 Maria Celeste Gauron
Maria Celeste Gauron Alexandra C. Newton2
Alexandra C. Newton2 María Isabel Colombo
María Isabel Colombo- 1Laboratorio de Mecanismos Moleculares Implicados en el Tráfico Vesicular y la Autofagia-Instituto de Histología y Embriología (IHEM)- Universidad Nacional de Cuyo, CONICET- Facultad de Ciencias Médicas, Mendoza, Argentina
- 2Department of Pharmacology, University of California San Diego, La Jolla, CA, United States
Hijacking the autophagic machinery is a key mechanism through which invasive pathogens such as Staphylococcus aureus replicate in their host cells. We have previously demonstrated that the bacteria replicate in phagosomes labeled with the autophagic protein LC3, before escaping to the cytoplasm. Here, we show that the Ca2+-dependent PKCα binds to S. aureus-containing phagosomes and that α-hemolysin, secreted by S. aureus, promotes this recruitment of PKCα to phagosomal membranes. Interestingly, the presence of PKCα prevents the association of the autophagic protein LC3. Live cell imaging experiments using the PKC activity reporter CKAR reveal that treatment of cells with S. aureus culture supernatants containing staphylococcal secreted factors transiently activates PKC. Functional studies reveal that overexpression of PKCα causes a marked inhibition of bacterial replication. Taken together, our data identify enhancing PKCα activity as a potential approach to inhibit S. aureus replication in mammalian cells.
Introduction
Autophagy is a cellular degradative process that not only plays an essential role in cellular homeostasis, but also in clearing infection by certain pathogens, a process known as xenophagy. Specifically, xenophagy is a selective defense mechanism by which, once microorganisms are recognized by the cell, proteins known as autophagic adaptors/receptors recruit LC3 and components of the autophagic machinery toward them (1–4). Pathogens sequestered in the interior of autophagosomes are degraded by the fusion of these vacuoles with lysosomes. Many invasive bacterial pathogens, including species of Salmonella, Shigella and Mycobacterium, induce an autophagic response in host cells, which leads to the degradation of these pathogens or partial restriction of their intracellular growth (5–7). However, the generated autophagic response is occasionally unsuccessful, as some microorganisms are able to manipulate this pathway for their own benefit in order to survive and replicate in the host cells (8). A clear example of this behavior is Staphylococcus aureus, a major hospital- and community-acquired pathogen associated with significant mortality (9). Understanding the mechanism by which S. aureus evades xenophagy has important therapeutic potential, particularly given the appearance of strains that manifest antibiotic resistance (10).
S. aureus exerts its effects both by the release of toxins and enzymes secreted by the bacterium, and by efficiently invading epithelial and endothelial cells (11–13). Once in the interior of the cells, S. aureus transits the phagosomal pathway, avoiding lysosomal degradation, to finally escape from phagosomes via a toxin-dependent mechanism to further replicate in the cytoplasm of the host cells (14, 15). It has been previously demonstrated that the S. aureus-containing phagosome is clearly marked by the autophagic protein LC3 (16). We have previously shown that, although S. aureus can localize into autophagosomes, their maturation is blocked and the fusion with lysosomes is inhibited, allowing bacterial replication inside this vacuole (16). Indeed, these S. aureus-containing compartments were also identified as autophagosomes by electron microscopy, due to their characteristic double membranes (17). Finally, S. aureus escapes from the autophagosomes and once in the cytoplasm, the bacterium induces apoptosis through a caspase-independent mechanism which allows the infection to spread (17).
S. aureus infection is accompanied by changes in the intracellular levels of second messengers. For example, it has been reported that when this pathogen invades cells, cAMP levels are decreased (18), leading to reduced activation of the protein Epac, in turn causing, higher levels of autophagy which are beneficial for the staphylococcal infection (19). On the other hand, it has also been reported that S. aureus invasion increases intracellular Ca2+ levels (20), suggesting that transducers of Ca2+ signals might also be involved in the regulation of S. aureus-induced autophagy.
A key transducer of Ca2+ signals is the subfamily of conventional isozymes of the Protein Kinase C (PKC) family of Ser/Thr kinases. PKC isozymes are involved in transduction of a wide range of extracellular signals that control important cellular functions, including the autophagic response (21). Conventional (PKCα, PKCβ and PKCγ) and novel (PKCδ, PKCϵ, PKCη and PKCθ) PKC family members are activated following receptor-mediated generation of diacylglycerol (DAG), which binds to their diacylglycerol-sensing C1 domains (22). Whereas novel PKC isozymes are activated by DAG alone, the conventional PKC isozymes have a lower affinity for DAG and require Ca2+-dependent targeting to membranes via their Ca2+-sensing C2 domain (22). It has been described that novel PKC isozymes participate both in xenophagy and in host cell responses against bacterial infection. For example, PKCϵ is implicated in innate immunity due to its role in the activation of macrophages in defense against S. aureus and E. coli infection (23). Also, it has been shown that both DAG and PKCδ have a role in the autophagy of Salmonella typhimurium (24). As for Ca2+-regulated conventional PKCs, PKCα impairs intracellular replication of Legionella pneumophila in macrophages (25), in addition, PKCα has been shown to be important in phagosomal maturation of latex beads phagosomes (26). However, the role of conventional PKC isozymes in xenophagy remains to be elucidated.
In the present report we have determined that DAG is present in the membranes of phagosomes containing S. aureus and that a conventional PKC isozyme, PKCα, is able to associate with these compartments in an α-hemolysin dependent manner. We have determined that the presence of PKCα in the S. aureus phagosome depends on Ca2+ concentration but is independent of the presence of DAG on the phagosomal membranes. Interestingly, in those phagosomes labelled by PKCα, the recruitment of the autophagic protein LC3 was hampered, indicating that the association of both proteins with the phagosomal membrane was mutually exclusive. In addition, we have found that overexpression of PKCα impaired the efficient intracellular replication of the bacteria. In summary, we present evidence that PKCα modulates the autophagic response induced by S. aureus in epithelial cells.
Materials And Methods
Materials
Cell culture media α-MEM was purchased from Gibco (Invitrogen, Argentina) and fetal bovine serum (FBS) (A15-101) was obtained from GE Healthcare Argentina S.A. Luria-Bertani (LB) broth and agar (Miller) were purchased from Merck (Merck S.A., Buenos Aires, Argentina). Chloramphenicol, gentamycin, U73122, 1-Butanol, Gö6976, PDBu (Phorbol 12,13-dibutirate) and Staphylococcus aureus α-hemolysin (H9395) were obtained from Sigma Aldrich. DNA markers Hoechst 33342 and Topro were from Molecular Probes (Buenos Aires, Argentina).
Cell Culture
Adherent epithelial CHO-K1 (Chinese Hamster Ovary) cells from ATCC were cultured in α-MEM supplemented with 10% of FBS, streptomycin (50μg/ml) and penicillin (50 U/ml).
Plasmids and Transfection
GFP-PKCα, GFP-PKCβII, GFP-PKCγ, and GFP-PKCη were provided by Dr. Yusuf Hannun (Medical University of South Carolina, USA), GFP-PKCδ and GFP-PKCϵ were a gift from Dr. Dominique Joubert (Universtités Montpellier, Francia). PKCδ-C1-GFP was kindly provided by Dr. Mauricio Terebiznik (University of Toronto, Canada). Cells were transfected with Lipofectamine 3000, Lipofectamine 2000 or JetPrime, following the manufacturer’s instructions.
Bacteria Strains, Culture, and Infection
S. aureus wild type, wt (8325–4), the mutant deficient for α-hemolysin: Hla (–) (DU1090) or the mutant Hla (–) complemented with an α-hemolysin plasmid (DU1090/pDU1212): Hla (–)+pHla were used. All strains were kindly provided by Dr. Claudia Sola (CBICI-CONICET, Córdoba, Argentina) and built by Dr. Richard J. O’Callaghan. Bacterial strains were cultured overnight at 37°C in 10 ml of a LB broth with the proper antibiotics for strain selection: streptomycin for the selection of the plasmid DU1090 and chloramphenicol for the selection of the plasmid pDU1212. For infection experiments, cells were resuspended in infection media (α-MEM supplemented with 10% FBS) to an OD=650 nm of 0.4 (~4x108 CFU) and diluted to a 10:1 multiplicity of infection (MOI). 1 hour post infection, the media was washed to eliminate extracellular bacteria and fresh infection media was added. Afterward, cells were incubated for an additional 3 hours at 37°C, washed with PBS, fixed with 4% paraformaldehyde, and processed for analysis by confocal microscopy.
Confocal Microscopy and Image Processing
Cells were analyzed with the microscope Olympus Confocal FV1000. The program FV10-ASW 3.0 was used for image acquisition and imaging configuration. The obtained images were processed by the deconvolution tool in ImageJ.
Real-Time Kinase Activity Monitoring
CHO-K1 cells co-transfected with mCherry-PKCα and CKAR2 (C Kinase Activity Reporter) were maintained in HBSS solution (ThermoFischer) during the experiment. 1 ml of LB broth was added to settle a baseline. After 5 minutes S. aureus culture supernatants were added and, after 10 minutes, 1 μM of the PKC inhibitor Gö6976 was added. To study the effect of α-hemolysin, 10 μg/ml or 30 μg/ml of pure protein was added to the cells, followed by 200 nM of the PKC activator PDBu and finally 1 μM Gö6976. Images were acquired using the microscope Zeiss Axiovert (CarlZeiss Microimaging, Inc.) with the digital camera MicroMax (Roper-Princeton Instruments) controlled by the software Metafluor (Universal Imaging, Corp.). FRET (Föster Resonance Energy Transfer) and CFP (Cyan Fluorescent Protein) were obtained every 25 seconds. YFP (Yellow Fluorescent Protein) and mCherry emissions were also obtained as transfection controls.
SDS-PAGE and Western Blot
CHO-K1 cells were treated as indicated and lysed with RIPA Buffer (150 mM NaCl, 1% Triton x-100, 50 mM Tris pH 7.5, 1% Sodium Deoxycholate, 0.1% SDS, 2 mM EDTA, 50 mM NaF). Bradford assays were performed to determine the protein concentration in the obtained samples. Samples were run in polyacrylamide gels and transferred to PVDF membranes (BioRad). Membranes were blocked during 30 minutes with a blocking solution (10% BSA, 0.05% Tween 20 in PBS) and washed twice with 0.05% Tween 20 in PBS. Afterward, they were incubated with specific antibodies diluted in 0.05% Tween 20 in PBS Tween: 1 μg/ml rabbit polyclonal anti-LC3 (Sigma-Aldrich), or 0.1 μg/ml rabbit monoclonal anti-actin (Developmental Studies Hybridoma Bank, University of Iowa, USA) overnight at 4°C. Membranes were incubated with secondary antibodies conjugated with peroxidase (Sigma-Aldrich). Detection of immunoreactive bands was performed by chemiluminescence in a FluorChem Q imaging system (Protein Simple).
Colony Forming Units (CFU) Assay
CHO-K1 cells were transfected with GFP empty vector (GFPv) or GFP-PKCα. Cells were subsequently infected for 2, 3 or 4 h with S. aureus wt. At 1 hour post infection (h.p.i.), cells were washed 3 times with 1X PBS, and incubated with gentamycin (100 μg/ml) for 30 minutes to eliminate all extracellular bacteria. After the infection period, cells were washed 3 times with 1X PBS, and once with HBSS. Afterward, cells were lysed with sterile distilled water for 10 minutes at room temperature. Cells were collected using a scraper, and lysates were diluted in 1X PBS, cultured in Brain Heart Infusion agar and incubated for 12 hours at 37°C. Colonies were counted in plates which showed between 50-100 visible colonies.
Statistical Analysis
All the data are shown as the mean ± standard error of the mean (SEM) and analyzed with GraphPad Prism version 5.01 (GraphPad Software Inc.) using the Student’s t test. Figures shown are representative of ≥3 experiments.
Results
Diacylglycerol Is Present in the S. aureus-Containing Phagosomes
Since DAG is required for activation of most members of the PKC family, we first asked whether the surface of the phagosomal membrane was conducive to PKC activation by assessing whether DAG was located in the S. aureus phagosomal membrane. CHO cells were transfected with the DAG sensor, PKCδ-C1-GFP, which consists of the C1 domain of PKCδ fused to green fluorescent protein (GFP). Cells were infected with S. aureus wild type (wt, labeled with Topro, shown in blue) and at 4 h.p.i. were fixed and processed for analysis by confocal microscopy. As depicted in Figure 1, DAG was clearly present in approximately 40% of the phagosomal membranes containing S. aureus (Figures 1A, B and Supplementary Figure 1A).
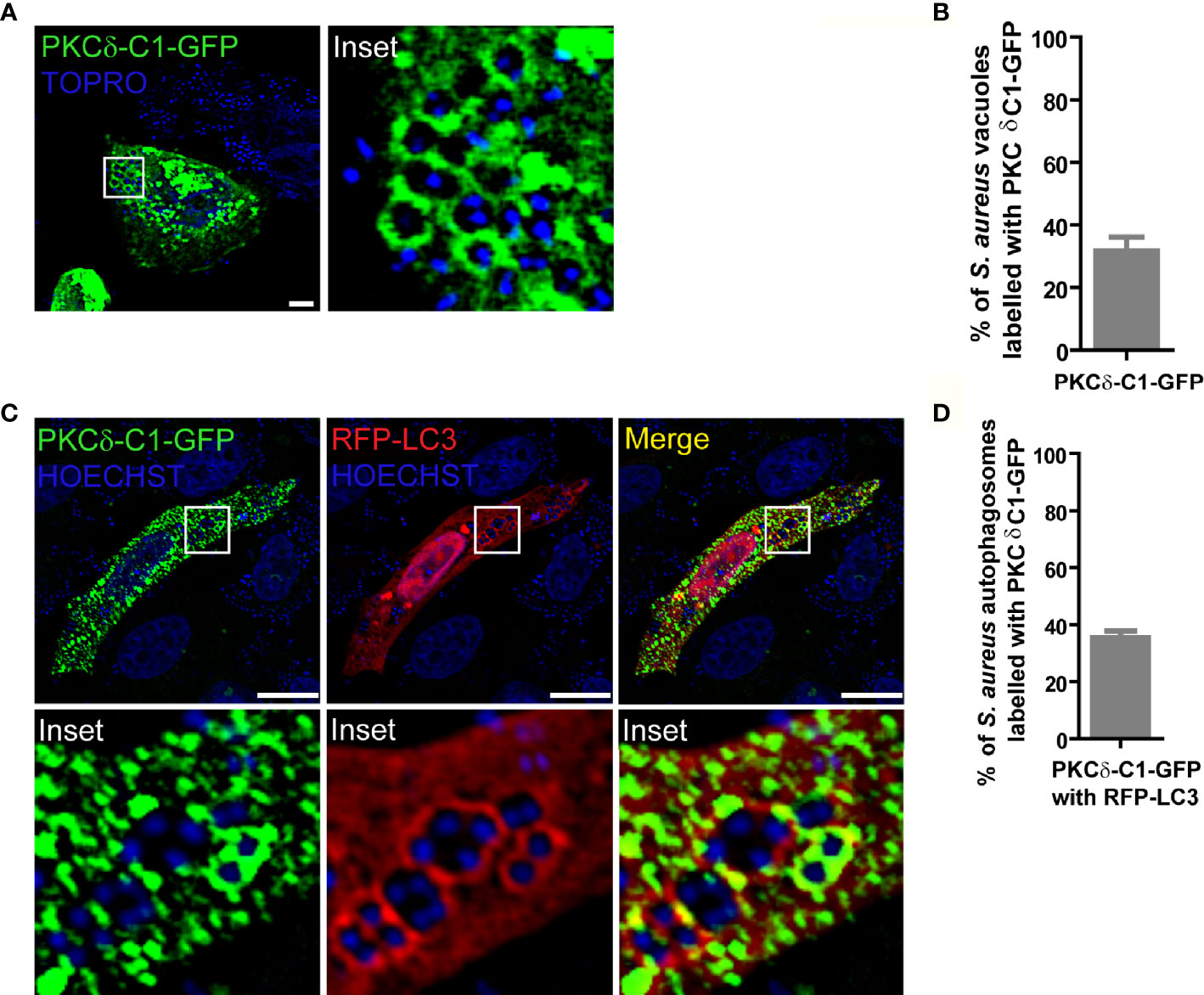
Figure 1 DAG is present in the S. aureus phagosomal membranes. (A) Confocal microscopy images of CHO cells overexpressing PKCδ-C1-GFP. Cells were infected with S. aureus wt for 4 hours as indicated under Material and Methods. Bacteria were labelled with Topro (shown in blue). Images are representative of three independent experiments. Bar: 10 μm. (B) Quantification of S. aureus vacuoles labelled with PKCδ-C1-GFP. Data are the mean ± SEM of three independent experiments. (C) Confocal microscopy images of CHO cells overexpressing PKCδ-C1-GFP together with RFP-LC3 and infected with S. aureus wt for 4 hours. Bacteria were labelled with Hoechst (shown in blue). Bar: 10 μm. Images are representative of three independent experiments. (D) Quantification of S. aureus vacuoles labelled with PKCδ-C1-GFP and RFP-LC3. Data are the mean ± SEM of three independent experiments.
Because it has been shown that internalized S. aureus resides in autophagosomes (17), we next asked whether DAG was also present in these autophagosomal membranes. For this purpose, CHO cells were cotransfected with the DAG sensor and the autophagic protein LC3 tagged with red fluorescent protein (RFP-LC3). Following infection with S. aureus wt, examination by confocal microscopy revealed that approximately 40% of the S. aureus-containing autophagosomes, characterized by the presence of LC3 in their membranes, were also labelled with the DAG probe (Figures 1C, D and Supplementary Figure 1B). These results demonstrate that the second messenger DAG is part of the vacuolar membranes where S. aureus resides.
PKCα Is Recruited to the S. aureus-Containing Phagosome
We next assessed whether the DAG-dependent PKC isozymes (conventional and novel family members) associated with phagosomes harboring S. aureus. For this purpose, CHO cells were transfected with the conventional PKC isozymes GFP-PKCα, GFP-PKCβII or GFP-PKCγ (Figure 2A), or with the novel PKC isozymes GFP-PKCδ, GFP-PKCϵ or GFP-PKCη (Figure 2B). Cells were infected with S. aureus wt and, at 4 h.p.i., were analyzed by confocal microscopy. As shown in Figure 2 (Supplementary Figure 2A), we observed that only the conventional isozyme PKCα was clearly recruited to the S. aureus-containing phagosomes. To confirm that the recruitment of the kinase was in fact to the phagosomal membranes containing S. aureus, GFP-PKCα was cotransfected with RFP-LAMP1, a late endosome marker, which has been proven to be present in the phagosomal membranes containing S. aureus (14). Cells were infected with S. aureus wt and at 4 h.p.i., confocal microscopy analysis showed that PKCα colocalized with LAMP1 at the membranes of phagosomes containing S. aureus (Supplementary Figures 2B and 2C).
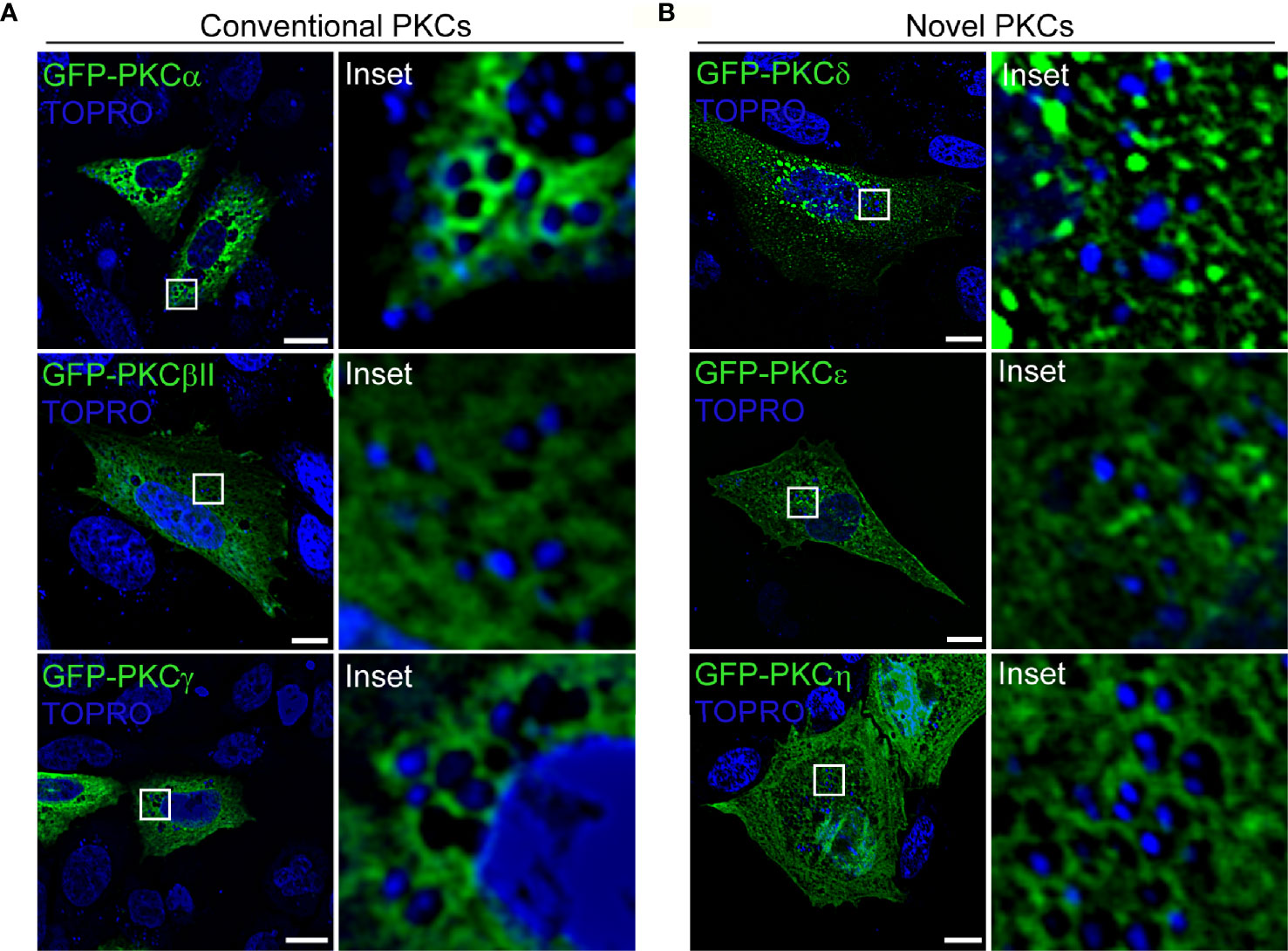
Figure 2 PKCα is recruited to S. aureus phagosomes. (A) Confocal microscopy images of CHO cells overexpressing each of the three members of the conventional PKCs family: GFP-PKCα, GFP-PKCβII, GFP-PKCγ. Cells were infected with S. aureus wt for 4 hours. Bacteria were labelled with Topro (shown in blue). Bar: 10 μm. (B) Confocal microscopy images of CHO cells transfected with members of the novel PKC family: GFP-PKCδ, GFP-PKCϵ or GFP-PKCη. Cells were infected with S. aureus wt for 4 hours. Bacteria were labelled with Topro (shown in blue). Bar: 10 μm. Figures are representative of five independent experiments.
PKCα Recruitment to S. aureus Phagosomes Does Not Depend on DAG
To assess whether the DAG present in S. aureus phagosomal membranes was necessary to anchor PKCα to these compartments, we examined the effect of DAG synthesis inhibition on PKCα localization to the S. aureus phagosomal membranes. CHO cells were transfected with PKCδ-C1-GFP and subsequently treated with inhibitors of DAG synthesis. First, the enzyme phospholipase C (PLC) which generates DAG by the hydrolysis of PIP2 (27) was inhibited using U73122. Treatment of S. aureus infected cells with the inhibitor U73122 had no significant effect on the amount of DAG present in the S. aureus phagosomal membranes (Figures 3A, B). Second, cells were treated with 1-butanol to inhibit phospholipase D (PLD). This enzyme catalyzes the hydrolysis of phosphatidylcholine to produce phosphatidic acid which is, in turn, converted into DAG by the action of the phosphatidic acid phosphatase (27). In contrast to the PLC inhibitor, treatment of S. aureus-infected cells with the PLD inhibitor caused a decrease in the amount of DAG present in phagosomal membranes (Figures 3A, B). These data revealed that PLD was the major pathway responsible for the production of DAG in the S. aureus-containing phagosomes.
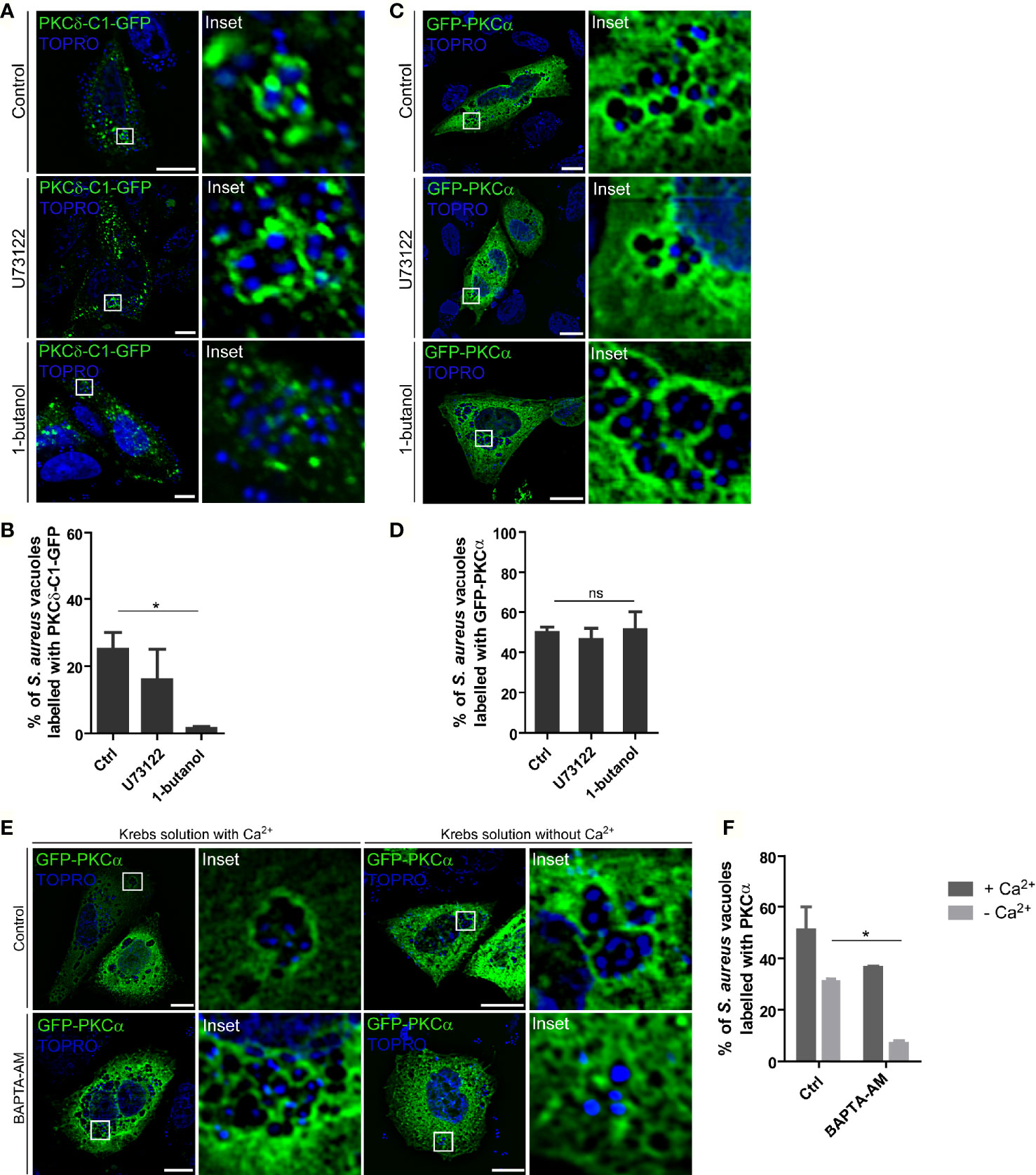
Figure 3 PKCα recruitment to the S. aureus phagosomes is independent of DAG but depends on Ca2+. (A) Confocal microscopy images of CHO cells overexpressing PKCδ-C1-GFP and treated with the PLC inhibitor, U73122 (1.5 µM), or the PLD inhibitor, 1-butanol (0.3% v/v), and infected for 4 hours with S. aureus wt. Bacteria were labelled with Topro (depicted in blue). Bar: 10µm. Images are representative of three independent experiments. (B) Quantification of S. aureus vacuoles decorated with PKCδ-C1-GFP. Data are the mean ± SEM of three independent experiments. *p ≤ 0.01. (C) Confocal microscopy images of CHO cells overexpressing GFP-PKCα and treated with the PLC inhibitor, U73122 (1.5 µM) and the PLD inhibitor, 1-butanol (0.3% v/v), and infected for 4 hours with S. aureus wt. Bacteria were labelled with Topro (shown in blue). Images are representative of three independent experiments. Bar: 10 μm. (D) Quantification of S. aureus vacuoles recruiting GFP-PKCα. Data are the mean ± SEM of three independent experiments. (E) Confocal microscopy images of CHO cells overexpressing GFP-PKCα treated with 10 μM BAPTA-AM in Krebs solution with (2.5 mM) or without Ca2+ and infected with S. aureus wt for 4 hours. Bacteria were labelled with Topro (shown in blue). Bar: 10 μm. Images are representative of three independent experiments. (F) Quantification of S. aureus vacuoles recruiting GFP-PKCα. Data are the mean ± SEM of three independent experiments. *p ≤ 0.05.
Once we determined that the inhibition of PLD caused a marked decrease in the presence of DAG in the phagosomal membranes, we asked whether suppressing PLD activity altered the recruitment of PKCα. CHO cells were transiently transfected with GFP-PKCα, treated with either the PLC or PLD inhibitors, and infected with S. aureus wt. After 4 h.p.i., cells were fixed and analyzed by confocal microscopy. Surprisingly, as shown in Figures 3C, D, there was no change in the presence of the kinase at the S. aureus-containing phagosomes. Thus, we concluded that PKCα is recruited to the phagosomal membranes in a DAG-independent manner.
Because PKCα activity and membrane localization is Ca2+-dependent, we next explored the dependence on Ca2+ in recruiting PKCα to S. aureus-containing phagosomes. CHO cells overexpressing GFP-PKCα were treated with the Ca2+ chelator BAPTA-AM. As we have previously described, S. aureus secretes cytotoxins that are able to generate pores in the plasma membrane allowing the entry of extracellular Ca2+ into the cellular cytosol, therefore, we assessed Ca2+ chelation in Krebs solution with or without Ca2+. In both conditions, cells were infected with S. aureus wt and after 4 h.p.i. they were analyzed by confocal microscopy. As shown in Figures 3E, F, cells treated with BAPTA-AM in a medium without Ca2+ showed a significant decrease of PKCα recruitment to the phagosomal membranes, indicating that this second messenger is required for the translocation and binding of PKCα to the S. aureus phagosomes.
PKCα Recruitment Depends on S. aureus α-hemolysin
We next sought to determine whether the observed PKCα recruitment was dependent on S. aureus viability. For this purpose, bacteria were first inactivated by incubation at 95°C for 10 minutes, and subsequently internalized by CHO cells overexpressing GFP-PKCα. As shown in Figures 4A, B, PKCα was not recruited to heat-inactivated S. aureus, indicating that the recruitment of PKCα only occurs when the bacteria enclosed in the vacuoles are alive. Next, we addressed whether bacterial protein synthesis was required for PKCα recruitment. S. aureus wt was pre-incubated with the inhibitor of bacterial protein synthesis, chloramphenicol. CHO cells were transfected with GFP-PKCα and infected with these bacteria. PKCα was no longer recruited to S. aureus when its protein synthesis was prevented (Figures 4A, B), confirming that the synthesis of bacterial products was required for PKCα’s association to the pathogen-containing phagosomes.
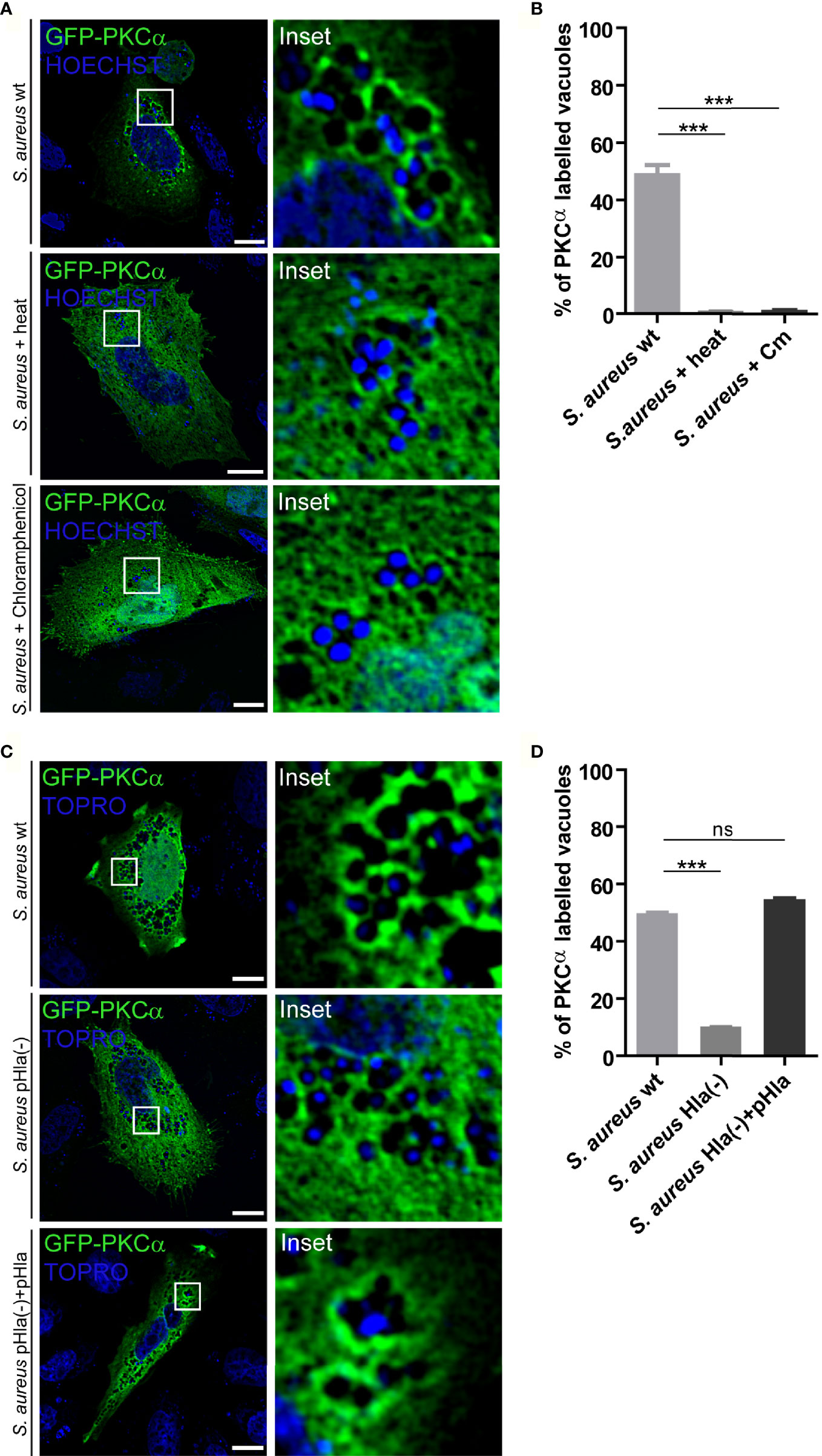
Figure 4 PKCα recruitment to S. aureus phagosomes depends on α-hemolysin. (A) Confocal microscopy images of CHO cells overexpressing GFP-PKCα, and then infected for 4 hours with S. aureus wt, S. aureus wt killed by heat or S. aureus wt treated with chloramphenicol. Bacteria were labelled with Hoechst (shown in blue). Bar: 10 μm. Images are representative of four independent experiments. (B) Quantification of S. aureus vacuoles labelled with GFP-PKCα in cells infected with S. aureus wt, S. aureus wt heat killed or S. aureus wt inactivated by chloramphenicol treatment. Data are the mean ± SEM of four independent experiments. ***p ≤ 0.0001. (C) Confocal microscopy images of CHO cells overexpressing GFP-PKCα, and then infected with S. aureus wt, S. aureus Hla (–), mutant deficient of α-hemolysin or S. aureus Hla (–)+pHla, mutant complemented with the α-hemolysin for 4 hours. Bacteria were labelled with Topro (shown in blue). Images are representative of five independent experiments. Bar: 10 μm. (D) Quantification of S. aureus vacuoles labelled with GFP-PKCα in cells infected with S. aureus wt, S. aureus Hla (–) or S. aureus Hla (–)+pHla. Data are the mean ± SEM of five independent experiments. ***p ≤ 0.0001; ns, non significant.
During staphylococcal infections, the bacteria produce a large amount of secreted virulence factors such as toxins and enzymes. One of the most important virulence factors secreted by S. aureus is α-hemolysin (Hla), a cytotoxin able to generate pores in cellular membranes (28). As we have previously demonstrated, this toxin is required for activation of the autophagic pathway during S. aureus cellular invasion (16). Having established that synthesis of bacterial proteins is necessary for the association of PKCα with pathogen-containing phagosomes (Figures 4A, B), we next addressed whether Hla was one of the bacterial secreted factors responsible for PKCα recruitment. CHO cells overexpressing GFP-PKCα were infected with S. aureus wt, the Hla deficient mutant strain S. aureus Hla (–), or the complemented mutant S. aureus Hla (–)+pHla, which overexpresses the toxin. The samples were processed and analyzed by confocal microscopy. As depicted in Figures 4C and 4D, cells infected with the Hla-deficient mutant showed a marked decrease in the recruitment of PKCα to the bacterial phagosomes. PKCα association was restored to values similar to those observed upon S. aureus wt infection when the cells were infected with the complemented strain S. aureus Hla (–)+pHla. Taken together, these results indicate that the bacterial production of Hla was critical for the recruitment of PKCα to the phagosomes harboring S. aureus.
S. aureus Secreted Factors Activate PKCα
Having established that PKCα was recruited to S. aureus-containing phagosomes by a mechanism that depended on the secreted toxin α-hemolysin, we next examined whether bacterial factors were able to activate PKCα. To this end, we used a genetically encoded C Kinase Activity Reporter (CKAR2), which consists of a PKC specific substrate flanked by mCerulean and a yellow fluorescent protein (YFP) (29). Phosphorylation of the reporter by PKC results in a change in the fluorescence resonance energy transfer (FRET) that functions as a read out for activity of the kinase (30). CHO cells cotransfected with CKAR2 and mCherry-PKCα, were treated with S. aureus culture supernatants, in order to stimulate cells with those virulence factors secreted by the bacterium. Since the LB broth used to grow bacteria is yellow and interferes with the CFP emission, in all the experiments cells were first treated with LB broth to establish a new baseline and then stimulated with S. aureus culture supernatants. Following stimulation with S. aureus supernatant, cells were treated with the PKC inhibitor Gö6976, which inhibits conventional PKC isozymes (31). Treatment of cells with the culture supernatant of S. aureus wt (red line), but not LB broth alone (green line) resulted in a transient activation of PKC, as assessed by the increase in FRET ratio (Figure 5A). These data suggested that factors secreted by S. aureus activate PKCα.
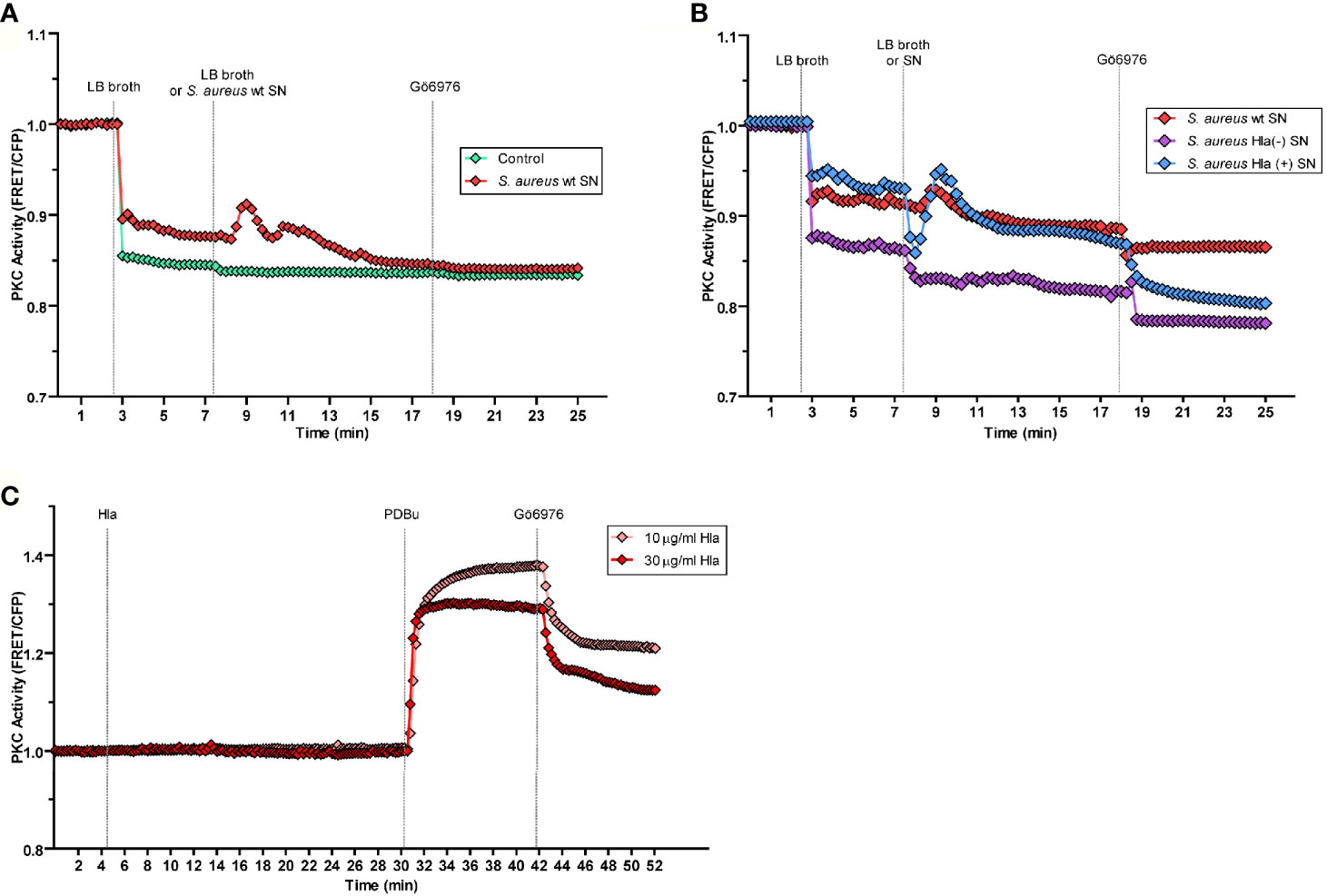
Figure 5 PKCα is activated by the virulence factors secreted by S. aureus. By the use of a genetically encoded biosensor, the C kinase activity reporter (CKAR2), PKC activity was assessed as indicated in Material and Methods. (A) CHO cells co-expressing CKAR2 and mCherry-PKCα were treated with LB broth to set a baseline. After 5 minutes of stable baseline, they were treated with S. aureus wt culture supernatant (red line) and then treated with the PKC inhibitor Gö6976 (1 µM). The control condition (green line) corresponds to LB broth. (B) CHO cells co-expressing CKAR and mCherry-PKCα were treated with LB broth to settle a baseline, then, after 5 minutes they were treated with S. aureus wt (red line), S. aureus Hla (–) (blue line) or S. aureus Hla (–)+pHla (violet line) culture supernatants and then treated with the inhibitor Gö6976 (1 µM). (C) CHO cells co-overexpressing CKAR2 and mCherry-PKCα were treated with 10 µg/ml (pink line) or 30 µg/ml (red line) of α-hemolysin pure protein. After 25 minutes, cells were treated with the PKC agonist PDBu (200 nM) and 12 minutes afterward, the PKC inhibitor Gö6976 (1 µM) was added. Data are the mean of three independent experiments.
We next addressed whether specifically α-hemolysin in the S. aureus supernatant was responsible for the observed PKCα activation. Cells overexpressing mCherry-PKCα and CKAR2 were treated with the supernatants from cultures of S. aureus wt, S. aureus Hla (–), or S. aureus Hla (–)+pHla. Whereas treatment of cells with S. aureus wt culture supernatant (red line) caused PKC activation, supernatant from the Hla-deficient mutant strain (violet line) did not cause activation of PKC (Figure 5B). As expected, activation was restored upon treatment of cells with the supernatant of the mutant complemented with the toxin, S. aureus Hla (–)+pHla (blue line) (Figure 5B). Indeed, the S. aureus Hla (–)+pHla’s supernatant caused an even greater PKC activation peak than the one produced by the wild type strain. Thus, we concluded that the secreted α-hemolysin is necessary for PKCα activation.
Next, in order to determine whether the α-hemolysin itself was able to activate PKCα, CHO cells cotransfected with mCherry-PKCα and CKAR2 were stimulated with two different concentrations of purified toxin, 10 µg/ml and 30 µg/ml. Neither concentration induced activation of the kinase (Figure 5C). To confirm that the system was active, cells were stimulated with the phorbol ester PDBu, a PKC agonist, and the expected activation peak was observed. Thus, α-hemolysin was necessary, but not sufficient, for the activation of PKCα. These data suggest that the presence of other virulence factors secreted by the bacterium likely contribute to the activation of PKCα.
PKCα Inhibits the Autophagy Induced by S. aureus Invasion
After demonstrating that the conventional isozyme PKCα is recruited to the phagosomes where S. aureus resides during its invasion, and that the secreted factors produced by the bacterium activate the kinase, we next examined whether this enzyme regulates the autophagic response that is induced during infection. First, we analyzed whether PKCα presence in phagosomes affected the recruitment of the autophagic protein LC3 to the phagosomal membrane. Cells were cotransfected with RFP-LC3 and GFP-PKCα, or RFP-LC3 and GFP empty vector as a control, infected with S. aureus wt and, after 4 hours, cells were fixed and analyzed by confocal microscopy. Surprisingly, the overexpression of PKCα caused around 40% decrease in the recruitment of the autophagic protein LC3 to the phagosomal membranes (Figures 6A, B). We also observed that PKCα and LC3 did not colocalize at the phagosomal membranes, but rather they were mutually exclusive: phagosomes in which PKCα was present had no detectable LC3 and vice versa (Figure 6C).
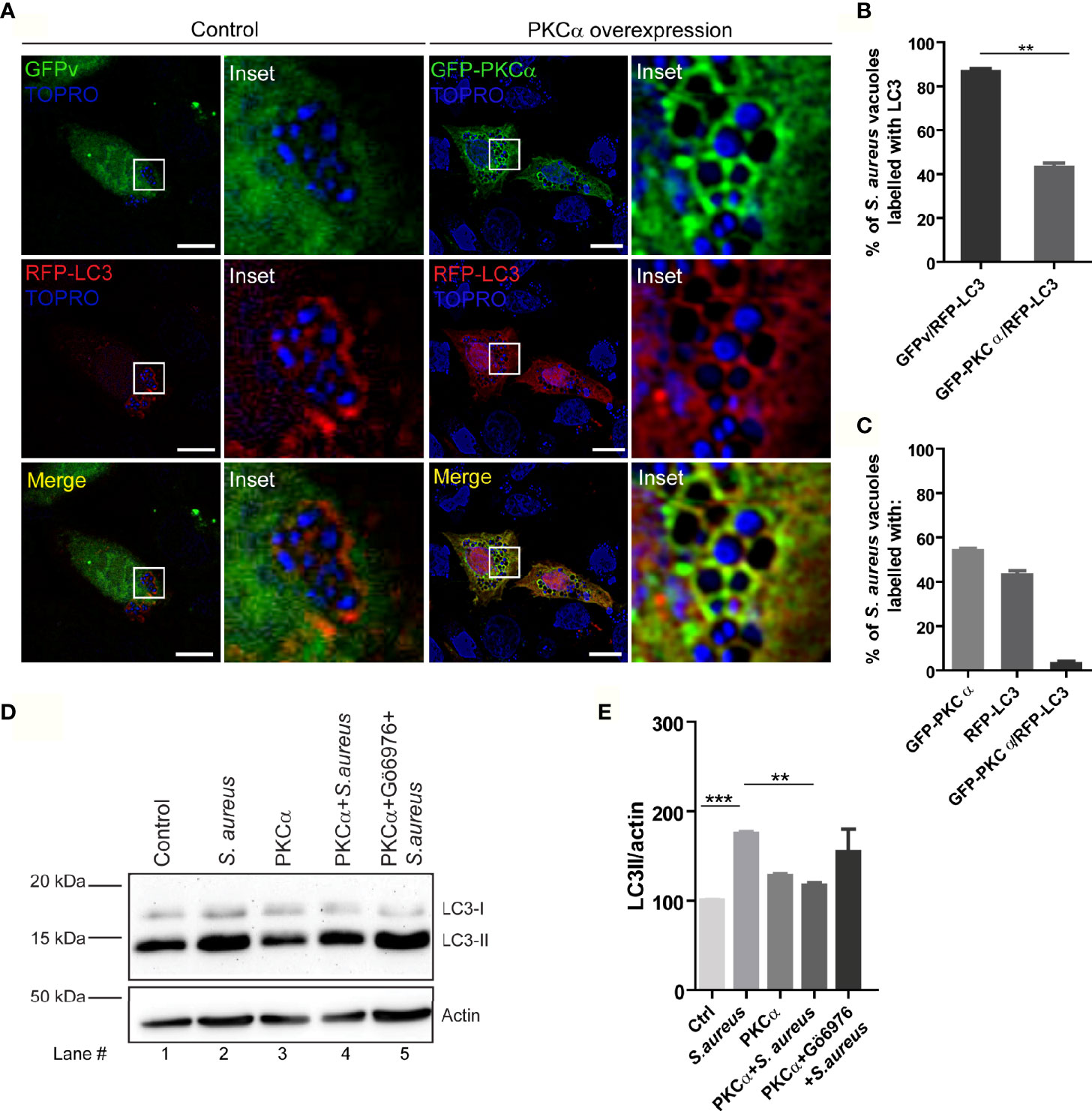
Figure 6 PKCα inhibits autophagy induced by S. aureus. (A) Confocal microscopy images of CHO cells co-overexpressing GFP-PKCα or GFP empty vector (GFPv) and RFP-LC3 and infected for 4 hours with S. aureus wt. Bacteria were labelled with Topro, shown in blue. Bar: 10µm. Images are representative of five independent experiments. (B) Quantification of S. aureus vacuoles recruiting RFP-LC3. **p ≤ 0.001. (C) Quantification of S. aureus vacuoles recruiting GFP-PKCα, RFP-LC3 or both. Data are the mean ± SEM of five independent experiments. (D) Image of a Western blot analysis corresponding to a membrane incubated with specific antibodies against LC3 and actin (as a loading control), of cell lysates obtained from CHO cells subjected to the following conditions: lane 1, control; lane 2, infected with S. aureus wt for 4 hours; lane 3, transfected with mCherry-PKCα; lane 4, transfected with mCherry-PKCα and infected with S. aureus wt for 4 hours; line 5, transfected with mCherry-PKCα, treated with the PKC inhibitor Gö6976 (250 nM) and infected with S. aureus wt for 4 hours. The figure is representative of four independent experiments. (E) Quantification of the Western blot bands intensities with ImageJ. Data are the mean ± SEM of four independent experiments **p ≤ 0.001, ***p ≤ 0.0001.
We next examined whether PKCα modulated autophagy by assessing one of the earliest events in autophagy: processing of the autophagic protein LC3 from the cytoplasmic form LC3-I to the lipidated form LC3-II, which is able to bind to autophagosomal membranes. The conversion of LC3-I to LC3-II as detected by Western blot can be used to measure the activation of autophagy. CHO cells were treated in the following conditions: i) control cells; ii) infected with S. aureus wt for 4 hours; iii) transfected with mCherry-PKCα; iv) transfected with mCherry-PKCα and infected with S. aureus wt; v) transfected with mCherry-PKCα, treated with the inhibitor Gö6976 and infected with S. aureus wt. Western blot analysis of cell lysates with a specific antibody for LC3 revealed that, as previously reported (19), LC3-II levels increased when the cells were infected with S. aureus wt (Figures 6D, E, lane 2). Interestingly, we found that when cells were transfected with PKCα and subsequently infected with S. aureus, LC3-II levels decreased significantly (Figures 6D, E, lane 4) compared to only infection and no kinase overexpression. Also, we observed that in cells treated with the inhibitor Gö6976 and infected with S. aureus, LC3-II levels were reestablished (Figures 6D, E, lane 5). Thus, PKCα overexpression results in reduced association of LC3 protein with phagosomes containing bacteria as assessed by confocal microscopy, and reduced LC3-II levels, as assessed by Western blot. Taken together, these results indicate that PKCα is able to inhibit the autophagic response induced during S. aureus infection.
PKCα Inhibits S. aureus Intracellular Replication
S. aureus is one of the pathogen microorganisms that modulates the autophagic pathway for its own benefit, utilizing the autophagosomes as a protective niche, where it actively replicates before escaping toward the cytoplasm (32). Given that PKCα inhibited the autophagic response induced by S. aureus, we reasoned that the ability of S. aureus to replicate in the interior of cells might be regulated by PKCα. To assess this, CHO cells were transfected with GFP-PKCα or GFP empty vector as a control, infected with S. aureus wt and lysed after 2, 3 or 4 hours. Samples were cultured in Brain Hearth Infusion Agar to allow quantification of bacterial colonies. As depicted in Figure 7, PKCα expression caused a significant decrease in the number of Colony Forming Units (CFU) compared to cells transfected with the empty vector. Therefore, we concluded that the overexpression of PKCα significantly impairs S. aureus intracellular replication.
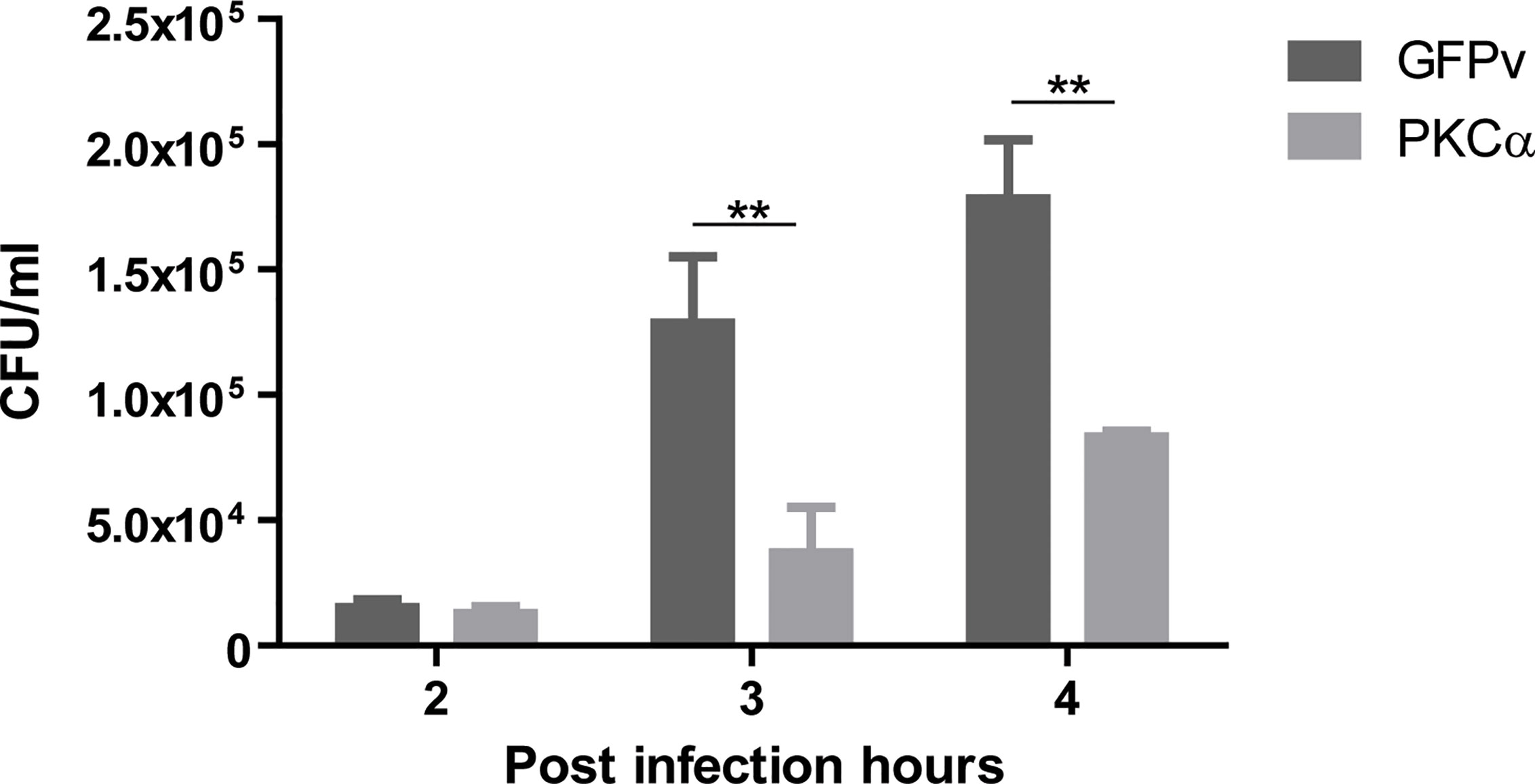
Figure 7 PKCα decreases intracellular replication of S. aureus. Colony forming units (CFU) quantification (see Materials and Methods) of CHO cells overexpressing GFP-PKCα or GFPv and infected for 4 hours with S. aureus wt. Data are the mean ± SEM of three independent experiments. **p ≤ 0.001.
Discussion
S. aureus is a pathogen responsible for a broad range of diseases that vary from local controlled infections to life-threatening systemic infections. The understanding of staphylococcal infections has gained importance given the high antibiotic resistance that the bacterium has developed in recent years. It is crucial to find new ways of impairing the ability of this bacterium to replicate in the interior of cells in order to modulate the damage that it causes.
During bacterial infection, several mechanisms are triggered. It is a battle between the survival mechanisms of the bacteria and the countless signaling pathways that the cells activate in order to avoid bacterial replication and promote their removal from the host cell. Xenophagy is meant to be a degradation pathway that contributes to the elimination of foreign pathogens, but S. aureus utilizes the autophagosomes as a replicative niche (33). Here, we have unraveled one novel signaling pathway that can be used to prevent S. aureus from taking advantage of the autophagic pathway for its replication. In this study, we have demonstrated that the overexpression of PKCα during S. aureus infection causes the recruitment of this kinase to the phagosomal membranes, hampering the autophagic response induced by the invasion of the bacterium, most likely by phosphorylation of the autophagy protein LC3 (34). Moreover, we have shown that overexpression of PKCα generates an important decrease in the intracellular replication of S. aureus in epithelial cells.
Our proposed model suggests that during S. aureus infection, after internalization, the bacteria follow the previously described intracellular pathway, residing in a phagosome whose membrane is disrupted by Hla causing the recruitment of the autophagic protein LC3. The bacteria use the double membrane autophagosomes to actively replicate and subsequently, escape toward the cytoplasm. However, when PKCα is overexpressed during S. aureus infection, the intracellular pathway that the bacteria transit is altered: the action of the bacterial Hla and other virulence factors secreted by S. aureus cause the activation of PKCα. Then, the kinase is recruited to the phagosomal membranes by action of the Hla. We hypothesize that PKCα phosphorylates LC3, preventing its association with the membranes of the compartments containing the bacteria, causing the inhibition of autophagy and in turn, inhibiting the intracellular replication of S. aureus (Figure 8).
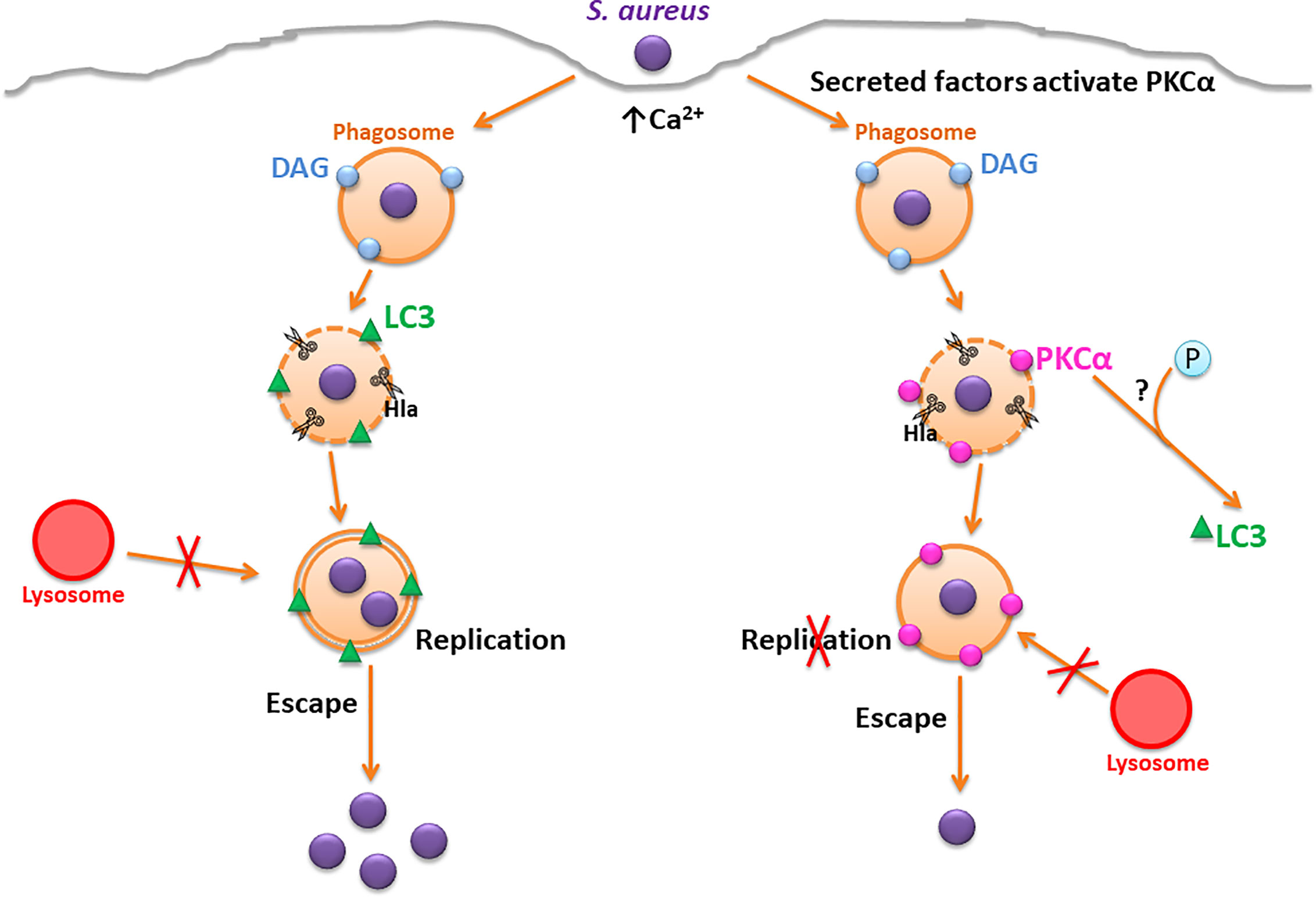
Figure 8 Proposed model. On the left, the canonical S. aureus intracellular traffic is depicted, where Hla causes the disruption of the phagosomal membrane and the activation of autophagic response. S. aureus replicates in the interior of autophagosomes where it resides and finally, escapes to the cytoplasm. On the right, the overexpression of PKCα causes a disruption in the regular S. aureus traffic. Hla causes the recruitment of PKCα to the phagosomal membranes, which inhibits autophagy and impairs S. aureus intracellular replication.
The lipid DAG is an important second messenger involved in a wide range of signaling pathways. Jongstra-Bilen and collaborators showed an accumulation of DAG in the phagosomal membranes containing opsonized latex beads. A burst of DAG was observed during phagosomal formation by the action of Bruton’s tyrosine kinase (Btk) (35). It has also been reported that the autophagosomes where Salmonella typhimurium resides present DAG in their membranes, which is required for the activation of antibacterial autophagy (36). Certain species of Listeria and S. aureus itself produce a phospholipase C enzyme that is able to generate DAG in the host cells (37). Indeed, S. aureus phospholipase C is considered an important virulence factor that contributes to lung injury during staphylococcal infections. In the present results, we have determined that DAG was present in 40% of phagosomes containing S. aureus (Figure 1), which were also labeled by the autophagic protein LC3 (i.e. autophagic compartments), a similar behavior as the one observed during S. typhimurium infection (36).
After screening the members of the PKC family that bind to DAG, we found that the conventional isozyme PKCα is recruited to the membranes of S. aureus containing-phagosomes (Figure 2 and Supplementary Figure 2). It is known that PKCϵ is present in the phagosomal membranes of opsonized beads (38); it has also been observed that PKCα is recruited to phagosomes containing latex beads in murine macrophages, an interaction that is crucial for the maturation of those phagosomes (26). However, to the best of our knowledge, our findings report the presence of this kinase in the phagosomes containing live bacteria for the first time.
The activation of conventional PKC isozymes requires the binding of the two second messengers DAG and Ca2+ to the C1 and C2 domains respectively (39). When DAG synthesis was inhibited, we observed that despite the lack of DAG in the S. aureus phagosomal membranes, PKCα still associated with these membranes (Figure 3), suggesting that the binding of the kinase to these compartments is independent of DAG. This behavior has been reported in the past, and it has been shown that PKCα can also be bound to membranes by protein-protein interactions (40, 41). Further experiments would be necessary to identify possible PKCα recruitment molecules to the vacuole membranes harboring S. aureus. In contrast, we have demonstrated that Ca2+ was required for the recruitment of PKCα to the phagosomal membrane. Eichstaedt and collaborators provided evidence that S. aureus alpha toxin (Hla) leads to an increase in the intracellular Ca2+ levels in a dose- and time-dependent manner (20). We have also demonstrated that the recruitment of PKCα to the phagosomes containing S. aureus is dependent on Hla, because in cells infected with the S. aureus mutant strain deficient for Hla, PKCα recruitment was lost (Figure 4). Taken together, it is likely that during S. aureus invasion, the pores made at the phagosomal membranes by the action of Hla, may cause a localized increase of Ca2+ concentration that promotes the association of this kinase to the phagosomes. In addition, we have established that the activation of PKCα needs the presence of Hla, although this toxin is not able to activate PKCα on its own, since other virulence factors produced and secreted by S. aureus seem to be required (Figure 5). The activation of PKC isozymes during bacterial infections has been described previously: E. coli activates PKCϵ during its invasion (23), Listeria monocytogenes activates PKC in order to be able to escape from phagosomes (42), and PKCδ is activated during S. typhimurium infection in order to activate the antibacterial autophagy (36). However, this is the first report showing that factors secreted by S. aureus activate PKCα.
Of note, we established that the overexpression of PKCα causes an inhibition in the autophagy induced by S. aureus (Figure 6). It has also been shown that PKCα has a role in the regulation of autophagy induced by other stimuli, for example, a pro-autophagy role has been assigned to PKCα in the autophagic response induced by palmitic acid (43). Additionally, it has been shown that PKCα can promote autophagy by mitochondrial disruption and ROS generation (44), but this is the first time that a role in the autophagy triggered by bacterial infection is given to PKCα. In the present report we have shown by confocal microscopy that the overexpression of PKCα during S. aureus infection caused a marked decrease in the recruitment of the autophagic protein LC3 to phagosomes containing the bacteria. We have also shown that PKCα negatively regulates the LC3-II levels by Western blot analysis when comparing cells infected with S. aureus with cells overexpressing the kinase and infected with the pathogen. Consistent with this, Jiang and collaborators have shown that PKC lead to inhibition of starvation-induced autophagy (34). It is likely that a similar mechanism is triggered during S. aureus infection, but further studies are needed to confirm this hypothesis.
We and others have previously demonstrated that transit of S. aureus via the autophagic pathway is beneficial for pathogen survival (16, 17, 32). The biological importance of all our findings is the fact that the overexpression of PKCα, through inhibition of autophagy, causes a marked hampering in the intracellular replication of S. aureus (Figure 7). It has been shown that PKCδ has a similar effect during S. typhimurium infection, causing the elimination of the bacterium, but in this case, the effect has been attributed to activation of autophagy (36). In this regard, it is important to note that autophagy is detrimental for S. typhimurium, but beneficial for S. aureus. Thus, both PKCs are important for bacterial degradation but by distinct mechanisms. It has been shown that PKCα has an important role in controlling infections in macrophages, since the overexpression of a dominant negative mutant of PKCα caused enhanced survival of Leishmania donovani and further replication of Legionella pneumophila (25). All these findings point to a new important focus in the study of antibacterial mechanisms, where PKCs play an essential role in these processes and deserve further studies as therapeutic targets as an alternative to antibiotic treatments.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author Contributions
MG: methodology and investigation. MG, AN, and MC: writing, review, and editing. MC and AN: funding acquisitions and resources. MC: supervision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by PICTs 2013-0305 and 2016-0443 from the ANPCYT and SIIIP 06/J470 (MC), NIH R35 GM122523 (AN). MG was supported by a CONICET (Consejo Nacional de Investigaciones Científicas y Técnicas de Argentina) Ph.D. Fellowship and in part by an IUBMB (International Union of Biochemistry and Molecular Biology) Wood-Whelan Fellowship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Alejandra Medero, Rodrigo Militello and Jorge Ibanez, for valuable technical assistance. We also thank Dr. Gema Lordén Losada and Dr. Maya Kunkel for help and guidance with CKAR experiments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.662987/full#supplementary-material
References
1. Mostowy S. Autophagy and bacterial clearance: A not so clear picture. Cell Microbiol (2013) 15:395–402. doi: 10.1111/cmi.12063
2. Sorbara MT, Girardin SE. Emerging themes in bacterial autophagy. Curr Opin Microbiol (2015) 23:163–70. doi: 10.1016/j.mib.2014.11.020
3. Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH. The Adaptor Protein p62/SQSTM1 Targets Invading Bacteria to the Autophagy Pathway. J Immunol (2009) 183:5909–16. doi: 10.4049/jimmunol.0900441
4. Thurston TLM, Wandel MP, von Muhlinen N, Foeglein A, Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature (2012) 482:414–8. doi: 10.1038/nature10744
5. Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J Biol Chem (2006) 281:11374–83. doi: 10.1074/jbc.M509157200
7. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell (2004) 119:753–66. doi: 10.1016/j.cell.2004.11.038
8. Kwon DH, Song HK. A Structural View of Xenophagy, a Battle between Host and Microbes. Mol Cells (2018) 41:27–34. doi: 10.14348/molcells.2018.2274
9. Lowy FD. Staphylococcus aureus infections. N Engl J Med (1998) 339:520–32. doi: 10.1056/NEJM199808203390806
10. Taylor TA, Unakal CG. Staphylococcus Aureus (2018). Available at: http://www.ncbi.nlm.nih.gov/28722898 (Accessed November 14, 2018).
11. Kintarak S, Whawell SA, Speight PM, Packer S, Nair SP. Internalization of Staphylococcus aureus by Human Keratinocytes. Infect Immun (2004) 72:5668–75. doi: 10.1128/IAI.72.10.5668-5675.2004
12. Edwards AM, Potter U, Meenan NAG, Potts JR, Massey RC. Staphylococcus aureus Keratinocyte Invasion Is Dependent upon Multiple High-Affinity Fibronectin-Binding Repeats within FnBPA. PLoS One (2011) 6:e18899. doi: 10.1371/journal.pone.0018899
13. Mongodin E, Bajolet O, Cutrona J, Bonnet N, Dupuit F, Puchelle E, et al. Fibronectin-binding proteins of Staphylococcus aureus are involved in adherence to human airway epithelium(2002). (Accessed November 14, 2018).
14. Horn J, Stelzner K, Rudel T, Fraunholz M. Inside job: Staphylococcus aureus host-pathogen interactions. Int J Med Microbiol (2018) 308:607–24. doi: 10.1016/j.ijmm.2017.11.009
15. Grosz M, Kolter J, Paprotka K, Winkler AC, Schäfer D, Chatterjee SS, et al. Cytoplasmic replication of Staphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin α. Cell Microbiol (2014) 16:451–65. doi: 10.1111/cmi.12233
16. Mestre MB, Fader CM, Sola C, Colombo MI. Alpha-hemolysin is required for the activation of the autophagic pathway in Staphylococcus aureus-infected cells. Autophagy (2010) 6:110–25. doi: 10.4161/auto.6.1.10698
17. Schnaith A, Kashkar H, Leggio SA, Addicks K, Krönke M, Krut O. Staphylococcus aureus subvert autophagy for induction of caspase-independent host cell death. J Biol Chem (2007) 282:2695–706. doi: 10.1074/jbc.M609784200
18. Mestre MB, Colombo MI. Staphylococcus aureus promotes autophagy by decreasing intracellular cAMP levels. Autophagy (2012) 8:1865–7. doi: 10.4161/auto.22161
19. Mestre MB, Colombo MI. cAMP and EPAC are key players in the regulation of the signal transduction pathway involved in the α-hemolysin autophagic response. PLoS Pathog (2012) 8:e1002664. doi: 10.1371/journal.ppat.1002664
20. Eichstaedt S, Gäbler K, Below S, Müller C, Kohler C, Engelmann S, et al. Effects of Staphylococcus aureus-hemolysin A on calcium signalling in immortalized human airway epithelial cells. Cell Calcium (2009) 45:165–76. doi: 10.1016/j.ceca.2008.09.001
21. Wang T, Liu C, Jia L. The roles of PKCs in regulating autophagy. J Cancer Res Clin Oncol (2018) 144:2303–11. doi: 10.1007/s00432-018-2731-4
22. Newton AC. Protein kinase C: poised to signal. Am J Physiol Endocrinol Metab (2010) 298:E395–402. doi: 10.1152/ajpendo.00477.2009
23. Castrillo A, Pennington DJ, Otto F, Parker PJ, Owen MJ, Boscá L. Protein kinase Cepsilon is required for macrophage activation and defense against bacterial infection(2001). (Accessed September 19, 2017).
24. Shahnazari S, Namolovan A, Klionsky DJ, Brumell JH. A role for diacylglycerol in antibacterial autophagy. Autophagy (2011) 7:331–3. doi: 10.4161/auto.7.3.14045
25. St-Denis A, Caouras V, Gervais F, Descoteaux A. Role of protein kinase C-α in the control of infection by intracellular pathogens in macrophages. J Immunol (1999) 163:5505–11.
26. Ng Yan Hing JD, Desjardins M, Descoteaux A. Proteomic analysis reveals a role for protein kinase C-alpha in phagosome maturation. Biochem Biophys Res Commun (2004) 319:810–6. doi: 10.1016/j.bbrc.2004.05.054
27. Carrasco S, Mérida I. Diacylglycerol, when simplicity becomes complex. Trends Biochem Sci (2007) 32:27–36. doi: 10.1016/j.tibs.2006.11.004
28. Berube B, Wardenburg J. Staphylococcus aureus α-Toxin: Nearly a Century of Intrigue. Toxins (Basel) (2013) 5:1140–66. doi: 10.3390/toxins5061140
29. Ross BL, Tenner B, Markwardt ML, Zviman A, Shi G, Kerr JP, et al. Single-color, ratiometric biosensors for detecting signaling activities in live cells. Elife (2018) 7:e35458. doi: 10.7554/elife.35458
30. Violin JD, Zhang J, Tsien RY, Newton AC. A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. J Cell Biol (2003) 161:899–909. doi: 10.1083/jcb.200302125
31. Wu-Zhang AX, Newton AC. Protein kinase C pharmacology: Refining the toolbox. Biochem J (2013) 452:195–209. doi: 10.1042/BJ20130220
32. López de Armentia MM, Gauron MC, Colombo MI. Staphylococcus aureus Alpha-Toxin Induces the Formation of Dynamic Tubules Labeled with LC3 within Host Cells in a Rab7 and Rab1b-Dependent Manner. Front Cell Infect Microbiol (2017) 7:431. doi: 10.3389/fcimb.2017.00431
33. López de Armentia M, Amaya C, Colombo M. Rab GTPases and the Autophagy Pathway: Bacterial Targets for a Suitable Biogenesis and Trafficking of Their Own Vacuoles. Cells (2016) 5:11. doi: 10.3390/cells5010011
34. Jiang H, Cheng D, Liu W, Peng J, Feng J. Protein kinase C inhibits autophagy and phosphorylates LC3. Biochem Biophys Res Commun (2010) 395:471–6. doi: 10.1016/j.bbrc.2010.04.030
35. Jongstra-Bilen J, Puig Cano A, Hasija M, Xiao H, Smith CIE, Cybulsky MI. Dual Functions of Bruton’s Tyrosine Kinase and Tec Kinase during Fcγ Receptor-Induced Signaling and Phagocytosis. J Immunol (2008) 181:288–98. doi: 10.4049/jimmunol.181.1.288
36. Shahnazari S, Yen WL, Birmingham CL, Shiu J, Namolovan A, Zheng YT, et al. A diacylglycerol-dependent signaling pathway contributes to regulation of antibacterial autophagy. Cell Host Microbe (2010) 8:137–46. doi: 10.1016/j.chom.2010.07.002
37. Daugherty S, Low MG. Cloning, expression, and mutagenesis of phosphatidylinositol-specific phospholipase C from Staphylococcus aureus: a potential staphylococcal virulence factor(1993). (Accessed September 19, 2017).
38. Larsen EC, Ueyama T, Brannock PM, Shirai Y, Saito N, Larsson C, et al. A role for PKC-epsilon in Fc gammaR-mediated phagocytosis by RAW 264.7 cells. J Cell Biol (2002) 159:939–44. doi: 10.1083/jcb.200205140
39. Antal CE, Newton AC. Tuning the signalling output of protein kinase C. Biochem Soc Trans (2014) 42:1477–83. doi: 10.1042/BST20140172
40. Mochly-Rosen D, Gordon AS. Anchoring proteins for protein kinase C: a means for isozyme selectivity(1998). (Accessed November 14, 2018).
41. Colón-González F, Kazanietz MG. C1 domains exposed: From diacylglycerol binding to protein-protein interactions. Biochim Biophys Acta - Mol Cell Biol Lipids (2006) 1761:827–37. doi: 10.1016/j.bbalip.2006.05.001
42. Poussin MA, Goldfine H. Involvement of Listeria monocytogenes Phosphatidylinositol-Specific Phospholipase C and Host Protein Kinase C in Permeabilization of the Macrophage Phagosome. Infect Immun (2005) 73:4410–3. doi: 10.1128/IAI.73.7.4410-4413.2005
43. Tan SH, Shui G, Zhou J, Li JJE, Bay BH, Wenk MR, et al. Induction of autophagy by palmitic acid via protein kinase C-mediated signaling pathway independent of mTOR (mammalian target of rapamycin). J Biol Chem (2012) 287:14364–76. doi: 10.1074/jbc.M111.294157
Keywords: Staphylococcus aureus, autophagy, xenophagy, Protein Kinase C, LC3
Citation: Gauron MC, Newton AC and Colombo MI (2021) PKCα Is Recruited to Staphylococcus aureus-Containing Phagosomes and Impairs Bacterial Replication by Inhibition of Autophagy. Front. Immunol. 12:662987. doi: 10.3389/fimmu.2021.662987
Received: 02 February 2021; Accepted: 03 March 2021;
Published: 18 March 2021.
Edited by:
Jere W. McBride, University of Texas Medical Branch at Galveston, United StatesReviewed by:
Marisa Mariel Fernandez, Institute of Studies on Humoral Immunity (IDEHU), ArgentinaEden Ramalho Ferreira, Federal University of São Paulo, Brazil
Copyright © 2021 Gauron, Newton and Colombo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María Isabel Colombo, bWNvbG9tYm9AZmNtLnVuY3UuZWR1LmFy