 Charu Rajput†‡
Charu Rajput†‡ Mingyuan Han‡Tomoko IshikawaJing LeiAdam M. Goldsmith
Mingyuan Han‡Tomoko IshikawaJing LeiAdam M. Goldsmith Seyedehzarifeh JazaeriClaudia C. StroupeJ. Kelley Bentley
Seyedehzarifeh JazaeriClaudia C. StroupeJ. Kelley Bentley Marc B. Hershenson*
Marc B. Hershenson*- Department of Pediatrics, University of Michigan Medical School, Ann Arbor, MI, United States
Rhinovirus C (RV-C) infection is associated with severe asthma exacerbations. Since type 2 inflammation is an important disease mechanism in asthma, we hypothesized that RV-C infection, in contrast to RV-A, preferentially stimulates type 2 inflammation, leading to exacerbated eosinophilic inflammation. To test this, we developed a mouse model of RV-C15 airways disease. RV-C15 was generated from the full-length cDNA clone and grown in HeLa-E8 cells expressing human CDHR3. BALB/c mice were inoculated intranasally with 5 x 106 ePFU RV-C15, RV-A1B or sham. Mice inoculated with RV-C15 showed lung viral titers of 1 x 105 TCID50 units 24 h after infection, with levels declining thereafter. IFN-α, β, γ and λ2 mRNAs peaked 24-72 hrs post-infection. Immunofluorescence verified colocalization of RV-C15, CDHR3 and acetyl-α-tubulin in mouse ciliated airway epithelial cells. Compared to RV-A1B, mice infected with RV-C15 demonstrated higher bronchoalveolar eosinophils, mRNA expression of IL-5, IL-13, IL-25, Muc5ac and Gob5/Clca, protein production of IL-5, IL-13, IL-25, IL-33 and TSLP, and expansion of type 2 innate lymphoid cells. Analogous results were found in mice treated with house dust mite before infection, including increased airway responsiveness. In contrast to Rorafl/fl littermates, RV-C-infected Rorafl/fl Il7rcre mice deficient in ILC2s failed to show eosinophilic inflammation or mRNA expression of IL-13, Muc5ac and Muc5b. We conclude that, compared to RV-A1B, RV-C15 infection induces ILC2-dependent type 2 airway inflammation, providing insight into the mechanism of RV-C-induced asthma exacerbations.
Introduction
First reported in 2006 (1, 2), rhinovirus C (RV-C)4 has been associated with severe respiratory illnesses in children and adults, often requiring hospitalization (3–15). Infections with RV-C are more likely to occur in children with a history of asthma or who develop asthma (6, 10–14). In addition, compared to RV-A, children with RV-C have been reported to have severe lower respiratory tract infections including wheezing, oxygen supplementation and intensive care unit admission (7, 8, 10, 11, 15).
Despite increasing recognition of RV-C as a cause of severe exacerbation, little is known about the pathogenesis of RV-C infections. Thus far, 55 RV-C genotypes have been reported which are believed to be synonymous with serotypes (16, 17). In contrast to major and minor group RV-A and RV-B viruses, RV-C does not utilize intercellular adhesion molecule (ICAM)-1 or low density lipoprotein family receptors (LDL-R). This is likely due to the fact that the hydrophobic pocket in VP1 is filled with multiple bulky residues (18). Instead, RV-C utilizes cadherin related family member 3 (CDHR3) as a receptor (19). Individuals with CDHR3 C529Y variants (AG and AA genotype) appear to be more susceptible to RV-C infection, as this variant is localized on the airway epithelial cell surface, where it is accessible to viral infection, in contrast to the more common GG genotype which is mostly localized to the cytoplasm (19–21).
RV-C has been refractory to study in part because it is difficult to grow in vitro. RV-C has been grown in primary mucociliary-differentiated human airway epithelial cells grown at air-liquid interface (22, 23) and HeLa cells transduced with the CDHR3 AA allele (HeLa-E8 cells) (24).
The mechanisms by which RV-C promotes severe respiratory illness are unknown. Since type 2 inflammation is an important disease mechanism in a large subgroup of individuals with asthma [reviewed in (25)], we hypothesized that RV-C infection, in contrast to RV-A infection, preferentially stimulates type 2 inflammation, leading to exacerbated eosinophilic inflammation. Mouse models have been utilized to study the host response against respiratory enteroviruses such as major group RV-A16 (26), minor group RV-A1B (26, 27) and enterovirus D68 (28). To study underlying mechanisms, we obtained cDNA encoding RV-C15 and HeLa-E8 cells from James Gern and Yury Bochkov (University of Wisconsin). We inoculated mature BALB/c mice with RV-C15, comparing inflammatory responses to those induced by RV-A1B. In addition, we compared the response of allergen-sensitized and challenged mice to the two viruses.
Material and Methods
Generation of RV-C15 and RV-A1B
Full length cDNA encoding RV-C15 and HeLa-E8 stable cells expressing human CDHR3 C529Y (19) were provided by James Gern and Yury Bochkov, University of Wisconsin. The cDNA was reverse transcribed and resulting full-length vRNA transfected into HeLa-H1 cells (ATCC, Manassas, VA) using lipofectamine (ThermoFisher Scientific, Waltham, MA). Virus was harvested from the HeLa-H1 cell supernatants and used to infect HeLa-E8 cells. Initial RV-C15 from transfected HeLa-H1 cells does not cause obvious cytopathic effects or form plaques in HeLa-E8 cells. However, upon close inspection, areas of cellular damage matched staining with Alexa Fluor 555-conjugated anti-mouse EV-D68 VP3 (Figure 1A). Anti EV-D68 VP3 (GeneTex, Irvine, CA) recognizes VP3 from EV-D68, RV-A1B and RV-C15 (Figure 1B). RV-C15 replicated in HeLa-E8 cells but not HeLa-H1 cells (Figure 1C). RV-A1B (ATCC), a minor group virus that infects mouse cells (29) was grown in Hela-H1 cells. The two viruses were propagated, concentrated and partially purified from infected HeLa cell lysates by means of ultrafiltration with a 100-kDa cutoff filter assay, as described previously (27). Similarly concentrated and purified HeLa cell lysates were used for sham infection. With propagation and concentration needed to produce sufficient quantities for animal experiments, we observed amplified cytopathic effects in RV-C15-infected HeLa-E8 cells. We took advantage of this result to test for live virus in the lungs of infected mice (see Results below).
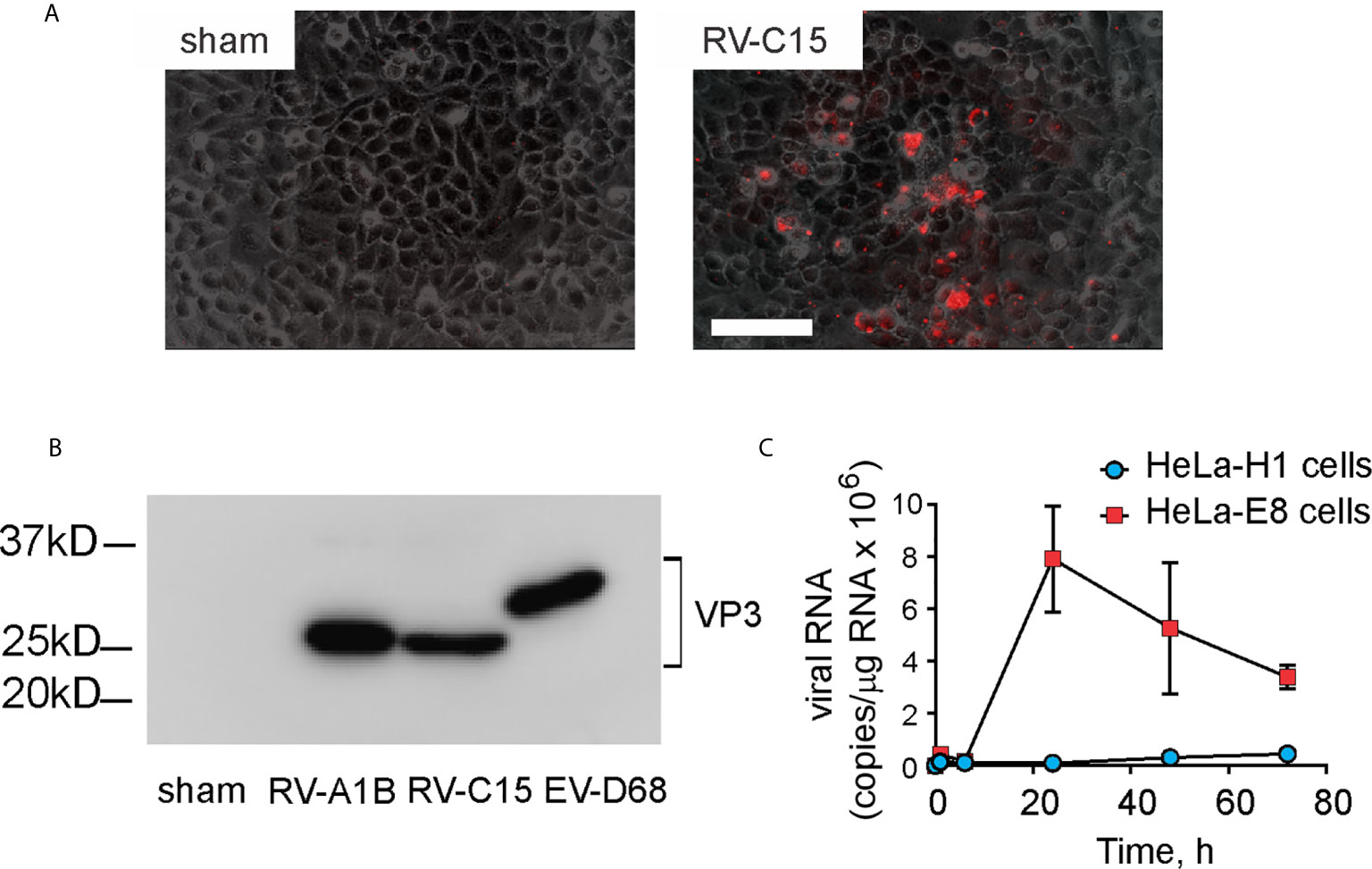
Figure 1 Generation of RV-C15. Full-length cDNA encoding RV-C15 was reverse transcribed, and the resulting vRNA transfected into HeLa-H1 cells. Virus was harvested from the cell supernatants and used to infect HeLa-H1 cells or HeLa-E8 cells expressing human CDHR3 C529Y. (A) RV-C15 induces cytopathic effects on HeLa-E8 cells. Areas of cell damage matched staining with AlexaFluor-labeled anti-VP3. The white bar is 100 µ (B) Anti-VP3 immunoblots of HeLa cell lysates infected with RV-A1B, RV-C15 and enterovirus D-68. Concentrated viral preparations were resolved by SDS-PAGE and probed with anti-VP3. (C) Infection of Hela-E8 and HeLa-H1 cells with RV-C15. RV copy number was determined by qPCR. Data shown are mean ± SD, n =3.
RV-C15 Infection and Treatment
All animal usage was approved by the Institutional Animal Care and Use Committee of University of Michigan and performed under National Institutes of Health guidelines. Eight-to-twelve-week-old female BALB/c mice (Jackson Laboratories, Bar Harbor, ME) were inoculated with 5 x 106 PFU equivalents (ePFU) of RV-C15 in 50 µL PBS by intranasal instillation. RV-C ePFU was calculated based on a calibrated standard curve for RV-A1B (18). Additional mice were inoculated with 5 x 106 ePFU RV-A1B (in 50 µL PBS) or an equal volume of sham HeLa cell lysate. In additional experiments, we examined RV-C15-induced mRNA expression in Rorafl/flIl7rCre mice and Rorafl/fl littermates (from Dr. Andrew McKenzie, MRC Laboratory of Molecular Biology, Cambridge, UK). Based on the requirement of RORα for ILC2 development (30), Rorafl/flIl7rCre mice are ILC2 deficient (31). Il7rCre mice were originally generated by Dr. Hans-Reimer Rodewald (Division of Cellular Immunology, German Cancer Research Center, Heidelberg) (32).
Using specific forward and reverse primers, RNA from passaged virus was used to produce random primed first strand cDNA and overlapping products using Phusion high fidelity DNA polymerase (New England Biolabs, Ipswich, MA) of about 2 kB each. These products were agarose gel purified and processed for Sanger sequencing (University of Michigan DNA Sequencing Core). Primers used for PCR production and sequencing are shown in the Supplemental Table. Sequencing through nucleotide 7042 of the W-10 RV-C15 reference sequence GU219984, we did not detect the previously described mutations in VP1 or 3A (24).
Model of Allergic Airways Disease
BALB/c mice were sensitized through the intranasal route with 100 μg D. pteronyssinus house dust mite (HDM) extract in 50 μl PBS (Greer Labs, Lenoir, NC) by intranasal installation on day 1 and challenged with 10 μg HDM on days 11 and 12 (33). On day 13, mice were inoculated through the intranasal route under Forane anesthesia with RV-C15, RV-A1B or sham, as noted above.
Real-Time Quantitative PCR
Lungs were harvested at different points and RNA was extracted with Trizol (Invitrogen, Carlsbad, CA). Lung RNA was isolated using an RNAeasy kit (Qiagen). cDNA was synthesized from 2 μg of RNA using high capacity cDNA synthases kit (Applied Biosystems, Foster City, CA) and subjected to quantitative real-time PCR using specific primers for mRNA (Table 1). The level of gene expression for each sample was normalized to GAPDH unless otherwise specified. RV copy number (vRNA) was determined by qPCR using previously published primers (34).
Generation of a Peptide Directed Anti-CDHR3 Antibody
The CDHR3 protein has extracellular calcium binding domains and a sialic-acid modified Asn186 important for RV-C15 binding (19). Hopp-Wood hydrophilicity analysis (DNASTAR, Madison WI) of the NIH BLAST sequence alignments of NP_689963.2 (human) and NP_001019649.1 (mouse) revealed a short conserved peptide (human amino acids 154-167, YQVEAFDPEDTSRN) in the second calcium binding domain representing a possible selective antigen for both human and mouse CDHR3. A rabbit polyclonal antibody was produced and purified using affinity chromatography (Genscript, Piscataway, NJ).
HeLa-H1 cells, HeLa-E8 cells, and mouse lungs were lysed, cellular proteins resolved by 10% SDS-PAGE, and proteins transferred to a nitrocellulose membrane. Membranes were probed with anti-CDHR3. Signals were amplified and visualized with horseradish peroxidase-conjugated secondary antibody (BioRad, Hercules, CA) and chemiluminescence solution (Pierce, Rockford, IL). To determine the specificity of the observed bands, primary antibody was incubated with cysteine-conjugated YQVEAFDPEDTSRN peptide. Anti-CDHR3 (1 µg/mL) recognized 100 kD bands in CDHR3-expressing HeLa-E8 cells and mouse lung lysate but not HeLa-H1 cells (Supplemental Figure 1A). Recognition of these bands was abolished by addition of YQVEAFDPEDTSRN peptide (10 µg/mL). Other bands, perhaps representing proteins with calcium binding domains, remained. HeLa-E8 cells, but not HeLa-H1 cells on glass coverslips stained positively with AF488-conjugated anti-CDHR3. Staining was blocked with addition of either unlabeled rabbit IgG or YQVEAFDPEDTSRN (Supplemental Figure 1B).
Lung Histology and Immunofluorescence
Lungs were harvested at different points, fixed with 10% formaldehyde overnight and paraffin embedded. Blocks were sectioned at 500 μm intervals at a thickness of 5 μm, and each section was deparaffinized, hydrated and stained. To visualize inflammation, sections were stained with H&E. Other lung sections were incubated with anti-CDHR3, anti-EV-D68 VP3, mouse anti-acetyl α-tubulin (MilliporeSigma, Burlington, MA), rat anti-IL-25 (Biolegend), goat anti-IL-33 (eBioscience), rat anti-TSLP (Biolegend), rat anti-CD123 (Biolegend) or isotypic IgG. Antibodies and IgGs were labeled with AlexaFluor NHS esters (ThermoFisher) according to manufacturer’s instructions. ILC2s were identified as IL-13- and GATA3-positive, and T cells were identified as IL-13-, GATA3- and CD3-positive. Images were visualized using an ApoTome microscope (Carl Zeiss, Thornwood, NY) or a Leica SP5 inverted laser confocal microscope (Buffalo Grove, IL).
Flow Cytometric Analysis
Lungs were harvested 2 days after sham, RV-C15 and RV-A1B treatment. Lungs were perfused with PBS containing EDTA and minced and digested in collagenase IV. Cells were filtered and washed with red blood cell lysis buffer, and dead cells were stained with Pacific Blue Live/Dead fixable dead staining dye (Invitrogen). Nonspecific binding was blocked by 10% FBS with 1% LPS-free BSA and 5-µg rat anti-mouse CD16/32 (BioLegend, San Diego, CA) added. To identify ILC2s, cells were then stained with FITC-conjugated antibodies for lineage markers [CD3ϵ, T-cell receptor- β (TCR β), B220/CD45R, Ter-119, Gr-1/Ly-6G/Ly-6C, CD11b, CD11c, F4/80, and FcϵRIα; all from BioLegend], anti-CD25-peridinin- chlorophyll-protein complex (PerCP)-Cy5.5 (eBioscience), anti-CD127-allophycocyanin (APC; eBioscience) and anti-ST2-phycoerythrin (PE)-Cy7 (BioLegend), as described (35). Cells were fixed, subjected to flow cytometry, and analyzed on a LSR Fortessa (BD Biosciences, San Jose, CA). Positive/negative staining was determined using fluorescence minus one (FMO) controls. Data were collected using FACSDiva software (BD Biosciences) and analyzed using FlowJo software (Tree Star, Ashland, OR).
Bronchoalveolar Lavage
Bronchoalveolar lavage (BAL) was performed using 1 ml PBS aliquots. Cytospins were stained with Diff-Quick (Dade Behring, Newark, DE) and differential counts determined from 200 cells (36).
ELISA
Mouse lungs were harvested, homogenized in PBS plus Roche Complete Protease Inhibitors (MilliporeSigma), and snap frozen in liquid nitrogen. After thawing and resuspension at 4°C, particles were centrifuged at 10,000 x g for 30 min, and the supernatant was diluted serially in the homogenization buffer for ELISA of IFN-β, IFN-λ, IL-5, IL-13, IL-25, IL-33 and TSLP according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN and eBioscience).
Measurement of Airway Responsiveness
Mice were anesthetized, intubated, and ventilated with a Buxco FinePointe System (Wilmington, NC). Mice were administered nebulized saline and increasing doses of nebulized methacholine to assess airways responsiveness (27).
Airway Epithelial Cell Culture
Mouse airway epithelial cells were purchased from Cell Biologics (Chicago, IL). Primary airway epithelial cells were cultured in Transwells at air-liquid interface as described previously, with some modifications (37). Briefly, airway epithelial cells were cultured under submerged conditions in complete PneumaCult-Ex Plus medium (Stemcell Technologies, Vancouver, CA) for 1 week. Cells were transferred to Transwells and cultured with complete medium in both basal and apical wells until confluence was reached. Cells were then maintained at air-liquid interface for three weeks in PneumaCult-ALI maintenance medium. Cells were infected with sham, RV-C15 or RV-A39 at a multiplicity of infection (MOI) of 2 for 12 hrs. RV-A39 was purchased from ATCC and purified from infected HeLa-H1 cell lysates by ultrafiltration with a 100-kDa cutoff filter. Selected cell cultures were fixed and stained with AlexaFluor 488-conjugated anti-acetyl α-tubulin (MilliporeSigma, Burlington, MA) and AlexaFluor 555-conjugated anti-VP3. Immunoreactivity was visualized with a NikonA1 laser confocal microscope.
Data Analysis
Normality was tested using the Shapiro-Wilk test. Group mean data are represented as mean ± SEM or median ± interquartile range as appropriate. Statistical significance was assessed by unpaired t-test, Mann-Whitney test, one-way analysis of variance (ANOVA) or Kruskal-Wallis test as appropriate. Group differences were pinpointed by the Tukey or Dunn’s multiple comparison test.
Results
RV-C15 Persists in Lungs at Similar Levels as RV-A1B
Female adult BALB/c mice were inoculated sham HeLa cell lysate, 5 x 106 PFU RV-C15 or 5 x 106 PFU RV-A1B (50 µl at 108 PFU/ml) by intranasal instillation. Mice were sacrificed, and the lungs were analyzed by qPCR at 0-96 h after infection for the presence of viral RNA. Viral RNA levels peaked 12 h after inoculation (Figure 2A). RV-A1B and RV-C15 infectivity was assessed by homogenizing lungs from virus- or sham-inoculated mice, overlying this material onto confluent monolayers of HeLa-H1 or HeLa-E8 cells, and assessing viral infectivity. Clarified supernatants from lung homogenates of RV-C15-infected mice caused cytopathic effects in HeLa-E8 cells. Fifty percent tissue culture infectivity doses (TCID50) of RV-A1B and RV-C15 were determined by the Spearman-Karber method (38). Lung RV-A1B and RV-C15 titers peaked 24 h after inoculation (Figure 2B, left panel). There was no difference in lung viral titers 48 h after inoculation (Figure 2B, right panel). In addition, lungs from RV-A1B- and RV-C15-infected mice showed significant increases in IFN-α, β and l2 mRNA expression (Figure 2C) and IFN-β and IFN-l protein expression (Figure 2D), consistent with the presence of viral RNA. Lung homogenates of RV-C15-infected mice also formed small plaques on HeLa-E8 cells up to 48 h after infection, but homogenates from sham-infected mice did not (Figure 2E).
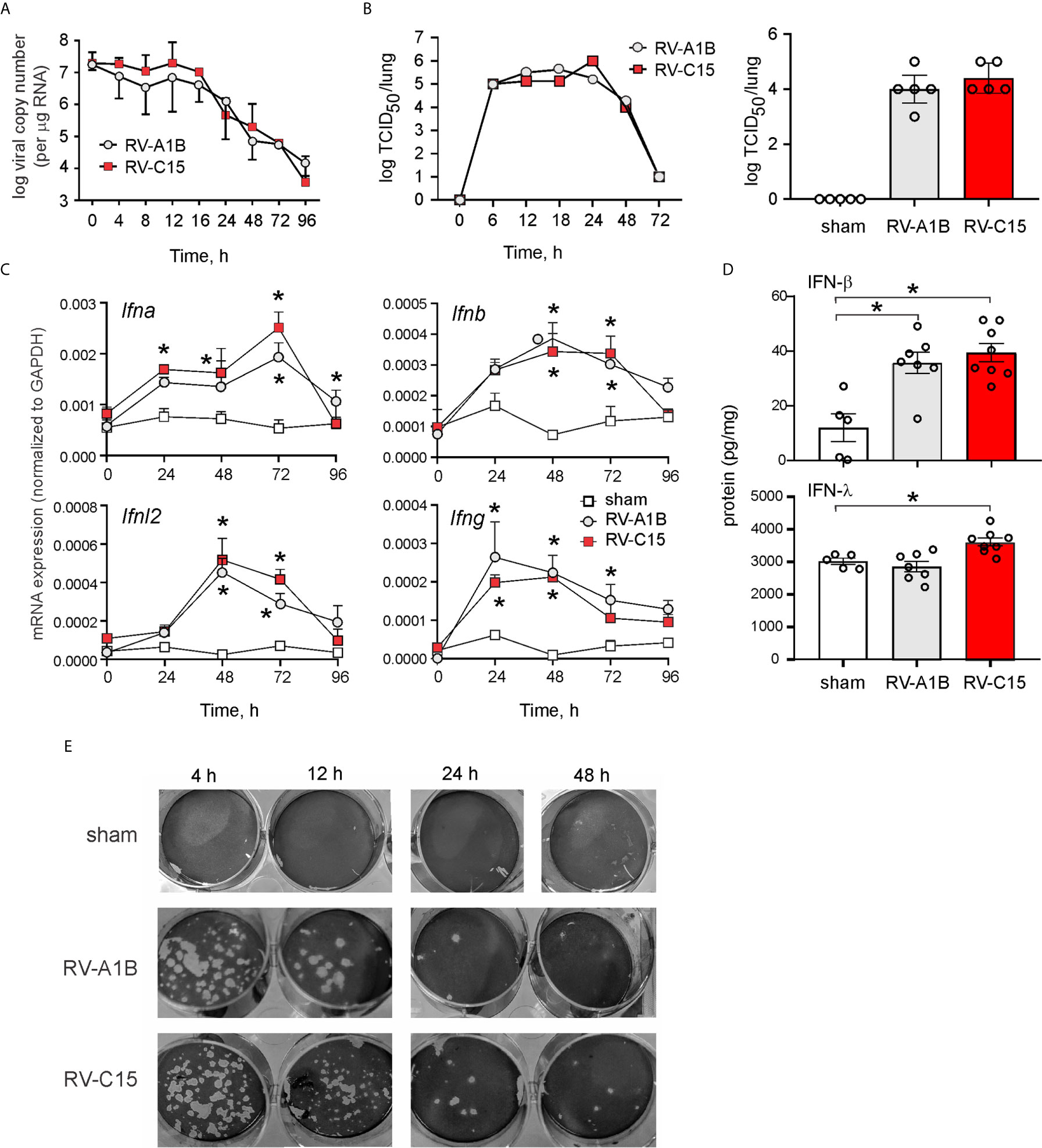
Figure 2 Viral RNA is detectable in the lungs of RV-C15 treated mice. (A) Female 8-10-week-old BALB/c mice were inoculated with 5 x 106 ePFU of RV-C15 or RV-A1B by intranasal instillation and lungs were examined by RT-PCR for viral RNA at the indicated time points. Graph showing RV-C15 and RV-A1B copy number at the indicated time points up to day 4. Data are mean ± SEM, n = 2-16 mice/group from five different experiments. (B) Left panel. Time course of lung viral titers in mice infected with RV-C15 and RV-A1B (7 mice per virus). Viral titer was assessed by TCID50. Right panel. Group mean data from an additional 5 mice at the 48 hr time point are also shown. Median ± interquartile range, n = 5 mice per group from one experiment are shown. (C) Graphs showing IFN mRNA expression analysis at indicated time points. The level of gene expression for each sample was normalized to GAPDH. Data represent mean ± SEM, n = 6 mice in each group from two different experiments, *P<0.05 by two-way ANOVA. (D) Graphs showing IFN expression analysis 48 hr after infection. Data represent mean ± SEM, n = 7 mice in each group from one experiment, *P<0.05 by one-way ANOVA. (E) Plaque assays show live virus in the lungs of RV-A1B and RV-C15-infected mice, as evidence by plaque formation in HeLa-H1 and HeLa-E8 cells, respectively.
Lungs were also formalin-fixed and paraffin-embedded 24 h after exposure, and sections stained with fluorescent tagged anti-VP3, anti-CDHR3 and acetyl-α-tubulin. RV-C15 was localized to CDHR3+ ciliated airway epithelial cells (Figure 3A). Confocal microscopy using anti-YQVEAFDPEDTSRN showed colocalization of CDHR3 with RV-C15 but not RV-A1B (Figure 3B). However, it is important to note that this antibody did not block RV-C15 replication in vitro or in vivo (not shown).
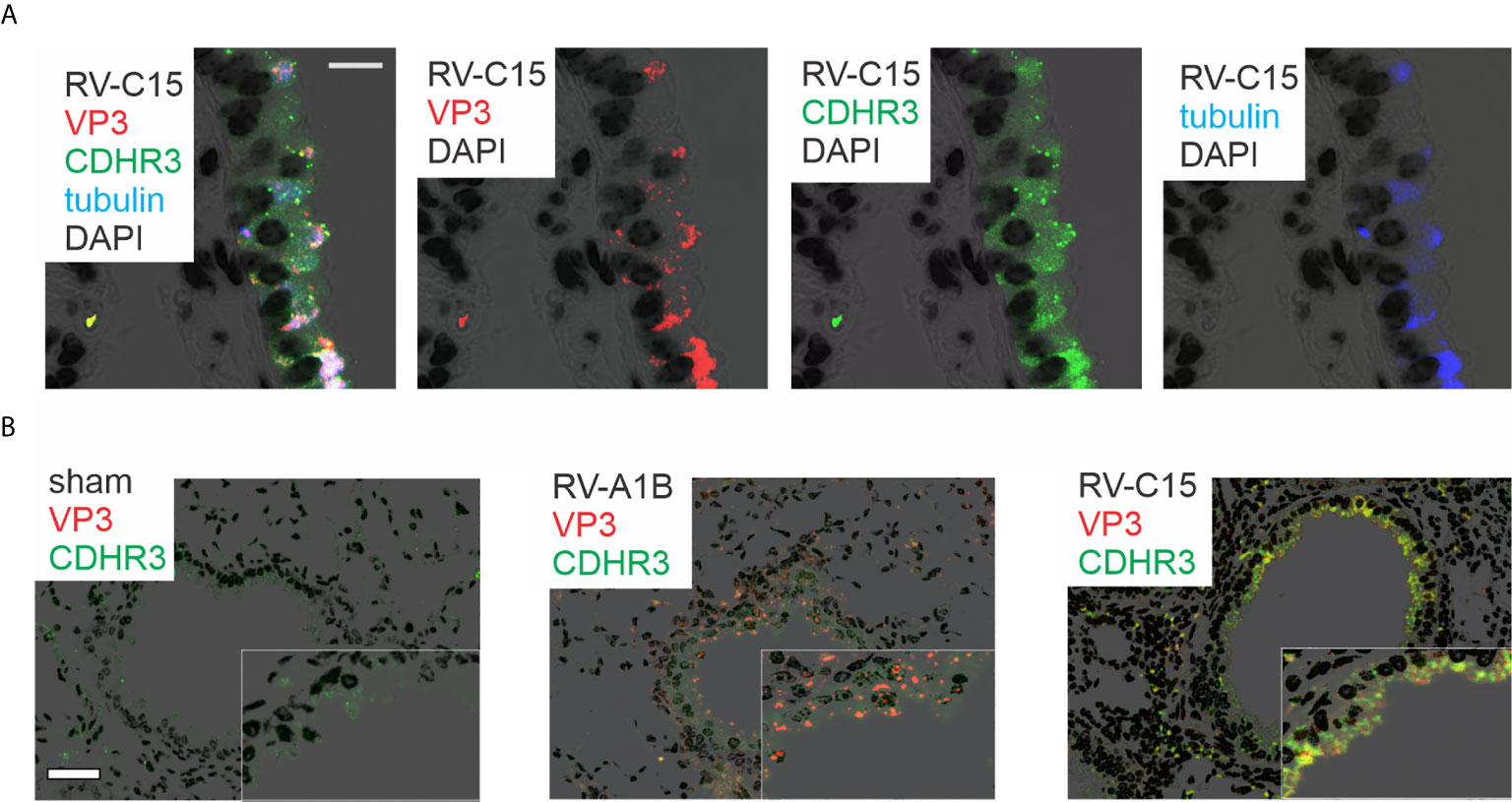
Figure 3 Colocalization of RV-C15 and CDHR3 in airway tissues of RV-C15-infected mice. (A) Airways from RV-C15-infected mice were stained with anti-VP3 (red), anti-acetyl α-tubulin (blue) and anti-CDHR3 (green). To stain mouse CDHR3, we identified a peptide (YQVEAFDPEDTSRN, human AAs 154-167) in the second extracellular calcium-binding domain identical in mouse and human. Polyclonal antiserum was generated and purified by affinity chromatography. Acetyl α-tubulin was localized to the epithelial cell apical surface. Colocalization is white. RV-C15 was localized to CDHR3+ ciliated airway epithelial cells. The white bar is 10 µ. (B) Airway sections from sham-, RV-A1B and RV-C15-infected mice stained for viral protein 3 (VP3, shown in orange/red) and CDHR3 (green). Colocalization is yellow. The white bar is 50 µ.
Infection of Cultured Mouse Airway Epithelial Cells With RV-C15
To confirm that RV-C15 infects mouse airway epithelial cells, we cultured differentiated mouse airway epithelial cells at air-liquid interface with sham, RV-C15 or RV-A39, a major group virus which does not infect mouse cells. Selected cell cultures were fixed and stained for AlexaFluor-conjugated anti-acetyl α-tubulin and anti-VP3. Cultures stained for acetylated tubulin indicating the presence of cilia (Figure 4, upper panel). Cultured inoculated with RV-A39 showed no VP3 present (middle panel). Cultures inoculated with RV-C15 showed colocalization of VP3 and acetyl α-tubulin, indicating infection of ciliated cells (lower panel).
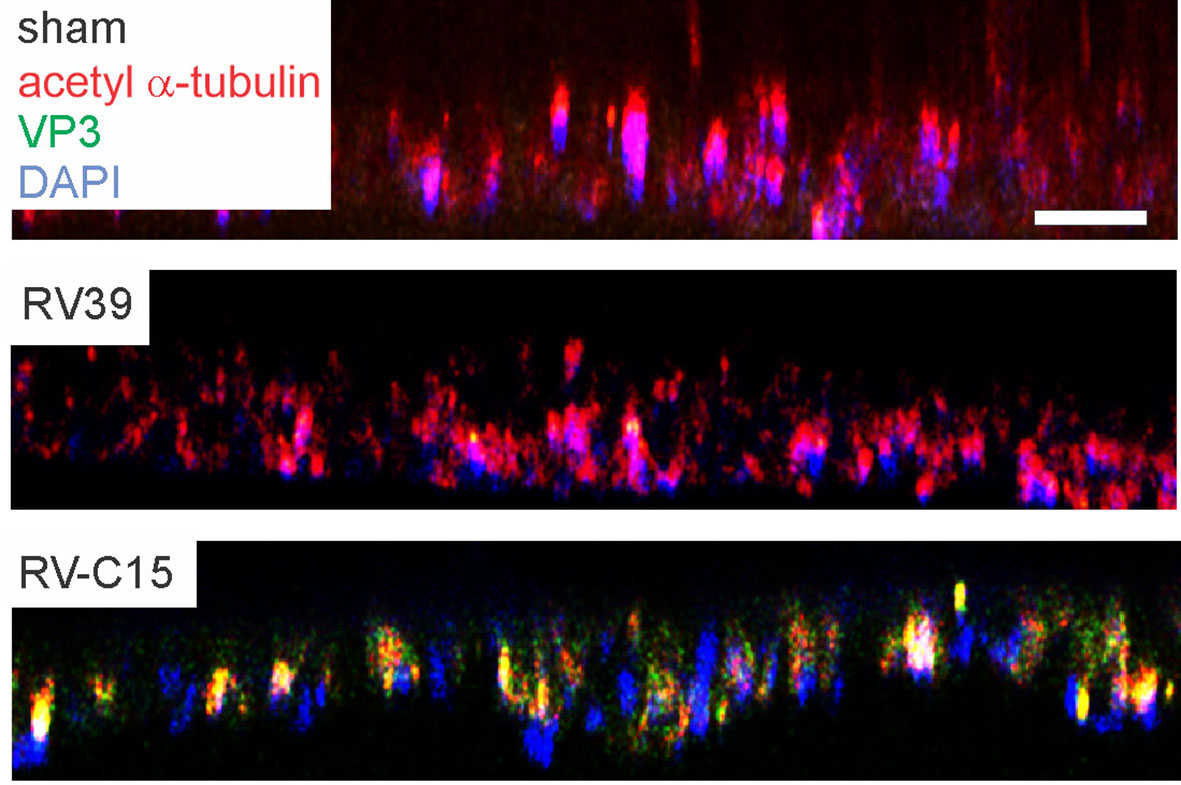
Figure 4 RV-C15 co-localizes with mouse epithelial cell acetyl-α-tubulin and induces expression of pro-inflammatory cytokines. Mouse airway epithelial cells were differentiated at air-liquid interface for 21 days. Cells were infected with sham HeLa cell lysate, RV-C15 or RV-A39 at MOI 2 and harvested 5 min after infection for immunofluorescence staining with anti-mouse acetyl α-tubulin (red) and anti-VP3 (green). Nuclei are stained with DAPI (blue). Confocal microscopy shows colocalization of cilia and RV-C15 (yellow). The white bar is 50 µ.
RV-C15 Induces Neutrophilic and Eosinophilic Inflammation and Expression of Type 2 Cytokines
Forty-eight h post-inoculation, lungs were stained with hematoxylin and eosin. Sham-inoculated mice showed no inflammation (Figure 5A). However, RV-C15 exposed mice showed leukocyte infiltration around large airways, which was similar to RV-A1B. Next, we determined bronchoalveolar lavage inflammatory cell counts in RV-C15- and sham-infected mice 24 and 48 h after treatment. We also examined the effects of replication-deficient UV-irradiated virus. Selected RV-C15 preparations were irradiated with ultraviolet (UV) light at 1200 mJ/cm2 for 30 mi using a UVB CL-1000 cross-linker (39). Forty-eight h after inoculation, RV-C15-infected mice had significantly greater monocyte, neutrophil, lymphocyte and eosinophil recruitment into the bronchoalveolar fluid than sham-treated mice (Figure 5B). Mice infected with UV-irradiated virus showed significantly reduced viral copy number and fewer neutrophils, lymphocytes and eosinophils in the airways (Figures 5B, C).
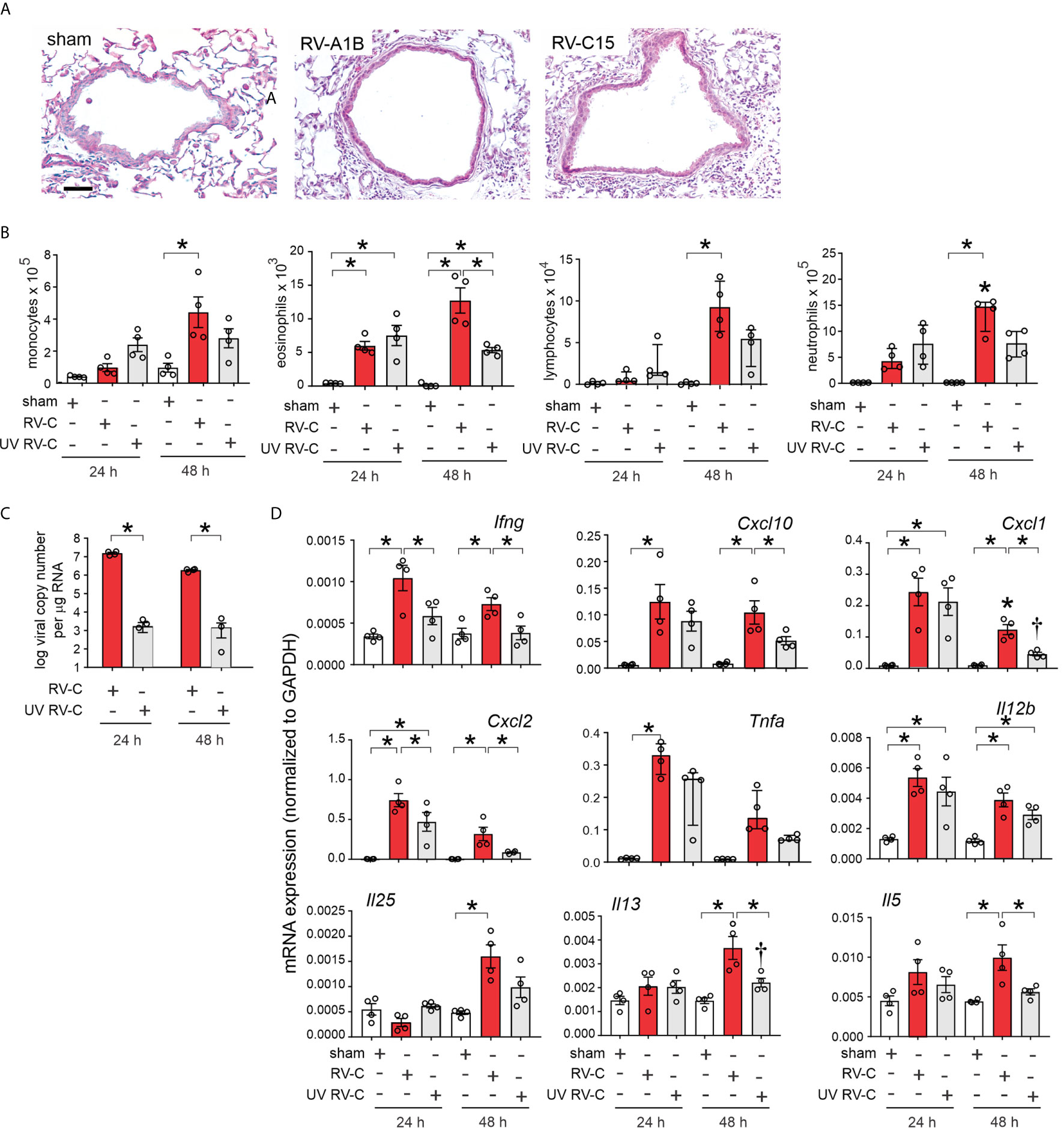
Figure 5 RV-C15 induces airway inflammation in naïve mice that is partially dependent on viral replication. Female 8-10-week-old BALB/c mice were treated with sham HeLa cell lysate, 5 x 106 ePFU RV-C15 or UV-irradiated RV-C15. (A) Hematoxylin- and eosin-stained lung tissue. Bar = 50 µ. (B) Lungs were harvested 24 or 48 h after inoculation and processed for BAL inflammatory cell counts. (C, D) Twenty-four or 48 hr after inoculation, lungs were harvested for vRNA and mRNA expression. For mRNA, the level of gene expression for each sample was normalized to GAPDH. (For panels B-D, data are mean ± SEM except for lymphocytes, neutrophilis and Tnfa, n = 3-4 mice in each group from one experiment, *P < 0.05 by one-way ANOVA; for lymphocytes, neutrophils and Tnfa, data are median ± IQR, *P < 0.05 by Kruskal-Wallis test).
Lungs were also harvested for mRNA expression 24 and 48 h after treatment, as measured by qPCR. Compared to sham treatment, RV-C15 infection significantly increased mRNA expression of IFN-γ, CXCL10, CXCL1, CXCL2, TNF-α, IL-12, IL-25, IL-13 and IL-5 (Figure 5D). UV irradiation significantly reduced RV-C15-induced IFN-γ, CXCL10, CXCL1, CXCL2, IL-13 and IL-5 transcript levels, and UV-irradiated virus failed to induce TNF-α or IL-25 mRNA expression.
Comparison of RV-C15 and RV-A1B Responses
We compared lung inflammatory responses 48 h after infection with RV-C15 and RV-A1B, a minor group virus which infects mouse cells (29, 40) and constitutes a well-established model of RV infection (26, 27). As noted above, RV-C induced significant eosinophilic inflammation, whereas RV-1B did not (Figure 6A). Compared to RV-A1B, RV-C15 infection induced significantly higher mRNA expression of CXCL1, CXCL2, IL-5 and IL-13 (Figure 6B). Only RV-C15 induced significant mRNA expression of the IL-13-responsive genes Muc5ac, Muc5b and Gob5/Clca1. Finally, only RV-C15 significantly increased protein expression of the type 2 cytokines IL-5 and IL-13 (Figure 7A).
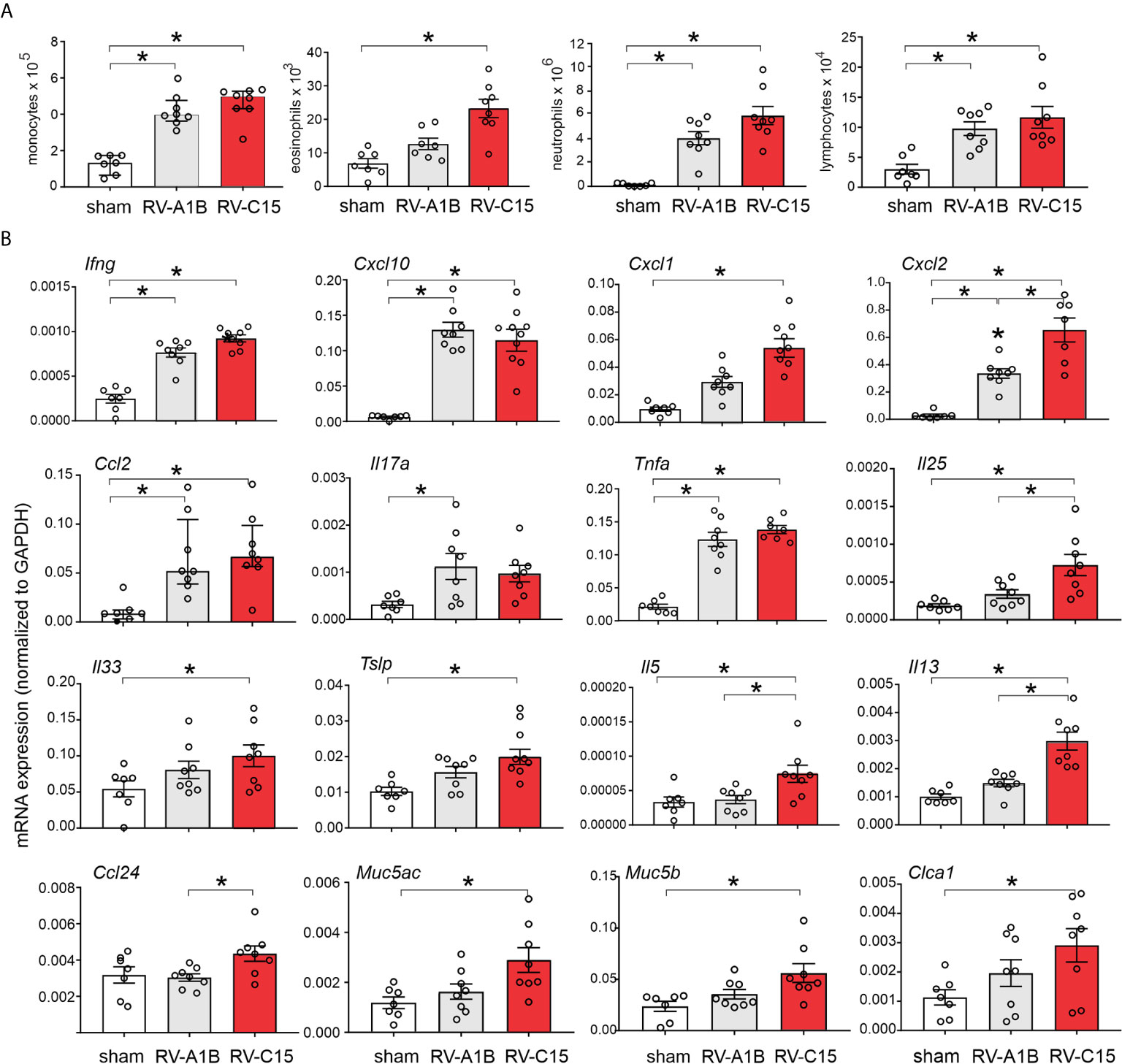
Figure 6 Comparison of RV-C15- and RV-A1B-induced airway inflammation in naïve mice. Female 8-10-week-old BALB/c mice were treated with sham HeLa cell lysate, 5 x 106 ePFU RV-C15 or 5 x 106 ePFU RV-A1B. (A) Lungs were harvested 48 hr after inoculation and processed for BAL inflammatory cell counts. (B) Forty-eight h after inoculation, lungs were harvested for mRNA expression. The level of gene expression for each sample was normalized to GAPDH. For panels (A, B) data are mean ± SEM except for monocytes and Ccl2, n = 7-8 mice in each group from two experiments, *P < 0.05 by one-way ANOVA; for monocytes and Ccl2, data are median ± IQR, *P < 0.05 by Kruskal-Wallis test).
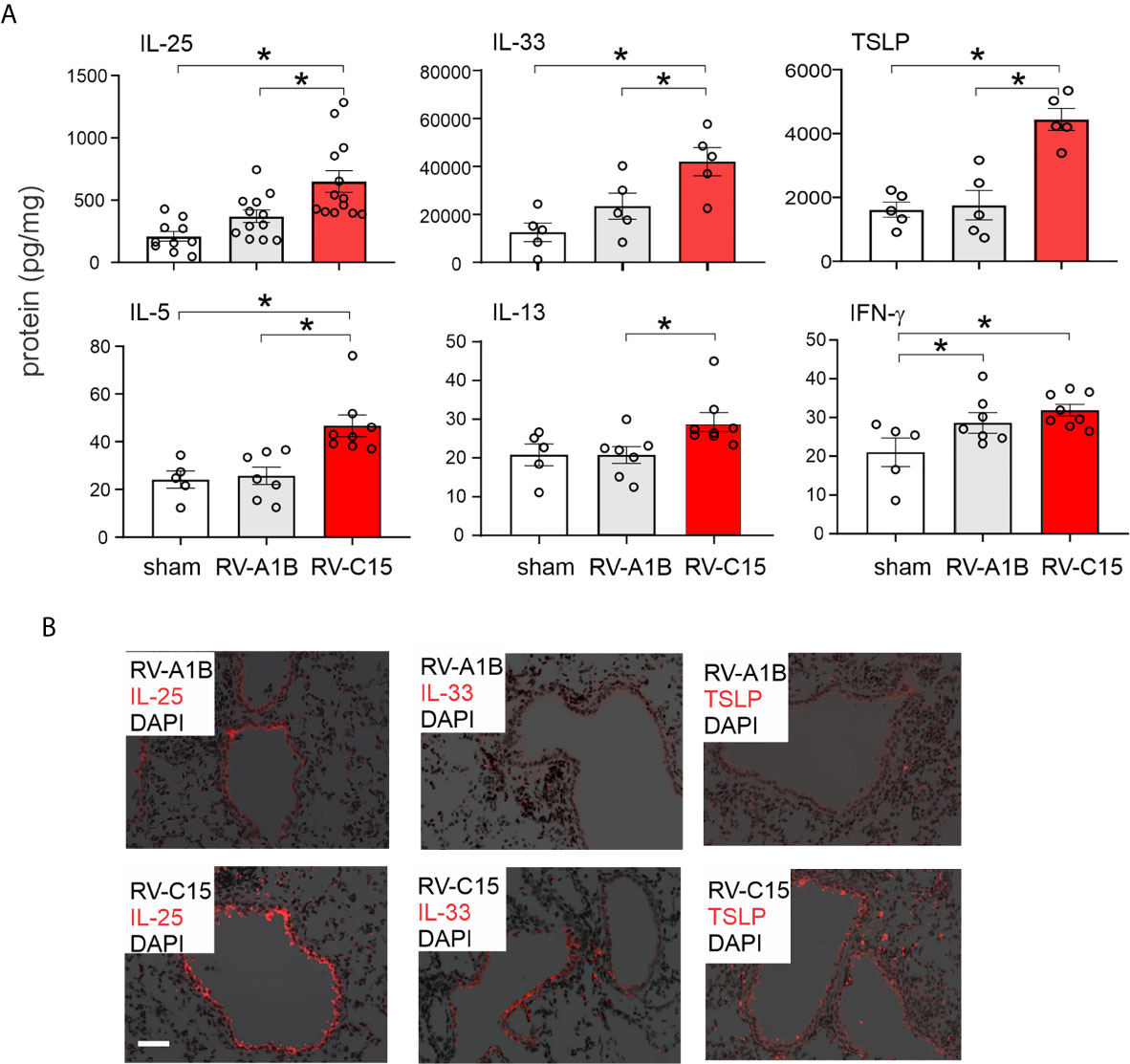
Figure 7 Comparison of RV-C15 and RV-A1B-induced cytokine expression in naïve mice. (A) Effects of RV-C15 and RV-A1B on cytokine expression measured by ELISA. Data are mean ± SEM, n = 5-12 mice in each group from one or two experiments, *P < 0.05 by one-way ANOVA. (B) Immunofluorescence images of mouse lungs infected with 5 x 106 ePFU RV-C15 or 5 x 106 ePFU RV-A1B. Staining for IL-25, IL-33 and TSLP is shown. Scale bar is 50 µm. These images are representative of four mice.
We also examined lungs for elaboration of IL-25, IL-33 and TSLP, innate cytokines that promote ILC2 expansion (41–47). Compared to RV-A1B, RV-C15 infection induced significantly higher mRNA expression of IL-25 (Figure 6B). Only RV-C15 significantly increased mRNA expression of IL-33 and TSLP. In addition, compared to RV-A1B, RV-C15 infection induced significantly higher protein expression of IL-25, IL-33 and TSLP (Figure 7A). Lungs were formalin-fixed and paraffin-embedded 2 days post-exposure, and sections stained for IL-25, IL-33 and TSLP immunofluorescence. Abundant staining for IL-25, IL-33 and TSLP was seen in the airways of mice inoculated with RV-C15 but not RV-A1B (Figure 7B). IL-25 and TSLP were localized to the airway epithelium whereas IL-33 was found in both airway epithelial and peribronchial cells.
RV-C15 Inoculation Enhances mRNA Expression of Type 2 Cytokines, Mucus Genes and Airway Responsiveness in Allergen-Challenged Mice
RV-C15-induced respiratory illnesses have been associated with a previous history of asthma (3–6, 11, 13). We therefore determined the response to RV-C15 infection in mice with allergic airways disease and compared the responses with RV-A1B. As described previously (33), wild-type BALB/c mice were sensitized with house dust mite (HDM) and challenged with HDM 10 and 11 days after sensitization. Two days later, mice were inoculated with sham, RV-C15 or RV-A1B. Forty-eight h after infection, mice were sacrificed for bronchoalveolar lavage and lung mRNA determination or anesthetized and endotracheally intubated for measurement of methacholine responsiveness. As noted above, RV-C15 infection of naïve mice increased BAL monocytes, neutrophils, eosinophils and lymphocytes (Figure 8A), while inducing mRNA expression of IFN-γ, CXCL10, IL-17A, IL-13 and IL-5 (Figure 8B). HDM sensitization and challenge induced lung infiltration with monocytes, lymphocytes and eosinophils (Figure 6A), and lung mRNA expression of the eosinophil chemoattractant CCL24, the type 2 cytokines IL-5 and IL-13 and the mucus-related genes Muc5AC and Gob5 (Figure 8B).
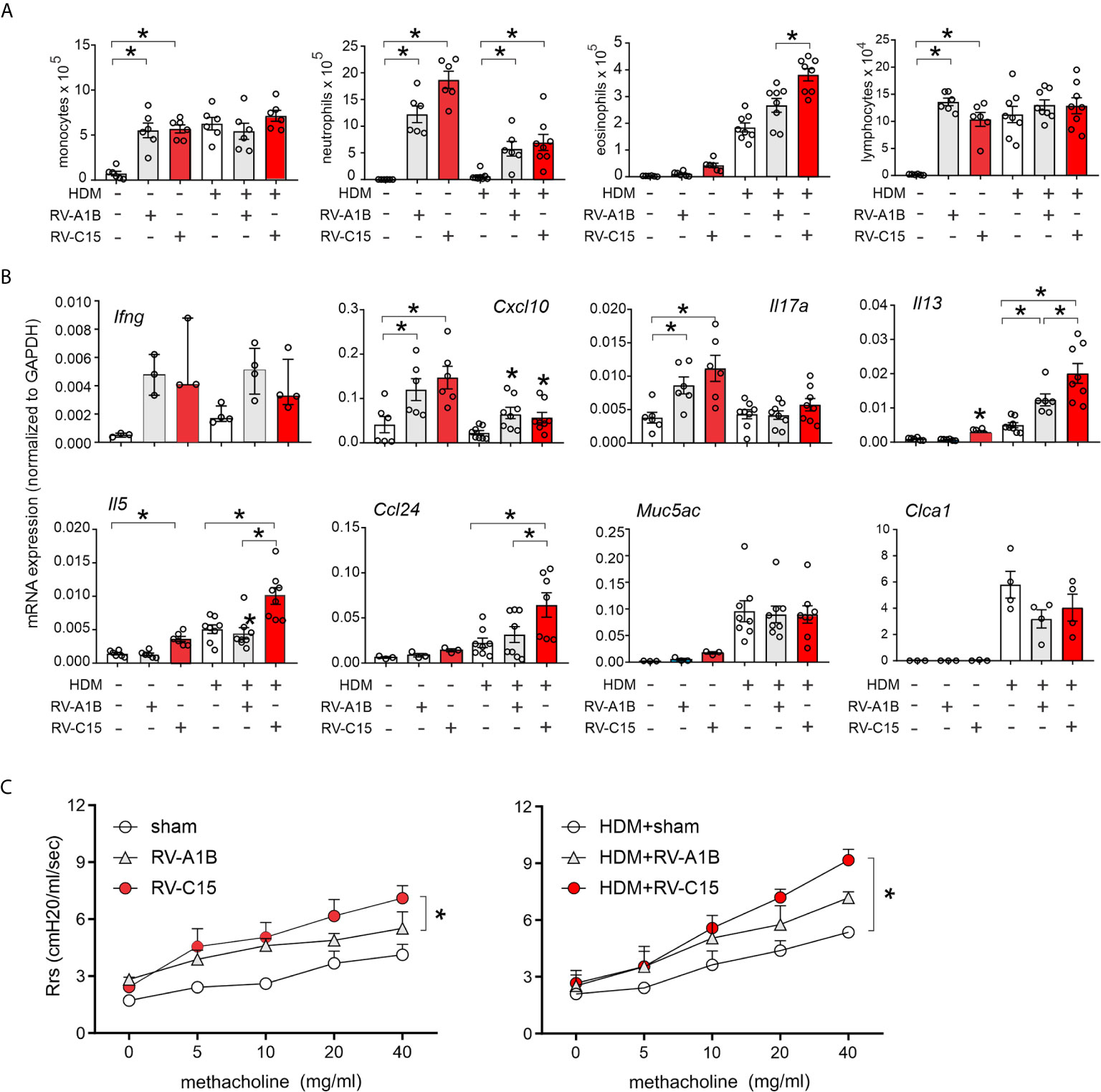
Figure 8 Comparison of RV-C15- and RV-A1B-induced airway inflammation in house dust mite-sensitized and -challenged mice. Female 8-12 week-old BALB/c mice were challenged with house dust mite (HDM) and treated one day after the last HDM treatment with sham HeLa cell lysate, 5 x 106 ePFU RV-C15 or 5 x 106 ePFU RV-A1B. Forty-eight hrs later lungs were harvested for BAL analysis and qPCR. A separate set of mice were similarly treated and were anesthetized and endotracheally intubated for measurement of airways responsiveness. (A) Graphs showing BAL cell counts. (B) Graphs showing qPCR analysis of lung mRNA expression. The level of gene expression for each sample was normalized to GAPDH. For panels (A, B), data are mean ± SEM except for Ifng, n = 3-8 mice/group for 1-3 experiments, *p<0.05, one-way ANOVA; for Ifng, data are median ± IQR. (C) Airways methacholine responsiveness of the indicated treatment groups. Data are mean ± SEM of 3-4 mice/group from two experiments, *P < 0.05, two-way ANOVA.
Infection of HDM-treated mice with either RV-A1B or RV-C15 had additive effects on airway inflammation (Figure 8A). RV-A1B and RV-C15 each increased neutrophilic and eosinophilic inflammation; however, the increase in eosinophils was significantly greater in mice infected with RV-C15. In addition, RV-C15 infection of mice with allergic airways disease had additive effects on BAL lung IL-13, IL-5 and CCL24 expression, which were greater than induced by RV-A1B (Figure 8B). Thus, RV-C15 infection enhanced allergen-induced type 2 inflammation to a greater extent than RV-A1B. On the other hand, RV-C15-induced neutrophilic inflammation and mRNA expression of CXCL10 and IL-17 tended to be lower in allergen-challenged mice.
Next, we examined the effects of RV infection on the airway responsiveness in naïve and HDM-sensitized and -challenged mice. Increasing doses of nebulized methacholine were given by inhalation and respiratory system resistance measured. In naïve mice, RV-C15 increased airways responsiveness compared to RV-A1B (Figure 8C). In HDM-treated mice, only RV-C15 increased airway responsiveness compared to HDM alone.
Potential Contribution of ILC2s to RV-C-Induced Airway Inflammation
Next, we examined lung ILC2s by flow cytometry. Cells were sorted for size, live/dead and surface markers for the various hematopoietic lineages (Figure 9A). Lungs from sham-treated mice showed a large number of lineage- CD25- CD127- cells, likely representing lung structural (epithelial and mesenchymal) cells. Two days after infection, RV-C15 infection was associated with a small but significant increase the number of lung lineage-negative, CD25-, CD127-double positive ILC2s (Figures 9B, C). There was no increase in the lungs of RV-A1B-infected mice. Identification of ILC2s was confirmed by co-staining with ST2, the IL-33 receptor (Figures 9D, E).
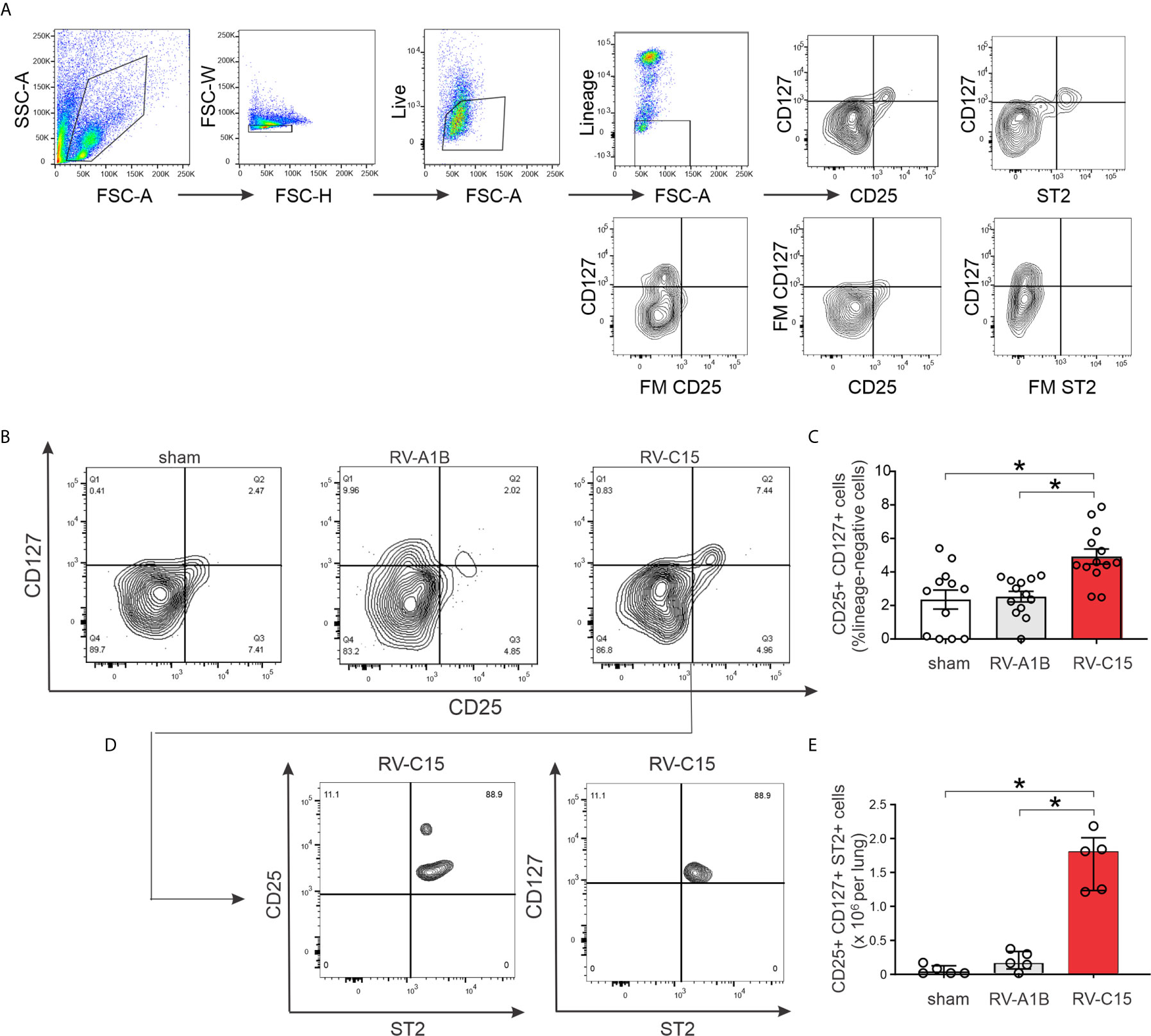
Figure 9 Flow cytometric analysis (A–E) for ILC2s was carried out in RV-A1B- and RV-C15-infected mice. For flow cytometry, ILC2s were identified as lineage-negative, CD25- and CD127-double positive cells (B, C) and lineage-negative, CD25+, CD127+, ST2+ cells (D, E). For (C), data are mean ± SEM, n = 12 mice in each group from three different experiments, *P < 0.05 by one-way ANOVA. For (E), data are median ± IQR, n = 5 mice in each group from one experiment, *P < 0.05 by Kruskal-Wallis test.
We examined RV-C15-induced cytokine responses in ILC2-deficient Rorafl/flIl7rCre mice and Rorafl/fl littermates. We previously found that six day-old Rorafl/flIl7rCre mice fail to show ILC2 expansion after RV-A1B infection despite a small increase in viral load, demonstrating the effectiveness of this knockout (48). RV-C15 infection increased airway eosinophilic inflammation (Figure 10A) and mRNA expression of IL-13, Muc5ac and Muc5b in Rorafl/fl mice but not Rorafl/flIl7rCre littermates (Figure 10B). In contrast, RV-C15-induced neutrophils, lymphocytes and mRNA expression of IFN-γ and CXCL10 were preserved.
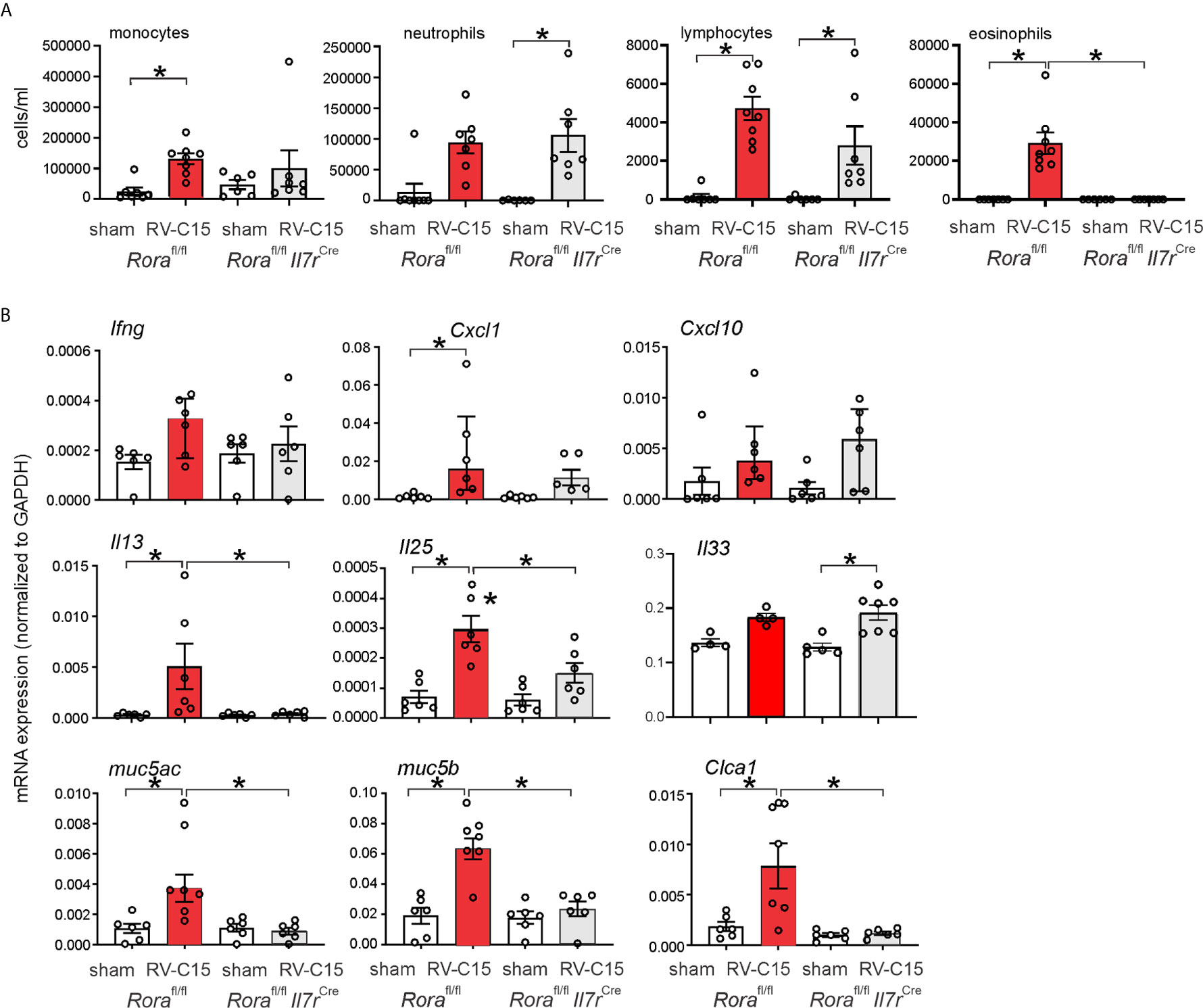
Figure 10 Requirement of ILC2s for RV-C15-induced airway inflammation and mRNA expression. (A) Rorafl/fl Il7rCre mice infected with RV-C show lower eosinophils compared to Rora-fl/fl littermates. Data are mean ± SEM, n = 6-8 mice/group from one experiment, *P < 0.05 by one-way ANOVA. (B) Rorafl/fl Il7rCre mice infected with RV-C show lower Il13, Muc5ac, Muc5b and Clca1 mRNA expression compared to Rora-fl/fl littermates. Data are mean ± SEM except for IFN-γ and CXCL10, n = 6 mice/group from three different experiments, *P < 0.05 by one-way ANOVA; for Ifng and Cxcl10, data are median ± IQR.
Discussion
Despite increasing recognition of RV-C as a cause of severe asthma exacerbation, little is known about the pathogenesis of RV-C infections. To accomplish this, we infected BALB/c mice with RV-C15, and compared responses to our previously established RV-A1B model (27). Following intranasal inoculation with RV-C15, we isolated positive-strand viral RNA from the lungs of mice up to four days after exposure. We detected RV-C15 protein in airway epithelial cells. We also showed that lung homogenate from RV-C15-exposed mice 48 hr after infection causes cytopathic effects and plaque formation in HeLa-E8 cells. RV-C exposure induced a robust type I and type III interferon response which peaked 48-72 hrs after infection, evidence of viral replication (49). UV-irradiated virus had significantly reduced effects on airway inflammation and cytokine expression. RV-C15 exposure induced airway inflammation, as demonstrated by lung histology, increased BAL cells, and increased cytokine expression. Airways inflammation was accompanied by a functional state of hyperresponsiveness. Finally, RV-C15 but not RV-A39 infected ciliary-differentiated mouse airway epithelial cells cultured at air-liquid interface. Together, these data suggest that mouse lower airways may be infected with RV-C15. However, the steep reduction in viral RNA we observed in our model, similar to that previously observed for RV-A16 (26), minor group RV-A1B (26, 27) and enterovirus D68 (28), speaks against a substantial replicative infection.
In addition, we found that RV-C15 infection induced quantitatively and qualitatively different airway responses than RV-A1B. RV-C15-infected mice showed significantly higher CXCL1 and CXCL2 mRNA expression than RV-A1B-infected mice. In addition, only RV-C-infected mice showed increases in lung mRNA expression of IL-5, IL-13 and CCL24, indicating a type 2 inflammatory response. While at times the differences between RV-C15 and RV-A1B responses were small, RV-C-induced type 2 cytokine responses were sufficient to generate robust airway eosinophilia and mucous gene expression. We also found increased mRNA expression and peribronchial deposition of the innate cytokines IL-25, IL-33 and TSLP. This response is distinctly different from the response to RV-A1B or RV-A16, which have been shown by five different laboratories to be a classic type 1 antiviral response (26, 27, 50–55). Only Tbet-deficient mice show IL-13 expression after RV-A1B-infection (56). Similarly, after human experimental RV-A16 infection, only subjects with asthma, but not controls, show elevation of IL-5, IL-13, IL-25 or IL-33 (51, 57, 58). Similar differential IL-25 and TSLP expression has been noted in asthmatic epithelial cells (51, 59). This pattern of increased type 2 and innate cytokine expression could explain why infections with RV-C are more likely to occur in children with a history of asthma or who develop asthma (6, 10, 11, 13, 14).
The precise mechanism by which RV-C15 induces greater type 2 and innate cytokine expression than RV-A1B is unclear. A previous study examining the effects of various respiratory viruses on airway epithelial cell replication kinetics, cell tropism, tissue integrity, and cytokine secretion showed no differences between RV-C15 and other RVs (23). Similarly, we did not notice more cell loss in cultures infected with RV-C15 (data not shown). We speculate that differential innate cytokine expression following RV-C15 reflects the receptor-linked signaling pathways that establish the inflammatory response. In support of this hypothesis, differences in macrophage gene expression between two rhinovirus serotypes, RV-A16 and RV-A1A, have been traced to differential kinase and transcription factor phosphorylation following initial RV binding (60). Also in support of this hypothesis are the robust increases in cytokine expression we observed in mice treated with UV-irradiated RV-C15, more than we observed previously with RV-A1B (27). Responses to replication-deficient virus are likely to reflect receptor binding and endocytosis of virus, rather than later replication-dependent responses. Previous in vitro studies have also shown responses to replication-deficient RV (61–63), suggesting that binding and endocytosis of RV is sufficient, and viral replication unnecessary, for a subset of inflammatory responses, to be followed by a second set of replication-dependent responses.
We found colocalization of RV-C and acetyl α-tubulin, a microtubule protein that is enriched in the axonemes of most cilia. Thus, these data confirm that RV-C binds to ciliated airway epithelial cells (64). We found that CDHR3 preferentially colocalizes with RV-C15 compared to RV-A1B. Mouse CDHR3 is highly homologous to human CDHR3 and includes the N186 glycosylation site (19) and EC-1 domain (65) required and sufficient for RVC15 binding to the human protein. However, we were unable to block RV-C binding or replication with anti-CDHR3 (not shown). We are therefore unable to state with certainty that CDHR3 is the RV-C receptor in mice. Since structural analysis suggests that, similar to enterovirus-D68, RV-C15 binds to a sialic acid moiety of a CDHR3-bound glycan (18), it is conceivable that RV-C binds to other glycan-binding proteins. A recent study in human airway epithelial cells showed that, while CDHR3 knockdown blocks RV-C replication, it does not affect binding of RV-C (66), suggesting that a co-receptor is required for binding of the virus.
As noted above, RV-C15 infection induced airway expression of IL-25, IL-33 and TSLP, innate cytokines responsible for activation of IL-5- and IL-13-producing ILC2s expansion (41–47). IL-25 production was mostly limited to airway epithelial cells, whereas IL-33 was noted in epithelial and subepithelial cells. A similar pattern of IL-33 deposition was observed in children with steroid-resistant asthma (67). Accordingly, we found increased lineage-negative, ST2+, CD25- and CD127 ILC2s in the lungs of RV-C15-, but not RV-A1B-infected mice. ILC2 depletion blocked RV-C15-induced mRNA expression of IL-13 and the mucus-related genes Muc5ac and Muc5b. Previously we noted expansion of ILC2s in RV-A1B-infected 6 day-old mice but not mature mice (35). Lung ILC2 expansion has also been shown after influenza infection (68, 69) and allergen-induced airway inflammation (44–46, 70–72).
A limitation of our study is the transient nature of viral infection in our model. Species differences restrict viral replication, requiring a high inoculum. However, we have previously shown that infection with RV-A1B increases lung type 1 IFN and negative-strand viral RNA expression (27), markers of viral replication. In addition, MDA5 is required for RV-A1B-induced inflammatory responses (73), inferring a role for double-stranded RNA, a form of viral RNA which is only made during viral replication. Inhibition of inflammasome activation (55) and corticosteroids (74) each increase viral load in RV-infected mice, consistent with the notion that antiviral immunity plays a significant role in our model. In addition, we have observed differences in the inflammatory response to RV-A1B, RV-C15 and EV-D68 (28) which are qualitative in nature and resemble responses in human subjects. Taken together, these results suggest that while replication of human RV is minimal in mice, the resulting host-induced innate immune response and immunopathology is worthy of study. Indeed, replication-deficient viral vectors are a useful tool for studying the innate immune response to acute viral infection without ongoing cytopathic effects (75).
We conclude that RV-C15 exposure induces airways inflammation in mice, binding to ciliated airway epithelial cells. Compared to RV-A1B infection, the inflammatory response to RV-C15 is characterized by greater eosinophils, epithelial-derived innate cytokines and IL-13-producing ILC2s. Further characterization of this model, combined with studies of human subjects, will provide insight into the pathogenesis of rhinovirus C infections.

Table 1 Primer sequences for real-time PCR.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of University of Michigan and performed under National Institutes of Health guidelines.
Author Contributions
CR performed experiments, analyzed the data, and drafted the manuscript. MYH performed experiments, analyzed data, and edited the manuscript. TI performed experiments and analyzed data, JL performed experiments. AG performed experiments and analyzed data. SJ performed experiments. CS performed experiments. JB performed experiments, analyzed data, and edited the manuscript. MBH supervised all aspects of the project, interpreted the data, and write the final draft of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported NIH grants R01 HL134369 and R01 AI155444 (to MBH).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank James Gern and Yury Bochkov, University of Wisconsin School of Medicine and Public Health, for providing cDNA encoding RV-C15 and HeLa-E8 cells expressing CDHR3 C529Y.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.649520/full#supplementary-material
References
1. Lamson D, Renwick N, Kapoor V, Liu Z, Palacios G, Ju J, et al. Masstag Polymerase-Chain-Reaction Detection of Respiratory Pathogens, Including a New Rhinovirus Genotype, That Caused Influenza-Like Illness in New York State During 2004-2005. J Infect Dis (2006) 194(10):1398–402. doi: 10.1086/508551
2. Arden KE, McErlean P, Nissen MD, Sloots TP, Mackay IM. Frequent Detection of Human Rhinoviruses, Paramyxoviruses, Coronaviruses, and Bocavirus During Acute Respiratory Tract Infections. J Med Virol (2006) 78(9):1232–40. doi: 10.1002/jmv.20689
3. Lee W-M, Kiesner C, Pappas T, Lee I, Grindle K, Jartti T, et al. A Diverse Group of Previously Unrecognized Human Rhinoviruses are Common Causes of Respiratory Illnesses in Infants. PloS One (2007) 2(10):e966. doi: 10.1371/journal.pone.0000966
4. Renwick N, Schweiger B, Kapoor V, Liu Z, Villari J, Bullmann R, et al. A Recently Identified Rhinovirus Genotype is Associated With Severe Respiratory-Tract Infection in Children in Germany. J Infect Dis (2007) 196(12):1754–60. doi: 10.1086/524312
5. Khetsuriani N, Lu X, Teague WG, Kazerouni N, Anderson LJ, Erdman DD. Novel Human Rhinoviruses and Exacerbation of Asthma in Children. Emerg Infect Dis J (2008) 14(11):1793. doi: 10.3201/eid1411.080386
6. Miller EK, Edwards KM, Weinberg GA, Iwane MK, Griffin MR, Hall CB, et al. A Novel Group of Rhinoviruses is Associated With Asthma Hospitalizations. J Allergy Clin Immunol (2009) 123(1):98–104.e1. doi: 10.1016/j.jaci.2008.10.007
7. Piralla A, Rovida F, Campanini G, Rognoni V, Marchi A, Locatelli F, et al. Clinical Severity and Molecular Typing of Human Rhinovirus C Strains During a Fall Outbreak Affecting Hospitalized Patients. J Clin Virol (2009) 45(4):311–7. doi: 10.1016/j.jcv.2009.04.016
8. Miller EK, Khuri-Bulos N, Williams JV, Shehabi AA, Faouri S, Al Jundi I, et al. Human Rhinovirus C Associated With Wheezing in Hospitalised Children in the Middle East. J Clin Virol (2009) 46(1):85–9. doi: 10.1016/j.jcv.2009.06.007
9. Calvo C, Casas I, García-García ML, Pozo F, Reyes N, Cruz N, et al. Role of Rhinovirus C Respiratory Infections in Sick and Healthy Children in Spain. Pediatr Infect Dis J (2010) 29(8):717–20. doi: 10.1097/INF.0b013e3181d7a708
10. Bizzintino J, Lee W-M, Laing IA, Vang F, Pappas T, Zhang G, et al. Association Between Human Rhinovirus C and Severity of Acute Asthma in Children. Eur Respir J (2011) 37(5):1037–42. doi: 10.1183/09031936.00092410
11. Cox DW, Bizzintino J, Ferrari G, Khoo SK, Zhang G, Whelan S, et al. Human Rhinovirus Species C Infection in Young Children With Acute Wheeze is Associated With Increased Acute Respiratory Hospital Admissions. Am J Respir Crit Care Med (2013) 188(11):1358–64. doi: 10.1164/rccm.201303-0498OC
12. Drysdale SB, Alcazar M, Wilson T, Smith M, Zuckerman M, Lauinger IL, et al. Respiratory Outcome of Prematurely Born Infants Following Human Rhinovirus A and C Infections. Eur J Pediatr (2014) 173(7):913–9. doi: 10.1007/s00431-014-2262-1
13. Moreno-Valencia Y, Hernandez-Hernandez VA, Romero-Espinoza JAI, Coronel-Tellez RH, Castillejos-Lopez M, Hernandez A, et al. Detection and Characterization of Respiratory Viruses Causing Acute Respiratory Illness and Asthma Exacerbation in Children During Three Different Seasons (2011–2014) in Mexico City. Influenza Other Respir Viruses (2015) 9(6):287–92. doi: 10.1111/irv.12346
14. Fawkner-Corbett DW, Khoo SK, Duarte CM, Bezerra PGM, Bochkov YA, Gern JE, et al. Rhinovirus-C Detection in Children Presenting With Acute Respiratory Infection to Hospital in Brazil. J Med Virol (2016) 88(1):58–63. doi: 10.1002/jmv.24300
15. Cox DW, Khoo S-K, Zhang G, Lindsay K, Keil AD, Knight G, et al. Rhinovirus is the Most Common Virus and Rhinovirus-C is the Most Common Species in Paediatric Intensive Care Respiratory Admissions. Eur Respir J (2018) 52(2):1800207. doi: 10.1183/13993003.00207-2018
16. Simmonds P, McIntyre C, Savolainen-Kopra C, Tapparel C, Mackay IM, Hovi T. Proposals for the Classification of Human Rhinovirus Species C Into Genotypically Assigned Types. J Gen Virol (2010) 91(10):2409–19. doi: 10.1099/vir.0.023994-0
17. McIntyre CL, Knowles NJ, Simmonds P. Proposals for the Classification of Human Rhinovirus Species a, B and C Into Genotypically Assigned Types. J Gen Virol (2013) 94(8):1791–806. doi: 10.1099/vir.0.053686-0
18. Liu Y, Hill MG, Klose T, Chen Z, Watters K, Bochkov YA, et al. Atomic Structure of a Rhinovirus C, a Virus Species Linked to Severe Childhood Asthma. Proc Natl Acad Sci USA (2016) 113(32):8997–9002. doi: 10.1073/pnas.1606595113
19. Bochkov YA, Watters K, Ashraf S, Griggs TF, Devries MK, Jackson DJ, et al. Cadherin-Related Family Member 3, a Childhood Asthma Susceptibility Gene Product, Mediates Rhinovirus C Binding and Replication. Proc Natl Acad Sci USA (2015) 112(17):5485–90. doi: 10.1073/pnas.1421178112
20. Bønnelykke K, Sleiman P, Nielsen K, Kreiner-Møller E, Mercader JM, Belgrave D, et al. A Genome-Wide Association Study Identifies CDHR3 as a Susceptibility Locus for Early Childhood Asthma With Severe Exacerbations. Nat Genet (2013) 46(1):51–5. doi: 10.1038/ng.2830
21. Bønnelykke K, Coleman AT, Evans MD, Thorsen J, Waage J, Vissing NH, et al. Cadherin-Related Family Member 3 Genetics and Rhinovirus C Respiratory Illnesses. Am J Respir Crit Care Med (2018) 197(5):589–94. doi: 10.1164/rccm.201705-1021OC
22. Ashraf S, Brockman-Schneider R, Gern JE. Propagation of Rhinovirus-C Strains in Human Airway Epithelial Cells Differentiated At Air-Liquid Interface. In: Jans DA, Ghildyal R, editors. Rhinoviruses: Methods and Protocols. New York, NY: Springer New York (2015). p. 63–70.
23. Essaidi-Laziosi M, Brito F, Benaoudia S, Royston L, Cagno V, Fernandes-Rocha M, et al. Propagation of Respiratory Viruses in Human Airway Epithelia Reveals Persistent Virus-Specific Signatures. J Allergy Clin Immunol (2018) 141(6):2074–84. doi: 10.1016/j.jaci.2017.07.018
24. Bochkov YA, Watters K, Basnet S, Sijapati S, Hill M, Palmenberg AC, et al. Mutations in VP1 and 3a Proteins Improve Binding and Replication of Rhinovirus C15 in Hela-E8 Cells. Virology (2016) 499:350–60. doi: 10.1016/j.virol.2016.09.025
25. Fahy JV. Type 2 Inflammation in Asthma – Present in Most, Absent in Many. Nat Rev Immunol (2015) 15(1):57–65. doi: 10.1038/nri3786
26. Bartlett NW, Walton RP, Edwards MR, Aniscenko J, Caramori G, Zhu J, et al. Mouse Models of Rhinovirus-Induced Disease and Exacerbation of Allergic Airway Inflammation. Nat Med (2008) 14(2):199–204. doi: 10.1038/nm1713
27. Newcomb DC, Sajjan US, Nagarkar DR, Wang Q, Nanua S, Zhou Y, et al. Human Rhinovirus 1B Exposure Induces Phosphatidylinositol 3-Kinase-Dependent Airway Inflammation in Mice. Am J Respir Crit Care Med (2008) 177(10):1111–21. doi: 10.1164/rccm.200708-1243OC
28. Rajput C, Han M, Bentley JK, Lei J, Ishikawa T, Wu Q, et al. Enterovirus D68 Infection Induces IL-17-dependent Neutrophilic Airway Inflammation and Hyperresponsiveness. JCI Insight (2018) 3(16):e121882. doi: 10.1172/jci.insight.121882
29. Tuthill TJ, Papadopoulos NG, Jourdan P, Challinor LJ, Sharp NA, Plumpton C, et al. Mouse Respiratory Epithelial Cells Support Efficient Replication of Human Rhinovirus. J Gen Virol (2003) 84(Pt 10):2829–36. doi: 10.1099/vir.0.19109-0
30. Wong SH, Walker JA, Jolin HE, Drynan LF, Hams E, Camelo A, et al. Transcription Factor Rorα is Critical for Nuocyte Development. Nat Immunol (2012) 13(3):229–36. doi: 10.1038/ni.2208
31. Barlow JL, Peel S, Fox J, Panova V, Hardman CS, Camelo A, et al. Il-33 is More Potent Than IL-25 in Provoking IL-13–producing Nuocytes (Type 2 Innate Lymphoid Cells) and Airway Contraction. J Allergy Clin Immunol (2013) 132(4):933–41. doi: 10.1016/j.jaci.2013.05.012
32. Schlenner SM, Madan V, Busch K, Tietz A, Läufle C, Costa C, et al. Fate Mapping Reveals Separate Origins of T Cells and Myeloid Lineages in the Thymus. Immunity (2010) 32(3):426–36. doi: 10.1016/j.immuni.2010.03.005
33. Toussaint M, Jackson DJ, Swieboda D, Guedan A, Tsourouktsoglou TD, Ching YM, et al. Host DNA Released by Netosis Promotes Rhinovirus-Induced Type-2 Allergic Asthma Exacerbation. Nat Med (2017) 23(11):1384–4. doi: 10.1038/nm1117-1384a
34. Contoli M, Message SD, Laza-Stanca V, Edwards MR, Wark PA, Bartlett NW, et al. Role of Deficient Type Iii Interferon-Lambda Production in Asthma Exacerbations. Nat Med (2006) 12:1023–6. doi: 10.1038/nm1462
35. Hong JY, Bentley JK, Chung Y, Lei J, Steenrod JM, Chen Q, et al. Neonatal Rhinovirus Induces Mucous Metaplasia and Airways Hyperresponsiveness Through IL-25 and Type 2 Innate Lymphoid Cells. J Allergy Clin Immunol (2014) 134(2):429–39. doi: 10.1016/j.jaci.2014.04.020
36. Tsai WC, Rodriguez ML, Young KS, Deng JC, Thannickal VJ, Tateda K, et al. Azithromycin Blocks Neutrophil Recruitment in Pseudomonas Endobronchial Infection. Am J Respir Crit Care Med (2004) 170(12):1331–9. doi: 10.1164/rccm.200402-200OC
37. Schneider D, Ganesan S, Comstock AT, Meldrum CA, Mahidhara R, Goldsmith AM, et al. Increased Cytokine Response of Rhinovirus-Infected Airway Epithelial Cells in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med (2010) 182(3):332–40. doi: 10.1164/rccm.200911-1673OC
38. Johnston SL, Tyrrell DAJ. Rhinoviruses. In: Lennette EH, Schmidt NJ, editors. Diagnostic Procedures for Viral, Rickettsial, and Chlamydial Infections. Washington D.C.: American Public Health Association (1997). p. 553–63.
39. Papi A, Johnston SL. Rhinovirus Infection Induces Expression of its Own Receptor Intercellular Adhesion Molecule 1 (ICAM-1) Via Increased NF-kappab-mediated Transcription. J Biol Chem (1999) 274(14):9707–20. doi: 10.1074/jbc.274.14.9707
40. Reithmayer M, Reischl A, Snyers L, Blaas D. Species-Specific Receptor Recognition by a Minor-Group Human Rhinovirus (HRV): HRV Serotype 1A Distinguishes Between the Murine and the Human Low-Density Lipoprotein Receptor. J Virol (2002) 76(14):6957–65. doi: 10.1128/JVI.76.14.6957-6965.2002
41. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TKA, et al. Nuocytes Represent a New Innate Effector Leukocyte That Mediates Type-2 Immunity. Nature (2010) 464(7293):1367–70. doi: 10.1038/nature08900
42. Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, et al. Systemically Dispersed Innate IL-13-expressing Cells in Type 2 Immunity. Proc Natl Acad Sci USA (2010) 107(25):11489–94. doi: 10.1073/pnas.1003988107
43. Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25-and Il-33-responsive Type 2 Innate Lymphoid Cells are Defined by Expression of Crth2 and Cd161. Nat Immunol (2011) 12(11):1055–U56. doi: 10.1038/ni.2104
44. Barlow JL, Bellosi A, Hardman CS, Drynan LF, Wong SH, Cruickshank JP, et al. Innate IL-13–producing Nuocytes Arise During Allergic Lung Inflammation and Contribute to Airways Hyperreactivity. J Allergy Clin Immunol (2012) 129(1):191–198.e4. doi: 10.1016/j.jaci.2011.09.041
45. Kim HY, Chang YJ, Subramanian S, Lee HH, Albacker LA, Matangkasombut P, et al. Innate Lymphoid Cells Responding to IL-33 Mediate Airway Hyperreactivity Independently of Adaptive Immunity. J Allergy Clin Immunol (2012) 129(1):216–27 e1-6. doi: 10.1016/j.jaci.2011.10.036
46. Halim TY, Krauss RH, Sun AC, Takei F. Lung Natural Helper Cells are a Critical Source of Th2 Cell-Type Cytokines in Protease Allergen-Induced Airway Inflammation. Immunity (2012) 36(3):451–63. doi: 10.1016/j.immuni.2011.12.020
47. Han M, Rajput C, Hong JY, Lei J, Hinde JL, Wu Q, et al. The Innate Cytokines IL-25, Il-33, and TSLP Cooperate in the Induction of Type 2 Innate Lymphoid Cell Expansion and Mucous Metaplasia in Rhinovirus-Infected Immature Mice. J Immunol (2017) 199(4):1308–18. doi: 10.4049/jimmunol.1700216
48. Rajput C, Han M, Ishikawa T, Lei J, Jazaeri S, Bentley JK, et al. Early Life Heterologous Rhinovirus Infections Induce an Exaggerated Asthma-Like Phenotype. J Allergy Clin Immunol (2020) 146(33):571–82.e3. doi: 10.1016/j.jaci.2020.03.039
49. Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, et al. Integration of Interferon-[Alpha]/[Beta] Signalling to p53 Responses in Tumour Suppression and Antiviral Defence. Nature (2003) 424(6948):516–23. doi: 10.1038/nature01850
50. Nagarkar DR, Bowman ER, Schneider D, Wang Q, Shim J, Zhao Y, et al. Rhinovirus Infection of Allergen-Sensitized and -Challenged Mice Induces Eotaxin Release From Functionally Polarized Macrophages. J Immunol (2010) 185(4):2525–35. doi: 10.4049/jimmunol.1000286
51. Beale J, Jayaraman A, Jackson DJ, Macintyre JDR, Edwards MR, Walton RP, et al. Rhinovirus-Induced IL-25 in Asthma Exacerbation Drives Type 2 Immunity and Allergic Pulmonary Inflammation. Sci Transl Med (2014) 6(256):256ra134. doi: 10.1126/scitranslmed.3009124
52. Chung Y, Hong JY, Lei J, Chen Q, Bentley JK, Hershenson MB. Rhinovirus Infection Induces interleukin-13 Production From CD11b-positive, M2-Polarized Exudative Macrophages. Am J Respir Cell Mol Biol (2015) 52(2):205–16. doi: 10.1165/rcmb.2014-0068OC
53. Girkin JL, Hatchwell LM, Collison AM, Starkey MR, Hansbro PM, Yagita H, et al. Trail Signaling is Proinflammatory and Proviral in a Murine Model of Rhinovirus 1B Infection. Am J Physiol: Lung Cell Mol Physiol (2017) 312(1):L89–99. doi: 10.1152/ajplung.00200.2016
54. Song DJ, Miller M, Beppu A, Rosenthal P, Das S, Karta M, et al. Rhinovirus Infection of ORMDL3 Transgenic Mice is Associated With Reduced Rhinovirus Viral Load and Airway Inflammation. J Immunol (2017) 199(7):2215–24. doi: 10.4049/jimmunol.1601412
55. Han M, Bentley JK, Rajput C, Lei J, Ishikawa T, Jarman CR, et al. Inflammasome Activation is Required for Human Rhinovirus-Induced Airway Inflammation in Naïve and Allergen-Sensitized Mice. Mucosal Immunol (2019) 12(4):958–68. doi: 10.1038/s41385-019-0172-2
56. Glanville N, Peel TJ, Schröder A, Aniscenko J, Walton RP, Finotto S, et al. Tbet Deficiency Causes T Helper Cell Dependent Airways Eosinophilia and Mucus Hypersecretion in Response to Rhinovirus Infection. PloS Pathog (2016) 12(9):e1005913. doi: 10.1371/journal.ppat.1005913
57. Hansel TT, Tunstall T, Trujillo-Torralbo M-B, Shamji B, del-Rosario A, Dhariwal J, et al. A Comprehensive Evaluation of Nasal and Bronchial Cytokines and Chemokines Following Experimental Rhinovirus Infection in Allergic Asthma: Increased Interferons (IFN-γ and IFN-λ) and Type 2 Inflammation (IL-5 and IL-13). EBioMedicine (2017) 19(Supplement C):128–38. doi: 10.1016/j.ebiom.2017.03.033
58. Jackson DJ, Makrinioti H, Rana BM, Shamji BW, Trujillo-Torralbo MB, Footitt J, et al. Il-33-dependent Type 2 Inflammation During Rhinovirus-Induced Asthma Exacerbations In Vivo. Am J Respir Crit Care Med (2014) 190(12):1373–82. doi: 10.1164/rccm.201406-1039OC
59. Lee H-C, Headley MB, Loo Y-M, Berlin A, Gale M, Debley JS, et al. Thymic Stromal Lymphopoietin is Induced by Respiratory Syncytial Virus–Infected Airway Epithelial Cells and Promotes a Type 2 Response to Infection. J Allergy Clin Immunol (2012) 130(5):1187–96.e5. doi: 10.1016/j.jaci.2012.07.031
60. Schuler BA, Schreiber MT, Li L, Mokry M, Kingdon ML, Raugi DN, et al. Major and Minor Group Rhinoviruses Elicit Differential Signaling and Cytokine Responses as a Function of Receptor-Mediated Signal Transduction. PloS One (2014) 9(4):e93897. doi: 10.1371/journal.pone.0093897
61. Griego SD, Weston CB, Adams JL, Tal-Singer R, Dillon SB. Role of p38 Mitogen-Activated Protein Kinase in Rhinovirus-Induced Cytokine Production by Bronchial Epithelial Cells. J Immunol (2000) 165(9):5211–20. doi: 10.4049/jimmunol.165.9.5211
62. Suzuki T, Yamaya M, Sekizawa K, Hosoda M, Yamada N, Ishizuka S, et al. Bafilomycin a(1) Inhibits Rhinovirus Infection in Human Airway Epithelium: Effects on Endosome and Icam-1. Am J Physiol: Lung Cell Mol Physiol (2001) 280(6):L1115–27. doi: 10.1152/ajplung.2001.280.6.L1115
63. Newcomb DC, Sajjan U, Nanua S, Jia Y, Goldsmith AM, Bentley JK, et al. Phosphatidylinositol 3-Kinase is Required for Rhinovirus-Induced Airway Epithelial Cell Interleukin-8 Expression. J Biol Chem (2005) 280(44):36952–61. doi: 10.1074/jbc.M502449200
64. Griggs TF, Bochkov YA, Basnet S, Pasic TR, Brockman-Schneider RA, Palmenberg AC, et al. Rhinovirus C Targets Ciliated Airway Epithelial Cells. Respir Res (2017) 18(1):84. doi: 10.1186/s12931-017-0567-0
65. Watters K, Palmenberg AC. CDHR Extracellular Domains EC1-3 Mediate Rhinovirus C Interaction With Cells and as Recombinant Derivatives, are Inhibitory to Virus Infection. PloS Pathog (2018) 14(12):e1007477. doi: 10.1371/journal.ppat.1007477
66. Everman JL, Sajuthi S, Saef B, Rios C, Stoner AM, Numata M, et al. Functional Genomics of CDHR3 Confirms its Role in HRV-C Infection and Childhood Asthma Exacerbations. J Allergy Clin Immunol (2019) 144(4):962–71. doi: 10.1016/j.jaci.2019.01.052
67. Saglani S, Lui S, Ullmann N, Campbell GA, Sherburn RT, Mathie SA, et al. Il-33 Promotes Airway Remodeling in Pediatric Patients With Severe Steroid-Resistant Asthma. J Allergy Clin Immunol (2013) 132(3):676–85.e13. doi: 10.1016/j.jaci.2013.04.012
68. Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, et al. Innate Lymphoid Cells Mediate Influenza-Induced Airway Hyper-Reactivity Independently of Adaptive Immunity. Nat Immunol (2011) 12(7):631–8. doi: 10.1038/ni.2045
69. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CGK, Doering TA, et al. Innate Lymphoid Cells Promote Lung-Tissue Homeostasis After Infection With Influenza Virus. Nat Immunol (2011) 12(11):1045–54. doi: 10.1031/ni.2131
70. Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. Il-33–responsive Lineage–CD25+CD44hi Lymphoid Cells Mediate Innate Type 2 Immunity and Allergic Inflammation in the Lungs. J Immunol (2012) 188(3):1503–13. doi: 10.4049/jimmunol.1102832
71. Hardman CS, Panova V, McKenzie ANJ. Il-33 Citrine Reporter Mice Reveal the Temporal and Spatial Expression of IL-33 During Allergic Lung Inflammation. Eur J Immunol (2013) 43(2):488–98. doi: 10.1002/eji.201242863
72. KleinJan A, Klein Wolterink RGJ, Levani Y, de Bruijn MJW, Hoogsteden HC, van Nimwegen M, et al. Enforced Expression of GATA3 in T Cells and Group 2 Innate Lymphoid Cells Increases Susceptibility to Allergic Airway Inflammation in Mice. J Immunol (2014) 192(4):1385–94. doi: 10.4049/jimmunol.1301888
73. Wang Q, Miller DJ, Bowman ER, Nagarkar DR, Schneider D, Zhao Y, et al. Mda5 and Tlr3 Initiate Pro-Inflammatory Signaling Pathways Leading to Rhinovirus-Induced Airways Inflammation and Hyperresponsiveness. PloS Pathog (2011) 7(5):e1002070. doi: 10.1371/journal.ppat.1002070
74. Singanayagam A, Glanville N, Girkin JL, Ching YM, Marcellini A, Porter JD, et al. Corticosteroid Suppression of Antiviral Immunity Increases Bacterial Loads and Mucus Production in COPD Exacerbations. Nat Commun (2018) 9(1):2229. doi: 10.1038/s41467-018-04574-1
Keywords: asthma, rhinovirus, innate cytokine, viral infection, exacerbation, ILC2
Citation: Rajput C, Han M, Ishikawa T, Lei J, Goldsmith AM, Jazaeri S, Stroupe CC, Bentley JK and Hershenson MB (2021) Rhinovirus C Infection Induces Type 2 Innate Lymphoid Cell Expansion and Eosinophilic Airway Inflammation. Front. Immunol. 12:649520. doi: 10.3389/fimmu.2021.649520
Received: 04 January 2021; Accepted: 07 April 2021;
Published: 22 April 2021.
Edited by:
Malcolm Ronald Starkey, Monash University, AustraliaReviewed by:
Taylor A. Doherty, University of California, San Diego, United StatesRoberto P. Garofalo, University of Texas Medical Branch at Galveston, United States
Copyright © 2021 Rajput, Han, Ishikawa, Lei, Goldsmith, Jazaeri, Stroupe, Bentley and Hershenson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marc B. Hershenson, bWhlcnNoZW5AdW1pY2guZWR1
†Present address: Charu Rajput, Department of Thoracic Medicine and Surgery, Temple University, Philadelphia, PA, United States
‡These authors share first authorship