 Barbara Rossi
Barbara Rossi Bruno Santos-Lima
Bruno Santos-Lima Eleonora Terrabuio
Eleonora Terrabuio Elena Zenaro
Elena Zenaro Gabriela Constantin1,2*
Gabriela Constantin1,2*- 1Section of General Pathology, Department of Medicine, University of Verona, Verona, Italy
- 2The Center for Biomedical Computing (CBMC), University of Verona, Verona, Italy
Neurodegenerative diseases are closely related to inflammatory and autoimmune events, suggesting that the dysregulation of the immune system is a key pathological factor. Both multiple sclerosis (MS) and Alzheimer's disease (AD) are characterized by infiltrating immune cells, activated microglia, astrocyte proliferation, and neuronal damage. Moreover, MS and AD share a common pro-inflammatory signature, characterized by peripheral leukocyte activation and transmigration to the central nervous system (CNS). MS and AD are both characterized by the accumulation of activated neutrophils in the blood, leading to progressive impairment of the blood–brain barrier. Having migrated to the CNS during the early phases of MS and AD, neutrophils promote local inflammation that contributes to pathogenesis and clinical progression. The role of circulating T cells in MS is well-established, whereas the contribution of adaptive immunity to AD pathogenesis and progression is a more recent discovery. Even so, blocking the transmigration of T cells to the CNS can benefit both MS and AD patients, suggesting that common adaptive immunity mechanisms play a detrimental role in each disease. There is also growing evidence that regulatory T cells are beneficial during the initial stages of MS and AD, supporting the link between the modulatory immune compartments and these neurodegenerative disorders. The number of resting regulatory T cells declines in both diseases, indicating a common pathogenic mechanism involving the dysregulation of these cells, although their precise role in the control of neuroinflammation remains unclear. The modulation of leukocyte functions can benefit MS patients, so more insight into the role of peripheral immune cells may reveal new targets for pharmacological intervention in other neuroinflammatory and neurodegenerative diseases, including AD.
Introduction
Multiple sclerosis (MS) and Alzheimer's disease (AD) are two of the most widely studied central nervous system (CNS) pathologies. MS is the most common inflammatory neurological disease in young adults, whereas AD is a neurodegenerative disorder that occurs more frequently in the elderly population and is the most common type of dementia. The number of MS and AD patients is growing continuously, highlighting the need to find new disease mechanisms and new therapeutic approaches (1, 2). MS and AD are both multifactorial diseases and the identification of their etiopathogenetic mechanisms is challenging. Genetic risk factors and environmental triggers are the principal risk factors for both MS and AD (3, 4).
From the neuropathological perspective, the early phases of MS, defined as relapsing-remitting MS (RRMS), are characterized by primary demyelination areas known as plaques, which are located in the white and gray matter. In contrast, the final phases (secondary progressive MS) are associated with axonopathy, neuronal death, and synaptic loss, correlating with the permanent motor disability classically shown by MS patients (5). AD neuropathology is characterized by two main hallmarks: amyloid β plaques and tau tangles. Both structures are formed from aggregated proteins, in one case due to the incorrect processing of amyloid precursor protein (APP), and in the other due to the hyperphosphorylation of tau, a microtubule-associated protein required to maintain neuronal architecture and function.
The involvement of immune and inflammatory reactions in the pathogenesis of MS has been understood for decades, but the same association was only recently identified in AD. The concept of CNS exclusion from surveillance and inflammatory responses mediated by peripheral immune cells was reconsidered more than 15 years ago describing how circulating immune cells enter into the brain for protective tissue immunosurveillance and has been recently reviewed following the discovery of the cerebral lymphatic system and its role in CNS physiology (6–8). Furthermore, strong evidence of peripheral immune cell trafficking into the CNS has been provided during the immune responses that occur during MS and its animal model, experimental autoimmune encephalomyelitis (EAE), confirming that the CNS is not an immune-privileged environment (9). More recently, immune cell trafficking has also been documented in AD and was shown to be detrimental in transgenic mice with AD-like disease (10, 11). Neuroinflammation in both MS and AD is also characterized by the activation of microglia and astrocytes, leading to the secretion of pro-inflammatory cytokines and chemokines that recruit more immune cells from the periphery to the CNS (12, 13). Interestingly, the specialized pro-resolving lipid mediators, which mediate inflammation resolution and reduce neutrophil and monocytes infiltration into the brain, are impaired in MS and AD patients and their levels correlate with disease severity (14–16). These defects in the resolution pathways further emphasize the common detrimental role of peripheral immune cells in the maintenance of neuroinflammatory process in MS and AD.
Inflammation during neurodegenerative disorders is not restricted to the CNS. Indeed, systemic inflammation has been confirmed in MS and AD, including the secretion of pro-inflammatory cytokines in peripheral domains such as the blood, cerebrospinal fluid (CSF), liver, and gut (12, 17–20). The involvement of peripheral inflammation mechanisms and immune cells in MS and AD provides strong evidence of immune dysregulation, but it is unclear whether this is a causal link in each disease or a secondary phenomenon triggered by brain injury.
Whereas, the role of humoral response has been reviewed elsewhere, here we discuss the common immune mechanisms in MS and AD and describe how neutrophils, monocytes and T cell subpopulations use similar mechanisms in MS and AD to migrate into the CNS and induce neuroinflammation and tissue damage (21). These insights suggest that interfering with shared cellular and molecular mechanisms may lead to common therapeutic approaches for MS and AD.
Neutrophils: The Emerging Players in MS and AD
Neutrophils are highly reactive leukocytes with a frontline role in the maintenance of tissue homeostasis during pathological conditions, including infections and tissue damage (22). Neutrophils are highly adaptable cells due to their remarkable plasticity, and can therefore adjust their phenotype and functions in response to various environmental stimuli, triggering acute inflammatory responses (23). However, when prolonged tissue stress and damage induce sterile inflammation, neutrophils play a more subtle detrimental role, leading to chronic tissue damage that can promote pathological conditions such as autoimmunity and neurodegenerative diseases if left uncontrolled (24). These heterogeneous cells have attracted significant interest given their ability to facilitate sterile and chronic inflammation (24, 25).
Infiltrating neutrophils have been detected in the brains of MS and AD patients (10, 26). Evidence for the early involvement of neutrophils in MS includes their correlation with hyperacute lesions and altered blood-brain barrier (BBB) permeability in humans, and their involvement in the preclinical phase of EAE and acute relapses in these animal models (26, 27). Studies in AD models also suggest that neutrophils may contribute to the initial disease stages, which are characterized by increased BBB permeability, neutrophil intravascular adhesion, and invasion of the CNS (10, 11). Indeed, in animal models of both MS and AD, neutrophils accumulate in the brain before clinical manifestation, representing a major source of inflammatory mediators during early disease stages (10, 28, 29). We and others have shown that blocking neutrophil recruitment at early disease stages reduces the disease burden and tissue damage in animal models of both MS and AD (10, 30, 31). However, neutrophils continue to accumulate in the CNS throughout the disease course, suggesting these cells also play a role in disease progression and chronicity (10, 28, 29).
In MS and AD patients, the neutrophil to lymphocyte ratio (NLR) is a classical blood marker of inflammation. In MS, the NLR increases during progression and relapses, whereas in AD it correlates with cognitive impairment (32–35). In both diseases, a large proportion of circulating neutrophils is primed, as shown by the induction of activation markers such as CD11b and CD177 (36–39). Interestingly, high levels of CD11b also coincide with relapses in MS patients and correlate with the severity of the cognitive deficit in AD, suggesting that circulating neutrophils with a primed phenotype may cross the cerebral vasculature to the CNS more readily (36, 37). A similar mechanism has been proposed for CD11a/CD18 (LFA-1 or αLβ2) in the recruitment of neutrophils in AD models (10). Furthermore, peripheral hyper-activated neutrophils secrete inflammatory mediators and intravascular neutrophil extracellular traps (NETs), thus contributing to BBB damage and disease development (10, 24, 40) (Figure 1, Table 1).
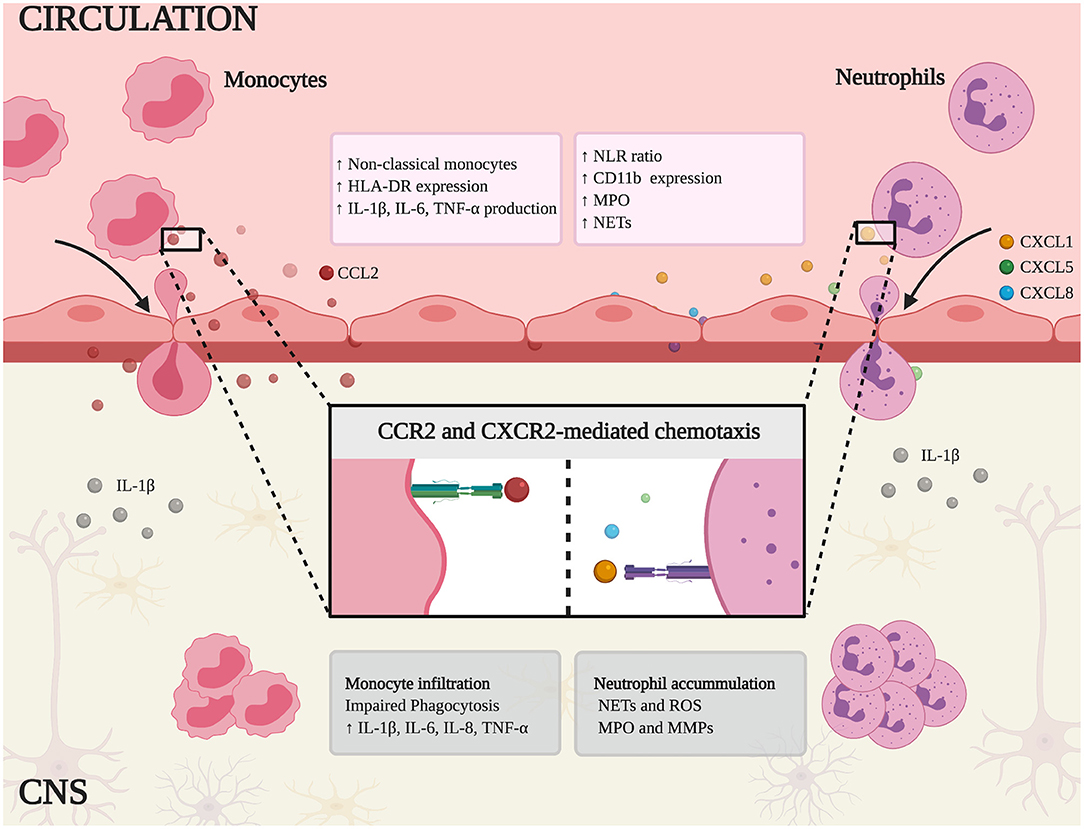
Figure 1. Schematic representation of innate immune mechanisms in MS and AD. Non-classical monocytes and neutrophils are expanded in peripheral blood and the higher neutrophil-lymphocyte ratio (NLR) correlates with the clinical symptoms of each disease. In addition, these cells express canonical activation markers (HLA-DR and CD11b) and release inflammatory mediators, such as pro-inflammatory cytokines (IL-1β, IL-6, TNF-α), myeloperoxidase (MPO), and neutrophil extracellular traps (NETs). Chemokine binding stimulates monocytes and neutrophils to infiltrate the CNS by chemotaxis (mediated by CCR2 and CXCR2, respectively). Within the CNS, monocytes show impaired phagocytosis and secrete the pro-inflammatory cytokines IL-1β, TNF-α, IL-6, and IL-8, thus fueling the inflammatory response. Similarly, infiltrating neutrophils contribute to neuroinflammation and tissue damage by releasing NETs, reactive oxygen species (ROS), MPO, and matrix metal proteinases (MMPs).

Table 1. Summary of common pathways in the innate immune system during the development of MS and AD.
During inflammation, the constitutive expression of CXCR2 (a chemokine receptor for the ELR+ chemokines CXCL1-3 and CXCL5-8) on mature neutrophils is strongly associated with neutrophil mobilization from the bone marrow to the bloodstream and their migration from the bloodstream to the site of injury (71). CXCL8, a CXCR2-dependent neutrophil chemoattractant, is more abundant in the plasma and CSF of MS and AD patients, and is linked to disease activity, suggesting that neutrophil migration is relevant in both diseases (44–48). Moreover, activated astrocytes produce CXCL1 (another CXCR2 ligand) at the lesion edges in EAE mice, and high levels of CXCL1, CXCL5, and CXCL8 are detected in the serum of MS patients, supporting a role for CXCR2 in the infiltration of neutrophils into the CNS in this disease (36, 44). CXCL1 is also produced by oligodendrocytes in EAE mice, attracting neutrophils into the CNS, exacerbating clinical impairment and enhancing BBB leakage (49). Interestingly, in the CNS of EAE mice, infiltrating T helper (Th) 17 cells stimulate the local release of CXCL1 and CXCL2, which leads to neutrophil recruitment (72). On the other hand, microglia in murine models of AD express CXCL1, and the levels of this chemokine in the CSF of AD patients correlate with cognitive impairment, suggesting that CXCL1 is also important in AD (50, 51) (Figure 1, Table 1). Restricting the infiltration of neutrophils using inhibitors of CXCR1 and CXCR2 has shown therapeutic efficacy in several experimental models of neuroinflammation including EAE, suggesting this may also be the case in animals with AD-like disease (73–75).
Amyloid β may also play a role in both AD and MS (76, 77). In AD brains, infiltrating neutrophils are closely associated with amyloid β deposits, and amyloid β peptides trigger the rapid integrin-dependent adhesion of neutrophils via G protein coupled receptors (10, 78). The non-random distribution of myeloperoxidase (MPO)-producing cells, presumably neutrophils, in the brain parenchyma of AD patients underlines the potential role of amyloid β as a chemoattractant that establishes a pro-inflammatory microenvironment to recruit circulating neutrophils (10). We speculate that the presence of amyloid β deposits in MS could also help to recruit neutrophils into the brain. The abundance of MPO and elastase in the blood and CNS of MS patients suggests that neutrophils may contribute to these pathological findings (56, 57). Moreover, neutrophil elastase is associated with the spread of MS lesions and clinical progression, whereas peripheral MPO activity is considered a predictor for executive function decline in AD patients (29, 46).
Neutrophil migration into the CNS during early or late phases of neuroinflammation plays a crucial role in BBB impairment. During migration, “outside-in” signaling generated following selectin and integrin engagement can induce ROS production by direct NADPH oxidase complex activation and release of other inflammation mediators such as cytokines (79–83). Moreover, it has been previously shown that intravascular neutrophil adhesion per se induces alterations in vascular permeability supporting a role for these cells in BBB breakdown (84–86). The inhibition of MPO and elastase in EAE mice reduced the number of infiltrating neutrophils, restored the integrity of the BBB, and attenuated the clinical symptoms (58, 59). MPO-producing cells were also identified in the brain parenchyma of AD patients and corresponding animal models (10). MPO and elastase are involved in the production of NETs, whose release in the CNS correlates with neuronal damage and BBB breakdown (57, 87). Indeed, the formation of NETs occurs in both MS and AD, strongly suggesting a role for neutrophils in the brain damage associated with both diseases (10, 40). Moreover, circulating neutrophils from MS and AD patients display a stronger oxidative burst, which may contribute to the formation of NETs, the activation of matrix metalloproteinases (MMPs), and therefore to BBB breakdown (40–42) (Figure 1, Table 1).
Taken together, these data suggest that activated circulating neutrophils mediate BBB damage and neurotoxicity in MS and AD by producing inflammatory mediators such as MPO and ROS, and by releasing NETs (88, 89). The blocking or inhibition of neutrophil activity could therefore achieve therapeutic benefits for both MS and AD patients (Figure 1).
The Role of Heterogeneous Monocytes in MS and AD
Circulating monocytes are heterogeneous and plastic innate immune cells that can promptly respond to changes in their environment. Traditionally, human monocytes are divided into three subsets: (i) classical (CD14+/CD16−), (ii) intermediate (CD14+/CD16+), and (iii) non-classical (CD14lo/CD16+). In mice, only two subclasses of monocytes have been identified: (i) classical (CCR2+/CX3CR1−/Ly6Chi), and (ii) non-classical (CCR2−/CX3CR1+/Ly6Clo) (90). In addition to the differential expression of surface markers, these subsets show transcriptional and functional differences: classical monocytes are the main subset recruited to sites of infection and injury (91), whereas non-classical monocytes circulate in the blood, patrolling the vasculature (92).
Systemic alterations in monocyte subsets have been reported in humans with neurodegenerative disorders. The dysregulation of monocyte subsets in patients with RRMS involves the expansion of non-classical and intermediate monocytes and the depletion of classical monocytes (61, 62). Similarly, AD patients with symptoms ranging from very mild to severe dementia accumulate non-classical and intermediate monocytes at the expense of classical monocytes (63) (Figure 1, Table 1). Although changes in circulating monocyte subsets have been confirmed across diverse CNS diseases, the meaning, causes and consequences of these alterations are still unclear. Whether different monocyte subsets correspond to developmental stages or whether each monocyte subset has a different developmental pathway remains to be determined. Given the plasticity of monocytes and their ability to respond rapidly to a wide variety of stimuli, the analysis of monocyte activation, and cytokine profiles should indicate their function and contribution to peripheral inflammation during neurodegeneration. Indeed, circulating monocytes in MS and AD patients shift toward a pro-inflammatory phenotype (63, 64) (Figure 1, Table 1). Particularly, in MS patients, isolated monocytes were shown to produce more IL-1β, TNF-α, IL-6, and IL-8 under basal conditions (61). Similarly, unstimulated monocytes from patients with dementia express higher levels of IL-6, IL-1β, and TNF-α mRNA (63) (Figure 1), suggesting a pro-inflammatory phenotype in AD. Interestingly, IL-8, and TNF-α mRNA levels also increase when human monocyte-like cell line THP-1 is incubated with plasma from AD patients or transgenic mice with AD-like disease, compared to plasma from healthy human controls or wild-type mice, respectively (65). This provides evidence that systemic inflammatory conditions affect the function of circulating monocytes.
Monocytes can migrate into tissues and differentiate into macrophages, making them important players in brain homeostasis. However, the correct identification of these cells in the CNS has proved challenging due to the similarities between microglia and monocyte-derived macrophages, especially during inflammation when the surface markers change. This has been addressed by advances in genome editing and cell tracing technology, leading to the identification of CCR2 and CX3CR1 as markers of murine classical monocytes and microglia, respectively (93). In this context, establishing how circulating monocytes are recruited to the inflamed brain can contribute significantly to the understanding of pathophysiology in MS and AD. Studies in murine models revealed the essential role of CCR2, a chemokine receptor involved in mononuclear trafficking at inflammation sites (94). In the EAE model of MS, classical monocytes were found to infiltrate the inflamed brain and the ablation of CCR2 blocked this process, suggesting a key role for this receptor in the recruitment of monocytes to the CNS during EAE (66). The same phenomenon has been observed in AD mice, where the loss of CCR2 reduces the number of monocytic phagocytes in the brain (67, 68) (Figure 1, Table 2). Together, these findings suggest that classical monocytes are the main subset that invades the CNS in neuroinflammatory conditions such as MS and AD and that CCR2 plays a fundamental role in the recruitment of these cells into the CNS. Interestingly, while CCR2 blockade in EAE has a beneficial effect, inhibition of this receptor in AD models increases Aβ deposition and worsens memory deficits, suggesting a decreased expression of CCR2 could play a potential role in the etiology of AD. Accordingly, the number of monocytes is lower in AD mouse models than controls, mostly due to the depletion of CCR2+ monocytes, suggesting these cells are severely impaired in AD (140). Also, in AD patients, CCR2 expression decreases in circulating monocytes whereas the levels of plasma CCL2 were increased, suggesting systemic immunologic dysfunction CCR2-CCL2 axis (141, 142) (Figure 1). Although mouse models of MS confirm the close involvement of monocytes in disease pathogenesis, clinical trials using CCR2 antagonists did not demonstrate efficacy (EU Clinical Trials: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2004-000073-64). Indeed, CCR2+ monocytes can be immunosuppressive, they replenish important macrophage populations, and they play pivotal roles during infection, potentially explaining the lack of positive results following a CCR2 therapeutic blockade in MS (68, 143, 144).

Table 2. Summary of common pathways in the adaptive immune system during the development of MS and AD.
Infiltrating monocytes and resident microglial cells can both react to inflammatory stimuli and mount an immune response in the CNS during neurodegenerative diseases (144). Indeed, monocytes not only infiltrate the CNS parenchyma but also colonize the meninges in EAE mice (145). Furthermore, monocyte infiltration begins at the onset of the disease and continues to increase until the disease peak, suggesting a role for these cells in disease induction and progression (145). Moreover, monocyte-derived cells infiltrating the CNS are major players in antigen presentation during EAE and recent studies identified several subtypes of infiltrating monocytes/myeloid cells in the CNS with different transcriptional landscapes during the acute and chronic stages of EAE (146). Similarly, single-cell studies revealed disease-specific transformations across several types of brain-associated phagocytes in murine models of AD, but the existence of common signatures between EAE/MS and AD is unclear (147). In AD mice, circulating monocytes have been shown to invade the brain and reduce amyloid β burden, suggesting a beneficial role for these cells in AD (148). Also, patrolling monocytes have been described to crawl onto the luminal walls of amyloid β-positive veins, suggesting their ability to target and clear amyloid β (149). The same applies to perivascular macrophages in another mouse model of AD, in which their depletion led to an increased accumulation of amyloid β deposits in blood vessels (150). Brain macrophages are not only involved in the clearance of CNS debris or amyloid β, but also play an important role in regulating iron levels. Extracellular accumulation of iron during neurodegeneration can be attributed to an array of processes including oligodendrocyte and myelin degeneration (151, 152). Indeed, increased iron deposits in white matter lesions have been shown in both MS and AD, and iron accumulation correlates with cognitive deficits (153, 154). Interestingly, the deposition of iron observed in MS was often co-localized with microglia/macrophages, which express the transferrin receptor, a main iron influx protein. By capturing iron and, therefore, preventing Fenton reactions and the creation of oxygen radicals, macrophages play an important regulatory function in the inflamed brain during MS (155, 156). In AD, however, there is limited information on iron uptake by macrophages, although recent evidence suggests that stimulation of microglia with Aβ increases the uptake of non-bound iron by these cells (157).
However, although monocytes infiltrate the brain and, to some degree, remove debris, iron and amyloid β, these cells in mice with AD-like disease are ineffective in clearing amyloid β in the diseased brain and their peripheral phenotype changes to a pro-inflammatory profile with limited phagocytic ability. Indeed, peripheral blood monocytes from AD patients cannot differentiate normally in vitro and have a lower capacity for the uptake of amyloid β uptake, eventually leading to apoptosis (70). Similarly, circulating monocytes in MS patients also adopt a pro-inflammatory profile with limited phagocytic ability, thus failing to promote remyelination and repair through the removal of myelin debris by phagocytosis (69) (Figure 1, Table 1). Collectively, these data suggest that chronic systemic inflammation in MS and AD leads to common pathological changes among the population of circulating monocytes. Understanding the role of these cells may provide insight into the disease mechanisms and lead to new therapeutic targets in neurodegenerative disorders.
The Role of T Cells in the Development of MS and AD
T lymphocytes are cells of the adaptive immune system that provide specific responses to eradicate pathogens or antigens that act as elicitors (158). Depending on their function, T lymphocytes can be subdivided into three main classes: (i) CD8+ cytotoxic T (Tc) lymphocytes responsible for the elimination of infected somatic cells, (ii) CD4+ T helper (Th) lymphocytes that assist and guide other immune cells, and (iii) regulatory T (Treg) lymphocytes associated with the attenuation and resolution of inflammation. The pathological dysregulation of the adaptive immune system promotes chronic and uncontrolled inflammatory reactions that may eventually lead to tissue damage. MS and AD are both characterized by a chronic neuroinflammatory pathology (159, 160). In MS, T cells are known to play an essential role in disease pathogenesis but the extent to which T cells contribute to the pathology of AD is less clear. In MS, T cells react against myelin autoantigens, migrate across a damaged BBB, accumulate in active lesions, and trigger damage to myelin and underlying axons, thus promoting all classical MS symptoms (161). Similarly, the post-mortem analysis of brains from AD patients revealed the accumulation of brain-infiltrating T cells, and recent experimental evidence from animal models of AD suggest a pathological role for these cells (100, 117). In support of this, the sequestration of T lymphocytes in lymphoid organs induced by fingolimod treatment decreases the number of circulating T cells and ameliorates disease in MS patients and animal models of AD (162–166).
CD4+ T Lymphocytes in MS and AD
CD4+ T cells infiltrate both the white and gray matter of MS patients, and similar observations have been reported in AD brains (95–100) (Table 2). Pathogenic CD4+ T cells in MS and EAE have been subtyped by cytokine profiling, revealing Th1 cells that produce IFN-γ and Th17 cells that produce IL-17. Both Th1 and Th17 cells are key components of the autoimmune inflammatory process during the development of MS, and they may fulfill analogous roles in AD. Following CNS invasion, Th1 and Th17 cells produce inflammatory mediators and cytokines to establish and/or maintain an inflammatory environment that promotes neuronal loss, a common feature of MS and AD that positively correlates with the disease course (167, 168). In particular, the infiltration of Th17 cells into the CNS of MS patients increases the concentration of IL-17 in the blood and CSF and the number of Th17 cells found in these compartments (101, 102, 169). Similarly, IL-17 also accumulates to higher levels in the serum of AD patients compared to healthy controls and has been proposed as part of a blood-based signature to distinguish individuals with AD from healthy subjects (104, 170). The population of circulating Th17 cells has also been shown to increase in MCI patients compared to both age-matched controls and AD patients, suggesting that Th17 cells may be involved in the early stages of AD (103) (Table 2). Intriguingly, EAE mice modified to abolish IL-17 production, as well as AD models treated with neutralizing antibodies against IL-17, show delayed clinical progression, confirming the harmful effects of IL-17 in EAE and suggesting that Th17 cells also contribute to the progression of AD (171).
CD4+ T cells appear to invade the CNS of AD and MS patients using common molecular pathways, emphasizing the common leukocyte recruitment mechanisms in these two diseases. For example, several studies in MS/EAE have shown that α4-integrins play a key role in the trafficking of Th cells (105, 106). EAE progression is delayed following the selective deletion of α4-integrin genes in T cells and MS progression is delayed by treatment with the α4-integrin-blocking humanized antibody natalizumab (172, 173). We and others recently demonstrated a similar molecular mechanism controlling the infiltration of Th cells in AD models (31, 107). We observed the stronger expression of α4-integrins on circulating CD4+ T cells in an AD mouse model compared to age-matched controls, along with an increase in the abundance of CD4+ cells in the brains of AD mice (31) (Figure 2, Table 2). Importantly, blocking α4-integrins inhibited the adhesion of circulating leukocytes in the brain microcirculation and reduced the neuropathological hallmarks of AD, highlighting the potential for a therapeutic approach that is similar in efficacy to the use of natalizumab in MS patients (172, 173).
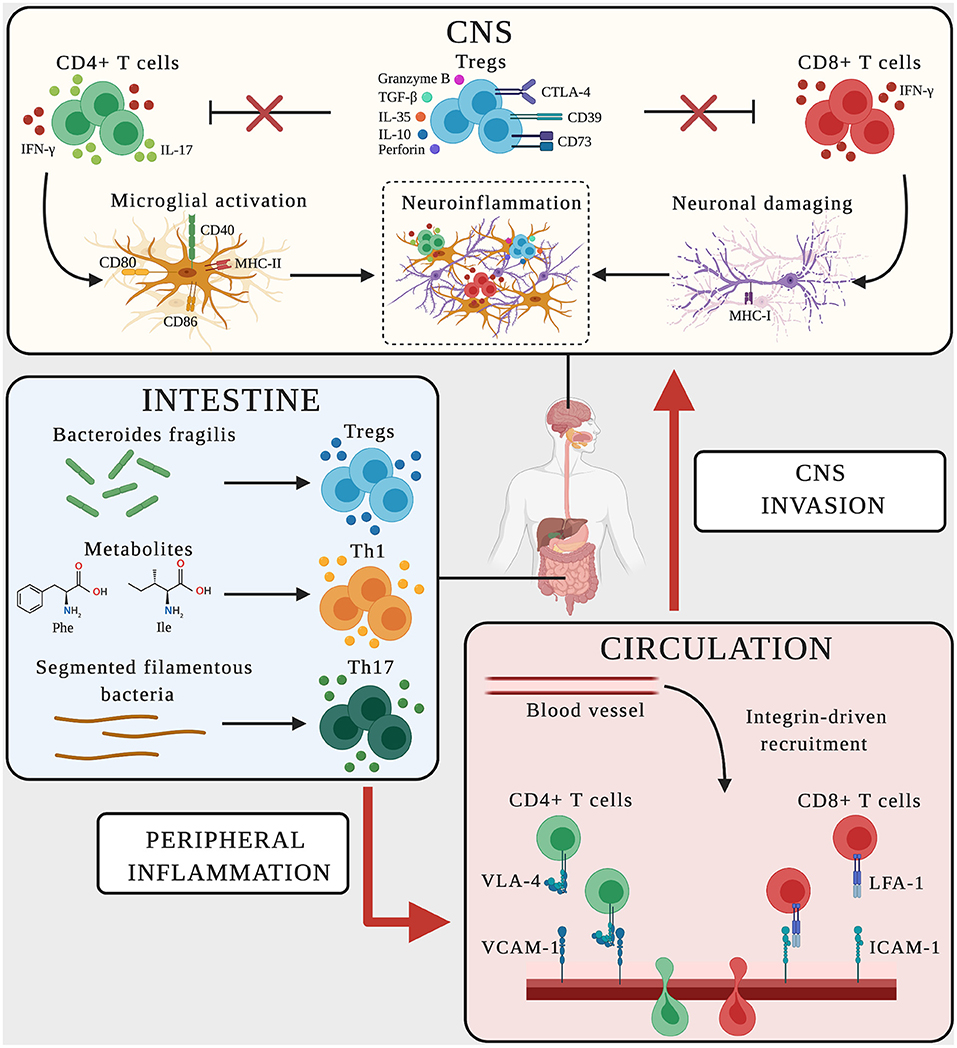
Figure 2. Schematic representation of adaptive immune mechanisms in MS and AD. In CNS, CD4+ T cells producing the pro-inflammatory mediators IFN-γ-and IL-17 promote microglial activation, upregulating the expression of CD40, MHC-II, CD80, and CD86 on the surface and favoring neuroinflammation. CD8+ T cells producing IFN-γ bind neurons expressing MHC-I, triggering neuronal damage and boosting neuroinflammation. Dysregulated Treg cells fail to suppress effector T cell functions (red crosses), thus sustaining the neuroinflammatory environment. In the intestine, the microbiome and its metabolites influence the polarization and activation of T cells. Bacteroides fragilis promotes the expansion of Treg cells, the amino acids phenylalanine and isoleucine induce the differentiation of Th1 cells, and segmented filamentous bacteria trigger Th17 cell polarization. Vascular endothelial cells express the LFA-1 and VLA-4 counter-ligands (ICAM-1 and VCAM-1) guiding the transmigration of peripherally activated T cells from the circulation to the CNS.
In EAE models, lymphocytes are presented with antigens in the periphery before CNS invasion. Indeed, T-cell priming begins in secondary lymphoid organs and leads to the activation and expansion of neuroantigen-reactive T cells that later infiltrate the CNS, where they re-encounter their cognate antigen (174). Within the CNS, microglial cells may promote the proliferation and activation of CNS-reactive T lymphocytes (174, 175). In both MS and AD, activated microglia express the main major histocompatibility complex class II molecule (MHC-II) as well as co-stimulatory molecules such as CD40, CD80, and CD86, which equip the microglia for antigen presentation to infiltrating T cells, creating a vicious cycle that promotes neuroinflammation and potentially antigen presentation (108–111) (Figure 2, Table 2). As a result, the pro-inflammatory environment that can activate CD4+ T cells is continuously boosted, and may promote neuronal damage in both MS and AD (Figure 2).
Although CD4+ T cells are considered pathogenic in several CNS disorders, they may also provide beneficial functions in AD, ranging from tissue protection to regeneration (176). In AD models, amyloid β-reactive T cells effectively target amyloid β plaques in the brain, enhancing phagocytosis by microglia and leading to neuronal repair (177). Furthermore, Th1 cells injected into the ventricles of AD mice were able to induce the differentiation of microglia (protective MHC-II+ subtype), boosting the capacity for amyloid β clearance (111). Despite these tantalizing results, a protective role for CD4+ T cells has yet to be confirmed in AD, and immunotherapeutic approaches based on amyloid β have not achieved efficacy in clinical trials (178).
Treg Cells and Their Failure to Control Inflammation in MS and AD
Treg cells fulfill an active regulatory role in peripheral tolerance mechanisms, preventing the onset of autoimmunity and limiting chronic inflammation. They downregulate the activities of various immune cells, including effector T cell functions and proliferation, by the secretion of immunosuppressive cytokines (including TGF-β, IL-10 and IL-35) and/or by direct cytotoxicity and the induction of apoptosis (through the release of granzyme B and perforin 1) (179–181). Treg cells also cause indirect immunosuppression via cytotoxic T lymphocyte antigen 4 (CTLA4), CD39, and CD73, and disrupt the metabolism of the effector T cells by modulating the maturation and/or function of the dendritic cells (DCs) required for their activation (182).
Dysfunctional Treg cells have been linked to neuroinflammatory conditions, and the analysis of peripheral blood demonstrates how Treg cells can contribute during neurodegenerative diseases. Treg cells have recently been shown to infiltrate the brain and suppress astrogliosis by producing amphiregulin in a model of ischemic stroke, but their role during AD and MS is unclear (183). Several studies have shown that the number of circulating Treg cells declines in both MS and AD patients compared to matched controls, suggesting that their dysregulation in the periphery reduces their immunosuppressive capacity and promotes uncontrolled inflammation (128–130). Indeed, Treg cells isolated from the peripheral blood of MS patients show an impaired ability to modulate CD4+ T cell proliferation and IFN-γ production (131). Compared to healthy controls, RRMS patients also produce more Th1-like Foxp3+ T cells that secrete IFN-γ and show a limited reduced immunosuppressive capacity (132). Similarly, the immunosuppressive functions of Treg cells in AD patients are compromised compared to both healthy controls and subjects with MCI (133, 134) (Table 2). These data are supported by animal models discussed below, where Treg cells appear to be important in both EAE and AD, especially during the early phases of both diseases, but at later stages they are depleted and/or dysfunctional and are therefore unable to control the inflammatory response.
The depletion of Treg cells by anti-CD25 antibodies in EAE mice increased the severity of the disease, boosting the production of IL-17 and T cell infiltration (135). In line with the protective role of Treg cells, the transplantation of neural stem cells in EAE mice induces remyelination by expanding the Treg cell population (136, 137). Similarly, the depletion of Treg cells in mice with AD-like disease is associated with the premature loss of cognitive functions and a worse outcome (138). Furthermore, low doses of IL-2 increased the number of Treg cells in the peripheral blood and lymphoid organs of these mice, restoring their cognitive ability (138). Similar results were reported by others, showing that Treg cell depletion for 4 months during the early stages of AD-like disease aggravated the cognitive deficits and increased the deposition of amyloid β plaques (139). In these studies, the adoptive transfer of purified Treg cells improved cognitive functions and reduced the amyloid β burden (139) (Table 2). Curiously, Treg cell depletion during the late disease stage in an aggressive model of amyloidosis also conferred a beneficial effect, presumably by boosting the immune response, suggesting that the effect of Treg cells on AD-like disease is stage-dependent (184). Taken together, these results suggest that Treg cells play a key role in controlling the development of neuroinflammation in both MS and AD (Figure 2). Therapeutic strategies aiming to exploit the immunosuppressive properties of Treg cells may therefore help to address both pathologies.
CD8+ T Lymphocytes in AD and MS
CD8+ T cells appear less heterogeneous than CD4+ T cells, but the functional classification of this population is not completely clear. Nevertheless, cytotoxic lymphocytes play a prominent role in the development of many viral and non-viral diseases by the direct killing of infected or otherwise modified cells. Following antigen recognition, cytotoxic T cells classically induce apoptosis in target cells via two alternative mechanisms: (i) FasL-CD95 (FasR) binding to activate caspase, and (ii) the release of granzyme B and perforin. CD8+ effector T cells may also cause cellular damage indirectly by secreting the pro-inflammatory cytokines TNF-α and IFN-γ (185). During immune responses, CD8+ T cells interact with CD4+ T cells to optimize the precision of CD8+ T cell effector functions after priming by enhancing their cytotoxicity and capacity for migration (186).
CD8+ T cells have received less attention than CD4+ T cells in MS and EAE, but it is now well-established that CD8+ T cells are more activated in the periphery and can infiltrate active lesions, contributing to the increasing severity of MS symptoms (115, 116, 187). Furthermore, clonally expanded cytotoxic T cells exacerbate brain inflammation in EAE initiated by CD4+ T cells, suggesting that CD8+ T cells may be primarily responsible for the observed cerebral alterations (187) (Table 2).
The role of CD8+ T cells in AD is less clear, although these cells were first detected in the brains of AD patients almost 20 years ago (100). The infiltration of CD8+ T cells into the brains of AD patients and corresponding mouse models also correlates with disease worsening, suggesting these cells may be involved in disease development (98, 100, 118). CD8+ T cells were shown to accumulate in an active state in the peripheral blood of MS patients and in AD patients with dementia (119–121) (Table 2). The comparison of blood samples from AD patients and healthy controls revealed the production of more pro-inflammatory cytokines by cytotoxic T cells in the AD patients and a greater proportion of activated HLA-DR+ CD8+ T cells (117, 125). Similarly, circulating CD8+ T cells from patients in the acute phase of RRMS showed increased adhesion to brain venules compared to control cells, further highlighting their activated phenotype (122). In line with this, the characterization of CD8+ T cells from the CSF and brain tissue of MS patients showed their activated/memory phenotype (123, 124) (Table 2). Notably, a higher frequency of CD8+ effector memory T cells was detected in the CSF of AD patients with dementia compared to controls, suggesting CD8+ T cells contribute to brain damage in both AD and MS with similar underlying mechanisms (117, 121).
The extravasation of cytotoxic T cells in the brain promotes the brain damage caused by CD8+ T cells and appears to be mediated by LFA-1 integrin (CD11a/CD18) in both MS and AD (Figure 2). LFA-1 is a marker of leukocyte activation that binds ICAM-1, which is overexpressed by endothelial cells in several neuroinflammatory conditions (10, 188). CD11a expression was shown to increase on clonally expanded CD8+ T cells in MS patients, promoting their transmigration into the brain (116). Without referring to specific T cell populations, previous studies have shown that the transfer of encephalitogenic CD11a−/− T cells to wild-type mice reduces the severity of EAE, suggesting that LFA-1 also facilitates the migration of CD8+ T cells in this disease (189). The presence of LFA-1+ T cells infiltrating the hippocampus of AD patients suggests a role for LFA-1 also in AD (100). This is consistent with the increase in Itgb2 (LFA-1) mRNA levels in the hippocampus of a mouse model of tauopathy (190). Finally, the strong upregulation of ICAM-1 was observed in cortical and hippocampal brain regions invaded by CD3+ T cells in mouse models of AD, suggesting that LFA-1 also promotes T cell migration into the AD brain (10, 191). Further studies are needed to confirm that LFA-1 is required for the trafficking of CD8+ T cells in AD, but given that LFA-1 is required for cytotoxic T cell activation, maturation, immuno-synapse stabilization and functioning, this integrin is likely to play a key role in driving CD8+ responses in AD (192).
Another important topic in the context of MS and AD is the antigenic specificity and clonal origin of CNS-infiltrating CD8+ T cells. The analysis of lymphocytes collected from the blood, CSF and brain lesions of many MS patients has shown that CD8+ T cells undergo clonal expansion, suggesting that they are activated by specific antigens (115, 123). Other studies in MS patients have identified a population of CD8+ T cells specific for myelin proteins, presenting an activated/memory phenotype due to in situ antigen presentation (96, 119, 120, 193). Tau protein may fulfill a similar role in AD, representing the potential link between cytotoxic T cells and disease development. Indeed, CD8+ T cells accumulate in the hippocampal regions of a mouse model of tau pathology, apparently via a tau-driven transmigration mechanism (190). A correlation between tau pathology and the infiltration of CD3+ T cells was revealed by the immunohistochemical analysis of post-mortem AD brains, further supporting the potential association between tau and CD8+ T cell-dependent pathology in AD (194).
Clonally expanded CD8+ TEMRA cells were recently identified in the CSF of AD patients, suggesting that antigen-experienced cytotoxic cells patrol the intrathecal space of AD patients. Interestingly, as already shown in MS patients, CD8+ T cells in the CSF were expanded predominantly against Epstein-Barr viral antigens, suggesting a new link between EBV, CD8+ T cells and AD (117, 127). CD8+ TEMRA cells accumulated not only in the CSF of AD patients, but also in the blood of RRMS and SPMS patients, suggesting these cells contribute to the progression of both AD and MS by promoting chronic inflammation (126) (Table 2). More detailed studies are required to determine precisely how this cell population fits into the complex pathogenesis of neurodegenerative disorders.
The pro-inflammatory nature of CD8+ TEMRA cells is strictly associated with IFN-γ secretion, which occurs mainly during active proliferation (195). One of the harmful functions of IFN-γ is the ability to favor neuronal killing by CD8+ T cells via the FasR-FasL pathway (185). Accordingly, previous studies have shown that the overexpression of MHC-I on neurons exposed to IFN-γ promotes neuronal damage by CD8+ T cells via TCR-MHC-I binding (196). Interestingly, MHC-I has been detected on neurons in both hippocampal and cortical brain regions, which are heavily damaged in AD (196). Similarly, CD8+ T cells in chronic and active MS plaques were found marginally in contact with oligodendrocytes, astrocytes and neurons expressing MHC-I at high levels (96, 96, 193). These results indicate that CD8+ T cells producing IFN-γ enhance the expression of MHC-I on neurons in MS and AD, and could therefore promote the brain alterations associated with the progression of both diseases (Figure 2). In conclusion, CD8+ T cells appear to drive the development of both MS and AD by sustaining chronic inflammation and directly causing CNS injury.
The Gut–Lymphocyte–Brain Axis in AD and MS
Several clinical and preclinical studies have shown that the course of MS and AD is influenced by the commensal gut microbiome, highlighting the interplay between the brain, gut microbes, intestinal barrier and immune system (197, 198). Indeed, fecal and mucosa-associated gastrointestinal tract microbes differ between AD patients and healthy controls, and recent studies comparing germ-free animals and those exposed to pathogenic bacteria, probiotics or antibiotics suggested a role for gut microbes in host cognitive functions and the development of AD-like neuropathological features (113, 199–202). Many studies have also focused on the role of gut microbes in MS and EAE (112, 203–208). These studies suggest that altering the gut microbiome with antibiotic cocktails or probiotics can attenuate the disease course by modulating regulatory immune responses (198). Although many of the microbes and metabolites in the gut–CNS axis have been identified, little is known about the underlying cellular and molecular immune mechanisms. In MS, the balance between pro-inflammatory myelin-reactive effector cells and anti-inflammatory immune elements controlling the formation of CNS lesions is continuously influenced by the gut. Indeed, both monocytes and gut-resident macrophages influence the gut-dependent activation of CD4+ T cells, which promotes Th1 polarization and IFN-γ secretion (209). Gut microbes and their metabolites also regulate T cell-mediated adaptive immune responses. For example, specific bacteria such as Akkermansia muciniphila and Acinetobacter calcoaceticus are associated with MS, inducing a pro-inflammatory T cell phenotype that perpetuates autoimmune responses, whereas segmented filamentous bacteria, symbiotic colonizers of the small intestine, induce Th17 differentiation and activation to promote the development of EAE (210, 211) (Figure 2). In contrast, CNS demyelination and inflammation during EAE is inhibited by gut flora rich in the commensal bacterium Bacteroides fragilis, which promotes the expansion of Treg cells expressing the ectonucleotidase CD39 and their migration into the CNS (212) (Figure 2). A recent innovative study in SPMS patients confirmed a role for the gut–brain axis in MS patients, showing the depletion of a subset of circulating memory CD4+ T cells expressing the gut-homing chemokine receptor CCR9 and the α4β7 adhesion molecule and a tendency to switch from a regulatory to a pro-inflammatory phenotype that produces more IFN-γ and IL-17 (213). Recently, gut dysbiosis in MCI patients was shown to increase phenylalanine and isoleucine levels, correlating with an increase in the number of circulating Th1 cells (114) (Figure 2). Interestingly, naive CD4+ T cells exposed to phenylalanine or isoleucine acquire an activated Th1 phenotype, and the oral treatment of mouse models of AD with the prebiotic oligosaccharide GV-971 (which suppresses gut dysbiosis) reduced the concentration of phenylalanine and isoleucine, resulting in the amelioration of neuroinflammation and cognitive impairment (114).
Collectively, these data show that gut dysbiosis contributes to peripheral immune cell dysregulation and triggers an enhanced inflammatory immune response. The gut–brain axis may therefore provide an appropriate target for immunomodulatory therapy in both diseases.
Conclusions
Neurodegenerative diseases are increasing in prevalence and socioeconomic impact. The identification of common cellular and molecular mechanisms involving the immune system may provide more insight into pathogenesis, leading to potential common therapeutic strategies. Several genome-wide association studies (GWAS) in both MS and AD patients have revealed associations between these diseases and gene expression in peripheral adaptive and innate immune cells, suggesting common immune mechanisms controlling neuroinflammation and neurodegeneration (214–216). Moreover, functional studies in vitro and in vivo have revealed shared detrimental molecular mechanisms in peripheral leukocytes, representing potential common therapeutic targets for the control of immune responses in MS and AD (217–219). One clear example of such a shared molecular mechanism is represented by α4 integrins, which can be targeted in both MS and AD. Indeed, the therapeutic effect of α4 integrins in EAE and MS has been corroborated by recent studies showing that targeting these adhesion molecules reduces neuroinflammation and the neuropathological hallmarks of AD (31). Moreover, reducing BBB breakdown that characterizes several neurodegenerative disorders, including MS and AD, could be considered as another common possible therapeutic strategy in reducing not only the influx of various neurotoxic agents, but also the recruitment of immune cells into the CNS (220, 221). By using animal models of both MS and AD, it was indeed demonstrated that targeting BBB pathways to preserve vascular integrity, ameliorates the course of brain pathology (222, 223). Therefore, a deeper understanding of the activation status of peripheral innate and adaptive immune cells in the blood, their trafficking mechanisms, CNS pathogenic signatures, and neurotoxic effects, may lead to the discovery of new common biomarkers of MS and AD that could facilitate the identification of common therapeutic strategies for both diseases.
Author Contributions
All authors contributed to the literature search and writing the review.
Funding
This work was supported in part by the European Research Council (ERC) advanced grant no. 695714 IMMUNOALZHEIMER and the ERC Proof of Concept grant no. 693606 IMPEDE (to GC); Alzheimer's Drug Discovery Foundation (ADDF), USA (to GC); Fondazione Italiana Sclerosi Multipla (FISM), Genoa, Italy [Cod. 2013/R/21 (to BR) and Cod. 2019/PR-single/044 (to EZ)]; University of Verona, Italy (FUR) (to GC); INVITE European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 754345 (to BS-L); Fondazione Umberto Veronesi post-doctoral fellowship 2019 and 2020 (to EZ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Rotstein DL, Chen H, Wilton AS, Kwong JC, Marrie RA, Gozdyra P, et al. Temporal trends in multiple sclerosis prevalence and incidence in a large population. Neurology. (2018) 90:e1435–41. doi: 10.1212/WNL.0000000000005331
2. Lopez OL, Kuller LH. Epidemiology of aging and associated cognitive disorders: prevalence and incidence of Alzheimer's disease and other dementias. Handb Clin Neurol. (2019) 167:139–48. doi: 10.1016/B978-0-12-804766-8.00009-1
3. Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol. (2017) 13:25–36. doi: 10.1038/nrneurol.2016.187
4. Stephenson J, Nutma E, van der Valk P, Amor S. Inflammation in CNS neurodegenerative diseases. Immunology. (2018) 154:204–19. doi: 10.1111/imm.12922
5. Rahmanzadeh R, Bruck W, Minagar A, Sahraian MA. Multiple sclerosis pathogenesis: missing pieces of an old puzzle. Rev Neurosci. (2018) 30:67–83. doi: 10.1515/revneuro-2018-0002
6. Yoles E, Hauben E, Palgi O, Agranov E, Gothilf A, Cohen A, et al. Protective autoimmunity is a physiological response to CNS trauma. J Neurosci. (2001) 21:3740–8. doi: 10.1523/JNEUROSCI.21-11-03740.2001
7. Schwartz M, Baruch K. The resolution of neuroinflammation in neurodegeneration: leukocyte recruitment via the choroid plexus. EMBO J. (2014) 33:7–22. doi: 10.1002/embj.201386609
8. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. (2015) 523:337–41. doi: 10.1038/nature14432
9. Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol. (2012) 12:623–35. doi: 10.1038/nri3265
10. Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S, et al. Neutrophils promote Alzheimer's disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med. (2015) 21:880–6. doi: 10.1038/nm.3913
11. Zenaro E, Piacentino G, Constantin G. The blood-brain barrier in Alzheimer's disease. Neurobiol Dis. (2017) 107:41–56. doi: 10.1016/j.nbd.2016.07.007
12. Prinz M, Priller J. The role of peripheral immune cells in the CNS in steady state and disease. Nat Neurosci. (2017) 20:136–44. doi: 10.1038/nn.4475
13. Vainchtein ID, Molofsky AV. Astrocytes and microglia: in sickness and in health. Trends Neurosci. (2020) 43:144–54. doi: 10.1016/j.tins.2020.01.003
14. Wang X, Zhu M, Hjorth E, Cortes-Toro V, Eyjolfsdottir H, Graff C, et al. Resolution of inflammation is altered in Alzheimer's disease. Alzheimers Dement. (2015) 11:40–50 e41–2. doi: 10.1016/j.jalz.2013.12.024
15. Kooij G, Troletti CD, Leuti A, Norris PC, Riley I, Albanese M, et al. Specialized pro-resolving lipid mediators are differentially altered in peripheral blood of patients with multiple sclerosis and attenuate monocyte and blood-brain barrier dysfunction. Haematologica. (2020) 105:2056–70. doi: 10.3324/haematol.2019.219519
16. Serhan CN, Gupta SK, Perretti M, Godson C, Brennan E, Li Y, et al. The atlas of inflammation resolution (AIR). Mol Aspects Med. (2020) 74:100894. doi: 10.1101/2020.01.27.921882
17. Mardiguian S, Ladds E, Turner R, Shepherd H, Campbell SJ, Anthony DC. The contribution of the acute phase response to the pathogenesis of relapse in chronic-relapsing experimental autoimmune encephalitis models of multiple sclerosis. J Neuroinflammation. (2017) 14:196. doi: 10.1186/s12974-017-0969-4
18. Giau VV, Wu SY, Jamerlan A, An SSA, Kim SY, Hulme J. Gut Microbiota and Their Neuroinflammatory Implications in Alzheimer's Disease. Nutrients. (2018) 10:1765. doi: 10.3390/nu10111765
19. Negi N, Das BK. CNS: Not an immunoprivilaged site anymore but a virtual secondary lymphoid organ. Int Rev Immunol. (2018) 37:57–68. doi: 10.1080/08830185.2017.1357719
20. Estrada LD, Ahumada P, Cabrera D, Arab JP. Liver dysfunction as a novel player in Alzheimer's progression: looking outside the brain. Front Aging Neurosci. (2019) 11:174. doi: 10.3389/fnagi.2019.00174
21. Sabatino JJ Jr, Probstel AK, Zamvil SS. B cells in autoimmune and neurodegenerative central nervous system diseases. Nat Rev Neurosci. (2019) 20:728–45. doi: 10.1038/s41583-019-0233-2
22. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. (2011) 11:519–31. doi: 10.1038/nri3024
23. Ng LG, Ostuni R, Hidalgo A. Heterogeneity of neutrophils. Nat Rev Immunol. (2019) 19:255–65. doi: 10.1038/s41577-019-0141-8
24. Rossi B, Constantin G, Zenaro E. The emerging role of neutrophils in neurodegeneration. Immunobiology. (2020) 225:151865. doi: 10.1016/j.imbio.2019.10.014
25. Silvestre-Roig C, Hidalgo A, Soehnlein O. Neutrophil heterogeneity: implications for homeostasis and pathogenesis. Blood. (2016) 127:2173–81. doi: 10.1182/blood-2016-01-688887
26. Aube B, Levesque SA, Pare A, Chamma E, Kebir H, Gorina R, et al. Neutrophils mediate blood-spinal cord barrier disruption in demyelinating neuroinflammatory diseases. J Immunol. (2014) 193:2438–54. doi: 10.4049/jimmunol.1400401
27. Kostic M, Dzopalic T, Zivanovic S, Zivkovic N, Cvetanovic A, Stojanovic I, et al. IL-17 and glutamate excitotoxicity in the pathogenesis of multiple sclerosis. Scand J Immunol. (2014) 79:181–6. doi: 10.1111/sji.12147
28. Wu F, Cao W, Yang Y, Liu A. Extensive infiltration of neutrophils in the acute phase of experimental autoimmune encephalomyelitis in C57BL/6 mice. Histochem Cell Biol. (2010) 133:313–22. doi: 10.1007/s00418-009-0673-2
29. Rumble JM, Huber AK, Krishnamoorthy G, Srinivasan A, Giles DA, Zhang X, et al. Neutrophil-related factors as biomarkers in EAE and MS. J Exp Med. (2015) 212:23–35. doi: 10.1084/jem.20141015
30. Miller NM, Wang J, Tan Y, Dittel BN. Anti-inflammatory mechanisms of IFN-gamma studied in experimental autoimmune encephalomyelitis reveal neutrophils as a potential target in multiple sclerosis. Front Neurosci. (2015) 9:287. doi: 10.3389/fnins.2015.00287
31. Pietronigro E, Zenaro E, Bianca VD, Dusi S, Terrabuio E, Iannoto G, et al. Blockade of alpha4 integrins reduces leukocyte-endothelial interactions in cerebral vessels and improves memory in a mouse model of Alzheimer's disease. Sci Rep. (2019) 9:12055. doi: 10.1038/s41598-019-48538-x
32. Kuyumcu ME, Yesil Y, Ozturk ZA, Kizilarslanoglu C, Etgul S, Halil M, et al. The evaluation of neutrophil-lymphocyte ratio in Alzheimer's disease. Dement Geriatr Cogn Disord. (2012) 34:69–74. doi: 10.1159/000341583
33. Demirci S, Demirci S, Kutluhan S, Koyuncuoglu HR, Yurekli VA. The clinical significance of the neutrophil-to-lymphocyte ratio in multiple sclerosis. Int J Neurosci. (2016) 126:700–6. doi: 10.3109/00207454.2015.1050492
34. Bisgaard AK, Pihl-Jensen G, Frederiksen JL. The neutrophil-to-lymphocyte ratio as disease actvity marker in multiple sclerosis and optic neuritis. Mult Scler Relat Disord. (2017) 18:213–7. doi: 10.1016/j.msard.2017.10.009
35. Dong X, Nao J, Shi J, Zheng D. Predictive value of routine peripheral blood biomarkers in Alzheimer's disease. Front Aging Neurosci. (2019) 11:332. doi: 10.3389/fnagi.2019.00332
36. Ziaber J, Pasnik J, Baj Z, Pokoca L, Chmielewski H, Tchorzewski H. The immunoregulatory abilities of polymorphonuclear neutrophils in the course of multiple sclerosis. Mediators Inflamm. (1998) 7:335–8. doi: 10.1080/09629359890857
37. Scali C, Prosperi C, Bracco L, Piccini C, Baronti R, Ginestroni A, et al. Neutrophils CD11b and fibroblasts PGE(2) are elevated in Alzheimer's disease. Neurobiol Aging. (2002) 23:523–30. doi: 10.1016/S0197-4580(01)00346-3
38. Cordiglieri C, Baggi F, Bernasconi P, Kapetis D, Faggiani E, Consonni A, et al. Identification of a gene expression signature in peripheral blood of multiple sclerosis patients treated with disease-modifying therapies. Clin Immunol. (2016) 173:133–46. doi: 10.1016/j.clim.2016.10.002
39. Le Page A, Lamoureux J, Bourgade K, Frost EH, Pawelec G, Witkowski JM, et al. Polymorphonuclear neutrophil functions are differentially altered in amnestic mild cognitive impairment and mild Alzheimer's disease patients. J Alzheimers Dis. (2017) 60:23–42. doi: 10.3233/JAD-170124
40. Naegele M, Tillack K, Reinhardt S, Schippling S, Martin R, Sospedra M. Neutrophils in multiple sclerosis are characterized by a primed phenotype. J Neuroimmunol. (2012) 242:60–71. doi: 10.1016/j.jneuroim.2011.11.009
41. Vitte J, Michel BF, Bongrand P, Gastaut JL. Oxidative stress level in circulating neutrophils is linked to neurodegenerative diseases. J Clin Immunol. (2004) 24:683–92. doi: 10.1007/s10875-004-6243-4
42. Dong Y, Lagarde J, Xicota L, Corne H, Chantran Y, Chaigneau T, et al. Neutrophil hyperactivation correlates with Alzheimer's disease progression. Ann Neurol. (2018) 83:387–405. doi: 10.1002/ana.25159
43. Ferretti G, Bacchetti T, Principi F, Di Ludovico F, Viti B, Angeleri VA, et al. Increased levels of lipid hydroperoxides in plasma of patients with multiple sclerosis: a relationship with paraoxonase activity. Mult Scler. (2005) 11:677–82. doi: 10.1191/1352458505ms1240oa
44. Lund BT, Ashikian N, Ta HQ, Chakryan Y, Manoukian K, Groshen S, et al. Increased CXCL8 (IL-8) expression in multiple sclerosis. J Neuroimmunol. (2004) 155:161–71. doi: 10.1016/j.jneuroim.2004.06.008
45. Bartosik-Psujek H, Stelmasiak Z. The levels of chemokines CXCL8, CCL2 and CCL5 in multiple sclerosis patients are linked to the activity of the disease. Eur J Neurol. (2005) 12:49–54. doi: 10.1111/j.1468-1331.2004.00951.x
46. Bawa KK, Krance SH, Herrmann N, Cogo-Moreira H, Ouk M, Yu D, et al. A peripheral neutrophil-related inflammatory factor predicts a decline in executive function in mild Alzheimer's disease. J Neuroinflammation. (2020) 17:84. doi: 10.1186/s12974-020-01750-3
47. Alsadany MA, Shehata HH, Mohamad MI, Mahfouz RG. Histone deacetylases enzyme, copper, and IL-8 levels in patients with Alzheimer's disease. Am J Alzheimers Dis Other Demen. (2013) 28:54–61. doi: 10.1177/1533317512467680
48. Correa JD, Starling D, Teixeira AL, Caramelli P, Silva TA. Chemokines in CSF of Alzheimer's disease patients. Arq Neuropsiquiatr. (2011) 69:455–9. doi: 10.1590/S0004-282X2011000400009
49. Tani M, Fuentes ME, Peterson JW, Trapp BD, Durham SK, Loy JK, et al. Neutrophil infiltration, glial reaction, and neurological disease in transgenic mice expressing the chemokine N51/KC in oligodendrocytes. J Clin Invest. (1996) 98:529–39. doi: 10.1172/JCI118821
50. Craig-Schapiro R, Kuhn M, Xiong C, Pickering EH, Liu J, Misko TP, et al. Multiplexed immunoassay panel identifies novel CSF biomarkers for Alzheimer's disease diagnosis and prognosis. PLoS ONE. (2011) 6:e18850. doi: 10.1371/journal.pone.0018850
51. Choi SH, Veeraraghavalu K, Lazarov O, Marler S, Ransohoff RM, Ramirez JM, et al. Non-cell-autonomous effects of presenilin 1 variants on enrichment-mediated hippocampal progenitor cell proliferation and differentiation. Neuron. (2008) 59:568–80. doi: 10.1016/j.neuron.2008.07.033
52. McGuinness MC, Powers JM, Bias WB, Schmeckpeper BJ, Segal AH, Gowda VC, et al. Human leukocyte antigens and cytokine expression in cerebral inflammatory demyelinative lesions of X-linked adrenoleukodystrophy and multiple sclerosis. J Neuroimmunol. (1997) 75:174–82. doi: 10.1016/S0165-5728(97)00020-9
53. Bauer J, Berkenbosch F, Van Dam AM, Dijkstra CD. Demonstration of interleukin-1 beta in Lewis rat brain during experimental allergic encephalomyelitis by immunocytochemistry at the light and ultrastructural level. J Neuroimmunol. (1993) 48:13–21. doi: 10.1016/0165-5728(93)90053-2
54. Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, et al. Brain interleukin 1 and S-100 immunoreactivity are elevated in down syndrome and Alzheimer disease. Proc Natl Acad Sci USA. (1989) 86:7611–5. doi: 10.1073/pnas.86.19.7611
55. Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, et al. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci. (2000) 20:5709–14. doi: 10.1523/JNEUROSCI.20-15-05709.2000
56. Minohara M, Matsuoka T, Li W, Osoegawa M, Ishizu T, Ohyagi Y, et al. Upregulation of myeloperoxidase in patients with opticospinal multiple sclerosis: positive correlation with disease severity. J Neuroimmunol. (2006) 178:156–60. doi: 10.1016/j.jneuroim.2006.05.026
57. Tillack K, Naegele M, Haueis C, Schippling S, Wandinger KP, Martin R, et al. Gender differences in circulating levels of neutrophil extracellular traps in serum of multiple sclerosis patients. J Neuroimmunol. (2013) 261:108–19. doi: 10.1016/j.jneuroim.2013.05.004
58. Zhang H, Ray A, Miller NM, Hartwig D, Pritchard KA, Dittel BN. Inhibition of myeloperoxidase at the peak of experimental autoimmune encephalomyelitis restores blood-brain barrier integrity and ameliorates disease severity. J Neurochem. (2016) 136:826–36. doi: 10.1111/jnc.13426
59. Yu G, Zheng S, Zhang H. Inhibition of myeloperoxidase by N-acetyl lysyltyrosylcysteine amide reduces experimental autoimmune encephalomyelitis-induced injury and promotes oligodendrocyte regeneration and neurogenesis in a murine model of progressive multiple sclerosis. Neuroreport. (2018) 29:208–13. doi: 10.1097/WNR.0000000000000948
60. Gellhaar S, Sunnemark D, Eriksson H, Olson L, Galter D. Myeloperoxidase-immunoreactive cells are significantly increased in brain areas affected by neurodegeneration in Parkinson's and Alzheimer's disease. Cell Tissue Res. (2017) 369:445–54. doi: 10.1007/s00441-017-2626-8
61. Waschbisch A, Schroder S, Schraudner D, Sammet L, Weksler B, Melms A, et al. Pivotal role for CD16+ monocytes in immune surveillance of the central nervous system. J Immunol. (2016) 196:1558–67. doi: 10.4049/jimmunol.1501960
62. Haschka D, Tymoszuk P, Bsteh G, Petzer V, Berek K, Theurl I, et al. Expansion of neutrophils and classical and nonclassical monocytes as a hallmark in relapsing-remitting multiple sclerosis. Front Immunol. (2020) 11:594. doi: 10.3389/fimmu.2020.00594
63. Thome AD, Faridar A, Beers DR, Thonhoff JR, Zhao W, Wen S, et al. Functional alterations of myeloid cells during the course of Alzheimer's disease. Mol Neurodegener. (2018) 13:61. doi: 10.1186/s13024-018-0293-1
64. Filion LG, Graziani-Bowering G, Matusevicius D, Freedman MS. Monocyte-derived cytokines in multiple sclerosis. Clin Exp Immunol. (2003) 131:324–34. doi: 10.1046/j.1365-2249.2003.02053.x
65. Wolfe H, Mela V, Minogue AM, Miller AM, McGuigan C, Williams L, et al. Monocytes exposed to plasma from patients with Alzheimer's disease undergo metabolic reprogramming. Neurosci Res. (2019) 148:54–60. doi: 10.1016/j.neures.2019.01.001
66. Mildner A, Mack M, Schmidt H, Bruck W, Djukic M, Zabel MD, et al. CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain. (2009) 132(Pt 9):2487–500. doi: 10.1093/brain/awp144
67. El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. (2007) 13:432–8. doi: 10.1038/nm1555
68. Naert G, Rivest S. CC chemokine receptor 2 deficiency aggravates cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer's disease. J Neurosci. (2011) 31:6208–20. doi: 10.1523/JNEUROSCI.0299-11.2011
69. Natrajan MS, Komori M, Kosa P, Johnson KR, Wu T, Franklin RJ, et al. Pioglitazone regulates myelin phagocytosis and multiple sclerosis monocytes. Ann Clin Transl Neurol. (2015) 2:1071–84. doi: 10.1002/acn3.260
70. Fiala M, Lin J, Ringman J, Kermani-Arab V, Tsao G, Patel A, et al. Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer's disease patients. J Alzheimers Dis. (2005) 7:221–32; discussion 255–62. doi: 10.3233/JAD-2005-7304
71. Wengner AM, Pitchford SC, Furze RC, Rankin SM. The coordinated action of G-CSF and ELR + CXC chemokines in neutrophil mobilization during acute inflammation. Blood. (2008) 111:42–9. doi: 10.1182/blood-2007-07-099648
72. Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. (2008) 205:1535–41. doi: 10.1084/jem.20080159
73. Kerstetter AE, Padovani-Claudio DA, Bai L, Miller RH. Inhibition of CXCR2 signaling promotes recovery in models of multiple sclerosis. Exp Neurol. (2009) 220:44–56. doi: 10.1016/j.expneurol.2009.07.010
74. Semple BD, Bye N, Ziebell JM, Morganti-Kossmann MC. Deficiency of the chemokine receptor CXCR2 attenuates neutrophil infiltration and cortical damage following closed head injury. Neurobiol Dis. (2010) 40:394–403. doi: 10.1016/j.nbd.2010.06.015
75. Hosking MP, Lane TE. ELR(+) chemokine signaling in host defense and disease in a viral model of central nervous system disease. Front Cell Neurosci. (2014) 8:165. doi: 10.3389/fncel.2014.00165
76. Chandra A. Role of amyloid from a multiple sclerosis perspective: a literature review. Neuroimmunomodulation. (2015) 22:343–6. doi: 10.1159/000375309
77. Stampanoni Bassi M, Garofalo S, Marfia GA, Gilio L, Simonelli I, Finardi A, et al. Amyloid-beta homeostasis bridges inflammation, synaptic plasticity deficits and cognitive dysfunction in multiple sclerosis. Front Mol Neurosci. (2017) 10:390. doi: 10.3389/fnmol.2017.00390
78. Baik SH, Cha MY, Hyun YM, Cho H, Hamza B, Kim DK, et al. Migration of neutrophils targeting amyloid plaques in Alzheimer's disease mouse model. Neurobiol Aging. (2014) 35:1286–92. doi: 10.1016/j.neurobiolaging.2014.01.003
79. Berton G, Laudanna C, Sorio C, Rossi F. Generation of signals activating neutrophil functions by leukocyte integrins: LFA-1 and gp150/95, but not CR3, are able to stimulate the respiratory burst of human neutrophils. J Cell Biol. (1992) 116:1007–17. doi: 10.1083/jcb.116.4.1007
80. Laudanna C, Constantin G, Baron P, Scarpini E, Scarlato G, Cabrini G, et al. Sulfatides trigger increase of cytosolic free calcium and enhanced expression of tumor necrosis factor-alpha and interleukin-8 mRNA in human neutrophils. Evidence for a role of L-selectin as a signaling molecule. J Biol Chem. (1994) 269:4021–6. doi: 10.1016/S0021-9258(17)41736-4
81. Waddell TK, Fialkow L, Chan CK, Kishimoto TK, Downey GP. Potentiation of the oxidative burst of human neutrophils. A signaling role for L-selectin. J Biol Chem. (1994) 269:18485–91. doi: 10.1016/S0021-9258(17)32335-9
82. Montresor A, Toffali L, Constantin G, Laudanna C. Chemokines and the signaling modules regulating integrin affinity. Front Immunol. (2012) 3:127. doi: 10.3389/fimmu.2012.00127
83. Fumagalli L, Campa CC, Germena G, Lowell CA, Hirsch E, Berton G. Class I phosphoinositide-3-kinases and SRC kinases play a nonredundant role in regulation of adhesion-independent and -dependent neutrophil reactive oxygen species generation. J Immunol. (2013) 190:3648–60. doi: 10.4049/jimmunol.1201951
84. Bolton SJ, Anthony DC, Perry VH. Loss of the tight junction proteins occludin and zonula occludens-1 from cerebral vascular endothelium during neutrophil-induced blood-brain barrier breakdown in vivo. Neuroscience. (1998) 86:1245–57. doi: 10.1016/S0306-4522(98)00058-X
85. Blamire AM, Anthony DC, Rajagopalan B, Sibson NR, Perry VH, Styles P. Interleukin-1beta -induced changes in blood-brain barrier permeability, apparent diffusion coefficient, and cerebral blood volume in the rat brain: a magnetic resonance study. J Neurosci. (2000) 20:8153–9. doi: 10.1523/JNEUROSCI.20-21-08153.2000
86. Gautam N, Herwald H, Hedqvist P, Lindbom L. Signaling via beta(2) integrins triggers neutrophil-dependent alteration in endothelial barrier function. J Exp Med. (2000) 191:1829–39. doi: 10.1084/jem.191.11.1829
87. Allen C, Thornton P, Denes A, McColl BW, Pierozynski A, Monestier M, et al. Neutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNA. J Immunol. (2012) 189:381–92. doi: 10.4049/jimmunol.1200409
88. Pietronigro EC, Della Bianca V, Zenaro E, Constantin G. NETosis in Alzheimer's disease. Front Immunol. (2017) 8:211. doi: 10.3389/fimmu.2017.00211
89. Manda-Handzlik A, Demkow U. The brain entangled: the contribution of neutrophil extracellular traps to the diseases of the central nervous system. Cells. (2019) 8:1477. doi: 10.3390/cells8121477
90. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. (2003) 19:71–82. doi: 10.1016/S1074-7613(03)00174-2
91. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. (2007) 204:3037–47. doi: 10.1084/jem.20070885
92. Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. (2007) 317:666–70. doi: 10.1126/science.1142883
93. Mizutani M, Pino PA, Saederup N, Charo IF, Ransohoff RM, Cardona AE. The fractalkine receptor but not CCR2 is present on microglia from embryonic development throughout adulthood. J Immunol. (2012) 188:29–36. doi: 10.4049/jimmunol.1100421
94. Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. (2005) 6:1182–90. doi: 10.1038/ni1275
95. Booss J, Esiri MM, Tourtellotte WW, Mason DY. Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. J Neurol Sci. (1983) 62:219–232. doi: 10.1016/0022-510X(83)90201-0
96. Traugott U, Reinherz EL, Raine CS. Multiple sclerosis: distribution of T cell subsets within active chronic lesions. Science. (1983) 219:308–10. doi: 10.1126/science.6217550
97. Hauser SL, Bhan AK, Gilles F, Kemp M, Kerr C, Weiner HL. Immunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions. Ann Neurol. (1986) 19:578–87. doi: 10.1002/ana.410190610
98. Rogers J, Luber-Narod J, Styren SD, Civin WH. Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer's disease. Neurobiol Aging. (1988) 9:339–49. doi: 10.1016/S0197-4580(88)80079-4
99. McGeer PL, Akiyama H, Itagaki S, McGeer EG. Immune system response in Alzheimer's disease. Can J Neurol Sci. (1989) 16:516–27. doi: 10.1017/S0317167100029863
100. Togo T, Akiyama H, Iseki E, Kondo H, Ikeda K, Kato M, et al. Occurrence of T cells in the brain of Alzheimer's disease and other neurological diseases. J Neuroimmunol. (2002) 124:83–92. doi: 10.1016/S0165-5728(01)00496-9
101. Durelli L, Conti L, Clerico M, Boselli D, Contessa G, Ripellino P, et al. T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-beta. Ann Neurol. (2009) 65:499–509. doi: 10.1002/ana.21652
102. Ashtari F, Madanian R, Shaygannejad V, Zarkesh SH, Ghadimi K. Serum levels of IL-6 and IL-17 in multiple sclerosis, neuromyelitis optica patients and healthy subjects. Int J Physiol Pathophysiol Pharmacol. (2019) 11:267–73.
103. Oberstein TJ, Taha L, Spitzer P, Hellstern J, Herrmann M, Kornhuber J, et al. Imbalance of circulating Th17 and regulatory T cells in Alzheimer's disease: a case control study. Front Immunol. (2018) 9:1213. doi: 10.3389/fimmu.2018.01213
104. Chen JM, Jiang GX, Li QW, Zhou ZM, Cheng Q. Increased serum levels of interleukin-18,−23 and−17 in Chinese patients with Alzheimer's disease. Dement Geriatr Cogn Disord. (2014) 38:321–9. doi: 10.1159/000360606
105. Vajkoczy P, Laschinger M, Engelhardt B. Alpha4-integrin-VCAM-1 binding mediates G protein-independent capture of encephalitogenic T cell blasts to CNS white matter microvessels. J Clin Invest. (2001) 108:557–65. doi: 10.1172/JCI12440
106. Perumal J, Fox RJ, Balabanov R, Balcer LJ, Galetta S, Makh S, et al. Outcomes of natalizumab treatment within 3 years of relapsing-remitting multiple sclerosis diagnosis: a prespecified 2-year interim analysis of STRIVE. BMC Neurol. (2019) 19:116. doi: 10.1186/s12883-019-1337-z
107. Manocha G, Ghatak A, Puig K, Combs C. Anti-alpha4beta1 integrin antibodies attenuated brain inflammatory changes in a mouse model of Alzheimer's disease. Curr Alzheimer Res. (2018) 15:1123–35. doi: 10.2174/1567205015666180801111033
108. Zrzavy T, Hametner S, Wimmer I, Butovsky O, Weiner HL, Lassmann H. Loss of 'homeostatic' microglia and patterns of their activation in active multiple sclerosis. Brain. (2017) 140:1900–13. doi: 10.1093/brain/awx113
109. Kawakami N, Nagerl UV, Odoardi F, Bonhoeffer T, Wekerle H, Flugel A. Live imaging of effector cell trafficking and autoantigen recognition within the unfolding autoimmune encephalomyelitis lesion. J Exp Med. (2005) 201:1805–14. doi: 10.1084/jem.20050011
110. Bossers K, Wirz KT, Meerhoff GF, Essing AH, van Dongen JW, Houba P, et al. Concerted changes in transcripts in the prefrontal cortex precede neuropathology in Alzheimer's disease. Brain. (2010) 133(Pt 12:3699–723. doi: 10.1093/brain/awq258
111. Mittal K, Eremenko E, Berner O, Elyahu Y, Strominger I, Apelblat D, et al. CD4 T cells induce a subset of MHCII-Expressing microglia that attenuates Alzheimer pathology. iScience. (2019) 16:298–311. doi: 10.1016/j.isci.2019.05.039
112. Ochoa-Reparaz J, Mielcarz DW, Ditrio LE, Burroughs AR, Foureau DM, Haque-Begum S, et al. Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. J Immunol. (2009) 183:6041–50. doi: 10.4049/jimmunol.0900747
113. Dodiya HB, Kuntz T, Shaik SM, Baufeld C, Leibowitz J, Zhang X, et al. Sex-specific effects of microbiome perturbations on cerebral Abeta amyloidosis and microglia phenotypes. J Exp Med. (2019) 216:1542–60. doi: 10.1084/jem.20182386
114. Wang X, Sun G, Feng T, Zhang J, Huang X, Wang T, et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer's disease progression. Cell Res. (2019) 29:787–803. doi: 10.1038/s41422-019-0216-x
115. Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. (2000) 192:393–404. doi: 10.1084/jem.192.3.393
116. Salou M, Garcia A, Michel L, Gainche-Salmon A, Loussouarn D, Nicol B, et al. Expanded CD8 T-cell sharing between periphery and CNS in multiple sclerosis. Ann Clin Transl Neurol. (2015) 2:609–22. doi: 10.1002/acn3.199
117. Gate D, Saligrama N, Leventhal O, Yang AC, Unger MS, Middeldorp J, et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer's disease. Nature. (2020) 577:399–404. doi: 10.1038/s41586-019-1895-7
118. Unger MS, Li E, Scharnagl L, Poupardin R, Altendorfer B, Mrowetz H, et al. CD8(+) T-cells infiltrate Alzheimer's disease brains and regulate neuronal- and synapse-related gene expression in APP-PS1 transgenic mice. Brain Behav Immun. (2020) 89:67–86. doi: 10.1016/j.bbi.2020.05.070
119. Zang YC, Li S, Rivera VM, Hong J, Robinson RR, Breitbach WT, et al. Increased CD8+ cytotoxic T cell responses to myelin basic protein in multiple sclerosis. J Immunol. (2004) 172:5120–7. doi: 10.4049/jimmunol.172.8.5120
120. Crawford MP, Yan SX, Ortega SB, Mehta RS, Hewitt RE, Price DA, et al. High prevalence of autoreactive, neuroantigen-specific CD8+ T cells in multiple sclerosis revealed by novel flow cytometric assay. Blood. (2004) 103:4222–31. doi: 10.1182/blood-2003-11-4025
121. Lueg G, Gross CC, Lohmann H, Johnen A, Kemmling A, Deppe M, et al. Clinical relevance of specific T-cell activation in the blood and cerebrospinal fluid of patients with mild Alzheimer's disease. Neurobiology of Aging. (2015) 36:81–9. doi: 10.1016/j.neurobiolaging.2014.08.008
122. Battistini L, Piccio L, Rossi B, Bach S, Galgani S, Gasperini C, et al. CD8+ T cells from patients with acute multiple sclerosis display selective increase of adhesiveness in brain venules: a critical role for P-selectin glycoprotein ligand-1. Blood. (2003) 101:4775–82. doi: 10.1182/blood-2002-10-3309
123. Jacobsen M, Cepok S, Quak E, Happel M, Gaber R, Ziegler A, et al. Oligoclonal expansion of memory CD8+ T cells in cerebrospinal fluid from multiple sclerosis patients. Brain. (2002) 125(Pt 3):538–50. doi: 10.1093/brain/awf059
124. Junker A, Ivanidze J, Malotka J, Eiglmeier I, Lassmann H, Wekerle H, et al. Multiple sclerosis: T-cell receptor expression in distinct brain regions. Brain. (2007) 130(Pt 11):2789–99. doi: 10.1093/brain/awm214
125. Pellicano M, Larbi A, Goldeck D, Colonna-Romano G, Buffa S, Bulati M, et al. Immune profiling of Alzheimer patients. J Neuroimmunol. (2012) 242:52–9. doi: 10.1016/j.jneuroim.2011.11.005
126. Nielsen BR, Ratzer R, Bornsen L, von Essen MR, Christensen JR, Sellebjerg F. Characterization of naive, memory and effector T cells in progressive multiple sclerosis. J Neuroimmunol. (2017) 310:17–25. doi: 10.1016/j.jneuroim.2017.06.001
127. Serafini B, Rosicarelli B, Veroni C, Mazzola GA, Aloisi F. Epstein-Barr Virus-Specific CD8 T cells selectively infiltrate the brain in multiple sclerosis and interact locally with virus-infected cells: clue for a virus-driven immunopathological mechanism. J Virol. (2019) 93:e00980–19. doi: 10.1128/JVI.00980-19
128. Bjerg L, Brosbol-Ravnborg A, Torring C, Dige A, Bundgaard B, Petersen T, et al. Altered frequency of T regulatory cells is associated with disability status in relapsing-remitting multiple sclerosis patients. J Neuroimmunol. (2012) 249:76–82. doi: 10.1016/j.jneuroim.2012.04.012
129. Kouchaki E, Salehi M, Reza Sharif M, Nikoueinejad H, Akbari H. Numerical status of CD4(+)CD25(+)FoxP3(+) and CD8(+)CD28(-) regulatory T cells in multiple sclerosis. Iran J Basic Med Sci. (2014) 17:250–5.
130. Ciccocioppo F, Lanuti P, Pierdomenico L, Simeone P, Bologna G, Ercolino E, et al. The characterization of regulatory T-cell profiles in Alzheimer's disease and multiple sclerosis. Sci Rep. (2019) 9:8788. doi: 10.1038/s41598-019-45433-3
131. Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. (2004) 199:971–9. doi: 10.1084/jem.20031579
132. Dominguez-Villar M, Baecher-Allan CM, Hafler DA. Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat Med. (2011) 17:673–5. doi: 10.1038/nm.2389
133. Rosenkranz D, Weyer S, Tolosa E, Gaenslen A, Berg D, Leyhe T, et al. Higher frequency of regulatory T cells in the elderly and increased suppressive activity in neurodegeneration. J Neuroimmunol. (2007) 188:117–27. doi: 10.1016/j.jneuroim.2007.05.011
134. Faridar A, Thome AD, Zhao W, Thonhoff JR, Beers DR, Pascual B, et al. Restoring regulatory T-cell dysfunction in Alzheimer's disease through ex vivo expansion. Brain Commun. (2020) 2:fcaa112. doi: 10.1093/braincomms/fcaa112
135. Akirav EM, Bergman CM, Hill M, Ruddle NH. Depletion of CD4(+)CD25(+) T cells exacerbates experimental autoimmune encephalomyelitis induced by mouse, but not rat, antigens. J Neurosci Res. (2009) 87:3511–9. doi: 10.1002/jnr.21981
136. McIntyre LL, Greilach SA, Othy S, Sears-Kraxberger I, Wi B, Ayala-Angulo J, et al. Regulatory T cells promote remyelination in the murine experimental autoimmune encephalomyelitis model of multiple sclerosis following human neural stem cell transplant. Neurobiol Dis. (2020) 140:104868. doi: 10.1016/j.nbd.2020.104868
137. Plaisted WC, Zavala A, Hingco E, Tran H, Coleman R, Lane TE, et al. Remyelination Is correlated with regulatory T cell induction following human embryoid body-derived neural precursor cell transplantation in a viral model of multiple sclerosis. PLoS ONE. (2016) 11:e0157620. doi: 10.1371/journal.pone.0157620
138. Dansokho C, Ait Ahmed D, Aid S, Toly-Ndour C, Chaigneau T, Calle V, et al. Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain. (2016) 139(Pt 4):1237–51. doi: 10.1093/brain/awv408
139. Baek H, Ye M, Kang GH, Lee C, Lee G, Choi DB, et al. Neuroprotective effects of CD4+CD25+Foxp3+ regulatory T cells in a 3xTg-AD Alzheimer's disease model. Oncotarget. (2016) 7:69347–57. doi: 10.18632/oncotarget.12469
140. Naert G, Rivest S. Age-related changes in synaptic markers and monocyte subsets link the cognitive decline of APP(Swe)/PS1 mice. Front Cell Neurosci. (2012) 6:51. doi: 10.3389/fncel.2012.00051
141. Zhang R, Miller RG, Madison C, Jin X, Honrada R, Harris W, et al. Systemic immune system alterations in early stages of Alzheimer's disease. J Neuroimmunol. (2013) 256:38–42. doi: 10.1016/j.jneuroim.2013.01.002
142. Lee WJ, Liao YC, Wang YF, Lin IF, Wang SJ, Fuh JL. Plasma MCP-1 and cognitive decline in patients with alzheimer's disease and mild cognitive impairment: a two-year follow-up study. Sci Rep. (2018) 8:1280. doi: 10.1038/s41598-018-19807-y
143. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. (2011) 11:762–74. doi: 10.1038/nri3070
144. Kierdorf K, Masuda T, Jordao MJC, Prinz M. Macrophages at CNS interfaces: ontogeny and function in health and disease. Nat Rev Neurosci. (2019) 20:547–62. doi: 10.1038/s41583-019-0201-x
145. Jordao MJC, Sankowski R, Brendecke SM, Sagar, Locatelli G, Tai YH, et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science. (2019) 363:eaat7554. doi: 10.1126/science.aat7554
146. Giladi A, Wagner LK, Li H, Dorr D, Medaglia C, Paul F, et al. Cxcl10(+) monocytes define a pathogenic subset in the central nervous system during autoimmune neuroinflammation. Nat Immunol. (2020) 21:525–34. doi: 10.1038/s41590-020-0661-1
147. Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, et al. High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity. (2018) 48:380–95.e6. doi: 10.1016/j.immuni.2018.01.011
148. Malm TM, Koistinaho M, Parepalo M, Vatanen T, Ooka A, Karlsson S, et al. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. (2005) 18:134–42. doi: 10.1016/j.nbd.2004.09.009
149. Michaud JP, Bellavance MA, Prefontaine P, Rivest S. Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid beta. Cell Rep. (2013) 5:646–53. doi: 10.1016/j.celrep.2013.10.010
150. Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc Natl Acad Sci USA. (2009) 106:1261–6. doi: 10.1073/pnas.0805453106
151. Connor JR, Menzies SL. Relationship of iron to oligodendrocytes and myelination. Glia. (1996) 17:83–93. doi: 10.1002/(SICI)1098-1136(199606)17:2<83::AID-GLIA1>3.0.CO;2-7
152. Ndayisaba A, Kaindlstorfer C, Wenning GK. Iron in neurodegeneration - cause or consequence? Front Neurosci. (2019) 13:180. doi: 10.3389/fnins.2019.00180
153. Bagnato F, Hametner S, Yao B, van Gelderen P, Merkle H, Cantor FK, et al. Tracking iron in multiple sclerosis: a combined imaging and histopathological study at 7 Tesla. Brain. (2011) 134(Pt 12):3602–15. doi: 10.1093/brain/awr278
154. Damulina A, Pirpamer L, Soellradl M, Sackl M, Tinauer C, Hofer E, et al. Cross-sectional and longitudinal assessment of brain iron level in Alzheimer disease using 3-T MRI. Radiology. (2020) 296:619–26. doi: 10.1148/radiol.2020192541
155. Mehta V, Pei W, Yang G, Li S, Swamy E, Boster A, et al. Iron is a sensitive biomarker for inflammation in multiple sclerosis lesions. PLoS ONE. (2013) 8:e57573. doi: 10.1371/journal.pone.0057573
156. Lee NJ, Ha SK, Sati P, Absinta M, Nair G, Luciano NJ, et al. Potential role of iron in repair of inflammatory demyelinating lesions. J Clin Invest. (2019) 129:4365–76. doi: 10.1172/JCI126809
157. Guerreiro C, Silva B, Crespo AC, Marques L, Costa S, Timoteo A, et al. xDecrease in APP and CP mRNA expression supports impairment of iron export in Alzheimer's disease patients. Biochim Biophys Acta. (2011) 1852(10 Pt A):2116–22. doi: 10.1016/j.bbadis.2015.07.017
158. Marshall JS, Warrington R, Watson W, Kim HL. An introduction to immunology and immunopathology. Allergy Asthma Clin Immunol. (2018) 14:49. doi: 10.1186/s13223-018-0278-1
159. Sommer A, Winner B, Prots I. The Trojan horse - neuroinflammatory impact of T cells in neurodegenerative diseases. Mol Neurodegener. (2017) 12:78. doi: 10.1186/s13024-017-0222-8
160. Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. (2019) 25:1822–32. doi: 10.1038/s41591-019-0675-0
161. Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol. (2010) 162:1–11. doi: 10.1111/j.1365-2249.2010.04143.x
162. Aytan N, Choi JK, Carreras I, Brinkmann V, Kowall NW, Jenkins BG, et al. Fingolimod modulates multiple neuroinflammatory markers in a mouse model of Alzheimer's disease. Sci Rep. (2016) 6:24939. doi: 10.1038/srep24939
163. Carreras I, Aytan N, Choi JK, Tognoni CM, Kowall NW, Jenkins BG, et al. Dual dose-dependent effects of fingolimod in a mouse model of Alzheimer's disease. Sci Rep. (2019) 9:10972. doi: 10.1038/s41598-019-47287-1
164. Fox E, Vieira MC, Johnson K, Peeples M, Bensimon AG, Signorovitch J, et al. Real-world durability of relapse rate reduction in patients with multiple sclerosis receiving fingolimod for up to 3years: a retrospective US claims database analysis. J Neurol Sci. (2019) 398:163–70. doi: 10.1016/j.jns.2019.01.036
165. Sica F, Centonze D, Buttari F. Fingolimod immune effects beyond its sequestration ability. Neurol Ther. (2019) 8:231–40. doi: 10.1007/s40120-019-00162-7
166. Hjorth M, Dandu N, Mellergard J. Treatment effects of fingolimod in multiple sclerosis: selective changes in peripheral blood lymphocyte subsets. PLoS ONE. (2020) 15:e0228380. doi: 10.1371/journal.pone.0228380
167. Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. (2007) 13:1173–5. doi: 10.1038/nm1651
168. Browne TC, McQuillan K, McManus RM, O'Reilly JA, Mills KH, Lynch MA. IFN-gamma Production by amyloid beta-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer's disease. J Immunol. (2013) 190:2241–51. doi: 10.4049/jimmunol.1200947
169. Brucklacher-Waldert V, Stuerner K, Kolster M, Wolthausen J, Tolosa E. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain. (2009) 132(Pt 12):3329–41. doi: 10.1093/brain/awp289
170. Doecke JD, Laws SM, Faux NG, Wilson W, Burnham SC, Lam CP, et al. Blood-based protein biomarkers for diagnosis of Alzheimer disease. Arch Neurol. (2012) 69:1318–25. doi: 10.1001/archneurol.2012.1282
171. Zhang J, Ke KF, Liu Z, Qiu YH, Peng YP. Th17 cell-mediated neuroinflammation is involved in neurodegeneration of abeta1–42-induced Alzheimer's disease model rats. PLoS ONE. (2013) 8:e75786. doi: 10.1371/journal.pone.0075786
172. Miller DH, Khan OA, Sheremata WA, Blumhardt LD, Rice GP, Libonati MA, et al. A controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. (2003) 348:15–23. doi: 10.1056/NEJMoa020696
173. Polman CH, O'Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. (2006) 354:899–910. doi: 10.1056/NEJMoa044397
174. Schetters STT, Gomez-Nicola D, Garcia-Vallejo JJ, Van Kooyk Y. Neuroinflammation: microglia and T cells get ready to tango. Front Immunol. (2017) 8:1905. doi: 10.3389/fimmu.2017.01905
175. Dong Y, Yong VW. When encephalitogenic T cells collaborate with microglia in multiple sclerosis. Nat Rev Neurol. (2019) 15:704–17. doi: 10.1038/s41582-019-0253-6
176. Evans FL, Dittmer M, de la Fuente AG, Fitzgerald DC. Protective and regenerative roles of T cells in central nervous system disorders. Front Immunol. (2019) 10:2171. doi: 10.3389/fimmu.2019.02171
177. Monsonego A, Nemirovsky A, Harpaz I. CD4 T cells in immunity and immunotherapy of Alzheimer's disease. Immunology. (2013) 139:438–46. doi: 10.1111/imm.12103
178. Karran E, De Strooper B. The amyloid cascade hypothesis: are we poised for success or failure? J Neurochem. (2016) 139(Suppl. 2):237–52. doi: 10.1111/jnc.13632
179. Lowther DE, Hafler DA. Regulatory T cells in the central nervous system. Immunol Rev. (2012) 248:156–69. doi: 10.1111/j.1600-065X.2012.01130.x
180. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine. (2015) 74:5–17. doi: 10.1016/j.cyto.2014.09.011
181. Duffy SS, Keating BA, Perera CJ, Moalem-Taylor G. The role of regulatory T cells in nervous system pathologies. J Neurosci Res. (2018) 96:951–68. doi: 10.1002/jnr.24073
182. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. (2008) 8:523–32. doi: 10.1038/nri2343
183. Ito M, Komai K, Mise-Omata S, Iizuka-Koga M, Noguchi Y, Kondo T, et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature. (2019) 565:246–50. doi: 10.1038/s41586-018-0824-5
184. Baruch K, Rosenzweig N, Kertser A, Deczkowska A, Sharif AM, Spinrad A, et al. Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer's disease pathology. Nat Commun. (2015) 6:7967. doi: 10.1038/ncomms8967
185. Verdon DJ, Mulazzani M, Jenkins MR. Cellular and molecular mechanisms of CD8(+) T cell differentiation, dysfunction and exhaustion. Int J Mol Sci. (2020) 21:7357. doi: 10.3390/ijms21197357
186. Ahrends T, Busselaar J, Severson TM, Babala N, de Vries E, Bovens A, et al. CD4(+) T cell help creates memory CD8(+) T cells with innate and help-independent recall capacities. Nat Commun. (2019) 10:5531. doi: 10.1038/s41467-019-13438-1
187. Wagner CA, Roque PJ, Mileur TR, Liggitt D, Goverman JM. Myelin-specific CD8+ T cells exacerbate brain inflammation in CNS autoimmunity. J Clin Invest. (2020) 130:203–13. doi: 10.1172/JCI132531
188. Piccio L, Rossi B, Scarpini E, Laudanna C, Giagulli C, Issekutz AC, et al. Molecular mechanisms involved in lymphocyte recruitment in inflamed brain microvessels: critical roles for P-selectin glycoprotein ligand-1 and heterotrimeric G(i)-linked receptors. J Immunol. (2002) 168:1940–9. doi: 10.4049/jimmunol.168.4.1940
189. Dugger KJ, Zinn KR, Weaver C, Bullard DC, Barnum SR. Effector and suppressor roles for LFA-1 during the development of experimental autoimmune encephalomyelitis. J Neuroimmunol. (2009) 206:22–7. doi: 10.1016/j.jneuroim.2008.10.006
190. Laurent C, Dorothee G, Hunot S, Martin E, Monnet Y, Duchamp M, et al. Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain. (2017) 140:184–200. doi: 10.1093/brain/aww270
191. Ferretti MT, Merlini M, Spani C, Gericke C, Schweizer N, Enzmann G, et al. T-cell brain infiltration and immature antigen-presenting cells in transgenic models of Alzheimer's disease-like cerebral amyloidosis. Brain Behav Immun. (2016) 54:211–25. doi: 10.1016/j.bbi.2016.02.009
192. Capece T, Walling BL, Lim K, Kim KD, Bae S, Chung HL, et al. A novel intracellular pool of LFA-1 is critical for asymmetric CD8(+) T cell activation and differentiation. J Cell Biol. (2017) 216:3817–29. doi: 10.1083/jcb.201609072
193. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Capello E, Mancardi GL, et al. Dendritic cells in multiple sclerosis lesions: maturation stage, myelin uptake, and interaction with proliferating T cells. J Neuropathol Exp Neurol. (2006) 65:124–41. doi: 10.1093/jnen/65.2.124
194. Merlini M, Kirabali T, Kulic L, Nitsch RM, Ferretti MT. Extravascular CD3+ T cells in brains of Alzheimer disease patients correlate with tau but not with amyloid pathology: an immunohistochemical study. Neurodegener Dis. (2018) 18:49–56. doi: 10.1159/000486200
195. Verma K, Ogonek J, Varanasi PR, Luther S, Bunting I, Thomay K, et al. Human CD8+ CD57− TEMRA cells: Too young to be called “old”. PLoS ONE. (2017) 12:e0177405. doi: 10.1371/journal.pone.0177405
196. Cebrian C, Loike JD, Sulzer D. Neuronal MHC-I expression and its implications in synaptic function, axonal regeneration and Parkinson's and other brain diseases. Front Neuroanat. (2014) 8:114. doi: 10.3389/fnana.2014.00114
197. Cerovic M, Forloni G, Balducci C. Neuroinflammation and the gut microbiota: possible alternative therapeutic targets to counteract Alzheimer's disease? Front Aging Neurosci. (2019) 11:284. doi: 10.3389/fnagi.2019.00284
198. Ma Q, Xing C, Long W, Wang HY, Liu Q, Wang RF. Impact of microbiota on central nervous system and neurological diseases: the gut-brain axis. J Neuroinflammation. (2019) 16:53. doi: 10.1186/s12974-019-1434-3
199. Cattaneo A, Cattane N, Galluzzi S, Provasi S, Lopizzo N, Festari C, et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol Aging. (2017) 49:60–8. doi: 10.1016/j.neurobiolaging.2016.08.019
200. Harach T, Marungruang N, Duthilleul N, Cheatham V, Mc Coy KD, Frisoni G, et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci Rep. (2017) 7:41802. doi: 10.1038/srep41802
201. Li B, He Y, Ma J, Huang P, Du J, Cao L, et al. Mild cognitive impairment has similar alterations as Alzheimer's disease in gut microbiota. Alzheimers Dement. (2019) 15:1357–66. doi: 10.1016/j.jalz.2019.07.002
202. Bonfili L, Cecarini V, Gogoi O, Gong C, Cuccioloni M, Angeletti M, et al. Microbiota modulation as preventative therapeutic approach in Alzheimer's disease. FEBS J. (2020). doi: 10.1111/febs.15571. [Epub ahead of print].
203. Ochoa-Reparaz J, Mielcarz DW, Haque-Begum S, Kasper LH. Induction of a regulatory B cell population in experimental allergic encephalomyelitis by alteration of the gut commensal microflora. Gut Microbes. (2010) 1:103–8. doi: 10.4161/gmic.1.2.11515
204. Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. (2011) 479:538–41. doi: 10.1038/nature10554
205. Wang Y, Begum-Haque S, Telesford KM, Ochoa-Reparaz J, Christy M, Kasper EJ, et al. A commensal bacterial product elicits and modulates migratory capacity of CD39(+) CD4 T regulatory subsets in the suppression of neuroinflammation. Gut Microbes. (2014) 5:552–61. doi: 10.4161/gmic.29797
206. Chen J, Chia N, Kalari KR, Yao JZ, Novotna M, Paz Soldan MM, et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci Rep. (2016) 6:28484. doi: 10.1038/srep28484
207. Berer K, Gerdes LA, Cekanaviciute E, Jia X, Xiao L, Xia Z, et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc Natl Acad Sci USA. (2017) 114:10719–24. doi: 10.1073/pnas.1711233114
208. Camara-Lemarroy CR, Metz L, Meddings JB, Sharkey KA, Wee Yong V. The intestinal barrier in multiple sclerosis: implications for pathophysiology and therapeutics. Brain. (2018) 141:1900–16. doi: 10.1093/brain/awy131
209. Schreiber HA, Loschko J, Karssemeijer RA, Escolano A, Meredith MM, Mucida D, et al. Intestinal monocytes and macrophages are required for T cell polarization in response to Citrobacter rodentium. J Exp Med. (2013) 210:2025–39. doi: 10.1084/jem.20130903
210. Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. (2011) 108(Suppl. 1):4615–22. doi: 10.1073/pnas.1000082107
211. Cekanaviciute E, Yoo BB, Runia TF, Debelius JW, Singh S, Nelson CA, et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc Natl Acad Sci USA. (2017) 114:10713–8. doi: 10.1073/pnas.1711235114
212. Wang Y, Telesford KM, Ochoa-Reparaz J, Haque-Begum S, Christy M, Kasper EJ, et al. An intestinal commensal symbiosis factor controls neuroinflammation via TLR2-mediated CD39 signalling. Nat Commun. (2014) 5:4432. doi: 10.1038/ncomms5432
213. Kadowaki A, Saga R, Lin Y, Sato W, Yamamura T. Gut microbiota-dependent CCR9+CD4+ T cells are altered in secondary progressive multiple sclerosis. Brain. (2019) 142:916–31. doi: 10.1093/brain/awz012
214. Gagliano SA, Pouget JG, Hardy J, Knight J, Barnes MR, Ryten M, et al. Genomics implicates adaptive and innate immunity in Alzheimer's and Parkinson's diseases. Ann Clin Transl Neurol. (2016) 3:924–33. doi: 10.1002/acn3.369
215. Hao S, Wang R, Zhang Y, Zhan H. Prediction of Alzheimer's disease-associated genes by integration of gwas summary data and expression data. Front Genet. (2018) 9:653. doi: 10.3389/fgene.2018.00653
216. International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science. (2019) 365:eaav7188. doi: 10.1126/science.aav7188
217. Sotgiu S, Murrighile MR, Constantin G. Treatment of refractory epilepsy with natalizumab in a patient with multiple sclerosis. Case report BMC Neurol. (2010) 10:84. doi: 10.1186/1471-2377-10-84
218. Angiari S, Constantin G. Selectins and their ligands as potential immunotherapeutic targets in neurological diseases. Immunotherapy. (2013) 5:1207–20. doi: 10.2217/imt.13.122
219. Fabene PF, Laudanna C, Constantin G. Leukocyte trafficking mechanisms in epilepsy. Mol Immunol. (2013) 55:100–4. doi: 10.1016/j.molimm.2012.12.009
220. Montagne A, Zhao Z, Zlokovic BV. Alzheimer's disease: a matter of blood-brain barrier dysfunction? J Exp Med. (2017) 214:3151–69. doi: 10.1084/jem.20171406
221. Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. (2018) 14:133–50. doi: 10.1038/nrneurol.2017.188
222. Van Skike CE, Jahrling JB, Olson AB, Sayre NL, Hussong SA, Ungvari Z, et al. Inhibition of mTOR protects the blood-brain barrier in models of Alzheimer's disease and vascular cognitive impairment. Am J Physiol Heart Circ Physiol. (2018) 314:H693–703. doi: 10.1152/ajpheart.00570.2017
Keywords: multiple sclerosis, Alzheimer's disease, neuroinflammation, neutrophils, monocytes, T cells
Citation: Rossi B, Santos-Lima B, Terrabuio E, Zenaro E and Constantin G (2021) Common Peripheral Immunity Mechanisms in Multiple Sclerosis and Alzheimer's Disease. Front. Immunol. 12:639369. doi: 10.3389/fimmu.2021.639369
Received: 08 December 2020; Accepted: 01 February 2021;
Published: 19 February 2021.
Edited by:
Philipp Albrecht, Heinrich Heine University of Düsseldorf, GermanyReviewed by:
Valerio Chiurchiù, Italian National Research Council, ItalyManuel Zeitelhofer, Karolinska Institutet (KI), Sweden
Copyright © 2021 Rossi, Santos-Lima, Terrabuio, Zenaro and Constantin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Barbara Rossi, YmFyYmFyYS5yb3NzaUB1bml2ci5pdA==; Gabriela Constantin, Z2FicmllbGEuY29uc3RhbnRpbkB1bml2ci5pdA==