 Kim Ohl
Kim Ohl Klaus Tenbrock
Klaus Tenbrock- Department of Pediatrics, Pediatric Rheumatology, Medical Faculty, RWTH Aachen University, Aachen, Germany
Oxidative stress is a major component of cellular damage in T cells from patients with systemic lupus erythematosus (SLE) resulting amongst others in the generation of pathogenic Th17 cells. The NRF2/Keap1 pathway is the most important antioxidant system protecting cells from damage due to oxidative stress. Activation of NRF2 therefore seems to represent a putative therapeutic target in SLE, which is nevertheless challenged by several findings suggesting tissue and cell specific differences in the effect of NRF2 expression. This review focusses on the current understanding of oxidative stress in SLE T cells and its pathophysiologic and therapeutic implications.
Introduction
Oxidative stress might be a central factor in the immunopathogenesis of Systemic Lupus Erythematosus (SLE) (1). There are data supporting the hypothesis that excessive reactive oxygen species (ROS) production is, along with many other factors, one of the factors that induce SLE (2). Indeed, ROS production associates with enhanced apoptosis and might delay clearance of apoptotic cells, both of which are hallmarks of SLE (3, 4). Apoptotic cells are a source of autoantigens (e.g., ribonucleoproteins, DNA), which can be recognized by auto-reactive T cells. Furthermore, apoptotic cells release danger-associated molecular patterns (DAMPs), DAMPs are danger signals, which induce inflammatory responses. Therefore, increased auto-antigenic exposure and decreased autoantigen removal might contribute to autoantibody production and autoimmunity. Peripheral blood mononuclear cells (PBMCs), T cells and neutrophils are exposed to increased concentrations of free radicals which leads to aberrant activation of these cells. In addition to this, mitochondrial dysfunction and ROS production of PBMCs themselves contribute to SLE pathogenesis, while mechanistic reasons that cause mitochondrial ROS production have not been identified (5). It is not clear if the chronic inflammation in SLE induces oxidative stress conditions or if ROS production is vice versa a cause of lupus pathology. Nevertheless, there is mounting evidence for a defective redox clearance in SLE. Patients with highly active disease show impaired activity of oxidative stress regulating enzymes, such as superoxide dismutase (SOD) and gluthathione peroxidase (GPX) in serum and saliva while PBMCs of active SLE patients reveal higher intracellular ROS than those of healthy controls (6–9). Increased oxygen intermediates have also been identified in macrophages of lupus mice (10). Therefore, studies even suggest considering assessment of redox status as a serological marker to evaluate disease activity and nephropathy in SLE (11–13).
On the other hand, ROS production has been associated with the prevention of autoimmune diseases. A genetically mediated deficiency of ROS production can contribute to initiation of autoimmunity and can facilitate disease progressing (14). A missense mutation in the p47phox (Neutrophil Cytosolic Factor 1, NCF1) subunit of NADPH oxidase (NOX) predisposes to SLE and other autoimmune diseases (15, 16). Mice with a mutation of Ncf1, which is associated with low ROS production, develop an accelerated lupus-like disease (16). In addition to this, point mutations in NCF2 are associated with increased SLE risk (17), since they reduce NOX activity and NCF-2 deficient or NCF-2 haplo-insufficient mice reveal accelerated lupus disease (18).
To sum up these findings, ROS play an ambivalent role in SLE, which depends on source, location, amount and most probably also time-point of their occurrence. As a consequence, balanced ROS levels are important to sustain an immune equilibrium which reduces tissue damage and prevents the development of autoimmunity (19, 20). But how is a redox balance achieved in our cells? To answer this question, we have to study NRF2, the main transcription factor of the anti-oxidative stress response.
NRF2
Oxidative stress can activate different transcriptional regulators, most importantly NRF2. In steady state conditions, NRF2 is bound in the cytosol by Kelch ECH associating protein 1 (KEAP1), which induces ubiquitinilation and degradation of NRF2 (21). Different cellular stimuli which induce oxidative stress result in conformational changes of KEAP1, which foster the release of NRF2 from KEAP1 followed by NRF2 translocation into the cellular nucleus and activation of genes containing an antioxidant response element (ARE) in their promoter regions (22). NRF2 thereby induces expression of phase II detoxifying enzymes and antioxidant proteins, which preserve cellular homeostasis by reducing chemical or oxidative stress molecules. These proteins include enzymes mediating glutathione (GSH) synthesis, the thioredoxin enzyme system and detoxifying enzymes such as heme oxygenases, or NAD(P)H: quinone oxidoreductase 1 (NQO1). In addition to induction of antioxidant genes, NRF2 also modulates immune responses by regulating transcription of several others including anti-inflammatory and metabolic genes in immune cells (23–26). So, being a critical regulator of cellular oxidative stress responses and inflammatory reactions, it is not surprising that NRF2 is indispensable to prevent cellular damage and subsequent inflammation. NRF2 deficient mice have problems to deal with inflammatory cues and therefore show a more sever phenotype in inflammation-mediated animal models like experimental asthma (27), acute lung injury (28), sepsis (29), T cell-mediated hepatitis (30), or dextran sulfate sodium-induced colitis (31) and arthritis (32). There are also several lines of evidence that NRF2 has a central role in the pathogenesis of SLE. Interestingly, aged female NRF2 deficient mice are prone to develop an autoimmune condition that closely resemble human SLE (33). Furthermore, NRF2 deficiency increased lupus nephritis and Th17 cells in B6/lpr mice (34). NRF2 furthermore suppresses Lupus nephritis by neutralizing ROS and preventing renal damage (35). A recent study revealed that NRF2 activation promotes resolution of chronic inflammation in lupus most probably by repolarization of macrophages and reduction of the IFN signature (36). In contrast, one study revealed a prolonged lifespan, improved autoimmune nephritis, and reduced lymphadenopathy of lupus mice in the absence of Nrf2 (37). However, these effects can be explained by a Nrf2 mediated suppression of the autoimmune accelerating gene lpr, which is used in this mouse model. It is therefore not clear, how this study can be transferred to the human SLE disease. With that regard, Gautam et al. found elevated ROS in SLE specific DCs, which might be due reduced clearance of ROS related to impaired levels of NRF2 (6). In addition, the association of the NRF2 -653G/A polymorphism with lupus nephritis in pediatric-onset SLE has been described (38). This polymorphism was only associated with nephritis, while there was no significant association between NRF2 -653G/A polymorphism and susceptibility to SLE. Another study identified three SNPs and one triplet polymorphism within the promotor and upstream regions of the NRF2 gene, but no association between risk of SLE and these polymorphisms. However, authors state that their analysis is preliminary and only a small number of patients were observed (39). Therefore, until now we can only assume the association between Nrf2 polymorphism and lupus nephritis.
In conclusion, there are several lines of evidence which support the conclusion that NRF2 activation is beneficial in SLE pathogenesis. However, it is not clear whether NRF2 - by induction of the antioxidant machinery - enhances cell survival and reduces apoptosis, which contributes to the control of systemic inflammation and limitation of autoimmune responses or by direct cellular mechanism including transcriptional regulation of inflammatory responses in immune cells.
A very interesting study addressing this question was performed by Suzuki et al., who analyzed NRF2 in the scurfy mouse model. These mice lack functional regulatory T cells (Tregs) and therefore develop mutiorgan-inflammation and autoimmunity at early ages. Interestingly, while systemic activation of NRF2 by Keap1 knockdown ameliorated tissue inflammation, NRF2 activation via cell lineage-specific Keap1 disruption (i.e., in T cells, myeloid cells, and dendritic cells) achieved only partial or no improvement in the inflammatory status of scurfy mice (40). This suggests NRF2 activation in multiple cell lineages appears to be required for sufficient anti-inflammatory effects. Our group even observed a pro-inflammatory effect of NRF2 when we specifically deleted NRF2 in Tregs (26). This raises the question if it is beneficial to use NRF2 activation in SLE as a therapeutic intervention. It is therefore important to first illuminate the exact role of NRF2 critically and ROS signaling in T cells in SLE.
NRF2 and Redox Metabolism in T Cells
Redox-dependent signaling pathways are of major importance in immune cells and specifically in T cells. They control different functions including T effector cell differentiation and migration, cell cycle progression and inflammation. The role of ROS in T cell signaling has been intensively reviewed elsewhere (41). We will therefore only give a short overview about the current knowledge in this field. The production of ROS is enhanced after T cell receptor stimulation and is necessary for clonal expansion. ROS which is produced in the mitochondrial electron transport chain (ETC) is important for the activation of NFAT, a T cell-specific transcription factor that facilitates interleukin-2 (IL-2) expression and thus cell cycle progression, which results in rapid proliferation of the T cells (42). However high ROS concentrations result in GSH depletion, since the murine Glutamate-Cysteine Ligase Catalytic Subunit (GCLC) is not expressed in murine T cells. GSH depletion negatively affects NFAT activity, mTOR signaling and MYC protein expression and thus inhibits T cell proliferation (43). The antioxidant GSH tightly regulates ROS activity, which thus controls cell cycle progression of T cells as well as their metabolic properties. The latter ones influence their differentiation program and as a consequence, the manner of protective immune responses. Thus, ROS influence metabolic programming; too much can be detrimental, but if applied correctly, it is important for protection of the host (19). For metabolic activity, Tregs rely on oxidative phosphorylation (OXPHOS), which produces ROS and which are also regulated by GSH. GSH generated by GCLC is indispensable for proper Treg function and lack of GCLC in Tregs results in autoimmunity (44).
Giving this critical role of ROS in T cells, it is obvious that NRF2, which regulates cellular redox homeostasis might also critically regulate T cell functions. It is known that NRF2 expression is increased in activated T cells in mice and men (26, 45, 46), however the exact role of NRF2 in T cells remains controversial. Constitutive activation of NRF2 in T cells was protective in a murine acute kidney injury model and involved higher frequencies but not total numbers of intrarenal Tregs, as well as reduced expression of inflammatory cytokines in CD4+ T cells (47). However constitutive ablation of NRF2 in T cells ameliorated graft-vs-host disease (GvHD) (46). Again, a study with human cells indicates that high expression of NRF2 in CD8+ T-cells might be protective against chronic GvHD (48). The role of oxidative stress in Tregs is controversely discussed. Mougiakakos et al. have been reported that Tregs are more resistant to oxidative stress-induced cell death than conventional T cells (49), while others have found higher toxicity of free oxygen species in Tregs to, which was attributed to a diminished NRF2-dependent antioxidant system (50). The weakness of the NRF2-associated antioxidant system facilitates Treg apoptosis but nevertheless causes enhanced immunosuppressive capacity within the tumor microenvironment. Our own study indicates that NRF2 is a negative regulator of Treg function and that Foxp3 specific activation of NRF2 results in a loss of Foxp3 expression and spontaneous accumulation of IFN-γ producing effector T cells and spontaneous inflammation (26).
Consequently, NRF2 has shown pro- and anti-inflammatory potential in T cells in different experimental inflammatory mouse models. In addition to this, there are differences between the role of NRF2 in human and murine T cells. Thus, the exact role of NRF2 in T cells and T cell subsets in autoimmunity is not clear.
Redox Metabolism in SLE T Cells
ROS production and cellular metabolism are intimately linked. More interestingly NRF2 is also involved in the regulation of different metabolic pathways. For immune cells, NRF2 increased glucose uptake and mitochondrial function, while reducing ROS in natural killer (NK) T cells and myeloid derived suppressor cells (MDSCs) (25, 51). Immune cells undergo metabolic reprogramming in autoimmune and autoinflammatory diseases. This is also the case in SLE, where T cells are hyper-oxidative (52). T cells from SLE patients differ from activated T cells from healthy persons (53, 54). Activated T cells mostly use glycolysis and pentose phosphate pathway to produce ATP and metabolic intermediates. SLE T cells show a chronically activated phenotype with increased TCA (trichloroacetic acid) cycle activity, they furthermore depend on OXPHOS to fulfill their energetic demands (55). It is suggested that SLE T cells therefore have lower antioxidant capacity with lower NADPH and glutathione levels (1, 56). This is further indicated by the fact that SLE T cells display high oxidative stress (57, 58). Although SLE T cells have an enlarged mitochondrial mass, the ATP generation from OXPHOS is insufficient compared to healthy person and a marked leakage of ROS is present in SLE T cells (58). ROS increase mammalian Target of Rapamycin (mTOR) activity, which is increased in CD4+ T cells in SLE patients (59). So reduced GSH levels induce a redox-dependent mTOR activation. CD4+ T cells from lupus patients show enhanced levels of mTOR activation which has been mechanistically linked with the disease process (60). As a consequence, therapeutic interventions with mTOR inhibitors in SLE patients normalized T cell activation and improved their clinical disease activity (61, 62).
There seem to be T cell subtype specific regulations that also need to be considered. Recently, Cailelli et al. identified CD4+ T cells that are induced by oxidized mitochondrial DNA-activated pDCs in SLE patients and accumulate ROS, secrete succinate and produce IL-10 and IFN-γ. By doing so, they provide superb B cell help (63).
This suggests that ROS accumulation plays a pathogenic role in SLE T cells. As a consequence, enhancing NRF2 activity and thereby reducing intracellular redox metabolism could be beneficial.
Discussion
Without a doubt redox metabolism critically regulates function, proliferation, and apoptosis of T cells. Furthermore, oxidative stress is a hallmark of SLE pathogenesis and T cells from SLE patients reveal metabolic aberrations that also involve higher ROS production. Therefore NRF2, as main transcriptional regulator of the anti-oxidative stress response and beyond this as regulator of metabolic and anti-inflammatory actions in immune cells, is most likely a central player in abnormal SLE T cell function. In line, 509 unique patent applications that define NRF2 pathway as molecular target and focus on medical conditions such as autoimmunity, liver, kidney, lung and neurodegenerative diseases have been filed since 2017 (64). Furthermore dimethyl fumarate (DMF), which activates Nrf2, is already used clinically to treat inflammatory diseases. DMF was approved for the treatment of patients with moderate and severe psoriasis as well as patients with relapsing remitting multiple sclerosis (65). In addition, there are clinical trials ongoing with DMF for treatment of inflammatory conditions, among others cutaneous lupus erythematosus (66). On the other hand, medication currently in use to treat patients with SLE clearly seems to affect Nrf2 signaling. In particular corticosteroids, which are first line treatment drugs downregulate Nrf2 transcriptional activation by direct and indirect means (67–69). Moreover, Cyclophosphamide, which is used in severe cases (lupus nephritis and CNS lupus) has severe side effects like hepatotoxicity and myelosuppression, which can be ameliorated by activation of NRF2 (70, 71). The same is true for methotrexate, which is used in lupus arthritis and downregulates the antoxidative Nrf2 response in the liver (72). On the other hand, mycophenolate mofetile is increasingly used in SLE and preserves the Nrf2 system in the liver as well as in the kidney, which might contribute to the broad therapeutic tolerability of the drug (73, 74). Finally, hydroxychloroquine also preserves the Nrf2 antioxidative system (75). Nevertheless, the described effects have mainly been shown in other systems than T cells and the immune system (liver, kidney and cancer models) and therefore we can only speculate on their effects on immune functions. Although it is well established that NRF2 has anti-inflammatory effects, the cellular mechanism and cell-specific actions are still not fully elucidated. Several controversies exist regarding the ambivalent role of ROS production in autoimmune diseases and the pathogenic effects of ROS are supposed to be dependent on threshold, location, and time. Furthermore, the cellular compartment seems to be of importance. While a global NRF2 activation is beneficial in autoimmune diseases, it is not clear if NRF2 has anti-inflammatory roles specifically in T cells (Figure 1). Future experiments might focus on T cells and T cell subtype specific roles of NRF2. Noticeable, SLE T cells display an altered redox metabolism. N-Acetylcysteine reversed glutathione depletion and thereby blocked mTOR activation in T cells and improved disease activity in SLE patients (76). One main problem of SLE patients is their susceptibility to infections. The use of glucocorticoids as immunosuppressive therapy increases this risk. Instead of completely blocking and suppressing the immune system a tight modulation of inflammation while preserving the cells’ overall functionality would be much more desirable to treat autoimmune diseases. Therefore, a therapeutic control of NRF2 activity might be a starting point to influence Redox metabolism in SLE T cells, but in-depth analysis of pathways and possible side-effects is absolutely necessary.
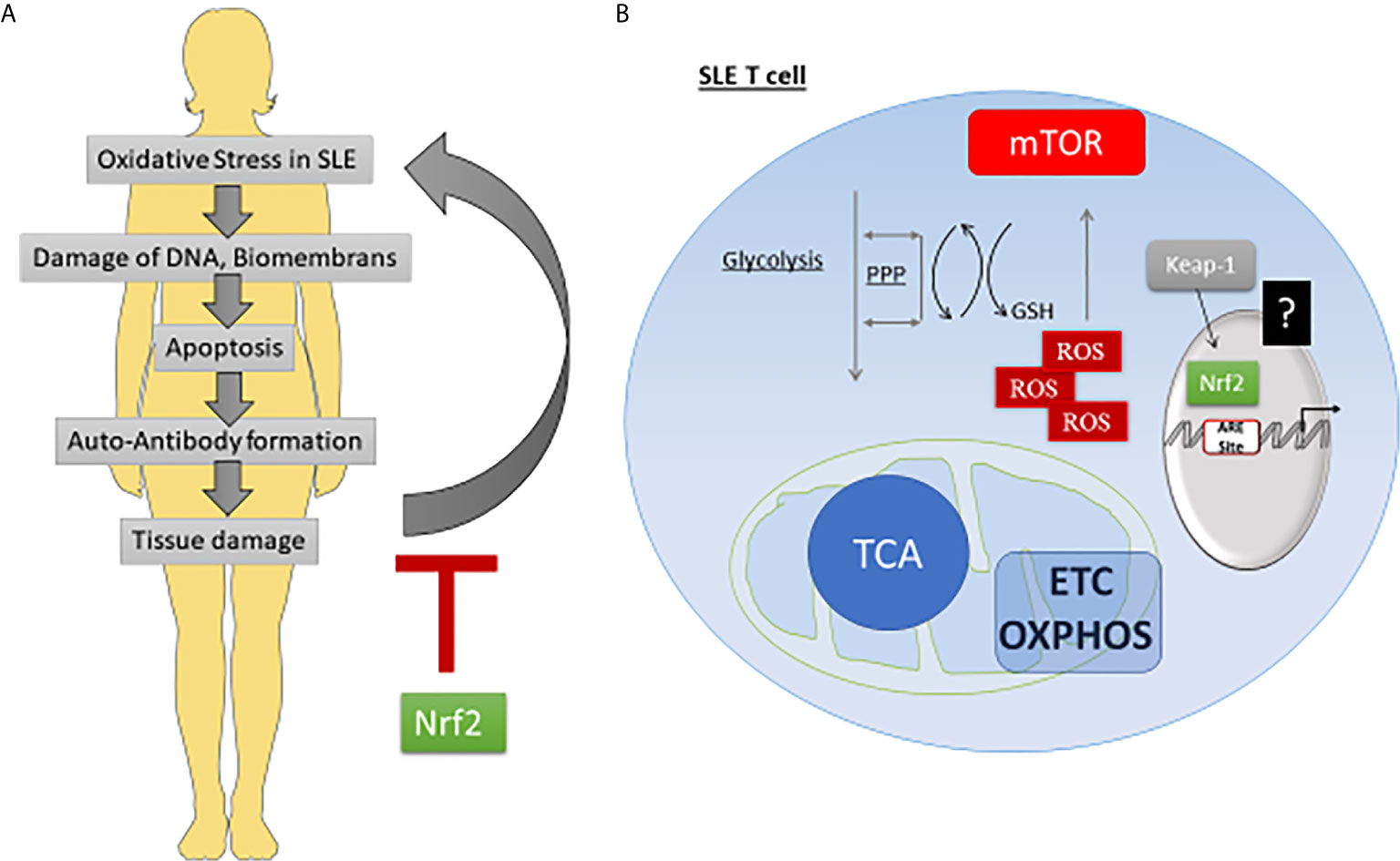
Figure 1 While systemic NRF2 activation can ameliorate pathogenesis, the exact role in T cells is not clear. (A) Oxidative stress is involved in SLE pathogenesis. Due to increased free radicals or a weak antioxidant system, SLE patients reveal high levels of oxidative stress which leads to cell damage and apoptosis. Subsequent release of autoantigens can enhance autoantibody formation and contribute to tissue damage. Tissue damage again can enhance oxidative stress. Several lines of evidence indicate that activated NRF2 can break the vicious circle by inducing anti-oxidative responses. (B) SLE T cell are hyper-oxidative. SLE T cells prefer the use of OXPHOS, which reduces NADPH and GSH pools, that are normally filled up during glycolysis and pentose phosphate pathway (PPP). In addition, they reveal a marked leakage of ROS during OXPHOS. High ROS levels induce mTOR activation. It is not clear, if NRF2 is not sufficiently activated or somehow fails to counteract high ROS levels in SLE T cells.
Author Contributions
KO and KT agree to be accountable for the content of the work. All authors contributed to the article and approved the submitted version.
Funding
This review was partially supported by the Deutsche Forschungsgemeinschaft (DFG OH 252/2‐1).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
DAMPs, danger-associated molecular patterns; ETC, electron transport chain; GCLC, Glutamate-Cysteine Ligase Catalytic Subunit; GPX, gluthathione peroxidase; GSH, gluthathione; GvHD, graft-vs-host disease; Keap1, Kelch ECH associating protein 1; MDSC, myeloid derived suppressor cells; mTOR, Mammalian Target of Rapamycin; NCF, Neutrophil Cytosolic Factor; NK, natural killer; NOX, NADPH oxidase; NQO1, NAD(P)H: quinone oxidoreductase 1; Nrf2, Nuclear factor erythroid 2-related factor 2; OXPHOS, oxidative phosphorylation; PBMCs, peripheral blood mononuclear cells; ROS, rective oxygen species; SLE, systemic lupus erythematosus; SOD, superoxid dismutase; TCA, trichloroacetic acid.
References
1. Perl A. Oxidative Stress in the Pathology and Treatment of Systemic Lupus Erythematosus. Nat Rev Rheumatol (2013) 9(11):674–86. doi: 10.1038/nrrheum.2013.147
2. Ahmad R, Ahsan H. Singlet Oxygen Species and Systemic Lupus Erythematosus: A Brief Review. J Immunoassay Immunochem (2019) 40(4):343–9. doi: 10.1080/15321819.2019.1616555
3. Kurien BT, Hensley K, Bachmann M, Scofield RH. Oxidatively Modified Autoantigens in Autoimmune Diseases. Free Radic Biol Med (2006) 41(4):549–56. doi: 10.1016/j.freeradbiomed.2006.05.020
4. Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The Role of Defective Clearance of Apoptotic Cells in Systemic Autoimmunity. Nat Rev Rheumatol (2010) 6(5):280–9. doi: 10.1038/nrrheum.2010.46
5. Lightfoot YL, Blanco LP, Kaplan MJ. Metabolic Abnormalities and Oxidative Stress in Lupus. Curr Opin Rheumatol (2017) 29(5):442–9. doi: 10.1097/BOR.0000000000000413
6. Gautam P, Kaur G, Tandon A, Sharma A, Bhatnagar A. Altered Redox Regulation by Nrf2-Keap1 System in Dendritic Cells of Systemic Lupus Erythematosus Patients. Lupus (2020) 29(12):1544–55. doi: 10.1177/0961203320950022
7. Zaieni SH, Derakhshan Z, Sariri R. Alternations of Salivary Antioxidant Enzymes in Systemic Lupus Erythematosus. Lupus (2015) 24(13):1400–5. doi: 10.1177/0961203315593170
8. Sam NB, Li BZ, Leng RX, Pan HF, Ye DQ. Circulating Antioxidant Levels in Systemic Lupus Erythematosus Patients: A Systematic Review and Meta-Analysis. Biomark Med (2019) 13(13):1137–52. doi: 10.2217/bmm-2019-0034
9. Tandon A, Anupam K, Kaushal J, Gautam P, Sharma A, Bhatnagar A. Altered Oxidative Stress Markers in Relation to T Cells, NK Cells & Killer Immunoglobulin Receptors That are Associated With Disease Activity in SLE Patients. Lupus (2020) 29(14):1831–44. doi: 10.1177/0961203320959441
10. Rokutan K, Hosokawa T, Nakamura K, Koyama K, Aoike A, Kawai K. Increased Superoxide Anion Production and Glutathione Peroxidase Activity in Peritoneal Macrophages From Autoimmune-Prone MRL/Mp-Ipr/lpr Mice. Int Arch Allergy Appl Immunol (1988) 87(2):113–9. doi: 10.1159/000234660
11. Elloumi N, Ben Mansour R, Marzouk S, Mseddi M, Fakhfakh R, Gargouri B, et al. Differential Reactive Oxygen Species Production of Neutrophils and Their Oxidative Damage in Patients With Active and Inactive Systemic Lupus Erythematosus. Immunol Lett (2017) 184:1–6. doi: 10.1016/j.imlet.2017.01.018
12. Bona N, Pezzarini E, Balbi B, Daniele SM, Rossi MF, Monje AL, et al. Oxidative Stress, Inflammation and Disease Activity Biomarkers in Lupus Nephropathy. Lupus (2020) 29(3):311–23. doi: 10.1177/0961203320904784
13. Lozovoy MA, Simao AN, Panis C, Rotter MA, Reiche EM, Morimoto HK, et al. Oxidative Stress is Associated With Liver Damage, Inflammatory Status, and Corticosteroid Therapy in Patients With Systemic Lupus Erythematosus. Lupus (2011) 20(12):1250–9. doi: 10.1177/0961203311411350
14. Kienhofer D, Boeltz S, Hoffmann MH. Reactive Oxygen Homeostasis - the Balance for Preventing Autoimmunity. Lupus (2016) 25(8):943–54. doi: 10.1177/0961203316640919
15. Zhao J, Ma J, Deng Y, Kelly JA, Kim K, Bang SY, et al. A Missense Variant in NCF1 is Associated With Susceptibility to Multiple Autoimmune Diseases. Nat Genet (2017) 49(3):433–7. doi: 10.1038/ng.3782
16. Olsson LM, Johansson AC, Gullstrand B, Jonsen A, Saevarsdottir S, Ronnblom L, et al. A Single Nucleotide Polymorphism in the NCF1 Gene Leading to Reduced Oxidative Burst is Associated With Systemic Lupus Erythematosus. Ann Rheum Dis (2017) 76(9):1607–13. doi: 10.1136/annrheumdis-2017-211287
17. Jacob CO, Eisenstein M, Dinauer MC, Ming W, Liu Q, John S, et al. Lupus-Associated Causal Mutation in Neutrophil Cytosolic Factor 2 (NCF2) Brings Unique Insights to the Structure and Function of NADPH Oxidase. Proc Natl Acad Sci U S A (2012) 109(2):E59–67. doi: 10.1073/pnas.1113251108
18. Jacob CO, Yu N, Yoo DG, Perez-Zapata LJ, Barbu EA, Kaplan MJ, et al. Haploinsufficiency of NADPH Oxidase Subunit Neutrophil Cytosolic Factor 2 Is Sufficient to Accelerate Full-Blown Lupus in NZM 2328 Mice. Arthritis Rheumatol (2017) 69(8):1647–60. doi: 10.1002/art.40141
19. Klein Geltink RI, O’Sullivan D, Pearce EL. Caught in the Crossfire: GSH Controls T Cell Metabolic Reprogramming. Immunity (2017) 46(4):525–7. doi: 10.1016/j.immuni.2017.03.022
20. Hoffmann MH, Griffiths HR. The Dual Role of Reactive Oxygen Species in Autoimmune and Inflammatory Diseases: Evidence From Preclinical Models. Free Radic Biol Med (2018) 125:62–71. doi: 10.1016/j.freeradbiomed.2018.03.016
21. Zhang DD, Hannink M. Distinct Cysteine Residues in Keap1 are Required for Keap1-dependent Ubiquitination of Nrf2 and for Stabilization of Nrf2 by Chemopreventive Agents and Oxidative Stress. Mol Cell Biol (2003) 23(22):8137–51. doi: 10.1128/MCB.23.22.8137-8151.2003
22. Kensler TW, Wakabayashi N, Biswal S. Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annu Rev Pharmacol Toxicol (2007) 47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046
23. Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, et al. Nrf2 Suppresses Macrophage Inflammatory Response by Blocking Proinflammatory Cytokine Transcription. Nat Commun (2016) 7:11624. doi: 10.1038/ncomms11624
24. Li W, Khor TO, Xu C, Shen G, Jeong WS, Yu S, et al. Activation of Nrf2-antioxidant Signaling Attenuates NFkappaB-inflammatory Response and Elicits Apoptosis. Biochem Pharmacol (2008) 76(11):1485–9. doi: 10.1016/j.bcp.2008.07.017
25. Ohl K, Fragoulis A, Klemm P, Baumeister J, Klock W, Verjans E, et al. Nrf2 Is a Central Regulator of Metabolic Reprogramming of Myeloid-Derived Suppressor Cells in Steady State and Sepsis. Front Immunol (2018) 9:1552. doi: 10.3389/fimmu.2018.01552
26. Klemm P, Rajendiran A, Fragoulis A, Wruck C, Schippers A, Wagner N, et al. Nrf2 Expression Driven by Foxp3 Specific Deletion of Keap1 Results in Loss of Immune Tolerance in Mice. Eur J Immunol (2020) 50(4):515–24. doi: 10.1002/eji.201948285
27. Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, et al. Disruption of Nrf2 Enhances Susceptibility to Severe Airway Inflammation and Asthma in Mice. J Exp Med (2005) 202(1):47–59. doi: 10.1084/jem.20050538
28. Reddy NM, Kleeberger SR, Kensler TW, Yamamoto M, Hassoun PM, Reddy SP. Disruption of Nrf2 Impairs the Resolution of Hyperoxia-Induced Acute Lung Injury and Inflammation in Mice. J Immunol (Baltimore Md 1950) (2009) 182(11):7264–71. doi: 10.4049/jimmunol.0804248
29. Thimmulappa RK, Scollick C, Traore K, Yates M, Trush MA, Liby KT, et al. Nrf2-dependent Protection From LPS Induced Inflammatory Response and Mortality by CDDO-Imidazolide. Biochem Biophys Res Commun (2006) 351(4):883–9. doi: 10.1016/j.bbrc.2006.10.102
30. Osburn WO, Yates MS, Dolan PD, Chen S, Liby KT, Sporn MB, et al. Genetic or Pharmacologic Amplification of Nrf2 Signaling Inhibits Acute Inflammatory Liver Injury in Mice. Toxicol Sci (2008) 104(1):218–27. doi: 10.1093/toxsci/kfn079
31. Khor TO, Huang MT, Kwon KH, Chan JY, Reddy BS, Kong AN. Nrf2-deficient Mice Have an Increased Susceptibility to Dextran Sulfate Sodium-Induced Colitis. Cancer Res (2006) 66(24):11580–4. doi: 10.1158/0008-5472.CAN-06-3562
32. Wruck CJ, Fragoulis A, Gurzynski A, Brandenburg LO, Kan YW, Chan K, et al. Role of Oxidative Stress in Rheumatoid Arthritis: Insights From the Nrf2-knockout Mice. Ann Rheum Dis (2011) 70(5):844–50. doi: 10.1136/ard.2010.132720
33. Ma Q, Battelli L, Hubbs AF. Multiorgan Autoimmune Inflammation, Enhanced Lymphoproliferation, and Impaired Homeostasis of Reactive Oxygen Species in Mice Lacking the Antioxidant-Activated Transcription Factor Nrf2. Am J Pathol (2006) 168(6):1960–74. doi: 10.2353/ajpath.2006.051113
34. Zhao M, Chen H, Ding Q, Xu X, Yu B, Huang Z. Nuclear Factor Erythroid 2-Related Factor 2 Deficiency Exacerbates Lupus Nephritis in B6/lpr Mice by Regulating Th17 Cell Function. Sci Rep (2016) 6:38619. doi: 10.1038/srep38619
35. Jiang T, Tian F, Zheng H, Whitman SA, Lin Y, Zhang Z, et al. Nrf2 Suppresses Lupus Nephritis Through Inhibition of Oxidative Injury and the NF-kappaB-mediated Inflammatory Response. Kidney Int (2014) 85(2):333–43. doi: 10.1038/ki.2013.343
36. Han S, Zhuang H, Lee PY, Li M, Yang L, Nigrovic PA, et al. Nf-E2-Related Factor 2 Regulates Interferon Receptor Expression and Alters Macrophage Polarization in Lupus. Arthritis Rheumatol (2020) 72(10):1707–20. doi: 10.1002/art.41383
37. Morito N, Yoh K, Hirayama A, Itoh K, Nose M, Koyama A, et al. Nrf2 Deficiency Improves Autoimmune Nephritis Caused by the Fas Mutation Lpr. Kidney Int (2004) 65(5):1703–13. doi: 10.1111/j.1523-1755.2004.00565.x
38. Cordova EJ, Velazquez-Cruz R, Centeno F, Baca V, Orozco L. The NRF2 Gene Variant, -653G/A, is Associated With Nephritis in Childhood-Onset Systemic Lupus Erythematosus. Lupus (2010) 19(10):1237–42. doi: 10.1177/0961203310367917
39. Yamamoto T, Yoh K, Kobayashi A, Ishii Y, Kure S, Koyama A, et al. Identification of Polymorphisms in the Promoter Region of the Human NRF2 Gene. Biochem Biophys Res Commun (2004) 321(1):72–9. doi: 10.1016/j.bbrc.2004.06.112
40. Suzuki T, Murakami S, Biswal SS, Sakaguchi S, Harigae H, Yamamoto M, et al. Systemic Activation of NRF2 Alleviates Lethal Autoimmune Inflammation in Scurfy Mice. Mol Cell Biol (2017) 37(15):e00063–17. doi: 10.1128/MCB.00063-17
41. Franchina DG, Dostert C, Brenner D. Reactive Oxygen Species: Involvement in T Cell Signaling and Metabolism. Trends Immunol (2018) 39(6):489–502. doi: 10.1016/j.it.2018.01.005
42. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are Required for Antigen-Specific T Cell Activation Through Reactive Oxygen Species Signaling. Immunity (2013) 38(2):225–36. doi: 10.1016/j.immuni.2012.10.020
43. Mak TW, Grusdat M, Duncan GS, Dostert C, Nonnenmacher Y, Cox M, et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunity (2017) 46(6):1089–90. doi: 10.1016/j.immuni.2017.06.009
44. Kurniawan H, Franchina DG, Guerra L, Bonetti L, Baguet LS, Grusdat M, et al. Glutathione Restricts Serine Metabolism to Preserve Regulatory T Cell Function. Cell Metab (2020) 31(5):920–36.e7. doi: 10.1016/j.cmet.2020.03.004
45. Morzadec C, Macoch M, Sparfel L, Kerdine-Romer S, Fardel O, Vernhet L. Nrf2 Expression and Activity in Human T Lymphocytes: Stimulation by T Cell Receptor Activation and Priming by Inorganic Arsenic and Tert-Butylhydroquinone. Free Radic Biol Med (2014) 71:133–45. doi: 10.1016/j.freeradbiomed.2014.03.006
46. Tsai JJ, Velardi E, Shono Y, Argyropoulos KV, Holland AM, Smith OM, et al. Nrf2 Regulates CD4(+) T Cell-Induced Acute Graft-Versus-Host Disease in Mice. Blood (2018) 132(26):2763–74. doi: 10.1182/blood-2017-10-812941
47. Noel S, Martina MN, Bandapalle S, Racusen LC, Potteti HR, Hamad AR, et al. T Lymphocyte-Specific Activation of Nrf2 Protects From AKI. J Am Soc Nephrol JASN (2015) 26(12):2989–3000. doi: 10.1681/ASN.2014100978
48. Karl F, Stoll A, Bottcher-Loschinski R, Bottcher M, Baur R, Jacobs B, et al. Impact of Nrf2 Expression in Reconstituting T-cells of Allogeneic Hematopoietic Stem Cell Transplanted Patients. Leukemia (2020) 35(3):910–5. doi: 10.1038/s41375-020-0956-0
49. Mougiakakos D, Johansson CC, Kiessling R. Naturally Occurring Regulatory T Cells Show Reduced Sensitivity Toward Oxidative Stress-Induced Cell Death. Blood (2009) 113(15):3542–5. doi: 10.1182/blood-2008-09-181040
50. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative Stress Controls Regulatory T Cell Apoptosis and Suppressor Activity and PD-L1-blockade Resistance in Tumor. Nat Immunol (2017) 18(12):1332–41. doi: 10.1038/ni.3868
51. Pyaram K, Kumar A, Kim YH, Noel S, Reddy SP, Rabb H, et al. Keap1-Nrf2 System Plays an Important Role in Invariant Natural Killer T Cell Development and Homeostasis. Cell Rep (2019) 27(3):699–707.e4. doi: 10.1016/j.celrep.2019.03.052
52. Teng X, Li W, Cornaby C, Morel L. Immune Cell Metabolism in Autoimmunity. Clin Exp Immunol (2019) 197(2):181–92. doi: 10.1111/cei.13277
53. Sharabi A, Tsokos GC. T Cell Metabolism: New Insights in Systemic Lupus Erythematosus Pathogenesis and Therapy. Nat Rev Rheumatol (2020) 16(2):100–12. doi: 10.1038/s41584-019-0356-x
54. Kono M, Yoshida N, Tsokos GC. Metabolic Control of T Cells in Autoimmunity. Curr Opin Rheumatol (2020) 32(2):192–9. doi: 10.1097/BOR.0000000000000685
55. Wahl DR, Petersen B, Warner R, Richardson BC, Glick GD, Opipari AW. Characterization of the Metabolic Phenotype of Chronically Activated Lymphocytes. Lupus (2010) 19(13):1492–501. doi: 10.1177/0961203310373109
56. Gergely P Jr., Grossman C, Niland B, Puskas F, Neupane H, Allam F, et al. Mitochondrial Hyperpolarization and ATP Depletion in Patients With Systemic Lupus Erythematosus. Arthritis Rheumatol (2002) 46(1):175–90. doi: 10.1002/1529-0131(200201)46:1<175::AID-ART10015>3.0.CO;2-H
57. Tsokos GC. Systemic Lupus Erythematosus. N Engl J Med (2011) 365(22):2110–21. doi: 10.1056/NEJMra1100359
58. Krishnan S, Farber DL, Tsokos GC. T Cell Rewiring in Differentiation and Disease. J Immunol (Baltimore Md 1950) (2003) 171(7):3325–31. doi: 10.4049/jimmunol.171.7.3325
59. Vukelic M, Kono M, Tsokos GC. T Cell Metabolism in Lupus. Immunometabolism (2020) 2(2):e200009. doi: 10.20900/immunometab20200009
60. Perl A. Systems Biology of Lupus: Mapping the Impact of Genomic and Environmental Factors on Gene Expression Signatures, Cellular Signaling, Metabolic Pathways, Hormonal and Cytokine Imbalance, and Selecting Targets for Treatment. Autoimmunity (2010) 43(1):32–47. doi: 10.3109/08916930903374774
61. Fernandez D, Bonilla E, Mirza N, Niland B, Perl A. Rapamycin Reduces Disease Activity and Normalizes T Cell Activation-Induced Calcium Fluxing in Patients With Systemic Lupus Erythematosus. Arthritis Rheumatol (2006) 54(9):2983–8. doi: 10.1002/art.22085
62. Perl A. Emerging New Pathways of Pathogenesis and Targets for Treatment in Systemic Lupus Erythematosus and Sjogren’s Syndrome. Curr Opin Rheumatol (2009) 21(5):443–7. doi: 10.1097/BOR.0b013e32832efe6b
63. Caielli S, Veiga DT, Balasubramanian P, Athale S, Domic B, Murat E, et al. A Cd4(+) T Cell Population Expanded in Lupus Blood Provides B Cell Help Through interleukin-10 and Succinate. Nat Med (2019) 25(1):75–81. doi: 10.1038/s41591-018-0254-9
64. Chartoumpekis DV, Fu CY, Ziros PG, Sykiotis GP. Patent Review (2017-2020) of the Keap1/Nrf2 Pathway Using Patseer Pro: Focus on Autoimmune Diseases. Antioxidants (Basel) (2020) 9(1):1138. doi: 10.3390/antiox9111138
65. Hennig P, Fenini G, Di Filippo M, Beer HD. Electrophiles Against (Skin) Diseases: More Than Nrf2. Biomolecules (2020) 10(2):271. doi: 10.3390/biom10020271
66. Kuhn A, Landmann A, Patsinakidis N, Ruland V, Nozinic S, Perusquia Ortiz AM, et al. Fumaric Acid Ester Treatment in Cutaneous Lupus Erythematosus (CLE): A Prospective, Open-Label, Phase II Pilot Study. Lupus (2016) 25(12):1357–64. doi: 10.1177/0961203316644335
67. Kratschmar DV, Calabrese D, Walsh J, Lister A, Birk J, Appenzeller-Herzog C, et al. Suppression of the Nrf2-dependent Antioxidant Response by Glucocorticoids and 11β-HSD1-mediated Glucocorticoid Activation in Hepatic Cells. PloS One (2012) 7(5):e36774. doi: 10.1371/journal.pone.0036774
68. Alam MM, Okazaki K, Nguyen LTT, Ota N, Kitamura H, Murakami S, et al. Glucocorticoid Receptor Signaling Represses the Antioxidant Response by Inhibiting Histone Acetylation Mediated by the Transcriptional Activator NRF2. J Biol Chem (2017) 292(18):7519–30. doi: 10.1074/jbc.M116.773960
69. Liang J, Zhang XY, Zhen YF, Chen C, Tan H, Hu J, et al. PGK1 Depletion Activates Nrf2 Signaling to Protect Human Osteoblasts From Dexamethasone. Cell Death Dis (2019) 10(12):888. doi: 10.1038/s41419-019-2112-1
70. Que L, He L, Yu C, Yin W, Ma L, Cao B, et al. Activation of Nrf2-ARE Signaling Mitigates Cyclophosphamide-Induced Myelosuppression. Toxicol Lett (2016) 262:17–26. doi: 10.1016/j.toxlet.2016.09.003
71. Aladaileh SH, Abukhalil MH, Saghir SAM, Hanieh H, Alfwuaires MA, Almaiman AA, et al. Galangin Activates Nrf2 Signaling and Attenuates Oxidative Damage, Inflammation, and Apoptosis in a Rat Model of Cyclophosphamide-Induced Hepatotoxicity. Biomolecules (2019) 9(8):346. doi: 10.3390/biom9080346
72. Mahmoud AM, Hussein OE, Hozayen WG, Abd El-Twab SM. Methotrexate Hepatotoxicity is Associated With Oxidative Stress, and Down-Regulation of Pparγ and Nrf2: Protective Effect of 18β-Glycyrrhetinic Acid. Chem Biol Interact (2017) 270:59–72. doi: 10.1016/j.cbi.2017.04.009
73. Tapia E, García-Arroyo F, Silverio O, Rodríguez-Alcocer AN, Jiménez-Flores AB, Cristobal M, et al. Mycophenolate Mofetil and Curcumin Provide Comparable Therapeutic Benefit in Experimental Chronic Kidney Disease: Role of Nrf2-Keap1 and Renal Dopamine Pathways. Free Radic Res (2016) 50(7):781–92. doi: 10.1080/10715762.2016.1174776
74. Serrya MS, Zaghloul MS. Mycophenolate Mofetil Attenuates Concanavalin A-induced Acute Liver Injury Through Modulation of TLR4/NF-κb and Nrf2/HO-1 Pathways. Pharmacol Rep (2020) 72(4):945–55. doi: 10.1007/s43440-019-00055-4
75. Shen H, Wu N, Wang Y, Zhao H, Zhang L, Li T, et al. Chloroquine Attenuates Paraquat-Induced Lung Injury in Mice by Altering Inflammation, Oxidative Stress and Fibrosis. Int Immunopharmacol (2017) 46:16–22. doi: 10.1016/j.intimp.2017.02.020
76. Lai ZW, Hanczko R, Bonilla E, Caza TN, Clair B, Bartos A, et al. N-Acetylcysteine Reduces Disease Activity by Blocking Mammalian Target of Rapamycin in T Cells From Systemic Lupus Erythematosus Patients: A Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol (2012) 64(9):2937–46. doi: 10.1002/art.34502
Keywords: Keap 1, ROS, mTOR, lupus, Tregs, Foxp3
Citation: Ohl K and Tenbrock K (2021) Oxidative Stress in SLE T Cells, Is NRF2 Really the Target to Treat? Front. Immunol. 12:633845. doi: 10.3389/fimmu.2021.633845
Received: 26 November 2020; Accepted: 08 April 2021;
Published: 23 April 2021.
Edited by:
Masayuki Mizui, Osaka University, JapanReviewed by:
Cheryl Rockwell, Michigan State University, United StatesChary Lopez-Pedrera, Maimonides Biomedical Research Institute of Cordoba (IMIBIC), Spain
Copyright © 2021 Ohl and Tenbrock. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Klaus Tenbrock, a3RlbmJyb2NrQHVrYWFjaGVuLmRl