 Jenny A. Nguyen
Jenny A. Nguyen Robin M. Yates
Robin M. Yates- 1Department of Biochemistry and Molecular Biology, Cumming School of Medicine, University of Calgary, Calgary, AB, Canada
- 2Department of Comparative Biology and Experimental Medicine, Faculty of Veterinary Medicine, University of Calgary, Calgary, AB, Canada
- 3Cumming School of Medicine, Snyder Institute of Chronic Disease, University of Calgary, Calgary, AB, Canada
Following phagocytosis, the nascent phagosome undergoes maturation to become a phagolysosome with an acidic, hydrolytic, and often oxidative lumen that can efficiently kill and digest engulfed microbes, cells, and debris. The fusion of phagosomes with lysosomes is a principal driver of phagosomal maturation and is targeted by several adapted intracellular pathogens. Impairment of this process has significant consequences for microbial infection, tissue inflammation, the onset of adaptive immunity, and disease. Given the importance of phagosome-lysosome fusion to phagocyte function and the many virulence factors that target it, it is unsurprising that multiple molecular pathways have evolved to mediate this essential process. While the full range of these pathways has yet to be fully characterized, several pathways involving proteins such as members of the Rab GTPases, tethering factors and SNAREs have been identified. Here, we summarize the current state of knowledge to clarify the ambiguities in the field and construct a more comprehensive phagolysosome formation model. Lastly, we discuss how other cellular pathways help support phagolysosome biogenesis and, consequently, phagocyte function.
Introduction
Professional phagocytes, such as macrophages, dendritic cells (DCs), and neutrophils, are critical to the innate immune response and the maintenance of homeostasis through their ability to ingest and degrade microbes, debris, and dying cells (1). The versatility of professional phagocytes stems from the expression of phagocytic receptors that can mediate the uptake of a vast array of cargoes via phagocytosis. Following uptake, the engulfed material is contained within the specialized vacuole called the phagosome, which initially has the lumenal characteristics of the extracellular space (2). The phagosome undergoes progressive maturation through multiple transient fusion-fission events with vesicles of the endolyososmal system leading to an increasingly acidic and hydrolytic environment within the phagosomal lumen (3, 4). The fusion of the phagosome with lysosomes forms the mature phagolysosome (PL) which has full degradative and microbicidal capacity. Heterotypic fusion between the phagosome and lysosome is imperative for phagocytes to carry out their functions in immunity and homeostasis and is a tightly regulated process. The importance of membrane fusion in phagocyte function is further highlighted by the myriad of mechanisms various pathogens have developed which impede fusion machinery in order to prevent PL formation, and the microbicidal environment that is established. Indeed, gram-negative bacteria Mycobacterium tuberculosis and Coxiella burnetii, parasites of the Leishmania genus and the fungi Aspergillus fumigatus can inhibit PL fusion to avoid death in phagosomes, among many others (5–9).
PL fusion involves the stepwise recruitment and coordinated action of a number of proteins. To date, 30 soluble N-ethylmaleimide-sensitive fusion factor attachment protein (SNAP)-receptors (SNAREs), 20 Rab GTPases, six multi-subunit tethering complexes, and four tethering proteins of the Sec1/Munc18 (SM) family have been identified on phagosomes (10, 11). The extensive number of fusion proteins and promiscuity among interacting components creates mechanistic redundancy of this crucial function of phagocytes. Given the overwhelming number of fusion proteins and the even larger pool of potential binding partners, it is unsurprising that the identity and roles of the fusion machinery, their regulatory signals, and their spatiotemporal organizations in PL fusion have yet to be fully elucidated. These gaps in the literature continue to be active areas of investigation, and here we highlight the progress made on this topic in the context of phagocyte biology. This review aims to summarize current and recent developments in understanding the fusion machinery modulating PL fusion and the consequences to phagocyte function when this machinery is impaired. Finally, we discuss how phagocytes can employ the related engulfment process of autophagy, to support the phagocytic pathway during homeostasis and defense against pathogens.
Phagosome-Lysosome Membrane Fusion: An Overview
Once the phagosome is formed and has undergone transient fusion-fission events with early and late endosomes, phagosomes migrate to the lysosome-rich perinuclear region of the phagocytic cell (12). Similar to many other types of cells, phagocytes employ a coordinated transport system consisting of the microtubule (MT) networks, the associated MT motors, and their effector/adaptor proteins to carry phagosomal vesicles to the perinuclear region (13, 14). Long-distance transport is initiated by the small Rab GTPases Rab5 and Rab7 which are recruited to early and late phagosomes, respectively. Rab5 and Rab7, in turn, coordinate the recruitment of necessary motor proteins required for the dynein-driven transport of phagosomes to the perinuclear region (13, 15). Once phagosomes and lysosomes are in close apposition, membrane fusion occurs through the concerted action of fusion proteins.
Membrane fusion is the process by which two separate lipid membranes combine to form one continuous bilayer (16). Within the endomembrane system, fusion requires the coordinated action between members of the Rab GTPase superfamily and their effectors, tethering factors, N-ethylmaleimide-sensitive factor (NSF), and SNAREs (17). Generally, these proteins are consecutively recruited during the following steps of membrane fusion: i) tethering; ii) SNARE assembly; iii) SNARE zippering and membrane fusion; and iv) SNARE disassembly and recycling (Figure 1). First, Rab GTPases are recruited to the membrane of vesicles pending fusion, to mark the position for fusion. Subsequently, Rabs, through the recruitment of their effectors bind to tethering factors from the cytosol (18, 19). Through this action, Rabs establish the site at which proteins assemble into membrane microdomains at the fusion site. Rab effector-mediated tethering brings adjacent membranes into proximity. Once tethered, SNARE proteins congregate at the fusion site and supply the energy necessary to overcome the electrostatic repulsion between two lipid membranes (20–22). Numerous SNAREs reside on endomembranes, yet only a finite number can form a stable SNARE complex in trans that consists of four SNARE domains (23). SNARE assembly is not a spontaneous process but requires SM proteins to form SNARE intermediates that await a missing cognate SNARE (24, 25). Once a trans-SNARE complex is formed, a conformational change brings the two membranes together, forming a cis-SNARE complex in a process called “zippering” (20–22, 26). The post-fusion cis-SNARE complex, where all SNARE proteins are located on the same membrane, is then disassembled by NSF along with its cofactor α-SNAP (27–29). The disassembly process is powered by NSF-mediated ATP hydrolysis which provides the energy for the dissociated SNARE proteins.
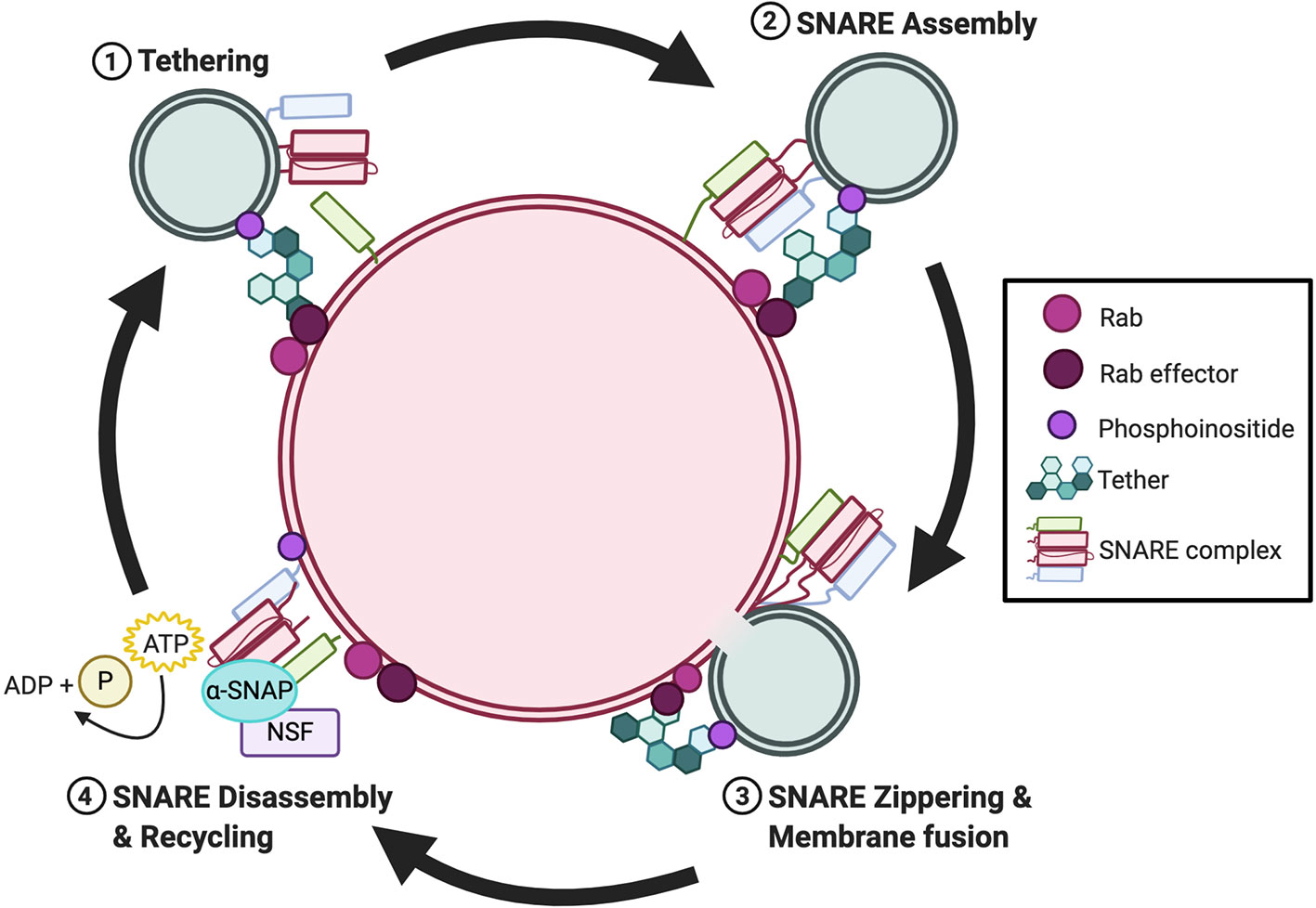
Figure 1 Overview of the cyclical nature of membrane fusion. Rab GTPases and their effectors recruit tethers to the membrane fusion site during the tethering step. Subsequently, SNAREs are recruited to opposing membranes and assemble in trans via the catalyzing activity of a SM protein. Once assembled, SNARE zippering drives membrane fusion. Post-fusion, NSF and α-SNAP bind to the cis-SNARE and ATP-driven complex disassembly recycles the individual SNAREs intothe cytosol for recycling.
Early studies on pathogen clearance and efferocytosis in mice and Caenorhabditis elegans identified specific Rab GTPases and their corresponding tethering complexes in PL biogenesis and phagocyte function (30–33). Despite their lack in professional phagocytes, C. elegans have phagocytic-like cells that have been used in many early and current studies to study phagosome maturation after efferocytosis due to their genetic conservation (34–36). Thus, C. elegans has been a powerful model to identify genes and mechanisms involved in PL fusion.
Identifying SNAREs involved in PL fusion has been more challenging due to their promiscuous nature and involvement in several cellular pathways crucial for cellular upkeep. Although, putative SNARE complexes have recently been identified using cell-free systems where subsequent investigations are beginning to unravel the functions of these proteins in PL fusion (37–40). Other insights into the proteins modulating PL fusion have been inferred from studies of the closely related process of endo-lysosome fusion in other cells. However, differences in the cargo and regulation between phagocytosis and endocytosis, maintain PL fusion as a distinct process compared to endosome-lysosome fusion (41). Given the large degree of heterogeneity in models and approaches to study this process, this review aims to consolidate the literature focusing on PL fusion proteins in phagocyte-specific studies, and where information is lacking in this context, identify potential candidate proteins and complexes, in order to generate a clearer picture of this fundamentally important aspect of phagocyte function. The key components of the PL fusion machinery are summarized in Supplementary Table 1.
Marking the Membranes: Small Rab GTPases
The Rab GTPases are peripheral membrane proteins that serve as molecular identifiers on membranes, and to regulate multiple steps during membrane fusion. Rab GTPases function as molecular switches that toggle between an inactive GDP-bound and an active GTP-bound state. Rabs are maintained in their inactive soluble GDP-bound state by a guanine dissociation inhibitor (GDI) (42, 43). At vesicular membranes, guanine exchange factors (GEFs) possess the dual capacity to recruit Rabs from the cytosol and catalyze the displacement of GDP with GTP, which both activates Rabs and displaces the GDI, thus allowing for Rabs to be inserted into the membrane (44, 45). Once activated, Rabs are able to bind to effector proteins on fusion targets in order to tether fusing vesicles, modulate the recruitment of fusion components, and mediate the process of PL biogenesis. Post-fusion, GTPase-activating proteins (GAPs) hydrolyze GTP to inactivate Rabs where they are solubilized by a GDI, returning them to the pool of inactive cytosolic Rabs (46). The numerous influences on GTPase regulation make Rabs a strong focal point for modulation of PL biogenesis.
It has long been established that small GTPase Rab7 is essential for the fusion between late-stage phagosomes and lysosomes, and consequently, for the function of phagocytes in both pathogen and apoptotic cell clearance (33, 47, 48). Several pathogens have been described to inhibit Rab7 recruitment in order to replicate and survive within phagosomes, and has been comprehensively reviewed (49). Early studies reported the inhibition of Rab7 recruitment to the phagosomes by M. tuberculosis and M. bovis J774 macrophages, preventing PL fusion (33). A more recent example describes the necessity of Rab7 in the clearance of Pseudomonas aeruginosa infection in J774A.1-derived macrophages (50). This study reported that sialylated P. aeruginosa remained viable in phagosomes by preventing the recruitment of Rab7. In addition to the clearance of microbes, Rab7 is essential to the degradation of apoptotic cells following phagocytosis. In a C. elegans model of apoptotic cell removal, the siRNA-mediated knockdown of Rab7 contributed to the accumulation of apoptotic bodies in phagosomes (34). Similarly, in an in vivo model of murine ulcerative colitis, inflammation and disease severity was exacerbated when Rab7 activation and recruitment was impaired in macrophages, reportedly due to an accumulation of apoptotic epithelial cells in the colon (48).
The mechanism by which Rab7 is recruited to phagosomes in professional phagocytes is not fully elucidated. However, studies in C. elegans demonstrated that Rab7 recruitment to late endosomes/lysosomes was facilitated by the cytosolic SAND1-CCZ1 complex (51–53). The mammalian orthologues of SAND1 and CCZ1, namely, Mon1a/b and Ccz1 display GEF activity and were found to form a complex which displaces the GDI associated with the GDP-bound form of Rab7, allowing GTP switching and integration of Rab7 into phagosomal membranes (Figure 2Ai) in C. elegans which is believed to be evolutionary conserved in mammals (52).
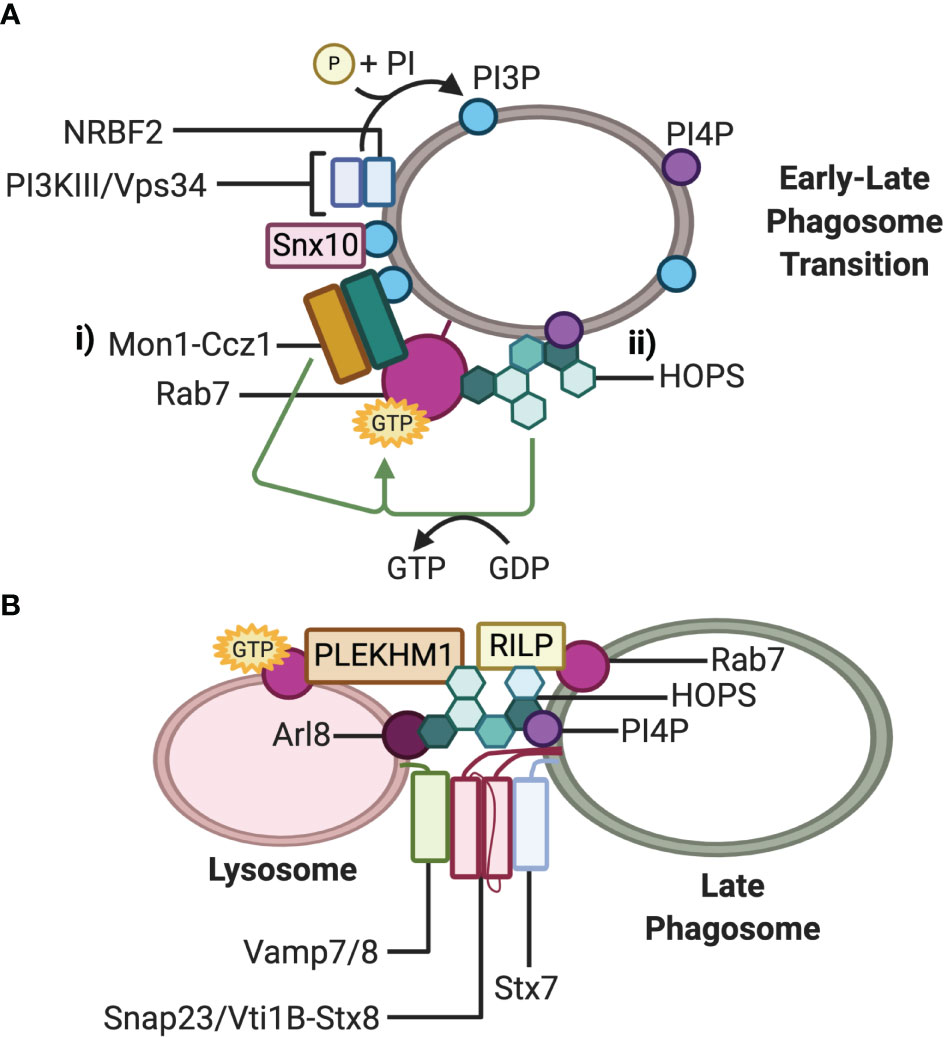
Figure 2 Fusion machinery involved in phagosome maturation. (A) Working models for Rab7 recruitment and activation at the phagosome membrane during maturation: i) The Mon1-Ccz1 complex is recruited directly by PI3P and indirectly by Snx10 and NRBF2-PI3KIII/Vps34. PI3P activates the GEF activity of Mon1-Ccz1; ii) The GEF activity of PI4P-recruited HOPS activates Rab7. (B) Working model of the trans-SNARE complex mediating fusion between the late phagosome and lysosome. The small GTPase Rab7 recruits effectors RILP and Plekhm1, and concurrently with Arl8, additionally recruits the HOPS tethering complex. HOPS docks the late phagosome to the lysosome and stabilizes the trans-SNARE Stx7-Snap23-Vamp7/8 or Stx7-Vti1b-Stx8-Vamp7/8 complex at the point of membrane fusion. Note that either Plekhm1, Arl8, PI4P, or RILP on either lysosomes or phagosomes bind to HOPS Vps41 at one time, while RILP can also bind HOPS Vps39.
Phosphatidylinositol-3-phosphates (PI3P) on early phagosomes have been proposed to both recruit Mon1-Ccz1 to early-to-late transitional phagosomes and activate the GEF activity of the complex (Figure 2Ai) (48, 53). Recently, nuclear receptor binding factor 2 (NRBF2), a component of the class III phosphoinositide 3-kinase (PI3KIII)/vacuolar sorting protein (Vps) 34 complex that generates PI3P on phagosomes, has been implicated to regulate the function of the Mon1-Ccz1 complex during apoptotic cell clearance in macrophages (48). NRBF2 is a binding partner of the catalytic subunit of the PI3KIII/Vps34 complex and can facilitate the interaction between PI3KIII/Vps34 and Mon1-Ccz1 (Figure 2Ai). Through this interaction, NRBF2 may bring the PI3KIII/Vps34 complex to phagosomes or support the activation of PI3KIII/Vps34, thereby generating PI3P to activate the GEF activity of the Mon1-Ccz1 complex. Further evidence that NRBF2 modulates the activity of the Mon1-Ccz1complex is provided by an observed loss of GTP-bound Rab7 around phagosomes as well as delayed Rab7 recruitment in NRBF2-knockout macrophages (48). Lou and colleagues (54) have proposed that another factor, sorting nexin 10 (Snx10), can promote the recruitment of Mon1-Ccz1 to early endosomes and phagosomes to mediate Rab7 recruitment (Figure 2Ai). Snx10 is upregulated in response to TLR signaling and phagocytosis of several different microbes, suggesting that Snx10 has an antimicrobial role (54). Indeed, depletion of Snx10 reduced Rab7 recruitment on bacteria-containing phagosomes. Taken together with the studies in C. elegans, Mon1-Ccz1 is likely a major Rab7 GEF on phagosomes in mammalian cells.
While there is ample evidence for the Mon1-Ccz1 complex as a Rab7 GEF on endosomes and phagosomes, it is potentially not the only one. Early insights from studies in yeast and C. elegans have implicated that the homotypic fusion and vacuole sorting (HOPS) complex can also act as a Rab7 GEF (Figure 2Aii) (52, 55). It was demonstrated by Barry and colleagues (56) that HOPS could function the same way in mammalian cells for Rab7 found on phagosomes in macrophages containing C. burnetii. Macrophages phagocytosing an avirulent strain of C. burnetti induce p38α-mitogen-activated protein kinase (MAPK)-dependent phosphorylation of the HOPS Vps41 subunit which was demonstrated to be necessary for Rab7 recruitment to endosomal and phagosomal membranes (56). In contrast, the lipopolysaccharide produced by the virulent strain of C. burnetti prevented p38α- MAPK activation and phosphorylation of Vps41, allowing bacterial persistence in Rab7-deficient phagosomes. Why Mon1-Ccz1 was unable to act as the Rab7 GEF instead of HOPs in this model is unknown; however, it is possible that Mon1-Ccz1 and the HOPS complex act independently depending on the phagosomal cargo.
Rab7 is vital for PL fusion and therefore phagocytic function, but it is not the only GTPase involved in PL fusion: Rab2 and Rab14 have been shown to possibly function redundantly to each other or in parallel to recruit lysosomes to phagosomes during apoptotic cell degradation in C. elegans (31, 32). Rab2 and Rab14 are transiently recruited to phagosomes prior to Rab7, and it is postulated that like Rab7, Rab2 and Rab14 may recruit tethering factors to initiate interaction between phagosomes and lysosomes (31). Interestingly, Rab2 is a direct binding partner of HOPS during autophagosome fusion with lysosomes in Drosophila melanogaster cells (57, 58), and thus, the HOPS complex may serve as the effector for both Rab2 and Rab14 to coordinate fusion of lysosomes to phagosomes. Additionally, proteomic studies in an oyster cell model of phagocytosis identified Rab2 and Rab14 in the phagosomal proteome (59). Although Rab2 has not been studied in a mammalian model, the role of Rab14 in PL fusion was partially elucidated in Raw264.7 and J774 murine macrophages infected with the fungal pathogen Candida albicans. Knockdown of Rab14 disrupted fusion of lysosomes to phagosomes which both delayed the acquisition and activation of hydrolytic proteases in PLs. This defect in lysosome fusion and protease activation resulted in increased susceptibility of macrophages to C. albicans (60).
Taken together, Rab2 and Rab14 may function redundantly in an early tethering step prior to Rab7 recruitment and the docking/fusion of lysosomes with phagosomes. The presence of Rab7 on the phagosome membrane sets the stage for PL fusion; however, the mere presence of Rab7 on phagosomes is not sufficient to enact PL fusion (61). Rather, it requires further interaction with effector proteins to tether the vesicles together to facilitate fusion.
Securing the Phagosomes to Lysosomes: Tethering Effectors
Tethering effectors, in the form of proteins and protein complexes, are recruited from the cytoplasm to specific membranes designated by small GTPases such as Rab or ADP-ribosylation factor-like protein (Arl) proteins (17). During the tethering step prior to fusion, tethering factors bind to Rab GTPases on the adjacent vesicles to provide an initial physical interaction (62). In addition to their function as linkages between two vicinal membranes, tethers can recruit SNARES as well as the proteins that catalyze the formation of the SNARE complex. Moreover, tethers can strengthen the association amongst fusion-relevant factors at the fusion site. Altogether, tethers ensure fusion fidelity and increase the efficiency of SNARE formation at the docking site.
Rab GTPases modulate fusion of vesicular membranes through their effector proteins. One of the most well-characterized Rab7 effectors is the Rab7-interacting lysosomal protein (RILP) which is found on endosomes, lysosomes, and phagosomes (47, 63). The role of RILP in PL formation lies in its interaction with the dynein-dynactin complex during centripetal migration of phagosomes (47). Further, RILP recruits other Rab effectors involved in vesicle tethering and Rab7 activation. Thus, RILP recruitment is an attractive target for intracellular pathogens to exploit. Indeed, live Mycobacteria were found to secrete a Rab7 deactivating factor that could disrupt the interaction between RILP and membrane-bound Rab7 in macrophages (30). This was later determined to be nucleoside diphosphate kinase (Ndk), which enacts GAP activity to lock Rab7 in a GDP-bound state, and permitted survival of Mycobacteria within the phagosome (64). While this effect is likely not specific to RILP-Rab7 interactions, it does highlight the importance of maintaining Rab7 in a GTP-bound state to facilitate effector protein function.
Pleckstrin homology domain-containing family M member 1 (Plekhm1) is another Rab7 adaptor that is implicated in modulating PL formation. After the uptake of Salmonella by macrophages, Plekhm1 has been shown to bind the multi-subunit HOPS complex which facilitates the delivery of late endosome and lysosomes to Salmonella-containing vacuoles, and this is essential to maintain the integrity of these phagosomes for bacterial persistence (65, 66).
The multi-subunit complex HOPS is a Rab7 effector that can function as an upstream Rab7 GEF and a downstream tethering effector (56, 67, 68). Mammalian HOPS is a well-characterized tethering complex that facilitates vesicle-lysosome fusion of multiple degradative pathways, including PL fusion. The complex consists of seven proteins: Vps11, Vps16, Vps18, Vps33A, Vps39, and Vps41 (56, 69, 70). Two subunits appear to be particularly crucial for tethering. Vps39 and Vps41, which are respectively recruited to the HOPs complex by Vps11 and Vps18, can each interact with RILP and this may bridge two RILP molecules on opposing vesicles (Figure 2B) (71). Vps41 can also bind directly to Plekhm1 on either lysosomes or phagosomes (66), lysosomal Arl8 (70, 72) and phospoinositide-4-phosphates (PI4P) on phagosomes (73). The capacity for HOPS to bind to multiple partners likely increases the efficiency of HOPS acquisition to the fusion site, improves tethering between vesicles and maintains fusion fidelity (73).
In addition to its function in vesicle tethering, the HOPS complex can promote trans-SNARE docking at the PL fusion site and guide the formation of SNARE complexes to promote SNARE-mediated PL fusion (73). The HOPS Vps33 subunit is a member of the SM-family of proteins (74, 75) that can interact directly with SNAREs (76) and is proposed to catalyze SNARE complex assembly. Insights from yeast models suggest a dual-binding capacity of Vps33, where it binds two SNAREs, each from a different membrane, and serve as a base for generating partially-formed SNARE intermediates (24, 25). Bach et al. (77) provided evidence that mammalian Vps33 has a similar function during PL fusion through host-pathogen studies in Mycobacterium-infected THP-1-derived macrophages. Protein phosphatase 2A (PtpA), produced by M. tuberculosis, can dephosphorylate and inactivate Vps33b, one of the two Vps33 proteins in higher eukaryotes, as evidenced by a reduction in PL fusion after the uptake of PtpA-coated experimental particles by macrophages (77). PL biogenesis was similarly abrogated in Vps33b-silenced macrophages with or without PtpA-coated experimental particles and re-introduction of active Vps33b to the Vps33b-silenced macrophages restored PL fusion. The observation that functional Vps33b is important in PL biogenesis and bacterial clearance is even further supported by the observation that the loss of Vps33b or its binding partner Vps16b in the Drosophila Oregon-R phagocytic cell line leads to defects in PL formation (78). Taken together, all seven HOPs subunits are necessary for phagosome maturation.
The HOPS complex is the most extensively characterized Rab7 tethering effector facilitating PL fusion. Since other Rabs have been implicated in PL biogenesis, such as, Rab2 and Rab14, non-Rab7-facilitated HOPS recruitment is possible, or other tethering factors are involved in PL biogenesis, which warrants further investigation. Additional Rab7 adaptors including oxysterol-binding protein-related protein 1L (ORP1L) (79), Vps34/p150 (80) and Rubicon (81) have been characterized in endosome maturation but have yet to be explored in the context of phagosome maturation. Overall, the multifunctional HOPS complex provides a base at the PL fusion site that allows for local SNARE assembly, stabilizing the intermediate complex as the SM protein activity of HOPS chaperones SNARE assembly.
Fusing the Phagosome and Lysosome Membranes: SNAREs
SNARE proteins compose the core machinery required to fuse phagosomes with lysosomes efficiently. Based on the contributing amino acid to the hydrophobic core in the assembled SNARE complex, SNAREs can be structurally categorized into R-SNAREs or Q-SNAREs (of which can be subclassified into Qa-, Qb-, and Qc-SNAREs) (82). During membrane fusion, an R-SNARE on one membrane forms a transient trans-SNARE complex with three Q-SNARES located on the partner membrane (82). The resulting complex consists of an R, Qa-, Qb-, and Qc-SNARE that aligns in a parallel four-helix bundle and fuses the juxtaposed membranes via a zipper model (22). Post-fusion, all of the SNAREs are located on the same membrane in a fully assembled cis-SNARE complex. The post-fusion cis-SNARE complex is then recognized and disassembled by NSF along with its cofactor α-SNAP (27).
Early studies performed in J774 macrophages and cell-free systems demonstrated that PL fusion is dependent on NSF, providing first experimental evidence that this process employs SNARE proteins (83, 84). According to several quantitative proteomic studies, the presence and abundance of different SNAREs found at the phagosome changes depending on the cell-type, stage of maturation and the phagocytosed cargo (11, 85–88). As many as 30 of the 38 known human SNARE proteins have been identified on phagosomes (10), but which of these members function in coordinating late-stage phagosome-lysosome fusion is not well described. Rather, there is a greater understanding of their involvement in phagocytosis, phagosomal trafficking and early phagosome maturation, than during the final stages of maturation. The SNAREs that have thus far been implicated in PL fusion and found to be enriched in lysosomes and phagosomes, include, Syntaxin (Stx) 7, Stx8, vesicle transport through interaction with T-SNAREs 1b (Vti1b), vesicle associated membrane protein 7 (Vamp7) and Vamp8 (37, 39, 89, 90). Initial evidence of the involvement of these SNAREs in PL fusion was provided in a cell-free study wherein SNARE function was inhibited by the addition of the soluble cytosolic domain of various SNAREs (37). These truncated SNARE fragments outcompete their endogenous counterparts for binding with their membrane-bound cognate partners and form a stable SNARE complex that cannot mediate fusion because they lack transmembrane anchors. In the presence of the truncated form of the Qabc-SNAREs Stx7, Vti1b and Stx8, and the R-SNAREs Vamp7 and Vamp8, PL fusion was reduced, as evidenced by decreased lumenal mixing between lysosomes and phagosomes (37). Stx7-Vti1b-Stx8 were found to form a stable quaternary complex with the lysosomal R-SNARE Vamp7 or Vamp8, suggesting these may constitute a possible SNARE complex in PL fusion (37). In summary, Stx7-Vti1b-Stx8 may form a ternary Qabc-SNARE intermediate on the late phagosome that can then mediate PL fusion together with Vamp7 or Vamp8 on the lysosome, although evidence is lacking to support that these particular SNARE complexes are occurring within living cells to facilitate the fusion of phagosomes with lysosomes.
Interestingly, several investigations into the molecular mechanism underlying gp91phox recruitment to phagosomes have provided insight into PL fusion, and identified synaptosomal-associated protein 23 (Snap23) as a potential Qbc-SNARE acting in this process (38, 39, 91). gp91phox is the integral membrane component of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 complex (Nox2) which is responsible for generating the reactive oxygen species (ROS) that facilitate pathogen killing and the modulation of phagosomal proteolysis for antigen processing (92–94). The phagosome accumulates gp91phox predominantly through its fusion with lysosomes (95–97). siRNA-mediated knockdown of Snap23, Stx7, and Vamp8 decreased phagosomal gp91phox recruitment and subsequent ROS production in J774 macrophages, presumably from inhibited PL fusion (39). In corroboration with this observation, silencing Snap23 in primary neutrophils (91) and DCs (38) also inhibited gp91phox trafficking to phagosomes for subsequent ROS production. Whether Snap23 can act as the Qbc-SNARE instead of Vti1b-Stx8 in the Stx7-Vti1b-Stx8-Vamp7/8 model described above was supported by immunofluorescence and cellular fractionation experiments which found that Snap23 colocalizes with Stx7 and Vamp8 and can interact with Stx7 in J774 macrophages (38, 39). Moreover, Snap23, Stx7, and Vamp8 can form a stable SNARE complex in vitro (39) and knockdown of any of these SNAREs decreases phagosomal acquisition of gp91phox (38, 98). In another study, Vamp8 contributed to the regulation of phagosomal oxidative activity via PL fusion in Leishmania-infected primary macrophages (99). It is worth noting that Snap23 can also pair with Vamp7 (39), thus Vamp7 and Vamp8 may have redundant roles as a cognate R-SNARE on lysosomes.
Further insight into the function of Snap23 in PL formation was generated from a recent study by Sakurai et al. (40) that reported Snap23 could regulate PL formation based on its phosphorylation status. It was found that the phosphorylation of Snap23 at Ser95 by the protein kinase inhibitor kappa B kinase 2 (IKK2), in response to interferon-γ (IFN-γ) treatment, decreased fusion between phagosomes and late-endosomes/lysosomes in J774 macrophages. Thus, Snap23 is implicated to have an important physiologic role in activated macrophages by delaying phagosome fusion with acidic lysosomes, thereby contributing to the development of a phagosomal environment that favors antigen processing in pro-inflammatory environments (40, 100).
Current evidence suggests that Vamp7/8 on lysosomes, paired with Stx7 and Snap23/Vti1B-Stx8 on late phagosomes, act as the R-, Qa- and Qbc-SNAREs respectively, to facilitate fusion of these vesicles (Figure 2B). However, the promiscuous nature of SNARE proteins likely allows multiple SNARE complex permutations to facilitate the fusion of phagosomes and lysosomes and is reflected in the studies examined (10, 37, 39, 40). Similarly, multiple levels of posttranslational regulation of SNARE proteins beyond Snap23 phosphorylation likely exist to prevent erroneous fusion of these vesicles yet remain to be characterized (40).
Other Factors Involved in PL Fusion
It is clear from the number of fusion proteins discussed thus far that PL fusion is a complex process. However, Rabs, tethers and SNAREs are not the only factors involved in this process. The lipid composition of phagosomes, the actin cytoskeleton, the vacuolar-type ATPase (V-ATPase) and calcium signaling play important roles during phagosome maturation. Here, we will only discuss the phosphoinositide lipids and the controversial role of actin and the V-ATPase in PL biogenesis, as calcium signaling has been extensively reviewed elsewhere (101).
Phosphoinositide Lipids and Their Kinases
Phosphoinositides (PIs) play a critical role in PL formation. These are a family of mono-, bi-, or tri- phosphorylated derivatives of the glycerophospholipid phosphatidylinositol. Phosphorylation of the third, fourth or fifth position of the PI inositol headgroup by their specific lipid kinases generates different PI variants that regulate the actin cytoskeleton, signal transduction and membrane fusion/fission through interactions with their respective effector proteins (102, 103). In the context of PL biogenesis, PI3P and PI4P together with their lipid kinases are the most well-characterized in the fusion of phagosomes and lysosomes (73).
P13P is enriched on early phagosomes and is necessary to bind Rab5 for fusion with early endosomes (5, 104). As the phagosome progresses to the early-to-late-stage transition, PI3P is lost while both PI4P and PI3,5P2 are acquired in tandem with Rab7 (105, 106). PI4P is associated with the recruitment of the HOPS complex (73), whereas the role of PI3,5P2 remains less understood and there is conflicting reports on whether this lipid is essential for PL fusion and the full activation of the phagosomal V-ATPase (107, 108). Reduced PI3,5P2 synthesis in Raw264.7 macrophages abrogated both PL fusion and degradative capacity but had little effect on phagosome acidification, and at least on lysosomes, the V-ATPase was still active (107, 109). However, in lower eukaryotic models and mammalian epithelial cells, PI3,5P2 was required for V-ATPase activity, efficient phagosome fusion with lysosomes and acquisition of a microbicidal phagosomal lumen (108, 110–112). Thus, these discrepancies are likely due to physiological differences between macrophages and other cell models/types. Interestingly, PI3,5P2 has also been implicated in regulating lysosome/phagosome calcium channels in Raw264.7 macrophages (113).
Deciphering the roles of PIs during each step of PL formation within phagocytes is challenging as PIs function in the early steps of phagocytosis, and interference of PIs also affects the maturation of the phagosome upstream of PL fusion (114). Thus, reconstitution of PL fusion in cell-free in vitro assays in this scenario provides an advantage over in vitro systems since each fusion “subreaction” in PL formation can be manipulated and studied in isolation, without interfering with the early maturation of the phagosome (73, 114). In particular, work by Jeschke and colleagues have shed additional light on the role of PI3P, PI4P and their lipid kinases. As found by Jeschke et al. (114), phagosomes containing opsonized latex beads isolated from J774 macrophages contained PI3P, PI4P, and the class II phosphatidylinositol 4-kinase α (PI4KIIα), one of the four lipid kinases that catalyze the formation of PIs to PI4P (114). Sequestration of PI3P by the mouse hepatocyte growth factor-regulated tyrosine kinase substrate 2xFYVE domain or PI4P by the P4C fragment of Legionella pneumophila protein SidC blocked PL formation (115, 116). Further, chemical inhibition of either PI4KIIα or PI3KIII/Vps34 respectively reduced the levels of PI4P or PI3P generated on phagosomes, also impeding PL formation. This corroborates earlier studies, wherein inhibition of PI3KIII/Vps34 in J774 and Raw264.7 macrophages precluded the acquisition of the late endosome/lysosome makers, LAMP1 and lysobisphosphatidic acid, by phagosomes due to a defect in PL fusion (5, 104). Hence, PI3P and PI4P are required for PL fusion in a cell-free system, and this process can be regulated by PI lipid kinases that modulate the levels of PI3P and PI4P generated on phagosomes (114).
Further investigation by Jeschke and Haas (73) into the role of PI3P and PI4P in the steps leading to PL fusion observed that these PIs were involved during the tethering step of membrane fusion. Both PI3P and PI4P were present on phagosomes bound to lysosomes and sequestration of PI4P with P4C inhibited phagosome-to-lysosome binding and hemi-fusion of vesicles, while sequestration of PI3P by the 2xFYVE domain inhibited PL fusion after vesicles had docked (73). Since PIs exert their functions by anchoring PI effector proteins to membranes, it was postulated that PI3P and PI4P were recruiting PL-fusion-relevant proteins to phagosomes and lysosomes (73, 117). Indeed, a lack of available PI3P due to sequestration decreased the presence of the HOPS component Vps41 on phagosomes and lysosomes, and similarly, PI4P sequestration decreased both Vps41 and Arl8 levels. Further, when P4C and 2xFYVE were added to fusion sub-reactions containing isolated phagosomes and lysosomes but in the absence of cytosol, (which contains free PIs), membrane-bound Vps41 and Arl8 were absent, thereby supporting the observation that PI3P and PI4P are required to anchor tethering effectors. In addition to tethering, PI3P and PI4P have an impact on the fusion step of PL biogenesis, and thus may directly or indirectly recruit SNARE proteins (73). Interestingly, Vamp8 can bind directly to PI3P on Salmonella-containing vacuoles (118). It is known that Vamp8 binds PI3P to support phagocytosis of Salmonella (118), but whether the Vamp8-PI3P interaction is required or able to facilitate SNARE complex formation or PL membrane fusion requires further investigation. PI4P is also a substrate for the formation of PI4,5P2 on late endosomes/phagosomes and lysosomes (119). PI4,5P2 is required for the nucleation of actin around phagosomes, wherein this process has been postulated to promote PL fusion by interacting with SNARE proteins and will be discussed below (119, 120).
The class IA PI3K (PI3KIA), a kinase commonly associated with phagocytosis of large particles (>4 µm) and early endosome-phagosome fusion, has also been implicated in PL fusion (121). Thi and colleagues (121) used THP-1-derived macrophages to report a novel role for the class IA PI3K in PL fusion that depended on its catalytic subunit p110α (121). In p110α knockdown cells, phagocytosis of M. smegmatis or experimental particles occurred normally, however, phagosomes displayed impaired procurement of LAMP1 and lysosomal hydrolases due to defective fusion with lysosomes. Interestingly, p110α knockdown did not prevent Rab7 recruitment to phagosomes, nor its subsequent activation, as the recruitment of the Rab7 effectors HOPS and RILP were unaffected. This suggests recruitment of Rab7 and its effectors is insufficient to drive PL fusion alone, and requires the additional action of PI3KIA to mediate the formation of the PL. Moreover, cellular and phagosomal levels of the SNAREs Vamp7 and Vti1b in p110α-silenced macrophages were similar to the control. Hence, in this experimental system, the blockage of PL fusion is not due to a lack of membrane fusion machinery, but an undetermined mechanism. PIK3IA generates PI3,4,5P3 by the phosphorylation of PI4,5P2 (122). PI3,4,5P3 is transiently enriched at the phagocytic cup and is known for its role in actin polymerization upon Fc-γ receptor and complement receptor-mediated phagocytosis (123). With the exception of Fc-γ receptor phagocytosis, PI3,4,5P3 reappears on the maturing phagosome and is implicated in activating a second wave of actin formation important for PL biogenesis (124, 125). Since PI3,4,5P3 levels are decreased upon depletion of p110α (121), it is plausible that this may interfere with PI3,4,5P3-regulated actin polymerization around phagosomes (124).
Overall, host-pathogen studies have unveiled the consequences to bacterial clearance when certain PIs are unavailable for PL fusion while cell-free assays have allowed us better to understand where and when certain PIs are involved in the formation of PLs. From these studies, PI3KIII/Vps34, PI4KIIα and their respective lipid products PI3P and PI4P, as well as PI3KIA, regulate the development of PLs.
The Actin Cytoskeleton
The actin cytoskeleton is a major player in regulating phagocytosis (126, 127) and the fusion/fission of endolysosomes with phagosomes (120, 125, 128–130). Filamentous actin (hereafter referred to as actin) has been observed to transiently polymerize around phagosomes in a process called “actin-flashing”. The function of this phenomena is unclear but has been proposed to prevent PL fusion (125). Numerous reports using phagocytes and cell-free systems have demonstrated that actin coats a subset of purified phagosomes and acts as a physical barrier to PL biogenesis (Figure 3A) (125, 131–133). Furthermore, when internalized, certain pathogens, including Legionella, Leishmania, and Mycobacteria, induce the assembly of actin on the early phagosome to prevent phagosomal maturation (133–136). However, conflicting reports propose that actin can have a stimulatory role in membrane fusion, thus lending uncertainty to the role of actin in PL fusion (Figure 3B) (120, 128). A plausible explanation is that actin has dual roles in membrane fusion whereby it can both inhibit and induce phagosome-lysosome fusion depending on the maturation state of the phagosome (120). Most studies reporting the inhibitory role of actin limited their investigation to early-stage phagosomes. In particular, transient accumulations of actin were observed to surround a subpopulation of purified immature phagosomes which prevented association with endolysosomal vesicles (125, 137). It was also demonstrated that de novo actin assembly on nascent phagosomes increases under cell stress conditions when the phagocytic or endocytic pathway is overloaded with digestive cargo (125). These actin “flashes” repeat in waves to physically block phagosome-lysosome fusion.
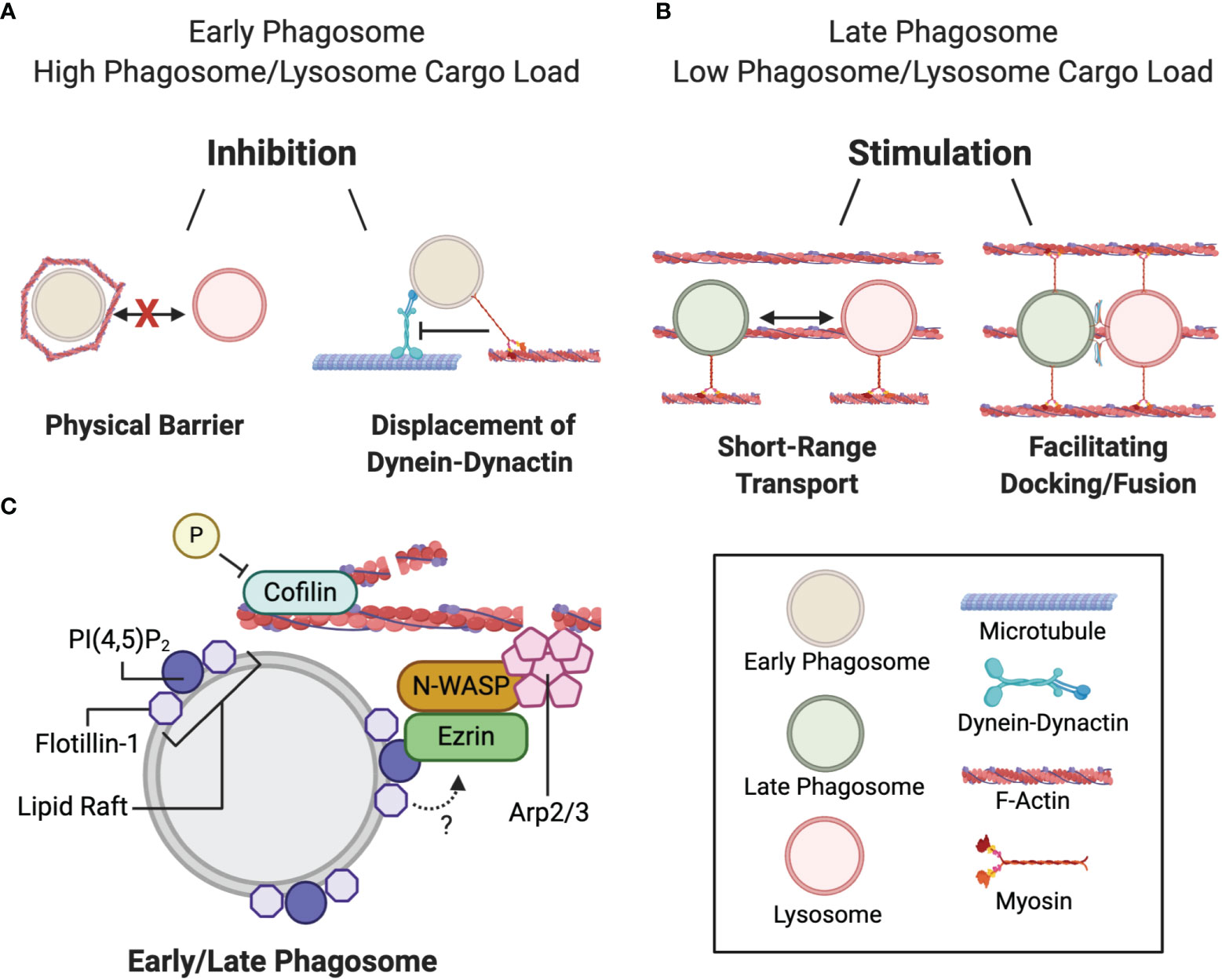
Figure 3 Duality of filamentous actin during phagosome maturation. (A) Actin delays phagolysosome biogenesis at the early phagosome maturation stage by: i) transiently assembling around nascent phagosomes when the early phagosome and/or lysosome system is overloaded with cargo thereby blocking phagosome-lysosome contact, or ii) by facilitating myosin-mediated displacement of the dynein-dynactin microtubule motor and preventing minus-end transport. (B) Actin stimulates phagolysosome biogenesis by: i) bringing the fusing organelles closer together, or ii) facilitating docking and fusion. (C) Actin polymerization at the phagosome is mediated by the ezrin-N-WASP-Arp2/3 complex binding to phagosomal PI(4,5)P2 at flotillin-1-associated lipid rafts. Cyclic actin polymerization and depolymerization is mediated by the respective phosphorylation and dephosphorylation of cofilin.
Not only can actin act as a physical barrier to fusion, it can also compete with dynein-dynactin-based binding to microtubules at the cellular periphery (Figure 3A). Phagosomes can bind to actin, in an ATP-dependent manner, through association with the actin motor protein myosin Va (131). Myosin Va interactions occur at defined regions of the phagosomal membrane, which dissociates the interactions with MT-associated proteins. It has been postulated that transient actin assembly on endolysosomal and phagosomal systems slows phagosome-lysosome fusion when these vesicles contain more cargo than they have the capacity to digest (125). Actomyosin association is a possible complimentary mechanism that can either delay phagosome migration to the perinuclear region to allow them extra time to preprocess internalized phagosomal content, and to not outpace the availability of lysosomes for fusion (131, 137).
In contrast, late-stage phagosomes and endolysosomes have been shown to nucleate actin locally around the phagosome-to-lysosome docking sites (128). Interestingly, numerous actin cytoskeleton-associated machinery regulating actin nucleation were localized around latex bead-containing late-phagosomes, such as ezrin, neural Wiskott-Aldrich syndrome protein (N-WASP) and the actin-related protein 2/3 (Arp2/3) complex (Figure 3C). Ezrin binds to PI4,5P2 on the phagosomal membrane, although it is unclear whether PI4,5P2 is sufficient to recruit ezrin (119, 138). Ezrin has been proposed to recruit N-WASP to the phagosome membrane, which subsequently activates Arp2/3 to initiate actin nucleation. The idea that actin promotes late-stage phagosome maturation was further supported by the observation that downregulating ezrin hindered PL fusion (120). In later studies, cell division control protein 42 homolog (CDC42) of the Rho-family of GTPases was found to be required for the activation of N-WASP (139, 140). The dynamic phosphorylation and dephosphorylation of actin-binding protein cofilin is also required to mediate cyclic actin polymerization and depolymerization that promotes PL fusion (141). Furthermore, flotillin-1, a membrane protein that associates with lipid rafts on late phagosomes is also implicated in actin nucleation since actin predominantly accumulates and polymerizes at these sites (142, 143). Dermine et al. (142) observed decreased flotillin-1 association with phagosomes containing Leishmania in J774 macrophages. Leishmania expresses a surface glycolipid lipophosphoglycan that impedes PL fusion. Since lipophosphoglycan preferentially anchors to phagosomal lipid rafts, it was proposed that this process may interfere with lipid raft formation, and subsequent flotillin-1 association and actin assembly (142). However, the molecular interactions between flotillin-1 and the other actin polymerizing machinery at phagosomal lipid microdomains were not addressed and remain largely obscure. How actin functions in tandem with membrane fusion machinery—whether by bringing fusing vesicles together via myosin motor proteins, or by facilitating the docking and tethering of lysosomes to the phagosome membrane (Figure 3B) (120, 128)—is unclear and warrants further investigation.
Interestingly, the same actin nucleation machinery is recruited to early and late phagosomes to exert dualistic stimulatory and inhibitory effects. Leishmania donovani promastigotes promote CDC42 retention at phagosomes thereby prolonging phagosomal actin accumulation to evade the host endomembrane system in Raw264.7 macrophages (135). It was recently found that CDC42 is unable to associate with its GAP and GDI in this context, thereby locking it in a GTP-bound state (144, 145). One of these studies demonstrated that Shigella flexneri exploits N-WASP and Arp2/3 recruitment to the nascent bacteria-containing vesicles to impede lysosomal fusion. Molecularly, the acyltransferase activity of the type 3 secretion system effector IcsB on CDC42 disrupts its association with its GDI (144), locking CDC42 in a GTP-bound state which retains CDC42 at phagosomes (145). Through this, IcsB clusters N-WASP and Arp2/3 around vesicles to nucleate thick actin coats, which both block the recruitment of fusion machinery and shield the bacterium from the endolysosomes. It is important to note that the studies by Liu et al. (144) and Kühn et al. (145) were conducted in epithelial cells that although are capable of phagocytosis, do not possess the repertoire of surface receptors of professional phagocytes.
In summary, actin networks are thought to have a dual role in phagosome-lysosome fusion depending on how far the phagosome has matured and the cargo load of phagosomes and lysosomes. Actin networks are inhibitory when phagosomes and lysosomes are saturated with digestive cargo by physically preventing early phagosomes from contacting endolysosomes. Actomyosin could potentially compete with dynein-dynactin motor proteins for binding to the phagosome to delay phagosome maturation. In contrast, actin networks can support late phagosome-lysosome fusion, either by bringing vesicles closer together or by facilitating vesicle docking—although the mechanisms by which actin achieves these functions and whether the involvement of actin is cargo-dependent is unclear. Moreover, the spatiotemporal control of actin that enables it to either block or enhance PL fusion remains uncharacterized. Indeed, the role of the actin-myosin network during phagosome maturation is only beginning to be understood, and further work in the area will help garner appreciation of such complex interactions.
The Vacuolar ATPase
Phagocytes acidify the phagosomal lumen by recruiting the V-ATPase, a multiprotein complex that consists of 14 different subunits (146). The cytosolic V1 domain mediates ATP hydrolysis, whereas the transmembrane V0 domain forms the proton channel (146). Phagosomal V-ATPase is largely considered to be derived from lysosomes; however, other data suggests that phagosomes acquire V-ATPases from multiple sources, including, the trans-Golgi, early endosomes, lysosomes and the plasma membrane (147–149).
Although it is well-known that V-ATPase-mediated phagosomal acidification is essential for antimicrobial defense and efficient digestion/processing of phagocytosed cargo, there is controversy surrounding the role of the V-ATPase in membrane fusion. The first evidence to demonstrate the requirement of the V-ATPase in homotypic vesicle fusion was from a study in yeast (150). Following trans-SNARE formation, the V0 domains from apposing vesicles were proposed to complex, leading to a conformational change which allows for lipid mixing and the formation of a fusion pore (150). An acidification-independent role of the V-ATPase in membrane fusion was observed in higher eukaryotes in subsequent studies (151–153).
The role of the V-ATPase in PL fusion was first observed in a zebrafish model, wherein knockdown of the V0 A1 subunit prevented PL fusion in microglial cells (154). Further, inhibiting the macrophage-specific V0 subunit D2 in murine bone marrow-derived macrophages impaired PL fusion and Salmonella clearance (155). Interestingly, by immunoprecipitation and glutathione S-transferase affinity isolation assays, subunit D2 was found to form a complex with the SNAREs Stx17 and Vamp8 important in autophagosome-lysosome fusion (155). Whether the V-ATPase could associate with fusion machinery involved in PL fusion was demonstrated in an earlier study in Mycobacterium-infected THP-1 macrophages (156). As mentioned above, the M. tuberculosis effector PtpA dephosphorylates the SM protein Vps33B to prevent PL fusion (77). Wong et al. (156) demonstrated that PtpA directly binds to the V1 subunit H to: i) block the trafficking of this subunit to the mycobacterial phagosome and ii) block its interaction with Vps33B (156). Moreover, this binding step is a prerequisite to dephosphorylate Vps33B. When macrophages were challenged with ptpA knockout strains, subunit H could localize to phagosomes and recruit Vps33B. Contrary to these findings implicating the V-ATPase in PL biogenesis, other reports dispute this contribution.
In mouse peritoneal macrophages, the loss of the V0 subunit A3 did not block PL formation during Fc-γ receptor phagocytosis of IgG-opsonized latex beads—phagosomes could acquire lysosomal characteristics including the mature forms of lysosomal cathepsins, and late-phagosome/lysosome markers Rab7 and Lamp2 (157). In corroboration with these findings, mouse bone marrow-derived macrophages deficient in the V0 subunits A1-3 had unaltered microbicidal activity since these cells could clear avirulent strains of Escherichia coli and Listeria innocua as efficiently as the control (158). Interestingly, through pulse-chase experiments with latex beads, the rate of PL fusion was elevated upon the loss of the V0 subunits A1-3.
Thus, whether the V-ATPase enhances PL fusion is still being debated. Different phagocytic cargoes and/or cell models leading to slightly altered maturation pathways may explain discrepancies in the works described. Moreover, it appears that specific subunits of the V-ATPase (V0 subunit D2 and V1 subunit H) have more important roles in supporting PL fusion than others (V0 subunits A1-3).
Autophagy to the Rescue: Supporting Phagocyte Function in the Context of PL Fusion
As noted earlier, several pathogens have evolved mechanisms to subvert canonical maturation of the phagosome. In response, alternative mechanisms to facilitate PL fusion have coevolved in professional phagocytes. Macroautophagy, or bulk degradation autophagy (herein referred to as autophagy), has been established as a fundamental homeostatic pathway that affects innate and adaptive immunity (159, 160). Similar to the phagocytic pathway for exogenous cargoes, autophagy delivers endogenous material to lysosomes for degradation. Low levels of basal autophagy is essential for all cells to clear unwanted cytosolic material and maintain cellular homeostasis (161–163). In phagocytes, autophagy is upregulated by several stress signals, including pro-inflammatory cytokines (164), TLR signaling (165), starvation (166), and microbial infection (167). The role of autophagy in infection, inflammation, and adaptive immunity has received increasing attention over the past 2 decades. Germane to this review, components of the autophagic pathway have been implicated in PL fusion—proposed as an alternate pathway to overcome pathogen inhibition of canonical phagosomal maturation and enhancing the presentation of exogenous antigens. In the following section, we will discuss how the autophagic pathway complements the phagocytic pathway with an emphasis on PL fusion.
Microbial Clearance
The best-understood pathways by which autophagy augments microbicidal capabilities of phagocytes are xenophagy (168) and microtubule-associated protein A1/B1-light chain 3 (LC3)-associated phagocytosis (LAP) (169–171). Xenophagy is the selective degradation of cytosolic pathogens or pathogen-containing vesicles that have been marked with ubiquitin through autophagic mechanisms (172–174). In contrast, LAP is a non-canonical form of autophagy that involves phagocytosis and uses some components of the autophagic pathway to create a more robust phagolysosome (169–171).
Xenophagy
Several intracellular pathogens have developed mechanisms to avoid PL-mediated destruction, and autophagy can be deployed to assist in their elimination (Figure 4). M. tuberculosis is well known to inhibit phagosomal maturation, but induction of autophagy is sufficient to eliminate the pathogen via autophagic consumption of the arrested bacteria-containing phagosome (175). The parasite Toxoplasma gondii can survive within macrophages by residing in vacuoles that avoid fusion with lysosomes (176). However, when macrophages are activated by a T cell ligand, cluster of differentiation 40 (CD40), xenophagy is induced to restrict T. gondii growth (177, 178). Alternatively, when activated by IFN-γ, macrophages use some components of the autophagic machinery to recruit members of the immunity-related GTPase (IRG) family to mediate the elimination of these parasites in a process that is not fully understood (179, 180). However, evidence from Mycobacteria-containing macrophages suggests that IRGs may promote the fusion of phagosomes to lysosomes since Irgm1 has been observed to interact with SNAP-associated protein (Snapin), which binds to SNAREs to mediate vesicular fusion (181, 182).
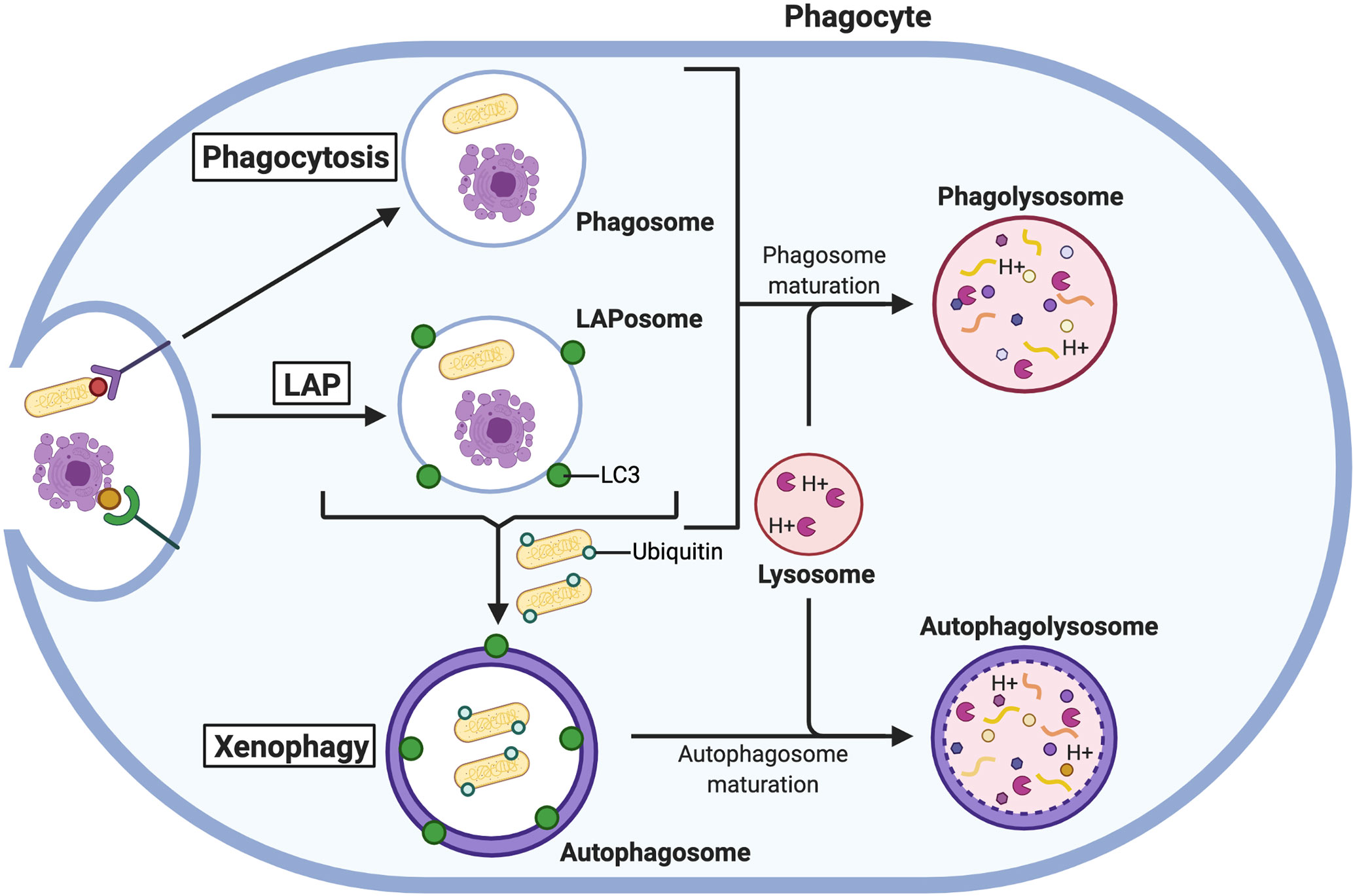
Figure 4 The autophagic response aids the phagocytic pathway. Phagocytosed microbes and apoptotic cells are degraded in PLs formed from the fusion of lysosomes with phagosomes. Under certain conditions, LC3 is recruited to phagosomes for LAP to create more robust PLs. Microbes that escape PL-mediated degradation are ubiquitinated in the cytosol and degraded in autophagolysosomes via xenophagy.
Although xenophagy functions as a second line of defense against pathogens that overcome PL-mediated killing in phagocytes, some pathogens have further evolved to evade xenophagic activity by inhibiting the autophagic response. For example, the virulence factors of M. tuberculosis early secretory antigenic target 6 (ESAT6) and secreted acid phosphatase M (SapM), block autophagosome-lysosome fusion in Raw264.7 macrophage-like cells; ESAT6 stimulates the negative regulator of autophagy mammalian target of rapamycin (MTOR), and SapM hydrolyzes PI3P to prevent Rab7 recruitment to phagosomes and autophagosomes (183–186). In another study using Raw264.7 cells, L. monocytogenes was found to hide from autophagic recognition through its virulence factor Inlk (187).
In summary, while xenophagy often serves as an effective second defense to pathogen PL arrest or escape in many cases, some well-adapted pathogens have evolved virulence factors that target these autophagic pathways.
LC3-Associated Phagocytosis
Autophagy can also augment the antimicrobial activity of phagocytes by supporting increased PL fusion through LAP (Figure 4). The process of LAP requires several members of the canonical autophagic machinery, namely autophagy-related (Atg) 5, Atg7, Atg12, Atg16L, Beclin1, and Vps34 (169), as well as Rubicon and ROS production by Nox2 (171, 188). Despite sharing components, LAP and autophagy are functionally distinct processes that are differentially regulated. For example, Rubicon inhibits autophagy but is required for efficient LAP (189). LAP can be stimulated when internalized particles activate certain surface receptors of phagocytes, including TLRs, Fc receptors, Dectin-1, and the apoptotic cell receptor T cell immunoglobin and mucin domain containing 4 (TIM4) (169–171). When these receptors are engaged, phagosomes recruit LC3 (now referred to as LAPosomes) which promotes rapid fusion with lysosomes and enhances cargo degradation (169, 188). Through LAP, phagocytes are better equipped to deal with microbial infection by bacteria and fungi. For example, PL fusion with phagosomes containing the bacteria L. monocytogenes (190) and Legionella dumoffi (191), and the fungi S. cerevisiae (169) and A. fumigatus (188) is promoted by LAP [reviewed by (192)]. In the absence of LC3 recruitment to phagosomes, macrophages could not efficiently kill these pathogens and were more susceptible to sustained infection.
It is unsurprising then that pathogens have also evolved strategies to avoid targeting by LAP. Some prominent examples are CpsA production by M. tuberculosis (193), surface metalloprotease GP63 production by Leishmania major (194), and melanin production by A. fumigatus (195), which all function to evade LAP by preventing the Nox2-mediated ROS production crucial for LC3-recruitment [reviewed by (192)].
It is worth noting that LAP is not the only process that can enhance PL fusion. Enhanced PL fusion was observed despite the absence of Nox2-mediated LC3 recruitment to phagosomes containing IgG-opsonized zymosan and sheep erythrocytes in macrophages (196). This suggests that under certain conditions, proteins other than those belonging to the autophagic pathway such as members of the IRG family mentioned above (181, 182), can also enhance membrane fusion. Alternatively, it has been recently demonstrated that the mechanical stress caused by the growth of some intraphagosomal pathogens can induce lysosome recruitment and fusion with the phagosome as a means to increase phagosomal surface area and maintain membrane integrity (197). Further, LAP has a dual function in antigen presentation depending on the cell type. LAP stimulates rapid PL fusion in murine macrophages, favoring cargo degradation over antigen presentation (169). In contrast, LAP delays recruitment of lysosomes in human macrophages and DCs to stabilize substrates for major histocompatibility complex class II (MHCII)-mediated presentation to the adaptive immunity (198). Thus, LC3-mediated phagosome maturation is host- and condition-dependent and is a dynamic process that is influenced by the activation of phagocytes.
How LAP promotes or inhibits PL fusion at the molecular level is only beginning to be discerned. One plausible mechanism was seen in neuronal cells, where LC3 binding of RILP to autophagosomes facilitated the recruitment of the dynein-dynactin motor complex for subsequent centripetal movement to the lysosome-rich perinuclear region (199). It would be interesting to investigate whether phagosomal LC3 can recruit RILP-dynein-dynactin to facilitate phagosome trafficking in a similar manner in phagocytes, particularly in the context of LAP. Another possibility is that LC3 plays a role in recruiting machinery to the phagosome that can assist in tethering lysosomes to phagosomes (192). Plekhm1 can be recruited by LC3 to autophagosomes in HeLa cells (200). By extension, LC3-recruited Plekhm1 on phagosomes could serve to tether HOPS- and RILP-containing vesicles during PL formation. Additionally, the LC3 isoforms γ-aminobutyric acid receptor-associated protein (Gabarap) and Gabarap-like 2 can recruit PI4KIIα (201), which generates PI4P on phagosomes and lysosomes to promote HOPS recruitment (114, 202). However, direct evidence to support these hypotheses in phagocyte LAPosomes has yet to be substantiated and awaits further investigation.
Homeostasis and Inflammation Dampening
Rapid and efficient clearance of apoptotic cells by phagocytes is critical for the regulation of tissue and immune homeostasis. Professional phagocytes are recruited by chemo-attractants released by apoptotic cells, and the “eat-me” cell-surface ligands on the dying cells mark them for phagocytotic engulfment by efferocytosis. It is now well-established that autophagy has a complex relationship with apoptosis in that autophagy can either be preventative or facilitative in programmed cell death [discussed comprehensively by (203)]. Most recently, LAP has been established as a crucial process in efferocytosis (Figure 4). Disrupting LAP in macrophages during TIM4-mediated engulfment delays degradation of apoptotic and necrotic cells in phagosomes (204). In the absence of LAP (created by deleting key autophagic genes), apoptotic bodies accumulate in phagosomes but are not digested due to a decrease in lysosomal fusion (205). Moreover, the accumulation in apoptotic cargo leads to secondary necrosis in macrophages. This was also observed in another study by Zhou et al. (206) in which autophagy had a cytoprotective effect on macrophages that phagocytosed apoptotic cells. Silencing a key autophagic protein, Beclin1, in macrophages displayed increased apoptotic cell cargo resulting in decreased viability and survival, and macrophage rupturing (206). When stimulated with apoptotic cells, macrophages deficient in LAP due to genetic knockout of Rubicon or Nox2 produced higher levels of pro-inflammatory cytokines (interleukin-(IL)-1β, IL-6, CXCL10) and lower levels of anti-inflammatory cytokines (IL-10) compared to LAP-sufficient cells (205). This increase in pro-inflammatory signals translates to autoimmunity and the development of systemic lupus erythematosus-like disease in in vivo mouse models when LAP is not functioning to clear apoptotic cells (205). Hence, LAP-enhanced PL fusion is fundamental to regulate pro-inflammatory and pro-death signals.
In summary, autophagy and phagocytosis are two complementary pathways that cooperate in PL-mediated elimination of dangerous exogenous and endogenous material to regulate infection and homeostasis. Canonical autophagic machinery is employed to either supplement or support phagosome maturation or to conduct microbicidal and homeostatic functions. Induction of the autophagic machinery enhances PL biogenesis via LAP, although the molecular mechanism has not yet been fully characterized. Current evidence suggests LC3-recruitment of phagosome trafficking and fusion machinery, yet this warrants further investigation.
Concluding Remarks
Here we have summarized recent findings regarding phagosome-lysosome fusion. The Rab GTPases Rab7, Rab2 and Rab14 participate in tethering phagosomes and lysosomes albeit through mechanisms that are less known for Rab2 and Rab14. Rab7 recruits tethering effectors, whereby the individual subunits of the HOPS complex give it the multifunctional capacity to serve as a Rab GEF, a tether and an SM protein that catalyzes SNARE assembly. The SNAREs Snap23, Stx7, Vamp8, Vti1b, and Stx8 have been shown to assemble into specific complexes to execute PL fusion. While the Rabs, tethering factors and SNAREs involved in PL biogenesis are best known, we know less about how phagosomal lipids, the actin network and the V-ATPase regulate PL fusion. PI3P and PI4P have been implicated to interact with fusion machinery, whereas PI4,5P2 is involved in actin nucleation, which exhibits dual function to either support or inhibit PL fusion. PI3,5P2 is required for V-ATPase activation and PL fusion in lower eukaryotes but this has yet to be shown in mammalian phagocytes. Acidification-independent roles of the V-ATPase in fusion is still under contention but some evidence suggests the involvement of specific subunits. Lastly, the autophagic pathway can bolster the phagocytic pathway through LAP-enhanced PL fusion, or xenophagy-mediated protection of phagocyte integrity.
Although there have been great advances in understanding the proceedings of the molecular events that mediate PL biogenesis, we have only scratched the surface in delineating the full extent of all the participating molecules and modulating signals. For those that have been identified, little is known regarding their spatiotemporal regulation. The greatest caveat to the existing studies that have furthered our understanding of the aforementioned fusion machinery is that they have been performed extensively in in vitro cell-free models or phagocyte-like cell lines which limits our ability to confidently extrapolate to primary phagocytes. To this degree, few studies have investigated the molecules that control phagosome-lysosome fusion in live primary phagocytes, generating a gap between experimental evidence and functional relevance to the management of infection and disease. For studies that have employed primary cell models, it is unclear whether the fusion molecules recruited to phagosomes is consistent for all cargo types. Phagocytic uptake of macromolecular cargos can be mediated by several cell-surface receptors that activate different signaling pathways based on the type of cargo. The activation/inflammation status of these cells can influence the rate and magnitude of the dynamic recruitment of fusion-mediating proteins. Even the type of phagocyte and particular species from which cells are sourced can influence which particular fusion proteins are involved. Thus, further studies are required to more comprehensively characterize this critical and often underappreciated aspect of phagocyte biology.
Phagosome fusion with lysosomes is crucial to the functional outcome of phagocytosis and efferocytosis. Impaired lysosome fusion—and by extension, cargo degradation—disrupts the immune response to microbial and apoptotic cell clearance, and can underlie autoimmune and inflammatory disorders, and persistence of infection. Deciphering the mechanisms that drive and regulate the fusion of phagosomes with lysosomes will not only contribute to the greater knowledge of the pathogenesis of many diseases, but also contribute to our understanding of health, growth, and homeostasis.
Author Contributions
JN wrote the manuscript with support from RY. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr. Catherine Greene and Dr. Corey Arnold and thank them for their critical review of the manuscript. The figures used in this review were created with BioRender.com. This work was supported by the Canadian Institutes of Health Research and Natural Sciences and Engineering Research Council of Canada.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.636078/full#supplementary-material
Abbreviations
Arl, ADP-ribosylation factor-like protein; Arp2/3, actin-related protein 2/3; Atg, autophagy-related; CDC42, cell division control protein 42 homolog; DC, dendritic cell; ESAT6, early secretory antigenic target 6; Gabarap, γ-aminobutyric acid receptor-associated protein; GAP, GTPase-activating protein; GDI, guanine dissociation inhibitor; GEF, guanine exchange factor; HOPS, homotypic fusion and vacuole sorting; IFN-γ, interferon-γ; IKK2, inhibitor kappa B kinase 2; IL, interleukin; IRG, immunity-related GTPase; LAP, LC3-associated phagocytosis; LC3, microtubule-associated protein A1/B1-light chain 3; MAPK, p38α-mitogen-activated protein kinase; MHCII, major histocompatibility complex class II; MT, microtubule; N-WASP, Wiskott-Aldrich syndrome protein; NADPH, nicotinamide adenine dinucleotide phosphate; Ndk, nucleoside diphosphate kinase; Nox2, NADPH oxidase 2 complex; NRBF2, nuclear receptor binding factor 2; NSF, N-ethylmaleimide-sensitive factor; ORP1L, oxysterol-binding protein-related protein 1L; PI, phosphoinositide; PI3K, phosphoinositide 3-kinase; PI3KIA, class IA PI3K; PI3KIII, class III phosphoinositide 3-kinase; PI4KIIα, class II phosphatidylinositol 4-kinase α; PI4P, phospoinositide-4-phosphate; PL, phagolysosome; Plekhm1, pleckstrin homology domain-containing family M member 1; PtpA, protein phosphatase 2A; RILP, Rab7-interacting lysosomal protein; ROS, reactive oxygen species; SapM, secreted acid phosphatase M; SM, Sec1/Munc18; SNAP, soluble N-ethylmaleimide-sensitive fusion factor attachment protein; Snap23, synaptosomal-associated protein 23; Snapin, SNAP-associated protein; SNARE, soluble NSF attachment protein receptor; Snx10, sorting nexin 10; Stx, syntaxin; TIM4, T cell immunoglobin and mucin domain containing 4; TLR, toll-like receptor; V-ATPase, vacuolar-type ATPase; Vamp, vesicle associated membrane protein; Vps, vacuolar protein sorting; Vti1b, vesicle transport though interaction with T-SNAREs 1b.
References
1. Turvey SE, Broide DH. Innate immunity. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S24–32. doi: 10.1016/j.jaci.2009.07.016
2. Vieira OV, Botelho RJ, Grinstein S. Phagosome maturation: aging gracefully. Biochem J (2002) 366(Pt 3):689–704. doi: 10.1042/BJ20020691
3. Desjardins M. Biogenesis of phagolysosomes: the ‘kiss and run’ hypothesis. Trends Cell Biol (1995) 5(5):183–6. doi: 10.1016/s0962-8924(00)88989-8
4. Desjardins M, Huber LA, Parton RG, Griffiths G. Biogenesis of phagolysosomes proceeds through a sequential series of interactions with the endocytic apparatus. J Cell Biol (1994) 124(5):677–88. doi: 10.1083/jcb.124.5.677
5. Fratti RA, Backer JM, Gruenberg J, Corvera S, Deretic V. Role of phosphatidylinositol 3-kinase and Rab5 effectors in phagosomal biogenesis and mycobacterial phagosome maturation arrest. J Cell Biol (2001) 154(3):631–44. doi: 10.1083/jcb.200106049
6. Vergne I, Fratti RA, Hill PJ, Chua J, Belisle J, Deretic V. Mycobacterium tuberculosis phagosome maturation arrest: mycobacterial phosphatidylinositol analog phosphatidylinositol mannoside stimulates early endosomal fusion. Mol Biol Cell (2004) 15(2):751–60. doi: 10.1091/mbc.e03-05-0307
7. Desjardins M, Descoteaux A. Inhibition of phagolysosomal biogenesis by the Leishmania lipophosphoglycan. J Exp Med (1997) 185(12):2061–8. doi: 10.1084/jem.185.12.2061
8. Graham JG, MacDonald LJ, Hussain SK, Sharma UM, Kurten RC, Voth DE. Virulent Coxiella burnetii pathotypes productively infect primary human alveolar macrophages. Cell Microbiol (2013) 15(6):1012–25. doi: 10.1111/cmi.12096
9. Thywissen A, Heinekamp T, Dahse HM, Schmaler-Ripcke J, Nietzsche S, Zipfel PF, et al. Conidial Dihydroxynaphthalene Melanin of the Human Pathogenic Fungus Aspergillus fumigatus Interferes with the Host Endocytosis Pathway. Front Microbiol (2011) 2:96. doi: 10.3389/fmicb.2011.00096
10. Dingjan I, Linders PTA, Verboogen DRJ, Revelo NH, Ter Beest M, van den Bogaart G. Endosomal and Phagosomal SNAREs. Physiol Rev (2018) 98(3):1465–92. doi: 10.1152/physrev.00037.2017
11. Rogers LD, Foster LJ. The dynamic phagosomal proteome and the contribution of the endoplasmic reticulum. Proc Natl Acad Sci U S A (2007) 104(47):18520–5. doi: 10.1073/pnas.0705801104
12. Knapp PE, Swanson JA. Plasticity of the tubular lysosomal compartment in macrophages. J Cell Sci (1990) 95( Pt 3):433–9.
13. Keller S, Berghoff K, Kress H. Phagosomal transport depends strongly on phagosome size. Sci Rep (2017) 7(1):17068. doi: 10.1038/s41598-017-17183-7
14. Li X, Rydzewski N, Hider A, Zhang X, Yang J, Wang W, et al. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat Cell Biol (2016) 18(4):404–17. doi: 10.1038/ncb3324
15. Rai A, Pathak D, Thakur S, Singh S, Dubey AK, Mallik R. Dynein Clusters into Lipid Microdomains on Phagosomes to Drive Rapid Transport toward Lysosomes. Cell (2016) 164(4):722–34. doi: 10.1016/j.cell.2015.12.054
16. Jahn R, Lang T, Sudhof TC. Membrane fusion. Cell (2003) 112(4):519–33. doi: 10.1016/s0092-8674(03)00112-0
17. Lurick A, Kummel D, Ungermann C. Multisubunit tethers in membrane fusion. Curr Biol (2018) 28(8):R417–R20. doi: 10.1016/j.cub.2017.12.012
18. Ali BR, Wasmeier C, Lamoreux L, Strom M, Seabra MC. Multiple regions contribute to membrane targeting of Rab GTPases. J Cell Sci (2004) 117(Pt 26):6401–12. doi: 10.1242/jcs.01542
19. Conibear E, Cleck JN, Stevens TH. Vps51p mediates the association of the GARP (Vps52/53/54) complex with the late Golgi t-SNARE Tlg1p. Mol Biol Cell (2003) 14(4):1610–23. doi: 10.1091/mbc.e02-10-0654
20. Fiebig KM, Rice LM, Pollock E, Brunger AT. Folding intermediates of SNARE complex assembly. Nat Struct Biol (1999) 6(2):117–23. doi: 10.1038/5803
21. Chen YA, Scales SJ, Scheller RH. Sequential SNARE assembly underlies priming and triggering of exocytosis. Neuron (2001) 30(1):161–70. doi: 10.1016/s0896-6273(01)00270-7
22. Melia TJ, Weber T, McNew JA, Fisher LE, Johnston RJ, Parlati F, et al. Regulation of membrane fusion by the membrane-proximal coil of the t-SNARE during zippering of SNAREpins. J Cell Biol (2002) 158(5):929–40. doi: 10.1083/jcb.200112081
23. Poirier MA, Xiao W, Macosko JC, Chan C, Shin YK, Bennett MK. The synaptic SNARE complex is a parallel four-stranded helical bundle. Nat Struct Biol (1998) 5(9):765–9. doi: 10.1038/1799
24. Baker RW, Jeffrey PD, Zick M, Phillips BP, Wickner WT, Hughson FM. A direct role for the Sec1/Munc18-family protein Vps33 as a template for SNARE assembly. Science (2015) 349(6252):1111–4. doi: 10.1126/science.aac7906
25. Song H, Orr AS, Lee M, Harner ME, Wickner WT. HOPS recognizes each SNARE, assembling ternary trans-complexes for rapid fusion upon engagement with the 4th SNARE. Elife (2020) 9. doi: 10.7554/eLife.53559
26. Ellena JF, Liang B, Wiktor M, Stein A, Cafiso DS, Jahn R, et al. Dynamic structure of lipid-bound synaptobrevin suggests a nucleation-propagation mechanism for trans-SNARE complex formation. Proc Natl Acad Sci U S A (2009) 106(48):20306–11. doi: 10.1073/pnas.0908317106
27. Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell (1993) 75(3):409–18. doi: 10.1016/0092-8674(93)90376-2
28. Hanson PI, Otto H, Barton N, Jahn R. The N-ethylmaleimide-sensitive fusion protein and alpha-SNAP induce a conformational change in syntaxin. J Biol Chem (1995) 270(28):16955–61. doi: 10.1074/jbc.270.28.16955
29. Ungermann C, Nichols BJ, Pelham HR, Wickner W. A vacuolar v-t-SNARE complex, the predominant form in vivo and on isolated vacuoles, is disassembled and activated for docking and fusion. J Cell Biol (1998) 140(1):61–9. doi: 10.1083/jcb.140.1.61
30. Sun J, Deghmane AE, Soualhine H, Hong T, Bucci C, Solodkin A, et al. Mycobacterium bovis BCG disrupts the interaction of Rab7 with RILP contributing to inhibition of phagosome maturation. J Leukoc Biol (2007) 82(6):1437–45. doi: 10.1189/jlb.10.1189
31. Guo P, Hu T, Zhang J, Jiang S, Wang X. Sequential action of Caenorhabditis elegans Rab GTPases regulates phagolysosome formation during apoptotic cell degradation. Proc Natl Acad Sci U S A (2010) 107(42):18016–21. doi: 10.1073/pnas.1008946107
32. Mangahas PM, Yu X, Miller KG, Zhou Z. The small GTPase Rab2 functions in the removal of apoptotic cells in Caenorhabditis elegans. J Cell Biol (2008) 180(2):357–73. doi: 10.1083/jcb.200708130
33. Via LE, Deretic D, Ulmer RJ, Hibler NS, Huber LA, Deretic V. Arrest of mycobacterial phagosome maturation is caused by a block in vesicle fusion between stages controlled by rab5 and rab7. J Biol Chem (1997) 272(20):13326–31. doi: 10.1074/jbc.272.20.13326
34. Kinchen JM, Doukoumetzidis K, Almendinger J, Stergiou L, Tosello-Trampont A, Sifri CD, et al. A pathway for phagosome maturation during engulfment of apoptotic cells. Nat Cell Biol (2008) 10(5):556–66. doi: 10.1038/ncb1718
35. Gumienny TL, Hengartner MO. How the worm removes corpses: the nematode C. elegans as a model system to study engulfment. Cell Death Differ (2001) 8(6):564–8. doi: 10.1038/sj.cdd.4400850
36. Zhou Z, Yu X. Phagosome maturation during the removal of apoptotic cells: receptors lead the way. Trends Cell Biol (2008) 18(10):474–85. doi: 10.1016/j.tcb.2008.08.002
37. Becken U, Jeschke A, Veltman K, Haas A. Cell-free fusion of bacteria-containing phagosomes with endocytic compartments. Proc Natl Acad Sci U S A (2010) 107(48):20726–31. doi: 10.1073/pnas.1007295107
38. Dingjan I, Linders PT, van den Bekerom L, Baranov MV, Halder P, Ter Beest M, et al. Oxidized phagosomal NOX2 complex is replenished from lysosomes. J Cell Sci (2017) 130(7):1285–98. doi: 10.1242/jcs.196931
39. Sakurai C, Hashimoto H, Nakanishi H, Arai S, Wada Y, Sun-Wada GH, et al. SNAP-23 regulates phagosome formation and maturation in macrophages. Mol Biol Cell (2012) 23(24):4849–63. doi: 10.1091/mbc.E12-01-0069
40. Sakurai C, Itakura M, Kinoshita D, Arai S, Hashimoto H, Wada I, et al. Phosphorylation of SNAP-23 at Ser95 causes a structural alteration and negatively regulates Fc receptor-mediated phagosome formation and maturation in macrophages. Mol Biol Cell (2018) 29(13):1753–62. doi: 10.1091/mbc.E17-08-0523
41. Fairn GD, Grinstein S. How nascent phagosomes mature to become phagolysosomes. Trends Immunol (2012) 33(8):397–405. doi: 10.1016/j.it.2012.03.003
42. Pfeffer SR, Dirac-Svejstrup AB, Soldati T. Rab GDP dissociation inhibitor: putting rab GTPases in the right place. J Biol Chem (1995) 270(29):17057–9. doi: 10.1074/jbc.270.29.17057
43. Sivars U, Aivazian D, Pfeffer SR. Yip3 catalyses the dissociation of endosomal Rab-GDI complexes. Nature (2003) 425(6960):856–9. doi: 10.1038/nature02057
44. Rybin V, Ullrich O, Rubino M, Alexandrov K, Simon I, Seabra MC, et al. GTPase activity of Rab5 acts as a timer for endocytic membrane fusion. Nature (1996) 383(6597):266–9. doi: 10.1038/383266a0
45. Blumer J, Rey J, Dehmelt L, Mazel T, Wu YW, Bastiaens P, et al. RabGEFs are a major determinant for specific Rab membrane targeting. J Cell Biol (2013) 200(3):287–300. doi: 10.1083/jcb.201209113
46. Rivera-Molina FE, Novick PJ. A Rab GAP cascade defines the boundary between two Rab GTPases on the secretory pathway. Proc Natl Acad Sci U S A (2009) 106(34):14408–13. doi: 10.1073/pnas.0906536106
47. Harrison RE, Bucci C, Vieira OV, Schroer TA, Grinstein S. Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol Cell Biol (2003) 23(18):6494–506. doi: 10.1128/mcb.23.18.6494-6506.2003
48. Wu MY, Liu L, Wang EJ, Xiao HT, Cai CZ, Wang J, et al. PI3KC3 complex subunit NRBF2 is required for apoptotic cell clearance to restrict intestinal inflammation. Autophagy (2020), 19:1–16. doi: 10.1080/15548627.2020.1741332
49. Mottola G. The complexity of Rab5 to Rab7 transition guarantees specificity of pathogen subversion mechanisms. Front Cell Infect Microbiol (2014) 4:180. doi: 10.3389/fcimb.2014.00180
50. Mukherjee K, Khatua B, Mandal C. Sialic Acid-Siglec-E Interactions During Pseudomonas aeruginosa Infection of Macrophages Interferes With Phagosome Maturation by Altering Intracellular Calcium Concentrations. Front Immunol (2020) 11:332. doi: 10.3389/fimmu.2020.00332
51. Nieto C, Almendinger J, Gysi S, Gomez-Orte E, Kaech A, Hengartner MO, et al. ccz-1 mediates the digestion of apoptotic corpses in C. elegans. J Cell Sci (2010) 123(Pt 12):2001–7. doi: 10.1242/jcs.062331
52. Kinchen JM, Ravichandran KS. Identification of two evolutionarily conserved genes regulating processing of engulfed apoptotic cells. Nature (2010) 464(7289):778–82. doi: 10.1038/nature08853
53. Poteryaev D, Datta S, Ackema K, Zerial M, Spang A. Identification of the switch in early-to-late endosome transition. Cell (2010) 141(3):497–508. doi: 10.1016/j.cell.2010.03.011
54. Lou J, Li X, Huang W, Liang J, Zheng M, Xu T, et al. SNX10 promotes phagosome maturation in macrophages and protects mice against Listeria monocytogenes infection. Oncotarget (2017) 8(33):53935–47. doi: 10.18632/oncotarget.19644
55. Wurmser AE, Sato TK, Emr SD. New component of the vacuolar class C-Vps complex couples nucleotide exchange on the Ypt7 GTPase to SNARE-dependent docking and fusion. J Cell Biol (2000) 151(3):551–62. doi: 10.1083/jcb.151.3.551
56. Barry AO, Boucherit N, Mottola G, Vadovic P, Trouplin V, Soubeyran P, et al. Impaired stimulation of p38alpha-MAPK/Vps41-HOPS by LPS from pathogenic Coxiella burnetii prevents trafficking to microbicidal phagolysosomes. Cell Host Microbe (2012) 12(6):751–63. doi: 10.1016/j.chom.2012.10.015
57. Lorincz P, Toth S, Benko P, Lakatos Z, Boda A, Glatz G, et al. Rab2 promotes autophagic and endocytic lysosomal degradation. J Cell Biol (2017) 216(7):1937–47. doi: 10.1083/jcb.201611027
58. Gillingham AK, Sinka R, Torres IL, Lilley KS, Munro S. Toward a comprehensive map of the effectors of rab GTPases. Dev Cell (2014) 31(3):358–73. doi: 10.1016/j.devcel.2014.10.007
59. Mao F, Mu H, Wong NK, Liu K, Song J, Qiu J, et al. Hemocyte phagosomal proteome is dynamically shaped by cytoskeleton remodeling and interorganellar communication with endoplasmic reticulum during phagocytosis in a marine invertebrate, Crassostrea gigas. Sci Rep (2020) 10(1):6577. doi: 10.1038/s41598-020-63676-3
60. Okai B, Lyall N, Gow NA, Bain JM, Erwig LP. Rab14 regulates maturation of macrophage phagosomes containing the fungal pathogen Candida albicans and outcome of the host-pathogen interaction. Infect Immun (2015) 83(4):1523–35. doi: 10.1128/IAI.02917-14
61. Vieira OV, Bucci C, Harrison RE, Trimble WS, Lanzetti L, Gruenberg J, et al. Modulation of Rab5 and Rab7 recruitment to phagosomes by phosphatidylinositol 3-kinase. Mol Cell Biol (2003) 23(7):2501–14. doi: 10.1128/mcb.23.7.2501-2514.2003
62. Wickner W, Rizo J. A cascade of multiple proteins and lipids catalyzes membrane fusion. Mol Biol Cell (2017) 28(6):707–11. doi: 10.1091/mbc.E16-07-0517
63. Wu M, Wang T, Loh E, Hong W, Song H. Structural basis for recruitment of RILP by small GTPase Rab7. EMBO J (2005) 24(8):1491–501. doi: 10.1038/sj.emboj.7600643
64. Sun J, Wang X, Lau A, Liao TY, Bucci C, Hmama Z. Mycobacterial nucleoside diphosphate kinase blocks phagosome maturation in murine RAW 264.7 macrophages. PloS One (2010) 5(1):e8769. doi: 10.1371/journal.pone.0008769
65. Sindhwani A, Arya SB, Kaur H, Jagga D, Tuli A, Sharma M. Salmonella exploits the host endolysosomal tethering factor HOPS complex to promote its intravacuolar replication. PloS Pathog (2017) 13(10):e1006700. doi: 10.1371/journal.ppat.1006700
66. McEwan DG, Richter B, Claudi B, Wigge C, Wild P, Farhan H, et al. PLEKHM1 regulates Salmonella-containing vacuole biogenesis and infection. Cell Host Microbe (2015) 17(1):58–71. doi: 10.1016/j.chom.2014.11.011
67. van der Kant R, Fish A, Janssen L, Janssen H, Krom S, Ho N, et al. Late endosomal transport and tethering are coupled processes controlled by RILP and the cholesterol sensor ORP1L. J Cell Sci (2013) 126(Pt 15):3462–74. doi: 10.1242/jcs.129270
68. Lin X, Yang T, Wang S, Wang Z, Yun Y, Sun L, et al. RILP interacts with HOPS complex via VPS41 subunit to regulate endocytic trafficking. Sci Rep (2014) 4:7282. doi: 10.1038/srep07282
69. Jiang P, Nishimura T, Sakamaki Y, Itakura E, Hatta T, Natsume T, et al. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol Biol Cell (2014) 25(8):1327–37. doi: 10.1091/mbc.E13-08-0447
70. Khatter D, Raina VB, Dwivedi D, Sindhwani A, Bahl S, Sharma M. The small GTPase Arl8b regulates assembly of the mammalian HOPS complex on lysosomes. J Cell Sci (2015) 128(9):1746–61. doi: 10.1242/jcs.162651
71. van der Kant R, Jonker CT, Wijdeven RH, Bakker J, Janssen L, Klumperman J, et al. Characterization of the Mammalian CORVET and HOPS Complexes and Their Modular Restructuring for Endosome Specificity. J Biol Chem (2015) 290(51):30280–90. doi: 10.1074/jbc.M115.688440
72. Garg S, Sharma M, Ung C, Tuli A, Barral DC, Hava DL, et al. Lysosomal trafficking, antigen presentation, and microbial killing are controlled by the Arf-like GTPase Arl8b. Immunity (2011) 35(2):182–93. doi: 10.1016/j.immuni.2011.06.009
73. Jeschke A, Haas A. Sequential actions of phosphatidylinositol phosphates regulate phagosome-lysosome fusion. Mol Biol Cell (2018) 29(4):452–65. doi: 10.1091/mbc.E17-07-0464
74. Seals DF, Eitzen G, Margolis N, Wickner WT, Price A. A Ypt/Rab effector complex containing the Sec1 homolog Vps33p is required for homotypic vacuole fusion. Proc Natl Acad Sci U S A (2000) 97(17):9402–7. doi: 10.1073/pnas.97.17.9402
75. Subramanian S, Woolford CA, Jones EW. The Sec1/Munc18 protein, Vps33p, functions at the endosome and the vacuole of Saccharomyces cerevisiae. Mol Biol Cell (2004) 15(6):2593–605. doi: 10.1091/mbc.e03-10-0767
76. Brocker C, Kuhlee A, Gatsogiannis C, Balderhaar HJ, Honscher C, Engelbrecht-Vandre S, et al. Molecular architecture of the multisubunit homotypic fusion and vacuole protein sorting (HOPS) tethering complex. Proc Natl Acad Sci U S A (2012) 109(6):1991–6. doi: 10.1073/pnas.1117797109
77. Bach H, Papavinasasundaram KG, Wong D, Hmama Z, Av-Gay Y. Mycobacterium tuberculosis virulence is mediated by PtpA dephosphorylation of human vacuolar protein sorting 33B. Cell Host Microbe (2008) 3(5):316–22. doi: 10.1016/j.chom.2008.03.008
78. Akbar MA, Tracy C, Kahr WH, Kramer H. The full-of-bacteria gene is required for phagosome maturation during immune defense in Drosophila. J Cell Biol (2011) 192(3):383–90. doi: 10.1083/jcb.201008119
79. Johansson M, Rocha N, Zwart W, Jordens I, Janssen L, Kuijl C, et al. Activation of endosomal dynein motors by stepwise assembly of Rab7-RILP-p150Glued, ORP1L, and the receptor betalll spectrin. J Cell Biol (2007) 176(4):459–71. doi: 10.1083/jcb.200606077
80. Stein MP, Feng Y, Cooper KL, Welford AM, Wandinger-Ness A. Human VPS34 and p150 are Rab7 interacting partners. Traffic (2003) 4(11):754–71. doi: 10.1034/j.1600-0854.2003.00133.x
81. Sun Q, Westphal W, Wong KN, Tan I, Zhong Q. Rubicon controls endosome maturation as a Rab7 effector. Proc Natl Acad Sci U S A (2010) 107(45):19338–43. doi: 10.1073/pnas.1010554107
82. Fasshauer D, Sutton RB, Brunger AT, Jahn R. Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc Natl Acad Sci U S A (1998) 95(26):15781–6. doi: 10.1073/pnas.95.26.15781
83. Mayorga LS, Bertini F, Stahl PD. Fusion of newly formed phagosomes with endosomes in intact cells and in a cell-free system. J Biol Chem (1991) 266(10):6511–7.
84. Funato K, Beron W, Yang CZ, Mukhopadhyay A, Stahl PD. Reconstitution of phagosome-lysosome fusion in streptolysin O-permeabilized cells. J Biol Chem (1997) 272(26):16147–51. doi: 10.1074/jbc.272.26.16147
85. Dill BD, Gierlinski M, Hartlova A, Arandilla AG, Guo M, Clarke RG, et al. Quantitative proteome analysis of temporally resolved phagosomes following uptake via key phagocytic receptors. Mol Cell Proteomics (2015) 14(5):1334–49. doi: 10.1074/mcp.M114.044594
86. Campbell-Valois FX, Trost M, Chemali M, Dill BD, Laplante A, Duclos S, et al. Quantitative proteomics reveals that only a subset of the endoplasmic reticulum contributes to the phagosome. Mol Cell Proteomics (2012) 11(7):M111 016378. doi: 10.1074/mcp.M111.016378
87. Duclos S, Clavarino G, Rousserie G, Goyette G, Boulais J, Camossetto V, et al. The endosomal proteome of macrophage and dendritic cells. Proteomics (2011) 11(5):854–64. doi: 10.1002/pmic.201000577
88. Goyette G, Boulais J, Carruthers NJ, Landry CR, Jutras I, Duclos S, et al. Proteomic characterization of phagosomal membrane microdomains during phagolysosome biogenesis and evolution. Mol Cell Proteomics (2012) 11(11):1365–77. doi: 10.1074/mcp.M112.021048
89. Collins RF, Schreiber AD, Grinstein S, Trimble WS. Syntaxins 13 and 7 function at distinct steps during phagocytosis. J Immunol (2002) 169(6):3250–6. doi: 10.4049/jimmunol.169.6.3250
90. Buschow SI, Lasonder E, Szklarczyk R, Oud MM, de Vries IJ, Figdor CG. Unraveling the human dendritic cell phagosome proteome by organellar enrichment ranking. J Proteomics (2012) 75(5):1547–62. doi: 10.1016/j.jprot.2011.11.024
91. Uriarte SM, Rane MJ, Luerman GC, Barati MT, Ward RA, Nauseef WM, et al. Granule exocytosis contributes to priming and activation of the human neutrophil respiratory burst. J Immunol (2011) 187(1):391–400. doi: 10.4049/jimmunol.1003112
92. Rybicka JM, Balce DR, Khan MF, Krohn RM, Yates RM. NADPH oxidase activity controls phagosomal proteolysis in macrophages through modulation of the lumenal redox environment of phagosomes. Proc Natl Acad Sci U S A (2010) 107(23):10496–501. doi: 10.1073/pnas.0914867107
93. Mantegazza AR, Savina A, Vermeulen M, Perez L, Geffner J, Hermine O, et al. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood (2008) 112(12):4712–22. doi: 10.1182/blood-2008-01-134791
94. Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell (2006) 126(1):205–18. doi: 10.1016/j.cell.2006.05.035
95. Arango Duque G, Fukuda M, Descoteaux A. Synaptotagmin XI regulates phagocytosis and cytokine secretion in macrophages. J Immunol (2013) 190(4):1737–45. doi: 10.4049/jimmunol.1202500
96. Casbon AJ, Allen LA, Dunn KW, Dinauer MC. Macrophage NADPH oxidase flavocytochrome B localizes to the plasma membrane and Rab11-positive recycling endosomes. J Immunol (2009) 182(4):2325–39. doi: 10.4049/jimmunol.0803476
97. Jancic C, Savina A, Wasmeier C, Tolmachova T, El-Benna J, Dang PM, et al. Rab27a regulates phagosomal pH and NADPH oxidase recruitment to dendritic cell phagosomes. Nat Cell Biol (2007) 9(4):367–78. doi: 10.1038/ncb1552
98. Dingjan I, Paardekooper LM, Verboogen DRJ, von Mollard GF, Ter Beest M, van den Bogaart G. VAMP8-mediated NOX2 recruitment to endosomes is necessary for antigen release. Eur J Cell Biol (2017) 96(7):705–14. doi: 10.1016/j.ejcb.2017.06.007
99. Matheoud D, Moradin N, Bellemare-Pelletier A, Shio MT, Hong WJ, Olivier M, et al. Leishmania evades host immunity by inhibiting antigen cross-presentation through direct cleavage of the SNARE VAMP8. Cell Host Microbe (2013) 14(1):15–25. doi: 10.1016/j.chom.2013.06.003
100. Yates RM, Hermetter A, Taylor GA, Russell DG. Macrophage activation downregulates the degradative capacity of the phagosome. Traffic (2007) 8(3):241–50. doi: 10.1111/j.1600-0854.2006.00528.x
101. Westman J, Grinstein S, Maxson ME. Revisiting the role of calcium in phagosome formation and maturation. J Leukoc Biol (2019) 106(4):837–51. doi: 10.1002/JLB.MR1118-444R
102. Balla T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev (2013) 93(3):1019–137. doi: 10.1152/physrev.00028.2012
103. Rusten TE, Stenmark H. Analyzing phosphoinositides and their interacting proteins. Nat Methods (2006) 3(4):251–8. doi: 10.1038/nmeth867
104. Vieira OV, Botelho RJ, Rameh L, Brachmann SM, Matsuo T, Davidson HW, et al. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J Cell Biol (2001) 155(1):19–25. doi: 10.1083/jcb.200107069
105. Samie M, Wang X, Zhang X, Goschka A, Li X, Cheng X, et al. A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev Cell (2013) 26(5):511–24. doi: 10.1016/j.devcel.2013.08.003
106. Levin R, Hammond GR, Balla T, De Camilli P, Fairn GD, Grinstein S. Multiphasic dynamics of phosphatidylinositol 4-phosphate during phagocytosis. Mol Biol Cell (2017) 28(1):128–40. doi: 10.1091/mbc.E16-06-0451
107. Kim GH, Dayam RM, Prashar A, Terebiznik M, Botelho RJ. PIKfyve inhibition interferes with phagosome and endosome maturation in macrophages. Traffic (2014) 15(10):1143–63. doi: 10.1111/tra.12199
108. Buckley CM, Heath VL, Gueho A, Bosmani C, Knobloch P, Sikakana P, et al. PIKfyve/Fab1 is required for efficient V-ATPase and hydrolase delivery to phagosomes, phagosomal killing, and restriction of Legionella infection. PloS Pathog (2019) 15(2):e1007551. doi: 10.1371/journal.ppat.1007551
109. Ho CY, Choy CH, Wattson CA, Johnson DE, Botelho RJ. The Fab1/PIKfyve phosphoinositide phosphate kinase is not necessary to maintain the pH of lysosomes and of the yeast vacuole. J Biol Chem (2015) 290(15):9919–28. doi: 10.1074/jbc.M114.613984
110. Gary JD, Wurmser AE, Bonangelino CJ, Weisman LS, Emr SD. Fab1p is essential for PtdIns(3)P 5-kinase activity and the maintenance of vacuolar size and membrane homeostasis. J Cell Biol (1998) 143(1):65–79. doi: 10.1083/jcb.143.1.65
111. Li SC, Diakov TT, Xu T, Tarsio M, Zhu W, Couoh-Cardel S, et al. The signaling lipid PI(3,5)P(2) stabilizes V(1)-V(o) sector interactions and activates the V-ATPase. Mol Biol Cell (2014) 25(8):1251–62. doi: 10.1091/mbc.E13-10-0563
112. Kerr MC, Wang JT, Castro NA, Hamilton NA, Town L, Brown DL, et al. Inhibition of the PtdIns(5) kinase PIKfyve disrupts intracellular replication of Salmonella. EMBO J (2010) 29(8):1331–47. doi: 10.1038/emboj.2010.28
113. Dayam RM, Saric A, Shilliday RE, Botelho RJ. The Phosphoinositide-Gated Lysosomal Ca(2+) Channel, TRPML1, Is Required for Phagosome Maturation. Traffic (2015) 16(9):1010–26. doi: 10.1111/tra.12303
114. Jeschke A, Zehethofer N, Lindner B, Krupp J, Schwudke D, Haneburger I, et al. Phosphatidylinositol 4-phosphate and phosphatidylinositol 3-phosphate regulate phagolysosome biogenesis. Proc Natl Acad Sci U S A (2015) 112(15):4636–41. doi: 10.1073/pnas.1423456112
115. Ragaz C, Pietsch H, Urwyler S, Tiaden A, Weber SS, Hilbi H. The Legionella pneumophila phosphatidylinositol-4 phosphate-binding type IV substrate SidC recruits endoplasmic reticulum vesicles to a replication-permissive vacuole. Cell Microbiol (2008) 10(12):2416–33. doi: 10.1111/j.1462-5822.2008.01219.x
116. Gillooly DJ, Morrow IC, Lindsay M, Gould R, Bryant NJ, Gaullier JM, et al. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J (2000) 19(17):4577–88. doi: 10.1093/emboj/19.17.4577
117. Jeschke A, Haas A. Deciphering the roles of phosphoinositide lipids in phagolysosome biogenesis. Commun Integr Biol (2016) 9(3):e1174798. doi: 10.1080/19420889.2016.1174798
118. Dai S, Zhang Y, Weimbs T, Yaffe MB, Zhou D. Bacteria-generated PtdIns(3)P recruits VAMP8 to facilitate phagocytosis. Traffic (2007) 8(10):1365–74. doi: 10.1111/j.1600-0854.2007.00613.x
119. Defacque H, Bos E, Garvalov B, Barret C, Roy C, Mangeat P, et al. Phosphoinositides regulate membrane-dependent actin assembly by latex bead phagosomes. Mol Biol Cell (2002) 13(4):1190–202. doi: 10.1091/mbc.01-06-0314
120. Marion S, Hoffmann E, Holzer D, Le Clainche C, Martin M, Sachse M, et al. Ezrin promotes actin assembly at the phagosome membrane and regulates phago-lysosomal fusion. Traffic (2011) 12(4):421–37. doi: 10.1111/j.1600-0854.2011.01158.x
121. Thi EP, Lambertz U, Reiner NE. Class IA phosphatidylinositol 3-kinase p110alpha regulates phagosome maturation. PloS One (2012) 7(8):e43668. doi: 10.1371/journal.pone.0043668
122. Hinchliffe KA. Cellular signalling: stressing the importance of PIP3. Curr Biol (2001) 11(9):R371–2. doi: 10.1016/s0960-9822(01)00197-x
123. Marshall JG, Booth JW, Stambolic V, Mak T, Balla T, Schreiber AD, et al. Restricted accumulation of phosphatidylinositol 3-kinase products in a plasmalemmal subdomain during Fc gamma receptor-mediated phagocytosis. J Cell Biol (2001) 153(7):1369–80. doi: 10.1083/jcb.153.7.1369
124. Bohdanowicz M, Cosio G, Backer JM, Grinstein S. Class I and class III phosphoinositide 3-kinases are required for actin polymerization that propels phagosomes. J Cell Biol (2010) 191(5):999–1012. doi: 10.1083/jcb.201004005
125. Liebl D, Griffiths G. Transient assembly of F-actin by phagosomes delays phagosome fusion with lysosomes in cargo-overloaded macrophages. J Cell Sci (2009) 122(Pt 16):2935–45. doi: 10.1242/jcs.048355
126. Schlam D, Bagshaw RD, Freeman SA, Collins RF, Pawson T, Fairn GD, et al. Phosphoinositide 3-kinase enables phagocytosis of large particles by terminating actin assembly through Rac/Cdc42 GTPase-activating proteins. Nat Commun (2015) 6:8623. doi: 10.1038/ncomms9623
127. Dewitt S, Tian W, Hallett MB. Localised PtdIns(3,4,5)P3 or PtdIns(3,4)P2 at the phagocytic cup is required for both phagosome closure and Ca2+ signalling in HL60 neutrophils. J Cell Sci (2006) 119(Pt 3):443–51. doi: 10.1242/jcs.02756
128. Kjeken R, Egeberg M, Habermann A, Kuehnel M, Peyron P, Floetenmeyer M, et al. Fusion between phagosomes, early and late endosomes: a role for actin in fusion between late, but not early endocytic organelles. Mol Biol Cell (2004) 15(1):345–58. doi: 10.1091/mbc.e03-05-0334
129. Gopaldass N, Patel D, Kratzke R, Dieckmann R, Hausherr S, Hagedorn M, et al. Dynamin A, Myosin IB and Abp1 couple phagosome maturation to F-actin binding. Traffic (2012) 13(1):120–30. doi: 10.1111/j.1600-0854.2011.01296.x
130. Jahraus A, Egeberg M, Hinner B, Habermann A, Sackman E, Pralle A, et al. ATP-dependent membrane assembly of F-actin facilitates membrane fusion. Mol Biol Cell (2001) 12(1):155–70. doi: 10.1091/mbc.12.1.155
131. Al-Haddad A, Shonn MA, Redlich B, Blocker A, Burkhardt JK, Yu H, et al. Myosin Va bound to phagosomes binds to F-actin and delays microtubule-dependent motility. Mol Biol Cell (2001) 12(9):2742–55. doi: 10.1091/mbc.12.9.2742
132. Lerm M, Brodin VP, Ruishalme I, Stendahl O, Sarndahl E. Inactivation of Cdc42 is necessary for depolymerization of phagosomal F-actin and subsequent phagosomal maturation. J Immunol (2007) 178(11):7357–65. doi: 10.4049/jimmunol.178.11.7357
133. Kolonko M, Geffken AC, Blumer T, Hagens K, Schaible UE, Hagedorn M. WASH-driven actin polymerization is required for efficient mycobacterial phagosome maturation arrest. Cell Microbiol (2014) 16(2):232–46. doi: 10.1111/cmi.12217
134. Franco IS, Shohdy N, Shuman HA. The Legionella pneumophila effector VipA is an actin nucleator that alters host cell organelle trafficking. PloS Pathog (2012) 8(2):e1002546. doi: 10.1371/journal.ppat.1002546
135. Lodge R, Descoteaux A. Leishmania donovani promastigotes induce periphagosomal F-actin accumulation through retention of the GTPase Cdc42. Cell Microbiol (2005) 7(11):1647–58. doi: 10.1111/j.1462-5822.2005.00582.x
136. Ghosh A, Tousif S, Bhattacharya D, Samuchiwal SK, Bhalla K, Tharad M, et al. Expression of the ARPC4 subunit of human Arp2/3 severely affects mycobacterium tuberculosis growth and suppresses immunogenic response in murine macrophages. PloS One (2013) 8(7):e69949. doi: 10.1371/journal.pone.0069949
137. Poirier MB, Fiorino C, Rajasekar TK, Harrison RE. F-actin flashes on phagosomes mechanically deform contents for efficient digestion in macrophages. J Cell Sci (2020) 133(12). doi: 10.1242/jcs.239384
138. Defacque H, Egeberg M, Habermann A, Diakonova M, Roy C, Mangeat P, et al. Involvement of ezrin/moesin in de novo actin assembly on phagosomal membranes. EMBO J (2000) 19(2):199–212. doi: 10.1093/emboj/19.2.199
139. Ho HY, Rohatgi R, Lebensohn AM, Le M, Li J, Gygi SP, et al. Toca-1 mediates Cdc42-dependent actin nucleation by activating the N-WASP-WIP complex. Cell (2004) 118(2):203–16. doi: 10.1016/j.cell.2004.06.027
140. Park H, Cox D. Cdc42 regulates Fc gamma receptor-mediated phagocytosis through the activation and phosphorylation of Wiskott-Aldrich syndrome protein (WASP) and neural-WASP. Mol Biol Cell (2009) 20(21):4500–8. doi: 10.1091/mbc.E09-03-0230
141. Akhter A, Caution K, Abu Khweek A, Tazi M, Abdulrahman BA, Abdelaziz DH, et al. Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity (2012) 37(1):35–47. doi: 10.1016/j.immuni.2012.05.001
142. Dermine JF, Duclos S, Garin J, St-Louis F, Rea S, Parton RG, et al. Flotillin-1-enriched lipid raft domains accumulate on maturing phagosomes. J Biol Chem (2001) 276(21):18507–12. doi: 10.1074/jbc.M101113200
143. Rozelle AL, Machesky LM, Yamamoto M, Driessens MH, Insall RH, Roth MG, et al. Phosphatidylinositol 4,5-bisphosphate induces actin-based movement of raft-enriched vesicles through WASP-Arp2/3. Curr Biol (2000) 10(6):311–20. doi: 10.1016/s0960-9822(00)00384-5
144. Liu W, Zhou Y, Peng T, Zhou P, Ding X, Li Z, et al. N(epsilon)-fatty acylation of multiple membrane-associated proteins by Shigella IcsB effector to modulate host function. Nat Microbiol (2018) 3(9):996–1009. doi: 10.1038/s41564-018-0215-6
145. Kuhn S, Bergqvist J, Gil M, Valenzuela C, Barrio L, Lebreton S, et al. Actin Assembly around the Shigella-Containing Vacuole Promotes Successful Infection. Cell Rep (2020) 31(6):107638. doi: 10.1016/j.celrep.2020.107638
146. Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol (2007) 8(11):917–29. doi: 10.1038/nrm2272
147. Fratti RA, Chua J, Vergne I, Deretic V. Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc Natl Acad Sci U S A (2003) 100(9):5437–42. doi: 10.1073/pnas.0737613100
148. Maxson ME, Grinstein S. The vacuolar-type H(+)-ATPase at a glance - more than a proton pump. J Cell Sci (2014) 127(Pt 23):4987–93. doi: 10.1242/jcs.158550
149. Kinchen JM, Ravichandran KS. Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol (2008) 9(10):781–95. doi: 10.1038/nrm2515
150. Peters C, Bayer MJ, Buhler S, Andersen JS, Mann M, Mayer A. Trans-complex formation by proteolipid channels in the terminal phase of membrane fusion. Nature (2001) 409(6820):581–8. doi: 10.1038/35054500
151. Liegeois S, Benedetto A, Garnier JM, Schwab Y, Labouesse M. The V0-ATPase mediates apical secretion of exosomes containing Hedgehog-related proteins in Caenorhabditis elegans. J Cell Biol (2006) 173(6):949–61. doi: 10.1083/jcb.200511072
152. Di Giovanni J, Boudkkazi S, Mochida S, Bialowas A, Samari N, Leveque C, et al. V-ATPase membrane sector associates with synaptobrevin to modulate neurotransmitter release. Neuron (2010) 67(2):268–79. doi: 10.1016/j.neuron.2010.06.024
153. Hiesinger PR, Fayyazuddin A, Mehta SQ, Rosenmund T, Schulze KL, Zhai RG, et al. The v-ATPase V0 subunit a1 is required for a late step in synaptic vesicle exocytosis in Drosophila. Cell (2005) 121(4):607–20. doi: 10.1016/j.cell.2005.03.012
154. Peri F, Nusslein-Volhard C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell (2008) 133(5):916–27. doi: 10.1016/j.cell.2008.04.037
155. Xia Y, Liu N, Xie X, Bi G, Ba H, Li L, et al. The macrophage-specific V-ATPase subunit ATP6V0D2 restricts inflammasome activation and bacterial infection by facilitating autophagosome-lysosome fusion. Autophagy (2019) 15(6):960–75. doi: 10.1080/15548627.2019.1569916
156. Wong D, Bach H, Sun J, Hmama Z, Av-Gay Y. Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification. Proc Natl Acad Sci U S A (2011) 108(48):19371–6. doi: 10.1073/pnas.1109201108
157. Sun-Wada GH, Tabata H, Kawamura N, Aoyama M, Wada Y. Direct recruitment of H+-ATPase from lysosomes for phagosomal acidification. J Cell Sci (2009) 122(Pt 14):2504–13. doi: 10.1242/jcs.050443
158. Kissing S, Hermsen C, Repnik U, Nesset CK, von Bargen K, Griffiths G, et al. Vacuolar ATPase in phagosome-lysosome fusion. J Biol Chem (2015) 290(22):14166–80. doi: 10.1074/jbc.M114.628891
159. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature (2011) 469(7330):323–35. doi: 10.1038/nature09782
160. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol (2013) 13(10):722–37. doi: 10.1038/nri3532
161. Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature (2006) 441(7095):885–9. doi: 10.1038/nature04724
162. Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature (2006) 441(7095):880–4. doi: 10.1038/nature04723
163. Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med (2007) 13(5):619–24. doi: 10.1038/nm1574
164. Matsuzawa T, Kim BH, Shenoy AR, Kamitani S, Miyake M, Macmicking JD. IFN-gamma elicits macrophage autophagy via the p38 MAPK signaling pathway. J Immunol (2012) 189(2):813–8. doi: 10.4049/jimmunol.1102041
165. Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J (2008) 27(7):1110–21. doi: 10.1038/emboj.2008.31
166. Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci U S A (2011) 108(12):4788–93. doi: 10.1073/pnas.1100844108
167. Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe (2009) 5(6):527–49. doi: 10.1016/j.chom.2009.05.016
168. Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol (2018) 20(3):233–42. doi: 10.1038/s41556-018-0037-z
169. Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature (2007) 450(7173):1253–7. doi: 10.1038/nature06421
170. Tam JM, Mansour MK, Khan NS, Seward M, Puranam S, Tanne A, et al. Dectin-1-dependent LC3 recruitment to phagosomes enhances fungicidal activity in macrophages. J Infect Dis (2014) 210(11):1844–54. doi: 10.1093/infdis/jiu290
171. Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA, et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci U S A (2009) 106(15):6226–31. doi: 10.1073/pnas.0811045106
172. Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, et al. The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe (2015) 17(6):811–9. doi: 10.1016/j.chom.2015.05.004
173. Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, et al. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature (2013) 501(7468):512–6. doi: 10.1038/nature12566
174. Franco LH, Nair VR, Scharn CR, Xavier RJ, Torrealba JR, Shiloh MU, et al. The Ubiquitin Ligase Smurf1 Functions in Selective Autophagy of Mycobacterium tuberculosis and Anti-tuberculous Host Defense. Cell Host Microbe (2017) 22(3):421–3. doi: 10.1016/j.chom.2017.08.005
175. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell (2004) 119(6):753–66. doi: 10.1016/j.cell.2004.11.038
176. Besteiro S. The role of host autophagy machinery in controlling Toxoplasma infection. Virulence (2019) 10(1):438–47. doi: 10.1080/21505594.2018.1518102
177. Andrade RM, Wessendarp M, Gubbels MJ, Striepen B, Subauste CS. CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J Clin Invest (2006) 116(9):2366–77. doi: 10.1172/JCI28796
178. Muniz-Feliciano L, Van Grol J, Portillo JA, Liew L, Liu B, Carlin CR, et al. Toxoplasma gondii-induced activation of EGFR prevents autophagy protein-mediated killing of the parasite. PloS Pathog (2013) 9(12):e1003809. doi: 10.1371/journal.ppat.1003809
179. Choi J, Park S, Biering SB, Selleck E, Liu CY, Zhang X, et al. The parasitophorous vacuole membrane of Toxoplasma gondii is targeted for disruption by ubiquitin-like conjugation systems of autophagy. Immunity (2014) 40(6):924–35. doi: 10.1016/j.immuni.2014.05.006
180. Sasai M, Sakaguchi N, Ma JS, Nakamura S, Kawabata T, Bando H, et al. Essential role for GABARAP autophagy proteins in interferon-inducible GTPase-mediated host defense. Nat Immunol (2017) 18(8):899–910. doi: 10.1038/ni.3767
181. Tiwari S, Choi HP, Matsuzawa T, Pypaert M, MacMicking JD. Targeting of the GTPase Irgm1 to the phagosomal membrane via PtdIns(3,4)P(2) and PtdIns(3,4,5)P(3) promotes immunity to mycobacteria. Nat Immunol (2009) 10(8):907–17. doi: 10.1038/ni.1759
182. MacMicking JD, Taylor GA, McKinney JD. Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science (2003) 302(5645):654–9. doi: 10.1126/science.1088063
183. Dong H, Jing W, Runpeng Z, Xuewei X, Min M, Ru C, et al. ESAT6 inhibits autophagy flux and promotes BCG proliferation through MTOR. Biochem Biophys Res Commun (2016) 477(2):195–201. doi: 10.1016/j.bbrc.2016.06.042
184. Hu D, Wu J, Wang W, Mu M, Zhao R, Xu X, et al. Autophagy regulation revealed by SapM-induced block of autophagosome-lysosome fusion via binding RAB7. Biochem Biophys Res Commun (2015) 461(2):401–7. doi: 10.1016/j.bbrc.2015.04.051
185. Zulauf KE, Sullivan JT, Braunstein M. The SecA2 pathway of Mycobacterium tuberculosis exports effectors that work in concert to arrest phagosome and autophagosome maturation. PloS Pathog (2018) 14(4):e1007011. doi: 10.1371/journal.ppat.1007011
186. Vergne I, Chua J, Lee HH, Lucas M, Belisle J, Deretic V. Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc Natl Acad Sci U S A (2005) 102(11):4033–8. doi: 10.1073/pnas.0409716102
187. Dortet L, Mostowy S, Samba-Louaka A, Gouin E, Nahori MA, Wiemer EA, et al. Recruitment of the major vault protein by InlK: a Listeria monocytogenes strategy to avoid autophagy. PloS Pathog (2011) 7(8):e1002168. doi: 10.1371/journal.ppat.1002168
188. Martinez J, Malireddi RK, Lu Q, Cunha LD, Pelletier S, Gingras S, et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol (2015) 17(7):893–906. doi: 10.1038/ncb3192
189. Wong SW, Sil P, Martinez J. Rubicon: LC3-associated phagocytosis and beyond. FEBS J (2018) 285(8):1379–88. doi: 10.1111/febs.14354
190. Gluschko A, Herb M, Wiegmann K, Krut O, Neiss WF, Utermohlen O, et al. The beta2 Integrin Mac-1 Induces Protective LC3-Associated Phagocytosis of Listeria monocytogenes. Cell Host Microbe (2018) 23(3):324–37 e5. doi: 10.1016/j.chom.2018.01.018
191. Hubber A, Kubori T, Coban C, Matsuzawa T, Ogawa M, Kawabata T, et al. Bacterial secretion system skews the fate of Legionella-containing vacuoles towards LC3-associated phagocytosis. Sci Rep (2017) 7:44795. doi: 10.1038/srep44795
192. Herb M, Gluschko A, Schramm M. LC3-associated phagocytosis - The highway to hell for phagocytosed microbes. Semin Cell Dev Biol (2020) 101:68–76. doi: 10.1016/j.semcdb.2019.04.016
193. Koster S, Upadhyay S, Chandra P, Papavinasasundaram K, Yang G, Hassan A, et al. Mycobacterium tuberculosis is protected from NADPH oxidase and LC3-associated phagocytosis by the LCP protein CpsA. Proc Natl Acad Sci U S A (2017) 114(41):E8711–E20. doi: 10.1073/pnas.1707792114
194. Matte C, Casgrain PA, Seguin O, Moradin N, Hong WJ, Descoteaux A. Leishmania major Promastigotes Evade LC3-Associated Phagocytosis through the Action of GP63. PloS Pathog (2016) 12(6):e1005690. doi: 10.1371/journal.ppat.1005690
195. Kyrmizi I, Ferreira H, Carvalho A, Figueroa JAL, Zarmpas P, Cunha C, et al. Calcium sequestration by fungal melanin inhibits calcium-calmodulin signalling to prevent LC3-associated phagocytosis. Nat Microbiol (2018) 3(7):791–803. doi: 10.1038/s41564-018-0167-x
196. Cemma M, Grinstein S, Brumell JH. Autophagy proteins are not universally required for phagosome maturation. Autophagy (2016) 12(9):1440–6. doi: 10.1080/15548627.2016.1191724
197. Westman J, Walpole GFW, Kasper L, Xue BY, Elshafee O, Hube B, et al. Lysosome Fusion Maintains Phagosome Integrity during Fungal Infection. Cell Host Microbe (2020) 28(6):798–812 e6. doi: 10.1016/j.chom.2020.09.004
198. Romao S, Gasser N, Becker AC, Guhl B, Bajagic M, Vanoaica D, et al. Autophagy proteins stabilize pathogen-containing phagosomes for prolonged MHC II antigen processing. J Cell Biol (2013) 203(5):757–66. doi: 10.1083/jcb.201308173
199. Khobrekar NV, Quintremil S, Dantas TJ, Vallee RB. The Dynein Adaptor RILP Controls Neuronal Autophagosome Biogenesis, Transport, and Clearance. Dev Cell (2020) 53(2):141–53 e4. doi: 10.1016/j.devcel.2020.03.011
200. McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D, et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell (2015) 57(1):39–54. doi: 10.1016/j.molcel.2014.11.006
201. Wang H, Sun HQ, Zhu X, Zhang L, Albanesi J, Levine B, et al. GABARAPs regulate PI4P-dependent autophagosome:lysosome fusion. Proc Natl Acad Sci U S A (2015) 112(22):7015–20. doi: 10.1073/pnas.1507263112
202. Lopez-Haber C, Levin-Konigsberg R, Zhu Y, Bi-Karchin J, Balla T, Grinstein S, et al. Phosphatidylinositol-4-kinase IIalpha licenses phagosomes for TLR4 signaling and MHC-II presentation in dendritic cells. Proc Natl Acad Sci U S A (2020). 117(45):28251–28262. doi: 10.1073/pnas.2001948117
203. Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol (2014) 15(2):81–94. doi: 10.1038/nrm3735
204. Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A (2011) 108(42):17396–401. doi: 10.1073/pnas.1113421108
205. Martinez J, Cunha LD, Park S, Yang M, Lu Q, Orchard R, et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature (2016) 533(7601):115–9. doi: 10.1038/nature17950
Keywords: phagosome-lysosome fusion, phagosome, phagocyte, lysosome, membrane fusion, microbial clearance, homeostasis, phagosome maturation
Citation: Nguyen JA and Yates RM (2021) Better Together: Current Insights Into Phagosome-Lysosome Fusion. Front. Immunol. 12:636078. doi: 10.3389/fimmu.2021.636078
Received: 30 November 2020; Accepted: 18 January 2021;
Published: 25 February 2021.
Edited by:
Gregory D. Fairn, St. Michael’s Hospital, CanadaReviewed by:
Johannes Westman, Hospital for Sick Children, CanadaAlbert Descoteaux, Université du Québec, Canada
Copyright © 2021 Nguyen and Yates. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robin M. Yates, cm15YXRlc0B1Y2FsZ2FyeS5jYQ==