleum, Citrobacter sp., and Francisella noatuensis. A patient with persistent pneumonia had grown C. lusitaniae from the lung tissue which was resistant to amphotericin B.
A wide spectrum of microorganisms that have been isolated in CGD patients with pneumonia suggests the importance of microbiological isolation of organism and appropriate targeting of anti-microbials. We suggest that patients with severe pneumonia in CGD must be managed in tertiary-care centers with facilities for advanced microbiological testing such as MALDI-TOF, 16S rRNA PCR, fungal cultures, and PCR.
Apart from signature organisms such as Aspergillus sp. and Staphylococcus sp., we also documented a high incidence of mycobacterial infections (18.5%) in our cohort. Most common presentation of mycobacterial infection was pneumonia followed by disseminated forms and lymphadenitis (Table 4). Localized or disseminated infections due to BCG have been documented in 8% of patients in our cohort. Abnormal BCG response has been documented at higher rates in other cohorts such as Iran (55.9%) (45), Turkey (22.5%) (43), Latin America (29.6%) (46), and China (64%) (41) that also administer BCG to all children. We do not investigate all patients with BCG adenitis for CGD. It is possible that milder forms of CGD may present only as BCG adenitis and that such patients may have been missed in our cohort. Incidence of Serratia infection in our cohort is very low (2/236, 0.8%) when compared to studies from North America and Europe (47). Possible reasons for this observation include tropical climate patterns and low microbiological isolation rates. Lee et al. have previously reported that infection due to Burkholderia pseudomallei and Chromobacterium violaceum in CGD have been predominantly noted in tropical countries (48). Melioidosis is common in Southern India, and we have also documented melioidosis in a patient from South India who had X-linked CGD (49). Septicemia due to C. violaceum has also been documented in a child with NCF1 defect who hailed from Central India. It is possible that many patients with melioidosis are not screened for CGD in the country and diagnosis of CGD may be missed in them.
Staphylococcus aureus was the commonest organism isolated in suppurative lymphadenitis and skin abscess. Other bacteria isolated in such infections included gram-negative organisms like Burkholderia sp., Klebsiella sp., and Pseudomonas sp. Fungi such as Candida sp. and Basidiobolus sp. were only occasionally isolated in such infections. The microbiological spectrum in these infections is similar to reports from other countries (1, 6, 43, 50). Among the BSI, the microbiological spectrum was almost similar—Staphylococcus sp. was the commonest, followed by Burkholderia sp. and other gram-negative bacteria. However, non-typhoidal Salmonella (8%) was isolated more frequently from blood as compared to other sites. This suggests the importance of frequent handwashing and avoidance of ingestion of raw food items in patients with CGD, as these factors increase the risk of acquiring non-typhoidal Salmonella. S. aureus is the only organism documented in patients with liver abscess. Oral prednisolone was successfully used in three children with liver abscess for enhanced resolution, and none of them required surgical intervention. Our experience is in line with recent evidence concerning use of corticosteroids in liver abscess in CGD (51).
Common non-infective manifestations documented in our cohort include inflammatory bowel disease-like colitis, visceral granuloma, and secondary HLH. We document a lower rate of colitis (5%) in our cohort compared to studies from North America, Europe and China. However, our rates are comparable to cohorts from Turkey, Iran, and Israel, where autosomal recessive forms of CGD are predominant compared to X-linked CGD (Supplementary Table 3) (43, 45). Two patients who had colitis since early infancy were managed as inflammatory bowel disease elsewhere, and were diagnosed to have CGD at 5 and 7 years, respectively. One of the children also developed fulminant pneumonia following infliximab therapy for IBD-like colitis, similar to the report by Uzel et al. (52). This suggests that work-up for CGD must be considered in children with early-onset colitis – pediatricians and gastroenterologists need to be aware of this presentation. Unlike reports from other studies (1, 53), children with AR-CGD had a higher incidence of colitis in our cohort compared to X-linked forms. The difference is probably due to predominance of AR forms of CGD in the present series. While four patients achieved remission with only oral prednisolone and mesalamine, three achieved partial control with azathioprine. Complete remission of colitis following HSCT was documented in a child with X-linked CGD. This shows that immunological reconstitution following HSCT is also beneficial for the inflammatory component of CGD.
Flow cytometry-based evaluation of NADPH oxidase components is a surrogate marker for identifying the genetic defect in CGD. However, this assay can be currently performed in only in two centers in India—PGIMER, Chandigarh and NIIH, Mumbai. We identified regional heterogeneity in molecular spectrum of CGD within India. Overall, NCF1 is the commonest molecular defect identified in patients with AR-CGD, similar to the cohorts from other parts of world. However, CYBA is the second common type of AR-CGD in patients from Southern and Western India, whereas, NCF2 defect is commoner in patients from North India. Moreover, a common dinucleotide deletion in NCF2 (c.835_836delAC) was observed in 10 patients from North India, suggesting a Founder effect in this population. Observed regional differences in molecular spectrum could be due to heterogeneity in genetic background of the population in India. Genescan is considered to be the preferred mode for identification of common dinucleotide deletion in Exon 2 of NCF1 (c.75_76delGT). This modality is, however, currently available only at Chandigarh and Mumbai. The preferred molecular approach for work-up of patients with CGD at Mumbai has been detailed in a previous publication. At Chandigarh, we follow a similar approach, except that Sanger sequencing for NCF2 c.835_836delAC variant is carried out first for patients who have decreased p67phox expression by flow cytometry, as this variant is commonly and exclusively observed in North Indian population. This is followed by NGS whenever required. We have not identified p40phox (NCF4) defect in our cohort. However, patients with NCF4 defect can have near normal neutrophil stimulation in the conventional DHR assay done using phorbol myristate acetate (PMA) as the stimulant (54). It is possible that this defect has been missed in many patients in India, as all patients suspected to have CGD were screened with DHR assay using stimulation with PMA. Recently, EROS (CYBC1) defect resulting in CGD has also been described (55). We have not screened for this defect in our cohort.
Most of the patients in our cohort were kept on antimicrobial prophylaxis—cotrimoxazole and itraconazole. Interferon-gamma was not used in any of our patients as it is not available in India. Number and severity of infections, especially those due to Staphylococcus sp., significantly came down after initiation of antimicrobial prophylaxis (Table 8). However, many patients had documented breakthrough infections even while being continued on prophylactic medications. Overall mortality in our cohort was 38.5% which is similar to reports from other countries such as Mexico and China (41, 50, 56, 57). Our mortality rates are, however, much higher than those reported from the USA, Iran, and European countries. Delays in diagnosis, severe infection at the presentation, and lack of widespread availability of pediatric HSCT services in the country accounted for high mortality rates in our cohort. However, we also document significant decrease in number of infections following initiation of antibiotic prophylaxis (Table 8). This suggests that patients with CGD in developing countries, especially AR forms (NCF1 defect), can have a reasonable quality of life provided they are diagnosed early and continued on long-term antimicrobial prophylaxis (Figure 5).
HSCT could be carried out in only 23 patients in our cohort. Most of the HSCTs have been carried out in Apollo Children’s Hospitals, Chennai. Only five other centers (BJ Wadia Hospitals for Children, Mumbai; PGIMER, Chandigarh; SGRH, New Delhi; Aster CMI Hospitals, Bangalore; Apollo Indraprastha Hospitals, New Delhi) have carried out HSCT for CGD until date. Successful engraftment was documented in 15 patients who received either bone marrow or peripheral blood-derived stem cells. A child who received cord-blood transplantation failed to develop early myeloid engraftment and succumbed to severe infection. This is similar to the experience reported by Morio et al. (58). Four out of six children who received haploidentical transplantation have had a successful engraftment, indicating that haploidentical transplantation is a potential life-saving option in children when completely matched donors are not available. We reiterate that establishment of dedicated pediatric HSCT centres, and government support for patients undergoing HSCT are essential for state-of-the-art management of CGD in developing countries.
To conclude, we document the first multicentric study on clinical and molecular features of 236 patients with CGD from India. To the best of our knowledge, ours is the third largest cohort of patients with CGD documented till date after the multicentric reports from the USA (n = 368) and Europe (n = 429). We have documented a wide spectrum of bacterial and fungal infections and a high incidence of mycobacterial infections in our cohort. Though AR-CGD predominates in our cohort, significant regional differences in molecular spectrum have been observed. Reasons for a high mortality rate (30%) include lack of awareness of CGD among internists and pediatricians, lack of easy access to immunological tests, and paucity of pediatric HSCT services in our country. Establishment of a nation-wide registry for a rare disease like CGD will be needed in future to decipher precise estimate of disease burden in the country and for better identification of barriers in the existing diagnostic and management strategies.
Data Availability Statement
The data sets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individuals for the publication of any potentially identifiable images or data included in this article.
Author Contributions
PV, MSu, DS, AJi, AnG, SL, PP, AK, PT, AP, VG, MD, SB, CG, HK, DM, MSi, RR, RU, FN, BG, HL, MKa, AS, SSe, TS, AmG, and SSi—clinical management of patients and follow-up and contribution of clinical data. AR, MSh, JS, GK, SC, BS, RM, VA, MKu, GH, UB, PK, MM, AJa, FN, KV, MD, KC, KI, OO, SN, and YL—laboratory work-up of patients and contribution of laboratory data. PV, MSu, MSh, and SL—preparation of first draft and literature review. AR, PV, and SSi—critical review and editing of manuscript. AR and PV—final approval of manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors gratefully acknowledge the support provided by the Indian Council of Medical Research and Department of Health Research, Government of India; Foundation of Primary Immunodeficiency Diseases (FPID), United States of America; Prof. Sudhir Gupta, Professor of Medicine, Pathology & Laboratory Medicine; and Microbiology & Molecular Genetics, University of California at Irvine, Irvine, CA, United States of America.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.625320/full#supplementary-material
References
1. Winkelstein JA, Marino MC, Johnston RBJr, Boyle J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Med (Baltimore) (2000) 79:155–69. doi: 10.1097/00005792-200005000-00003
3. Wallace J. Chronic granulomatous disease. Lancet (1972) 1:1024. doi: 10.1016/s0140-6736(72)91205-6
4. Roos D. The genetic basis of chronic granulomatous disease. Immunol Rev (1994) 138:121–57. doi: 10.1111/j.1600-065x.1994.tb00850.x
5. Schäppi MG, Jaquet V, Belli DC, Krause KH. Hyperinflammation in chronic granulomatous disease and anti-inflammatory role of the phagocyte NADPH oxidase. Semin Immunopathol (2008) 30:255–71. doi: 10.1007/s00281-008-0119-2
6. Van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. Chronic granulomatous disease: the European experience. PloS One (2009) 4:e5234. doi: 10.1371/journal.pone.0005234
7. Park BH, Holmes BM, Rodey GE, Good RA. Nitroblue-tetrazolium test in children with fatal granulomatous disease and newborn infants. Lancet (1969) 1:157. doi: 10.1016/S0140-6736(69)91172-6
8. Baehner RL, Nathan DG. Quantitative nitroblue tetrazolium test in chronic granulomatous disease. N Engl J Med (1968) 278:971–6. doi: 10.1056/NEJM196805022781801
9. Emmendörffer A, Hecht M, Lohmann-Matthes ML, Roesler J. A fast and easy method to determine the production of reactive oxygen intermediates by human and murine phagocytes using dihydrorhodamine 123. J Immunol Methods (1990) 131:269–75. doi: 10.1016/0022-1759(90)90198-5
10. Roesler J, Hecht M, Freihorst J, Lohmann-Matthes ML, Emmendörffer A. Diagnosis of chronic granulomatous disease and of its mode of inheritance by dihydrorhodamine 123 and flow microcytofluorometry. Eur J Pediatr (1991) 150:161–5. doi: 10.1007/BF01963557
11. Dimitrova G, Bunkall C, Lim D, Kendrick C. Comparison of two methods for the diagnosis of chronic granulomatous disease – neutrophil oxidative burst measured by the nitroblue tetrazolium slide test versus the dihydrorhodamine 123 flow cytometric assay. N Z J Med Lab Sci (2013) 67:45–51.
12. Meshaal S, El Hawary R, Abd Elaziz D, Alkady R, Galal N, Boutros J, et al. Chronic granulomatous disease: Review of a cohort of Egyptian patients. Allergol Immunopathol (Madr) (2015) 43:279–85. doi: 10.1016/j.aller.2014.11.003
13. Kulkarni M, Desai M, Gupta M, Dalvi A, Taur P, Terrance A, et al. Clinical, Immunological, and Molecular Findings of Patients with p47phox Defect Chronic Granulomatous Disease (CGD) in Indian Families. J Clin Immunol (2016) 36:774–84. doi: 10.1007/s10875-016-0333-y
14. Madkaikar MR, Shabrish S, Kulkarni M, Aluri J, Dalvi A, Kelkar M, et al. Application of Flow Cytometry in Primary Immunodeficiencies: Experience From India. Front Immunol (2019) 10:1248. doi: 10.3389/fimmu.2019.01248
15. Rawat A, Singh S, Suri D, Gupta A, Saikia B, Minz RW, et al. Chronic granulomatous disease: two decades of experience from a tertiary care centre in North West India. J Clin Immunol (2014) 34:58–67. doi: 10.1007/s10875-013-9963-5
16. Rawat A, Vignesh P, Sharma A, Shandilya JK, Sharma M, Suri D, et al. Infection Profile in Chronic Granulomatous Disease: a 23-Year Experience from a Tertiary Care Center in North India. J Clin Immunol (2017) 37:319–28. doi: 10.1007/s10875-017-0382-x
17. Rawat A, Arora K, Shandilya J, Vignesh P, Suri D, Kaur G, et al. Flow Cytometry for Diagnosis of Primary Immune Deficiencies-A Tertiary Center Experience From North India. Front Immunol (2019) 10:2111. doi: 10.3389/fimmu.2019.02111
18. Kulkarni M, Hule G, de Boer M, van Leeuwen K, Kambli P, Aluri J, et al. Approach to Molecular Diagnosis of Chronic Granulomatous Disease (CGD): An Experience from a Large Cohort of 90 Indian Patients. J Clin Immunol (2018) 38:898–916. doi: 10.1007/s10875-018-0567-y
19. Pilania RK, Rawat A, Vignesh P, Guleria S, Jindal AK, Das G, et al. Liver Abscess in Chronic Granulomatous Disease—Two Decades of Experience from a Tertiary Care Centre in North-West India. J Clin Immunol (2021) doi: 10.1007/s10875-020-00938-9
20. Vignesh P, Bhattad S, Shandilya JK, Vyas S, Garg R, Rawat A. Vertebral Osteomyelitis and Acinetobacter Spp. Paravertebral Soft Tissue Infection in a 4-Year-Old Boy With X-Linked Chronic Granulomatous Disease. Pediatr Infect Dis J (2016) 35:1043–5. doi: 10.1097/INF.0000000000001221
21. Vignesh P, Loganathan SK, Sudhakar M, Chaudhary H, Rawat A, Sharma M, et al. Hemophagocytic Lymphohistiocytosis in Children with Chronic Granulomatous Disease-Single-Center Experience from North India. J Allergy Clin Immunol Pract (2020) 28:S2213–2198(20)31263-0. doi: 10.1016/j.jaip.2020.11.041
22. Roos D, Kuhns DB, Maddalena A, Roesler J, Lopez JA, Ariga T, et al. Hematologically important mutations: X-linked chronic granulomatous disease (third update). Blood Cells Mol Dis (2010) 45:246–65. doi: 10.1016/j.bcmd.2010.07.012
23. Roos D, Kuhns DB, Maddalena A, Bustamante J, Kannengiesser C, de Boer M, et al. Hematologically important mutations: the autosomal recessive forms of chronic granulomatous disease (second update). Blood Cells Mol Dis (2010) 44:291–9. doi: 10.1016/j.bcmd.2010.01.009
24. Kannengiesser C, Gérard B, El Benna J, Henri D, Kroviarski Y, Chollet-Martin S, et al. Molecular epidemiology of chronic granulomatous disease in a series of 80 kindreds: identification of 31 novel mutations. Hum Mutat (2008) 29:E132–49. doi: 10.1002/humu.20820
25. Wolach B, Gavrieli R, de Boer M, van Leeuwen K, Berger-Achituv S, Stauber T, et al. Chronic granulomatous disease: Clinical, functional, molecular, and genetic studies. The Israeli experience with 84 patients. Am J Hematol (2017) 92:28–36. doi: 10.1002/ajh.24573
26. National Center for Biotechnology Information. Available at: www.ncbi.nlm.nih.gov/clinvar/variation/VCV000426990.7 (Accessed Oct. 29, 2020).
27. Jacob CO, Eisenstein M, Dinauer MC, Ming W, Liu Q, John S, et al. Lupus-associated causal mutation in neutrophil cytosolic factor 2 (NCF2) brings unique insights to the structure and function of NADPH oxidase. Proc Natl Acad Sci U S A (2012) 10(109):E59–67. doi: 10.1073/pnas.1113251108
28. GnomAd Browser. ENST00000367536.1. Available at: https://gnomad.broadinstitute.org/variant/1-183532568-C-T?dataset=gnomad_r2_1 (Accessed Oct. 29, 2020).
29. Rae J, Noack D, Heyworth PG, Ellis BA, Curnutte JT, Cross AR. Molecular analysis of 9 new families with chronic granulomatous disease caused by mutations in CYBA, the gene encoding p22(phox). Blood (2000) 96(3):1106–12. doi: 10.1182/blood.V96.3.1106
30. Yamada M, Ariga T, Kawamura N, Ohtsu M, Imajoh-Ohmi S, Ohshika E, et al. Genetic studies of three Japanese patients with p22-phox-deficient chronic granulomatous disease: detection of a possible common mutant CYBA allele in Japan and a genotype-phenotype correlation in these patients. Br J Haematol (2000) 108:511–7. doi: 10.1046/j.1365-2141.2000.01857.x
31. Vignesh P, Rawat A, Kumar A, Suri D, Gupta A, Lau YL, et al. Chronic Granulomatous Disease Due to Neutrophil Cytosolic Factor (NCF2) Gene Mutations in Three Unrelated Families. J Clin Immunol (2017) 37:109–12. doi: 10.1007/s10875-016-0366-2
32. Arunachalam AK, Maddali M, Aboobacker FN, Korula A, George B, Mathews V, et al. Primary Immunodeficiencies in India: Molecular Diagnosis and the Role of Next-Generation Sequencing. J Clin Immunol (2021) 41(2):393–413. doi: 10.1007/s10875-020-00923-2
33. Prabhat N, Chakravarty K, Pattnaik SN, Takkar A, Ray S, Lal V. Systemic lupus erythematosus with autoimmune neurological manifestations in a carrier of chronic granulomatous disease - a rare presentation. J Neuroimmunol (2020) 343:577229. doi: 10.1016/j.jneuroim.2020.577229
34. Janeway CA, Craig J, Davidson M, Downey W, Gitlin D, Sullivan JC. Hypergammaglobulinemia associated with severe, recurrent and chronic non-specific infection. Am J Dis Child (1954) 88:388–92.
35. Berendes H, Bridges RA, Good RA. A fatal granulomatosus of childhood: the clinical study of a new syndrome. Minn Med (1957) 40:309–12.
36. Baehner RL, Karnovsky ML. Deficiency of reduced nicotinamide-adenine dinucleotide oxidase in chronic granulomatous disease. Science (1968) 162:1277–9. doi: 10.1126/science.162.3859.1277
37. Segal AW. Absence of both cytochrome b-245 subunits from neutrophils in X-linked chronic granulomatous disease. Nature (1987) 326(6108):88–91. doi: 10.1038/326088a0
38. Baehner RL. Chronic granulomatous disease of childhood: clinical, pathological, biochemical, molecular, and genetic aspects of the disease. Pediatr Pathol (1990) 10(1-2):143–53. doi: 10.3109/15513819009067103
39. Vignesh P, Sharma M, Pilania RK, Shandilya JK, Kaur A, Goel S, et al. Myriad Faces of Chronic Granulomatous Disease: All in an Indian Family with Novel CYBB Defect. J Clin Immunol (2019) 39:611–5. doi: 10.1007/s10875-019-00661-0
40. Loganathan SK, Vignesh P, Pilania RK, Rawat A, Singh S. Chronic Granulomatous Disease: A Perspective from a Developing Nation. Int Arch Allergy Immunol (2020) 15:1–4. doi: 10.1159/000511149
41. Zhou Q, Hui X, Ying W, Hou J, Wang W, Liu D, et al. A Cohort of 169 Chronic Granulomatous Disease Patients Exposed to BCG Vaccination: a Retrospective Study from a Single Centre in Shanghai, China (2004-2017). J Clin Immunol (2018) 38:260–72. doi: 10.1007/s10875-018-0486-y
42. Marciano BE, Spalding C, Fitzgerald A, Mann D, Brown T, Osgood S, et al. Common severe infections in chronic granulomatous disease. Clin Infect Dis (2015) 60(8):1176–83. doi: 10.1093/cid/ciu1154
43. Köker MY, Camcıoğlu Y, van Leeuwen K, Kılıç SŞ, Barlan I, Yılmaz M, et al. Clinical, functional, and genetic characterization of chronic granulomatous disease in 89 Turkish patients. J Allergy Clin Immunol (2013) 132(5):1156–1163.e5. doi: 10.1016/j.jaci.2013.05.039
44. Jones LB, McGrogan P, Flood TJ, Gennery AR, Morton L, Thrasher A, et al. Special article: chronic granulomatous disease in the United Kingdom and Ireland: a comprehensive national patient-based registry. Clin Exp Immunol (2008) 152(2):211–8. doi: 10.1111/j.1365-2249.2008.03644.x
45. Fattahi F, Badalzadeh M, Sedighipour L, Movahedi M, Fazlollahi MR, Mansouri SD, et al. Inheritance pattern and clinical aspects of 93 Iranian patients with chronic granulomatous disease. J Clin Immunol (2011) 31(5):792–801. doi: 10.1007/s10875-011-9567-x
46. de Oliveira-Junior EB, Zurro NB, Prando C, Cabral-Marques O, Pereira PV, Schimke LF, et al. Clinical and Genotypic Spectrum of Chronic Granulomatous Disease in 71 Latin American Patients: First Report from the LASID Registry. Pediatr Blood Cancer (2015) 62(12):2101–7. doi: 10.1002/pbc.25674
47. Bhattarai D, Gupta A, Vignesh P, Rao H, Angrup A, Rawat A. Infection Due to Serratia sp. in Chronic Granulomatous Disease-Is the Incidence Low in Tropical Countries? J Clin Immunol (2020) 41(2):486–90. doi: 10.1007/s10875-020-00919-y
48. Lee PP, Lau YL. Endemic infections in Southeast Asia provide new insights to the phenotypic spectrum of primary immunodeficiency disorders. Asian Pac J Allergy Immunol (2013) 31(3):217–26.
49. Saravu K, Mukhopadhyay C, Vishwanath S, Valsalan R, Docherla M, Vandana KE, et al. Melioidosis in southern India: epidemiological and clinical profile. Southeast Asian J Trop Med Public Health (2010) 41:401–9.
50. Blancas-Galicia L, Santos-Chávez E, Deswarte C, Mignac Q, Medina-Vera I, León-Lara X, et al. Genetic, Immunological, and Clinical Features of the First Mexican Cohort of Patients with Chronic Granulomatous Disease. J Clin Immunol (2020) 40(3):475–93. doi: 10.1007/s10875-020-00750-5
51. Leiding JW, Freeman AF, Marciano BE, Anderson VL, Uzel G, Malech HL, et al. Corticosteroid therapy for liver abscess in chronic granulomatous disease. Clin Infect Dis (2012) 54(5):694–700. doi: 10.1093/cid/cir896
52. Uzel G, Orange JS, Poliak N, Marciano BE, Heller T, Holland SM. Complications of tumor necrosis factor-α blockade in chronic granulomatous disease-related colitis. Clin Infect Dis (2010) 51:1429–34. doi: 10.1086/657308
53. Marciano BE, Rosenzweig SD, Kleiner DE, Anderson VL, Darnell DN, Anaya-O’Brien S, et al. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics (2004) 114(2):462–8. doi: 10.1542/peds.114.2.462
54. van de Geer A, Nieto-Patlán A, Kuhns DB, Tool AT, Arias AA, Bouaziz M, et al. Inherited p40phox deficiency differs from classic chronic granulomatous disease. J Clin Invest (2018) 128(9):3957–75. doi: 10.1172/JCI97116
55. Arnadottir GA, Norddahl GL, Gudmundsdottir S, Agustsdottir AB, Sigurdsson S, Jensson BO, et al. A homozygous loss-of-function mutation leading to CYBC1 deficiency causes chronic granulomatous disease. Nat Commun (2018) 9(1):4447. doi: 10.1038/s41467-018-06964-x
56. Gao LW, Yin QQ, Tong YJ, Gui JG, Liu XY, Feng XL, et al. Clinical and genetic characteristics of Chinese pediatric patients with chronic granulomatous disease. Pediatr Allergy Immunol (2019) 30(3):378–86. doi: 10.1111/pai.13033
57. Wang S, Wang T, Xiang Q, Xiao M, Cao Y, Xu H, et al. Clinical and Molecular Features of Chronic Granulomatous Disease in Mainland China and a XL-CGD Female Infant Patient After Prenatal Diagnosis. J Clin Immunol (2019) 39(8):762–75. doi: 10.1007/s10875-019-00680-x
Keywords: Chronic Granulomatous Disease, India, Mycobacterium tuberculosis, Bacillus Calmette Guerin
Citation: Rawat A, Vignesh P, Sudhakar M, Sharma M, Suri D, Jindal A, Gupta A, Shandilya JK, Loganathan SK, Kaur G, Chawla S, Patra PK, Khadwal A, Saikia B, Minz RW, Aggarwal V, Taur P, Pandrowala A, Gowri V, Desai M, Kulkarni M, Hule G, Bargir U, Kambli P, Madkaikar M, Bhattad S, Ginigeri C, Kumar H, Jayaram A, Munirathnam D, Sivasankaran M, Raj R, Uppuluri R, Na F, George B, Lashkari HP, Kalra M, Sachdeva A, Seth S, Sabui T, Gupta A, van Leeuwen K, de Boer M, Chan KW, Imai K, Ohara O, Nonoyama S, Lau YL and Singh S (2021) Clinical, Immunological, and Molecular Profile of Chronic Granulomatous Disease: A Multi-Centric Study of 236 Patients From India. Front. Immunol. 12:625320. doi: 10.3389/fimmu.2021.625320
Received: 02 November 2020; Accepted: 06 January 2021;
Published: 25 February 2021.
Edited by:
Sudhir Gupta, University of California, Irvine, United StatesReviewed by:
Reinhard Alexander Seger, University of Zurich, SwitzerlandAntonio Condino-Neto, University of São Paulo, Brazil
Copyright © 2021 Rawat, Vignesh, Sudhakar, Sharma, Suri, Jindal, Gupta, Shandilya, Loganathan, Kaur, Chawla, Patra, Khadwal, Saikia, Minz, Aggarwal, Taur, Pandrowala, Gowri, Desai, Kulkarni, Hule, Bargir, Kambli, Madkaikar, Bhattad, Ginigeri, Kumar, Jayaram, Munirathnam, Sivasankaran, Raj, Uppuluri, Na, George, Lashkari, Kalra, Sachdeva, Seth, Sabui, Gupta, van Leeuwen, de Boer, Chan, Imai, Ohara, Nonoyama, Lau and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amit Rawat, cmF3YXRhbWl0QHlhaG9vLmNvbQ==
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share second authorship