 Fabian A. Fischer
Fabian A. Fischer Kaiwen W. Chen
Kaiwen W. Chen Jelena S. Bezbradica
Jelena S. Bezbradica- 1Kennedy Institute of Rheumatology, NDORMS, University of Oxford, Oxford, United Kingdom
- 2Immunology Programme and Department of Microbiology & Immunology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore, Singapore
Pyroptosis is a proinflammatory form of cell death, mediated by membrane pore-forming proteins called gasdermins. Gasdermin pores allow the release of the pro-inflammatory cytokines IL-1β and IL-18 and cause cell swelling and cell lysis leading to release of other intracellular proteins that act as alarmins to perpetuate inflammation. The best characterized, gasdermin D, forms pores via its N-terminal domain, generated after the cleavage of full length gasdermin D by caspase-1 or -11 (caspase-4/5 in humans) typically upon sensing of intracellular pathogens. Thus, gasdermins were originally thought to largely contribute to pathogen-induced inflammation. We now know that gasdermin family members can also be cleaved by other proteases, such as caspase-3, caspase-8 and granzymes, and that they contribute to sterile inflammation as well as inflammation in autoinflammatory diseases or during cancer immunotherapy. Here we briefly review how and when gasdermin pores are formed, and then focus on emerging endogenous mechanisms and therapeutic approaches that could be used to control pore formation, pyroptosis and downstream inflammation.
Introduction: Gasdermin-Mediated Cell Death as a Driver of Inflammation
Apoptosis is traditionally viewed as a non-inflammatory form of caspase-dependent programmed cell death (1). During apoptosis, caspase-mediated inactivation of innate immune signaling molecules and the preservation of membrane integrity ensures that apoptotic cells remain immunologically silent (albeit in some cases, cells can transition from apoptosis to other, more inflammatory forms of cell death) (2). Pyroptosis and necroptosis are inflammatory types of programmed cell death, driven by dedicated membrane pore-forming proteins, gasdermins (pyroptosis) or MLKL (necroptosis), respectively. During pyroptosis and necroptosis, the cell loses plasma membrane integrity, ruptures and uncontrollably releases the cytosolic content including cytosolic alarmins that drive inflammation (3).
Pyroptosis can drive both microbe-induced and sterile inflammation and can be beneficial or pathological. For example, during infection, gasdermin D (GSDMD) is cleaved by caspase-1, caspase-8 and caspase-11 (caspase 4/5 in humans), to release its membrane pore-forming fragment and induce pyroptotic death of infected cells (4–10). This is beneficial, as pyroptosis releases alarmins and destroys the cellular niche for pathogen replication (11, 12). But if excessive, pyroptosis can cause immunopathology, and in fact, caspase-1/11, caspase-11 and GSDMD-deficient mice are protected from mouse models of lethal LPS- and TNF-induced shock and polymicrobial sepsis (4, 13–20). Gasdermins can also drive sterile inflammation, which can also be beneficial or pathological. For example, gasdermin B (GSMDB) and gasdermin E (GSDME) are cleaved into their membrane pore-forming fragments by the enzymes granzyme A and B, respectively, when granzymes are delivered directly into the tumor cell during the attack by cytotoxic T cells. Once cleaved, GSDMB and GSMDE induce the death of tumor cells by pyroptosis, resulting in low-grade local inflammation that is essential for successful clearance of tumors by myeloid cells (21, 22). If however GSDME-mediated pyroptosis of tumor cells is excessive, as seen in response to chimeric antigen receptor (CAR) T cell therapy, dying cells release alarmins, activate caspase-1/GSDMD pathway in recruited macrophages, and cause systemic cytokine release syndrome, a common complication of CAR T cell therapy (23). Similarly, several chemotherapy drugs that were designed to induce non-inflammatory apoptosis of tumors, end up causing systemic pathology, by activating the apoptotic caspase-3, which can cleave GSDME into its pore-forming fragment. These systemic drugs thus induce pyroptosis not only in GSDME-positive tumors, but also in other GSDME-expressing healthy cells, leading to wide-spread inflammation, tissue damage and weight loss (24). Select chemotherapeutic agents have also been described to activate caspase-8-dependent GSDMC activation in tumor cells (25), or GSDMD activation in myeloid cells (10), however, whether myeloid GSDMD activation promotes or dampens tumor growth in vivo remains unclear. Finally, GSDMD deletion is protective in several mouse models of inherited and acquired sterile inflammatory diseases, such as Familial Mediterranean Fever, Neonatal-Onset Multisystem Inflammatory Disease, Experimental Autoimmune Encephalomyelitis, or liver damage (26–31). Therefore, understanding how and where gasdermins are activated and how pyroptosis can be regulated, will provide new opportunities for the control of inflammation.
Many excellent reviews have discussed in detail events leading to gasdermin activation (32, 33). Here we will focus briefly on how and when gasdermin pores are formed, and then focus on emerging endogenous posttranslational mechanisms and therapeutic approaches that could be used to control gasdermin pore formation, pyroptosis and downstream inflammation.
Gasdermins: Expression, Function and Localization
Gasdermins are a family of newly described proteins that are emerging as key players in inflammation. Humans express six gasdermin family proteins: GSDMA, GSDMB, GSDMC, GSDMD, GSDME (formerly called DFNA5) and PJVK. In contrast, mice and rats do not express Gsdmb, but instead, have three GSDMA homologs (Gsdma1–Gsdma3) and four GSDMC homologs (Gsdmc1–Gsdmc4) (34). Gasdermin family proteins are differentially expressed in various tissues and we are only beginning to understand the biological functions of these proteins (33). Of which, the pore-forming properties of GSDMD and GSDME in myeloid cells and tumors have gained considerable attention recently and will be the focus of this review.
Gasdermins contain a cytotoxic N-terminal domain (GSDM-NT) and an autoinhibitory C-terminal domain (GSDM-CT), connected by a linker region that harbors a protease cleavage site (33). Microbial infection or cellular stress promotes the assembly of a cytosolic multiprotein inflammasome complex, which serves as a platform to activate inflammatory caspases, caspase-1 and -11 (caspase-4/5 in humans). These activated caspases as well as the proteases neutrophil elastase and cathepsin G cleave GSDMD within its linker region and liberate the cytotoxic GSDMD-NT to trigger plasma membrane damage and cell lysis by pyroptosis (Figure 1A) (4–6, 35, 36). Interestingly, the apoptotic executioner caspases-3 and-7 can cleave gasdermin D outside of the linker region at Asp87 leading to its inactivation and thereby negative regulation of pyroptosis (37). More recently, blockade of NF-κB and MAPK signaling, or perturbation in RIPK1 post-translational modifications by bacterial effectors or chemotherapeutic drugs were also demonstrated to promote GSDMD cleavage (8–10). Surprisingly, GSDMD cleavage under such circumstances occurred largely independently of inflammatory caspase-1/11, but instead, is mediated via apoptotic caspase-8. While new and exciting functions of GSDMD are mainly characterized in myeloid cells, the role of GSDME in host defense remains poorly characterized and is mainly characterized in tumor cells, where cleavage of GSDME by apoptotic caspase-3 and -7 or granzyme B switches tumor cell apoptosis to pyroptosis (7, 22, 24).
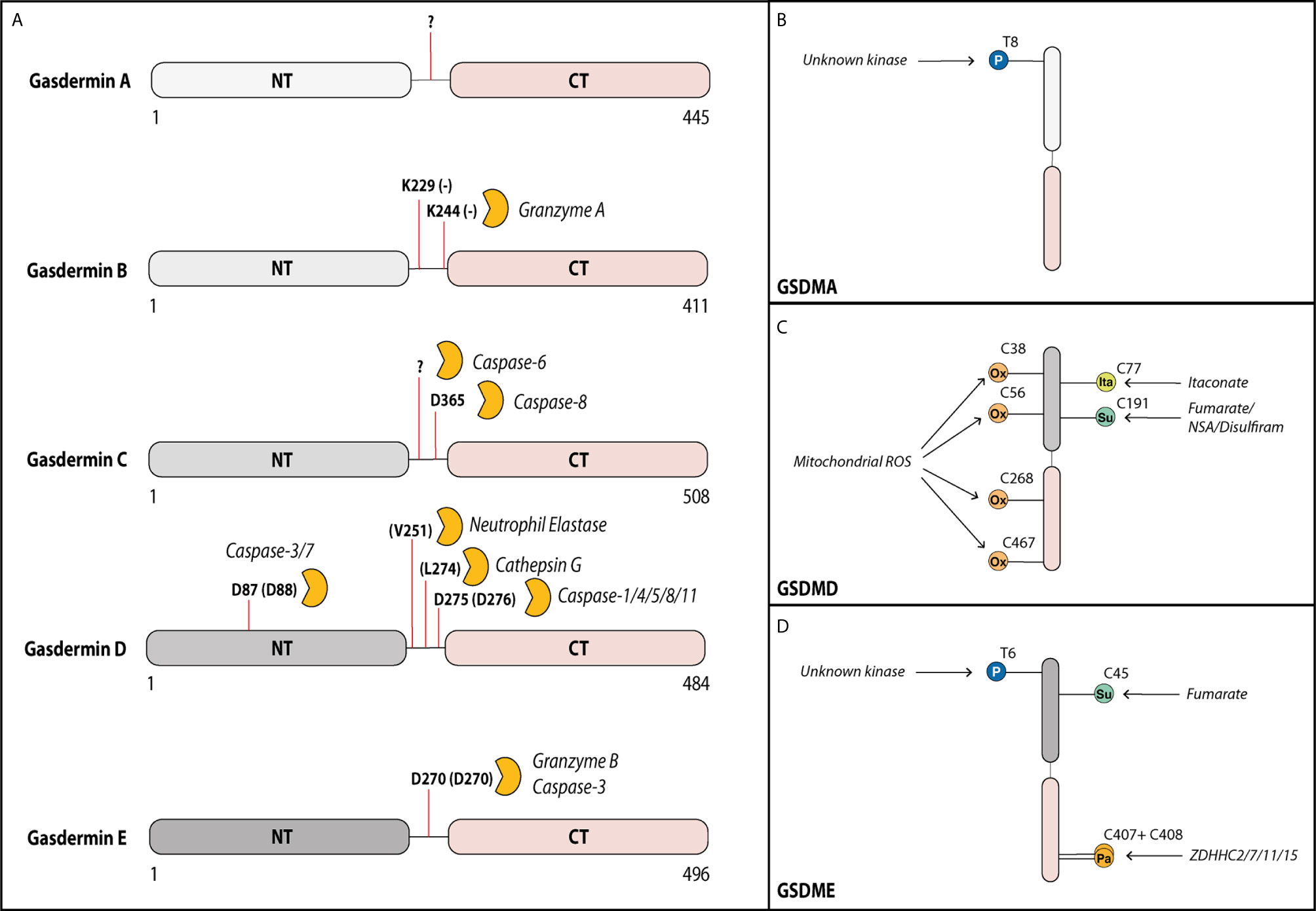
Figure 1 Gasdermins can be cleaved by various proteases in their linker region. Activity of gasdermins is regulated by cleavage and by post-translational modifications. (A) Gasdermin A can form membrane pores after cleavage of the linker domain, but the cleaving protease remains unknown. In tumor cells, Gasdermin B can be cleaved by granzyme A from cytotoxic T cells at Lys229 and Lys244 into the pore forming fragment. In several cell types including most myeloid cells, gasdermin D can be cleaved by multiple proteases at Asp275 (mouse Asp276) leading to its activation, but it can additionally be cleaved at Asp87 (mouse Asp88) by caspase-3 and -7 inactivating it during apoptosis. In neutrophils, gasdermin D can also be cleaved by neutrophil elastase and cathepsin D. In response to some chemotherapy drugs, gasdermin C can be cleaved by caspase-6 (at unknown site) and caspase-8 (at Asp365) into the pore forming fragment. Gasdermin E can be cleaved by granzyme B or caspase-3 at Asp270 leading to activation. Activity of gasdermins is also regulated by several post-translational modifications. (B) Gasdermin A can be phosphorylated (P) by an unknown kinase at Thr8, supporting its pore-forming capacity. (C) Gasdermin D is oxidized (Ox) at multiple residues (Cys38, Cys56, Cys268 and Cys467) by reactive oxygen species from the mitochondria promoting its activation. Prolonged LPS exposure of macrophages results in binding of itaconate at Cys77 preventing gasdermin D cleavage. Gasdermin D can also be succinated (Su) at Cys191 by the metabolic product fumarate or by covalent binding of the cysteine-reactive drugs necrosulfonamide (NSA) or disulfiram, which prevents its oligomerization. (D) Similar to gasdermin A, gasdermin E is phosphorylated at Thr6 promoting its pore formation. Gasdermin E is inhibited by succination at Cys45. During the activation, the palmitoyltransferases ZDHHC2, -7, -11 and -15 palmitoylate (Pa) gasdermin E at Cys407 and Cys408 promoting the dissociation of the GSDME-NT from GSDME-CT.
Gasdermin Pore Formation, Repair and Cell Lysis Are Distinct and Regulated Events
A caspase-1-dependent cell death was initially described during Salmonella Typhimurium infection. Infected cells displayed cell death hallmarks such as release of the intracellular enzyme LDH, uptake of cell-impermeable dyes and exposure of phosphatidylserine from the inner to the outer leaflet of the plasma membrane (3, 38). The discovery of GSDMD as the cell death executioner explained all these features, owing to its ability to form pores in the plasma membrane leading to activation of several intracellular processes and resulting in cell swelling and death (4–6). However, the exact steps leading to pore formation and death, and the mechanisms in place to control these steps, remained poorly understood. Recent literature suggests that pyroptosis is a highly chronological and fine-tuned process that can be separated into sequential, highly regulated events. The main events in the process are GSDMD pore formation, ion fluxes, cellular death, and cell rupture. The following section dissects pyroptosis into these steps.
Gasdermin Pore Formation
After cleavage of GSDMD at Asp275 (human) or Asp276 (mouse) by caspases, the N-terminal fragment (GSDMD-NT) localizes to the plasma membrane where it can bind to phospholipids, such as phosphatidylinositol phosphates or phosphatidyl serine on the inner leaflet of the plasma membrane (39, 40). Once at the membrane, the N-terminal fragments oligomerize, in a process dependent on a cysteine residue at position 192 in humans (Cys191 in mice), to form a functional pore (39). An elegant genetic screen by Evavold et al. (41), designed to identify regulators of pyroptosis downstream of GSDMD cleavage, showed that the GSDMD oligomerization is not a passive event but is regulated downstream of the Ragulator-Rag complex, typically known for its metabolic control of mTORC1 pathway. The components of the Ragulator-Rag complex, such as RagA or RagC, were dispensable for GSDMD trafficking to the cell membrane, but were essential for GSDMD-NT oligomerization and pore formation. During pyroptosis, GSDMD pore formation is typically followed by the loss of mitochondrial function (42), cell ballooning (42) and finally cell rupture (all discussed below). GSDMD-NT translocated to the plasma membrane in RagA-deficient cells but maintained membrane permeability (measured by propidium iodide (PI) uptake), mitochondrial function, and cell morphology, suggesting a role of the Ragulator-Rag complex in GSDMD pore formation itself and not in other, downstream events leading to cell death (41). It remains to be investigated whether any component of the Ragulator-Rag-mTORC1 pathway directly binds to GSDMD-NT to support oligomerization or if an intermediate interaction partner exists that exerts this action. LPS priming has been shown so activate mTORC1; hence, we speculate that the inflammasome priming step already puts the cell in a state in which it is prepared to commit cell death if needed (43).
Ion Fluxes and Membrane Repair
Assembled GSDMD pores can measure up to 20 nm in diameter depending on the study and on the cellular system used (39, 44–47). These pores allow secretion of smaller intracellular proteins such as IL-1β (17 kDa) but do not permit the secretion of larger proteins such as a LDH (140 kDa) or the inflammation mediator HMGB1 (tetramer of 150 kDa), which were previously thought to be released via the gasdermin pores (44, 48, 49). These bigger mediators are instead released after cell lysis and the cellular content released (50). GSDMD pores not only function as protein secretion channels but also allow trafficking of nucleotides and act as non-selective ion channels. Shortly after pore assembly, extracellular Ca2+-ions enter the cell through the pore (42, 48, 51, 52). This ion influx triggers several processes in the cell. First, it activates the Endosomal Sorting Complexes Required for Transport (ESCRT) proteins I and III, which assemble at the plasma membrane to remove gasdermin pores by encapsulating them into vesicles. When successful, membrane integrity is restored, and cell lysis and IL-1β secretion are prevented (51). Intriguingly, recent analyses of ESCRT-produced vesicles during necroptosis revealed that they contain the pore-forming MLKL, active caspases and both full length and cleaved IL-1β, among other proteins (53, 54). This suggests that a similar repair mechanism and subsequent vesicle release might exist during Gasdermin-mediated pore formation in order to release these pro-inflammatory mediators, but this remains to be tested.
Contrary to the membrane repair and rescue role, Ca2+-ion influx through GSDMD pores can also have a pathologic consequence for the cell and the organism. For example, Ca2+-ion influx through GSDMD pores activates the Ca2+-dependent transmembrane protein 16F (TMEM16F), a membrane phospholipid scramblase, which enhances the presence of phosphatidylserine (PS) in the outer leaflet of the plasma membrane (52, 55). Once exposed, PS activates the initiator of coagulation called tissue factor, leading to life-threatening disseminated intravascular coagulation often seen in bacterial endotoxemia (52, 56). TMEM16F activation also causes a change in the cellular ion currents, at least in part due to the efflux of Cl- ions (55), further contributing to the loss of ion homeostasis and cell death. Ca2+-ion influx during bacterial endotoxemia also activates STING (TMEM173) on the ER membrane. Activated STING then binds to and activates the calcium channel ITPR1 to trigger further Ca2+-release from ER stores. Elevated Ca2+ contributes to the activation of inflammatory caspases-1/11 or -8 (depending on the pathogen), leading to further GSMDM cleavage, activation of tissue factor, and lethal coagulation in bacterial sepsis (57). Finally, elevated Ca2+-levels trigger lipid peroxidation of cytoplasmic membrane lipids, by the enzyme PLCγ1, contributing to progression to pyroptosis and inflammation in polymicrobial sepsis (20, 52). While Ca2+-ions are taken up, nucleotides like ATP are released through the pore (48). Next to having an impact on the cellular energetic status, it was suggested that ATP release activates the ion channel P2X7 leading to increased uptake of Ca2+ and further progression to pyroptosis (58). Interestingly, both Ca2+- and ATP-mediated pyroptosis appeared to be blocked by extracellular Mg2+-ions, which are known to chelate ATP and prevent increased Ca2+ influx and pyroptosis. Consistent with this notion, treatment of mice with solutions containing high levels of Mg2+ was sufficient to protect mice from LPS-induced septic shock, the exact mechanism, however, still remains unexplored (58).
The above described STING (TMEM173)-dependent Ca2+-flux during bacterial endotoxemia occurred independently of the canonical cGAS pathway (57.) The canonical, dsDNA-activated cGAS-STING pathway, can induce NLRP3-Caspase-1 inflammasome activation as well, as a result of STING mediated lysosomal damage and K+ efflux (59). However this activation route is dependent on signal strength and cell type. It is functional in primary human monocytes, but not in macrophages and mouse embryonic fibroblasts, most likely as a way to limit large amount of pro-inflammatory cell death during antiviral responses (59).
Cellular Death
The next step of pyroptosis is the cell committing to die. Pore formation is correlated with a loss of mitochondrial membrane potential (MMP), which is not due to cellular rupture as the loss of MMP still occurs in cells where lysis is prevented with the osmoprotectant glycine. This process is dependent on GSDMD pores as GSDMD knock-out cells do not lose mitochondrial viability (42, 60). The loss in viability is likely caused by ion influx or a general loss of membrane potential rather than proteins leaving or entering the cell, as Vasconcelos et al. (42) described in a time-resolved single-cell analysis of pyroptotic cells that loss of MMP and Ca2+-influx occurred much earlier than uptake of small molecules like PI (670 Da). In their study, loss of mitochondrial viability was quickly followed by cellular swelling, a loss of lysosome stability and finally loss of nuclear integrity seen as nuclear rounding and condensation (42). Hence, cells undergoing pyroptosis are already dead before rupturing.
Membrane Rupture
Cellular swelling is one of the features observed during pyroptosis, and hypertonic solutions have been described to rescue GSDMD-dependent cell death (61). This supports a model, in which a cell is losing its integrity and is passively rupturing due to osmotic pressure, resulting in the release of proteins such as LDH or the alarmins HMGB1 and galectin-1 (50, 62). Intriguingly, a study by Kayagaki et al. found that in reality, cell lysis is a process regulated by dedicated proteins (63). Cells deficient in the cell adhesion protein Ninjurin 1 (NINJ1) did not rupture even after mitochondrial death, despite formation of GSDMD pores, release of IL-1β, and display of the typical balloon morphology. NINJ1 is not an exclusive regulator of pyroptosis as LDH release by apoptotic triggers is also impaired in NINJ1-deficient cells, implicating NINJ1 as a key cell lysis regulator in several cell death pathways. NINJ1 expression was recently shown to be stimulated by oxidative stress, making loss of mitochondrial viability and mitochondrial ROS production potential triggers of its activation (64). How exactly NINJ1 mediates cell rupture and whether NINJ1 is activated downstream of all gasdermins need further study. Interestingly, membrane rupture is not only important for the release of inflammatory mediators but also for the clearance of intracellular bacteria. Of note, pyroptotic cells trap viable bacteria within ruptured cellular debris, and these structures are termed pore-induced cellular traps (PITs) (65). Subsequently, PITs are removed by recruited phagocytes resulting in the clearance of the dead cell as well as the contained bacteria (66).
One open question remains, how some cells maintain IL-1β secretion without undergoing pyroptosis (67). For example, dendritic cells activated with certain oxidized lipids generated during tissue injury, can maintain IL-1β secretion for several days without cellular rupture (68). Neutrophils can also maintain IL-1β secretion without undergoing pyroptosis (69). It will be interesting to see in future studies whether this cellular integrity is maintained by increased ESCRT-mediated membrane repair, prevention of mitochondrial damage or other processes.
Posttranslational Modifications Regulate Gasdermin Activity
Phosphorylation of Gasdermins
Pyroptosis is a highly inflammatory and extremely rapid process and, hence, gasdermins need to be tightly regulated in their activity. One effective means of regulating fast cellular processes is by post-translational modifications (PTM), which have been shown to also regulate other, upstream steps of inflammasome activation (70). Phosphorylation is the best studied PTM. It could theoretically control the activity of gasdermins directly by modifying them or indirectly by modifying their interacting partners. An unbiased proteomic screen showed that phosphorylation of substrates can alter their cleavage by caspases-3, -7 and -8, which are all enzymes that can also cleave GSDMD and GSMDE (71–73). Evidence so far for phosphorylation exists only for Thr8 and Thr6 of human GSDMA and GSDME, respectively (Figures 1B, D). Both phosphorylation sites block gasdermin oligomerization and pore formation (74). Mechanistically, they likely block interaction between gasdermin monomers, via changing the charge of the first alpha-helix of the N-terminal domain that is critical for oligomerization (40). The serine-threonine kinase Polo-like kinase 1 (PLK1) mediates GSDMA phosphorylation, but whether it phosphorylates Thr6 in GSDME remains unexplored (74, 75). Whether the functions of other gasdermins are regulated via direct phosphorylation is unknown although all contain at least one serine or threonine residue in the first alpha-helix. Additionally, a phosphorylation site prediction tool has pointed to several potential phosphorylation sites for gasdermin D and E located in their linker regions. Future studies will ascertain whether the predicted sites are indeed phosphorylated and how these PTMs influence the nearby cleavage sites. The necroptosis-associated pore-forming protein MLKL shows a similar dependency on phosphorylation via RIPK3-dependent signaling before pore formation and subsequent necroptosis. Hence, phosphorylation of pore-forming proteins could be a common mechanism by which cells regulate pore formation via of death effector proteins (76).
Phosphorylation of gasdermin-interacting partners also may play an important, additional role as a means to regulate gasdermin activity. Phosphorylation of the apoptosis-associated caspases-3, -7 and -8 has been shown in several reports to regulate their activation and/or substrate recognition (77–80). Interestingly, the only known phosphorylation site on caspase-1 at Ser376 is also needed for its activation; whether caspase-4, -5 or -11 are phosphorylated is unknown (81). This raises the question whether the threshold of activation for caspase-4, -5 or -11 is set low, or the phosphorylation of these pyroptosis-associated caspases remains unexplored in sufficient detail. The N-terminal domain of gasdermins inserts into membranes via binding to lipids such as phosphatidylinositol phosphate on the inner membrane leaflet or cardiolipin present on the inner mitochondrial membrane or plasma membrane in mammalian and bacterial cells respectively (39, 40). Interestingly, GSDMD-NT as well as GSDMA-NT and GSDMA3-NT are only capable of binding to membrane lipids when phosphorylated, identifying membrane lipid composition and phospho-modifications as another control mechanism in the process of pyroptosis (45).
Other Posttranslational Modifications of Gasdermins
Macrophages and dendritic cells stimulated with inflammatory stimuli such as LPS are known to switch their metabolic profile from oxidative phosphorylation to aerobic glycolysis (82). A study by Humphries et al. (83) recently discovered that one metabolic intermediate of this pathway, fumarate, can irreversibly bind to GSDMD at Cys191 (human)/Cys192 (mouse) and GSDME at Cys45 (mouse) in a process termed succination (Figures 1C, D). GSDMD Cys191 is located next to Leu192, which is the contact point for the C-terminal GSDMD domain responsible for autoinhibition (45). As mutation of Leu192 blocks binding of GSDMD-NT to membrane lipids, succination at Cys191 likely confers a similar effect on GSDMD. This prediction was confirmed by cysteine-modifying drugs, which blocked pyroptosis and death in an animal model of lethal endotoxemia (13, 83). Although speculative, a model emerges whereby the metabolic switch—i.e., oxidative phosphorylation to aerobic glycolysis—specifically blocks gasdermin pore formation by succination. Quite likely, this is only a part of a much broader effect caused by the reactive nature of fumarate, as multiple cysteine residues are modified in both GSDMD and GSDME (83). A model that metabolic switch influences gasdermin-mediated death was also supported by a recent study in which prolonged LPS stimulation of macrophages lead to an accumulation of the cell metabolite itaconate. Mass spectrometry analysis revealed, that itaconate directly bound to GSDMD at Cys77. This modification blocked caspase-1-dependent GSDMD cleavage and conferred tolerance to extended periods of LPS exposure (84). Another study observed that chemotherapy modified human GSDME by palmitoylation at Cys407 and Cys408 via the palmitoyltransferases ZDHHC2, 7, 11 and 15 (Figure 1D) (85). These modifications led to a decreased interaction of GSDME-NT with GSDME-CT after GSMDE cleavage by caspase-3, which facilitated the release of cleaved GSDME-NT and, thereby, pore formation (85). Inflammasome stimuli are known inducers of mitochondrial ROS, and ROS production is required for pyroptosis (86). One recently described mechanism suggested regulation of GSDMD by ROS via direct oxidation of human GSDMD at Cys38, Cys56, Cys268 and Cys467 (Figure 1C) (87). Indeed, mutation of these residues reduced GSDMD cleavage by caspase-1 and pore formation, supporting the idea that GSDMD oxidation is an important regulator of its function.
Only a few PTMs are currently described to influence gasdermin activity, although a high amount of phosphorylations and ubiquitylations are predicted. The necroptosis-associated protein MLKL, its upstream activators RIPK1 and RIPK3 as well as multiple inflammasome components are known to be ubiquitylated (88, 89). This, in combination with a wide range of predicted ubiquitylation sites, makes it likely that future studies will uncover gasdermin regulation via ubiquitin by influencing either its activation or degradation. Currently, the PTM landscape of gasdermins is sparsely described, with only few interaction partners and locations known. An increasing amount of research on PTMs will hopefully help to answer which upstream processes lead to these modifications, which enzymes catalyze the addition or removal of modifications, and how these modifications are hierarchically ordered to inhibit or activate gasdermin-mediated pore formation.
Therapeutic Strategies to Block Gasdermin-Mediated Cell Death
Current therapies against aberrant inflammasome activation use IL-1-targeting drugs, such as the IL-1 receptor antagonist Anakinra (90), or direct inhibition of NLRP3 by specific inhibitors, such as MCC950 (91), and several others (92). As additional pro-inflammatory alarmins are released via the gasdermin pores and multiple inflammasomes can lead to gasdermin activation, gasdermin-specific drugs are highly desirable therapeutics. The extracellular addition of the amino acid glycine can inhibit gasdermin-mediated cell rupture, but it cannot prevent intracellular processes leading to cell death or IL-1β secretion (Figure 2) (38, 49, 60). The protective effect of glycine is dependent on its carboxyl group but is relatively unspecific, as glycine acts as an osmoprotectant and not as a specific inhibitor of gasdermin pore formation (93). Other described inhibitors such as lanthanides, Mg2+-ions and hypertonic solutions likely act via similar unspecific mechanisms, although Mg2+ ions block gasdermin oligomerization by blocking Ca2+-influx needed for pore formation (48, 58, 61). It is noteworthy, that the inhibitory concentration of magnesium needed to inhibit pyroptosis in macrophages exceeds the physiological extracellular concentration of magnesium by 10-fold, and that inhibitory effects were seen with nearly all divalent cations (58). Rathkey et al. (94) reported that the cysteine-modifying drug necrosulfonamide (NSA) blocked pyroptosis in human and mouse cells by binding to Cys191 (human)/Cys192 (mouse) of GSDMD and, thereby, blocked its oligomerization. NSA inhibition is not selective to gasdermins, as NSA blocks necroptosis by a similar mechanism: NSA binds to Cys86 of the pore-forming protein MLKL in humans to prevent oligomerization (95). Other groups found that cysteine-reactive drugs such as NSA and Bay 11-7082 rather inhibit pyroptosis by modifying upstream caspase-1 cleavage and not GSDMD itself, and that NSA can block IL-1β secretion even further upstream by inhibiting LPS-mediated priming (13, 96). Hu and colleagues (13) discovered in their compound screening that another cysteine-reactive drug, disulfiram (used in people to treat alcohol abuse), potently blocked GSDMD-mediated pyroptosis by the same mechanism as NSA, with no activity on other gasdermins or MLKL-mediated cell death (13). Disulfiram was described much earlier to oxidize thiols present in caspases and, thereby, blocked their substrate cleavage activity (97). And in a more recent study, Wang et al. (98) showed that disulfiram’s inhibitory effect is not restricted to GSDMD only, but can also inhibit GSDME. Although disulfiram is an attractive anti-pyroptotic drug, particularly because it is widely used in humans, the exact targets of disulfiram need to be better defined owing to its highly cysteine-reactive nature. Regardless, both NSA and disulfiram block GSDMD oligomerization via modifying Cys191, making either this site or the first alpha-helix (needed for pore formation) an attractive target for designing a more specific drug in the future.
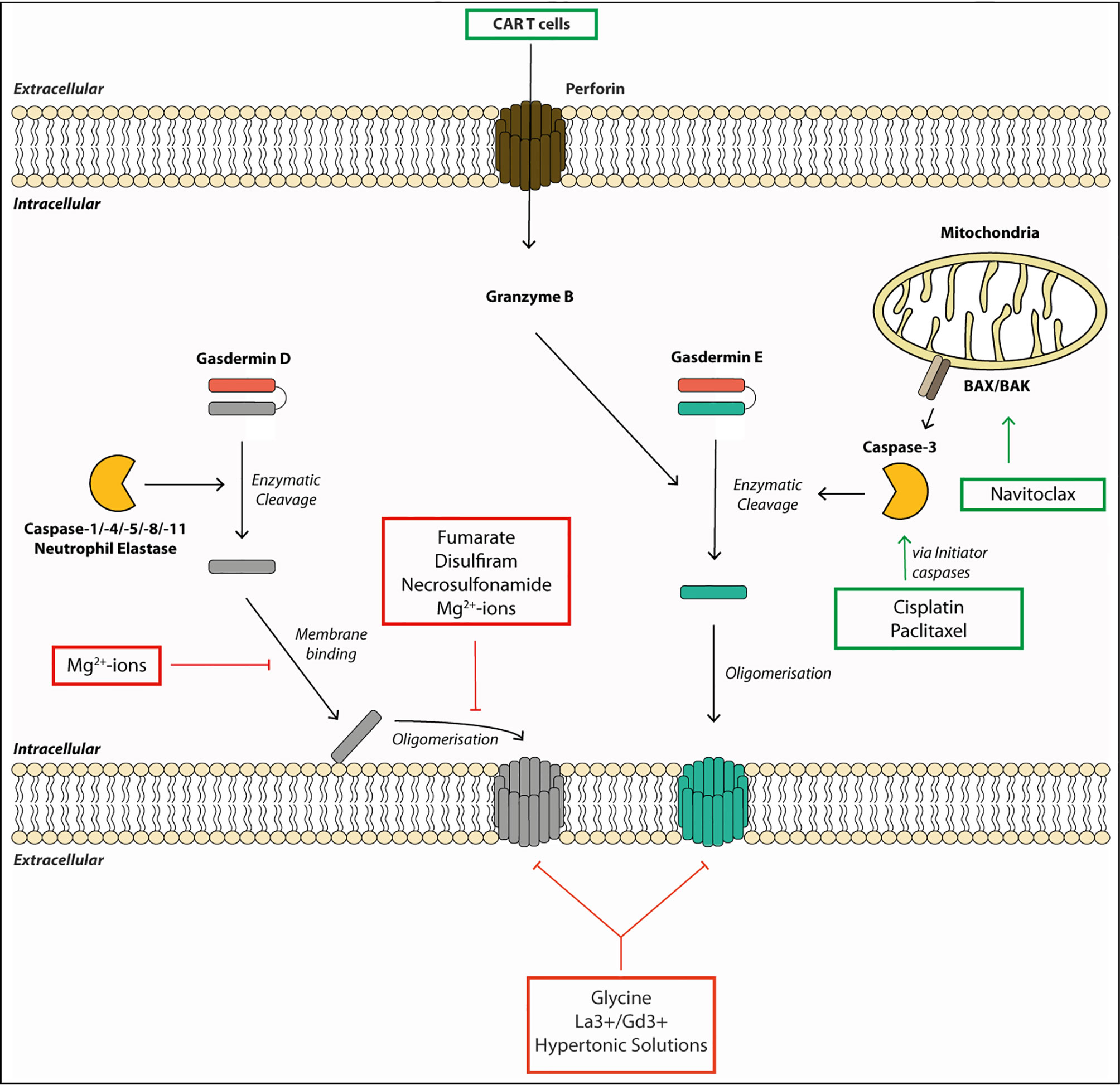
Figure 2 Gasdermin pore formation and cell lysis require multiple steps and can be targeted by therapeutics. Gasdermins are activated by enzymatic cleavage by proteases such as caspases or neutrophil elastase. This liberates the pore-forming N-terminal fragment (GSDM-NT). The GSDM-NT binds to phospholipids on the inner membrane leaflet. GSDM-NT then oligomerizes to form a membrane pore allowing the efflux of small proteins and ions across the membrane. Gasdermin pores eventually lead to cell death and membrane rupture. Some drugs promote gasdermin-mediated cell death. For example, chemotherapeutic drugs, such as Cisplatin, Paclitaxel or Navitoclax can initiate gasdermin E cleavage. They activate the initiator caspases, which, in turn, lead to gasdermin E cleavage by activating the executioner caspase-3. Other drugs or small molecules can block gasdermin-mediated cell death. For example, the membrane binding and the oligomerization step of gasdermin D can be blocked by Mg2+-ions by an unknown mechanism. Fumarate, Necrosulfonamide and Disulfiram can block oligomerization of gasdermin D by modifying Cys191. Finally, membrane rupture can be blocked by the osmoprotectant glycine, hypertonic solutions or the lanthanide ions La3+ and Gd3+.
While most drugs focus on blocking detrimental pyroptosis mediated by GSDMD, in some cases, boosting pyroptosis can be beneficial. For example, induced activation of GSDME has beneficial effect in treating various cancers because it promotes a more inflammatory tumor milieu (24). In a synthetic approach, Wang et al. (99) showed that tumor cells treated with engineered nanoparticle-GSDMA3 conjugates led to increased cell death and a significant reduction in tumor burden upon release of GSDMA3 from the nanoparticle. Chemotherapeutic drugs such as Cisplatin or Paclitaxel can induce activation of the initiator caspases-8 and -9, resulting in caspase-3 activation. Caspase-3 activation drives GSDME cleavage and pyroptosis, and makes this pathway an attractive target for tumor treatments (24, 85, 100). These are systemic drugs and should be used with caution, as systemic GSDME activation can have detrimental effects. For example, cytokine-release syndrome in patients treated with CAR T cells was GSDME-dependent. Granzymes released from these cells led to caspase-3 and -7 activation, and ultimately GSDME cleavage (23). As granzyme A and B directly cleave GSDMB and E, respectively, targeted delivery of these enzymes poses an interesting therapeutic option to treat immunosuppressive tumors, yet their administration will have to be tightly titrated or targeted to avoid detrimental side effects (21, 22). In conclusion, gasdermins are newly emerging targets for therapeutic targeting for positive and negative modulation of cell death and the resulting immune responses.
Author Contributions
FF, KC, and JB drafted the manuscript. KC and JB supervised and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
JB is supported by Kennedy Trust, KTRR start-up fellowship (KENN 15 16 06), and MRC New Investigator Grant (MR/S000623/1). FF is supported by a Kennedy Trust, KTPS Studentship. KC is supported by National University of Singapore Start Up grant and a Ministry of Education Inauguration Grant.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to express their gratitude to Dr. Dave Boucher for his critical reading of the manuscript and contributing valuable feedback. Further, we would like to thank Prof Sebastian Joyce and Laura Loporcaro for careful proof-reading of this manuscript.
References
1. Nagata S. Apoptosis and Clearance of Apoptotic Cells. Annu Rev Immunol (2018) 36:489–517. doi: 10.1146/annurev-immunol-042617-053010
2. White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell (2014) 159:1549–62. doi: 10.1016/j.cell.2014.11.036
3. Fink SL, Cookson BT. Pyroptosis and host cell death responses during Salmonella infection. Cell Microbiol (2007) 9:2562–70. doi: 10.1111/j.1462-5822.2007.01036.x
4. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature (2015) 526:666–71. doi: 10.1038/nature15541
5. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
6. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res (2015) 25:1285–98. doi: 10.1038/cr.2015.139
7. Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun (2017) 8:14128. doi: 10.1038/ncomms14128
8. Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen blockade of TAK1 triggers caspase-8–dependent cleavage of gasdermin D and cell death. Science (2018) 362:1064–9. doi: 10.1126/science.aau2818
9. Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci USA (2018) 115:E10888–97. doi: 10.1073/pnas.1809548115
10. Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, et al. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J (2019) 38. doi: 10.15252/embj.2019101638
11. Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev (2017) 277:61–75. doi: 10.1111/imr.12534
12. Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol (2017) 17:151–64. doi: 10.1038/nri.2016.147
13. Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol (2020) 21:736–45. doi: 10.1038/s41590-020-0669-6
14. Demarco B, Grayczyk JP, Bjanes E, Le Roy D, Tonnus W, Assenmacher C-A, et al. Caspase-8–dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF-induced lethality. Sci Adv (2020) 6:eabc3465. doi: 10.1126/sciadv.abc3465
15. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science (2013) 341:1250–3. doi: 10.1126/science.1240988
16. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science (2013) 341:1246–9. doi: 10.1126/science.1240248
17. Wang S, M M, Jung YK, H Z, Li E, Yuan J. Murine Caspase-11, an ICE-Interacting Protease, Is Essential for the Activation of ICE. Cell (1998) 92:501–9. doi: 10.1016/S0092-8674(00)80943-5
18. Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, et al. Mice Deficient in IL-lp-Converting Enzyme Are Defective in Production of Mature IL-lp and Resistant to Endotoxic Shock. Cell (1995) 80:401–11. doi: 10.1016/0092-8674(95)90490-5
19. Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol (2010) 185:4385–92. doi: 10.4049/jimmunol.1000803
20. Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, et al. Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe (2018) 24:97–108 e104. doi: 10.1016/j.chom.2018.05.009
21. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science (2020) 368:eaaz7548. doi: 10.1126/science.aaz7548
22. Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature (2020) 579:415–20. doi: 10.1038/s41586-020-2071-9
23. Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, et al. Gasdermin E–mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci Immunol (2020) 5:eaax7969. doi: 10.1126/sciimmunol.aax7969
24. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature (2017) 547:99–103. doi: 10.1038/nature22393
25. Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol (2020) 22:1264–75. doi: 10.1038/s41556-020-0575-z
26. Kanneganti A, Malireddi RKS, Saavedra PHV, Vande Walle L, Van Gorp H, Kambara H, et al. GSDMD is critical for autoinflammatory pathology in a mouse model of Familial Mediterranean Fever. J Exp Med (2018) 215:1519–29. doi: 10.1084/jem.20172060
27. Xiao J, Wang C, Yao JC, Alippe Y, Xu C, Kress D, et al. Gasdermin D mediates the pathogenesis of neonatal-onset multisystem inflammatory disease in mice. PloS Biol (2018) 16:e3000047. doi: 10.1371/journal.pbio.3000047
28. Li S, Wu Y, Yang D, Wu C, Ma C, Liu X, et al. Gasdermin D in peripheral myeloid cells drives neuroinflammation in experimental autoimmune encephalomyelitis. J Exp Med (2019) 216:2562–81. doi: 10.1084/jem.20190377
29. Khanova E, Wu R, Wang W, Yan R, Chen Y, French SW, et al. Pyroptosis by caspase11/4-gasdermin-D pathway in alcoholic hepatitis in mice and patients. Hepatology (2018) 67:1737–53. doi: 10.1002/hep.29645
30. Xu B, Jiang M, Chu Y, Wang W, Chen D, Li X, et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J Hepatol (2018) 68:773–82. doi: 10.1016/j.jhep.2017.11.040
31. Yang C, Sun P, Deng M, Loughran P, Li W, Yi Z, et al. Gasdermin D protects against noninfectious liver injury by regulating apoptosis and necroptosis. Cell Death Dis (2019) 10:481. doi: 10.1038/s41419-019-1719-6
32. Orning P, Lien E, Fitzgerald KA. Gasdermins and their role in immunity and inflammation. J Exp Med (2019) 216:2453–65. doi: 10.1084/jem.20190545
33. Broz P, Pelegrin P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol (2020) 20:143–57. doi: 10.1038/s41577-019-0228-2
34. Feng S, Fox D, Man SM. Mechanisms of Gasdermin Family Members in Inflammasome Signaling and Cell Death. J Mol Biol (2018) 430:3068–80. doi: 10.1016/j.jmb.2018.07.002
35. Burgener SS, Leborgne NGF, Snipas SJ, Salvesen GS, Bird PI, Benarafa C. Cathepsin G Inhibition by Serpinb1 and Serpinb6 Prevents Programmed Necrosis in Neutrophils and Monocytes and Reduces GSDMD-Driven Inflammation. Cell Rep (2019) 27:3646–56 e3645. doi: 10.1016/j.celrep.2019.05.065
36. Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, et al. Gasdermin D Exerts Anti-inflammatory Effects by Promoting Neutrophil Death. Cell Rep (2018) 22:2924–36. doi: 10.1016/j.celrep.2018.02.067
37. Taabazuing CY, Okondo MC, Bachovchin DA. Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem Biol (2017) 24:507–514 e504. doi: 10.1016/j.chembiol.2017.03.009
38. Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol (2006) 8:1812–25. doi: 10.1111/j.1462-5822.2006.00751.x
39. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature (2016) 535:153–8. doi: 10.1038/nature18629
40. Chen X, He WT, Hu L, Li J, Fang Y, Wang X, et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res (2016) 26:1007–20. doi: 10.1038/cr.2016.100
41. Evavold CL, Hafner-Bratkovič I, Kagan JC. Downstream of gasdermin D cleavage, a Ragulator-Rag-mTORC1 pathway promotes pore formation and pyroptosis. bioRxiv (2020). doi: 10.1101/2020.11.02.362517
42. de Vasconcelos NM, Van Opdenbosch N, Van Gorp H, Parthoens E, Lamkanfi M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ (2019) 26:146–61. doi: 10.1038/s41418-018-0106-7
43. Schmitz F, Heit A, Dreher S, Eisenacher K, Mages J, Haas T, et al. Mammalian target of rapamycin (mTOR) orchestrates the defense program of innate immune cells. Eur J Immunol (2008) 38:2981–92. doi: 10.1002/eji.200838761
44. Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J (2016) 35:1766–78. doi: 10.15252/embj.201694696
45. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535:111–6. doi: 10.1038/nature18590
46. Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA (2016) 113:7858–63. doi: 10.1073/pnas.1607769113
47. Ruan J, Xia S, Liu X, Lieberman J, Wu H. Cryo-EM structure of the gasdermin A3 membrane pore. Nature (2018) 557:62–7. doi: 10.1038/s41586-018-0058-6
48. Russo HM, Rathkey J, Boyd-Tressler A, Katsnelson MA, Abbott DW, Dubyak GR. Active Caspase-1 Induces Plasma Membrane Pores That Precede Pyroptotic Lysis and Are Blocked by Lanthanides. J Immunol (2016) 197:1353–67. doi: 10.4049/jimmunol.1600699
49. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity (2018) 48:35–44 e36. doi: 10.1016/j.immuni.2017.11.013
50. Volchuk A, Ye A, Chi L, Steinberg BE, Goldenberg NM. Indirect regulation of HMGB1 release by gasdermin D. Nat Commun (2020) 11:4561. doi: 10.1038/s41467-020-18443-3
51. Rühl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science (2018) 362:956–60. doi: 10.1126/science.aar7607
52. Yang X, Cheng X, Tang Y, Qiu X, Wang Y, Kang H, et al. Bacterial Endotoxin Activates the Coagulation Cascade through Gasdermin D-Dependent Phosphatidylserine Exposure. Immunity (2019) 51:983–996 e986. doi: 10.1016/j.immuni.2019.11.005
53. Yoon S, Kovalenko A, Bogdanov K, Wallach D. MLKL, the Protein that Mediates Necroptosis, Also Regulates Endosomal Trafficking and Extracellular Vesicle Generation. Immunity (2017) 47:51–65 e57. doi: 10.1016/j.immuni.2017.06.001
54. Shlomovitz I, Yanovich-Arad G, Erlich Z, Edry-Botzer L, Zargarian S, Cohen H, et al. Proteomic analysis of necroptotic extracellular vesicles. bioRxiv (2020). doi: 10.1101/2020.04.11.037192
55. Ousingsawat J, Wanitchakool P, Schreiber R, Kunzelmann K. Contribution of TMEM16F to pyroptotic cell death. Cell Death Dis (2018) 9:300. doi: 10.1038/s41419-018-0373-8
56. Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, et al. Inflammasome Activation Triggers Blood Clotting and Host Death through Pyroptosis. Immunity (2019) 50:1401–11 e1404. doi: 10.1016/j.immuni.2019.04.003
57. Zhang H, Zeng L, Xie M, Liu J, Zhou B, Wu R, et al. TMEM173 Drives Lethal Coagulation in Sepsis. Cell Host Microbe (2020) 27:556–70 e556. doi: 10.1016/j.chom.2020.02.004
58. Wang D, Zheng J, Hu Q, Zhao C, Chen Q, Shi P, et al. Magnesium protects against sepsis by blocking gasdermin D N-terminal-induced pyroptosis. Cell Death Differ (2020) 27:466–81. doi: 10.1038/s41418-019-0366-x
59. Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S, et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell (2017) 171:1110–24 e1118. doi: 10.1016/j.cell.2017.09.039
60. DiPeso L, Ji DX, Vance RE, Price JV. Cell death and cell lysis are separable events during pyroptosis. Cell Death Discov (2017) 3:17070. doi: 10.1038/cddiscovery.2017.70
61. Zheng J, Wang D, Chen Q, Liu Q, Lin Z, Hu Q, et al. Hypertonic saccharide solution delays pyroptosis in murine macrophages regardless of the membrane binding of gasdermin D N-terminal. Eur J Immunol (2020) 50:464–7. doi: 10.1002/eji.201948230
62. Russo AJ, Vasudevan SO, Mendez-Huergo SP, Kumari P, Menoret A, Duduskar S, et al. Intracellular immune sensing promotes inflammation via gasdermin D-driven release of a lectin alarmin. Nat Immunol (2021) 22:154–65. doi: 10.1038/s41590-020-00844-7
63. Kayagaki N, Kornfeld OS, Lee BL, Stowe IB, O'Rourke K, Li Q, et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature (2021) 591:131–6. doi: 10.21203/rs.3.rs-62714/v1
64. Toma L, Sanda GM, Raileanu M, Stancu CS, Niculescu LS, Sima AV. Ninjurin-1 upregulated by TNFalpha receptor 1 stimulates monocyte adhesion to human TNFalpha-activated endothelial cells; benefic effects of amlodipine. Life Sci (2020) 249:117518. doi: 10.1016/j.lfs.2020.117518
65. Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med (2016) 213:2113–28. doi: 10.1084/jem.20151613
66. Jorgensen I, Lopez JP, Laufer SA, Miao EA. IL-1beta, IL-18, and eicosanoids promote neutrophil recruitment to pore-induced intracellular traps following pyroptosis. Eur J Immunol (2016) 46:2761–6. doi: 10.1002/eji.201646647
67. Monteleone M, Stow JL, Schroder K. Mechanisms of unconventional secretion of IL-1 family cytokines. Cytokine (2015) 74:213–8. doi: 10.1016/j.cyto.2015.03.022
68. Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science (2016) 352:1232–6. doi: 10.1126/science.aaf3036
69. Chen KW, Gross CJ, Sotomayor FV, Stacey KJ, Tschopp J, Sweet MJ, et al. The neutrophil NLRC4 inflammasome selectively promotes IL-1beta maturation without pyroptosis during acute Salmonella challenge. Cell Rep (2014) 8:570–82. doi: 10.1016/j.celrep.2014.06.028
70. Gong T, Jiang W, Zhou R. Control of Inflammasome Activation by Phosphorylation. Trends Biochem Sci (2018) 43:685–99. doi: 10.1016/j.tibs.2018.06.008
71. Turowec JP, Zukowski SA, Knight JD, Smalley DM, Graves LM, Johnson GL, et al. An unbiased proteomic screen reveals caspase cleavage is positively and negatively regulated by substrate phosphorylation. Mol Cell Proteomics (2014) 13:1184–97. doi: 10.1074/mcp.M113.037374
72. Demarco B, Chen KW, Broz P. Pannexin-1 channels bridge apoptosis to NLRP3 inflammasome activation. Mol Cell Oncol (2019) 6:1610324. doi: 10.1080/23723556.2019.1610324
73. Dix MM, Simon GM, Wang C, Okerberg E, Patricelli MP, Cravatt BF. Functional interplay between caspase cleavage and phosphorylation sculpts the apoptotic proteome. Cell (2012) 150:426–40. doi: 10.1016/j.cell.2012.05.040
74. Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun (2019) 10:1689. doi: 10.1038/s41467-019-09397-2
75. Santamaria A, Wang B, Elowe S, Malik R, Zhang F, Bauer M, et al. The Plk1-dependent phosphoproteome of the early mitotic spindle. Mol Cell Proteomics (2011) 10:M110 004457. doi: 10.1074/mcp.M110.004457
76. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell (2014) 54:133–46. doi: 10.1016/j.molcel.2014.03.003
77. Alvarado-Kristensson M, Melander F, Leandersson K, Ronnstrand L, Wernstedt C, Andersson T. p38-MAPK signals survival by phosphorylation of caspase-8 and caspase-3 in human neutrophils. J Exp Med (2004) 199:449–58. doi: 10.1084/jem.20031771
78. Jia SH, Parodo J, Kapus A, Rotstein OD, Marshall JC. Dynamic regulation of neutrophil survival through tyrosine phosphorylation or dephosphorylation of caspase-8. J Biol Chem (2008) 283:5402–13. doi: 10.1074/jbc.M706462200
79. Li X, Wen W, Liu K, Zhu F, Malakhova M, Peng C, et al. Phosphorylation of caspase-7 by p21-activated protein kinase (PAK) 2 inhibits chemotherapeutic drug-induced apoptosis of breast cancer cell lines. J Biol Chem (2011) 286:22291–9. doi: 10.1074/jbc.M111.236596
80. Voss OH, Kim S, Wewers MD, Doseff AI. Regulation of monocyte apoptosis by the protein kinase Cdelta-dependent phosphorylation of caspase-3. J Biol Chem (2005) 280:17371–9. doi: 10.1074/jbc.M412449200
81. Basak C, Pathak SK, Bhattacharyya A, Mandal D, Pathak S, Kundu M. NF-kappaB- and C/EBPbeta-driven interleukin-1beta gene expression and PAK1-mediated caspase-1 activation play essential roles in interleukin-1beta release from Helicobacter pylori lipopolysaccharide-stimulated macrophages. J Biol Chem (2005) 280:4279–88. doi: 10.1074/jbc.M412820200
82. O’Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med (2016) 213:15–23. doi: 10.1084/jem.20151570
83. Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, et al. Succination inactivates gasdermin D and blocks pyroptosis. Science (2020) 369:1633–7. doi: 10.1126/science.abb9818
84. Bambouskova M, Potuckova L, Paulenda T, Kerndl M, Mogilenko DA, Lizotte K, et al. Itaconate confers tolerance to late NLRP3 inflammasome activation. Cell Rep (2021) 34:108756. doi: 10.1016/j.celrep.2021.108756
85. Hu L, Chen M, Chen X, Zhao C, Fang Z, Wang H, et al. Chemotherapy-induced pyroptosis is mediated by BAK/BAX-caspase-3-GSDME pathway and inhibited by 2-bromopalmitate. Cell Death Dis (2020) 11:281. doi: 10.1038/s41419-020-2476-2
86. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469:221–5. doi: 10.1038/nature09663
87. Wang Y, Shi P, Chen Q, Huang Z, Zou D, Zhang J, et al. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J Mol Cell Biol (2019) 11:1069–82. doi: 10.1093/jmcb/mjz020
88. Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D'Cruz AA, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun (2015) 6:6282. doi: 10.1038/ncomms7282
89. Lopez-Castejon G. Control of the inflammasome by the ubiquitin system. FEBS J (2020) 287:11–26. doi: 10.1111/febs.15118
90. Cavalli G, Dinarello CA. Anakinra Therapy for Non-cancer Inflammatory Diseases. Front Pharmacol (2018) 9:1157. doi: 10.3389/fphar.2018.01157
91. Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med (2015) 21:248–55. doi: 10.1038/nm.3806
92. Zahid A, Li B, Kombe AJK, Jin T, Tao J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front Immunol (2019) 10:2538. doi: 10.3389/fimmu.2019.02538
93. Loomis WP, den Hartigh AB, Cookson BT, Fink SL. Diverse small molecules prevent macrophage lysis during pyroptosis. Cell Death Dis (2019) 10:326. doi: 10.1038/s41419-019-1559-4
94. Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol (2018) 3:eaat2738. doi: 10.1126/sciimmunol.aat2738
95. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell (2012) 148:213–27. doi: 10.1016/j.cell.2011.11.031
96. Rashidi M, Simpson DS, Hempel A, Frank D, Petrie E, Vince A, et al. The Pyroptotic Cell Death Effector Gasdermin D Is Activated by Gout-Associated Uric Acid Crystals but Is Dispensable for Cell Death and IL-1beta Release. J Immunol (2019) 203:736–48. doi: 10.4049/jimmunol.1900228
97. Stefan MKC, Nobel I, Nicholson DW, Orrenius S, Andrew F, Slater G. Disulfiram Is a Potent Inhibitor of Proteases of the Caspase Family. Chem Res Toxicol (1997) 10:1319–24. doi: 10.1021/tx970131m
98. Wang C, Yang T, Xiao J, Xu C, Alippe Y, Sun K, et al. Activation of GSDME compensates for GSDMD deficiency in a mouse model of NLRP3 inflammasomopathy. bioRxiv (2021). doi: 10.1101/2021.01.06.425634
99. Wang Q, Wang Y, Ding J, Wang C, Zhou X, Gao W, et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature (2020) 579:421–6. doi: 10.1038/s41586-020-2079-1
Keywords: pyroptosis, gasdermins, cell death, post-translational modifications, therapeutics, phosphorylation, inflammation
Citation: Fischer FA, Chen KW and Bezbradica JS (2021) Posttranslational and Therapeutic Control of Gasdermin-Mediated Pyroptosis and Inflammation. Front. Immunol. 12:661162. doi: 10.3389/fimmu.2021.661162
Received: 30 January 2021; Accepted: 19 March 2021;
Published: 02 April 2021.
Edited by:
Ioannis Kourtzelis, University of York, United KingdomReviewed by:
Etienne Meunier, UMR5089 Institut de Pharmacologie et de Biologie Structurale (IPBS), FranceEva Bartok, Institute of Tropical Medicine Antwerp, Belgium
Copyright © 2021 Fischer, Chen and Bezbradica. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jelena S. Bezbradica, amVsZW5hLmJlemJyYWRpY2FAa2VubmVkeS5veC5hYy51aw==; Kaiwen W. Chen, a2Fpd2VuLmNoZW5AbnVzLmVkdS5zZw==
†ORCIDKaiwen W. Chen, orcid.org/0000-0001-5903-3251