 Tiffany Davia Ricketts
Tiffany Davia Ricketts Nestor Prieto-Dominguez
Nestor Prieto-Dominguez Pramod Sreerama Gowda
Pramod Sreerama Gowda Eric Ubil
Eric Ubil- Department of Microbiology, University of Alabama at Birmingham, Birmingham, AL, United States
Macrophages are a specialized class of innate immune cells with multifaceted roles in modulation of the inflammatory response, homeostasis, and wound healing. While developmentally derived or originating from circulating monocytes, naïve macrophages can adopt a spectrum of context-dependent activation states ranging from pro-inflammatory (classically activated, M1) to pro-wound healing (alternatively activated, M2). Tumors are known to exploit macrophage polarization states to foster a tumor-permissive milieu, particularly by skewing macrophages toward a pro-tumor (M2) phenotype. These pro-tumoral macrophages can support cancer progression by several mechanisms including immune suppression, growth factor production, promotion of angiogenesis and tissue remodeling. By preventing the adoption of this pro-tumor phenotype or reprogramming these macrophages to a more pro-inflammatory state, it may be possible to inhibit tumor growth. Here, we describe types of tumor-derived signaling that facilitate macrophage reprogramming, including paracrine signaling and activation of innate immune checkpoints. We also describe intervention strategies targeting macrophage plasticity to limit disease progression and address their implications in cancer chemo- and immunotherapy.
Introduction
Macrophages represent one of the most phenotypically diverse innate immune cell populations. They are key homeostatic regulators that activate and modulate the innate and, subsequent adaptive immune response to infectious agents and host-derived components. Much like other innate immune cells, they are hard-wired to respond to cues rather than being “educated” to elicit a response, as is the case of adaptive immune cells (1). Macrophages are equipped with a variety of Pattern Recognition Receptors (PRRs) that, once activated, trigger pre-determined programs in response to environmental stimuli. Some pro-inflammatory stimuli include Pathogen-Associated Molecular Patterns (PAMPs), cellular or chemical moieties derived from pathogens, or Damage-Associated Molecular Patterns (DAMPs) which are released by damaged cells and malignancies. These signatures permit macrophage adoption of the appropriate functional phenotype to restore physiological equilibrium.
During infections, macrophage polarization to the proinflammatory state is crucial for the production of type 1 cytokines such as interferon-γ (IFNγ), tumor necrosis factor-α (TNFα) and interleukin 12 (IL-12) for host resistance (2–4). This is similar to the response following injury. Cells in damaged tissues undergo necrosis and release their contents in an uncontrolled manner (5–7). Contrary to apoptosis, which is a highly organized program for cell death, necrosis is more immunogenic and induces a macrophage pro-inflammatory response. Cellular components released during necrosis act as DAMPs that, when bound to PRRs like Toll-like Receptors (TLRs), initiate pro-inflammatory signaling in resident and extravasated monocyte-derived macrophages. Activation of PRRs, and other sensors, facilitate the adoption of a pre-programmed pro-inflammatory state, also termed M1 or “classically activated” (Figure 1). This occurs through increased activation of signaling pathways involving NFκB, p38, MAPK, and others, which regulate the expression of pro-inflammatory cytokines (e.g., IL-1, IL-6, IL-12 (8, 9)) (Figure 2). These macrophage-secreted signals recruit a variety of other immune cells that pioneer the clearance of infected and damaged material.
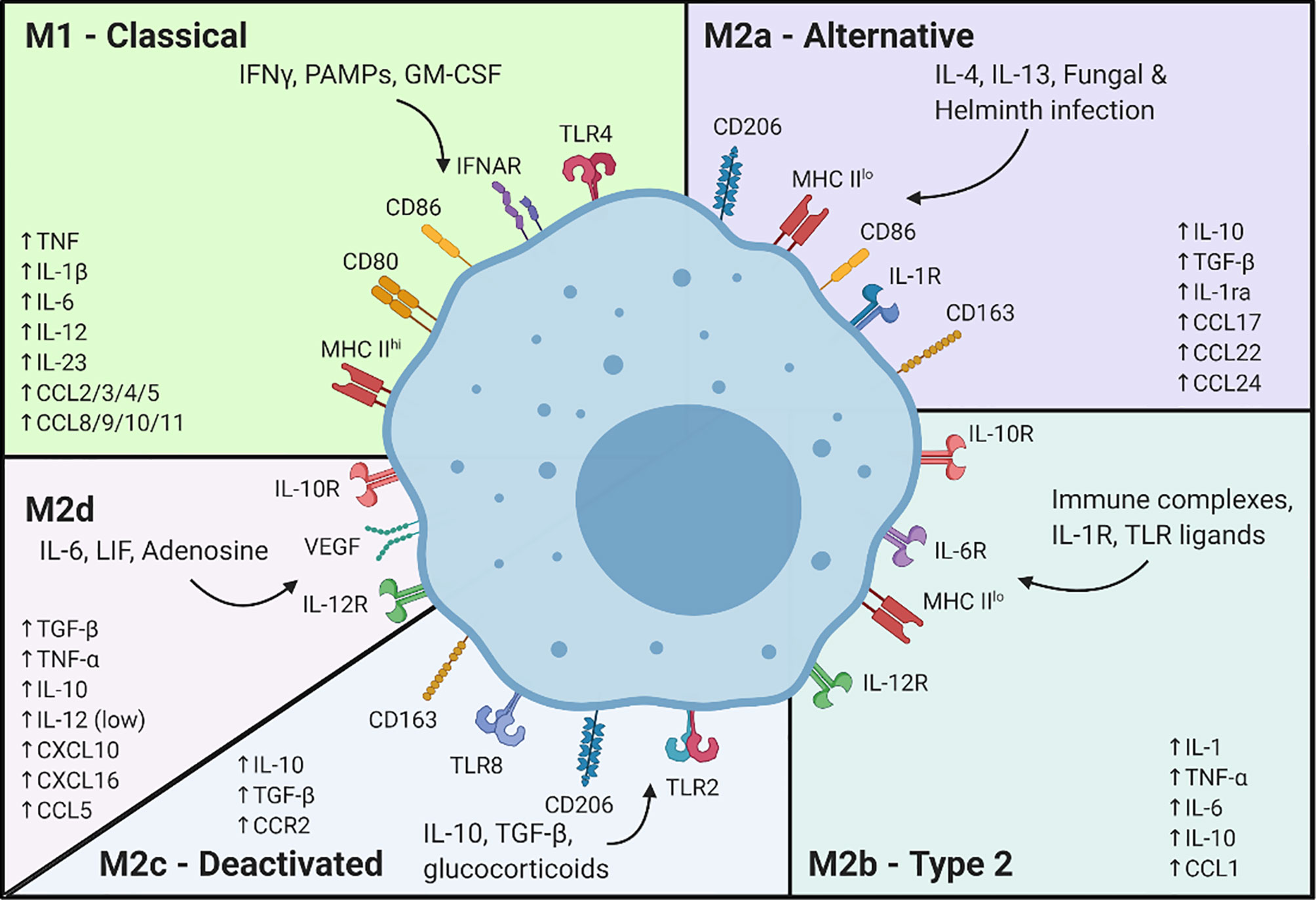
Figure 1 Signals associated with macrophage differentiation to the classically and alternatively activated subsets. Created with BioRender.
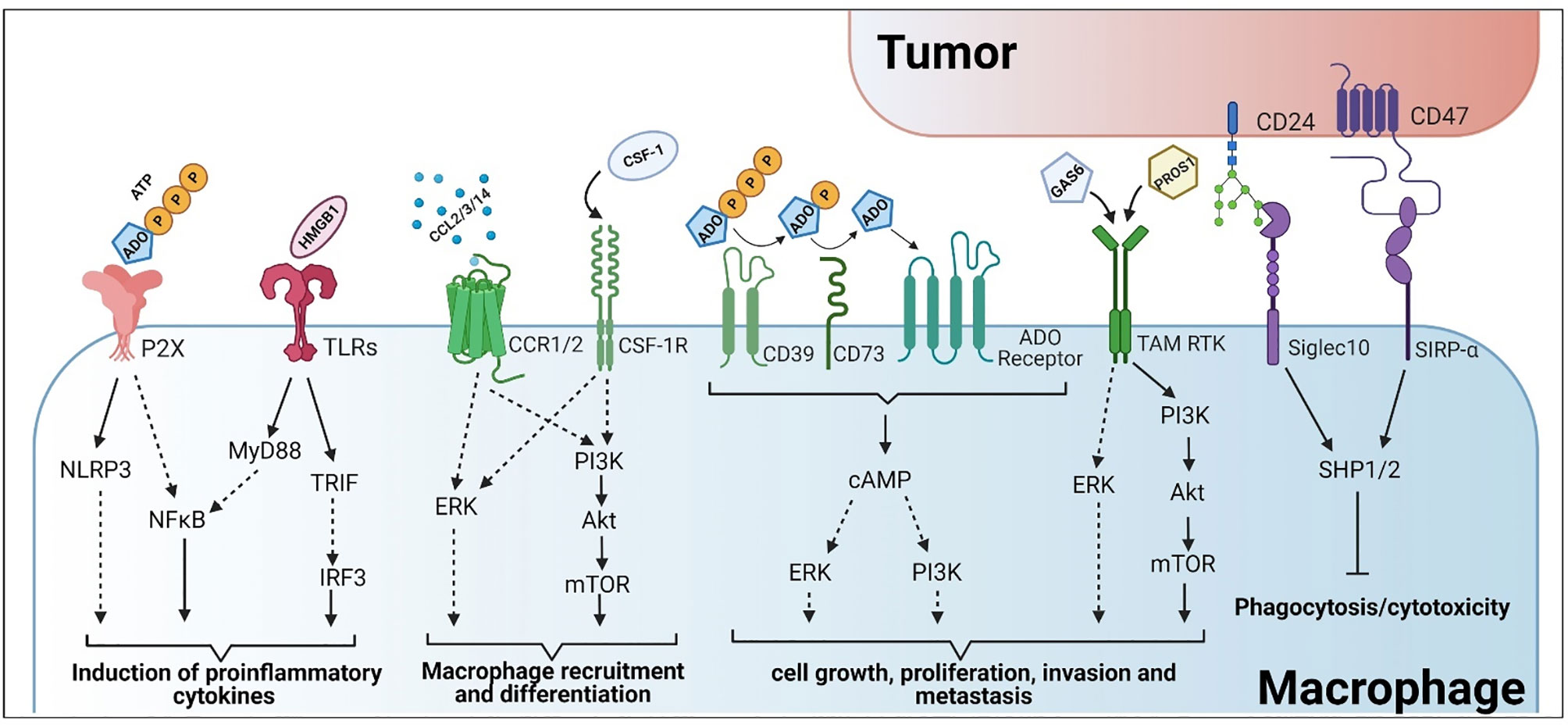
Figure 2 Tumor-macrophage interactions and their subsequent roles in immune evasion and activation. Created with BioRender.
A hallmark of the pro-inflammatory response is the destruction of damaged cells and those in the immediate vicinity. This creates a need for wound healing to restore tissue integrity. Upon removal of damaged tissue, the aggregate population of macrophages at the site of injury transitions to a pro-wound healing phenotype, also referred to as M2 (Figure 1). This transition is triggered by anti-inflammatory mediators following the loss of pro-inflammatory signals, like DAMPs. These pro-wound healing macrophages coordinate the proliferation of key cell types including vascular endothelial cells, which promote recellularization by delivering oxygen and nutrients to the site of repair, and fibroblasts which drive scar formation (10–12). Macrophages also dampen the local inflammatory response, fostering a more hospitable environment for continued repair, cellular proliferation and the prevention of extensive or persistent inflammation that might contribute to further tissue damage (13–16).
While macrophage plasticity is beneficial during the wound healing process, the macrophage response is subverted during cancer. Often termed “a wound that does not heal” (17), tumors manipulate and reshape the immune response to promote and sustain tumor growth. Presumably, due to the inhospitable nature of the tumor microenvironment (e.g., hypoxia, nutrient starvation), cancer cells undergo necrotic death which should induce the macrophage pro-inflammatory response, ultimately leading to further immune activation and reduced tumor growth. However, in many tumors, the pro-wound healing phenotype is predominant, which actually supports cancer progression. This review outlines strategies employed by tumors to mitigate macrophage pro-inflammatory activation or engage the pro-wound healing response. Current therapeutic interventions that alter the intra-tumoral M1/M2 balance and shift it towards a more pro-inflammatory/anti-tumor response are also described. We also explore potential conceptual flaws in the current pro-inflammatory/pro-wound healing paradigm in cancer, based on recent single-cell RNA-seq findings, and implications these could have in the manipulation of macrophage activation state to reduce tumor growth.
The Role of Macrophages in the Anti-Tumor Response
During tumorigenesis, genetic mutations can be acquired through exposure to chemical carcinogens (18), radiation (19) or viral infections (20, 21). Alternatively, inherited mutations (22, 23) or those accumulated during chronic inflammation (24–26) may also drive carcinogenesis. Cell intrinsic tumor suppressive mechanisms, like DNA repair, senescence or apoptosis (27), often fail to contain tumor cell proliferation, promoting the need for immune-mediated elimination of the aberrant cells. Ideally, early responding immune cells, like macrophages, will detect and eliminate tumor cells. Much like during wound healing, macrophages may detect DAMPs, possibly from hypoxia-induced tumor cell death or dysregulated cellular processes (28), to trigger a pro-inflammatory response and pave the way for true wound healing or a return to homeostasis. Alternatively, macrophages or dendritic cells, as antigen presenting cells, may engulf tumor neo-antigens, process them and present antigenic peptides to tissue resident CD8+ or CD4+ T cells, or in the case of dendritic cells, transit to the draining lymph node to activate T cells (29–31). Whether for tissue resident or T cells transiting from the lymph nodes, pro-inflammatory macrophages provide co-stimulatory signals such as CD40 (32) or CD80/86 (33), secrete activating cytokines (34), and generate nitric oxide to increase vascular permeability and immune cell infiltrate. T cells with the cognate receptor matching the tumor neo-antigen, in the presence of co-stimulation, should eradicate tumor cells unless they encounter other immuno-suppressive signals.
While many early-stage tumors are presumably destroyed through these mechanisms, the immune response to cancer is clearly not effective. Rather, based on the immune-editing hypothesis (35), the pro-inflammatory response applies a selective pressure, forcing tumors to “evolve” to avoid detection (e.g., through reduced antigenic protein expression, reduction in antigen presentation (35) or suppression of the local immune response (36)). Alternatively, nascent tumors may undergo a period of dormancy, and may later be reactivated by acquired secondary or tertiary mutations that allow for reduced immunogenicity or increased immune suppression. Collectively, this evolution is thought to allow tumor cells to reach an equilibrium with the immune response. Following this equilibrium state, tumors may effectively “escape” the immune response by utilizing mechanisms to prevent immune activation, allowing them to grow largely unchecked.
Consequently, these immuno-editing processes may limit macrophage responsiveness to DAMPs and tumor neo-antigens, effectively abrogating their ability to transition to an M1 phenotype (37) and promote T cell activation. In many tumors, there is a promotion of the M2 phenotype which fosters tumor growth. Presumably, either acquired through the equilibrium/escape processes of immuno-editing or because tumors provide contextual cues similar to those that promote the pro-wound healing response. These M2 macrophages are pro-tumorigenic and are often denoted as tumor-associated macrophages (TAMs). Akin to the wound healing response, macrophages facilitate cellular proliferation through production of growth factors like Wnts (38), CXCL8 (39) or IL-6 (40, 41). However, instead of promoting the re-growth of tissue resident cells, these factors drive tumor growth. Likewise, macrophages also secrete key effectors of vascularization, like the vascular endothelial growth factor (VEGF) (42, 43), platelet-derived growth factor (PDGF) (44) and transforming growth factor β (TGFβ) (45) to promote angiogenesis (Figure 1). These physiologic processes are hijacked to increase blood flow to the tumor, increasing tumor cell access to oxygen and nutrients for continued cell proliferation. M2 macrophages may also maintain tumor growth through the remodeling of the extracellular matrix (ECM) through secretion of matrix metalloproteases (MMPs) and other factors (45, 46) (Figure 1).
In the tumor context, pro-inflammatory macrophages are considered a positive prognostic marker (47–49). Pro-inflammatory macrophages are thought to positively regulate the immune response and kill tumor cells directly. These polarized macrophages prevent tumor growth by generating factors such as reactive oxygen and nitrogen species, or other secreted factors like TNFα, that lead to tumor cell death (50–53). Macrophages can be induced to a pro-inflammatory state by other immune cells, such as through the secretion of IFNγ by T cells, or directly by tumor cells. Alternatively, DAMPs can be released by necrotic or necroptotic tumor cell death due to hypoxia or nutrient deprivation within the tumor microenvironment (54, 55). These DAMPs, whether they be nucleic acids, ATP, stress-related proteins such as heat shock proteins (HSPs) (56–58), or transcription factors such as HMGB1, HMGN1 (59–65), bind to and activate two major classes of PRRs including the TLRs or the NOD-like receptor (NLR) family. Interestingly, several TLRs that recognize pathogenic signatures also recognize DAMPs. For instance, TLR4, which is activated by the binding of bacterial lipopolysaccharide (LPS) also recognizes HSPs and transcription factors (66).
Conversely, the presence of M2 pro-wound healing macrophages in tumors is generally a negative prognostic marker, with patients with high numbers of intra-tumoral M2 macrophages showing decreased survival (67). Tumor cells are known to secrete, or induce the secretion of, factors like IL-4, IL-10 or IL-13 that polarize macrophages toward an M2 phenotype (44, 68). Some pro-wound healing properties of M2 macrophages foster tumor growth and prepare a tumor-friendly milieu (Figure 1). M2 macrophages can act to directly increase tumor growth by secretion of growth factors like endothelial growth factor (EGF), VEGF and TGFβ (69–73), and can reduce the hypoxia inherent in most tumors while allowing the delivery of nutrients to sustain tumor growth. M2 macrophages also assist in the remodeling of the tumor microenvironment. Regulation of fibroblast ECM placement, degradation of existing ECMs through MMPs and chemotactic migration signals, allow continued tumor growth and metastasis. In some cases, live cell imaging has shown tumor cells utilizing accessory macrophages to travel to blood vessels and allow entry into the vasculature (74–76).
Macrophage-Directed Therapeutic Strategies for Cancer Treatment
Based on knowledge garnered from the study of macrophage activation states in tumors, as well as associated signaling affecting polarization, several strategies have been developed to mitigate tumor progression by altering macrophage infiltration or by activating/re-activating them to a pro-inflammatory state. While a limited number of macrophage-directed therapeutics are currently in use in clinical trials, continued identification and pharmacological targeting of macrophages is expected to bolster the use of macrophage targeted agents.
Macrophage Depletion to Reduce Pro-Tumoral Activity
Since higher numbers of TAMs are associated with worse cancer prognosis, research has focused on reducing their numbers by targeting their tumor recruitment and differentiation (77–79). As a result, some of the subsequent strategies are being tested for clinical use and may be broadly available soon.
Macrophages, similar to other phagocytes, can be selectively targeted by complexing cellular pro-apoptotic substances, such as bisphosphonates, into nanoparticles (80) (Table 1). The deletion of TAMs by using clodronate encapsulated in liposomes (clodrolip) leads to reduced teratocarcinoma and rhabdomyosarcoma tumor growth in pre-clinical murine studies (144). This inhibition was coupled with a decrease in tumor microvascular density, suggesting its potential combination with VEGF-neutralizing agents to maximize its effect (144).
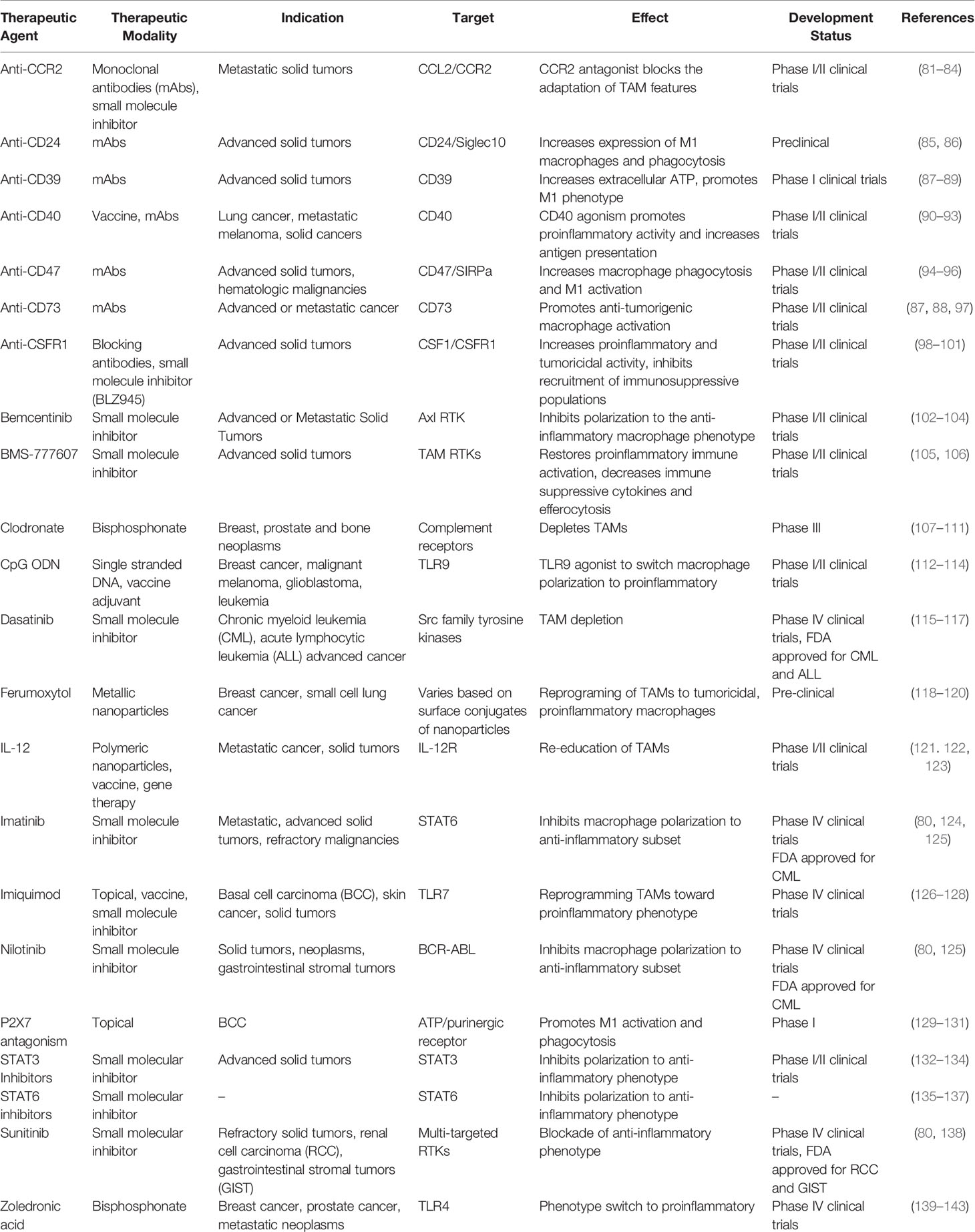
Table 1 Summary of preclinical, clinical and current therapeutic approaches targeting macrophages for the treatment of various malignancies.
Alternatively, inhibition of the chemotactic axis CCL2-CCR2 may prevent the accumulation of circulating macrophages within the tumor microenvironment. Indeed, several monotherapy or combinational clinical trials are currently underway with positive results (81). However, CCL2-CCR2 inhibitors should be carefully administered since the sudden interruption of therapeutic regimens could dramatically increase tumor progression and metastasis (145).
Additionally, targeting the monocyte/macrophage colony stimulating factor (CSF-1) and its receptor (CSF-1R) is a tractable strategy for macrophage depletion. In the absence of this signal, bloodborne monocytes are unable to differentiate into macrophages, preventing macrophage tumoral accumulation (146). Accordingly, several CSF-1R/CSF-1 targeted therapies, such as PLX3397, JNJ-40346527 and BLZ945, are currently being tested in clinical trials either alone or in combination for the treatment of several cancers (98, 147–149). However, these inhibitors can also stimulate the recruitment of tumor-promoting granulocytes to the site of the tumor, resulting in therapy failure (150). Therefore, combination of CSF-1R repressor with adaptive immune checkpoint inhibitors may be an interesting strategy to mitigate this unexpected effect (150).
Finally, the antineoplastic agent, trabectedin, also depletes TAMs to induce pro-inflammatory T cell recruitment in pancreatic ductal adenocarcinoma preclinical models (151). Therefore, it could also be a potential new strategy for TAM depletion during cancer treatment.
Manipulating Macrophage Activation State to Improve the Anti-Tumor Response
Using in vitro models of macrophage polarization, it has been shown that responses to respective M1/M2 stimuli are transient. Treatment with M1 inducing agents, like LPS and IFNγ, induce a pro-inflammatory response within 2-4 hours, which may subside within 24-48 hours (51, 152). After this transient activation, macrophages return to a “resting” state akin to the naïve (M0) polarization. Likewise, activation with one stimulus does not preclude the ability to adopt a subsequent, alternative polarization. A notable example is when stimulating conditions are switched from IFNγ to IL-4 or vice versa, macrophages adopt the profile of the most current cytokine microenvironment (153). Gao and colleagues utilized M-CSF and IL-4 to induce human monocyte differentiation to the M2 phenotype. Following M2 polarization, macrophages were treated with lactoferrin-containing IgG immunocomplex (LTF-IC), which promotes M1-like activation and is an immune activator in rheumatoid arthritis (154). After M1 stimulation, M2 marker expression was reduced while M1 markers were increased. In a similar experiment, Cheng et al. induced M2 polarization in murine RAW264.7 cells using IL-4 and IL-13. Subsequent treatment of M2 macrophages with a β-1,6-glucan (AAMP-A70) caused a reduction of M2 polarization concurrently with increased M1 marker expression (155). These findings are particularly important in the context of cancer treatment, as they clearly demonstrate the plasticity of macrophages depend on the environmental stimuli.
Considering the transient and plastic nature of macrophages, paired with the negative prognosis of intra-tumoral M2 macrophage accumulation, several approaches have been developed to repolarize M2 macrophages to an M1 phenotype. Macrophages, much like T cells, also have immune checkpoints. The prevention of tumors from activating innate immune checkpoints, is another approach in preventing the suppression of macrophage anti-tumor responses. Alternative approaches that manipulate the plasticity of macrophages are being heavily explored. Several of these strategies are described in the following sections.
Pro-Inflammatory Stimulation via TLR Agonism
The activation of TLRs, surface or endosomal proteins able to detect cellular damage and induce a proinflammatory immune response, have been broadly used therapeutically to alter macrophage activation in several diseases, including cancer (156–158) (Figure 2). The rationale is that the stimulation of these receptors, particularly within the tumor environment, may activate the pro-inflammatory response seen during the early stages of wound healing and infection, leading to the eradication of tumor cells (159, 160). Moreover, the release of tumor-derived DAMPs and neo-antigens during this process should generate a positive feedback loop to further increase the anti-tumor response (75, 159). A potential drawback of this form of therapy is tolerization, a state of unresponsiveness that appears after repetitive exposure to the same inductor, characterized by the release of anti-inflammatory factors that mask TLR activation (161).
Components of pathogenic organisms, such as LPS, derived mainly from Eschericia coli, are commonly used tools to activate macrophages and induce a pro-inflammatory state, often in combination with IFNγ to maximize the effects (162). However, LPS administration in humans produces severe toxicity and multiple exposures rapidly lead to tolerance, thus new strategies to improve its clinical use are currently being investigated (162). More recently, TLR3, TLR7/8 and TLR9 agonists have risen as new therapeutic alternatives to induce a TLR-dependent, tumor-localized pro-inflammatory response (163). For instance, the TLR7 agonist, Imiquimod, induces a robust rejection of skin primary malignancies and metastases by generating a pro-inflammatory tumor microenvironment in human patients (164) (Table 1). Similarly, polyinosinic-polycytidylic acid (poly-IC), a TLR3 agonist, triggers T cell tumor infiltration and Th1 responses, which should in turn activate macrophages through IFNγ signaling, to reduce malignant growth (165). Finally, the TLR9 agonist family CpG oligodeoxynucleotides (CpG ODN) have also shown strong cancer cytotoxic effects by exerting a potent tumor-localized immunostimulatory action (166) (Table 1). Based on early successes, these TLR agonists are currently in Phase 1/2/3 clinical trials (162, 163).
To target macrophages more specifically, nanoparticles that take advantage of the phagocytic properties of macrophages are being developed. After injection, nanoparticles are trafficked to the tumor where they are engulfed by macrophages. Techniques are being developed to package TLR agonists into nanoparticles for more specific activation of these immune cells (167). This novel approach would reduce the off-target effects of TLR agonists on other immune cells, such as lymphocytes, as well as to reduce their tolerizing effects (168). Furthermore, injected nanoparticles tend to accumulate in the tumor because of often ill-formed and leaky tumor vasculature, leading to a therapy more targeted to intra-tumoral macrophages (169). Loading β-cyclodextrin nanoparticles with the TLR7/8 agonist R484 has surfaced as one of the most promising techniques to restrain tumor growth by shifting TAM behavior to the M1 state (170).
Activating ATP NOD-Like Receptors to Promote M1 Polarization
Purinergic activation of macrophages plays a crucial role for the secretion of the pro-inflammatory cytokines, IL-1β and IL-18, and can be mediated through the activation of the NLRP3 inflammasome (171–173) (Figure 2). Cellular stress (e.g., exposure to chemotherapeutics, toxins, and radiation) and tissue damage are key contributors to ATP release into the extracellular environment (174). Release of ATP is one of the most potent DAMPs for immune activation, promoting M1 macrophage polarization and increasing macrophage tumoricidal potential (87, 129, 175), (Figure 2). However, to maintain the cellular ATP equilibrium, tumor cells, macrophages, and other immune cells, express ectonucleotidases to maintain the concentration gradient. CD39 and CD73 are ectonucleotidases that are involved in the formation of the metabolite adenosine (ADO). CD39 sequentially hydrolyzes ATP and ADP to form AMP, whereas CD73 hydrolyzes AMP to form ADO (Figure 2). This shift in the concentration gradient also acts as a switch to a more M2-like functional program and attenuates the anti-tumor response. Adenosine activates ADO/purinergic G-coupled protein receptors on tumor and immune cells, such as macrophages, to induce immunosuppression (176). Likewise, ADO also functions to inhibit TLR signaling and the secretion of proinflammatory cytokines such as TNFα, IL-6, and IL-8 from activated human monocytes (177). Given the contrasting nature of ATP versus ADO signaling for macrophage activation in tumor immunity, this interface serves as a potential target for the clearance of tumor cells. Inhibition of CD39 in preclinical models have shown significant promise in diminishing the immunosuppressive activity of TAMs, whereas inhibition of CD73 proved effective in controlling metastatic growth (178) (Table 1). Furthermore, combinational therapeutic strategies employing innate immune checkpoint inhibitors and anti-CD39 or anti-CD73 promoted antitumor immunity (88). Lastly, antagonism of the ATP receptors (P2X7) increases tumor infiltrating immune effector populations and decreases tumor burden (130) (Table 1).
Macrophage Polarization by Targeting Intracellular Signaling Mechanisms
In addition to mimicking extracellular pro-inflammatory stimuli, intracellular signaling pathways are also being targeted to reduce the prevalence of M2 signaling in tumors. This has been observed in the tumor-mediated manipulation of macrophage PI3Kγ signaling to reduce the pro-inflammatory response (179). Actually, targeting PI3Kγ pharmacologically has effectively “flipped the switch” from M2 to M1 in preclinical models (179, 180). PI3K is a family of phosphorylation enzymes that act on the 3’ end of phosphatidylinositol (PI) and work in conjunction with the Akt family of serine/threonine kinases and the mechanistic target of rapamycin complex (mTORC) 2 to switch the activation status of TLR-stimulated macrophages to a less pro-inflammatory program (181, 182) (Figure 2). PI3K/Akt signaling is involved in migration and diapedesis of innate immune effectors such as neutrophils and monocytes/macrophages and is associated with the upregulation and stabilization of hypoxia-induced transcription factors in macrophages (183). Induction of these transcription factors is associated with the hypoxic tumor microenvironment and stimulates M2-like characteristics in macrophages, thus supporting tumorigenesis and metastasis (184–186). Moreover, the PI3K/Akt pathway also promotes macrophage-mediated remodeling of the ECM, angiogenesis and immunosuppression of the adaptive immune response. Inhibition of PI3K signaling has shown considerable effects in regulating VEGF expression, a known factor that stimulates the adoption of the M2 functional program (183). There are several preclinical and clinical studies aimed at manipulating PI3K signaling to improve tumor outcomes. Inhibition of this pathway has been shown to increase macrophage infiltration and production of proinflammatory cytokines and chemokines (187). Akt signaling has differential downstream effects and deficiencies in Akt1 induced M1 activation (188). Consequently, inhibition of Akt signaling disrupts mTORC2 aggregation which diminished macrophage viability and proliferation (189).
The signal transducer and activator of transcription (STAT) signaling pathway is also of clinical interest. Downstream of several receptor tyrosine kinases, the STAT family communicates signals from the cytosolic face of the plasma membrane to the nucleus, where STAT dimers act as transcription factors and transcriptional modulators. STAT1 is recognized as a pro-inflammatory mediator and signaling can be initiated by type I and II interferons, growth factors, TLR activity and cytokine release. STAT1 signaling has broad effects on cancer and can either be antitumoral or pro-tumoral. Antitumoral STAT1 signaling is usually attributed to the tumoricidal activity of M1 macrophages while the pro-tumoral action is affiliated with the enrichment of STAT1-dependent genes that protect against genotoxic damage or promote tumor growth (190). Conversely, STAT3 is broadly recognized as an anti-inflammatory regulator, stimulating M2-like macrophage polarization. STAT3 phosphorylation can be triggered by interleukins such as IL-8, IL-10, IL-35 and growth factors such as EGF. Following activation, STAT3 signaling promotes a myriad of pro-tumoral outcomes such as the inhibition of apoptosis, cell proliferation, metastasis, angiogenesis and therapeutic resistance (41, 191). Studies targeting the activation of STAT1 or the suppression of STAT3 may be crucial for manipulating the balance of M1/M2 signaling.
Other transcription factors are also under study for potential roles in M1/M2 plasticity. These include KLF6, Zeb1 and NFAT1. KLF6 is a transcriptional regulator of macrophage polarization that serves as a phenotypic switch to transform M2-polarized TAMs to M1, effectively inhibiting tumor proliferation and migration (192). Contrariwise, ZEB1 is associated with TAM pro-tumoral activity, indicated by its ability to pioneer epithelial to mesenchymal transition to maintain tumor progression and initiate metastasis (8). Nuclear factor of activated T cell (NFAT) also supports the M2-like phenotype of TAMs through the regulation of interleukins (IL-6, IL-10, IL-12) and multiple TLR-induced genes such as iNOS (193). NFAT1 is overexpressed in TAMs and promotes tumor cell proliferation, invasion and metastasis and facilitates the recruitment of macrophage populations that are associated with poorer outcomes (194, 195). Given the role of NFAT signaling in regulating immune homeostasis, NFAT inhibition may effectively suppress anti-inflammatory cytokine production while subsequently initiating pro-inflammatory and tumoricidal programs within these tumor-associated macrophage populations.
Unfortunately, because individual transcription factors tend to be involved in transcriptional regulation throughout the genome, specifically targeting them to selectively target individual regulatory programs remains elusive. However, as time goes on, it may be possible to more selectively target individual immune cell types or add co-factors to increase specificity, yielding more robust anti-tumor efficacy.
Manipulating Macrophage Metabolism to Increase M1 Polarization
The metabolic changes associated with M1/M2 polarization may also regulate activation state (196, 197). Much like the distinct glutaminase-dependent differentiations of Th17 and Th1 T cells to regulate the immune response (198), direct metabolic changes in macrophages, or the output of altered metabolism, can affect M1/M2 polarization.
Arginase is essential for amino acid metabolism and has potent immunomodulatory effects through the catalysis of L-arginine. L-arginine is involved in nitric oxide synthesis which contributes to the tumoricidal activity of macrophages (199). However, the catabolism of L-arginine by arginase results in the formation of L-ornithine and its decomposition product, putrescine, which are known to support the cell growth and proliferation of tumor cells (199–202). Furthermore, increased production by TAMs impairs the antitumor immune response (203). Likewise, putrescine induces macrophage efferocytosis to prevent inflammation and promote tissue repair (204), a hallmark of tumor progression. Catabolism of L-arginine also has devastating consequences for other immune effectors, such as cell cycle arrest and anergy (203). Inhibition of arginase I expression reduces tumor burden and subsequently increases lymphocyte infiltration within the tumor microenvironment (205, 206) indicating significant potential for clinical testing.
Like arginase, indoleamine 2,3-dioxygenase (IDO1) is an immunosuppressive molecule secreted by TAMs. IDO1 catabolizes tryptophan to kynurenine which binds to the aryl hydrocarbon receptor to trigger a myriad of immunoregulatory mechanisms in immune cells (207). The signaling cascade triggered by IDO1 enzymatic activity facilitates immune evasion by diminishing lymphocyte responsiveness and anticancer immunosurveillance (208–210). IDO1 activity is also suggested to increase tolerance in macrophages, downregulate antigen presentation molecules (HLA-DR) and decreased macrophage phagocytic activity (211). Furthermore, IDO has also been shown to increase M2 polarization and recruitment while inhibition of IDO activity increases M1 populations (212). IDO1 inhibition prevents tryptophan depletion and subsequently blocks the associated downstream immunosuppressive signals (213, 214). This suggests that targeting IDO enzymatic activity in tumors that overexpress this enzyme may improve macrophage polarization to M1, immune activation and immunotherapeutic efficacy.
Targeting Innate Immune Checkpoints to Improve Therapeutic Outcomes
Much like the adaptive immune response, immune checkpoints have been discovered and characterized for innate immune cells. One example is the Tyro3/Axl/Mer (TAM) family of receptor tyrosine kinases, (Figure 2). During normal physiological processes, this family of receptors is instrumental in apoptotic cell engulfment and degradation (efferocytosis). The TAM family of receptors has 5 known ligands, Gas6 (215), Pros1 (216), Gal3 (217), Tubby and Tulp1 (218). As cells undergo apoptosis, phosphatidylserine that has flipped from the cytosolic face of the plasma membrane to the extracellular region is recognized by these ligands to form a bridge to the TAM receptors. However, these ligands can also activate the TAM receptors in the absence of phosphatidylserine (219), though activation is reduced. Lastly, kinase inhibition or genetic loss of Mer prevents internalization of apoptotic material (220, 221).
In addition to its role in efferocytosis, genetic lack of Mer is associated with hypersensitivity to TLR activation (222, 223), suggesting its role in limiting the innate immune response and preventing autoimmunity. More recently, it was shown by Lemke and Rothlin, in dendritic cells, that activation of Mer initiates an anti-inflammatory program involving upregulation of Socs1/2 (224). Later, Cook et al., demonstrated, in the context of cancer, that genetic deletion of Mer was associated with reduced M2 macrophage polarization with increased M1 (225). Ubil et al. later showed that tumor-secreted Pros1, acting on Mer and Tyro3 induces the downregulation of pro-inflammatory gene expression (51). Mice bearing tumors with genetic deletion of Pros1 showed increased intra-tumoral macrophages that were skewed towards the M1 phenotype. This was associated with increased adaptive immune infiltrate with approximately 5-fold more CD4+ and CD8+ T cells as well as a ~50% reduction in Tregs. Mice with Pros1 deficient tumors lived ~30% longer than mice with parental tumors. Furthermore, addition of the TLR7/8 agonist, Resiquimod, did not improve survival in mice bearing Pros1 replete tumors whereas survival duration was doubled for mice whose tumors lacked Pros1. Taken together, these findings demonstrate that tumor secretions can dampen the innate, macrophage, response and subsequently the adaptive immune response. TAM kinase inhibitors are currently in Phase I clinical trials for the treatment of human cancers.
Another marker involved in immune checkpoints and expressed by intra-tumoral macrophages is PD-L1. PD-L1 is generally associated with expression by tumors, particularly in response to IFNγ. When tumor expressed PD-L1 binds to PD-1 on T cells, it leads to T cell inactivation and facilitates tumor immune evasion. Tumors are also able to induce expression of PD-L1 in macrophages to similarly limit the action of effector T cells (226). Macrophage PD-L1 - T cell PD-1 interactions are, therefore, at the interface of innate and adaptive immune responses.
Several PD-1 and PD-L1 targeted therapeutics are currently in the clinic for treatment of various forms of cancer (227). In addition to the direct effects of blocking PD-1/PD-L1 interactions, PD-1 targeted treatments also induce secondary effects, such as the increased polarization of macrophages from a pro-wound healing phenotype to a more anti-tumor, pro-inflammatory, state. Xiong et al. characterized intra-tumoral macrophage polarization states of MC38 tumor bearing mice after anti-PD-1 treatment. They observed an increase in the numbers of M1-like and M1/M2 intermediate macrophages with a decrease in M2-like phenotypes. Using IFNγ depletion of supernatants from tumors which had either been treated with vehicle or anti-PD-1 antibody, they determined that IFNγ was a primary driver of macrophage polarization (228). Presumably, anti-PD-1 treatment of tumor bearing mice led to increased T cell activation, including IFNγ secretion. In turn, polarization of intra-tumoral macrophages were skewed towards an M1 state, including increased antigen presentation and expression of pro-inflammatory cytokines. Activated M1 macrophages increased T cell activation in a self-reinforcing cycle, ultimately leading to reduced tumor growth. This study succinctly demonstrates the importance and inter-relatedness of the innate and adaptive immune functions in limiting tumor progression.
Targeting “Don’t Eat Me” Signaling to Improve Macrophage Activation and Antitumor Immunity
A crucial aspect of macrophage activity is phagocytosis, the internalization of cells, pathogens, and other particles for tissue homeostasis. As key endocytosing immune cells, macrophages are the primary phagocytic population and should be able to recognize aberrant cells and clear them using this process. However, tumor cells express anti-phagocytic ligands or “don’t eat me” signals similar to healthy cells in order to avoid elimination.
CD47 is an immunoglobulin that is crucial in self recognition for the maintenance of immune tolerance and homeostasis. It complexes with the signal regulatory protein α (SIRPα) on phagocytic cells to inhibit uptake and subsequent immune activation (229). However, this molecule is also expressed on the surface of many tumor cells and plays a key role in immune evasion (Figure 2). CD47/SIRPα signaling leads to the phosphorylation of the SIRPα cytoplasmic immunoreceptor tyrosine-based inhibition motifs (ITIM) resulting in the recruitment of the tyrosine phosphatases SHP1/2. This signaling mechanism prevents the accumulation of myosin at the phagocytic synapse, effectively inhibiting phagocytosis (230–232). This process is crucial in preventing uncontrolled clearance of healthy cells but becomes a detriment based on its role in facilitating immune evasion in cancer. As such, these signals are also targeted to improve the antitumor response. CD47 blockade has shown significant efficacy in the treatment of several hematological cancers and solid tumors which may be mediated by innate immune effector populations such as macrophages (94, 95, 233, 234) (Table 1). Furthermore, preclinical models of the CD47/SIRPα signaling axis are highly efficacious for treating multiple cancer types and are currently being probed in clinical trials.
CD24 is another “don’t eat me” signal that is expressed by many tumor types (Figure 2). CD24 is a glycosylphosphatidylinositol anchored protein that is known to complex with Siglec10 on macrophages and other innate immune cells for the suppression of the inflammatory response in many conditions including sepsis, liver damage and infection (85, 235, 236). Like CD47 signaling, the CD24/Siglec10 signaling axis results in the recruitment of SHP1/2 at the ITIMs of Siglec10, inhibiting the TLR-mediated inflammatory response and the cytoskeleton rearrangement required for phagocytosis (85). As such, the CD24/Siglec10 complex is a potent inhibitor of macrophage phagocytic activity and is protective of cancer cells. Inhibition of the CD24/Siglec10 signaling axis restores the macrophage-mediated antitumor response by enhancing phagocytic clearance of tumor cells (85, 86). Moreover, increased uptake of antigenic materials is also associated with increased immune activation and infiltration within the tumor microenvironment (85).
The importance of these signaling cascades in regulating macrophage plasticity are extensively studied and new models are currently being probed to increase innate immune activation and improve current immunotherapeutic approaches. A summary of these targets and their effect on macrophage activity within the tumor microenvironment, along with their development status, are described in Table 1.
Current Experimental Modeling of M1/M2 Phenotypes May Not Accurately Represent Intra-Tumoral Macrophage Polarization States
To model macrophage responses, the M1/M2 paradigm was developed and dates back more than 20 years (237). In early models, naïve macrophages were induced to adopt two known polarization states (238). Since then, through decades of research, multiple in vitro models of M1 and M2 polarization have been developed in which various exogenous stimuli can induce activation states that mimic physiological conditions (e.g., pathogenic infection (239–241), pro-inflammatory activation by T cells (242, 243), etc.). At present, experimental macrophage models have been delineated into 5 core subsets: M1, M2a, M2b, M2c and M2d (244), (Figure 1).
Historically, activation of the M1 state has been modeled using stimuli such as LPS, IFNγ (a pro-inflammatory signal derived from activated T cells) or both in combination. While LPS induces TLR4 activation and downstream NFκB signaling, IFNγ binds the IFNgR1/2 complex, leading to STAT1 phosphorylation and nuclear translocation to mediate pro-inflammatory gene expression (245, 246). Alternatively, addition of TNFα (247) to naïve macrophages yields a similar activation state. TNFα binds to TNFR1 and TNFR2, leading to activation of downstream signaling cascades including p38 (248, 249) and others (250–253). The pro-inflammatory signaling pathways tend to converge on NFκB, STAT1 and MAPK pathways, with significant crosstalk effectively leading to similar outcomes in terms of gene expression changes and activation states.
M2 activation states are comparatively more complicated with at least 4 different subsets being identified, including M2a, M2b, M2c and the relatively newer M2d phenotype (152, 254, 255) (Figure 1). Induced by IL-4, IL-13 or the combination thereof, M2a has been described as an anti-inflammatory and pro-wound healing subset (256–258). M2b, which is induced by addition of IL-1β, has shown immuno-regulatory properties and associated gene expression (244, 259). M2c macrophages, induced by treatment with IL-10, show increased expression of immune suppressive and tissue remodeling markers (260). Some indications also suggest efferocytosis is increased in M2c macrophages (261). Finally, in an attempt to create a model of TAMs (M2d), it was discovered that treatment with IL-6 could cause upregulation of tumor growth and angiogenesis markers (262).
At this point, there is not one clearly prevailing macrophage M2 subset that best represents tumor associated macrophages. Instead, researchers often combine multiple stimuli, such as IL-4 (M2a), IL-13 (M2a) and IL-10 (M2c), which are present in the tumor microenvironment, to mimic tumor associated macrophages (263, 264).
While continually improving, our understanding of intra-tumoral macrophage activation states have led to an iterative improvement in models. However, newer and better methodologies are currently being utilized to disaggregate our current population-level understanding. Specifically, single cell RNA-seq (sc-RNA-seq) has refined our understanding of intra-tumoral macrophage heterogeneity and called into question some of our existing paradigms on “either/or” M1/M2 polarization.
Single-Cell RNA-Seq Data Sheds New Light on Intra-Tumoral Macrophage Polarization
Based on established in vitro models of macrophage polarization (M1/M2), early characterization of intra-tumoral macrophages focused on a few pro-inflammatory or pro-wound healing markers (e.g., iNOS, IL-1, CD206, etc.) to identify activation states. As more nuanced models of polarization have been developed, additional markers have been identified, demonstrating that rather than adhering to distinct polarized types, macrophages exhibit a spectrum of overlapping activation states. Further complicating the ability to describe tumor associated macrophages is that spatial location and microenvironmental factors can have major impacts on polarity, causing macrophages in one part of the tumor to have very different activation states than those in adjacent locations. The advent of single cell RNA-seq has opened new venues for understanding intra-tumoral macrophage activation and may identify misconceptions about how macrophages behave in the tumor microenvironment. This new technique allows for the characterization of individual cells within the tumor resident immune cell subset. Depending on the process flow, immune cell subtypes may be enriched prior to single-cell RNA-seq analysis (265, 266) or bioinformatically identified based on expression patterns (267). Several variations of single-cell RNA-seq exist, some of which also incorporate locational data.
Characterization of Macrophage Activation State in Tumors
Using single-cell RNA-seq to characterize immune subset in primary breast cancer samples, Chung et al. found that macrophages tend toward the M2 phenotype (265), confirming previous findings that breast cancer tends to foster M2 polarization (46, 268). Of the 515 cells from 11 patients characterized, most non-carcinoma cells in the cancer samples were identified as immune cells based on their gene expression signatures. TAMs were primarily found to have pro-wound healing M2-associated profiles (269, 270). A key finding of this paper is that it supports the notion that in breast cancer, many macrophages and other innate and adaptive cell populations have an immune suppressive phenotype.
Recognizing that there is robust heterogeneity of intra-tumoral macrophage polarization states, single cell RNA-seq is also being used to determine whether there are discrete activation states or whether there is a contiguous spectrum driven by local microenvironmental conditions. Azizi et al., employed a large-scale, high-dimensional analysis platform to characterize the immune profiles of more than 45,000 cells from eight breast carcinomas, matched with normal breast tissue, blood and lymph nodes using single-cell RNA-seq (271). To do so, they collected CD45 positive cells from treatment-naïve breast cancer patients including estrogen receptor (ER+) and progesterone receptor (PR+) positive, human epidermal growth factor receptor 2 amplified (HER2+) and triple negative (TNBC) tumors. These CD45+ cells were isolated by fluorescence-activated cell sorting (FACS) and subjected to single-cell RNA-seq using the inDrop platform (272, 273). Data was preprocessed using the SEQC pipeline with the Bayesian clustering and normalization method, Biscuit, utilized for data analysis. One of the key findings of the study is that intra-tumoral macrophages have higher numbers, diversity and activation relative to those derived from normal tissues or lymph nodes. Somewhat surprisingly, the authors of this study found a positive correlation between M1 and M2 gene expression, with simultaneous co-expression of markers associated with both activation states. This is in direct contrast to previous results from in vitro model studies, in which one or more agents used to activate macrophages led to one aggregate activation state, either M1 or M2.
A different study, characterizing the heterogeneity of macrophages activation states in gliomas using single-cell RNA-seq made a similar observation on the simultaneous co-expression of M1 and M2 markers in TAMs. This study, conducted by Muller et al. (274), compared marker expression of two macrophage populations – brain-resident microglia, derived from progenitors that migrated to the central nervous system (CNS) and bone marrow-derived monocytes that extravasate through the blood brain barrier and differentiate into macrophages. Similar to Azizi et al., Muller et al., found that macrophages could co-express M1 and M2 markers simultaneously with 66% of tumor associated macrophages co-expressing the canonical M2 marker, IL-10, while also expressing the M1 marker, TNFα. They confirmed their results by using flow cytometry of tumor derived macrophages to show that CD11b+ cells could co-express the M1 co-stimulatory marker, CD86, while also expressing CD206.
Taken together, these studies call the M1/M2 polarization paradigm into question. While, to some extent, supporting the notion that a spectrum of intra-tumoral macrophage activation states exist (275, 276), the finding of simultaneous M1 and M2 associated markers by macrophages is quite novel. Perhaps historical use of conventional models coupled with aggregate analyses of pooled macrophage populations fail to detect a more widespread phenomenon of M1/M2 marker co-expression in tumors. Further experiments and analysis will be required to confirm these finding. Also, development of model systems that better recapitulate the dual activation states observed in vivo may yield better understanding of how intra-tumoral macrophages will respond to targeted therapeutics. Perhaps most importantly, these findings suggest that activating, or re-activating, the M1 phenotype in tumors may consequently lead to concurrent increased M2 polarization, thereby confounding outcomes.
Using Single Cell RNA-Seq Based Methods to Characterize Macrophage Activation While Incorporating Spatial Localization Within the Tumor
Conventional large-scale characterization of macrophage polarization loses spatial resolution. As such, novel single-cell RNA-seq/bioinformatic approaches are being developed that provide contextual identity. One such technique involves the use of spatial transcriptomics (277). This method performs unbiased mapping of transcripts over entire tissue sections using spatially barcoded oligo-deoxythymidine microarrays. Individual microarray spots capture transcriptome information from between 10-200 cells and the data is integrated with single cell RNA-seq data to provide both cellular context and transcription data at the single cell level. Using this approach, Moncada and colleagues performed multimodal intersection analysis on patient pancreatic ductal adenocarcinoma (PDAC) tumors (278). One of their key findings was that macrophages seem to adhere to the M1/M2 paradigm and exist in two main subpopulations. The first was a pro-inflammatory M1 subset, which expressed IL-1β, and a second subset, which expressed M2 associated genes like CD163 (278). Likewise, the two subpopulations were differentially localized, with M1 macrophages enriched in the cancerous regions or the stroma, while M2-like macrophages were enriched in the ducts. This data demonstrates that two opposing macrophage polarizations can exist in the same tumor, though their activation state is driven by local micro-environmental conditions. These findings suggest that, fundamentally, treatments may be more effective if they can be selectively targeted to regions where they will make the biggest change. Conversely, systemic treatment with an M1 inducing agent could disrupt essential processes and induce off-target effects.
Derivation of M2 Macrophage Subpopulations
Circulating monocytes are recruited to tumors by the expression of chemoattractants such as CCL2 (279–281), S100A8 and S100A9 (282, 283). Once monocytes extravasate, they are thought to differentiate into M1 or M2 macrophages based on signals from the tumor microenvironment. In a recent study, Song et al. used single-cell RNA-seq to characterize the differentiation process of extravasating monocytes. 11,485 cells from Non-Small Cell Lung Cancer (NSCLC) patients were used to develop a model of divergent monocyte differentiation into M1 or M2 macrophages. While there were differences between patients, on average, a substantially larger proportion of the recruited monocytes adopted the M2 phenotype (283). In CD14+ cells derived from in NSCLC samples, expression of polarization markers was stratified along a continuum effectively providing a snapshot of macrophage differentiation states. Work by Song et al., may enable the identification of specific lineage markers that will allow prediction of future differentiation states. They also identified signals from tumor-derived epithelial cells that skew differentiation to the M2 phenotype. By better understanding the process through which tumor resident M2 macrophages are derived, it may be possible to develop specific interventions that prevent accumulation of M2 macrophages.
Open Questions in Macrophage Plasticity During Cancer
Macrophages are a highly plastic innate immune cell subset. Depending on contextual cues from their local environment, they adopt phenotypes across a spectrum of activation states, ranging from pro-inflammatory (M1) to pro-wound healing (M2). Further, macrophages, both individually and in aggregate, can readily transition from one polarization state to the next depending on the most recent signals prevailing in their environment. This plasticity allows them to effectively adapt to the changing environments associated with infection and wound healing and facilitate the return to immune homeostasis. Unfortunately, in the context of cancer, macrophage plasticity is subverted to benefit continued tumor progression. Either by tumor-mediated suppression of M1 polarization or through the evolved lack of pro-inflammatory cues associated with cancer, intra-tumoral macrophages are generally of the pro-wound healing (M2) phenotype. The pro-wound healing properties which would be beneficial during injury repair, such as production of growth factors or promotion of angiogenesis, support continued tumor cell proliferation and tumor expansion.
Recognizing the inherent plasticity of macrophages, several therapeutics have been developed to either reduce the number of intra-tumoral macrophages, thereby reducing the M2 pool, or alter the M1/M2 balance to favor a more pro-inflammatory/anti-tumor response. Numerous clinical trials have demonstrated that increasing M1-associated polarization or effector functions can improve clinical outcomes. This is, perhaps, not surprising since a pro-inflammatory milieu is associated with better patient outcomes for many cancer types. However, to realize the promise of these new treatment modalities, several factors still need to be considered. As we have learned from adaptive immune targeted treatments, activation or checkpoint blockade alone are not likely to be sufficient to generate durable responses in several cancer types. Rather, macrophage targeted therapies will likely require co-treatments targeting the cancer directly (e.g., chemotherapy) or the adaptive immune response (e.g., checkpoint directed therapeutics) or both. Also, for the most part, M1 polarization is thought to reduce tumor growth. However, chronic and persistent local inflammatory conditions are also known to induce tumor formation (284–287). A prime example is that increased inflammation associated with obesity can actually increase the likelihood of tumor progression (288). Several other preclinical models of inflammation, such as colitis-induced colon cancer (72–76), have shown that persistent inflammation exacerbates tumor progression. As an illustration, in a high-fat diet induced inflammation model, prostate cancer progression was substantially increased (289). The rationale is that persistent cell damaging conditions may elicit genetic mutation or cell signaling alterations that foster tumor growth. While the current paradigm is that “more inflammation is better”, there is likely to be an optimal amount of inflammation so as not to induce secondary tumor formation.
Another key question to be addressed, in addition to finding optimal combinations, is how to limit potential engagement of the autoimmune response. Even if a macrophage targeted therapy is successful in generating an anti-tumor response, what are the best ways to ensure it is targeted strictly to the tumor and not surrounding healthy tissues or organ systems? While some delivery systems, like nanoparticles, favor intra-tumoral macrophages, many require systemic delivery, increasing the potential for off-target effects. Potentially compounding the likelihood of off-target effects is reliance upon the bystander effect to generate an anti-tumor response. For example, TLR agonists mimic PAMPs and DAMPs that would be released during infection or injury. However, the resulting immune activation does not target tumor-intrinsic moieties, but rather utilize the destructive potential of pro-inflammatory macrophages to either kill neighboring tumor cells or activate other local immune cells. This lack of tumor specificity opens the greater possibility of non-specific cellular damage or even autoimmunity based on the release of cryptic epitopes.
In addition to questions of developing targeted therapeutics, some basic scientific questions also remain unanswered about macrophages in the tumor environment. While several models have shown, in vitro, that macrophages can move from one polarization state to the next, it is unclear whether this is also true in tumors. For instance, lack of lineage tracing prevents the accurate monitoring of individual intra-tumoral macrophages to determine what happens after treatment. Are macrophages that are present in the tumor prior to treatment adopting another phenotype or is macrophage turnover the cause for an aggregate shift in polarization? Development and use of lineage tracing models would provide a more expansive knowledge of macrophage activation during treatment.
Other questions that have arisen with the advent of single-cell RNA-seq include whether there is a previously unknown macrophage state the possesses elements of both the diametrically opposed M1 and M2 phenotypes. Can both activation states co-exist in one cell or group of cells? What environmental or cell intrinsic factors would allow for dual expression of pro- and anti-inflammatory markers? Do these dual activation macrophages also exist during wound healing or response to pathogenic infection or are they a cancer-specific phenomenon? Are there ways in which these specialized cells can be modeled in vitro? Perhaps most importantly, how do pro-inflammatory inducing treatments affect dual M1/M2 macrophages? Does their presence confound treatments focusing on M1 induction? For instance, if a TLR agonist is utilized for treatment, does it also increase the expression of M2 associated markers, simultaneously activating and inactivating the immune response? Further analysis of single-cell RNA-seq data may answer these questions. However, it may be possible, using flow cytometry or other techniques, to isolate these cells and characterize them using more traditional biochemical methods.
While there is a more comprehensive understanding of macrophage biology now than in the past, development of macrophage targeted therapeutics has trailed behind those promoting the adaptive immune response. Continuing to address the unanswered questions presented here, as well continued testing, both alone and in combination with other therapeutics, may bridge the gap, providing new hope for improved survival of cancer patients.
Author Contributions
TR - wrote the manuscript, prepared figures, and edited final work. NP-D - wrote manuscript and edited final work. PG - wrote manuscript. EU - conceptualized the work, wrote manuscript, and edited final work. All authors contributed to the article and approved the submitted version.
Funding
NIH/NCI K22 Transition Career Development Award (1 K22 CA237742-01) - Funding for EU, and University of Alabama at Birmingham Development Funds - Funding for EU.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
2. Wang J, Wakeham J, Harkness R, Xing Z. Macrophages are a Significant Source of Type 1 Cytokines During Mycobacterial Infection. J Clin Invest (1999) 103(7):1023–9. doi: 10.1172/JCI6224
3. Verreck FAW, De Boer T, Langenberg DML, Hoeve MA, Kramer M, Vaisberg E, et al. Human IL-23-producing Type 1 Macrophages Promote But IL-10-producing Type 2 Macrophages Subvert Immunity to (Myco)Bacteria. Proc Natl Acad Sci USA (2004) 101(13):4560–5. doi: 10.1073/pnas.0400983101
4. Verreck FAW, de Boer T, Langenberg DML, van der Zanden L, Ottenhoff THM. Phenotypic and Functional Profiling of Human Proinflammatory Type-1 and Anti-Inflammatory Type-2 Macrophages in Response to Microbial Antigens and IFN-γ- and CD40L-mediated Costimulation. J Leuk Biol (2006) 79(2):285–93. doi: 10.1189/jlb.0105015
5. Cocco RE, Ucker DS. Distinct Modes of Macrophage Recognition for Apoptotic and Necrotic Cells are Not Specified Exclusively by Phosphatidylserine Exposure. Mol Biol Cell (2001) 12(4):919–30. doi: 10.1091/mbc.12.4.919
6. Sachet M, Liang YY, Oehler R. The Immune Response to Secondary Necrotic Cells. Apoptosis Int J Programmed Cell Death (2017) 22(10):1189–204. doi: 10.1007/s10495-017-1413-z
7. Atanasov G, Dietel C, Feldbrügge L, Benzing C, Krenzien F, Brandl A, et al. Tumor Necrosis and Infiltrating Macrophages Predict Survival After Curative Resection for Cholangiocarcinoma. Oncoimmunology (2017) 6(8):e1331806–e1331806. doi: 10.1080/2162402X.2017.1331806
8. Cortés M, Sanchez-Moral L, de Barrios O, Fernández-Aceñero MJ, Martínez-Campanario M, Esteve-Codina A, et al. Tumor-Associated Macrophages (Tams) Depend on ZEB1 for Their Cancer-Promoting Roles. EMBO J (2017) 36(22):3336–55. doi: 10.15252/embj.201797345
9. Zhang R, Qi F, Zhao F, Li G, Shao S, Zhang X, et al. Cancer-Associated Fibroblasts Enhance Tumor-Associated Macrophages Enrichment and Suppress NK Cells Function in Colorectal Cancer. Cell Death Dis (2019) 10(4):273. doi: 10.1038/s41419-019-1435-2
10. Polverini PJ, Cotran PS, Gimbrone MA, Jr., Unanue ER. Activated Macrophages Induce Vascular Proliferation. Nature (1977) 269(5631):804–6. doi: 10.1038/269804a0
11. Danon D, Kowatch MA, Roth GS. Promotion of Wound Repair in Old Mice by Local Injection of Macrophages. Proc Natl Acad Sci (1989) 86(6):2018–20. doi: 10.1073/pnas.86.6.2018
12. Hunt TK, Knighton DR, Thakral KK, Goodson WH. 3rd, and Andrews, W.s., Studies on Inflammation and Wound Healing: Angiogenesis and Collagen Synthesis Stimulated In Vivo by Resident and Activated Wound Macrophages. Surgery (1984) 96(1):48–54.
13. Guth AM, Janssen WJ, Bosio CM, Crouch EC, Henson PM, Dow SW. Lung Environment Determines Unique Phenotype of Alveolar Macrophages. Am J Physiol Lung Cell Mol Physiol (2009) 296(6):L936–46. doi: 10.1152/ajplung.90625.2008
14. Hashimoto D, Chow A, Noizat C, Teo P, Beasley, Mary B, Leboeuf M, et al. Tissue-Resident Macrophages Self-Maintain Locally Throughout Adult Life With Minimal Contribution From Circulating Monocytes. Immunity (2013) 38(4):792–804. doi: 10.1016/j.immuni.2013.04.004
15. Heidt T, Courties G, Dutta P, Sager HB, Sebas M, Iwamoto Y, et al. Differential Contribution of Monocytes to Heart Macrophages in Steady-State and After Myocardial Infarction. Circ Res (2014) 115(2):284–95. doi: 10.1161/CIRCRESAHA.115.303567
16. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-Resident Macrophage Enhancer Landscapes are Shaped by the Local Microenvironment. Cell (2014) 159(6):1312–26. doi: 10.1016/j.cell.2014.11.018
17. Dvorak HF. Tumors: Wounds That do Not Heal-Redux. Cancer Immunol Res (2015) 3(1):1–11. doi: 10.1158/2326-6066.CIR-14-0209
18. Innes JRM, Ulland BM, Valerio MG, Petrucelli L, Fishbein L, Hart ER, et al. Bioassay of Pesticides and Industrial Chemicals for Tumorigenicity in Mice: A Preliminary Note23. JNCI: J Natl Cancer Inst (1969) 42(6):1101–14. doi: 10.1093/jnci/42.6.1101
19. Green AES, Findley GB Jr, Klenk KF, Wilson WM, Mo T. The Ultraviolet Dose Dependence of Non-Melanoma Skin Cancer Incidence. Photochem Photobiol (1976) 24(4):353–62. doi: 10.1111/j.1751-1097.1976.tb06836.x
20. Baba AI, Câtoi C. Carcinogenesis. In: Comparative Oncology. Bucharest (RO): The Publishing House of the Romanian Academy (2007).
21. Yabe Y, Trentin JJ, Taylor G. Cancer Induction in Hamsters by Human Type 12 Adenovirus. Effect of Age and of Virus Dose. Proc Soc Exp Biol Med (1962) 111(2):343–4. doi: 10.3181/00379727-111-27786
22. Corso G, Intra M, Trentin C, Veronesi P, Galimberti V. CDH1 Germline Mutations and Hereditary Lobular Breast Cancer. Fam Cancer (2016) 15(2):215–9. doi: 10.1007/s10689-016-9869-5
23. Rusak B, Kluźniak W, Wokołorczykv D, Stempa K, Kashyap A, Gronwald J, et al. Inherited NBN Mutations and Prostate Cancer Risk and Survival. Cancer Res Treat (2019) 51(3):1180–7. doi: 10.4143/crt.2018.532
24. Fishbein A, Wang W, Yang H, Yang J, Hallisey VM, Deng J, et al. Resolution of Eicosanoid/Cytokine Storm Prevents Carcinogen and Inflammation-Initiated Hepatocellular Cancer Progression. Proc Natl Acad Sci (2020) 117(35):21576–87. doi: 10.1073/pnas.2007412117
25. Ntp Carcinogenesis Bioassay of Melamine (Cas No. 108-78-1) in F344/N Rats and B6C3F1 Mice (Feed Study). Natl Toxicol Program Tech Rep Ser (1983) 245:1–171.
26. Saffiotti U, Cefis F, Kolb LH. A Method for the Experimental Induction of Bronchogenic Carcinoma. Cancer Res (1968) 28(1):104–24. doi: 10.1097/00043764-196902000-00016
27. Sherr CJ. Principles of Tumor Suppression. Cell (2004) 116(2):235–46. doi: 10.1016/S0092-8674(03)01075-4
28. Solari JIG, Filippi-Chiela E, Pilar ES, Nunes V, Gonzalez EA, Figueiró F, et al. Damage-Associated Molecular Patterns (Damps) Related to Immunogenic Cell Death are Differentially Triggered by Clinically Relevant Chemotherapeutics in Lung Adenocarcinoma Cells. BMC Cancer (2020) 20(1):474. doi: 10.1186/s12885-020-06964-5
29. Wang S, He Z, Wang X, Li H, Liu X-S. Antigen Presentation and Tumor Immunogenicity in Cancer Immunotherapy Response Prediction. eLife (2019) 8:e49020. doi: 10.7554/eLife.49020
30. McKean DJ, Infante AJ, Nilson A, Kimoto M, Fathman CG, Walker E, et al. Major Histocompatibility Complex-Restricted Antigen Presentation to Antigen-Reactive T Cells by B Lymphocyte Tumor Cells. J Exp Med (1981) 154(5):1419–31. doi: 10.1084/jem.154.5.1419
31. Shimizu J, Suda T, Yoshioka T, Kosugi A, Fujiwara H, Hamaoka T. Induction of Tumor-Specific In Vivo Protective Immunity by Immunization With Tumor Antigen-Pulsed Antigen-Presenting Cells. J Immunol (1989) 142(3):1053–9. doi: 10.1097/00008390-199309002-00068
32. Vitale LA, Thomas LJ, He L-Z, O’Neill T, Widger J, Crocker A, et al. Development of CDX-1140, an Agonist CD40 Antibody for Cancer Immunotherapy. Cancer Immunol Immunother (2019) 68(2):233–45. doi: 10.1007/s00262-018-2267-0
33. Slavik JM, Hutchcroft JE, Bierer BE. CD80 and CD86 are Not Equivalent in Their Ability to Induce the Tyrosine Phosphorylation of CD28. J Biol Chem (1999) 274(5):3116–24. doi: 10.1074/jbc.274.5.3116
34. Hoves S, Ooi C-H, Wolter C, Sade H, Bissinger S, Schmittnaegel M, et al. Rapid Activation of Tumor-Associated Macrophages Boosts Preexisting Tumor Immunity. J Exp Med (2018) 215(3):859–76. doi: 10.1084/jem.20171440
35. Dunn GP, Old LJ, Schreiber RD. The Three Es of Cancer Immunoediting. Annu Rev Immunol (2004) 22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803
36. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-Regulation of PD-L1, IDO, and T(regs) in the Melanoma Tumor Microenvironment is Driven by CD8(+) T Cells. Sci Trans Med (2013) 5(200):200ra116–200ra116. doi: 10.1126/scitranslmed.3006504
37. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. Ifngamma and Lymphocytes Prevent Primary Tumour Development and Shape Tumour Immunogenicity. Nature (2001) 410(6832):1107–11. doi: 10.1038/35074122
38. Yang Y, Ye YC, Chen Y, Zhao JL, Gao CC, Han H, et al. Crosstalk Between Hepatic Tumor Cells and Macrophages Via Wnt/β-Catenin Signaling Promotes M2-like Macrophage Polarization and Reinforces Tumor Malignant Behaviors. Cell Death Dis (2018) 9(8):793. doi: 10.1038/s41419-018-0818-0
39. Fang W, Ye L, Shen L, Cai J, Huang F, Wei Q, et al. Tumor-Associated Macrophages Promote the Metastatic Potential of Thyroid Papillary Cancer by Releasing CXCL8. Carcinogenesis (2014) 35(8):1780–7. doi: 10.1093/carcin/bgu060
40. Weng YS, Tseng HY, Chen YA, Shen PC, Al Haq AT, Chen LM, et al. Mct-1/miR-34a/IL-6/IL-6R Signaling Axis Promotes EMT Progression, Cancer Stemness and M2 Macrophage Polarization in Triple-Negative Breast Cancer. Mol Cancer (2019) 18(1):42. doi: 10.1186/s12943-019-0988-0
41. Yin Z, Ma T, Lin Y, Lu X, Zhang C, Chen S, et al. Il-6/STAT3 Pathway Intermediates M1/M2 Macrophage Polarization During the Development of Hepatocellular Carcinoma. J Cell Biochem (2018) 119(11):9419–32. doi: 10.1002/jcb.27259
42. Lacal PM, Graziani G. Therapeutic Implication of Vascular Endothelial Growth Factor Receptor-1 (VEGFR-1) Targeting in Cancer Cells and Tumor Microenvironment by Competitive and non-Competitive Inhibitors. Pharmacol Res (2018) 136:97–107. doi: 10.1016/j.phrs.2018.08.023
43. Lai Y-S, Wahyuningtyas R, Aui S-P, Chang K-T. Autocrine VEGF Signalling on M2 Macrophages Regulates PD-L1 Expression for Immunomodulation of T Cells. J Cell Mol Med (2019) 23(2):1257–67. doi: 10.1111/jcmm.14027
44. Yang F, Li W-B, Qu Y-W, Gao J-X, Tang Y-S, Wang D-J, et al. Bone Marrow Mesenchymal Stem Cells Induce M2 Microglia Polarization Through PDGF-AA/MANF Signaling. World J Stem Cells (2020) 12(7):633–58. doi: 10.4252/wjsc.v12.i7.633
45. O’Meara T, Marczyk M, Qing T, Yaghoobi V, Blenman K, Cole K, et al. Immunological Differences Between Immune-Rich Estrogen Receptor–Positive and Immune-Rich Triple-Negative Breast Cancers. JCO Precis Oncol (2020) 4):767–79. doi: 10.1200/PO.19.00350
46. Sousa S, Brion R, Lintunen M, Kronqvist P, Sandholm J, Mönkkönen J, et al. Human Breast Cancer Cells Educate Macrophages Toward the M2 Activation Status. Breast Cancer Res (2015) 17(1):101. doi: 10.1186/s13058-015-0621-0
47. López-Janeiro Á, Padilla-Ansala C, de Andrea CE, Hardisson D, Melero I. Prognostic Value of Macrophage Polarization Markers in Epithelial Neoplasms and Melanoma. A Systematic Review and Meta-Analysis. Mod Pathol (2020) 33(8):1458–65. doi: 10.1038/s41379-020-0534-z
48. Zeiner PS, Preusse C, Golebiewska A, Zinke J, Iriondo A, Muller A, et al. Distribution and Prognostic Impact of Microglia/Macrophage Subpopulations in Gliomas. Brain Pathol (2019) 29(4):513–29. doi: 10.1111/bpa.12690
49. Zhao Y, Ge X, Xu X, Yu S, Wang J, Sun L. Prognostic Value and Clinicopathological Roles of Phenotypes of Tumour-Associated Macrophages in Colorectal Cancer. J Cancer Res Clin Oncol (2019) 145(12):3005–19. doi: 10.1007/s00432-019-03041-8
50. Saha D, Martuza RL, Rabkin SD. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell (2017) 32(2):253–267.e5. doi: 10.1016/j.ccell.2017.07.006
51. Ubil E, Caskey L, Holtzhausen A, Hunter D, Story C, Earp HS. Tumor-Secreted Pros1 Inhibits Macrophage M1 Polarization to Reduce Antitumor Immune Response. J Clin Invest (2018) 128(6):2356–69. doi: 10.1172/JCI97354
52. Wu JY, Huang TW, Hsieh YT, Wang YF, Yen CC, Lee GL, et al. Cancer-Derived Succinate Promotes Macrophage Polarization and Cancer Metastasis Via Succinate Receptor. Mol Cell (2020) 77(2):213–27.e5. doi: 10.1016/j.molcel.2019.10.023
53. Hori K, Ehrke MJ, Mace K, Maccubbin D, Doyle MJ, Otsuka Y, et al. Effect of Recombinant Human Tumor Necrosis Factor on the Induction of Murine Macrophage Tumoricidal Activity. Cancer Res (1987) 47(11):2793–8.
54. Fauskanger M, Haabeth OAW, Skjeldal FM, Bogen B, Tveita AA. Tumor Killing by CD4+ T Cells Is Mediated Via Induction of Inducible Nitric Oxide Synthase-Dependent Macrophage Cytotoxicity. Front Immunol (2018) 9(1684):1–13. doi: 10.3389/fimmu.2018.01684
55. Feng M, Chen JY, Weissman-Tsukamoto R, Volkmer J-P, Ho PY, McKenna KM, et al. Macrophages Eat Cancer Cells Using Their Own Calreticulin as a Guide: Roles of TLR and Btk. Proc Natl Acad Sci (2015) 112(7):2145. doi: 10.1073/pnas.1424907112
56. Fauconneau B, Petegnief V, Sanfeliu C, Piriou A, Planas AM. Induction of Heat Shock Proteins (Hsps) by Sodium Arsenite in Cultured Astrocytes and Reduction of Hydrogen Peroxide-Induced Cell Death. J Neurochem (2002) 83(6):1338–48. doi: 10.1046/j.1471-4159.2002.01230.x
57. Park YH, Seo JH, Park J-H, Lee HS, Kim K-W. Hsp70 Acetylation Prevents Caspase-Dependent/Independent Apoptosis and Autophagic Cell Death in Cancer Cells. Int J Oncol (2017) 51(2):573–8. doi: 10.3892/ijo.2017.4039
58. Son K-J, Choi KR, Ryu C-K, Lee SJ, Kim HJ, Lee H. Induction of Immunogenic Cell Death of Tumors by Newly Synthesized Heterocyclic Quinone Derivative. PloS One (2017) 12(3):e0173121–e0173121. doi: 10.1371/journal.pone.0173121
59. Dong XD, Ito N, Lotze MT, DeMarco RA, Popovic P, Shand SH, et al. High Mobility Group Box I (Hmgb1) Release From Tumor Cells After Treatment: Implications for Development of Targeted Chemoimmunotherapy. J Immunother (2007) 30(6):596–606. doi: 10.1097/CJI.0b013e31804efc76
60. Zhang XY, Guo ZQ, Ji SQ, Zhang M, Jiang N, Li XS, et al. Small Interfering RNA Targeting HMGN5 Induces Apoptosis Via Modulation of a Mitochondrial Pathway and Bcl-2 Family Proteins in Prostate Cancer Cells. Asian J Androl (2012) 14(3):487–92. doi: 10.1038/aja.2012.18
61. Bell CW, Jiang W, Charles F. Reich I, Pisetsky DS. The Extracellular Release of HMGB1 During Apoptotic Cell Death. Am J Physiol Cell Physiol (2006) 291(6):C1318–25. doi: 10.1152/ajpcell.00616.2005
62. Tang D, Loze MT, Zeh HJ, Kang R. The Redox Protein HMGB1 Regulates Cell Death and Survival in Cancer Treatment. Autophagy (2010) 6(8):1181–3. doi: 10.4161/auto.6.8.13367
63. Dai Y, Bae K, Pampo C, Siemann DW. Impact of the Small Molecule Met Inhibitor BMS-777607 on the Metastatic Process in a Rodent Tumor Model With Constitutive c-Met Activation. Clin Exp Metastasis (2012) 29(3):253–61. doi: 10.1007/s10585-011-9447-z
64. Elkon KB. Review: Cell Death, Nucleic Acids, and Immunity: Inflammation Beyond the Grave. Arthritis Rheum (2018) 70(6):805–16. doi: 10.1002/art.40452
65. Maelfait J, Liverpool L, Rehwinkel J. Nucleic Acid Sensors and Programmed Cell Death. J Mol Biol (2020) 432(2):552–68. doi: 10.1016/j.jmb.2019.11.016
66. He M, Bianchi ME, Coleman TR, Tracey KJ, Al-Abed Y. Exploring the Biological Functional Mechanism of the HMGB1/TLR4/MD-2 Complex by Surface Plasmon Resonance. Mol Med (2018) 24(1):21. doi: 10.1186/s10020-018-0030-9
67. Petrillo M, Zannoni GF, Martinelli E, Pedone Anchora L, Ferrandina G, Tropeano G, et al. Polarisation of Tumor-Associated Macrophages Toward M2 Phenotype Correlates With Poor Response to Chemoradiation and Reduced Survival in Patients With Locally Advanced Cervical Cancer. PloS One (2015) 10(9):e0136654. doi: 10.1371/journal.pone.0136654
68. Lin L, Chen Y-S, Yao Y-D, Chen J-Q, Chen J-N, Huang S-Y, et al. CCL18 From Tumor-Associated Macrophages Promotes Angiogenesis in Breast Cancer. Oncotarget (2015) 6(33):34758–73. doi: 10.18632/oncotarget.5325
69. Gurevich DB, Severn CE, Twomey C, Greenhough A, Cash J, Toye AM, et al. Live Imaging of Wound Angiogenesis Reveals Macrophage Orchestrated Vessel Sprouting and Regression. EMBO J (2018) 37(13):1–23. doi: 10.15252/embj.201797786
70. Fang W, Zhou T, Shi H, Yao M, Zhang D, Qian H, et al. Progranulin Induces Immune Escape in Breast Cancer Via Up-Regulating PD-L1 Expression on Tumor-Associated Macrophages (Tams) and Promoting CD8+ T Cell Exclusion. J Exp Clin Cancer Res (2021) 40(1):4. doi: 10.1186/s13046-020-01786-6
71. Qin T, Zeng Y-d, Qin G, Xu F, Lu J-b, Fang W-f, et al. High PD-L1 Expression was Associated With Poor Prognosis in 870 Chinese Patients With Breast Cancer. Oncotarget (2015) 6(32):33972–81. doi: 10.18632/oncotarget.5583
72. Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT, et al. Neoantigen-Directed Immune Escape in Lung Cancer Evolution. Nature (2019) 567(7749):479–85. doi: 10.1038/s41586-019-1032-7
73. Schoppmann SF, Birner P, Stöckl J, Kalt R, Ullrich R, Caucig C, et al. Tumor-Associated Macrophages Express Lymphatic Endothelial Growth Factors and are Related to Peritumoral Lymphangiogenesis. Am J Pathol (2002) 161(3):947–56. doi: 10.1016/S0002-9440(10)64255-1
74. Kadioglu E, De Palma M. Cancer Metastasis: Perivascular Macrophages Under Watch. Cancer Discovery (2015) 5(9):906–8. doi: 10.1158/2159-8290.CD-15-0819
75. Wang J, Cao Z, Zhang XM, Nakamura M, Sun M, Hartman J, et al. Novel Mechanism of Macrophage-Mediated Metastasis Revealed in a Zebrafish Model of Tumor Development. Cancer Res (2015) 75(2):306–15. doi: 10.1158/0008-5472.CAN-14-2819
76. Yamauchi K, Yang M, Jiang P, Xu M, Yamamoto N, Tsuchiya H, et al. Development of Real-time Subcellular Dynamic Multicolor Imaging of Cancer-Cell Trafficking in Live Mice With a Variable-Magnification Whole-Mouse Imaging System. Cancer Res (2006) 66(8):4208–14. doi: 10.1158/0008-5472.CAN-05-3927
77. Bonde A-K, Tischler V, Kumar S, Soltermann A, Schwendener RA. Intratumoral Macrophages Contribute to Epithelial-Mesenchymal Transition in Solid Tumors. BMC Cancer (2012) 12(1):35. doi: 10.1186/1471-2407-12-35
78. Fu X-T, Dai Z, Song K, Zhang Z-J, Zhou Z-J, Zhou S-L, et al. Macrophage-Secreted IL-8 Induces Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma Cells by Activating the JAK2/STAT3/Snail Pathway. Int J Oncol (2015) 46(2):587–96. doi: 10.3892/ijo.2014.2761
79. Hughes R, Qian B-Z, Rowan C, Muthana M, Keklikoglou I, Olson OC, et al. Perivascular M2 Macrophages Stimulate Tumor Relapse After Chemotherapy. Cancer Res (2015) 75(17):3479–91. doi: 10.1158/0008-5472.CAN-14-3587
80. Ngambenjawong C, Gustafson HH, Pun SH. Progress in Tumor-Associated Macrophage (TAM)-Targeted Therapeutics. Adv Drug Deliv Rev (2017) 114:206–21. doi: 10.1016/j.addr.2017.04.010
81. Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting Tumour-Associated Macrophages With CCR2 Inhibition in Combination With FOLFIRINOX in Patients With Borderline Resectable and Locally Advanced Pancreatic Cancer: A Single-Centre, Open-Label, Dose-Finding, non-Randomised, Phase 1b Trial. Lancet Oncol (2016) 17(5):651–62. doi: 10.1016/S1470-2045(16)00078-4
82. Flores-Toro JA, Luo D, Gopinath A, Sarkisian MR, Campbell JJ, Charo IF, et al. CCR2 Inhibition Reduces Tumor Myeloid Cells and Unmasks a Checkpoint Inhibitor Effect to Slow Progression of Resistant Murine Gliomas. Proc Natl Acad Sci (2020) 117(2):1129–38. doi: 10.1073/pnas.1910856117
83. Teng K-Y, Han J, Zhang X, Hsu S-H, He S, Wani NA, et al. Blocking the CCL2–CCR2 Axis Using Ccl2-Neutralizing Antibody is an Effective Therapy for Hepatocellular Cancer in a Mouse Model. Mol Cancer Ther (2017) 16(2):312–22. doi: 10.1158/1535-7163.MCT-16-0124
84. Tu MM, Abdel-Hafiz HA, Jones RT, Jean A, Hoff KJ, Duex JE, et al. Inhibition of the CCL2 Receptor, CCR2, Enhances Tumor Response to Immune Checkpoint Therapy. Commun Biol (2020) 3(1):720. doi: 10.1038/s42003-020-01441-y
85. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 Signalling Through Macrophage Siglec-10 is a Target for Cancer Immunotherapy. Nature (2019) 572(7769):392–6. doi: 10.1038/s41586-019-1456-0
86. Liu H, Hai L, Tian J, Xiang J, Fan Y, Zhang H, et al. Anti-CD24 Neutralizing Antibody Exacerbates Concanavalin a-Induced Acute Liver Injury in Mice Via Liver M1 Macrophages. Immunol Lett (2017) 181:87–93. doi: 10.1016/j.imlet.2016.11.016
87. Allard B, Longhi MS, Robson SC, Stagg J. The Ectonucleotidases CD39 and CD73: Novel Checkpoint Inhibitor Targets. Immunol Rev (2017) 276(1):121–44. doi: 10.1111/imr.12528
88. Perrot I, Michaud H-A, Giraudon-Paoli M, Augier S, Docquier A, Gros L, et al. Blocking Antibodies Targeting the CD39/CD73 Immunosuppressive Pathway Unleash Immune Responses in Combination Cancer Therapies. Cell Rep (2019) 27(8):2411–25.e9. doi: 10.1016/j.celrep.2019.04.091
89. Hayes GM, Cairns B, Levashova Z, Chinn L, Perez M, Theunissen JW, et al. CD39 is a Promising Therapeutic Antibody Target for the Treatment of Soft Tissue Sarcoma. Am J Transl Res (2015) 7(6):1181–8. doi: 10.1016/j.ccell.2017.10.005
90. Vonderheide RH, Flaherty KT, Khalil M, Stumacher MS, Bajor DL, Hutnick NA, et al. Clinical Activity and Immune Modulation in Cancer Patients Treated With CP-870,893, a Novel CD40 Agonist Monoclonal Antibody. J Clin Oncol (2007) 25(7):876–83. doi: 10.1200/JCO.2006.08.3311
91. Bensinger W, Maziarz RT, Jagannath S, Spencer A, Durrant S, Becker PS, et al. A Phase 1 Study of Lucatumumab, a Fully Human anti-CD40 Antagonist Monoclonal Antibody Administered Intravenously to Patients With Relapsed or Refractory Multiple Myeloma. Br J Haematol (2012) 159(1):58–66. doi: 10.1111/j.1365-2141.2012.09251.x
92. Byrd JC, Kipps TJ, Flinn IW, Cooper M, Odenike O, Bendiske J, et al. Phase I Study of the anti-CD40 Humanized Monoclonal Antibody Lucatumumab (HCD122) in Relapsed Chronic Lymphocytic Leukemia. Leuk Lymphoma (2012) 53(11):2136–42. doi: 10.3109/10428194.2012.681655
93. Johnson P, Challis R, Chowdhury F, Gao Y, Harvey M, Geldart T, et al. Clinical and Biological Effects of an Agonist Anti-CD40 Antibody: A Cancer Research Uk Phase I Study. Clin Cancer Res (2015) 21(6):1321–8. doi: 10.1158/1078-0432.CCR-14-2355
94. Tseng D, Volkmer JP, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, et al. Anti-CD47 Antibody-Mediated Phagocytosis of Cancer by Macrophages Primes an Effective Antitumor T-cell Response. Proc Natl Acad Sci USA (2013) 110(27):11103–8. doi: 10.1073/pnas.1305569110
95. Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47-signal Regulatory Protein Alpha (Sirpa) Interaction is a Therapeutic Target for Human Solid Tumors. Proc Natl Acad Sci USA (2012) 109(17):6662–7. doi: 10.1158/1078-0432.ccr-07-1443
96. Liu X, Kwon H, Li Z. And Fu, Y.-X., Is CD47 an Innate Immune Checkpoint for Tumor Evasion? J Hematol Oncol (2017) 10(1):12. doi: 10.1186/s13045-016-0381-z
97. Antonioli L, Yegutkin GG, Pacher P, Blandizzi C, Haskó G. Anti-CD73 in Cancer Immunotherapy: Awakening New Opportunities. Trends Cancer (2016) 2(2):95–109. doi: 10.1016/j.trecan.2016.01.003
98. Tap WD, Gelderblom H, Palmerini E, Desai J, Bauer S, Blay J-Y, et al. Pexidartinib Versus Placebo for Advanced Tenosynovial Giant Cell Tumour (ENLIVEN): A Randomised Phase 3 Trial. Lancet (2019) 394(10197):478–87. doi: 10.1016/S0140-6736(19)30764-0
99. Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, et al. Orally Administered Colony Stimulating Factor 1 Receptor Inhibitor PLX3397 in Recurrent Glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium Phase II Study. Neuro Oncol (2016) 18(4):557–64. doi: 10.1093/neuonc/nov245
100. Gomez-Roca CA, Italiano A, Le Tourneau C, Cassier PA, Toulmonde M, D’Angelo SP, et al. Phase I Study of Emactuzumab Single Agent or in Combination With Paclitaxel in Patients With Advanced/Metastatic Solid Tumors Reveals Depletion of Immunosuppressive M2-like Macrophages. Ann Oncol (2019) 30(8):1381–92. doi: 10.1093/annonc/mdz163
101. Papadopoulos KP, Gluck L, Martin LP, Olszanski AJ, Tolcher AW, Ngarmchamnanrith G, et al. First-in-Human Study of AMG 820, a Monoclonal Anti-Colony-Stimulating Factor 1 Receptor Antibody, in Patients With Advanced Solid Tumors. Clin Cancer Res (2017) 23(19):5703–10. doi: 10.1158/1078-0432.CCR-16-3261
102. Felip E, Brunsvig P, Vinolas N, Aix SP, Costa EC, Gomez MD, et al. A Phase II Study of Bemcentinib (BGB324), a First-in-Class Highly Selective AXL Inhibitor, With Pembrolizumab in Pts With Advanced NSCLC: OS for Stage I and Preliminary Stage II Efficacy. J Clin Oncol (2019) 37(15_suppl):9098–8. doi: 10.1200/JCO.2019.37.15_suppl.9098
103. Lorens J, Arce-Lara CE, Arriola E, Brunsvig P, Costa EC, Domine M, et al. Phase II Open-Label, Multi-Centre Study of Bemcentinib (BGB324), a First-in-Class Selective AXL Inhibitor, in Combination With Pembrolizumab in Patients With Advanced NSCLC. J Clin Oncol (2018) 36(15_suppl):3078–8. doi: 10.1200/JCO.2018.36.15_suppl.3078
104. Holland SJ, Pan A, Franci C, Hu Y, Chang B, Li W, et al. R428, a Selective Small Molecule Inhibitor of Axl Kinase, Blocks Tumor Spread and Prolongs Survival in Models of Metastatic Breast Cancer. Cancer Res (2010) 70(4):1544–54. doi: 10.1158/0008-5472.CAN-09-2997
105. Holtzhausen A, Harris W, Ubil E, Hunter DM, Zhao J, Zhang Y, et al. Tam Family Receptor Kinase Inhibition Reverses Mdsc-Mediated Suppression and Augments Anti-Pd-1 Therapy in Melanoma. Cancer Immunol Res (2019) 7(10):1672–86. doi: 10.1158/2326-6066.CIR-19-0008
106. Kasikara C, Davra V, Calianese D, Geng K, Spires TE, Quigley M, et al. Pan-Tam Tyrosine Kinase Inhibitor Bms-777607 Enhances Anti–PD-1 Mab Efficacy in a Murine Model of Triple-Negative Breast Cancer. Cancer Res (2019) 79(10):2669. doi: 10.1158/0008-5472.CAN-18-2614
107. Delanian S, Chatel C, Porcher R, Depondt J, Lefaix JL. Complete Restoration of Refractory Mandibular Osteoradionecrosis by Prolonged Treatment With a Pentoxifylline-Tocopherol-Clodronate Combination (PENTOCLO): A Phase II Trial. Int J Radiat Oncol Biol Phys (2011) 80(3):832–9. doi: 10.1016/j.ijrobp.2010.03.029
108. Delanian SE, Lenglet T, Maisonobe T, Resche-Rigon M, Pradat PF. Randomized, Placebo-Controlled Clinical Trial Combining Pentoxifylline-Tocopherol and Clodronate in the Treatment of Radiation-Induced Plexopathy. Int J Radiat Oncol Biol Phys (2020) 107(1):154–62. doi: 10.1016/j.ijrobp.2020.01.002
109. Ha TC, Li H. Meta-Analysis of Clodronate and Breast Cancer Survival. Br J Cancer (2007) 96(12):1796–801. doi: 10.1038/sj.bjc.6603661
110. Opperman KS, Vandyke K, Clark KC, Coulter EA, Hewett DR, Mrozik KM, et al. Clodronate-Liposome Mediated Macrophage Depletion Abrogates Multiple Myeloma Tumor Establishment In Vivo. Neoplasia (2019) 21(8):777–87. doi: 10.1016/j.neo.2019.05.006
111. Francian A, Mann K, Kullberg M. Complement C3-dependent Uptake of Targeted Liposomes Into Human Macrophages, B Cells, Dendritic Cells, Neutrophils, and Mdscs. Int J Nanomed (2017) 12:5149–61. doi: 10.2147/IJN.S138787
112. Milas L, Mason KA, Ariga H, Hunter N, Neal R, Valdecanas D, et al. Cpg Oligodeoxynucleotide Enhances Tumor Response to Radiation. Cancer Res (2004) 64(15):5074–7. doi: 10.1158/0008-5472.CAN-04-0926
113. Otsuka T, Nishida S, Hamaguchi M, Shibahara T, Shiroyama T, Kimura K, et al. Phase I Study of CpG ODN(K3), a Novel Toll-Like Receptor 9 Agonist, for Advanced Lung Cancer: Interim Analyses of Safety and Immunity in Subcutaneous Injection Cohort. J Clin Oncol (2019) 37(8_suppl):126–6. doi: 10.1200/JCO.2019.37.8_suppl.126
114. Sommariva M, de Cesare M, Meini A, Cataldo A, Zaffaroni N, Tagliabue E, et al. High Efficacy of CpG-ODN, Cetuximab and Cisplatin Combination for Very Advanced Ovarian Xenograft Tumors. J Trans Med (2013) 11(1):25. doi: 10.1186/1479-5876-11-25
115. Finn RS, Bengala C, Ibrahim N, Roché H, Sparano J, Strauss LC, et al. Dasatinib as a Single Agent in Triple-Negative Breast Cancer: Results of an Open-Label Phase 2 Study. Clin Cancer Res (2011) 17(21):6905–13. doi: 10.1158/1078-0432.CCR-11-0288
116. Twardowski PW, Beumer JH, Chen CS, Kraft AS, Chatta GS, Mitsuhashi M, et al. A Phase II Trial of Dasatinib in Patients With Metastatic Castration-Resistant Prostate Cancer Treated Previously With Chemotherapy. Anti-cancer Drugs (2013) 24(7):743–53. doi: 10.1097/CAD.0b013e328361feb0
117. Zhang M, Tian J, Wang R, Song M, Zhao R, Chen H, et al. Dasatinib Inhibits Lung Cancer Cell Growth and Patient Derived Tumor Growth in Mice by Targeting Limk1. Front Cell Dev Biol (2020) 8(1361):4159–8. doi: 10.3389/fcell.2020.556532
118. Ponzoni M, Pastorino F, Di Paolo D, Perri P, Brignole C. Targeting Macrophages as a Potential Therapeutic Intervention: Impact on Inflammatory Diseases and Cancer. Int J Mol Sci (2018) 19(7):1953. doi: 10.3390/ijms19071953
119. Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, et al. Iron Oxide Nanoparticles Inhibit Tumour Growth by Inducing Pro-Inflammatory Macrophage Polarization in Tumour Tissues. Nat Nanotechnol (2016) 11(11):986–94. doi: 10.1038/nnano.2016.168
120. Mody VV, Siwale R, Singh A, Mody HR. Introduction to Metallic Nanoparticles. J Pharm Bioallied Sci (2010) 2(4):282–9. doi: 10.4103/0975-7406.72127
121. Grohmann U, Belladonna ML, Vacca C, Bianchi R, Fallarino F, Orabona C, et al. Positive Regulatory Role of IL-12 in Macrophages and Modulation by IFN-γ. J Immunol (2001) 167(1):221. doi: 10.4049/jimmunol.167.1.221
122. Koneru M, O’Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ. A Phase I Clinical Trial of Adoptive T Cell Therapy Using IL-12 Secreting MUC-16(ecto) Directed Chimeric Antigen Receptors for Recurrent Ovarian Cancer. J Transl Med (2015) 13:102. doi: 10.1186/s12967-015-0460-x
123. Sands BE, Sandborn WJ, Panaccione R, O’Brien CD, Zhang H, Johanns J, et al. Ustekinumab as Induction and Maintenance Therapy for Ulcerative Colitis. N Engl J Med (2019) 381(13):1201–14. doi: 10.1056/NEJMoa1900750
124. Yao Z, Zhang J, Zhang B, Liang G, Chen X, Yao F, et al. Imatinib Prevents Lung Cancer Metastasis by Inhibiting M2-like Polarization of Macrophages. Pharmacol Res (2018) 133:121–31. doi: 10.1016/j.phrs.2018.05.002
125. Blay J-Y, von Mehren M. Nilotinib: A Novel, Selective Tyrosine Kinase Inhibitor. Semin Oncol (2011) 38 Suppl 1(0 1):S3–9. doi: 10.1053/j.seminoncol.2011.01.016
126. Pachman DR, Barton DL, Clayton AC, McGovern RM, Jefferies JA, Novotny PJ, et al. Randomized Clinical Trial of Imiquimod: An Adjunct to Treating Cervical Dysplasia. Am J Obstet Gynecol (2012) 206(1):42.e1–7. doi: 10.1158/1541-7786.mcr-13-0682
127. Rozenblit M, Hendrickx W, Heguy A, Chiriboga L, Loomis C, Ray K, et al. Transcriptomic Profiles Conducive to Immune-Mediated Tumor Rejection in Human Breast Cancer Skin Metastases Treated With Imiquimod. Sci Rep (2019) 9(1):8572. doi: 10.1038/s41598-019-42784-9
128. Salazar LG, Lu H, Reichow JL, Childs JS, Coveler AL, Higgins DM, et al. Topical Imiquimod Plus Nab-paclitaxel for Breast Cancer Cutaneous Metastases: A Phase 2 Clinical Trial. JAMA Oncol (2017) 3(7):969–73. doi: 10.1001/jamaoncol.2016.6007
129. Gilbert SM, Oliphant CJ, Hassan S, Peille AL, Bronsert P, Falzoni S, et al. ATP in the Tumour Microenvironment Drives Expression of nfP2X7, a Key Mediator of Cancer Cell Survival. Oncogene (2019) 38(2):194–208. doi: 10.1038/s41388-018-0426-6
130. De Marchi E, Orioli E, Pegoraro A, Sangaletti S, Portararo P, Curti A, et al. The P2X7 Receptor Modulates Immune Cells Infiltration, Ectonucleotidases Expression and Extracellular ATP Levels in the Tumor Microenvironment. Oncogene (2019) 38(19):3636–50. doi: 10.1038/s41388-019-0684-y
131. Ruiz-Ruiz C, Calzaferri F, García AG. P2x7 Receptor Antagonism as a Potential Therapy in Amyotrophic Lateral Sclerosis. Front Mol Neurosci (2020) 13(93):9517–22. doi: 10.3389/fnmol.2020.00093
132. Brown JR, Walker SR, Heppler LN, Tyekucheva S, Nelson EA, Klitgaard J, et al. Targeting Constitutively Active STAT3 in Chronic Lymphocytic Leukemia: A Clinical Trial of the STAT3 Inhibitor Pyrimethamine With Pharmacodynamic Analyses. Am J Hematol (2020) 4:1–15. doi: 10.1002/ajh.26084
133. Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, et al. AZD9150, a Next-Generation Antisense Oligonucleotide Inhibitor of STAT3 With Early Evidence of Clinical Activity in Lymphoma and Lung Cancer. Sci Trans Med (2015) 7(314):314ra185–314ra185. doi: 10.1126/scitranslmed.aac5272
134. Wang S, Lin Y, Xiong X, Wang L, Guo Y, Chen Y, et al. Low-Dose Metformin Reprograms the Tumor Immune Microenvironment in Human Esophageal Cancer: Results of a Phase Ii Clinical Trial. Clin Cancer Res (2020) 26(18):4921–32. doi: 10.1158/1078-0432.CCR-20-0113
135. Mendoza-Rodríguez MG, Sánchez-Barrera CÁ, Callejas BE, García-Castillo V, Beristain-Terrazas DL, Delgado-Buenrostro NL, et al. Use of STAT6 Phosphorylation Inhibitor and Trimethylglycine as New Adjuvant Therapies for 5-Fluorouracil in Colitis-Associated Tumorigenesis. Int J Mol Sci (2020) 21(6):2130. doi: 10.3390/ijms21062130
136. Tariq M, Zhang JQ, Liang GK, He QJ, Ding L, Yang B. Gefitinib Inhibits M2-like Polarization of Tumor-Associated Macrophages in Lewis Lung Cancer by Targeting the STAT6 Signaling Pathway. Acta Pharmacol Sin (2017) 38(11):1501–11. doi: 10.1038/aps.2017.124
137. Xiao H, Guo Y, Li B, Li X, Wang Y, Han S, et al. M2-Like Tumor-Associated Macrophage-Targeted Codelivery of STAT6 Inhibitor and Ikkβ Sirna Induces M2-to-M1 Repolarization for Cancer Immunotherapy With Low Immune Side Effects. ACS Cent Sci (2020) 6(7):1208–22. doi: 10.1021/acscentsci.9b01235
138. Le Tourneau C, Raymond E, Faivre S. Sunitinib: A Novel Tyrosine Kinase Inhibitor. A Brief Review of its Therapeutic Potential in the Treatment of Renal Carcinoma and Gastrointestinal Stromal Tumors (GIST). Ther Clin Risk Manage (2007) 3(2):341–8. doi: 10.2147/tcrm.2007.3.2.341
139. Zhu W, Xu R, Du J, Fu Y, Li S, Zhang P, et al. Zoledronic Acid Promotes TLR-4-mediated M1 Macrophage Polarization in Bisphosphonate-Related Osteonecrosis of the Jaw. FASEB J (2019) 33(4):5208–19. doi: 10.1096/fj.201801791RR
140. Bercovici N, Guérin MV, Trautmann A, Donnadieu E. The Remarkable Plasticity of Macrophages: A Chance to Fight Cancer. Front Immunol (2019) 10:1563–3. doi: 10.3389/fimmu.2019.01563
141. Cartenì G, Bordonaro R, Giotta F, Lorusso V, Scalone S, Vinaccia V, et al. Efficacy and Safety of Zoledronic Acid in Patients With Breast Cancer Metastatic to Bone: A Multicenter Clinical Trial. Oncologist (2006) 11(7):841–8. doi: 10.1634/theoncologist.11-7-841
142. Okegawa T, Higaki M, Matsumoto T, Kase H, Murata A, Noda K, et al. Zoledronic Acid Improves Clinical Outcomes in Patients With Bone Metastatic Hormone-Naïve Prostate Cancer in a Multicenter Clinical Trial. Anticancer Res (2014) 34(8):4415–20. doi: 10.1016/j.cell.2018.10.001
143. Uemura H, Yanagisawa M, Ikeda I, Fujinami K, Iwasaki A, Noguchi S, et al. Possible Anti-Tumor Activity of Initial Treatment With Zoledronic Acid With Hormonal Therapy for Bone-Metastatic Prostate Cancer in Multicenter Clinical Trial. Int J Clin Oncol (2013) 18(3):472–7. doi: 10.1007/s10147-012-0406-8
144. Zeisberger SM, Odermatt B, Marty C, Zehnder-Fjällman AHM, Ballmer-Hofer K, Schwendener RA. Clodronate-Liposome-Mediated Depletion of Tumour-Associated Macrophages: A New and Highly Effective Antiangiogenic Therapy Approach. Br J Cancer (2006) 95(3):272–81. doi: 10.1038/sj.bjc.6603240
145. Bonapace L, Coissieux M-M, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 Inhibition Accelerates Breast Cancer Metastasis by Promoting Angiogenesis. Nature (2014) 515(7525):130–3. doi: 10.1038/nature13862
146. Hamilton JA. Colony-Stimulating Factors in Inflammation and Autoimmunity. Nat Rev Immunol (2008) 8(7):533–44. doi: 10.1038/nri2356
147. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. Csf-1R Inhibition Alters Macrophage Polarization and Blocks Glioma Progression. Nat Med (2013) 19(10):1264–72. doi: 10.1038/nm.3337
148. Rosenbaum E, Kelly C, D’Angelo SP, Dickson MA, Gounder M, Keohan ML, et al. A Phase I Study of Binimetinib (Mek162) Combined With Pexidartinib (PLX3397) in Patients With Advanced Gastrointestinal Stromal Tumor. Oncol (2019) 24(10):1309–e983. doi: 10.1634/theoncologist.2019-0418
149. von Tresckow B, Morschhauser F, Ribrag V, Topp MS, Chien C, Seetharam S, et al. Multicenter, Phase I/Ii Study of JNJ-40346527, a CSF-1R Inhibitor, in Patients With Relapsed or Refractory Hodgkin Lymphoma. Clin Cancer Res (2015) 21(8):1843–50. doi: 10.1158/1078-0432.CCR-14-1845
150. Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla-Alonso S, et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing Pmn-Mdsc Infiltration of Tumors. Cancer Cell (2017) 32(5):654–68.e5. doi: 10.1016/j.ccell.2017.10.005
151. Borgoni S, Iannello A, Cutrupi S, Allavena P, D’Incalci M, Novelli F, et al. Depletion of Tumor-Associated Macrophages Switches the Epigenetic Profile of Pancreatic Cancer Infiltrating T Cells and Restores Their Anti-Tumor Phenotype. Oncoimmunology (2018) 7(2):e1393596. doi: 10.1080/2162402X.2017.1393596
152. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The Chemokine System in Diverse Forms of Macrophage Activation and Polarization. Trends Immunol (2004) 25(12):677–86. doi: 10.1016/j.it.2004.09.015
153. Davis MJ, Tsang TM, Qiu Y, Dayrit JK, Freij JB, Huffnagle GB, et al. Macrophage M1/M2 Polarization Dynamically Adapts to Changes in Cytokine Microenvironments in Cryptococcus Neoformans Infection. mBio (2013) 4(3):e00264–13. doi: 10.1128/mBio.00264-13
154. Gao CH, Dong HL, Tai L, Gao XM. Lactoferrin-Containing Immunocomplexes Drive the Conversion of Human Macrophages From M2- Into M1-like Phenotype. Front Immunol (2018) 9:37. doi: 10.3389/fimmu.2018.00037
155. Cheng H, Sun L, Shen D, Ren A, Ma F, Tai G, et al. Beta-1,6 Glucan Converts Tumor-Associated Macrophages Into an M1-like Phenotype. Carbohydr Polymers (2020) 247:116715. doi: 10.1016/j.carbpol.2020.116715
156. Dudek AZ, Yunis C, Harrison LI, Kumar S, Hawkinson R, Cooley S, et al. First in Human Phase I Trial of 852A, a Novel Systemic Toll-Like Receptor 7 Agonist, to Activate Innate Immune Responses in Patients With Advanced Cancer. Clin Cancer Res (2007) 13(23):7119–25. doi: 10.1158/1078-0432.CCR-07-1443
157. Dummer R, Hauschild A, Becker JC, Grob JJ, Schadendorf D, Tebbs V, et al. An Exploratory Study of Systemic Administration of the Toll-Like Receptor-7 Agonist 852A in Patients With Refractory Metastatic Melanoma. Clin Cancer Res (2008) 14(3):856–64. doi: 10.1158/1078-0432.CCR-07-1938
158. Vollmer J, Weeratna R, Payette P, Jurk M, Schetter C, Laucht M, et al. Characterization of Three CpG Oligodeoxynucleotide Classes With Distinct Immunostimulatory Activities. Eur J Immunol (2004) 34(1):251–62. doi: 10.1002/eji.200324032
159. Miura K, Ishioka M, Minami S, Horie Y, Ohshima S, Goto T, et al. Toll-Like Receptor 4 on Macrophage Promotes the Development of Steatohepatitis-related Hepatocellular Carcinoma in Mice. J Biol Chem (2016) 291(22):11504–17. doi: 10.1074/jbc.M115.709048
160. Gamrekelashvili J, Kapanadze T, Sablotny S, Ratiu C, Dastagir K, Lochner M, et al. Notch and TLR Signaling Coordinate Monocyte Cell Fate and Inflammation. eLife (2020) 9:e57007. doi: 10.7554/eLife.57007
161. Butcher SK, O’Carroll CE, Wells CA, Carmody RJ. Toll-Like Receptors Drive Specific Patterns of Tolerance and Training on Restimulation of Macrophages. Front Immunol (2018) 9:933. doi: 10.3389/fimmu.2018.00933
162. Shetab Boushehri MA, Lamprecht A. Tlr4-Based Immunotherapeutics in Cancer: A Review of the Achievements and Shortcomings. Mol Pharm (2018) 15(11):4777–800. doi: 10.1021/acs.molpharmaceut.8b00691
163. Kumar S, Ramesh A, Kulkarni A. Targeting Macrophages: A Novel Avenue for Cancer Drug Discovery. Expert Opin Drug Discovery (2020) 15(5):561–74. doi: 10.1080/17460441.2020.1733525
164. Adams S, Kozhaya L, Martiniuk F, Meng T-C, Chiriboga L, Liebes L, et al. Topical TLR7 Agonist Imiquimod can Induce Immune-Mediated Rejection of Skin Metastases in Patients With Breast Cancer. Clin Cancer Res (2012) 18(24):6748–57. doi: 10.1158/1078-0432.CCR-12-1149
165. Sultan H, Salazar AM, Celis E. Poly-ICLC, a Multi-Functional Immune Modulator for Treating Cancer. Semin Immunol (2020) p:101414. doi: 10.1016/j.smim.2020.101414
166. Jahrsdörfer B, Weiner GJ. Cpg Oligodeoxynucleotides as Immunotherapy in Cancer. Update Cancer Ther (2008) 3(1):27–32. doi: 10.1016/j.uct.2007.11.003
167. Heuking S, Rothen-Rutishauser B, Raemy DO, Gehr P, Borchard G. Fate of TLR-1/TLR-2 Agonist Functionalised pDNA Nanoparticles Upon Deposition At the Human Bronchial Epithelium In Vitro. J Nanobiotechnol (2013) 11:29–9. doi: 10.1186/1477-3155-11-29
168. Heidegger S, Gößl D, Schmidt A, Niedermayer S, Argyo C, Endres S, et al. Immune Response to Functionalized Mesoporous Silica Nanoparticles for Targeted Drug Delivery. Nanoscale (2016) 8(2):938–48. doi: 10.1039/C5NR06122A
169. Blanco E, Shen H, Ferrari M. Principles of Nanoparticle Design for Overcoming Biological Barriers to Drug Delivery. Nat Biotechnol (2015) 33(9):941–51. doi: 10.1038/nbt.3330
170. Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, et al. TLR7/8-Agonist-Loaded Nanoparticles Promote the Polarization of Tumour-Associated Macrophages to Enhance Cancer Immunotherapy. Nat BioMed Eng (2018) 2:578–88. doi: 10.1038/s41551-018-0236-8
171. Barberà-Cremades M, Baroja-Mazo A, Pelegrín P. Purinergic Signaling During Macrophage Differentiation Results in M2 Alternative Activated Macrophages. J Leukoc Biol (2016) 99(2):289–99. doi: 10.1189/jlb.1A0514-267RR
172. Zanin RF, Braganhol E, Bergamin LS, Campesato LFI, Filho AZ, Moreira JCF, et al. Differential Macrophage Activation Alters the Expression Profile of NTPDase and Ecto-5’-Nucleotidase. PloS One (2012) 7(2):e31205–5. doi: 10.1371/journal.pone.0031205
173. Dosch M, Zindel J, Jebbawi F, Melin N, Sanchez-Taltavull D, Stroka D, et al. Connexin-43-Dependent ATP Release Mediates Macrophage Activation During Sepsis. Elife (2019) 8:321–34. doi: 10.7554/eLife.42670
174. Martins I, Tesniere A, Kepp O, Michaud M, Schlemmer F, Senovilla L, et al. Chemotherapy Induces ATP Release From Tumor Cells. Cell Cycle (2009) 8(22):3723–8. doi: 10.4161/cc.8.22.10026
175. Feng L-l, Cai Y-q, Zhu M-c, Xing L-j, Wang X. The Yin and Yang Functions of Extracellular ATP and Adenosine in Tumor Immunity. Cancer Cell Int (2020) 20(1):110. doi: 10.1186/s12935-020-01195-x
176. Bouma MG, Stad RK, van den Wildenberg FA, Buurman WA. Differential Regulatory Effects of Adenosine on Cytokine Release by Activated Human Monocytes. J Immunol (1994) 153(9):4159–68. doi: 10.18632/oncotarget.16547
177. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic Cell Death and DAMPs in Cancer Therapy. Nat Rev Cancer (2012) 12(12):860–75. doi: 10.1038/nrc3380
178. Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, et al. Targeting CD73 in the Tumor Microenvironment With MEDI9447. OncoImmunology (2016) 5(8):e1208875. doi: 10.1080/2162402X.2016.1208875
179. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. Pi3kγ is a Molecular Switch That Controls Immune Suppression. Nature (2016) 539(7629):437–42. doi: 10.1038/nature19834
180. Amano MT, Castoldi A, Andrade-Oliveira V, Latancia MT, Terra FF, Correa-Costa M, et al. The Lack of PI3Kγ Favors M1 Macrophage Polarization and Does Not Prevent Kidney Diseases Progression. Int Immunopharmacol (2018) 64:151–61. doi: 10.1016/j.intimp.2018.08.020
181. Lin F, Yin HB, Li XY, Zhu GM, He WY, Gou X. Bladder Cancer Cell−Secreted Exosomal miR−21 Activates the PI3K/AKT Pathway in Macrophages to Promote Cancer Progression. Int J Oncol (2020) 56(1):151–64. doi: 10.3892/ijo.2019.4933
182. Zhao S-J, Kong F-Q, Jie J, Li Q, Liu H, Xu A-D, et al. Macrophage MSR1 Promotes BMSC Osteogenic Differentiation and M2-like Polarization by Activating PI3K/AKT/GSK3β/β-Catenin Pathway. Theranostics (2020) 10(1):17–35. doi: 10.7150/thno.36930
183. Joshi S, Singh AR, Zulcic M, Durden DL. A Macrophage-Dominant Pi3k Isoform Controls Hypoxia-Induced Hif1α and HIF2α Stability and Tumor Growth, Angiogenesis, and Metastasis. Mol Cancer Res (2014) 12(10):1520–31. doi: 10.1158/1541-7786.MCR-13-0682
184. Attri KS, Mehla K, Singh PK. Evaluation of Macrophage Polarization in Pancreatic Cancer Microenvironment Under Hypoxia. In: Huang LE, editor. Hypoxia: Methods and Protocols. New York, NY: Springer New York (2018). p. 265–76.
185. Guo X, Xue H, Shao Q, Wang J, Guo X, Zhang J, et al. Hypoxia Promotes Glioma-Associated Macrophage Infiltration Via Periostin and Subsequent M2 Polarization by Upregulating TGF-beta and M-CSFR. Oncotarget (2016) 7(49):1129–38. doi: 10.18632/oncotarget.11825
186. Qi L, Chen J, Yang Y, Hu W. Hypoxia Correlates With Poor Survival and M2 Macrophage Infiltration in Colorectal Cancer. Front Oncol (2020) 10(2491):312–22. doi: 10.3389/fonc.2020.566430
187. Usman MW, Gao J, Zheng T, Rui C, Li T, Bian X, et al. Macrophages Confer Resistance to PI3K Inhibitor GDC-0941 in Breast Cancer Through the Activation of NF-κb Signaling. Cell Death Dis (2018) 9(8):809–9. doi: 10.1038/s41419-018-0849-6
188. Arranz A, Doxaki C, Vergadi E, Martinez de la Torre Y, Vaporidi K, Lagoudaki ED, et al. Akt1 and Akt2 Protein Kinases Differentially Contribute to Macrophage Polarization. Proc Natl Acad Sci (2012) 109(24):9517–22. doi: 10.1073/pnas.1119038109
189. Byles V, Covarrubias AJ, Ben-Sahra I, Lamming DW, Sabatini DM, Manning BD, et al. The TSC-mTOR Pathway Regulates Macrophage Polarization. Nat Commun (2013) 4(1):2834. doi: 10.1038/ncomms3834
190. Ohmori Y, Hamilton TA. Requirement for STAT1 in LPS-induced Gene Expression in Macrophages. J Leukoc Biol (2001) 69(4):598–604. doi: 10.1111/j.1365-2141.2012.09251.x
191. Mu X, Shi W, Xu Y, Xu C, Zhao T, Geng B, et al. Tumor-Derived Lactate Induces M2 Macrophage Polarization Via the Activation of the ERK/STAT3 Signaling Pathway in Breast Cancer. Cell Cycle (2018) 17(4):428–38. doi: 10.1080/15384101.2018.1444305
192. Zhao Y, Yu Z, Ma R, Zhang Y, Zhao L, Yan Y, et al. Lncrna-Xist/Mir-101-3p/KLF6-C/Ebpα Axis Promotes TAMs Polarization to Regulate Cancer Cells Proliferation and Migration. Mol Ther Nucleic Acids (2020) 21:1321–8. doi: 10.1016/j.omtn.2020.12.005
193. Elloumi HZ, Maharshak N, Rao KN, Kobayashi T, Ryu HS, Mühlbauer M, et al. A Cell Permeable Peptide Inhibitor of NFAT Inhibits Macrophage Cytokine Expression and Ameliorates Experimental Colitis. PloS One (2012) 7(3):e34172. doi: 10.1371/journal.pone.0034172
194. Tripathi MK, Deane NG, Zhu J, An H, Mima S, Wang X, et al. Nuclear Factor of Activated T-cell Activity is Associated With Metastatic Capacity in Colon Cancer. Cancer Res (2014) 74(23):6947–57. doi: 10.1158/0008-5472.CAN-14-1592
195. Tran Quang C, Leboucher S, Passaro D, Fuhrmann L, Nourieh M, Vincent-Salomon A, et al. The Calcineurin/NFAT Pathway is Activated in Diagnostic Breast Cancer Cases and is Essential to Survival and Metastasis of Mammary Cancer Cells. Cell Death Dis (2015) 6(2):e1658–8. doi: 10.1038/cddis.2015.14
196. Colegio OR, Chu N-Q, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional Polarization of Tumour-Associated Macrophages by Tumour-Derived Lactic Acid. Nature (2014) 513(7519):559–63. doi: 10.1038/nature13490
197. Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, et al. Immune-Responsive Gene 1 Protein Links Metabolism to Immunity by Catalyzing Itaconic Acid Production. Proc Natl Acad Sci (2013) 110(19):7820–5. doi: 10.1073/pnas.1218599110
198. Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell (2018) 175(7):1780–95.e19. doi: 10.1200/jco.2019.37.15_suppl.9098
199. Chang CI, Liao JC, Kuo L. Macrophage Arginase Promotes Tumor Cell Growth and Suppresses Nitric Oxide-Mediated Tumor Cytotoxicity. Cancer Res (2001) 61(3):1100–6. doi: 10.1200/jco.2018.36.15_suppl.3078
200. Kung JT, Brooks SB, Jakway JP, Leonard LL, Talmage DW. Suppression of In Vitro Cytotoxic Response by Macrophages Due to Induced Arginase. J Exp Med (1977) 146(3):665–72. doi: 10.1084/jem.146.3.665
201. Hardbower DM, Asim M, Luis PB, Singh K, Barry DP, Yang C, et al. Ornithine Decarboxylase Regulates M1 Macrophage Activation and Mucosal Inflammation Via Histone Modifications. Proc Natl Acad Sci (2017) 114(5):E751–60. doi: 10.1073/pnas.1614958114
202. Sahin E, Haubenwallner S, Kuttke M, Kollmann I, Halfmann A, Dohnal AB, et al. Macrophage PTEN Regulates Expression and Secretion of Arginase I Modulating Innate and Adaptive Immune Responses. J Immunol (2014) 193(4):1717–27. doi: 10.4049/jimmunol.1302167
203. de Boniface J, Mao Y, Schmidt-Mende J, Kiessling R, Poschke I. Expression Patterns of the Immunomodulatory Enzyme Arginase 1 in Blood, Lymph Nodes and Tumor Tissue of Early-Stage Breast Cancer Patients. Oncoimmunology (2012) 1(8):1305–12. doi: 10.4161/onci.21678
204. Yurdagul A, Jr, Subramanian M, Wang X, Crown SB, Ilkayeva OR, Darville L, et al. Macrophage Metabolism of Apoptotic Cell-Derived Arginine Promotes Continual Efferocytosis and Resolution of Injury. Cell Metab (2020) 31(3):518–33.e10. doi: 10.1016/j.ijrobp.2010.03.029
205. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I Production in the Tumor Microenvironment by Mature Myeloid Cells Inhibits T-Cell Receptor Expression and Antigen-Specific T-Cell Responses. Cancer Res (2004) 64(16):5839–49. doi: 10.1158/0008-5472.CAN-04-0465
206. Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. Inhibition of Arginase by CB-1158 Blocks Myeloid Cell-Mediated Immune Suppression in the Tumor Microenvironment. J ImmunoTher Cancer (2017) 5(1):101. doi: 10.1186/s40425-017-0308-4
207. Labadie BW, Bao R, Luke JJ. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy Via Downstream Focus on the Tryptophan–Kynurenine–Aryl Hydrocarbon Axis. Clin Cancer Res (2019) 25(5):1462–71. doi: 10.1158/1078-0432.CCR-18-2882
208. Miyasato Y, Takashima Y, Takeya H, Yano H, Hayano A, Nakagawa T, et al. The Expression of PD-1 Ligands and IDO1 by Macrophage/Microglia in Primary Central Nervous System Lymphoma. J Clin Exp Hematopathol JCEH (2018) 58(2):95–101. doi: 10.3960/jslrt.18001
209. Su S, Zhao J, Xing Y, Zhang X, Liu J, Ouyang Q, et al. Immune Checkpoint Inhibition Overcomes Adcp-Induced Immunosuppression by Macrophages. Cell (2018) 175(2):442–57.e23. doi: 10.1200/jco.2019.37.8_suppl.126
210. Toulmonde M, Penel N, Adam J, Chevreau C, Blay J-Y, Le Cesne A, et al. Use of PD-1 Targeting, Macrophage Infiltration, and IDO Pathway Activation in Sarcomas: A Phase 2 Clinical Trial. JAMA Oncol (2018) 4(1):93–7. doi: 10.1001/jamaoncol.2017.1617
211. Mei J, Chang KK, Sun HX. Immunosuppressive Macrophages Induced by IDO1 Promote the Growth of Endometrial Stromal Cells in Endometriosis. Mol Med Rep (2017) 15(4):2255–60. doi: 10.3892/mmr.2017.6242
212. Jiang N, Zhang L, Zhao G, Lin J, Wang Q, Xu Q, et al. Indoleamine 2,3-Dioxygenase Regulates Macrophage Recruitment, Polarization and Phagocytosis in Aspergillus Fumigatus Keratitis. Invest Ophthalmol Visual Sci (2020) 61(8):28–8. doi: 10.1167/iovs.61.8.28
213. Hou D-Y, Muller AJ, Sharma MD, DuHadaway J, Banerjee T, Johnson M, et al. Inhibition of Indoleamine 2,3-Dioxygenase in Dendritic Cells by Stereoisomers of 1-Methyl-Tryptophan Correlates With Antitumor Responses. Cancer Res (2007) 67(2):792–801. doi: 10.1158/0008-5472.CAN-06-2925
214. Metz R, Rust S, DuHadaway JB, Mautino MR, Munn DH, Vahanian NN, et al. IDO Inhibits a Tryptophan Sufficiency Signal That Stimulates Mtor: A Novel IDO Effector Pathway Targeted by D-1-Methyl-Tryptophan. OncoImmunology (2012) 1(9):1460–8. doi: 10.4161/onci.21716
215. Prieto AL, Weber JL, Tracy S, Heeb MJ, Lai C. Gas6, a Ligand for the Receptor Protein-Tyrosine Kinase Tyro-3, is Widely Expressed in the Central Nervous System. Brain Res (1999) 816(2):646–61. doi: 10.1016/S0006-8993(98)01159-7
216. Al Kafri N, Hafizi S, Tumour-Secreted Protein S. (Pros1) Activates a Tyro3-Erk Signalling Axis and Protects Cancer Cells From Apoptosis. Cancers (Basel) (2019) 11(12):282–9. doi: 10.3390/cancers11121843
217. Al Kafri N, Hafizi S. Galectin-3 Stimulates Tyro3 Receptor Tyrosine Kinase and Erk Signalling, Cell Survival and Migration in Human Cancer Cells. Biomolecules (2020) 10(7):1035. doi: 10.3390/biom10071035
218. Caberoy NB, Zhou Y, Li W. Tubby and Tubby-Like Protein 1 are New MerTK Ligands for Phagocytosis. EMBO J (2010) 29(23):3898–910. doi: 10.1038/emboj.2010.265
219. Lew ED, Oh J, Burrola PG, Lax I, Zagórska A, Través PG, et al. Differential TAM Receptor–Ligand–Phospholipid Interactions Delimit Differential TAM Bioactivities. eLife (2014) 3:e03385. doi: 10.7554/eLife.03385
220. Sandahl M, Hunter DM, Strunk KE, Earp HS, Cook RS. Epithelial Cell-Directed Efferocytosis in the Post-Partum Mammary Gland is Necessary for Tissue Homeostasis and Future Lactation. BMC Dev Biol (2010) 10(1):122. doi: 10.1186/1471-213X-10-122
221. Nishi C, Yanagihashi Y, Segawa K, Nagata S. MERTK Tyrosine Kinase Receptor Together With TIM4 Phosphatidylserine Receptor Mediates Distinct Signal Transduction Pathways for Efferocytosis and Cell Proliferation. J Biol Chem (2019) 294(18):7221–30. doi: 10.1074/jbc.RA118.006628
222. Camenisch TD, Koller BH, Earp HS, Matsushima GK. A Novel Receptor Tyrosine Kinase, Mer, Inhibits TNF-alpha Production and Lipopolysaccharide-Induced Endotoxic Shock. J Immunol (1999) 162(6):3498–503. doi: 10.1016/j.ajog.2011.06.105
223. Guttridge KL, Luft JC, Dawson TL, Kozlowska E, Mahajan NP, Varnum B, et al. Mer Receptor Tyrosine Kinase Signaling: Prevention of Apoptosis and Alteration of Cytoskeletal Architecture Without Stimulation or Proliferation. J Biol Chem (2002) 277(27):24057–66. doi: 10.1074/jbc.M112086200
224. Rothlin CV, Ghosh S, Zuniga EI, Oldstone MBA, Lemke G. Tam Receptors are Pleiotropic Inhibitors of the Innate Immune Response. Cell (2007) 131(6):1124–36. doi: 10.1016/j.cell.2007.10.034
225. Cook RS, Jacobsen KM, Wofford AM, DeRyckere D, Stanford J, Prieto AL, et al. MerTK Inhibition in Tumor Leukocytes Decreases Tumor Growth and Metastasis. J Clin Invest (2013) 123(8):3231–42. doi: 10.1172/JCI67655
226. Wen ZF, Liu H, Gao R, Zhou M, Ma J, Zhang Y, et al. Tumor Cell-Released Autophagosomes (Traps) Promote Immunosuppression Through Induction of M2-like Macrophages With Increased Expression of PD-L1. J Immunother Cancer (2018) 6(1):151. doi: 10.1186/s40425-018-0452-5
227. Tang J, Pearce L, O’Donnell-Tormey J, Hubbard-Lucey VM. Trends in the Global Immuno-Oncology Landscape. Nat Rev Drug Discovery (2018) 17(11):783–4. doi: 10.1038/nrd.2018.167
228. Xiong H, Mittman S, Rodriguez R, Moskalenko M, Pacheco-Sanchez P, Yang Y, et al. Anti-Pd-L1 Treatment Results in Functional Remodeling of the Macrophage Compartment. Cancer Res (2019) 79(7):1493–506. doi: 10.1158/0008-5472.CAN-18-3208
229. Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-Surface Calreticulin Initiates Clearance of Viable or Apoptotic Cells Through trans-Activation of LRP on the Phagocyte. Cell (2005) 123(2):321–34. doi: 10.1016/j.cell.2005.08.032
230. Weiskopf K, Jahchan NS, Schnorr PJ, Cristea S, Ring AM, Maute RL, et al. CD47-Blocking Immunotherapies Stimulate Macrophage-Mediated Destruction of Small-Cell Lung Cancer. J Clin Invest (2016) 126(7):2610–20. doi: 10.1172/JCI81603
231. Tsai RK, Discher DE. Inhibition of “Self” Engulfment Through Deactivation of myosin-II At the Phagocytic Synapse Between Human Cells. J Cell Biol (2008) 180(5):989–1003. doi: 10.1083/jcb.200708043
232. Liu R, Wei H, Gao P, Yu H, Wang K, Fu Z, et al. CD47 Promotes Ovarian Cancer Progression by Inhibiting Macrophage Phagocytosis. Oncotarget (2017) 8(24):341–48. doi: 10.18632/oncotarget.16547
233. Manna PP, Frazier WA. CD47 Mediates Killing of Breast Tumor Cells Via Gi-dependent Inhibition of Protein Kinase a. Cancer Res (2004) 64(3):1026–36. doi: 10.1158/0008-5472.CAN-03-1708
234. Yang H, Shao R, Huang H, Wang X, Rong Z, Lin Y. Engineering Macrophages to Phagocytose Cancer Cells by Blocking the CD47/SIRPа Axis. Cancer Med (2019) 8(9):4245–53. doi: 10.1002/cam4.2332
235. Chen G-Y, Chen X, King S, Cavassani KA, Cheng J, Zheng X, et al. Amelioration of Sepsis by Inhibiting Sialidase-Mediated Disruption of the CD24-SiglecG Interaction. Nat Biotechnol (2011) 29(5):428–35. doi: 10.1038/nbt.1846
236. Chen G-Y, Tang J, Zheng P, Liu Y. CD24 and Siglec-10 Selectively Repress Tissue Damage–Induced Immune Responses. Science (2009) 323(5922):1722–5. doi: 10.1126/science.1168988
237. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J Immunol (2000) 164(12):6166–73. doi: 10.4049/jimmunol.164.12.6166
238. Martinez FO, Gordon S. The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment. F1000Prime Rep (2014) 6:13. doi: 10.12703/P6-13
239. Nau GJ, Richmond JFL, Schlesinger A, Jennings EG, Lander ES, Young RA. Human Macrophage Activation Programs Induced by Bacterial Pathogens. Proc Natl Acad Sci USA (2002) 99(3):1503–8. doi: 10.1073/pnas.022649799
240. Blériot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M. Liver-Resident Macrophage Necroptosis Orchestrates Type 1 Microbicidal Inflammation and type-2-mediated Tissue Repair During Bacterial Infection. Immunity (2015) 42(1):145–58. doi: 10.1016/j.immuni.2014.12.020
241. Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, et al. The Jmjd3-Irf4 Axis Regulates M2 Macrophage Polarization and Host Responses Against Helminth Infection. Nat Immunol (2010) 11(10):936–44. doi: 10.1038/ni.1920
242. Ji Y, Sun S, Xu A, Bhargava P, Yang L, Lam KSL, et al. Activation of Natural Killer T Cells Promotes M2 Macrophage Polarization in Adipose Tissue and Improves Systemic Glucose Tolerance Via Interleukin-4 (Il-4)/Stat6 Protein Signaling Axis in Obesity. J Biol Chem (2012) 287(17):13561–71. doi: 10.1074/jbc.M112.350066
243. Koh TJ, DiPietro LA. Inflammation and Wound Healing: The Role of the Macrophage. Expert Rev Mol Med (2011) 13:e23. doi: 10.1017/S1462399411001943
244. Wang LX, Zhang SX, Wu HJ, Rong XL, Guo J. M2b Macrophage Polarization and its Roles in Diseases. J Leukoc Biol (2019) 106(2):345–58. doi: 10.1002/JLB.3RU1018-378RR
245. Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted Disruption of the Mouse Stat1 Gene Results in Compromised Innate Immunity to Viral Disease. Cell (1996) 84(3):443–50. doi: 10.1016/S0092-8674(00)81289-1
246. Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, et al. Targeted Disruption of the Stat1 Gene in Mice Reveals Unexpected Physiologic Specificity in the JAK-STAT Signaling Pathway. Cell (1996) 84(3):431–42. doi: 10.1016/S0092-8674(00)81288-X
247. Ait-Lounis A, Laraba-Djebari F. TNF-Alpha Modulates Adipose Macrophage Polarization to M1 Phenotype in Response to Scorpion Venom. Inflammation Res (2015) 64(11):929–36. doi: 10.1007/s00011-015-0876-z
248. Campbell J, Ciesielski CJ, Hunt AE, Horwood NJ, Beech JT, Hayes LA, et al. A Novel Mechanism for TNF-alpha Regulation by P38 MAPK: Involvement of NF-Kappa B With Implications for Therapy in Rheumatoid Arthritis. J Immunol (2004) 173(11):6928–37. doi: 10.4049/jimmunol.173.11.6928
249. Waetzig GH, Seegert D, Rosenstiel P, Nikolaus S, Schreiber S. p38 Mitogen-Activated Protein Kinase is Activated and Linked to TNF-alpha Signaling in Inflammatory Bowel Disease. J Immunol (2002) 168(10):5342–51. doi: 10.4049/jimmunol.168.10.5342
250. Brinkman BM, Telliez JB, Schievella AR, Lin LL, Goldfeld AE. Engagement of Tumor Necrosis Factor (TNF) Receptor 1 Leads to ATF-2- and p38 Mitogen-Activated Protein Kinase-Dependent TNF-alpha Gene Expression. J Biol Chem (1999) 274(43):30882–6. doi: 10.1074/jbc.274.43.30882
251. Chiang CF, Chao TT, Su YF, Hsu CC, Chien CY, Chiu KC, et al. Metformin-Treated Cancer Cells Modulate Macrophage Polarization Through AMPK-NF-κb Signaling. Oncotarget (2017) 8(13):20706–18. doi: 10.18632/oncotarget.14982
252. Yang Y, Wang X, Moore DR, Lightfoot SA, Huycke MM. Tnf-α Mediates Macrophage-Induced Bystander Effects Through Netrin-1. Cancer Res (2012) 72(20):5219–29. doi: 10.1158/0008-5472.CAN-12-1463
253. Lukic A, Larssen P, Fauland A, Samuelsson B, Wheelock CE, Gabrielsson S, et al. Gm-CSF- and M-CSF-primed Macrophages Present Similar Resolving But Distinct Inflammatory Lipid Mediator Signatures. FASEB J (2017) 31(10):4370–81. doi: 10.1096/fj.201700319R
254. Cao W, Peters JH, Nieman D, Sharma M, Watson T, Yu J. Macrophage Subtype Predicts Lymph Node Metastasis in Oesophageal Adenocarcinoma and Promotes Cancer Cell Invasion In Vitro. Br J Cancer (2015) 113(5):738–46. doi: 10.1038/bjc.2015.292
255. Wang Q, Ni H, Lan L, Wei X, Xiang R, Wang Y. Fra-1 Protooncogene Regulates IL-6 Expression in Macrophages and Promotes the Generation of M2d Macrophages. Cell Res (2010) 20(6):701–12. doi: 10.1038/cr.2010.52
256. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 Potently Enhances Murine Macrophage Mannose Receptor Activity: A Marker of Alternative Immunologic Macrophage Activation. J Exp Med (1992) 176(1):287–92. doi: 10.1084/jem.176.1.287
257. Jetten N, Verbruggen S, Gijbels MJ, Post MJ, De Winther MP, Donners MM. Anti-Inflammatory M2, But Not Pro-Inflammatory M1 Macrophages Promote Angiogenesis In Vivo. Angiogenesis (2014) 17(1):109–18. doi: 10.1007/s10456-013-9381-6
258. Shiratori H, Feinweber C, Luckhardt S, Linke B, Resch E, Geisslinger G, et al. THP-1 and Human Peripheral Blood Mononuclear Cell-Derived Macrophages Differ in Their Capacity to Polarize In Vitro. Mol Immunol (2017) 88:58–68. doi: 10.1016/j.molimm.2017.05.027
259. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages That Have Ingested Apoptotic Cells In Vitro Inhibit Proinflammatory Cytokine Production Through Autocrine/Paracrine Mechanisms Involving TGF-beta, PGE2, and PAF. J Clin Invest (1998) 101(4):890–8. doi: 10.1172/JCI1112
260. Atri C, Guerfali FZ, Laouini D. Role of Human Macrophage Polarization in Inflammation During Infectious Diseases. Int J Mol Sci (2018) 19(6):890–8. doi: 10.3390/ijms19061801
261. Lai YS, Putra R, Aui SP, Chang KT. M2(C) Polarization by Baicalin Enhances Efferocytosis Via Upregulation of MERTK Receptor. Am J Chin Med (2018) 46(8):1899–914. doi: 10.1142/S0192415X18500957
262. Huang X, Li Y, Fu M, Xin HB. Polarizing Macrophages In Vitro. Methods Mol Biol (2018) 1784:119–26. doi: 10.1007/978-1-4939-7837-3_12
263. Benner B, Scarberry L, Suarez-Kelly LP, Duggan MC, Campbell AR, Smith E, et al. Generation of Monocyte-Derived Tumor-Associated Macrophages Using Tumor-Conditioned Media Provides a Novel Method to Study Tumor-Associated Macrophages In Vitro. J ImmunoTher Cancer (2019) 7(1):140. doi: 10.1186/s40425-019-0622-0
264. Binnemars-Postma K, Bansal R, Storm G, Prakash J. Targeting the Stat6 Pathway in Tumor-Associated Macrophages Reduces Tumor Growth and Metastatic Niche Formation in Breast Cancer. FASEB J (2018) 32(2):969–78. doi: 10.1096/fj.201700629R
265. Chung W, Eum HH, Lee H-O, Lee K-M, Lee H-B, Kim K-T, et al. Single-Cell RNA-seq Enables Comprehensive Tumour and Immune Cell Profiling in Primary Breast Cancer. Nat Commun (2017) 8:15081. doi: 10.1038/ncomms15081
266. He D, Wang D, Lu P, Yang N, Xue Z, Zhu X, et al. Single-Cell RNA Sequencing Reveals Heterogeneous Tumor and Immune Cell Populations in Early-Stage Lung Adenocarcinomas Harboring EGFR Mutations. Oncogene (2020) 8:1–12. doi: 10.1038/s41388-020-01528-0
267. Yu X, Chen YA, Conejo-Garcia JR, Chung CH, Wang X. Estimation of Immune Cell Content in Tumor Using Single-Cell RNA-seq Reference Data. BMC Cancer (2019) 19(1):715. doi: 10.1186/s12885-019-5927-3
268. Valeta-Magara A, Gadi A, Volta V, Walters B, Arju R, Giashuddin S, et al. Inflammatory Breast Cancer Promotes Development of M2 Tumor-Associated Macrophages and Cancer Mesenchymal Cells Through a Complex Chemokine Network. Cancer Res (2019) 79(13):3360–71. doi: 10.1158/0008-5472.CAN-17-2158
269. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional Profiling of the Human Monocyte-to-Macrophage Differentiation and Polarization: New Molecules and Patterns of Gene Expression. J Immunol (2006) 177(10):7303–11. doi: 10.4049/jimmunol.177.10.7303
270. Singh JK, Simões BM, Howell SJ, Farnie G, Clarke RB. Recent Advances Reveal IL-8 Signaling as a Potential Key to Targeting Breast Cancer Stem Cells. Breast Cancer Res (2013) 15(4):210. doi: 10.1186/bcr3436
271. Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell (2018) 174(5):1293–308.e36. doi: 10.1186/bcr3436
272. Klein, Allon M, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, et al. Droplet Barcoding for Single-Cell Transcriptomics Applied to Embryonic Stem Cells. Cell (2015) 161(5):1187–201. doi: 10.1016/j.cell.2015.04.044
273. Zilionis R, Nainys J, Veres A, Savova V, Zemmour D, Klein AM, et al. Single-Cell Barcoding and Sequencing Using Droplet Microfluidics. Nat Protoc (2017) 12(1):44–73. doi: 10.1038/nprot.2016.154
274. Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, et al. Single-Cell Profiling of Human Gliomas Reveals Macrophage Ontogeny as a Basis for Regional Differences in Macrophage Activation in the Tumor Microenvironment. Genome Biol (2017) 18(1):234. doi: 10.1186/s13059-017-1362-4
275. Allard B, Panariti A, Martin JG. Alveolar Macrophages in the Resolution of Inflammation, Tissue Repair, and Tolerance to Infection. Front Immunol (2018) 9(1777):1–14. doi: 10.3389/fimmu.2018.01777
276. Mosser DM, Hamidzadeh K, Goncalves R. Macrophages and the Maintenance of Homeostasis. Cell Mol Immunol (2020) 9:1–7. doi: 10.1038/s41423-020-00541-3
277. Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and Analysis of Gene Expression in Tissue Sections by Spatial Transcriptomics. Science (2016) 353(6294):78–82. doi: 10.1126/science.aaf2403
278. Moncada R, Barkley D, Wagner F, Chiodin M, Devlin JC, Baron M, et al. Integrating Microarray-Based Spatial Transcriptomics and Single-Cell RNA-seq Reveals Tissue Architecture in Pancreatic Ductal Adenocarcinomas. Nat Biotechnol (2020) 38(3):333–42. doi: 10.1038/s41587-019-0392-8
279. Chen C, He W, Huang J, Wang B, Li H, Cai Q, et al. LNMAT1 Promotes Lymphatic Metastasis of Bladder Cancer Via CCL2 Dependent Macrophage Recruitment. Nat Commun (2018) 9(1):3826. doi: 10.1038/s41467-018-06152-x
280. De la Fuente López M, Landskron G, Parada D, Dubois-Camacho K, Simian D, Martinez M, et al. The Relationship Between Chemokines CCL2, CCL3, and CCL4 With the Tumor Microenvironment and Tumor-Associated Macrophage Markers in Colorectal Cancer. Tumour Biol (2018) 40(11):1010428318810059. doi: 10.1177/1010428318810059
281. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 Recruits Inflammatory Monocytes to Facilitate Breast-Tumour Metastasis. Nature (2011) 475(7355):222–5. doi: 10.1038/nature10138
282. Lim SY, Yuzhalin AE, Gordon-Weeks AN, Muschel RJ. Tumor-Infiltrating Monocytes/Macrophages Promote Tumor Invasion and Migration by Upregulating S100A8 and S100A9 Expression in Cancer Cells. Oncogene (2016) 35(44):5735–45. doi: 10.1038/onc.2016.107
283. Song Q, Hawkins GA, Wudel L, Chou PC, Forbes E, Pullikuth AK, et al. Dissecting Intratumoral Myeloid Cell Plasticity by Single Cell RNA-Seq. Cancer Med (2019) 8(6):3072–85. doi: 10.1002/cam4.2113
284. Abdalla SI, Sanderson IR, Fitzgerald RC. Effect of Inflammation on Cyclooxygenase (COX)-2 Expression in Benign and Malignant Oesophageal Cells. Carcinogenesis (2005) 26(9):1627–33. doi: 10.1093/carcin/bgi114
285. Perry CJ, Muñoz-Rojas AR, Meeth KM, Kellman LN, Amezquita RA, Thakral D, et al. Myeloid-Targeted Immunotherapies Act in Synergy to Induce Inflammation and Antitumor Immunity. J Exp Med (2018) 215(3):877–93. doi: 10.1084/jem.20171435
286. Takada Y, Kobayashi Y, Aggarwal BB. Evodiamine Abolishes Constitutive and Inducible Nf-κb Activation by Inhibiting Iκbα Kinase Activation, Thereby Suppressing Nf-κb-Regulated Antiapoptotic and Metastatic Gene Expression, Up-Regulating Apoptosis, and Inhibiting Invasion*. J Biol Chem (2005) 280(17):17203–12. doi: 10.1074/jbc.M500077200
287. Kiraly O, Gong G, Olipitz W, Muthupalani S, Engelward BP. Inflammation-Induced Cell Proliferation Potentiates Dna Damage-Induced Mutations In Vivo. PloS Genet (2015) 11(2):e1004901. doi: 10.1371/journal.pgen.1004901
288. Iyengar NM, Gucalp A, Dannenberg AJ, Hudis CA. Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation. J Clin Oncol (2016) 34(35):4270–6. doi: 10.1200/JCO.2016.67.4283
Keywords: cancer, macrophage, plasticity, therapy, tumor, inflammation
Citation: Ricketts TD, Prieto-Dominguez N, Gowda PS and Ubil E (2021) Mechanisms of Macrophage Plasticity in the Tumor Environment: Manipulating Activation State to Improve Outcomes. Front. Immunol. 12:642285. doi: 10.3389/fimmu.2021.642285
Received: 15 December 2020; Accepted: 16 April 2021;
Published: 07 May 2021.
Edited by:
Jose A. Garcia-Sanz, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Alexandre Corthay, Oslo University Hospital, NorwayGuilan Shi, University of South Florida, United States
Michael Rückert, University Hospital Erlangen, Germany
Copyright © 2021 Ricketts, Prieto-Dominguez, Gowda and Ubil. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric Ubil, ZXJpY3ViaWxAdWFiLmVkdQ==