 Anno Saris1,2†
Anno Saris1,2† Juulke Steuten1†
Juulke Steuten1† David P. Schrijver1
David P. Schrijver1 Gijs van Schijndel1
Gijs van Schijndel1 Jaap Jan Zwaginga3,4
Jaap Jan Zwaginga3,4 S. Marieke van Ham1,5
S. Marieke van Ham1,5 Anja ten Brinke1*
Anja ten Brinke1*- 1Department of Immunopathology, Sanquin Research and Landsteiner Laboratory, Amsterdam University Medical Center, University of Amsterdam, Amsterdam, Netherlands
- 2Department of Infectious Disease, Leiden University Medical Center, Leiden, Netherlands
- 3Center for Clinical Transfusion Research, Sanquin Research, Leiden, Netherlands
- 4Department of Hematology, Leiden University Medical Center, Leiden, Netherlands
- 5Swammerdam Institute for Life Sciences, University of Amsterdam, Amsterdam, Netherlands
Platelet transfusions are a frequently administered therapy for especially hemato-oncological patients with thrombocytopenia. Next to their primary function in hemostasis, currently there is increased attention for the capacity of platelets to affect the function of various cells of the immune system. Here, we investigate the capacity of platelets to immuno-modulate monocyte-derived dendritic cells (moDC) as well as primary dendritic cells and effects on subsequent T cell responses. Platelets significantly inhibited pro-inflammatory (IL-12, IL-6, TNFα) and increased anti-inflammatory (IL-10) cytokine production of moDCs primed with toll-like receptor (TLR)-dependent and TLR-independent stimuli. Transwell assays and ultracentrifugation revealed that a soluble factor secreted by platelets, but not microvesicles, inhibited DC activation. Interestingly, platelet-derived soluble mediators also inhibited cytokine production by human ex vivo stimulated myeloid CD1c+ conventional DC2. Moreover, platelets and platelet-derived soluble mediators inhibited T cell priming and T helper differentiation toward an IFNγ+ Th1 phenotype by moDCs. Overall, these results show that platelets are able to inhibit the pro-inflammatory properties of DCs, and may even induce an anti-inflammatory DC phenotype, with decreased T cell priming capacity by the DC. The results of this study provide more insight in the potential role of platelets in immune modulation, especially in the context of platelet transfusions.
Introduction
Up to three million platelet transfusions are administered every year in Europe, and the majority (up to 65%) of these transfusions are administered to hemato-oncological patients (1–7). Prophylactic administration of platelet transfusions in patients not at direct risk of bleeding is currently subject of debate (6–9). Next to the fact that platelet transfusions are costly and involve complicated logistics, they are associated with adverse reactions like alloimmunization, febrile non-hemolytic transfusion reactions (FNHTR), transfusion related acute lung injury (TRALI), as well as the still somewhat elusive transfusion related immune modulation (TRIM) (8, 10–13). TRIM associates with increased risk of infections, decreased graft rejection and recurrence of tumor growth in patients receiving transfusions (14–16). Even though a number of studies have investigated TRIM, the underlying mechanisms remain largely unresolved (17–20).
Platelets are anucleate megakaryocyte-derived cell fragments packed with secretory granules containing many hemostatic and angiogenic factors, but also a collection of mediators that affect the immune system, ranging from chemokines (e.g. chemokine ligand 5 (CCL5)) to microbicidal molecules (e.g. thrombocidin-1 and 2) (21, 22). Platelets also express many molecules such as toll-like receptors (TLRs), integrins and costimulatory molecules through which they can sense pathogen-associated molecular patterns and interact with leukocytes (23–26). Moreover, platelets are the main source of (s)CD40L in plasma, and in CD40L deficient mice transfer of wildtype platelets increased virus-specific IgG titers (27, 28). In contrast, platelet derived active transforming growth factor β (TGF-β), together with lactate, was found to inhibit T cell anti-tumor functions (29). Due to increased understanding of the potential immune functions of platelets, the question arises to what extent transfused platelets will affect the recipient’s immune system (12, 17, 20, 30, 31), which would be especially relevant for hematological patients that are frequently strongly immune-compromised (7).
Dendritic cells (DCs) are instrumental for the initiation and skewing of the adaptive immune response. DCs are equipped with many pattern recognition receptors (PRRs) that recognize various pathogen- and danger- associated molecular patterns (PAMP/DAMP). Upon encounter of pathogenic antigens, DCs can engulf and process these antigens and present antigenic peptides in the context of MHC molecules to T cells. The upregulation of costimulatory molecules and secretion of cytokines is directed by the signaling pathways of the engaged PRR(s), whereby DCs prime and skew the antigen-specific T cells toward a particular immune response (32). Due to the central role of DCs in the adaptive immune response it is important to study the potential immune modulatory effect of platelets on DC effector functions. Previous studies describe variable effects of platelets on DC effector function depending mostly on the presence or absence of an additional activating stimulus (33–43). Although some studies have shown that platelets may reduce DC activation, lipopolysaccharide (LPS) was almost exclusively used for DC stimulation (33, 34, 36–38). Therefore, questions remain on the generalizability and consistency of these effects and more importantly what are the consequences on T cell activation and priming.
In the present study we aimed to advance our understanding of platelet-mediated inhibition of human DCs and how this affects subsequent T cell responses. Our studies revealed that upon activation platelets release soluble factors (platelet releasate) that affect both moDC and primary myeloid CD1c+ conventional DC2 (cDC2) cytokine production induced by a range of TLR-dependent and TLR-independent stimuli with minimal effects on co-stimulatory marker expression. Furthermore, moDCs stimulated in presence of platelets or platelet releasate show less ability to induce T cell proliferation and have decreased capacity to skew naïve T cells toward a Th1 IFNγ+ phenotype. Overall, we show here that mediators derived from activated platelets directly impair DC effector functions, which may contribute to the occurrence of TRIM upon transfusion of platelets.
Materials and Methods
Human Blood Samples
Leukapheresis products or buffy coats obtained from anonymized Sanquin blood donors were used to isolate monocytes and T cells after written informed consent by donors. Citrated whole blood was obtained to isolate fresh platelets and platelet releasate. All procedures were approved by the Sanquin Ethical Advisory Board and are in accordance with the declaration of Helsinki and Dutch regulations.
Isolation of Human Monocytes and Differentiation to Immature moDC
Monocytes were isolated using the Elutra Cell Separation System (Gambro, Lakewood, CO, USA) from fresh leukapheresis material or from PBMCs isolated from buffy coats using CD14+ magnetic cell separation (Miltenyi Biotech, Leiden, Netherlands) and subsequently frozen until further use as previously described (13, 44). Purity of monocytes was >90% as determined using flow cytometry with APC labeled anti-CD14 (clone MϕP9, BD biosciences). Upon initiation of culture, monocytes were thawed and differentiated into immature dendritic cells (DCs) as previously described (13, 44).
Platelet Isolation and Production and Ultracentrifugation of Platelet Releasate
Platelets were isolated from citrated whole blood by centrifugation at 200g for 10 min and supernatant platelet rich plasma was washed (5 min, 1650g) 1:2 with sequestrine buffer (17.5 mM Na2HPO4, 8.9 mM Na2EDTA, 154 mM NaCl, pH 6.9, containing 0.1% [wt/vol] bovine serum albumin, all obtained from Merck Millipore, Amsterdam, Netherlands). After washing, platelets were resuspended in GMP DC serum-free medium (Cellgenix, Freiburg, Germany) containing 100 U/ml penicillin and 100 U/ml streptomycin (Gibco). To collect platelet releasate, platelets were incubated 30 min with 250 µM thrombin receptor-activating peptide 6 amide (TRAP-6) (Bachem, Bubendorf, Switzerland) after which platelets were removed by centrifugation: first 5 min at 1,650 g and subsequently 10 min at 4,000g. If indicated, platelet releasate was hereafter centrifuged to remove microparticles at 100,000g for 1 h in Optima L-100 XP Ultracentrifuge (Beckman Coulter, IN, United States). Experiments that directly compared whole platelets and releasate used releasate and whole platelets from the same donor. Otherwise, releasate was pooled from four or five whole blood donors, aliquoted and stored at −20°C until further use.
Stimulation of DCs
Immature moDCs were stimulated in the absence or presence of platelets or platelet releasate with a platelet:DC ratio ranging from 80:1 to 1:1 (or equivalent amount of releasate) in GMP DC serum-free medium (Cellgenix, Freiburg, Germany) containing 100 U/ml penicillin and 100 U/ml streptomycin (Gibco). When indicated, DCs were stimulated using 2.5 µg/ml monophosporyl lipid A (MPLA) (Sigma Aldrich, Zwijndrecht, Netherlands), 5 µg/ml pam3CysSerLys4 (PAM3CSK4) (Invivogen, San Diego, CA, USA), 2.5 µg/ml resiquimod (R848) (Alexis Biochemical, Paris, France) or 0.25 µg/ml flagellin bacillus subtilis (Invivogen) in combination with 1,000 U/ml IFNγ (Boehringer Ingelheim, Alkmaar, Netherlands) or a cytokine maturation cocktail consisting of 10 ng/ml TNFα, 10 ng/ml IL-1β (both Cellgenix, Freiburg, Germany), and 1 µg/ml prostaglandin E2 (PGE2) (Sigma Aldrich, Zwijndrecht, Netherlands). When indicated, releasate or recombinant human TGF-β (R&D Systems, Minneapolis, MN, United States) was pre-incubated with blocking antibody against TGF-β (Clone 1D11, 10 µg/ml, BioXCell, NH, United States) for 30’ at 37°C, 5% CO2. For transwell experiments, moDCs were stimulated with MPLA/IFNγ in a 96-well transwell system (Corning, pore size ≤0.3 µM) in absence or presence of TRAP-6 activated platelets (platelet:DC ratio 40:1) that were either below the transwell (i.e. in the same compartment as the DCs) or separated by the transwell. After 24 or 48 h at 37°C, 5% CO2 culture supernatant was harvested and frozen at −20°C until cytokine measurements.
Cytokine Measurements
The levels of IL-6 and IL-10 in culture supernatants were measured using compact PeliKine Cytokine ELISA kits (Sanquin Reagents, Amsterdam, Netherlands); the level of TNF-α was determined with Human TNF-α ELISA set (Diaclone, Besançon, France). To determine production of IL-12p40, a combination of two anti-IL-12p40 antibodies was used (coat: clone C11.79 and detection: clone C8.6, Sanquin Reagents, Amsterdam, Netherlands) and recombinant IL-12p40 was used as calibration. The level of human TGF-β1 was measured using DuoSet ELISA kit (R&D Systems, Minneapolis, MN, United States) either after or without chemical activation of the samples according to manufacturer’s protocol. Absorbance was measured at 540nm with SynergyTM 2 (BioTek, Winooski, VT, USA).
Phenotyping DCs
After 48 h of stimulation, DCs were harvested with 0.4% trisodium citrate (Merck Millipore, Amsterdam, Netherlands) in PBS supplemented with 4 mg/ml human albumin (Albuman, Sanquin Reagents, Amsterdam, Netherlands). DCs were stained 30 min at room temperature with LIVE/DEAD Fixable near-IR Dead Cell Stain Kit (Thermo Fisher Scientific) and subsequently with either PE-labeled anti-HLA-DR (Clone G46-6) or with FITC-labeled anti-CD80 (Clone L307.4), PE-labeled anti-CD40 (Clone 5C3), APC-labeled anti-CD83 (Clone HB15e) and BV421-labeled anti-CD86 (Clone 2331) (all from BD Biosciences) in the presence of 3 mg/ml human γ-globulin (Nanogam, Sanquin, Amsterdam, Netherlands). Samples were measured with BD FACSCanto II and analyzed using Flowjo VX (Beckton, Dickinson & Company, Ashland, OR, United States). Gating strategy involved selection of DCs on FSC/SSC, single cells and viable cells.
Isolation of Human T Cells
Total human lymphocytes were isolated using the Elutra Cell Separation System (Gambro, Lakewood, CO, USA) from fresh leukapheresis material and frozen until further use. Naïve human CD4+ T cells were isolated from buffy coats. First, the PBMC fraction was isolated by density gradient centrifugation (Lymphoprep, Axis-shield, Alere Technologies AS, Oslo, Norway). From PBMC fraction, CD4+ cells were isolated by negative selection using magnetic separation followed by negative selection of CD45RA population (both Miltenyi Biotech, Leiden, Netherlands). Purity of isolated naïve CD4+ T cells ≥95% as determined by staining with FITC-labeled anti-CD45RA (Clone HI100, eBioscience) and BV421-labeled anti-CD45RO (Clone UCHL1, BD Bioscience).
Isolation and Stimulation of Primary Blood DCs
Primary blood DCs were isolated from buffy coats as previously described (45). In short, PBMCs were enriched for blood DCs by elutriation with JE-5.0 elutriator (Beckman Coulter, Brea, CA, USA), followed by sorting on FACSAria IIIu or III cell sorter (BD Bioscience, San Jose, CA, USA). For sorting, the cells were stained with APC-labeled anti-CD3 (clone HIT3a), anti-CD14 (clone MϕP9), anti-CD19 (clone SJ25-C1) (all BD Biosciences), PE-Cy7-labeled anti-CD1c (clone L161, Biolegend) and FITC-labeled 6-sulfo LacNAc (Slan)-specific antibody (clone M-DC8, Miltenyi Biotech) and LIVE/DEAD fixable Near-IR Dead Cell Stain Kit (Invitrogen). Different DC subtypes were gated as follows: Slan+ non-classical monocytes as CD3-CD14-CD19-M-DC8+ and myeloid cDC2 as CD3-CD14-CD19-CD1c+ (see Supplementary Figure S3 for gating strategy). Purity after sort was >90% as tested by flow cytometry. Primary DCs were rested overnight in GMP DC serum-free medium (Cellgenix) containing 100 U/ml penicillin and 100 U/ml streptomycin (Gibco) at 37°C and 5% CO2. After overnight rest, DCs were stimulated with R848 (Alexis Biochemical, Paris, France) at a concentration of 5 µg/ml in absence or presence of platelets or platelet releasate in a 50:1 platelet:DC ratio.
T Cell Proliferation Assay
After thawing and 1 h resting total T cells or naïve CD4+ CD45RA+ T cells were washed twice with PBS and incubated with 5µM CFSE (Invitrogen) for 20 min at room temperature. After washing, T cells were resuspended in IMDM+5%HS medium and incubated 10:1 (T:DC ratio) with autologous (TT-loaded) or allogeneic DCs. If indicated, DCs were first incubated 1 h at 37°C with 5 µg/ml tetanus toxoid (AJVaccines, Copenhagen, Denmark) before stimulation with MPLA/IFNγ. After maturation with MPLA/IFNγ or IL-1β/TNFα/PGE2, moDCs were washed twice with IMDM + 5%HS medium to remove potential remaining platelets or platelet-derived factors. After 8 days, total T cells were harvested, labeled with LIVE/DEAD Fixable near-IR Dead Cell Stain Kit (Thermo Fisher Scientific) and subsequently with PE-labeled anti-CD4 (Clone SK3) and APC-labeled anti-CD8 (Clone SK1) (both from BD biosciences) in FACS buffer (PBS + 0.5% BSA + 0.02% Azide). After 9-10 days, naïve T cells were stimulated with PMA (100 ng/ml) and ionomycin (1 µg/ml) in presence of Brefeldin A (10 µg/ml) (all from Sigma Aldrich) for 5 h. T cells were harvested and washed, labeled with LIVE/DEAD Fixable near-IR Dead Cell Stain Kit (Thermo Fisher Scientific) at RT and subsequently with BUV395-labeled anti-CD4 (clone SK3, BD Bioscience). Hereafter, cells were fixed for 15 min with 4% paraformaldehyde (Merck, Darmstadt, Germany) after which cells were stained with PECy7-labeled anti-IFNγ (Clone B27, BD Bioscience), APC-labeled anti-TNFα (clone Mab11, Biolegend) in Brilliant Stain buffer (BD Bioscience) in permeabilization buffer containing 0.5% Saponin, 1%BSA, and 0.02% azide. Cells were washed and measured on Canto II (total T cells) or LSR Fortessa (both from BD Bioscience). Data were analyzed in Flowjo VX (Beckton, Dickinson & Company, Ashland, OR, United States), gating involved selection of lymphocytes based on FSC/SSC, single cells, live CD4+ cells, proliferation dye dilution and positivity for specific cytokines.
Statistical Analysis
Graphical presentation and statistical analyses were performed using GraphPad prism v8.02 (GraphPad Software Inc, La Jolla, California, USA). Paired t-tests, repeated measures one-way ANOVA and two-way ANOVA with Tukey post test were used to determine statistical differences in DC and T cell cytokine production, DC co-stimulatory marker expression and viability, and T cell proliferation. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Data Sharing Statement
For original data, please contactYS50ZW5icmlua2VAc2FucXVpbi5ubA==
Results
Platelets Decrease moDC Pro-Inflammatory Cytokine Production Upon DC Activation With Different Stimuli
To investigate if and to what extent platelets affect human DC function, monocyte-derived DCs (moDCs) were stimulated with several stimuli to mimic various types of physiological activation, including MPLA (TLR4; Figure 1A), PAM3CSK4 (TLR1/2; Figure 1C), R848 (TLR7/8; Figure 1D) or flagellin (TLR5; Figure 1E) in the presence of IFNγ or with a cytokine maturation cocktail containing IL-1β, TNF-α; and PGE2 (Figure 1B, TNF-α not measured as this was added with stimulation cocktail). After stimulation, moDCs produced pro-inflammatory cytokines TNF-α, IL-12, and IL-6. In the presence of platelets however, pro-inflammatory cytokine production reduced significantly after all stimulations with an average residual cytokine production of 18.9%, 19.7%, and 43.4% for TNFα, IL-12p40, and IL-6 respectively after MPLA/IFNγ stimulation. Platelet-mediated inhibition of cytokine production was not observed for IL-6 production after R848/IFNγ stimulation and IL-12p40 production upon flagellin/IFNγ or cytokine cocktail stimulation, which both failed to induce a strong IL-12p40 response. The decrease in cytokine production was dependent on the number of platelets present (Supplementary Figure S1A). Additionally, in presence of platelets anti-inflammatory IL-10 production was induced for R848/IFNγ and IL-1β/TNFα/PGE2 stimulated moDCs (Figures 1B, D). MoDCs stimulated with MPLA/IFNγ and PAM3CSK/IFNγ display a non-significant trend of IL-10 induction in the presence of platelets (Figures 1A, C), whereas IL-10 production was not detectable for moDCs stimulated with Flagellin/IFNγ (Figure 1E). When immature moDCs were cultured with platelets without additional maturation stimulus, some donors showed an increase in cytokine production. This non-significant trend was most clear for IL-6 and IL-10 production by immature moDCs in presence of platelets (Supplementary Figure 1B).
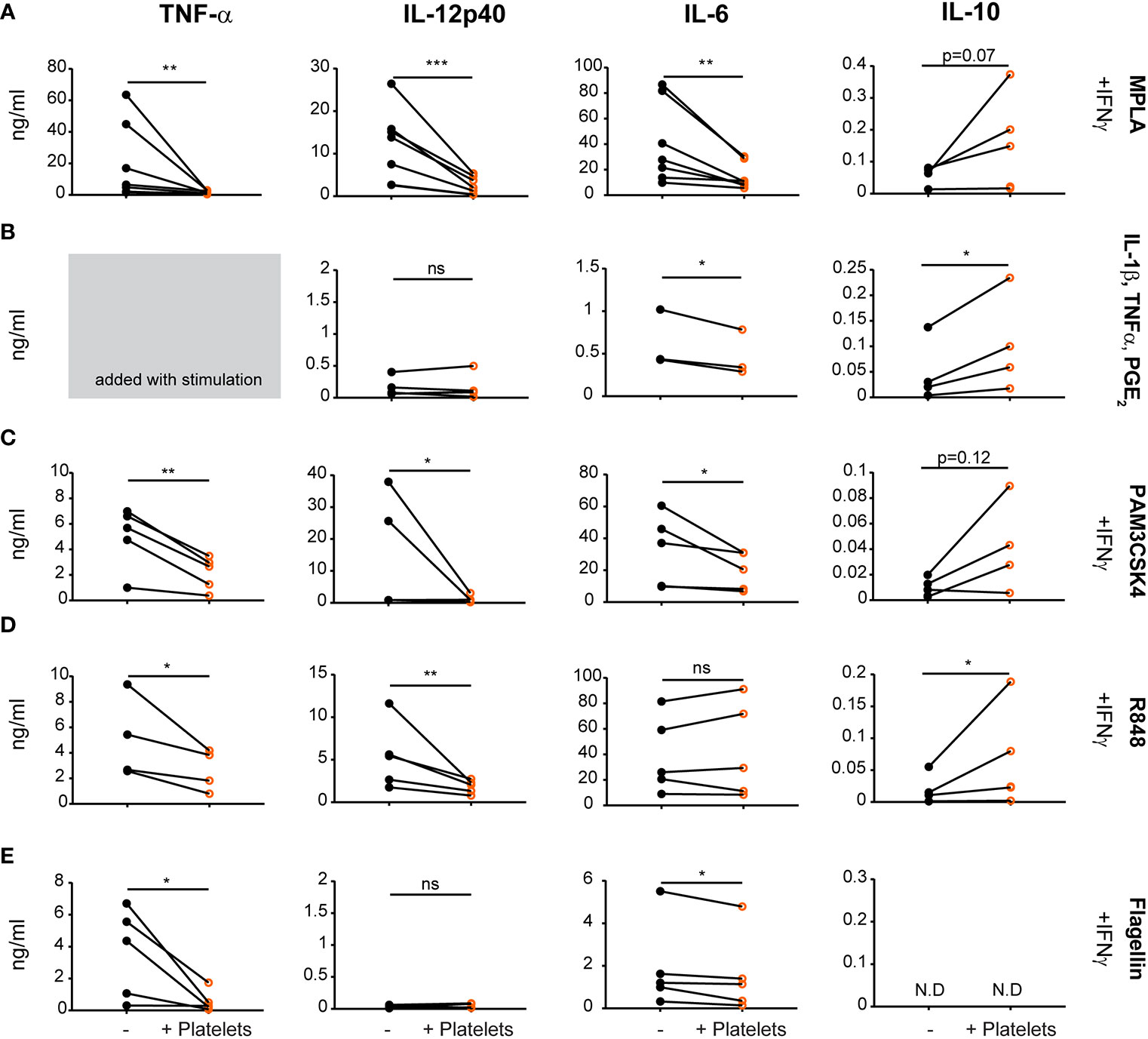
Figure 1 Platelets decrease monocyte-derived dendritic cells (moDC) pro-inflammatory cytokine production upon activation with different stimuli. moDCs were stimulated with (A) monophosporyl lipid A (MPLA), (B) a cytokine cocktail containing IL-1β, TNFα and prostaglandin E2 (PGE2), (C) pam3CysSerLys4 (PAM3CSK), (D) R848, or (E) Flagellin (A, C–E) in the presence of IFNγ (A–E) in absence or presence of platelets (platelet:DC ratio 40:1). After 48 h, supernatant was harvested and production of TNF-α, IL-12p40, IL-6, and IL-10 was determined by ELISA. Differences in cytokine production (mean ± SEM) were determined by paired t-tests, (A, C–E) n=5 donors; five independent experiments (B) n=4 donors; two independent experiments. (A–E) IL-6 panel B and IL-10 panel E fewer replicates due to values below detection limit. N.D. not detectable. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Stimulation with MPLA/IFNγ or IL-1β/TNFα/PGE2 induced the highest expression of co-stimulatory molecules as measured by flow cytometry. Interestingly, platelets minimally affected the phenotype of MPLA/IFNγ or IL-1β/TNFα/PGE2 stimulated DCs, with only limited changes in expression of CD83 or CD40 and CD86, respectively (Supplementary Figure S1C; for gating strategy see Supplementary Figure S2A). In summary, platelets reduced moDC pro-inflammatory cytokine production after various DAMP/PAMP stimulations, despite minimal effects on co-stimulatory molecule expression.
The Effect of Platelets on moDC Cytokine Production Is Mediated by a Soluble Factor
Next, the contact dependency of platelet mediated inhibition of DC cytokine production was investigated. When platelets were cultured with DCs in a transwell system to prevent direct cell-cell contact, IL-12p40, IL-6, and TNF-α production by stimulated moDCs was also reduced although to a slightly lower extent, which possibly can be explained by a concentration gradient-induced effect of the transwell itself (Figure 2A). Hence platelets inhibited pro-inflammatory cytokine production by moDCs also without direct cell-cell contact.
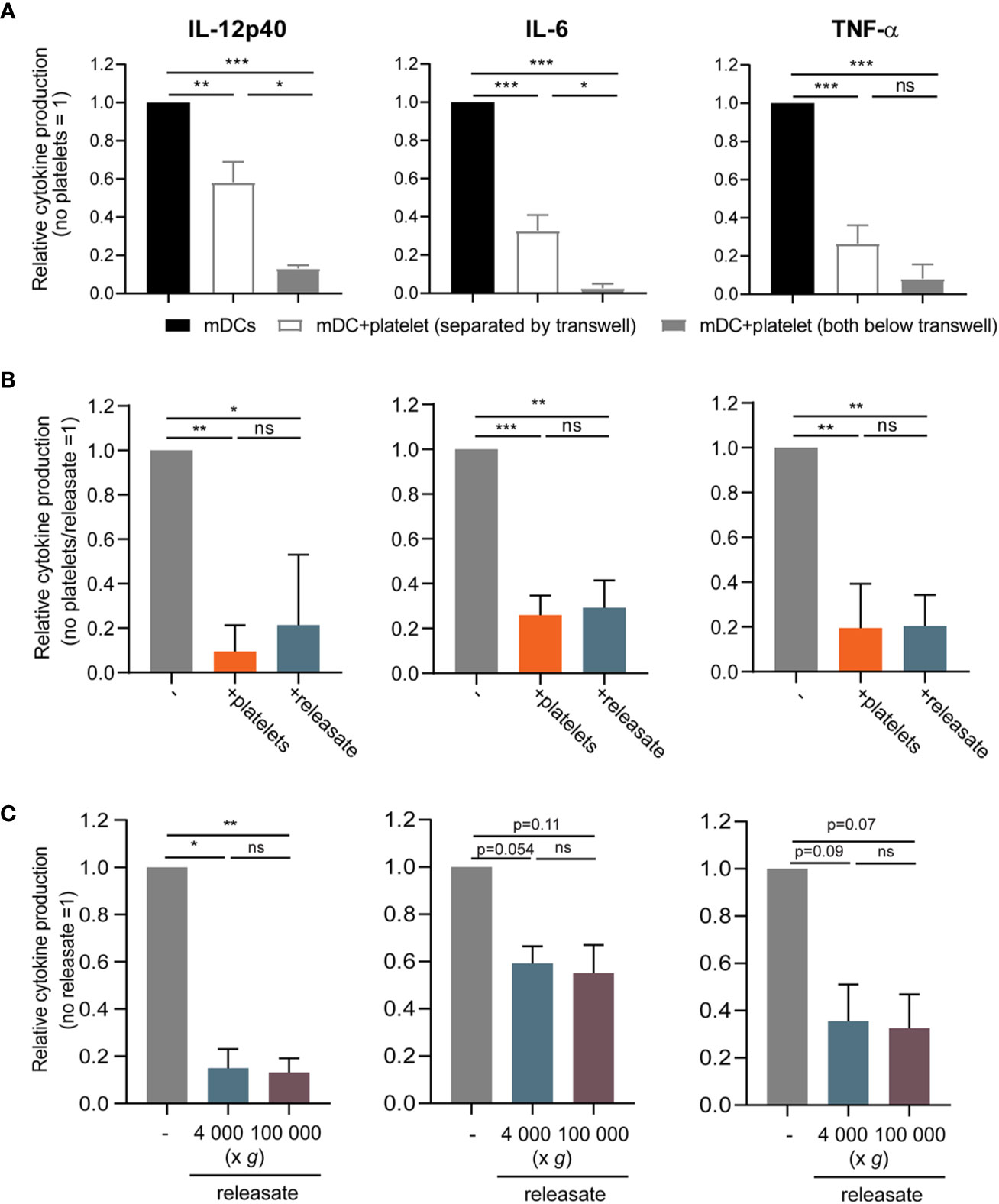
Figure 2 The effect of platelets on monocyte-derived dendritic cells (moDC) cytokine production is mediated by a soluble factor. (A) moDCs were stimulated with monophosporyl lipid A (MPLA)/IFNγ in a transwell system (pore size ≤ 0.3 µm) in absence or presence of thrombin receptor-activating peptide (TRAP)-activated platelets (platelet:DC ratio 40:1) that were either below the transwell (i.e. in the same compartment as the DCs) or separated by the transwell (n=4 donors; four independent experiments) (no platelets: IL-12p40 211-110714 pg/ml mean 23,068 pg/ml; IL-6 927.0 -127446 pg/ml mean 34,503 pg/ml; TNFα 352-44501 pg/ml mean 14,471 pg/ml). (B, C) moDCs were stimulated with MPLA/IFNγ (B) in absence or presence of TRAP-activated whole platelets or platelet releasate centrifuged at 4,000 × g for 10 min ((equivalent) platelet:DC ratio 50:1 (n=4 donors; two independent experiments)) (no platelets: IL-12p40 24568-495930 pg/ml mean 233,399 pg/ml; IL-6 35500-62984 pg/ml mean 51,077 pg/ml; TNFα 6670-42400 pg/ml mean 24,768 pg/ml) or (C) in absence or presence of platelet releasate (equivalent platelet:DC ratio 50:1) centrifuged at 4000 g for 10 min (microvesicles in suspension) or additionally at 100 000 g for 1 h (no microvesicles in suspension) (n=3 donors; three independent experiments) (no platelets: IL-12p40 3370-10800 pg/ml mean 6,220 pg/ml; IL-6 5230-34900 pg/ml mean 17,168 pg/ml; TNFα 7540-32400 pg/ml mean 15,427 pg/ml). (A–C) After 48 h, supernatant was harvested and production of TNF-α, IL-12p40, and IL-6 was determined by ELISA. Differences in relative cytokine production (mean ± SEM) were determined using repeated measures one-way ANOVA with Tukey post testing. *p ≤ 0.05, **p ≤ 0.01.
To further explore the involvement of platelet-derived soluble mediators, the releasate secreted by activated platelets was tested. For this, isolated platelets were activated with the PAR1 agonist thrombin receptor activator peptide-6 (TRAP-6). After activation, the platelet suspension was centrifuged twice to remove whole platelets and cell fragments. Whole platelets and platelet releasate from the same platelet donor induced a comparable decrease of pro-inflammatory cytokine production by MPLA/IFNγ stimulated moDCs (Figure 2B), while TRAP-6 itself did not affect moDC viability or cytokine production (Supplementary Figures S2B, C). Lastly, to exclude involvement of microvesicles, the releasate was ultracentrifuged at 100,000 g. Releasate with or without ultracentrifugation equally inhibited pro-inflammatory cytokine production by stimulated moDCs (Figure 2C). Overall, platelets inhibit MPLA/IFNγ-stimulated moDC pro-inflammatory cytokine production via a soluble factor, and not through release of microvesicles.
As platelets are a major source of DC-tolerizing active TGF-β in plasma (46–50) and platelet-derived TGF-β was shown to inhibit the anti-tumor T cell response in a murine model (29, 51), we investigated the involvement of TGF-β in platelet-mediated DC inhibition. Both platelets and DCs possess machinery to activate latent TGF-β and release the mature 25-kDa dimer (29, 48). Upon chemical activation, inducing dissociation of the mature TGF-β dimer from latent TGF-β binding proteins (LTBPs), we could detect active TGF-β1 in platelet releasate (Supplementary Figure S3A), indicating that latent TGF-β is present and may potentially be activated in our culture system. However, blocking TGF-β in platelet releasate could not restore cytokine production by moDCs (Supplementary Figure S3B-C), indicating that TGF-β is not the responsible factor in the releasate for the inhibition of cytokine production by moDC.
Platelet Releasate Affects Myeloid Conventional DC2 TNFα Production
In human blood, two types of myeloid or conventional DCs (myeloid cDC1 (CD141+) and cDC2 (CD1c+)) can be distinguished (32) and there is a subset of slan+ non-classical monocytes that show DC-like characteristics (52). To investigate to what extent our findings translate to human primary blood DCs, we isolated human myeloid CD1c+ conventional DC2 and slan+ non-classical monocytes from blood (Supplementary Figure S4).
After overnight rest, isolated myeloid cDC2’s and slan+ non-classical monocytes were stimulated with R848 as primary DCs were found to not respond to stimulation with MPLA/IFNγ (data not shown; (45)). Interestingly, platelet releasate significantly decreased TNF-α production by myeloid cDC2 and a trend for IL-12p40 inhibition was observed (Figure 3A), whereas cytokine production by slan+ non-classical monocytes was not affected (Figure 3B). Unfortunately, IL-10 production could not be detected for any of the primary DC subsets tested (data not shown). These findings confirm that the platelet-mediated effects found on moDC cytokine production could partly be validated in a subset of primary human blood DCs, namely myeloid cDC2s.
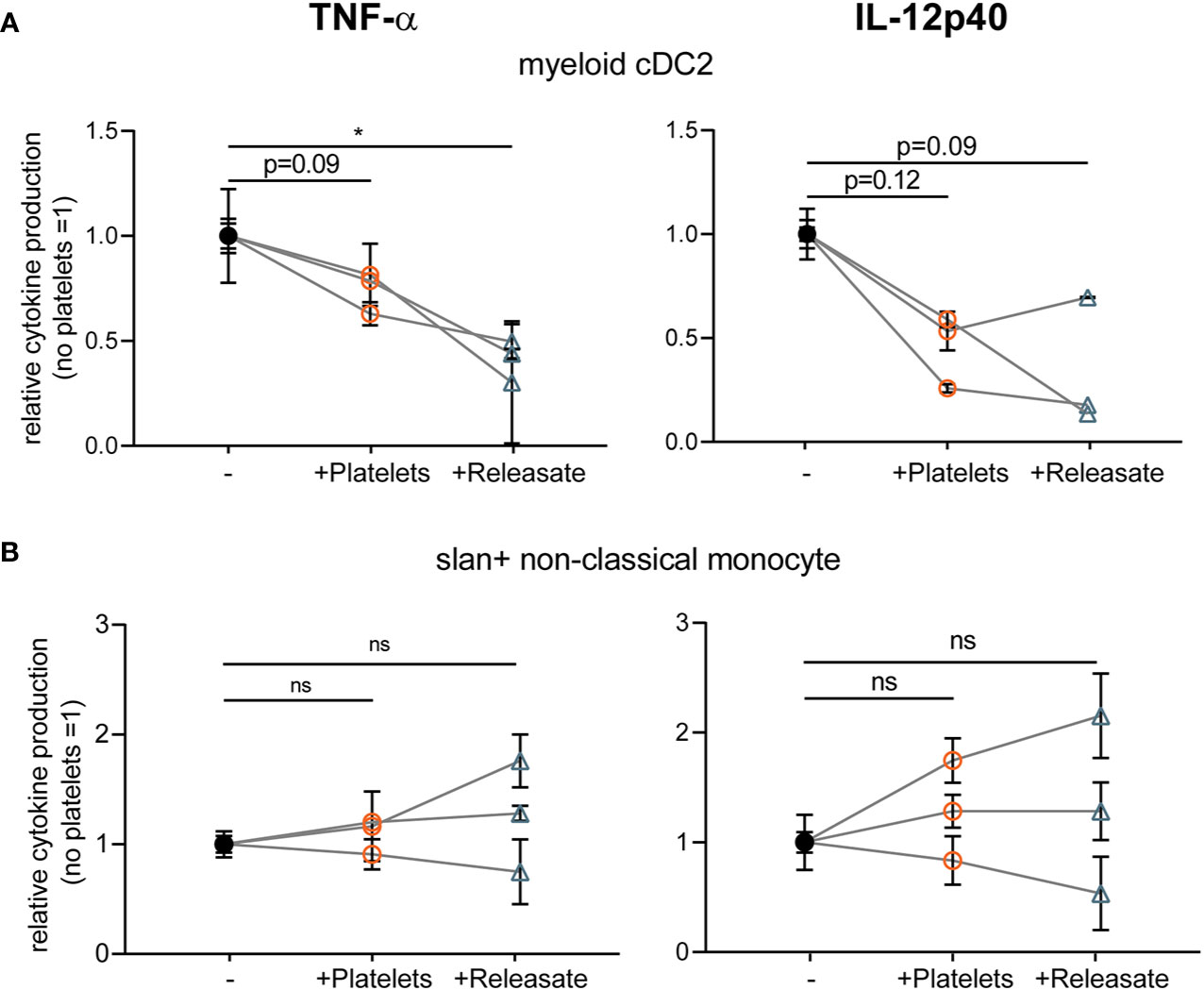
Figure 3 Platelet releasate affects myeloid conventional DC2 TNFα production. Primary human blood (A) myeloid CD1c+ conventional DC2 and (B) slan+ non-classical monocytes isolated from buffy coats were (A, B) stimulated with R848 in absence or presence of platelets or platelet releasate (centrifuged 4000 × g 10’) ((equivalent) platelet: dendritic cells DC ratio 50:1). After 24 h, supernatant was harvested and production of TNF-α (left; no platelets myeloid cDC2 141-364 pg/ml mean 284.7 pg/ml; slanDC 959-10100 pg/ml mean 4,306.5 pg/ml) and IL-12p40 (right; no platelets myeloid cDC2 111-335 pg/ml mean 226 pg/ml; slanDC 930-1530 pg/ml mean 1,238.3 pg/ml) was determined by ELISA (n=3 donors; three independent experiments). Differences in relative cytokine production (mean ± SEM) were determined using repeated measures one-way ANOVA with Tukey post testing. *p ≤ 0.05.
Platelets and Platelet Releasate Inhibit T Cell Activation and Skewing of Naïve T Cells Toward an IFNγ+ Th1 Phenotype by moDCs
Above we have shown that platelets and platelet releasate affect pro-inflammatory cytokine production by DCs, but the effect on subsequent T cell responses remains largely unresolved. To investigate the effect of platelets on DC-mediated T cell stimulation, moDCs were loaded with tetanus toxoid (TT) and subsequently stimulated with MPLA/IFNγ in presence of platelets. MoDCs stimulated in presence of platelets induced less TT-specific autologous CD4+ and CD8+ T cell proliferation (Figure 4A and Supplementary Figure S5A). A similar decrease in the ability of moDCs to induce allogeneic CD4+ but not CD8+ proliferation under influence of platelets or platelet releasate was found (Figure 4B and Supplementary Figures S5B, C).
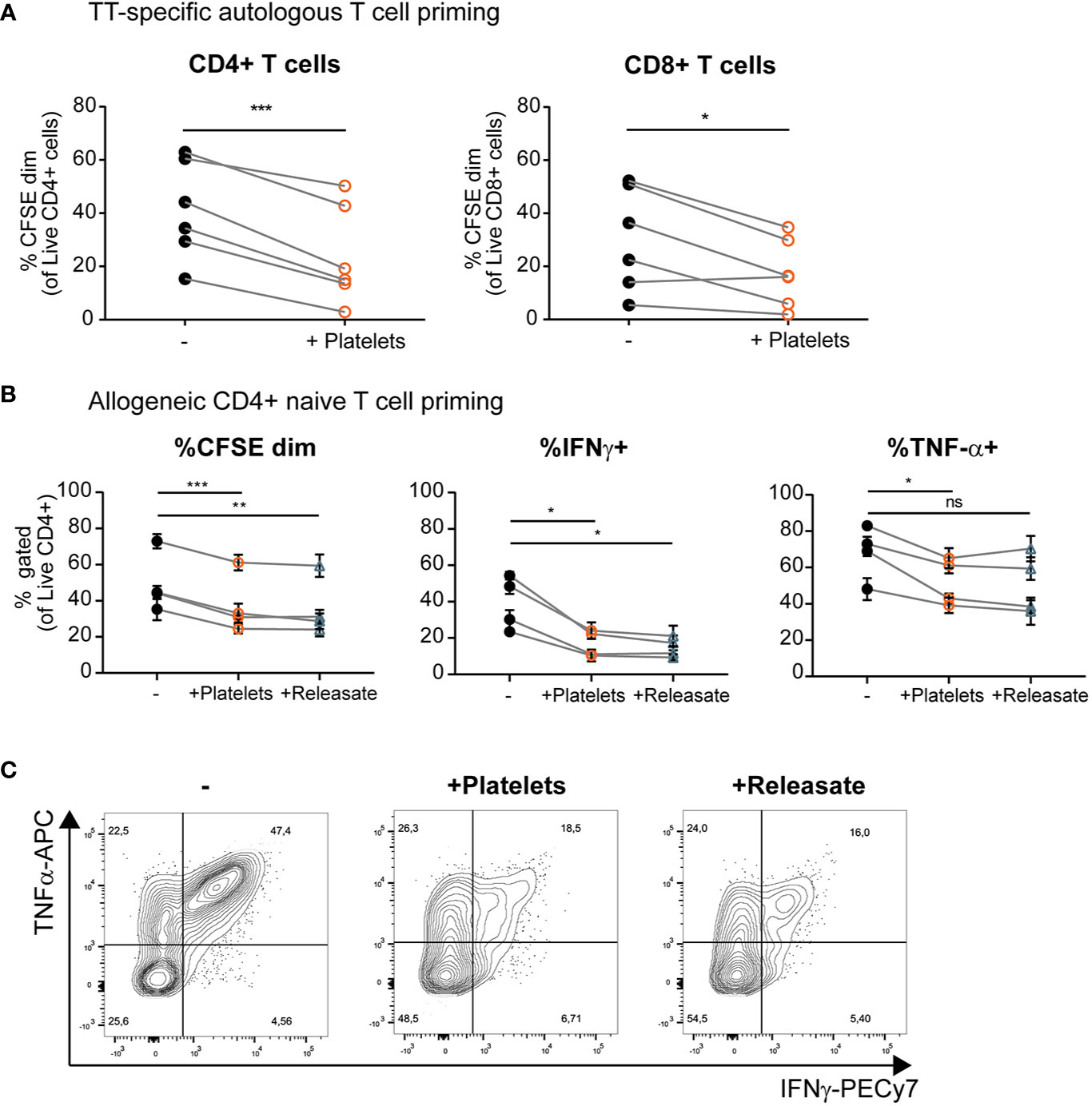
Figure 4 Platelets and platelet releasate inhibit T cell activation and skewing of naïve T cells toward an IFNγ+ Th1 phenotype by monocyte-derived dendritic cells (moDCs). (A) After 1 h of pre-incubation with tetanus toxoid (TT), moDCs were stimulated with monophosporyl lipid A (MPLA)/IFNγ in absence or presence of whole platelets (platelet: dendritic cells DC ratio 40:1). After 48 h, DCs were harvested and incubated 1:10 with CFSE-labeled autologous lymphocytes. After 8 days, T cell proliferation was determined using flow cytometry (n=6 donors; six independent experiments) (B, C) moDCs were stimulated with MPLA/IFNγ in absence or presence of whole platelets or platelet releasate (centrifuged 4000 x g 10’)((equivalent) platelet:DC ratio 50:1). After 48 h, DCs were harvested and incubated 1:10 with CFSE labeled allogeneic naïve CD4+ CD45RA+ T cells. After 10 days, T cell proliferation and intracellular cytokine production was determined by flow cytometry (n=4 donors; two independent experiments). (C) Representative plots of intracellular TNFα and IFNγ production by naïve CD4+ CD45RA+ T cells stimulated as described above. (A) Differences in proliferation were determined using paired t-tests. (B) Differences in proliferation and cytokine production (mean ± SEM) were determined by repeated measures one-way ANOVA with Tukey post testing. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Another interesting aspect of DC biology is their ability to skew naïve T cells toward a specific T helper phenotype upon the initiation of an immune response. MoDCs were cultured with MPLA/IFNγ in presence of whole platelets and platelet releasate. In a subsequent co-culture with naïve CD4+ CD45RA+ T cells, moDCs stimulated in presence of platelets or platelet releasate induced less allogeneic proliferation of naïve T cells (Figure 4B, left panel, gating strategy in Supplementary Figure S6A). Additionally, divided T cells (CFSEdim) displayed decreased IFNγ production compared to control co-cultures where moDCs were stimulated in the absence of platelets or platelet releasate (Figures 4B, middle panel, and 4C). Furthermore, the divided T cells produced less TNFα compared to control co-cultures where moDCs were stimulated in the absence of platelets but not platelet releasate (Figures 4B, right panel, and 4C). No significant production of IL-10 was detected in any of the DC-T cell co-cultures with or without platelet releasate (data not shown).
To determine whether effects on T cell skewing are dependent on the type of stimulation, cultures were performed with moDCs stimulated with TNFα, IL-1β; and PGE2. We observed an increase in moDC anti-inflammatory IL-10 production when stimulated in presence of platelet releasate (Supplementary Figure S6B). Simultaneously, we observed a decrease in IL-6 production (Supplementary Figure S6B; TNFα could not be analyzed as it was present in the stimulation cocktail) and also a decreased ability to induce naïve T cell proliferation as measured by CFSE dilution by flow cytometry (Supplementary Figure S6C, left panel). No effect was found on the IFNγ production of these naïve T cells compared to controls (Supplementary Figure S6C, right panel). All in all, platelets and platelet releasate can decrease the ability of moDCs to induce both memory and naïve T cell proliferation. Additionally, the ability of MPLA/IFNγ stimulated moDCs to induce IFNγ+ Th1 responses was reduced when stimulated in presence of platelets or platelet releasate.
Discussion
The mechanisms underlying TRIM are hard to investigate, but the role platelets themselves may play in this immune modulatory phenomenon after transfusion is increasingly being acknowledged (12, 31). Mechanisms of immune suppression as a result of TRIM are worth elucidating as these might be both beneficial (e.g. decreased graft rejection) or detrimental (e.g. recurrence resected malignancies) for the patient (14, 15, 18). However, much is still unclear about platelet-mediated immune modulation on DCs and whether platelets also affect subsequent ability of DCs to prime naïve and re-activate memory T cells. Here we show that a soluble factor secreted by platelets affects both moDC and primary myeloid CD1c+ conventional DC2 cytokine production under different stimulation conditions. Furthermore, these platelet-modulated moDCs are less capable to induce T cell proliferation and to skew naïve T cells toward a Th1 IFNγ+ phenotype.
Activation of transfused platelets could be induced by the so-called platelet storage lesion (53) or after transfusion by vascular damage in the recipient, which is frequently observed in hemato-oncological patients (7). Our observation of activated platelets decreasing pro-inflammatory responses at a potential physiological site of tissue damage makes sense when considering that here platelet-mediated immune suppression could favor wound healing over an excessive inflammatory response. In the setting of TRIM however, interaction of activated platelets with DCs might also occur in absence of tissue injury. This is an important mechanism to consider especially in thrombopenic and immune-compromised hemato-oncological patients whom are the primary recipients of platelet transfusions (7). Within the vasculature, transfused activated platelets might come across blood DCs (53–55) or encounter APCs while passing through the white and red pulp of the spleen (56).
Previous studies have reported contrasting results on the effect of platelets on DC activation. Platelets have generally been found to increase activation of DCs in absence of an additional stimulus (36, 39–42), while in presence of TLR-4 stimulus (i.e. LPS) platelets suppress DCs pro-inflammatory capacity or even induce an anti-inflammatory phenotype (33, 34, 37, 38, 43). In line, here platelets are shown to decrease pro-inflammatory cytokine production by DCs after stimulation with various TLR-dependent as well as TLR-independent agonists, implicating a more generic effect. Hagihara and colleagues described previously that moDCs activated with LPS in presence of platelets showed increased IL-10 production (33). Here, in agreement with a general immune suppressive effect, we found an increased IL-10 production for R848/IFNγ- and IL-1β/TNFα/PGE2- stimulated moDCs in the presence of platelets.
Despite the profound effect of platelet-derived mediators on moDC cytokine production, the effect on costimulatory marker expression was limited. Previous studies have shown varying effects of platelets on co-stimulatory marker expression by DCs (33–36, 38, 43). We have previously observed that inhibition of cytokine production by moDCs is not always linked to reduced co-stimulatory marker expression (57). To gain more insight in the mechanism behind reduced cytokine expression with limited effects on co-stimulatory marker expression, it is important to investigate at which level of the DC activation signaling cascade platelets or platelet-derived factors interfere. Irrespectively, we have shown that moDCs stimulated in presence of platelets showed decreased ability to activate a TT-specific memory response as well as induce proliferation and Th1 IFNγ+ skewing of naïve T cells in an allogeneic setting.
A number of studies have also reported on direct effects of platelets on T cell proliferation and cytokine production (29, 51, 58). Despite extensive washing, complex formation between platelets and DCs might occur during DC stimulation (13, 41). However, our experiments using platelet releasate where platelet-derived factors were washed away before co-culture of DCs and T cells show very comparable effects on T cell proliferation and cytokine production. This demonstrates that the effects of platelets on T cell proliferation and cytokine production are highly likely to be mediated through a direct effect of platelets on DC effector function.
Interestingly, experiments performed with platelet releasate showed that the platelet-mediated effects on DC cytokine production and T cell priming capacity are primarily mediated by soluble factors and are not mediated by microvesicles. We are the first to show that the platelet-mediated effects on DC cytokine production are not mediated by microvesicles. Microvesicles were previously shown to affect HLA II and CD80 expression as well as phagocytic capacity of immature moDCs differentiated in the presence of microparticles shed by stored platelets (38), and were shown to inhibit human plasmacytoid DC pro-inflammatory cytokine production (59).
The immune modulatory factor(s) secreted by platelets that affect DC function remain(s) to be identified. Platelets are capable of secreting a wide range of mediators that could influence DC biology (12, 30). For example, TGF-β is activated and expressed by platelets and is known to alter DC effector functions by directing toward a tolerance-inducing phenotype (46–49). Here we show for the first time that platelet-derived TGF-β is unlikely to be involved in platelet-induced inhibition of DC. Next to TGF-β, platelets are able to express and secrete (soluble) CD40L, but as this molecule usually induces DC maturation and upregulation of cytokine production (40, 60, 61) (s)CD40L is an unlikely mediator of the results described here. For CXCL4 (platelet factor 4) contrasting literature exists on its effects on DC function, with reports documenting anti-inflammatory effects (62) as well as potentiated response to TLR ligands upon culture of moDCs in presence of CXCL4 (63, 64). In addition, multiple factors could act together to mediate suppression of pro-inflammatory cytokine production by moDC.
Further research is warranted to elucidate which platelet-derived factors affect DC activation. We propose for upcoming studies to first validate whether the platelet-derived factor is (partly) of a proteinaceous nature, or whether e.g. platelet metabolites are responsible for the observed effects. An unbiased approach using liquid chromatography, potentially in combination with mass spectrometry (as described in (29)) would be interesting to identify the responsible platelet-derived factor(s) involved in the inhibition of DC effector functions.
Platelet-mediated inhibition was not only found for moDC but also observed for myeloid conventional DC2’s isolated from the blood, in line with previous research using platelet concentrates (36, 37). However, as these studies were performed in a whole blood culture, indirect effects on DCs via other cells could not be excluded. Here we show that platelet-derived soluble mediators inhibit pro-inflammatory cytokine production of purified myeloid cDC2, thus proving a direct effect of platelet (releasate) on primary DCs. As myeloid cDC2s have an extensive array of PRRs and a widespread tissue distribution, platelet-mediated inhibition could affect immunity against a wide range of pathogens (32). Interestingly, cytokine production of slan+ non-classical monocytes was not affected, which may be explained by their intrinsic capacity of higher production of IL-12 and TNFα compared to myeloid cDCs (65, 66).
In summary, we have shown that platelets reduce pro-inflammatory cytokine production of both moDCs and primary myeloid CD1c+ conventional DC2s. This inhibition is mediated by a soluble factor, and was observed in the presence of a range of both TLR-dependent as well as TLR-independent stimuli. Moreover, moDCs stimulated in the presence of platelets show a decreased ability to induce (antigen-specific) T cell proliferation as well as naïve T cell skewing toward a Th1 IFNγ+ phenotype. Together, these data show that platelets can influence the adaptive T cell response by inhibition of DC activation, with potential implications in platelet transfusions.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethical Advisory Council of Sanquin Blood Supply Foundation and are in accordance with the declaration of Helsinki and Dutch regulations. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
AS and JS conceptualized and designed the study, acquired and interpreted data, wrote and revised the manuscript. DS and GS acquired data. JZ, SH, and AB conceptualized and designed the study, interpreted data and critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Sanquin Blood Supply Foundation (grant PPOC 17-44).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the central facility at Sanquin for help with operation of the ELUTRA system and isolation of primary DCs.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.631285/full#supplementary-material
Abbreviations
CFSE, Carboxyfluoresceïne succinimidyl ester; DAMP, damage-associated molecular pattern; FNHTR, febrile non-hemolytic transfusion reactions; IFNγ; interferon γ; IL-1β; interleukin 1β; LTBP, latent TGF-β binding protein; MPLA, monophosphoryl lipid A; PAM3CSK4, pam3CysSerLys4; PAMP, pathogen-associated molecular pattern; PGE2, prostaglandin E2; PMA, phorbol myristate acetate; TGFβ; transforming growth factor β; TNFα; tumor necrosis factor α; TRALI, transfusion related acute lung injury; TRAP-6, thrombin receptor-activating peptide 6; TRIM, transfusion related immune modulation; TT, tetanus toxoid.
References
1. Stroncek D, Rebulla P. Platelet transfusions. Lancet (2017) 370:1391–401. doi: 10.1007/978-3-319-47462-5_94
2. Cameron B, Rock G, Olberg B, Neurath D. Evaluation of platelet transfusion triggers in a tertiary-care hospital. Transfusion (2007) 47:206–11. doi: 10.1111/j.1537-2995.2007.01090.x
3. Charlton A, Wallis J, Robertson J, Watson D, Iqbal A, Tinegate H. Where did platelets go in 2012? A survey of platelet transfusion practice in the North of England. Transfus Med (2014) 24:213–8. doi: 10.1111/tme.12126
4. Greeno E, McCullough J, Weisdorf D. Platelet utilization and the transfusion trigger: A prospective analysis. Transfusion (2007) 47:201–5. doi: 10.1111/j.1537-2995.2007.01089.x
5. Mayr WR. Blood transfusion in Europe-The White Book 2005: The patchwork of transfusion medicine in Europe. Transfus Clin Biol (2005) 12:357–8. doi: 10.1016/j.tracli.2005.10.006
6. Estcourt L, Stanworth S, Doree C, Hopewell S, Murphy MF, Tinmouth A, et al. Prophylactic platelet transfusion for prevention of bleeding in patients with haematological disorders after chemotherapy and stem cell transplantation. Cochrane Database Syst Rev (2012) 5:CD004269. doi: 10.1002/14651858.CD004269.pub3
7. Wandt H, Schäfer-Eckart K, Greinacher A. Platelet transfusion in hematology, oncology and surgery. Dtsch Arztebl Int (2014) 111:809–15. doi: 10.3238/arztebl.2014.0809
8. Katus MC, Szczepiorkowski ZM, Dumont LJ, Dunbar NM. Safety of platelet transfusion: Past, present and future. Vox Sang (2014) 107:103–13. doi: 10.1111/vox.12146
9. Eder AF, Moroff G. Platelet storage and adverse transfusion outcomes: Old platelets? Transfusion (2010) 50:2288–91. doi: 10.1111/j.1537-2995.2010.02901.x
10. Semple JW, Rebetz J, Kapur R. Transfusion-Associated Circulatory Overload and Transfusion-Related Acute Lung Injury. Blood Rev (2019) 33:767–79. doi: 10.1016/j.hoc.2019.05.003
11. Muszynski JA, Spinella PC, Cholette JM, Acker JP, Hall MW, Juffermans NP, et al. Transfusion-related immunomodulation: review of the literature and implications for pediatric critical illness. Transfusion (2017) 57:195–206. doi: 10.1111/trf.13855
12. Sut C, Tariket S, Aubron C, Aloui C, Hamzeh-Cognasse H, Berthelot P, et al. The non-hemostatic aspects of transfused platelets. Front Med (2018) 5:42. doi: 10.3389/fmed.2018.00042
13. Saris A, Peyron I, van der Meer PF, Stuge TB, Zwaginga JJ, van Ham SM, et al. Storage-induced platelet apoptosis is a potential risk factor for alloimmunization upon platelet transfusion. Front Immunol (2018) 9:1251. doi: 10.3389/fimmu.2018.01251
14. Blumberg N, Heal JM. Effects of transfusion on immune function. Cancer recurrence and infection. Arch Pathol Lab Med (1994) 118:371–9.
15. Heiss MM, Mempel W, Delanoff C, Jauch KW, Gabka C, Mempel M, et al. Blood transfusion-modulated tumor recurrence: first results of a randomized study of autologous versus allogeneic blood transfusion in colorectal cancer surgery. J Clin Oncol (1994) 12:1859–67. doi: 10.1200/JCO.1994.12.9.1859
16. Jensen LS, Kissmeyer-Nielsen P, Wolff B, Qvist N. Randomised comparison of leucocyte-depleted versus buffy-coat-poor blood transfusion and complications after colorectal surgery. Lancet (London England) (1996) 348:841–5. doi: 10.1016/S0140-6736(96)06168-5
17. Vamvakas EC, Blajchman MA. Transfusion-related immunomodulation (TRIM): An update. Blood Rev (2007) 21:327–48. doi: 10.1016/j.blre.2007.07.003
18. Aslam R, Speck ER, Kim M, Freedman J, Semple JW. Transfusion-related immunomodulation by platelets is dependent on their expression of MHC Class I molecules and is independent of white cells. Transfusion (2008) 48:1778–86. doi: 10.1111/j.1537-2995.2008.01791.x
19. Geiger TL. Transfusion-associated immune modulation: a reason to TRIM platelet transfusions? Transfusion (2008) 48:1772–3. doi: 10.1111/j.1537-2995.2008.01860.x
20. Nelson KA, Aldea GS, Warner P, Latchman Y, Gunasekera D, Tamir A, et al. Transfusion-related immunomodulation: gamma irradiation alters the effects of leukoreduction on alloimmunization. Transfusion (2019) 59:3396–404. doi: 10.1111/trf.15555
21. Frojmovic MM, Milton JG. Human platelet size, shape, and related functions in health and disease. Physiol Rev (1982) 62:185–261. doi: 10.1152/physrev.1982.62.1.185
22. Blair P, Flaumenhaft R. Platelet α–granules: Basic biology and clinical correlates. Blood Rev (2009) 23:177–89. doi: 10.1016/j.blre.2009.04.001.Platelet
23. Cognasse F, Nguyen KA, Damien P, McNicol A, Pozzetto B, Hamzeh-Cognasse H, et al. The inflammatory role of platelets via their TLRs and Siglec receptors. Front Immunol (2015) 6:83. doi: 10.3389/fimmu.2015.00083
24. Semple JW, Italiano JE, Freedman J. Platelets and the immune continuum. Nat Rev Immunol (2011) 11:264–74. doi: 10.1038/nri2956
25. Duerschmied D, Bode C, Ahrens I. Immune functions of platelets. Thromb Haemost (2014) 112:678–91. doi: 10.1160/TH14-02-0146
26. Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, et al. The role of platelets in mediating a response to human influenza infection. Nat Commun (2019) 10:1780. doi: 10.1038/s41467-019-09607-x
27. Elzey BD, Tian J, Jensen RJ, Swanson AK, Lees JR, Lentz SR, et al. Platelet-mediated modulation of adaptive immunity: a communication link between innate and adaptive immune compartments. Immunity (2003) 28:555–60. doi: 10.1016/j.smim.2016.10.008
28. Elzey BD, Grant JF, Sinn HW, Nieswandt B, Waldschmidt TJ, Ratliff TL. Cooperation between platelet-derived CD154 and CD4+ T cells for enhanced germinal center formation. J Leukoc Biol (2005) 78:80–4. doi: 10.1189/jlb.1104669
29. Rachidi S, Metelli A, Riesenberg B, Wu BX, Nelson MH, Wallace C, et al. Platelets subvert T cell immunity against cancer via GARP-TGF axis. Sci Immunol (2017) 2:7911. doi: 10.1126/sciimmunol.aai7911
30. Kapur R, Zufferey A, Boilard E, Semple JW. Nouvelle Cuisine: Platelets Served with Inflammation. J Immunol (2015) 194:5579–87. doi: 10.4049/jimmunol.1500259
31. Stolla M, Refaai MA, Heal JM, Spinelli SL, Garraud O, Phipps RP, et al. Platelet transfusion - The new immunology of an old therapy. Front Immunol (2015) 6:28. doi: 10.3389/fimmu.2015.00028
32. Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology (2018) 154:3–20. doi: 10.1111/imm.12888
33. Hagihara M, Higuchi A, Tamura N, Ueda Y, Hirabayashi K, Ikeda Y, et al. Platelets, after Exposure to a High Shear Stress, Induce IL-10-Producing, Mature Dendritic Cells In Vitro. J Immunol (2004) 172:5297–303. doi: 10.4049/jimmunol.172.9.5297
34. Kissel K, Berber S, Nockher A, Santoso S, Bein G, Hackstein H. Human platelets target dendritic cell differentiation and production of proinflammatory cytokines. Transfusion (2006) 46:818–27. doi: 10.1111/j.1537-2995.2006.00802.x
35. Singh MV, Suwunnakorn S, Simpson SR, Weber EA, Singh VB, Kalinski P, et al. Monocytes complexed to platelets differentiate into functionally deficient dendritic cells. J Leukoc Biol (2020) 2020:1–14. doi: 10.1002/JLB.3A0620-460RR
36. Ki KK, Faddy HM, Flower RL, Dean MM. Platelet concentrates modulate myeloid dendritic cell immune responses. Platelets (2018) 29:373–82. doi: 10.1080/09537104.2017.1306045
37. Perros AJ, Christensen AM, Flower RL, Dean MM. Soluble Mediators in Platelet Concentrates Modulate Dendritic Cell Inflammatory Responses in an Experimental Model of Transfusion. J Interf Cytokine Res (2015) 35:821–30. doi: 10.1089/jir.2015.0029
38. Sadallah S, Eken C, Martin PJ, Schifferli JA. Microparticles (Ectosomes) Shed by Stored Human Platelets Downregulate Macrophages and Modify the Development of Dendritic Cells. J Immunol (2011) 186:6543–52. doi: 10.4049/jimmunol.1002788
39. Ki KK, Johnson L, Faddy HM, Flower RL, Marks DC, Dean MM. Immunomodulatory effect of cryopreserved platelets: altered BDCA3+ dendritic cell maturation and activation in vitro. Transfusion (2017) 57:2878–87. doi: 10.1111/trf.14320
40. Czapiga M, Kirk AD, Lekstrom-Himes J. Platelets deliver costimulatory signals to antigen-presenting cells: A potential bridge between injury and immune activation. Exp Hematol (2004) 32:135–9. doi: 10.1016/j.exphem.2003.11.004
41. Hamzeh-cognasse H, Cognasse F, Palle S, Chavarin P, Olivier T, Delézay O, et al. Direct contact of platelets and their released products exert different effects on human dendritic cell maturation. BMC Immunol (2008) 15:1471–2172. doi: 10.1186/1471-2172-9-54
42. Nguyen XF, Müller-Berghaus J, Kälsch T, Schadendorf D, Borggrefe M, Klüter H. Differentiation of monocyte-derived dendritic cells under the influence of platelets. Cytotherapy (2008) 10:720–9. doi: 10.1080/14653240802378912
43. Tešić N, Pekle Simonič I, Roškar K, Rožman P, Švajger U. Dendritic Cells Generated in the Presence of Platelet Lysate Have a Reduced Type 1 Polarization Capacity. Immunol Invest (2019) 0139:215–31. doi: 10.1080/08820139.2019.1624768
44. ten Brinke A, Karsten ML, Dieker MC, Zwaginga JJ, van Ham SM. The clinical grade maturation cocktail monophosphoryl lipid A plus IFNγ generates monocyte-derived dendritic cells with the capacity to migrate and induce Th1 polarization. Vaccine (2007) 25:7145–52. doi: 10.1016/j.vaccine.2007.07.031
45. Zaal A, Dieker M, Oudenampsen M, Turksma AW, Lissenberg-Thunnissen SN, Wouters D, et al. Anaphylatoxin C5a regulates 6-Sulfo-LacNAc dendritic cell function in human through crosstalk with toll-like receptor-induced CREB signaling. Front Immunol (2017) 8:818. doi: 10.3389/fimmu.2017.00818
46. Blakytny R, Ludlow A, Martin GEM, Ireland G, Lund LR, Ferguson MWJ, et al. Latent TGF-beta1 activation by platelets. J Cell Physiol (2004) 199:67–76. doi: 10.1002/jcp.10454
47. Esebanmen GE, Langridge WHR. The role of TGF-beta signaling in dendritic cell tolerance. Immunol Res (2017) 65:987–94. doi: 10.1007/s12026-017-8944-9
48. Travis MA, Sheppard D. TGF-β Activation and Function in Immunity. Annu Rev Immunol (2014) 32:51–82. doi: 10.1146/annurev-immunol-032713-120257
49. Strobl H, Knapp W. TGF-β1 regulation of dendritic cells. Microbes Infect (1999) 1:1283–90. doi: 10.1016/S1286-4579(99)00256-7
50. Ramalingam R, Larmonier CB, Thurston RD, Midura-Kiela MT, Zheng SG, Ghishan FK, et al. Dendritic Cell-Specific Disruption of TGF-β Receptor II Leads to Altered Regulatory T Cell Phenotype and Spontaneous Multiorgan Autoimmunity. J Immunol (2012) 189:3878–93. doi: 10.4049/jimmunol.1201029
51. Riesenberg BP, Ansa-Addo EA, Gutierrez J, Timmers CD, Liu B, Li Z. Cutting Edge: Targeting Thrombocytes to Rewire Anticancer Immunity in the Tumor Microenvironment and Potentiate Efficacy of PD-1 Blockade. J Immunol (2019) 203:1105–10. doi: 10.4049/jimmunol.1900594
52. Calzetti F, Tamassia N, Micheletti A, Finotti G, Bianchetto-Aguilera F, Cassatella MA. Human dendritic cell subset 4 (DC4) correlates to a subset of CD14dim/–CD16++ monocytes. J Allergy Clin Immunol (2018) 141:2276–2279.e3. doi: 10.1016/j.jaci.2017.12.988
53. Ng MSY, Tung J-P, Fraser JF. Platelet Storage Lesions: What More Do We Know Now? Transfus Med Rev (2018) 32:144–54. doi: 10.1016/j.tmrv.2018.04.001
54. Langer HF, Daub K, Braun G, Schönberger T, May AE, Schaller M, et al. Platelets recruit human dendritic cells via Mac-1/JAM-C interaction and modulate dendritic cell function in vitro. Arterioscler Thromb Vasc Biol (2007) 27:1463–70. doi: 10.1161/ATVBAHA.107.141515
55. Maître B, Mangin PH, Eckly A, Heim V, Cazenave JP, Lanza F, et al. Immature myeloid dendritic cells capture and remove activated platelets from preformed aggregates. J Thromb Haemost (2010) 8:2262–72. doi: 10.1111/j.1538-7836.2010.03983.x
56. Villadangos JA, Heath WR. Life cycle, migration and antigen presenting functions of spleen and lymph node dendritic cells: limitations of the Langerhans cells paradigm. Semin Immunol (2005) 17:262–72. doi: 10.1016/j.smim.2005.05.015
57. Kolanowski STHM, Dieker MC, Lissenberg-thunnissen SN, Van Schijndel GMW, Van Ham SM. TLR4-mediated pro-inflammatory dendritic cell differentiation in humans requires the combined action of MyD88 and TRIF. Innate Immun (2013) 0:1–8. doi: 10.1177/1753425913498626
58. Zimmer N, Krebs FK, Zimmer S, Mitzel-rink H, Kumm EJ, Jurk K, et al. Platelet-Derived GARP Induces Peripheral Regulatory T Cells — Potential Impact on T Cell Suppression in Patients with. Cancers (Basel) (2020) 12:3653. doi: 10.3390/cancers12123653
59. Ceroi A, Delettre FA, Marotel C, Gauthier T, Asgarova A, Biichlé S, et al. The anti-inflammatory effects of platelet-derived microparticles in human plasmacytoid dendritic cells involve liver X receptor activation. Haematologica (2016) 101:72–6. doi: 10.3324/haematol.2015.135459
60. Sprague DL, Elzey BD, Crist SA, Waldschmidt TJ, Jensen RJ, Ratliff TL. Platelet-mediated modulation of adaptive immunity: Unique delivery of CD154 signal by platelet-derived membrane vesicles. Blood (2008) 111:5028–36. doi: 10.1182/blood-2007-06-097410
61. Nishat S, Wuescher LM, Worth RG. Platelets enhance dendritic cell responses against Staphylococcus aureus through CD40-CD40L. Infect Immun (2018) 86:00186–18. doi: 10.1128/IAI.00186-18
62. Xia C-Q, Kao K-J. Effect of CXC chemokine platelet factor 4 on differentiation and function of monocyte-derived dendritic cells. Int Immunol (2003) 15:1007–15. doi: 10.1093/intimm/dxg100
63. Silva-Cardoso SC, Tao W, Malvar Fernandez B, Boes M, Radstake TRDJ, Pandit A. CXCL4 suppresses tolerogenic immune signature of monocyte-derived dendritic cells. Eur J Immunol (2020) 50:1–9. doi: 10.1002/eji.201948341
64. Silva-Cardoso SC, Affandi AJ, Spel L, Cossu M, van Roon JAG, Boes M, et al. CXCL4 Exposure Potentiates TLR-Driven Polarization of Human Monocyte-Derived Dendritic Cells and Increases Stimulation of T Cells. J Immunol (2017) 199:253–62. doi: 10.4049/jimmunol.1602020
65. Ahmad F, Döbel T, Schmitz M, Schäkel K. Current Concepts on 6-sulfo LacNAc Expressing Monocytes (slanMo). Front Immunol (2019) 10:948. doi: 10.3389/fimmu.2019.00948
Keywords: platelet immunomodulation, transfusion-related immune modulation, platelet releasate, monocyte-derived dendritic cells, primary dendritic cell, T cell priming
Citation: Saris A, Steuten J, Schrijver DP, van Schijndel G, Zwaginga JJ, van Ham SM and ten Brinke A (2021) Inhibition of Dendritic Cell Activation and Modulation of T Cell Polarization by the Platelet Secretome. Front. Immunol. 12:631285. doi: 10.3389/fimmu.2021.631285
Received: 19 November 2020; Accepted: 20 January 2021;
Published: 25 February 2021.
Edited by:
Alexandre Corthay, Oslo University Hospital, NorwayReviewed by:
Kitti Pázmándi, University of Debrecen, HungaryNiels Schaft, University Hospital Erlangen, Germany
Jurjen Tel, Eindhoven University of Technology, Netherlands
Copyright © 2021 Saris, Steuten, Schrijver, van Schijndel, Zwaginga, van Ham and ten Brinke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anja ten Brinke, YS50ZW5icmlua2VAc2FucXVpbi5ubA==
ORCID: Anno Saris, orcid.org/0000-0003-0493-9501
†These authors have contributed equally to this work and share first authorship