 Zuzanna Rydzynska
Zuzanna Rydzynska Bartlomiej Pawlik
Bartlomiej Pawlik Damian Krzyzanowski
Damian Krzyzanowski Wojciech Mlynarski1†
Wojciech Mlynarski1† Joanna Madzio
Joanna Madzio- 1Department of Pediatrics, Oncology and Hematology, Medical University of Lodz, Lodz, Poland
- 2Postgraduate School of Molecular Medicine, Medical University of Warsaw, Warsaw, Poland
- 3Laboratory of Epigenetics, Institute of Medical Biology, Polish Academy of Sciences, Lodz, Poland
Severe congenital neutropenia (SCN) is a rare hematological condition with heterogenous genetic background. Neutrophil elastase (NE) encoded by ELANE gene is mutated in over half of the SCN cases. The role of NE defects in myelocytes maturation arrest in bone marrow is widely investigated; however, the mechanism underlying this phenomenon has still remained unclear. In this review, we sum up the studies exploring mechanisms of neutrophil deficiency, biological role of NE in neutrophil and the effects of ELANE mutation and neutropenia pathogenesis. We also explain the hypotheses presented so far and summarize options of neutropenia therapy.
Introduction
Functional neutrophils are the most abundant subpopulation of human leukocytes, representing the first line of the innate immune system defense against a broad spectrum of microorganisms (1). They exhibit an advanced antimicrobial mechanisms, from which we distinguish phagocytosis with, i.e. oxidative burst, release of nuclear material in the neutrophil extracellular traps (NETs) or degranulation of neutrophil granules (2).
Neutrophils are produced in hematopoietic cords inside venous sinuses in the bone marrow. Development of neutrophils, referred to granulopoiesis, begins with hematopoietic stem cells, (HSC) which may differentiate into common myeloid progenitor cells (3). These cells transform to granulocyte-monocyte progenitors, which in turn differentiate to neutrophils by intermediate stages of promyelocytes, myelocytes, metamyelocytes, band cells and segmented, polymorphonuclear cells (4). The main factor regulating both proliferation of neutrophil precursors and mature neutrophils release form the bone marrow is granulocyte colony-stimulating factor (G-CSF) (5). Disturbed production of neutrophils during hematopoiesis results in neutropenia in the peripheral blood, which leads to immunodeficiency. Among many causes of neutropenia, numerous genetic defects are described causative for congenital neutropenia (6). Mutations of neutrophil elastase gene (ELANE) are ones of the most commonly observed in patients suffering from congenital neutropenia (7). Nowadays, pathogenesis of these defects is still arguable and controversial, thus, it attracts a great scientific focus.
Physiological Functions
Neutrophil elastase is one of the four serine proteases stored in the azurophil granules of neutrophils. It shows a relatively broad elastase specificity, preferring aliphatic amino acids, Ala, Val, and Ile, in the P1 position of substrates (8).
Serine proteases have been found to be a degradative non-specific enzymes as they are all able to cleave extracellular matrix proteins such as fibronectin, elastin, proteoglycans, collagens, the platelet IIb/IIIa receptor, cadherins and a wide range of plasma proteins (9–14). NE is also capable of degrading soluble proteins such as coagulation factors, complements immunoglobulins and many protease inhibitors (15). In addition to its role in degradation of extracellular matrix, neutrophil elastase functions as a negative regulator of the inflammation process. It degrades proinflammatory mediators such as IL-1β, TNF-α (16). Neutrophil elastase participates in the intracellular pathogen destruction with a potent antimicrobial activity against Gram-negative bacteria (17), spirochetes (18) and fungi (19). Bellaaouaj et al. generating strains of mice deficient in NE by targeted mutagenesis have shown that NE-/- mice are more susceptible to sepsis and death due to infection with Klebsiella pneumoniae and Escherichia coli, both Gram-negative bacteria, as compared to wild type siblings (17). It has been proven that neutrophil elastase works directly through degradation of the outer membrane protein A (OmpA) located on the surface of E. coli. Additionally, in vitro incubation of NE with E. coli leads to a loss of bacterial membrane integrity or local weakening of the cell wall by degradation of Om protein A followed by osmotic lysis of bacteria (20). Alternatively, loss of wall integrity by OmpA cleavage may lead to NE entrance and intracellular protein degradation resulting in proteolysis and bacterial death. Hirche et al. have also shown that NE mediates innate host protection against Pseudomonas aeruginosa by degradation of the major outer membrane protein F (OmpF), a protein with important functions, including maintenance of membrane integrity, porin activity and sensing the host immune system activation (21).
Another direct function of human neutrophil elastase is the ability to cleave virulence factors such as flagellin, Gram-negative bacteria virulence factor with a strong proinflammatory activity on epithelial cells and other cell types (13). There is strong evidence that purified flagellin from P. aeruginosa is effectively degraded by neutrophil elastase and cathepsin G in the inhibitor dependent manner. What is more, López-Boado et al. have also shown in experimental conditions that none of the tested metalloproteinases (MMP-1, MMP-7 and MMP-8, which have a similar ability as NE) cleaved flagellin proving the role of NE (22).
Weinrauch et al. have shown that NE degrades Shigella virulence factors such as IpaA, IpaB and IpaC, as well as proteins secreted by Salmonella and Yersinia (SipA, SipB, SipC, HAPS and YopB, YopD, YopE, respectively) (23). Other in vitro studies have shown that NE plays a role in the neutrophil adhesion. The integrin Mac-1 binds to ligands such as fibrinogen and intercellular adhesion molecule 1 (ICAM-1) and mediates adhesion of neutrophils to the endothelial surface (24). Authors postulate that neutrophil elastase performs a proteolytic function by ICAM-1 cleavage in a manner dependent on the α1-antitrypsin (α1-AT) and N-methoxysuccinyl-Ala-Ala-Pro-Val-chloromethyl ketone (MSAAPVCK) – NE inhibitors (25).
Neutrophil proteases may also act indirectly through cleavage of serum proteins of the complement and coagulation systems to generate anti-microbial peptides. NE cleaves the central complement protein C3 to generate a peptide that imitates the natural C3a anaphylatoxin. Similarly to C3a, NE-derived C3 shows antimicrobial activity against P. aeruginosa and E. faecalis (26).
Apart from the intracellular degradation activity, neutrophil elastase demonstrates a broad spectrum of extracellular functions of which the best-known is connective tissue digestion in inflammation to clear the path for migrating neutrophils to reach sites of infection. It is capable of digesting many types of matrix proteins, including fibronectin, (denatured) collagen, proteoglycans, laminin and many others (27). Moreover, neutrophil elastase acts on cell‐surface ligands and receptors like CD14 (28) or TLR4 receptor and adhesion molecules like integrins. NE may activate TLR-4 receptors, which leads to the activation of Nuclear Factor κB (NFKB) and consequently, to the production of IL-8 (29, 30).
ELANE Gene and NE Protein Structure
ELANE gene (also known as ELA2, HLE, HNE, NE, SCN1, according to OMIM 130130) encodes human neutrophil elastase (31). ELANE consists of five exons and six introns, and resides on chromosome 19 (19p13.3), the locus for several serine proteases (32, 33) ELANE is highly expressed at mRNA level in the bone marrow in promyelocytes and then it is downregulated with maturation of neutrophil.
NE is synthesized as an inactive form of pro-pre-enzyme (zymogen) of 267 amino acids, containing N-terminal, single pro-dipeptide and a C-terminal pro-peptide. For neutrophil elastase to be active it has to undergo four consecutive post-translational protein modifications. First, 29 amino acid signal peptide is removed by a signal peptidase cleavage. Then, NE proform is glycosylated on asparagine residues at position 109 and 159. Further, N-terminal dipeptide – SerGlu is removed by cysteine protease – cathepsin C. Subsequently, the cleavage of dipeptide results in structural rearrangement of N-terminal region, which becomes inserted into the protein core. Finally, the C-terminal pro-peptide is removed, which altogether leads to formation of catalytically active enzyme comprising 218 amino acid residues and a mass of 29 to 34 kDa (EC 3.4.21.37) (32) (Figure 1B). The enzyme responsible for cleaving off the C-terminal pro-peptide has not been identified so far.
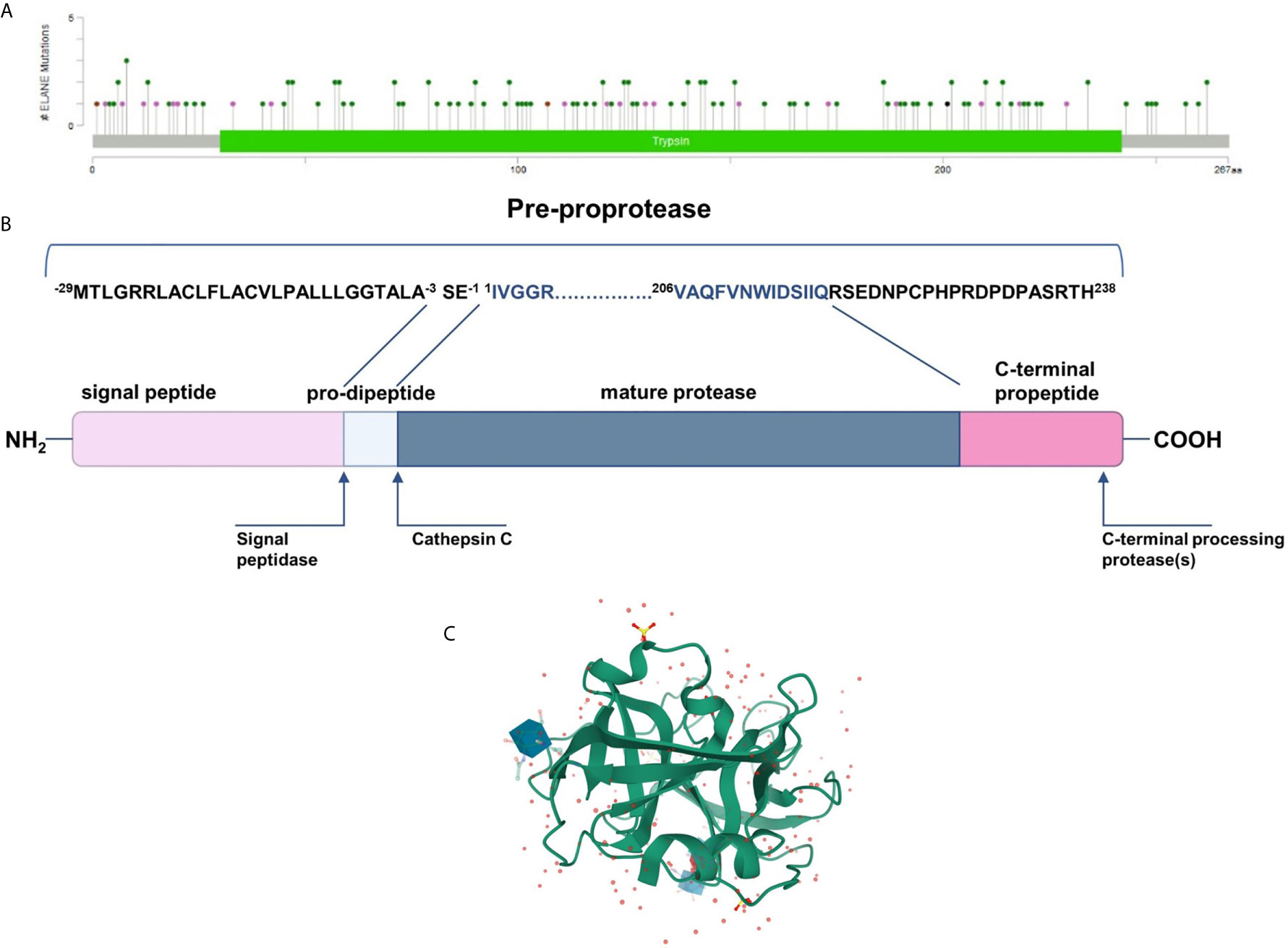
Figure 1 (A) Mutational spectrum of the neutrophil elastase (ClinVar database; RefSeq NM_001972.4) (34). The lolliplot reports mutations projected on the schematic Trypsin domain of the human NE protein. The lolliplot was produced using the MutationMapper software, freely available through the Cbioportal (https://www.cbioportal.org/mutation_mapper) (35, 36). In the scheme, mutations are reported as circles (green for missense mutations; black for duplication; brown for inframe deletions and insertions; violet for all the other types. (B) 267-residue preproprotein scheme and post-translational modification at both ends. (C) 3D structure of human neutrophil elastase 3Q76 (uncomplexed). Image created using Mol* from RCSB PDB (37, 38).
Mature neutrophil elastase, like all chymotrypsin-like serine proteases, forms a fold of two β-barrels, each made of six anti-parallel β-sheets connected through a linker segment, and a carboxyl-terminal α-helical domain (Figure 1C) (39). The active-site residues are located in a crevice between the two β-barrels. This single polypeptide chain has two glycosylation sites, it is stabilized by four disulfide bridges and has no lysine or tyrosine residues which, together with the surface distribution of the positively charged arginine restudies, make it extremely cationic. This asymmetric formation gives the protein a very high isoelectric point of about 10.5 (40).
ELANE Mutations in Pathogenesis of Congenital Neutropenia
ELANE-induced neutropenia is not related to a NE deficit itself, but rather to a dysfunction of theisprotease. Heterozygous mutations identified in ELANE gene result in a SCN, which might be a life threatening condition, or a cyclic neutropenia (CyN) with moderate to mild clinical features. Pathogenesis of SCN and CyN is a subject of many studies and scientists are still not able to unequivocally explain how dysfunctions in NE lead to maturation arrest of granulocytic differentiation. The first reports have presented a theory that mutations in ELANE lead to dysregulated vesicular sorting and trafficking. Another, an equally popular theory, indicates the role of the unfolded protein response (UPR) system, which is responsible for stress response in the endoplasmic reticulum (ER) by improperly folded proteins. As there is a wide spectrum of ELANE mutations, over the years of research, other hypotheses for the pathogenesis of congenital neutropenia have been proposed.
ELANE Mutations
ELANE mutation was first mentioned in the Horwitz report on neutrophil elastase as a cause for cyclic neutropenia, and the next year, the same team identified ELANE mutations in patients with SCN. The findings suggested that both forms are molecularly closer to each other than it was previously assumed (33, 41). The general pattern of mutations in the SCN versus CyN seems to be different, nevertheless, up to a dozen mutations overlap in both groups (42, 43). Based on genetic databases, a total of more than 230 ELANE mutations have been described so far (44), all the mutations mentioned in this article are according to RefSeq NM_001972.4 and their distribution is shown in Figure 1A (34). The vast majority of pathogenic defects are single amino acid substitutions, but there are also some frameshifts, nonsense, inframe indels as well as splice junction loss (42). The entire ELANE deletions are not described in the context of neutropenia, and the few cases such as chromosome 19p terminal deletion including this gene phenotypically do not show neutrophil count deficiency (45). Interestingly, humans (45) and mice (46) with ELANE biallelic loss of function defect produce functional neutrophils with normal complete blood counts. Moreover, knock-out of the mutant allele in hematopoietic stem cells derived from SCN patients restores neutrophils maturation (47). This excludes the NE haploinsufficiency as a pathomechanism of SCN, and it is likely that the cause of neutropenia is not the lack of neutrophil elastase itself, but protease malfunction.
To understand the role of NE in neutrophil dysfunction, distribution of mutations across EL-ANE structure seems to be more crucial than the type of defects. Although no clear link between genotype and phenotype has been described so far, the latest research substantiates nonsense-mediated decay (NMD) as the explanation of diversity in ELANE variants pathogenicity. Mutations introducing premature termination codons (PTCs) upstream of the 50th nucleotide position in early exons of ELANE (1–4 exons) trigger NMD – the mechanism that eliminates defective mRNA transcripts and prevents truncated protein translations. In contrast, the mutation downstream of the 50th nucleotide position within a late exon (4th and 5th) of ELANE avoids NMD, thus, produces defective NE that impairs neutrophil maturation and manifests as severe neutropenia (48). It is consistent with previous observations in neutropenic patients, exons 4 and 5 are enriched in nonsense and frameshift mutations, which result in disruption of the disulphide bond domain in C-terminus of elastase that is essential for its correct intracellular localization (39, 43, 49). An example is p.Gly214Arg mutation that leads to NE mislocalization predominantly to the nuclear and plasma membranes, which accelerates apoptosis of differentiating cells and manifests itself as a severe neutropenia phenotype (50).
An alternative mechanism is described based on observation of the mutations in AUG initiation codon p.Met1Ile and p.Met1Val (c.3G>A and c.1A>G) and the noncoding Kozak sequence (c.-3A>T). Disruption of the start signal forces translation from internal methionine codons, thus, produces N-terminus truncated protein that cannot be targeted toward ER and is probably accumulated in the nucleus (51). Hypothetically, mutations in the signal peptide such as A25V may also be responsible for nuclear localization (43).
A negative synergistic effect on NE protein structure has been described based on the case of a double mutation of ELANE. The authors suggest that not only the range of the molecular damage but also synergistic effect may contribute to distinct pathomechanisms (52). Depending on various amino acids that could be substituted in Ala57 site, different neutropenia phenotypes can be manifested. Bioinformatics predictions have shown that a hydrophilic amino acid causes a clash with the side chain of adjacent amino acids, changes tertiary structure of neutrophil elastase and leads to its malfunction (53).
Opinions are divided as to the risk of malignant transformation; however, the most recent evidence indicates that the development of acute myeloid leukemia is increased in patients harboring ELANE mutant (42, 43, 54). Mutations such as p.Gly214Arg and p.Cys151Tyr are related to a poor response to G−CSF and a more severe phenotype, whereas variants p.Ser126Leu and p.Pro139Leu are associated with a good prognosis (42, 55, 56)
Neutrophil Elastase Mistrafficking
Neutrophil elastase belongs to the family of serine proteases located mainly in the azurophilic granules such as zymogens. Normally, the inactive serine protease undergoes post-translational modifications at the N-terminus and C-terminus. Although intact C-terminal does not affect enzymatic activity (57), its processing is crucial for recognition of NE by µ3a subunit of adaptor protein-3 (58, 59). AP-3 complex facilitates intracellular trafficking of NE from trans-Golgi network to the granule lumen (Figure 2A).
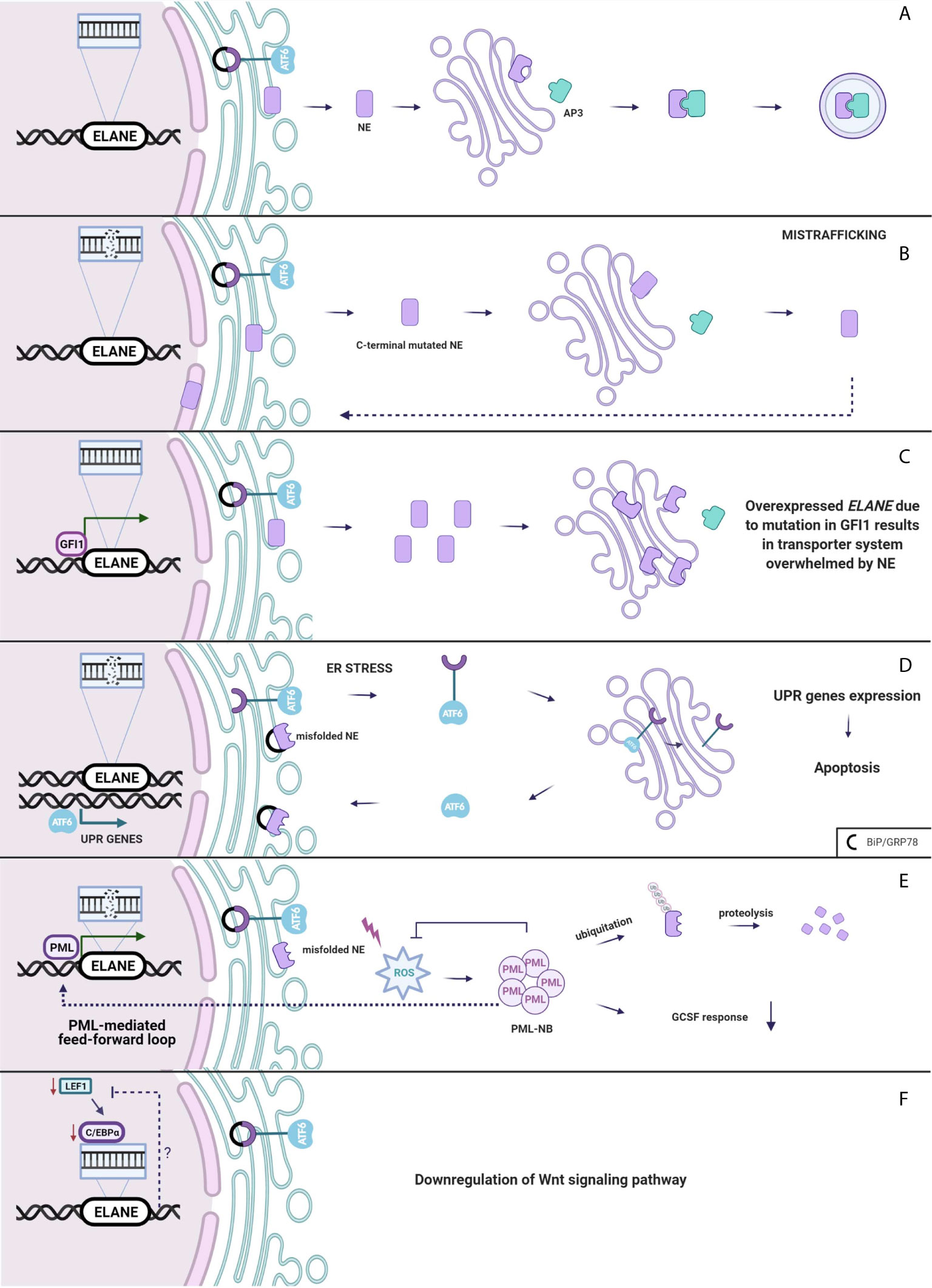
Figure 2 (A) Neutrophil elastase is trafficked from Golgi to the granules. (B) Mistrafficking. Mutation in ELANE results in intact C-terminal of NE disrupting an interaction between AP3 which mislocates it to the cytoplasm, plasma and nuclear membrane. (C) Overexpressed ELANE due to mutation in GFI1 results in the transporter system overwhelmed by NE. (D) Unfolded protein response. Misfolded NE accumulation in ER leads to the ER stress and molecular chaperones upregulation and a subsequent induction of the UPR. (E) PML-mediated feed-forward loop. PML binds to ELANE promoter inducing misfolded NE expression thus increases ROS production and PML-NBs formation. (F) Either Lef-1 downregulates ELANE downstream or the Wnt pathway is downregulated through a negative feedback loop from mutated ELANE. Created with BioRender.com.
Absence of AP-3 significantly reduces the amount of neutrophil elastase in the granules, leading to SCN in humans (60) and CyN in dogs (58). Mutations in ELANE cause a disruption of the interaction between NE and AP-3 resulting in mistrafficking and excessive routing of the enzyme to the plasma membrane (61), and the nuclear membrane (50) (Figure 2B). Additionally Köllner et al. described accumulation of NE in cytoplasm of primary granules in cells derived from congenital neutropenia patients (62). However, not the lack of NE in granule itself leads to neutropenia but the incorrect location of mutant protease provokes elastase aberrant behavior resulting in abnormal neutrophil maturation.
This leads to a hypothesis that the mutant NE does not completely lose its proteolytic properties and, through mislocalization, it spontaneously degrades molecules that are essential for promyelocyte development. Mutated, though still active, NE present in the cytoplasm and cell membrane may defectively interact with potential substrates such as granulocyte colony stimulating factor receptor (G-CSF-R) (63) and Notch-family proteins (64), which regulate granulopoiesis. Intriguingly, according to the Garg et al. article, neutrophil elastase is inactive, but significantly sequesters expressions of hematopoietic transcription factors like Gfi1, Cebpd, Cebpe, and Spi1, cell surface receptors Csf3r and Gr1 and Mpo granule protein, which all are markers of granulocyte differentiation (65).
GFI1 Overexpression Leads to NE Mislocalization
Interestingly, growth factor independence 1 (GFI1) has been found to have an indirect impact on NE mistrafficking. GFI1 encodes a nuclear transcription factor repressor GFI1, and it is widely expressed in the immune cells, playing a role in the development of hematopoietic cells (66). It has been found that Gfi1-deficient mice develop severe neutropenia and fail to produce mature granulocytes (67, 68). In normal conditions, GFI1 binds to the ELANE promoter, thus, repressing it. Depending on the mutation in GFI1, ELANE expression may either be increased or remain unchanged (69). Overexpression of neutrophil elastase leads to accumulation of the enzyme in multiple subcellular locations (70). Functional consequences of NE mislocalization are associated with impaired granulocytic differentiation (Figure 2C). NE is predominantly localized within granules but was also detected in the nucleus (71, 72). NE and GFI1 both were tested to bind to the nuclear protein PFAAP5, which silenced in HSC impaired myeloid differentiation. Thus, it is very likely that mutations of NE may influence the NE-PFAAP5-GFI1 interactions and result in abnormal modulations of transcription (73).
Misfolded NE Triggers the Unfolded Protein Response
Canonical UPR
Neutrophil elastase, like most eukaryotic proteins, is transported to the endoplasmic reticulum, where it folds and matures. Protein folding is a process strictly controlled by genes encoding ER-resident chaperones. Excessive unfolded proteins accumulation in ER leads to ER stress and a subsequent induction of the regulatory pathway called the unfolded protein response. UPR can respond in three ways. Firstly, it diminishes cell protein synthesis. Secondly, it enhances transcriptional activation of chaperones, which helps in the proper folding of proteins. If the first two systems fail, ER-stress continues and homeostasis cannot be evoked and as a final response, a cell death pathway is induced (74). There is much evidence which proves that overwhelmed UPR system aims towards apoptosis and gives rise to diseases such as: hepatic fibrosis (75), neurodegenerative diseases (76) and cancer (77). Induction of apoptosis through the UPR demands an extremely high accumulation of misfolded proteins at the site of ER. In SCN, at the promyelocytic stage, when majority of granulocytes are arrested, mutated NE is highly expressed, thus, contributing to the cell death through apoptosis (78). Mutations in ELANE may cause neutrophil elastase misfolding that consequently aggregates in the promyelocyte cytoplasm and induces apoptosis through the UPR system (62). Indeed, diverse ELANE mutations are associated with upregulation of BiP/GRP78 – molecular chaperone, synthetized during UPR activation (79). Additionally, studies indicate that mutations causing cyclic neutropenia activate UPR in myeloid-derived cell lines to a lesser extent than ELANE mutations specific only for SCN (79, 80). In cyclic neutropenia, hematopoietic stem cell differentiation is not fully arrested, thus, some of the HSC are able to escape the UPR-induced ER stress, respond to G-CSF and generate neutrophils. However, most of the HSC carrying mutated NE will suffer from the UPR stress, thereby leading to cell death (81). Despite accelerated apoptosis, in vitro and in vivo studies (where one of the most severe NE mutation p.Gly214Arg was expressed) demonstrate that in SCN, neutrophil differentiation itself is not disrupted (50, 82). Due to the unsuccessful protein folding, the UPR system kills neutrophils before G-CSF induction, during the highest mutated NE expression, at the stage of promyelocyte (Figure 2D).
ROS-Induced Misfolded Protein Degradation
Abundant evidence indicates that UPR is one of the crucial mechanisms in SCN and CyN pathogenesis related to ELANE mutations. However, there are studies that support alternative scenarios. In a cellular model of HL-60, human promyeloblast cell line transfected with mutant NE, none of the studied UPR markers, neither BiP/GRP78 nor ATF6 were expressed (83). Likewise, in a similar in vitro model of granulopoiesis was impaired in ELANE-mutated cell line, albeit the UPR was not induced (51). In a study by Olofsen et al., there was no upregulation of aforementioned UPR-related genes in hematopoietic progenitor cells (HPC) derived from SCN patients with ELANE and HAX1 mutations, but elevated levels of reactive oxygen species (ROS) were detected. However, only in HPC with ELANE mutations causing NE misfolding, levels of promyelocytic nuclear bodies (PML-NBs) were increased (84). PML-NBs are produced under excessive oxidative stress and target misfolded proteins for ubiquitination and further proteasomal degradation (85, 86). Moreover, misfolded NE activates ROS production that induces formation of PML-NBs. PML binds at the ELANE promoter and therefore, enhances the mutated ELANE transcription and misfolded NE expression that is described as a feed-forward mechanism. In addition, PML impairs the cell’s response to G-CSF. This leads to an alternative mechanism, where NE degradation is determined by the type of ELANE mutation and its influence on NE structure and folding, therefore, would proceed either through PML-NBs or by inducing the canonical UPR response triggered by ER-stress (Figure 2E) (84). However, as this is a newly described mechanism in terms of both pathogenesis and neoplastic transformation, more research on misfolded-related mutations and PML-NB needs to be performed.
WNT Signaling in SCN Pathogenesis
In 2006, Skokowa et al., analyzed mRNA expression of various transcription factors in CD33+ cells derived from SCN patients or healthy individuals. The most significant difference between the two groups with respect to expression among transcription factors was detected in lymphoid enhancer-binding factor 1 (LEF-1). In promyelocytes of the patients with SCN, LEF-1 mRNA expression was 20-fold downregulated as compared to the healthy controls. LEF-1 was found to be a decisive transcription factor which mediates granulocyte differentiation into mature neutrophils (87). It belongs to the family of proteins included in the Wnt3a pathway. In the induced pluripotent stem cells (iPSCs) isolated from SCN patient with ELANE gene mutation, most of the genes related to the Wnt3a pathway including LEF-1 and C/EBP-α were down-regulated. Moreover, addition of the Wnt3a protein to the cultured NE-mutated iPSCs resulted in a dose-dependent increase of mature neutrophils, which was comparable with the results obtained after addition of G-CSF (88). Disruptions in the Wnt3a pathway appear to represent another mechanism of SN development (Figure 2F).
Clinical Features and Therapy of SCN With ELANE Mutations
Congenital neutropenia is usually diagnosed in very young children and is linked with recurrent infectious incidences including recurrent pneumonia, mucositis, gingivitis, skin abscesses or otitis media. These bacterial infiltrates might result in life-threatening conditions including sepsis. Mainly bacterial infections are present in congenital neutropenia. All of these occurring within the first months of life (89). In general, the clinical presentation of congenital neutropenia is mainly classified as cyclic neutropenia (CyN) and severe congenital neutropenia (SCN), depending on intermittent or continuous lack of neutrophil-granulocytes in the peripheral blood, which corresponds with the occurrence of infections typical for neutropenia.
There are no specific clinical descriptions, which are associated with mutations of the ELANE gene (43). The neutrophil elastase gene is the only one identified so far as causative for cyclic neutropenia. Carriers of ELANE mutation suffered from pure neutropenia as a cause of immunodeficiency. No other clinical features apart from immunodeficiency are presented. The distinction between cyclic and severe forms is firstly based on clinical features and serial absolute neutrophil counts (ANC) measurement (90). CyN has an estimated prevalence of one per million in the population and is characterized by neutrophil counts fluctuating with generally 21-day periodicity between nearly normal and 0.2 × 109/l. Similar intervals of recurrence of clinical presentation as fever, skin and oropharyngeal infections are observed (41, 90, 91). However, exact time intervals may vary even between patients with the same ELANE mutation ((42). Interestingly, the same ELANE mutation present within the family might be associated with a clinical phenotype of CyN and severe congenital neutropenia suggesting that also other factors affect the clinical manifestation of the disease (92). SCN is estimated to be 3-4 per million births and is defined as a chronic reduction in the absolute number of neutrophils circulating in the blood below 0.5 × 109/l. SCN is manifested by more severe infectious episodes as CyN. The majority of symptoms are managed with antibiotics, specific glucocorticoids, anti-inflammatory drugs; however, the patient outcome depends on the G-CSF response (89). The G-CSF response in CyN patients is revealed as a shortening of the period of the cycle length from 21 days to approximately 14 days and, thus, improves treatment outcome (81, 83). In the case of ELANE-mutated SCN, bone marrow aspiration always shows a hematopoietic arrest at the promyelocytic stage of immature neutrophil precursors (93).
Contemporary Therapy of ELANE SCN With G-CSF or Hematopoietic Stem Cells Transplantation
G-CSF
Treatment of choice for SCN is the administration of granulocyte-colony stimulating factor, which increases the neutrophil number and improves survival and quality of life (94). Filgrastim, as a human, recombinant G-CSF, stimulates the bone marrow to produce and release granulocytes into the bloodstream, thus ≥10-fold increases the neutrophil and significantly reduces the severity of infections (5). Most of the patients with ELANE-related congenital neutropenia require exogenous G-CSF treatment. Only a few cases who have no history of severe infection might be subjected to antibiotic prophylaxis and wait-and-watch therapeutic strategy.
HSCT
In approximately 20% of SCN cases, the ANCs cannot be achieved in patients considered as non-responders, when G-CSF dosage is more than 50 µg/kg/24h, whilst the neutrophil count still remains at the level of <0.5x109 cells per l (95). In such cases, an allogeneic hematopoietic stem cell transplantation (HSCT) should be considered (96). Nevertheless, the optimal time point for transplantation in patients not responding to G-CSF is still debatable (97). A recent report from the French cohort has revealed that not only G-CSF unresponding patients but also patients with a high dose G-CSF requirement (>15 μg/kg/day) have benefited from HSCT by reducing the risk of the MDS and/or acute myeloid leukemia (AML) transformation (98). Another critical indication for HSCT is a presence of a mutation in CSF3R or RUNX1 genes within the bone marrow of patients with SCN, which significantly increases the risk of development of MDS/AML (99).
MDS/AML Transformation
The patients with CyN have no increased risk of myelodysplasia or leukemic transformation and patients with SCN have. Patients suffering from SCN have approximately 10% increased risk of development of myelodysplastic syndrome (MDS) and/or acute myelogenous leukemia (89, 100, 101). Some data suggest that particular mutations in ELANE and a high dose of G-CSF are responsible for MDS/leukemia risk (42). About 70% of MDS/AML patients acquire nonsense mutations affecting the cytoplasmic domain of CSF3R (the G-CSF receptor). This might evade the proapoptotic activity within myeloid precursor cells, which become prone to accumulate further genetic defects leading to leukemia. Importantly, mutations in the RUNX1 gene are by far the most frequent somatic secondary mutations in SCN-MDS/AML and preferentially occurred in CSF3R mutation clones. Additionally, mutations in SUZ12, ASXL1 and EP300 genes were also identified (102). Recent studies on the mice model have shown tertiary mutations in CXXC4 gene suggesting that TET2 dysfunction is also involved in MDS/AML evolution in SCN (103).
One possible explanation of leukemic risk among ELANE carriers is an increased ROS level induced by misfolded proteins leads to oxidative damage, which may be responsible for 2nd hits and initiates malignant transformation, so AML or MDS development. PML-NBs via MYC and mTOR signaling induce metabolism and cell cycling that promotes malignant cell proliferation (Figure 3) (84). However, looking at the clinical perspective, since approximately 70% of SCN patients with AML harbor additional mutations in the RUNX1 gene, patients with SCN who are positive for CSF3R and/or RUNX1 mutation should be also transplanted to minimize the risk of MDS/AML occurrence.
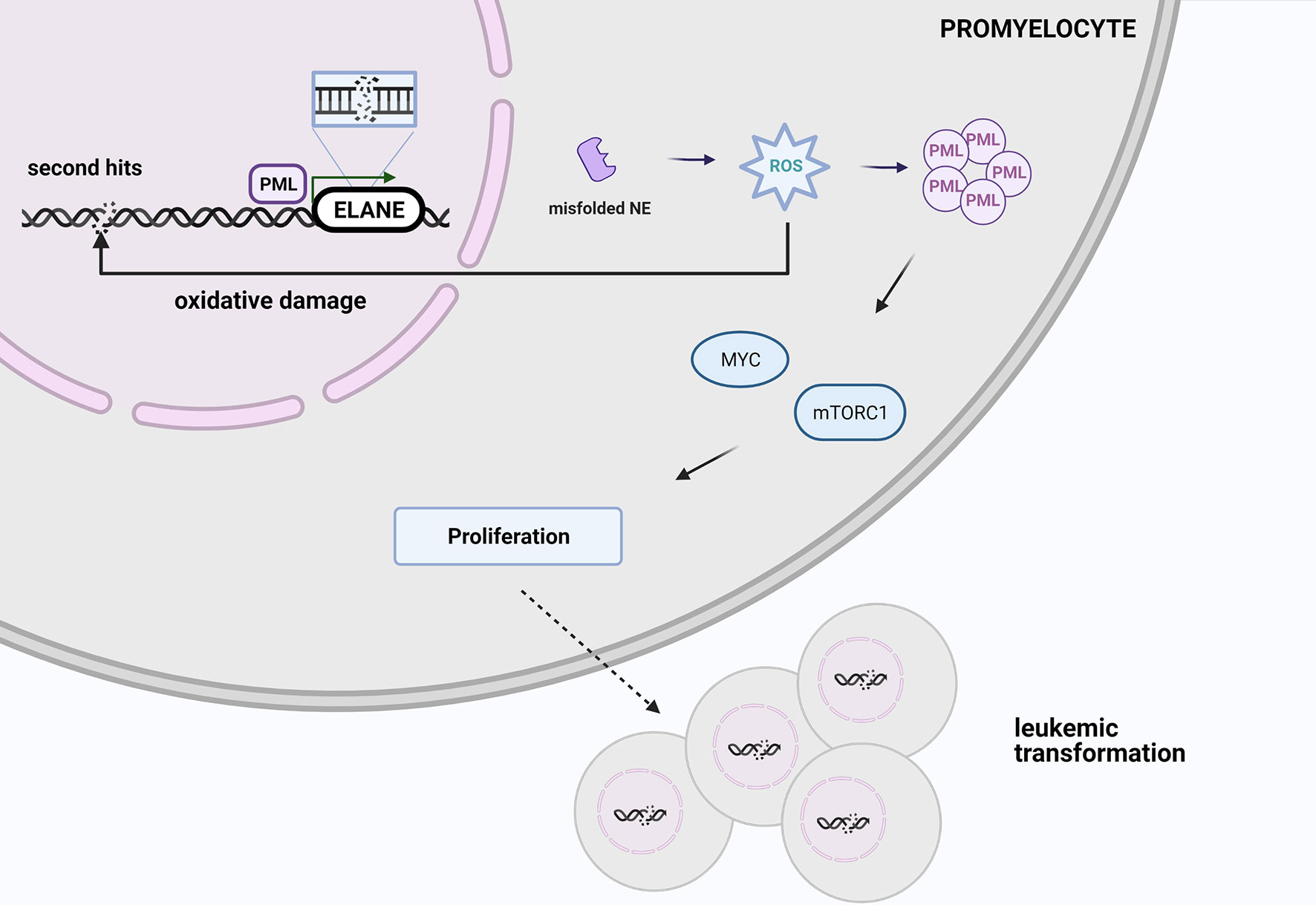
Figure 3 At the stage of promyelocyte the increased ROS level triggered by misfolded elastase leads to oxidative damage, which may be responsible for 2nd hits and initiate malignant transformation. PML-NBs via MYC and mTOR signaling promotes malignant cell proliferation. Created with BioRender.com.
Future Therapeutic Perspectives
So far, no effective treatment has been developed for patients considered to be non-responders or resistant to G-CSF. Below, we present current efforts and potential implementation of novel methods based on NE inhibition, enhancing G-CSF functions or CRISPR/Cas9 ELANE gene edition that may help these patients in future studies. However, it should be noted, that the described methods do not yet have clinical application.
Neutrophil Elastase Inhibitors
To treat neutropenia an administration of endogenous recombinant or synthetic inhibitors of NE has been studied in preclinical or clinical settings. The first effort was focused on a small molecular weight compound, sivelestat sodium hydrate, which is a specific inhibitor of polymorphonuclear neutrophil elastase. Dokai et al. have shown decreasing apoptotic effects of neutrophil elastase after neutralization of its enzymatic activity by sivelestat in K562 and MEG-01 cell lines (104). In 2017, Makaryan et al. presented a report on four β-lactam-derived compounds, i.e. MK0339, L-910, L-538 and L-635. They also selected four ELANE mutations, p.Pro139Leu, p.Cys151Tyr, p.V174_C181del and p.Gly214Arg, on the model of patient-derived iPSC and HL60 cell line. One of the drugs, MK0339, showed not only improved survival of HL-60 cells and iPSc, but also their potential to differentiate to mature neutrophils. Pharmacokinetics studies of MK0339 have revealed that this drug achieves a plasma concentration of less than 0.5 mg/ml after an oral dosage of 10 mg/kg (83). Based on these preclinical studies, inhibition of neutrophil elastase might be a therapeutic option in future. However, some data also suggest that enzyme activity of elastase is not really linked with neutropenia, thus the further studies are needed in this field.
Nicotinamide
A small form of vitamin B3, nicotinamide (NAM), was previously described as responsible for the upregulation of G-CSF and G-CSF receptors expression, through NAD+/SIRT1 deacetylation in cellular models (105). Interestingly, to date, NAM at doses far above those recommended for vitamins is suggested to be an effective and safe therapeutic option for a wide spectrum of diseases and conditions, including neurological dysfunctions, psychological disorders and inflammatory diseases (106). In 2021, Deordieva et al. have shown, that combined therapy of G-CSF with NA may have a beneficial impact on increasing neutrophil count. Within 18 analyzed patients including 14 who harbored ELANE mutation, who received NA orally (20 mg/kg/day) and G-CSF (0.6-50.8 µg/kg/day), 78% showed a gradual increase of ANC after 3 months of therapy. Moreover, in half of the patients, treatment with NA reduced G-CSF effective dose, with complete NA replacement in one patient (107). These data have the strong and promising perspective for a reduction of G-CSF clinical effectiveness, especially in the case of patients with neoplastic predispositions.
CRISPR/Cas9 Gene Editing
Recent studies reveal a potential application of CRISPR/Cas9 mediated gene therapy in patients with ELANE mutated SCN. In 2019 Ritter et al. published a report on efficient correction of ELANE mutations in primary hematopoietic stem and progenitor cells (HSPCs). They selected four ELANE mutations in two exons (p.Ala57Val and p.Ala57Thr in exon 2 and p.Gly214Arg and p.Gly214Val in exon 5) that are hot spot mutations in non-responders to G-CSF. Using CRISPR/Cas 9 to edit these mutations and adeno-associated virus 6 (rAAV6) to deliver the template for repair the structure by homology directed repair (HDR), they successfully achieved an increasing number of mature neutrophils with NET formation and chemotaxis improved. At the same time, there were no changes in functionality of the produced cells (108). In 2020, another team supervised by Skokowa successfully restored neutrophil “maturation arrest” in SCN cells by complete knockout of the ELANE gene (ELANE KO). They observed granulocytic differentiation as well as phagocytic ability, reactive oxygen species production and chemotaxis by neutrophils without ELANE in SCN patient-derived iPSc, primary hematopoietic stem cells and HL-60 cell line with ELANE KO. It suggests an alternative method of SCN treatment using ex vivo CRISPR/Cas9 ribonucleoprotein mediated ELANE KO (47). An alternated approach to repair ELANE mutations was performed using two different single guide RNA (sgRNA) targeting both mutant allele and exon 4. Using this method, a group headed by Chu successfully repaired ELANE mutation in SCN patient-derived hematopoietic stem and progenitor cells together with restored neutrophil differentiation and normal elastase expression level (109).
Moreover, the group headed by Daniel E. Bauer confirmed, that complete loss of NE is not associated with SCN using a new approach of ELANE gene edition within early exons, which elicited nonsense-mediated decay (NMD). They found that -1 frame insertions or deletions that produce premature termination codons escaped from NMD and were responsible for neutrophil maturation arrest, whereas -2 frame indels are tied with translation repression and neutrophil maturation (48). Those findings may be useful in therapeutic gene editing of human HSPC that trigger NMD and can restore normal neutrophil production, at the same time lay the groundwork for a new and universal therapeutic strategy for ELANE-mutant SCN.
All the above-mentioned studies focus on promising data to further analyze a neutrophil elastase inhibitor or even gene editing by CRISPR/Cas9 method and may pave the way to the novel therapies for SCN.
Conclusion
In light of the current literature and presented data on neutrophil elastase amount, localization, and activity there is no versatile model, which could explain how dysfunctions in NE lead to maturation arrest of granulocytic differentiation. Several hypotheses have emerged, the most popular of which indicates the role of dysregulated vesicular sorting and mistrafficking of mutated NE. The other theory points out the role of ER stress and unfolded protein response. In addition, genetic and epigenetic factors modulate these processes.
Although no clear genotype-phenotype correlation exists, the spectrum of mutations identified in neutropenic patients indicates that distribution of mutations across ELANE structure appears to be crucial. Recent studies have shown that ELANE whole gene deletion mutation does not cause neutropenia in humans and mice (45). Interestingly, ELANE knockout restores the granulopoiesis and increases differentiation into functional mature neutrophils in vitro (47). Therefore, we expect the neutrophil elastase gene to be a potential gene therapy target in patients with ELANE mutations, especially those who do not respond to G-CSF or who are at high risk of malignant transformation.
Author Contributions
ZR initiated the review article, reviewed the literature and drew the figures. BP reviewed the literature and prepared drafts of the manuscript. DK evaluated the literature, drew the figures and edited drafts of the manuscript. WM contributed to the conception and provided input throughout. JM designed and edited drafts of the manuscript and finalized the manuscript for submission. All authors contributed to the article and approved the submitted version.
Funding
The Foundation for Polish Science (FNP) TEAM NET Programme, POIR.04.04.00-00-1603/18. Project title: Fix Neutropenia (FIXNET): focusing on neutrophil proteases defects which serve as novel diagnostic and therapeutic options.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Borregaard N. Neutrophils, From Marrow to Microbes. Immunity (2010) 33:657–70. doi: 10.1016/j.immuni.2010.11.011
2. Rosales C. Neutrophil: A Cell With Many Roles in Inflammation or Several Cell Types? Front Physiol (2018) 9:1–17. doi: 10.3389/fphys.2018.00113
3. Hong CW. Current Understanding in Neutrophil Differentiation and Heterogeneity. Immune Netw (2017) 17:298–306. doi: 10.4110/in.2017.17.5.298
4. Cowland JB, Borregaard N. Granulopoiesis and Granules of Human Neutrophils. Immunol Rev (2016) 273:11–28. doi: 10.1111/imr.12440
5. Mehta HM, Malandra M, Corey SJ. G-CSF and GM-CSF in Neutropenia. J Immunol (2015) 195:1341–9. doi: 10.4049/jimmunol.1500861
6. Skokowa J, Dale DC, Touw IP, Zeidler C, Welte K. Severe Congenital Neutropenias. Nat Rev Dis Prim (2017) 3:17032. doi: 10.1038/nrdp.2017.32
7. Arun AK, Senthamizhselvi A, Hemamalini S, Edison ES, Korula A, Fouzia NA, et al. Spectrum of Elane Mutations in Congenital Neutropenia: A Single-Centre Study in Patients of Indian Origin. J Clin Pathol (2018) 71:1046–50. doi: 10.1136/jclinpath-2018-205235
8. Fu Z, Thorpe M, Akula S, Chahal G, Hellman LT. Extended Cleavage Specificity of Human Neutrophil Elastase, Human Proteinase 3, and Their Distant Ortholog Clawed Frog Pr3-Three Elastases With Similar Primary But Different Extended Specificities and Stability. Front Immunol (2018) 9:2387. doi: 10.3389/fimmu.2018.02387
9. Rao NV, Wehner NG, Marshall BC, Gray WR, Gray BH, Hoidal JR. Characterization of Proteinase-3 (Pr-3), a Neutrophil Serine Proteinase: Structural and Functional Properties. J Biol Chem (1991) 266:9540–8. doi: 10.1016/S0021-9258(18)92854-1
10. Pipoly DJ, Crouch EC. Degradation of Native Type IV Procollagen by Human Neutrophil Elastase. Implications for Leukocyte-Mediated Degradation of Basement Membranes. Biochemistry (1987) 26:5748–54. doi: 10.1021/bi00392a025
11. Heusinkveld LE, Majumdar S, Gao JL, McDermott DH, Murphy PM. Whim Syndrome: From Pathogenesis Towards Personalized Medicine and Cure. J Clin Immunol (2019) 39:532–56. doi: 10.1007/s10875-019-00665-w
12. Taylor JC, Crawford IP, Hugli TE. Limited Degradation of the Third Component (C3) of Human Complement by Human Leukocyte Elastase (Hle): Partial Characterization of C3 Fragments. Biochemistry (1977) 16:3390–6. doi: 10.1021/bi00634a016
13. Anderssen T, Halvorsen H, Bajaj SP, Osterud B. Human Leukocyte Elastase and Cathepsin G Inactivate Factor VII by Limited Proteolysis. Thrombosis Haemostasis (1993) 70(3):414–7. doi: 10.1055/s-0038-1649596
14. Si-Tahar M, Pidard D, Balloy V, Moniatte M, Kieffer N, Van Dorsselaer A, et al. Human Neutrophil Elastase Proteolytically Activates the Platelet Integrin A(Iib)B3 Through Cleavage of the Carboxyl Terminus of the A(Iib) Subunit Heavy Chain. Involvement in the Potentiation of Platelet Aggregation. J Biol Chem (1997) 272:11636–47. doi: 10.1074/jbc.272.17.11636
15. Epstein FH, Weiss SJ. Tissue Destruction by Neutrophils. N Engl J Med (1989) 320:365–76. doi: 10.1056/NEJM198902093200606
16. Owen CA, Campbell MA, Boukedes SS, Campbell EJ. Cytokines Regulate Membrane-Bound Leukocyte Elastase on Neutrophils: A Novel Mechanism for Effector Activity. Am J Physiol - Lung Cell Mol Physiol (1997) 272:L385–L93. doi: 10.1152/ajplung.1997.272.3.L385
17. Belaaouaj A, Mccarthy R, Baumann M, Gao Z, Ley TJ, Abraham SN, et al. Mice Lacking Neutrophil Elastase Reveal Impaired Host Defense Against Gram Negative Bacterial Sepsis. Nat Med (1998) 4:615–8. doi: 10.1038/nm0598-615
18. Garcia R, Gusmani L, Murgia R, Guarnaccia C, Cinco M, Rottini G, et al. Elastase is the Only Human Neutrophil Granule Protein That Alone is Responsible for in Vitro Killing of Borrelia Burgdorferi. Infect Immun (1998)66(4):1408–12. doi: 10.1128/IAI.66.4.1408-1412
19. Tkalcevic J, Novelli M, Phylactides M, Iredale JP, Segal AW, Roes J. Impaired Immunity and Enhanced Resistance to Endotoxin in the Absence of Neutrophil Elastase and Cathepsin G. Immunity (2000) 12:201–10. doi: 10.1016/S1074-7613(00)80173-9
20. Belaaouaj AA, Kwang SK, Shapiro SD. Degradation of Outer Membrane Protein a in Escherichia Coli Killing by Neutrophil Elastase. Sci (80- ) (2000) 289:1185–7. doi: 10.1126/science.289.5482.1185
21. Hirche TO, Benabid R, Deslee G, Gangloff S, Achilefu S, Guenounou M, et al. Neutrophil Elastase Mediates Innate Host Protection Against Pseudomonas Aeruginosa. J Immunol (2008) 181:4945–54. doi: 10.4049/jimmunol.181.7.4945
22. López-Boado YS, Espinola M, Bahr S, Belaaouaj A. Neutrophil Serine Proteinases Cleave Bacterial Flagellin, Abrogating Its Host Response-Inducing Activity. J Immunol (2004) 172:509–15. doi: 10.4049/jimmunol.172.1.509
23. Weinrauch Y, Drujan D, Shapiro SD, Weiss J, Zychlinsky A. Neutrophil Elastase Targets Virulence Factors of Enterobacteria. Nature (2002) 417:91–4. doi: 10.1038/417091a
24. Cai TQ, Wright SD. Human Leukocyte Elastase is an Endogenous Ligand for the Integrin Cr3 (Cd11b/Cd18, Mac-1 A(M)B2) and Modulates Polymorphonuclear Leukocyte Adhesion. J Exp Med (1996) 184:1213–23. doi: 10.1084/jem.184.4.1213
25. Champagne B, Tremblay P, Cantin A, St Pierre Y. Proteolytic Cleavage of ICAM-1 by Human Neutrophil Elastase. J Immunol (1998) 161(11):6398–405.
26. Nordahl EA, Rydengård V, Nyberg P, Nitsche DP, Mörgelin M, Malmsten M, et al. Activation of the Complement System Generates Antibacterial Peptides. Proc Natl Acad Sci USA (2004) 101:16879–84. doi: 10.1073/pnas.0406678101
27. Chua F, Laurent GJ. Neutrophil elastase: mediator of extracellular matrix destruction and accumulation. Proc Am Thorac Soc. (2006) 3(5):424–7. doi: 10.1513/pats.200603-078AW
28. Le-Barillec K, Si-Tahar M, Balloy V, Chignard M. Proteolysis of Monocyte CD14 by Human Leukocyte Elastase Inhibits Lipopolysaccharide-Mediated Cell Activation. J Clin Invest (1999) 103:1039–46. doi: 10.1172/JCI5779
29. Walsh DE, Greene CM, Carroll TP, Taggart CC, Gallagher PM, O’Neill SJ, et al. Interleukin-8 Up-Regulation by Neutrophil Elastase is Mediated by Myd88/IRAK/TRAF-6 in Human Bronchial Epithelium. J Biol Chem (2001) 276:35494–9. doi: 10.1074/jbc.M103543200
30. Devaney JM, Greene CM, Taggart CC, Carroll TP, O’Neill SJ, McElvaney NG. Neutrophil Elastase Up-Regulates Interleukin-8 Via Toll-Like Receptor 4. FEBS Lett (2003) 544:129–32. doi: 10.1016/S0014-5793(03)00482-4
31. Fouret P, Du Bois RM, Bernaudin JF, Takahashi H, Ferrans VJ, Crystal RG. Expression of the Neutrophil Elastase Gene During Human Bone Marrow Cell Differentiation. J Exp Med (1989) 169:833–45. doi: 10.1084/jem.169.3.833
32. Takahashi H, Nukiwa T, Yoshimura K, Quick CD, States DJ, Holmes MD, et al. Structure of the Human Neutrophil Elastase Gene. J Biol Chem (1988) 263:14739–47. doi: 10.1016/S0021-9258(18)68099-8
33. Dale DC, Person RE, Bolyard AA, Aprikyan AG, Bos C, Bonilla MA, et al. Mutations in the Gene Encoding Neutrophil Elastase in Congenital and Cyclic Neutropenia. Blood (2000) 96:2317–22. doi: 10.1182/blood.V96.7.2317.h8002317_2317_2322
34. Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. Clinvar: Improving Access to Variant Interpretations and Supporting Evidence. Nucleic Acids Res (2018) 46:D1062–7. doi: 10.1093/nar/gkx1153
35. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal (2013) 6. doi: 10.1126/scisignal.2004088
36. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov (2012) 2:401–4. doi: 10.1158/2159-8290.CD-12-0095
37. Sehnal D, Rose AS, Koča J, Burley SK, Velankar S. Mol*: Towards a Common Library and Tools for Web Molecular Graphics. In: Workshop on Molecular Graphics and Visual Analysis of Molecular Data Molva ‘18 (2018). Goslar, DEU: Eurographics Association. p. 29–33. doi: 10.2312/molva.20181103
38. Hansen G, Gielen-Haertwig H, Reinemer P, Schomburg D, Harrenga A, Niefind K, et al. Unexpected active-site flexibility in the structure of human neutrophil elastase in complex with a new dihydropyrimidone inhibitor. J Mol Biol (2011) 409:681–91. doi: 10.1016/j.jmb.2011.04.047
39. Horwitz MS, Corey SJ, Grimes HL, Tidwell T. Elane Mutations in Cyclic and Severe Congenital Neutropenia. Genetics and Pathophysiology. Hematol Oncol Clin North Am (2013) 27:19–41. doi: 10.1016/j.hoc.2012.10.004
40. Korkmaz B, Moreau T, Gauthier F. Neutrophil Elastase, Proteinase 3 and Cathepsin G: Physicochemical Properties, Activity and Physiopathological Functions. Biochimie (2008) 90:227–42. doi: 10.1016/j.biochi.2007.10.009
41. Horwitz M, Benson KF, Person RE, Aprikyan AG, Dale DC. Mutations in ELA2, Encoding Neutrophil Elastase, Define a 21-Day Biological Clock in Cyclic Haematopoiesis. Nat Genet (1999) 23:433–6. doi: 10.1038/70544
42. Makaryan V, Zeidler C, Bolyard AA, Skokowa J, Rodger E, Kelley ML, et al. The Diversity of Mutations and Clinical Outcomes for ELANE-Associated Neutropenia. Curr Opin Hematol (2015) 22:3–11. doi: 10.1097/MOH.0000000000000105
43. Germeshausen M, Deerberg S, Peter Y, Reimer C, Kratz CP, Ballmaier M. The Spectrum of ELANE Mutations and Their Implications in Severe Congenital and Cyclic Neutropenia. Hum Mutat (2013) 34:905–14. doi: 10.1002/humu.22308
44. Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NST, et al. Human Gene Mutation Database (Hgmd®): 2003 Update. Hum Mutat (2003) 21:577–81. doi: 10.1002/humu.10212
45. Horwitz MS, Laurino MY, Keel SB. Normal Peripheral Blood Neutrophil Numbers Accompanying Elane Whole Gene Deletion Mutation. Blood Adv (2019) 3:2470–3. doi: 10.1182/bloodadvances.2019000498
46. Martinod K, Witsch T, Farley K, Gallant M, Remold-O’Donnell E, Wagner DD. Neutrophil Elastase-Deficient Mice Form Neutrophil Extracellular Traps in an Experimental Model of Deep Vein Thrombosis. J Thromb Haemost (2016) 14:551–8. doi: 10.1111/jth.13239
47. Nasri M, Ritter M, Mir P, Dannenmann B, Aghaallaei N, Amend D, et al. Crispr/Cas9-Mediated ELANE Knockout Enables Neutrophilic Maturation of Primary Hematopoietic Stem and Progenitor Cells and Induced Pluripotent Stem Cells of Severe Congenital Neutropenia Patients. Haematologica (2020) 105:598–609. doi: 10.3324/haematol.2019.221804
48. Rao S, Yao Y, Soares de Brito J, Yao Q, Shen AH, Watkinson RE, et al. Dissecting ELANE Neutropenia Pathogenicity by Human Hsc Gene Editing. Cell Stem Cell (2021) S1934–5909(20)30599–3. doi: 10.1016/j.stem.2020.12.015
49. Nagy E, Maquat LE. A Rule for Termination-Codon Position Within Intron-Containing Genes: When Nonsense Affects RNA Abundance. Trends Biochem Sci (1998) 23:198–9. doi: 10.1016/S0968-0004(98)01208-0
50. Massullo P, Druhan LJ, Bunnell BA, Hunter MG, Robinson JM, Marsh CB, et al. Aberrant Subcellular Targeting of the G185R Neutrophil Elastase Mutant Associated With Severe Congenital Neutropenia Induces Premature Apoptosis of Differentiating Promyelocytes. Blood (2005) 105:3397–404. doi: 10.1182/blood-2004-07-2618
51. Tidwell T, Wechsler J, Nayak RC, Trump L, Salipante SJ, Cheng JC, et al. Neutropenia-Associated ELANE Mutations Disrupting Translation Initiation Produce Novel Neutrophil Elastase Isoforms. Blood (2014) 123:562–9. doi: 10.1182/blood-2013-07-513242
52. Lanciotti M, Caridi G, Rosano C, Pigullo S, Lanza T, Dufour C. Severe Congenital Neutropenia: A Negative Synergistic Effect of Multiple Mutations of ELANE (Ela2) Gene. Br J Haematol (2009) 146:578–80. doi: 10.1111/j.1365-2141.2009.07787.x
53. Park CH, Park S, Kim YJ, Kim SH, Kim HJ. Cyclic Neutropenia From a Novel Mutation Ala57Asp of ELANE: Phenotypic Variability in Neutropenia From Mutated Ala57 Residue. J Pediatr Hematol Oncol (2018) 42(4):e231–4. doi: 10.1097/MPH.0000000000001353
54. Rosenberg PS, Alter BP, Link DC, Stein S, Rodger E, Bolyard AA, et al. Neutrophil Elastase Mutations and Risk of Leukaemia in Severe Congenital Neutropenia. Br J Haematol (2008) 140(2):210–3. doi: 10.1111/j.1365-2141.2007.06897.x
55. Bellanné-Chantelot C, Clauin S, Leblanc T, Cassinat B, Rodrigues-Lima F, Beaufils S, et al. Mutations in the ELA2 Gene Correlate With More Severe Expression of Neutropenia: A Study of 81 Patients From the French Neutropenia Register. Blood (2004) 103:4119–25. doi: 10.1182/blood-2003-10-3518
56. Rosenberg PS, Zeidler C, Anna Bolyard A, Alter BP, Ann Bonilla M, Boxer LA, et al. Stable Long-Term Risk of Leukaemia in Patients With Severe Congenital Neutropenia Maintained on G-CSF Therapy. Br J Haematol (2010) 150:196–9. doi: 10.1111/j.1365-2141.2010.08216.x
57. Li FQ, Horwitz M. Characterization of Mutant Neutrophil Elastase in Severe Congenital Neutropenia. J Biol Chem (2001) 276:14230–41. doi: 10.1074/jbc.M010279200
58. Benson KF, Li FQ, Person RE, Albani D, Duan Z, Wechsler J, et al. Mutations Associated With Neutropenia in Dogs and Humans Disrupt Intracellular Transport of Neutrophil Elastase. Nat Genet (2003) 35:90–6. doi: 10.1038/ng1224
59. Simpson F, Peden AA, Christopoulou L, Robinson MS, Darnell RB, Robinson MS, et al. Characterization of the Adaptor-Related Protein Complex, Ap-3. J Cell Biol (1997) 137(4):835–45. doi: 10.1083/jcb.137.4.835
60. Dell’Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered Trafficking of Lysosomal Proteins in Hermansky-Pudlak Syndrome Due to Mutations in the B3a Subunit of the AP-3 Adaptor. Mol Cell (1999) 3:11–21. doi: 10.1016/S1097-2765(00)80170-7
61. Horwitz M, Benson KF, Duan Z, Li F-Q, Person RE. Hereditary Neutropenia: Dogs Explain Human Neutrophil Elastase Mutations. Trends Mol Med (2004) 10:163–70. doi: 10.1016/j.molmed.2004.02.002
62. Köllner I, Sodeik B, Schreek S, Heyn H, Von Neuhoff N, Germeshausen M, et al. Mutations in Neutrophil Elastase Causing Congenital Neutropenia Lead to Cytoplasmic Protein Accumulation and Induction of the Unfolded Protein Response. Blood (2006) 108:493–500. doi: 10.1182/blood-2005-11-4689
63. Hunter MG, Druhan LJ, Massullo PR, Avalos BR. Proteolytic Cleavage of Granulocyte Colony-Stimulating Factor and Its Receptor by Neutrophil Elastase Induces Growth Inhibition and Decreased Cell Surface Expression of the Granulocyte Colony-Stimulating Factor Receptor. Am J Hematol (2003) 74:149–55. doi: 10.1002/ajh.10434
64. Duan Z, Li F-Q, Wechsler J, Meade-White K, Williams K, Benson KF. Horwitz M. A Novel Notch Protein, N2n, Targeted by Neutrophil Elastase and Implicated in Hereditary Neutropenia. Mol Cell Biol (2004) 24:58–70. doi: 10.1128/MCB.24.1.58-70.2004
65. Garg B, Mehta HM, Wang B, Kamel R, Marshall S, Corey SJ, et al. Inducible Expression of a Disease-Associated Elane Mutation Impairs Granulocytic Differentiation, Without Eliciting an Unfolded Protein Response. J Biol Chem (2020) 295(21):7492–500. doi: 10.1074/jbc.RA120.012366
66. Hock H, Hamblen MJ, Rooke HM, Traver D, Bronson RT, Cameron S, et al. Intrinsic Requirement for Zinc Finger Transcription Factor Gfi-1 in Neutrophil Differentiation. Immunity (2003) 18:109–20. doi: 10.1016/S1074-7613(02)00501-0
67. Karsunky H, Zeng H, Schmidt T, Zevnik B, Kluge R, Schmid KW, et al. Inflammatory Reactions and Severe Neutropenia in Mice Lacking the Transcriptional Repressor Gfi1. Nat Genet (2002) 30:295–300. doi: 10.1038/ng831
68. Geissler S, Textor M, Stumpp S, Seitz S, Lekaj A, Brunk S, et al. Loss of Murine Gfi1 Causes Neutropenia and Induces Osteoporosis Depending on the Pathogen Load and Systemic Inflammation. PloS One (2018) 13:1–21. doi: 10.1371/journal.pone.0198510
69. Muench DE, Olsson A, Ferchen K, Pham G, Serafin RA, Chutipongtanate S, et al. Mouse Models of Neutropenia Reveal Progenitor-Stage-Specific Defects. Nature (2020) 582:109–14. doi: 10.1038/s41586-020-2227-7
70. Person RE, Li F, Duan Z, Benson KF, Papadaki HA, Eliopoulos G, et al. Mutations in proto-oncogene GFI1 cause human neutropenia and target ELA2. Nat Genet (2003) 34(3):308–12. doi: 10.1038/ng1170
71. Lane AA, Ley TJ. Neutrophil Elastase Cleaves PML-Raralpha and is Important for the Development of Acute Promyelocytic Leukemia in Mice. Cell (2003) 115:305–18. doi: 10.1016/S0092-8674(03)00852-3
72. Armistead PM, Wieder E, Akande O, Alatrash G, Quintanilla K, Liang S, et al. Cyclic Neutropenia Associated With T Cell Immunity to Granulocyte Proteases and a Double De Novo Mutation in GFI1, a Transcriptional Regulator of ELANE. Br J Haematol (2010) 150:716–9. doi: 10.1111/j.1365-2141.2010.08274.x
73. Salipante SJ, Rojas MEB, Korkmaz B, Duan Z, Wechsler J, Benson KF, et al. Contributions to Neutropenia From PFAAP5 (N4BP2L2), a Novel Protein Mediating Transcriptional Repressor Cooperation Between Gfi1 and Neutrophil Elastase. Mol Cell Biol (2009) 29:4394–405. doi: 10.1128/MCB.00596-09
74. Ron D, Walter P. Signal Integration in the Endoplasmic Reticulum Unfolded Protein Response. Nat Rev Mol Cell Biol (2007) 8:519–29. doi: 10.1038/nrm2199
75. Maiers JL, Malhi H. Endoplasmic Reticulum Stress in Metabolic Liver Diseases and Hepatic Fibrosis. Semin Liver Dis (2019) 39:235–48. doi: 10.1055/s-0039-1681032
76. Shah SZA, Zhao D, Khan SH, Yang L. Unfolded Protein Response Pathways in Neurodegenerative Diseases. J Mol Neurosci (2015) 57:529–37. doi: 10.1007/s12031-015-0633-3
77. Ojha R, Amaravadi RK. Targeting the Unfolded Protein Response in Cancer. Pharmacol Res (2017) 120:258–66. doi: 10.1016/j.phrs.2017.04.003
78. Pham CTN. Neutrophil Serine Proteases: Specific Regulators of Inflammation. Nat Rev Immunol (2006) 6:541–50. doi: 10.1038/nri1841
79. Grenda DS, Murakami M, Ghatak J, Xia J, Boxer LA, Dale D, et al. Mutations of the ELA2 Gene Found in Patients With Severe Congenital Neutropenia Induce the Unfolded Protein Response and Cellular Apoptosis. Blood (2007) 110:4179–87. doi: 10.1182/blood-2006-11-057299
80. Nustede R, Klimiankou M, Klimenkova O, Kuznetsova I, Zeidler C, Welte K, et al. Elane Mutant-Specific Activation of Different Upr Pathways in Congenital Neutropenia. Br J Haematol (2016) 172:219–27. doi: 10.1111/bjh.13823
81. Mir P, Klimiankou M, Findik B, Hähnel K, Mellor-Heineke S, Zeidler C, et al. New Insights Into the Pathomechanism of Cyclic Neutropenia. Ann N Y Acad Sci (2020)1466(1):83–92. doi: 10.1111/nyas.14309
82. Wiesmeier M, Gautam S, Kirschnek S, Häcker G. Characterisation of Neutropenia-Associated Neutrophil Elastase Mutations in a Murine Differentiation Model in Vitro and In Vivo. PloS One (2016) 11:1–20. doi: 10.1371/journal.pone.0168055
83. Makaryan V, Kelley ML, Fletcher B, Bolyard AA, Aprikyan AA, Dale DC. Elastase Inhibitors as Potential Therapies for ELANE -Associated Neutropenia. J Leukoc Biol (2017) 102:1143–51. doi: 10.1189/jlb.5A1016-445R
84. Olofsen PA, Bosch DA, Roovers O, van Strien PMH, de Looper HWJ, Hoogenboezem RM, et al. Pml-Controlled Responses in Severe Congenital Neutropenia With ELANE-Misfolding Mutations. Blood Adv (2021) 5:775–86. doi: 10.1182/bloodadvances.2020003214
85. Guo L, Giasson BI, Glavis-Bloom A, Brewer MD, Shorter J, Gitler AD, et al. A Cellular System That Degrades Misfolded Proteins and Protects Against Neurodegeneration. Mol Cell (2014) 55:15–30. doi: 10.1016/j.molcel.2014.04.030
86. Becker S, Steinemann G, Karle W, Roos K, Liem CH, Muralikumar S, et al. Stability of Smyd1 in Endothelial Cells is Controlled by PML-Dependent Sumoylation Upon Cytokine Stimulation. Biochem J (2021) 478:217–34. doi: 10.1042/BCJ20200603
87. Skokowa J, Cario G, Uenalan M, Schambach A, Germeshausen M, Battmer K, et al. LEF-1 is Crucial for Neutrophil Granulocytopoiesis and Its Expression is Severely Reduced in Congenital Neutropenia. Nat Med (2006) 12:1191–7. doi: 10.1038/nm1474
88. Hiramoto T, Ebihara Y, Mizoguchi Y, Nakamura K, Yamaguchi K, Ueno K, et al. Wnt3a Stimulates Maturation of Impaired Neutrophils Developed From Severe Congenital Neutropenia Patient-Derived Pluripotent Stem Cells. Proc Natl Acad Sci USA (2013) 110:3023–8. doi: 10.1073/pnas.1217039110
89. Dale DC, Cottle TE, Fier CJ, Bolyard AA, Bonilla MA, Boxer LA, et al. Severe Chronic Neutropenia: Treatment and Follow-Up of Patients in the Severe Chronic Neutropenia International Registry. Am J Hematol (2003) 72:82–93. doi: 10.1002/ajh.10255
90. Dale DC, Hammond WP IV. Cyclic Neutropenia: A Clinical Review. Blood Rev (1988) 2:178–85. doi: 10.1016/0268-960X(88)90023-9
91. Dale DC, Bolyard AA, Aprikyan A. Cyclic Neutropenia. Semin Hematol (2002) 39:89–94. doi: 10.1053/shem.2002.31917
92. Newburger PE, Pindyck TN, Zhu Z, Bolyard AA, Aprikyan AAG, Dale DC, et al. Cyclic Neutropenia and Severe Congenital Neutropenia in Patients With a Shared Elane Mutation and Paternal Haplotype: Evidence for Phenotype Determination by Modifying Genes. Pediatr Blood Cancer (2010) 55:314–7. doi: 10.1002/pbc.22537
93. Liu Q, Sundqvist M, Li W, Holdfeldt A, Zhang L, Björkman L, et al. Functional Characteristics of Circulating Granulocytes in Severe Congenital Neutropenia Caused by ELANE Mutations. BMC Pediatr (2019) 19:189. doi: 10.1186/s12887-019-1556-x
94. Bonilla MA, Gillio AP, Ruggeiro M, Kernan NA, Brochstein JA, Abboud M, et al. Effects of Recombinant Human Granulocyte Colony-Stimulating Factor on Neutropenia in Patients With Congenital Agranulocytosis. N Engl J Med (1989) 320:1574–80. doi: 10.1056/NEJM198906153202402
95. Dale DC, Bonilla MA, Davis MW, Nakanishi AM, Hammond WP, Kurtzberg J, et al. A Randomized Controlled Phase Iii Trial of Recombinant Human Granulocyte Colony-Stimulating Factor (Filgrastim) for Treatment of Severe Chronic Neutropenia. Blood (1993) 81:2496–502. doi: 10.1182/blood.V81.10.2496.bloodjournal81102496
96. Ferry C, Ouachée M, Leblanc T, Michel G, Notz-Carrére A, Tabrizi R, et al. Hematopoietic Stem Cell Transplantation in Severe Congenital Neutropenia: Experience of the French Scn Register. Bone Marrow Transplant (2005) 35:45–50. doi: 10.1038/sj.bmt.1704718
97. Choi SW, Boxer LA, Pulsipher MA, Roulston D, Hutchinson RJ, Yanik GA, et al. Stem Cell Transplantation in Patients With Severe Congenital Neutropenia With Evidence of Leukemic Transformation. Bone Marrow Transplant (2005) 35:473–7. doi: 10.1038/sj.bmt.1704813
98. Rotulo GA, Beaupain B, Rialland F, Paillard C, Nachit O, Galambrun C, et al. Hsct May Lower Leukemia Risk in ELANE Neutropenia: A Before–After Study From the French Severe Congenital Neutropenia Registry. Bone Marrow Transplant (2020) 55(8):1614–22. doi: 10.1038/s41409-020-0800-1
99. Skokowa J, Steinemann D, Katsman-Kuipers JE, Zeidler C, Klimenkova O, Klimiankou M, et al. Cooperativity of RUNX1 and CSF3R Mutations in Severe Congenital Neutropenia: A Unique Pathway in Myeloid Leukemogenesis. Blood (2014) 123:2229–37. doi: 10.1182/blood-2013-11-538025
100. Berliner N. Lessons From Congenital Neutropenia: 50 Years of Progress in Understanding Myelopoiesis. Blood (2008) 111:5427–32. doi: 10.1182/blood-2007-10-077396
101. Bejjani N, Beaupain B, Bertrand Y, Bellanne-Chantelot C, Donadieu J. How to Differentiate Congenital From Noncongenital Chronic Neutropenia At the First Medical Examination? Proposal of Score: A Pilot Study From the French Severe Chronic Neutropenia Registry. Pediatr Blood Cancer (2017) 64(12). doi: 10.1002/pbc.26722
102. Beekman R, Valkhof MG, Sanders MA, Van Strien PMH, Haanstra JR, Broeders L, et al. Sequential Gain of Mutations in Severe Congenital Neutropenia Progressing to Acute Myeloid Leukemia. Blood (2012) 119:5071–7. doi: 10.1182/blood-2012-01-406116
103. Olofsen PA, Fatrai S, van Strien PMH, Obenauer JC, de Looper HWJ, Hoogenboezem RM, et al. Malignant Transformation Involving CXXC4 Mutations Identified in a Leukemic Progression Model of Severe Congenital Neutropenia. Cell Rep Med (2020) 1(5):100074. doi: 10.1016/j.xcrm.2020.100074
104. Dokai M, Madoiwa S, Yasumoto A, Kashiwakura Y, Ishiwata A, Sakata A, et al. Local Regulation of Neutrophil Elastase Activity by Endogenous A1-Antitrypsin in Lipopolysaccharide-Primed Hematological Cells. Thromb Res (2011) 128:283–92. doi: 10.1016/j.thromres.2011.04.024
105. Skokowa J, Lan D, Thakur BK, Wang F, Gupta K, Cario G, et al. NAMPT is Essential for the G-CSF-Induced Myeloid Differentiation Via a NAD+-Sirtuin-1-Dependent Pathway. Nat Med (2009) 15:151–8. doi: 10.1038/nm.1913
106. Hwang ES, Song SB. Possible Adverse Effects of High-Dose Nicotinamide: Mechanisms and Safety Assessment. Biomolecules (2020) 10(5):687. doi: 10.3390/biom10050687
107. Deordieva E, Shvets O, Voronin K, Maschan A, Welte K, Skokowa J, et al. Nicotinamide (Vitamin B3) Treatment Improves Response to G-CSF in Severe Congenital Neutropenia Patients. Br J Haematol (2021) 192(4):788–92. doi: 10.1111/bjh.17313
108. Ritter MU, Secker B, Klimiankou M, Nasri M, Aghaallaei N, Dannenmann B, et al. Efficient Correction of ELANE Mutations in Primary Hspcs of Severe Congenital Neutropenia Patients Using CRISPR/Cas9 and Ravv6 Hdr Repair Templates. Blood (2019) 134:1036–6. doi: 10.1182/blood-2019-131708
Keywords: severe congenital neutropenia, cyclic neutropenia, neutrophil elastase, ELANE mutations, unfolded protein response, mistrafficking, mislocalization
Citation: Rydzynska Z, Pawlik B, Krzyzanowski D, Mlynarski W and Madzio J (2021) Neutrophil Elastase Defects in Congenital Neutropenia. Front. Immunol. 12:653932. doi: 10.3389/fimmu.2021.653932
Received: 15 January 2021; Accepted: 06 April 2021;
Published: 22 April 2021.
Edited by:
Hans AR Bluyssen, Adam Mickiewicz University, PolandReviewed by:
Ivo Paul Touw, Erasmus University Rotterdam, NetherlandsSeth Corey, Cleveland Clinic, United States
Jean Donadieu, Hôpital Armand Trousseau, France
Copyright © 2021 Rydzynska, Pawlik, Krzyzanowski, Mlynarski and Madzio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joanna Madzio, am9hbm5hLm1hZHppb0B1bWVkLmxvZHoucGw=
†These authors have contributed equally to this work and share last authorship