 Mohamed Jeljeli1,2
Mohamed Jeljeli1,2 Charlotte Chêne1Sandrine Chouzenoux1Marine Thomas1Benjamin Segain1
Charlotte Chêne1Sandrine Chouzenoux1Marine Thomas1Benjamin Segain1 Ludivine Doridot1
Ludivine Doridot1 Carole Nicco1†
Carole Nicco1† Frédéric Batteux1,2*†
Frédéric Batteux1,2*†- 1Département 3I «Infection, Immunité et Inflammation», Institut Cochin, INSERM U1016, Université de Paris, Paris, France
- 2Université de Paris, Faculté de Médecine, AP-HP-Centre Université de Paris, Hôpital Cochin, Service d’immunologie biologique, Paris, France
Despite significant therapeutic advances, graft-versus-host disease (GvHD) remains the main life-threatening complication following allogeneic hematopoietic stem cell transplantation. The pathogenesis of GvHD is dominated by a dysregulated allogeneic immune response that drives fibrosis and autoimmunity in chronic forms. A multitude of cell therapy approaches, including infusion of myeloid cells, has been proposed to prevent GvHD through tolerance induction but yielded variable results. Myeloid cells like macrophages can be reprogrammed to develop adaptive-like features following antigenic challenge to reinforce or inhibit a subsequent immune response; a phenomenon termed ‘trained immunity’. Here we report that, whereas LPSlow-trained macrophages elicit a suppressor effect on allogeneic T cell proliferation and function in vitro in an IL-10-dependent manner, Bacille Calmette et Guérin (BCG)-trained macrophages exert an opposite effect. In a murine model of sclerodermatous chronic GvHD, LPSlow-trained macrophages attenuate clinical signs of GvHD with significant effects on T cell phenotype and function, autoantibodies production, and tissue fibrosis. Furthermore, infusion of LPSlow-macrophages significantly improves survival in mice with acute GvHD. Importantly, we also provide evidence that LPSlow-macrophages do not accelerate A20-lymphoma tumor growth, which is significantly reduced upon transfer of BCG-macrophages. Collectively, these data indicate that macrophages can be trained to significantly inhibit in vitro and in vivo allo-reactive T cell proliferation without exhibiting pro-tumoral effect, thereby opening the way to promising clinical applications.
Introduction
Graft-versus-host disease (GvHD) constitutes the leading cause of morbidity and non-relapse related mortality after allogeneic hematopoietic stem cell transplantation (HSCT) (1, 2). From the standpoint of hematological malignancies, GvHD treatment remains challenging as allo-reactive T cell depletion by aggressive immunosuppressive therapies can dampen the graft-versus-leukemia effect with a significantly increased risk of relapse of the primary malignancy (3, 4). Despite the dualistic classification of GvHD into acute and chronic forms, based on a cutoff of 100 days of clinical manifestation(s) following HSCT, involvement of the skin and the gastro-intestinal (GI) tract is the constitutive hallmark of these disorders (5). Of note, a patho-physiological continuum exists between the acute and chronic forms of GvHD with inflammation as a trigger for the dysregulated immune response against host tissues (6). Indeed, inflammatory processes play a key role in the initiation and maintenance of immunological mechanisms that underlie alloantigen recognition of host antigens and subsequent tissue destruction by effector T cells (7, 8). The cellular damage induced by conditioning regimen leads to the massive release of damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) triggered by the leakage of gut bacteria and activation of tissue dendritic cells and macrophages (6). The subsequent production of pro-inflammatory cytokines such as Interleukin (IL)-6, IL-1β and Interferon (IFN)-α favors the differentiation of donor CD4+ T cells into Th1 and Th17 inflammatory effector subsets (9). This balance towards a hyperinflammatory status, triggered by the innate immune response, increases the activation of the donor allogeneic CD4+ T cells and enhances their recognition of host allo-antigens presented by activated dendritic cells and macrophages (7). Recently, an emerging concept termed ‘trained immunity’ has been demonstrated whereby innate immune cells like macrophages and NK cells display adaptive features with a rudimentary memory to past antigenic challenges (10–12). Numerous studies conducted by Netea and colleagues have demonstrated that macrophages exposed to BCG or β-Glucan from Candida albicans show a heightened capacity to produce pro-inflammatory cytokines and an upregulation of activation markers and recognition receptors following re-stimulation with pathogens or inflammatory stimuli (13–15). In contrast, macrophages can also acquire a hypo-responsive memory and mount an immunosuppressive response upon exposure to certain stimuli such as helminth products or chronic stimulation with bacterial endotoxin (LPS) (16–19). Trained immunity is accompanied by profound changes in cellular metabolic pathways that regulate chromatin-modifying enzymes leading to long lasting epigenetic modifications (20). Studies targeting trained immunity have mostly focused on the enhanced activation of the immune cells and inflammation to target impaired immune responses and to improve vaccination scheme (21, 22). We recently reported that macrophages trained with low dose of LPS (LPSlow-macrophages) were able to reduce inflammation-induced fibrosis in a murine model of systemic sclerosis, while BCG-trained macrophages (BCG-macrophages) worsened it (23). Clinical strategies to reduce GvHD have focused on the use of immunosuppressive agents that non-specifically affect T-cell function and may be responsible for compromising T-cell immunity by reducing absolute T-cell numbers or inhibiting the function of existing donor T cells (24). Based on these observations, we assessed the effects of trained macrophages, especially the LPSlow-trained macrophages on the allogeneic activity of T cells on the development of acute and chronic GvHD in a murine model. In parallel, we sought to analyze whether the effects observed could influence tumor growth/progression in the A20 B lymphoma murine model as a proof-of-concept for potential cellular therapeutic strategy.
Materials and Methods
Reagents and Chemicals
All chemicals were from Sigma Aldrich (Saint Quentin Fallavier, France). LPS was from E. coli serotype 0127: B8 (Sigma Aldrich); BCG vaccine was purchased from Sanofi Pasteur and composed of 0.5 mg of the Brazilian strain (BCG Biomed-Lublin Laboratory).
Mice
For graft-versus-host disease experiments, eight-week-old female Balb/c (H2d) and C57BL6 (H2b), weighing 15–17 g, were purchased from Janvier Laboratory (Le Genest Saint Isle, France). Male B10.D2 (H2d) mice were kindly offered by Colette Kanellopoulos-Langevin, CDTA-CNRS-Orléans, France). For the A20 lymphoma induction, eight-week-old Balb/c AnN strain mice (H2d) weighing 15–17 g were purchased from Janvier Laboratory (Le Genest Saint Isle, France). All mice were housed in ventilated cages with free access to food and water and maintained under standard 12-h photoperiod. Animals were treated humanely in compliance with the institutional guidelines. The protocols and all experimental procedures of this study were approved by the Ethics Committee from Paris Descartes University (N° CEEA34.CN.017.12).
CD4+ T-Cell Purification and CFSE Labeling
The purification of CD4+ T cells derived from B10.D2 mice (set as responder cells) was performed using the magnetic MACS separator from Miltenyi (130-110-443, Bergisch Gladbach, Germany) and compbeads CD4+ (01-1111-42, Invitrogen) for positive cell selection. Purity of the CD4+ T cell suspension obtained was verified by flow cytometry and was typically >95%. Splenic cells from Balb/c mice extracted as described above were irradiated at an intensity of 30 Gy to render them non-proliferative and constitute the stimulator cells. For CFSE labeling, irradiated splenocytes and CD4+ T cells were stained with 5 µM of CFSE (Ref C34554, Invitrogen) and incubated for 15 min at 37°C in a CO2 incubator protected from light. Labeling was stopped by adding complete MLR medium [RPMI with 10% FBS, 1% HEPES, 1% antibiotic and antimycotic (Gibco)].
Bone Marrow Derived Macrophage Isolation and Innate Immune Training
Bone marrow (BM) cells were collected from the femurs and tibias of female Balb/c mice and differentiated into BMDM using BMDM medium (RPMI with 20% L929 cell supernatant, 10% FBS, 1% antibiotic/antimycotic, and 1% HEPES), as previously described (Manzanero, Methods Mol. Biol 2012; Weischenfeldt, CSH Protoc. 2008). At day 7, cells were washed in warm Ca2+/Mg2+-free PBS and collected by brief trypsin-EDTA (Ref 25200-056, Gibco) treatment. Cells were spun at 1,200 rpm for 7 min to form a pellet and then resuspended in complete RPMI medium. In vitro immune training of BMDMs was performed as previously described (23, 25). BCG treatment was achieved by 48 h incubation of macrophages with dry BCG vaccine (BCG Biomed-Lublin Laboratory) diluted in sterile PBS (Ref CS1PBS01-01, Eurobio) at a ratio of one bacillus for one cell followed by a resting phase of 3 days. LPSlow macrophage training was obtained by adding one daily dose of 10 ng/ml of LPS from E. coli serotype 0127:B8 (Sigma Aldrich) from day 1 to day 3 with daily change of culture medium to avoid cumulative dose toxicity. Control macrophages were obtained by daily treatment with 100 µl of sterile PBS. All cells were cultured in complete DMEM (Ref D0819-500, Sigma Aldrich) with 10% FCS and incubated at 37°C in an atmosphere containing 5% CO2. At day 7, cells were collected by brief trypsin-EDTA treatment, counted and used for either the MLR or for intraperitoneal injection of mice.
Mixed Lymphocyte Reaction
Both the responder and the stimulator cells were adjusted at a density of 105 per 200 µl of complete MLR medium and seeded in U-bottom 96-well microtiter plates (Ref 353077, Falcon). PBS, BCG, or LPSlow-trained macrophages were obtained from Balb/c mice as described above and added at a concentration of 105 to the respective wells as indicated. The assay included appropriate negative controls (CD4+ purified T cells and irradiated Balb/c splenocytes seeded alone) and positive control cultures (CD4+ T cells stimulated with 5 µg/ml of anti-CD3 (Ref 14-0032-86, eBioscience) and 2 µg/ml anti-CD28 (Ref 14-0281-86, eBioscience). Each condition of co-culture was performed in 10-plicate, and plates were incubated for 4 days at 37°C in 5% CO2.
Macrophages IL-10 Invalidation With Small Interfering RNA
Differentiated bone marrow derived macrophages obtained from Balb/c mice were plated at 2 × 105 cells per well in a six-well plate in 2 ml of antibiotic free normal growth medium supplemented with 10% of FBS (Gibco) for 24 h. The siRNA IL-10 (sc-39635) and control (scrambled sequence) siRNA (sc-37007) obtained from Santa Cruz Biotechnology were used to obtain IL-10 KO macrophages by using siRNA transfection medium (sc-36868) and siRNA transfection reagent (sc-29528) (Santa Cruz Biotechnology) according to the instructions of the manufacturer. Macrophages IL-10 invalidation was evaluated by Western blotting and ELISA measurement of IL-10 in stimulated LPSlow-trained IL-10 KO macrophages.
In Vitro Stimulation With Mouse Recombinant IL-10 and STAT3 Inhibition
The specific STAT3 Inhibitor V, Stattic sc-202818 (Santa Cruz Biotechnology, Catalog # 19983-44-9), was used as described before (26). A working solution of 500 µM was obtained after DMSO reconstitution and a 1/100 dilution in PBS. The CD4+ T cells were treated with 10 µM of the STAT3 Inhibitor V in 2 ml of complete RPMI medium (Ref R8758-500, Sigma Aldrich) supplemented with 10% heat inactivated Fetal Bovine Serum (FBS, Ref 10270-106, Gibco), 2 mM L-glutamine, 1% streptomycin–penicillin (Ref 15140-122, Gibco), 1 mM sodium pyruvate (Ref CSTVAT00-0U, Eurobio) and 10 mM HEPES (Ref 15630-056, Gibco). A control vehicle condition was composed of allogeneic CD4+ cells cultured in complete DMEM medium with PBS/DMSO (final concentration of 1% DMSO). Then, allogeneic CD4+ T cells were stimulated with the mouse recombinant IL-10 from BioLegend (Ref #575802, Ozyme France, 78180 Montigny-le-Bretonneux) at a concentration of 100 ng/ml and incubated for 24 h at 37°C 5% CO2. All the conditions were performed in six-plicate and the experiments were carried out twice. Cells were then collected to perform further analysis (Western blot and RT-qPCR).
Clinical Assessment of GvHD
The severity of systemic GvHD in mice was assessed according to a mouse clinical GvHD scoring system inspired by Anderson et al. (27). We set the scoring system for chronic GvHD to five clinical criteria (maximum index = 5) representing the sum of the recorded values. Weight loss of <10% was scored 0 and of >20% was scored as 1. Regarding the GI symptoms, the scores were 0 for mice with no diarrhea and 1 for mice suffering from diarrhea. For posture, the scoring system denoted 0 for normal, 0.5 for hunching at rest, and 1 for severe hunching. For activity, the scoring represented 0 for normal, 0.5 for mild to moderate decrease, and 1 for severe decrease activity. For signs of vasculitis and cutaneous involvement, the scoring system denoted 0 for normal, 0.5 for mild lesions on ears or tails, and 1 for eyelid and severe alopecia. Total clinical GvHD score of each mouse was measured twice a week by a scientist blinded to the different groups.
Isolation and Stimulation of Spleen Cells, Splenic Macrophages, and Skin-Derived Immune Cells
Mice were sacrificed by cervical dislocation. Spleens were aseptically removed, and cellular splenic suspensions were prepared after hypotonic lysis of erythrocytes in potassium acetate solution and three washes in complete RPMI medium supplemented with 10% heat inactivated Fetal Bovine Serum, 2 mM L-glutamine, 1% streptomycin–penicillin, 1 mM sodium pyruvate, and 10 mM HEPES. For each mouse, splenocytes were enumerated using a Malassez counting chamber. Skin-derived immune cell isolation was achieved as described before (28). Briefly, 8 mm-calibrated dermal punches of tissue were collected from the shaved back of each mouse and minced into small pieces in a 60 mm Petri dish. Tissue was then lysed with 1 ml of Liver Digest Medium (Gibco) solution and 1 mg/ml of collagenase (Ref C2674-1G, Sigma-Aldrich) in 9 ml of complete RPMI medium for 3 h at 37°C in an atmosphere of 5% CO2. Cell suspension was then filtered through a 70 µm cell strainer, washed in complete RPMI medium and counted with Malassez counting chamber for further analysis.
Induction of cGvHD in Balb/c Mice and Trained Macrophage Infusion
Sclerodermatous chronic GvHD (cGvHD) was induced in female Balb/c mice (H2d) by grafting cells from 9- to 10-week-old male B10.D2 mice (H2d), as previously described (29). Briefly, Balb/c mice were lethally irradiated with 750 cGy from a Gammacel [137Cs] source. Graft was prepared by adding 2 × 106 spleen cells previously purified from red cells with a hypotonic solution of potassium acetate to 1 × 106 of bone marrow cells resuspended in sterile PBS. Graft was injected in the retro-orbital vein 3 h after the irradiation of recipient mice. A group composed of (n = 5) lethally irradiated mice but non-transplanted was included to validate the irradiation source. A control syngeneic group (n = 5) of Balb/c recipient mice grafted with Balb/c spleen and bone marrow cells was added. GvHD mice were divided into four groups. One group (cGvHD; n = 5 mice) did not receive any macrophage injection. The three remaining groups (n = 10 mice) received a weekly infusion of 2 × 106 intra-peritoneal Balb/c PBS-, LPSlow-, or BCG-trained BMDM, prepared as described above. A total of six injections was performed on D1, D7, D14, D21, D28, and D35 of the experiment. During the early post-irradiation and graft period (day <10), twice mice from the cGvHD-PBS, one mouse from the cGvHD-BCG and two mice from the cGvHD-LPSlow group either died spontaneously or were humanly sacrificed because of evident clinical suffering. The remaining animals were sacrificed by cervical dislocation at day 40 after the graft for further analysis.
Induction of aGvHD in Balb/c Mice and Trained Macrophage Infusion
Acute GvHD (aGvHD) was induced in female Balb/c mice (H-2d; Janvier Laboratory) by grafting cells from 9- to 10-week-old female fully mismatched major histocompatibility complex (MHC) C57BL/6 mice (H-2b; Janvier Laboratory), as previously described (30). Balb/c mice were lethally irradiated with 750 cGy from a Gammacel [137Cs] source and grafted 3 h later with 5 × 106 RBC-free spleen cells and 1 × 106 BM cells in sterile PBS. Control groups were as follows: a group (n = 5 mice) composed of lethally irradiated mice but non-transplanted and a control syngeneic group (n = 5 mice) of Balb/c recipient mice grafted with Balb/c spleen and BM cells. Acute GvHD mice were divided in four groups (n = 8 mice each). One group (aGvHD mice) did not receive any macrophage injection. The three remaining groups received a weekly intraperitoneal infusion of 2 × 106 Balb/c PBS-, LPSlow- or BCG-trained bone marrow derived macrophages as previously described. A total of four injections was performed on D1, D7, D14, and D21 of the experiment. Animals were monitored daily for survival and three times a week for weight measurement.
Graft-Versus-Host Disease Monitoring
Monitoring of graft recipients was based on daily observation and measurement of clinical evidence of GvHD (weight loss, manifestations of skin erythema, alopecia, hunching, and diarrhea) and survival during the observation period for both cGvHD and aGvHD experiments. Animals were humanely sacrificed if they had lost more than 40% of their original weight or if they showed evident signs of morbidity and suffering.
Detection of Anti-DNA Topoisomerase I Autoantibodies in Sera
Levels of anti-DNA topoisomerase I IgG antibodies were assayed by the Scl-70 IgG ELISA kit. Diluted (1/4) mouse serum was distributed into the wells of a microtiter plate (Abnova, Taipei City, Taiwan) coated with purified calf thymus DNA topoisomerase I. The conjugated anti-mice Ig horseradish peroxidase (Dako, Glostrup, Denmark) secondary antibody (1/100) was then added, and the absorbance was read at 450 nm. Results were expressed as antibody index using the cutoff value derived from the Calibrator optical density as previously described (29).
Serum Alanine Transferase Level
Serum level of ALT was used as a marker of hepatocyte cytolysis and was quantified using a standard clinical automatic analyzer (Modular PP, Roche Diagnostics, Meylan, France).
Subcutaneous Injection of A20 Cells in Balb/c Mice and Trained Macrophage Infusion
The A20 cell line, a Balb/c AnN mouse (H-2d haplotype, female genotype) B lymphoma cell line derived from a spontaneous reticulum cell neoplasm (31), was purchased from the American Type Culture Collection (ATTC TIB-208; 1999). A20 cells were expanded in 20 ml culture Flask (Ref 353110, Falcon) in RPMI 1640 culture medium supplemented with 10% FCS, 5 µM β-mercapto-ethanol (Ref 31350-010, Gibco) and 1% streptomycin–penicillin.
For tumor challenge, A20 cells were washed twice in RPMI medium and resuspended at a concentration of 2 × 106 cells in 200 µl of PBS and injected in the shaved back of mice. Mice were divided into four groups of n = 7 mice per group. One group (A20-CTRL) did not receive any macrophage injection. The three remaining groups (A20-PBS, A20-LPSlow, and A20-BCG, respectively) received a weekly intraperitoneal infusion of 2 × 106 Balb/c AnN PBS-, LPSlow- or BCG-trained BMDM as described above. A total of five injections was performed on D1, D7, D14, D21, and D28 of the experiment. Animals were monitored daily for survival and three times a week for weight and tumor measurement. Tumors were measured three times a week with a microcaliper in three dimensions by a scientist blinded to the different treatments, and tumor volumes were calculated as length × width × depth × 0.5236, as previously described (32). Mice that extremely suffered from the tumors and expected to die within the same day of observation (signs including lack of eating/drinking/moving activity under mild stimulation, shivering, eye lids shut, etc.) were humanely euthanized on the days of observation. Mice were sacrificed at day 34 of the experiment for further analysis.
Reverse Transcription-Quantitative PCR
Total mRNA was extracted from crushed samples with TRIzol reagent (Ref 15596018, Ambion). QuantiTect SYBR® Green RT-PCR Kit (Ref 04053228014782, Qiagen) on a LightCycler 480 II instrument (Roche Applied Science, France) was used to perform one-step RT-qPCR. Samples were normalized to mRNA expression of housekeeping genes (GAPDH), and results were expressed as fold increase using the formula 2−˄˄Ct. Primers used for PCR are listed in Supplementary Table S1.
Flow Cytometric Analysis of Splenic and Skin-Derived Immune Cells
Cell suspensions (spleen cells, skin-derived immune cells and cell suspensions of the MLR) were incubated for 20 min with 10 µg/ml anti-CD16/CD32 antibody (clone 93, eBiosciences) for Fc receptor saturation and then stained with the appropriate labeled antibody at 4°C for 30 min in the dark in PBS with 2% FBS. The FACS Fortessa II flow cytometer (BD Biosciences) was used to perform flow cytometry according to standard techniques. For spleen and skin characterization of immune cells, the monoclonal antibodies used were B220-Pacific Blue (RA3-6B2, catalog#103230, dilution 1/200), CD4-BV711 (RM4-5, catalog#563726, dilution 1/100), CD8-PE-Cy7 (QA17A07, catalog#155017, dilution 1/200), CD62L-PE-Cy5 (MEL-14, catalog# 104410, dilution 1/200), CD44-APC (IM7, catalog#103012, dilution 1/200), CD44-PeCy7 (IM7, catalog#103029, dilution 1/200), CD69-PercPCy5.5 (H1.2F3, catalog#104522, dilution 1/100), CD11b-BV510 (M1/70C, catalog#101245, dilution 1/200), F4/80-BV711 (BM8, catalog#123147, dilution 1/200), CCR4-BV421 (2G12, catalog#131217, dilution 1/100), CCR5-PercP-Cy5.5 (HM-CCR5, catalog#107015, dilution 1/200), (iCOS ligand) CD278-PeCy5 (15F9, catalog#107708, dilution 1/200), CXCR3-PE Dazzle 594 (CXCR3-173, catalog#126533, dilution 1/100), from BioLegend (Ozyme France, 78180 Montigny-le-Bretonneux), and CCR10-Alexa Fluor 700 (conjugated, catalog#FAB2815N, dilution 1/100) from R&D Systems (RD-BIOTECH, Besancon, France). Data were analyzed with FlowJo software (Tree Star, Ashland, OR).
ELISA Cytokine Detection
Measurements of IL-2, IL-6, IL-10, IL-13, IL-17A, TNF-α,and IFN-γ were performed by specific mouse ELISA kits from eBiosciences (Thermo Fisher Scientific, Villebon-Sur-Yvette, France). Concentrations were calculated from a standard curve according to the protocol of the manufacturer.
Western Blotting of Cell Lysates
Cells were lysed in ice-cold RIPA buffer (10 mmol/L Tris-HCl, pH 7.5; NaCl, 150 mmol/L; 1% Triton X-100; 0.1% sodium dodecyl sulfate) supplemented with 25 mmol/L sodium fluoride, anti-protease 1%, and 0.5 mmol/L sodium orthovanadate. Equal amounts of protein (30 μg) were loaded and separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Biorad Marnes-la-Coquette, France). After transfer and blocking with 5% of fat-free milk and 0.1% Tween in PBS, nitrocellulose membrane was incubated overnight at 4°C with the appropriate dilution of each antibody used according to the guidelines of the manufacturer. The antibodies used in the Western blotting analysis are listed in Supplementary Table S2. Specific unconjugated proteins were detected using a 1:10,000 dilution of a horseradish peroxidase-conjugated goat anti-rabbit IgG (Life Technologies, Catalog# 31490) and visualized by an enhanced chemiluminescence system (Advansta, Diagomics, France).
Histopathological Analysis
Liver, lung, and skin pieces fixed in formol were embedded in paraffin. Tissue sections of 6 mm thickness were made and stained with Sirius red or H&E (hematoxylin and eosin). The slides were examined by standard bright field microscopy (Nikon Eclispe 80i) by a pathologist who was blinded to the experimental group assignment.
Statistical Analysis
The results were analyzed with the GraphPad Prism5 program. The one-way test ANOVA (Kruskal–Wallis test) was used to verify that the groups were comparable to each other. The Student’s t-test (Mann–Whitney test) was used to determine the differences between two experimental groups. A difference <0.05 was considered as significant.
Results
LPSlow-Trained Macrophages Inhibit T Cell Proliferation in a Mixed Lymphocyte Reaction
Trained macrophages can exhibit opposite phenotype and function according to the nature of the first antigenic stimulation (23). Here, we performed a mixed lymphocyte reaction mimicking in vitro the allo-immune response involved in the in vivo GvHD to evaluate the impact of trained macrophages on the proliferation and cytokine secretion of allogenic CD4+ T cells from B10.D2 mice. The proliferative response was detected by CFSE dilution, and activation of the responder cells was assessed by CD44 surface expression. The CD4+ T responder cells showed a strong proliferative response when cultured with irradiated splenic cells from Balb/c mice compared to CD4+ T responder cells cultured alone. As a positive and a negative control respectively, CD4+ T cells stimulated with anti-CD3/CD28 antibodies showed a strong proliferation index and a high activation status while irradiated Balb/c splenic cells barely proliferated (Supplementary Figures S1A, B). The addition of PBS-macrophages to the allogeneic reaction did not influence the proliferative response and activation of the allogeneic CD4+ T responder cells. By contrast, CD4+ T responder cells showed an enhanced proliferative index (p = 0.01), associated with increased CD44 expression (p = 0.0002) when cultured with BCG-macrophages, while their proliferation as well as activation was significantly reduced when cultured with LPSlow-macrophages (p = 0.0009 and p = 0.003, respectively, Figures 1A, B). We then assessed the impact of trained macrophages on the function of allogeneic CD4+ T responder cells by measuring the levels of IL-2, IFN-γ, TNF-α, IL-6, and IL-10 in the supernatant of the different MLR conditions. Basal cytokine production of the irradiated Balb/c splenocytes and the B10.D2 purified CD4+ T cells (either unstimulated or stimulated with anti-CD3/CD28 antibodies) is shown in Supplementary Figure S1C. Of note, analysis of cytokine production in the supernatant of MLR with PBS-trained macrophages condition did not show any difference compared to the MLR condition alone. Addition of BCG-macrophages was associated with a significant increase in IL-2 (p < 0.0001), IL-6 (p = 0.009), TNF-α (p = 0.01), and IFN-γ (p = 0.01) in the supernatant, compared to the allogeneic MLR condition (Figure 1C). On the contrary, the addition of LPSlow-macrophages in the MLR significantly reduced the levels of IL-2 (p < 0.0001), IL-6 (p < 0.0001), TNF-α (p = 0.0008), and IFN-γ (p = 0.02) while significantly increasing IL-10 in the culture supernatants (p = 0.0004, Figure 1C). Collectively, these data suggest that appropriately trained macrophages can act as either an enhancer or a suppressor of allogeneic CD4+ T cell proliferation, activation, and cytokine response in the setting of MLR.
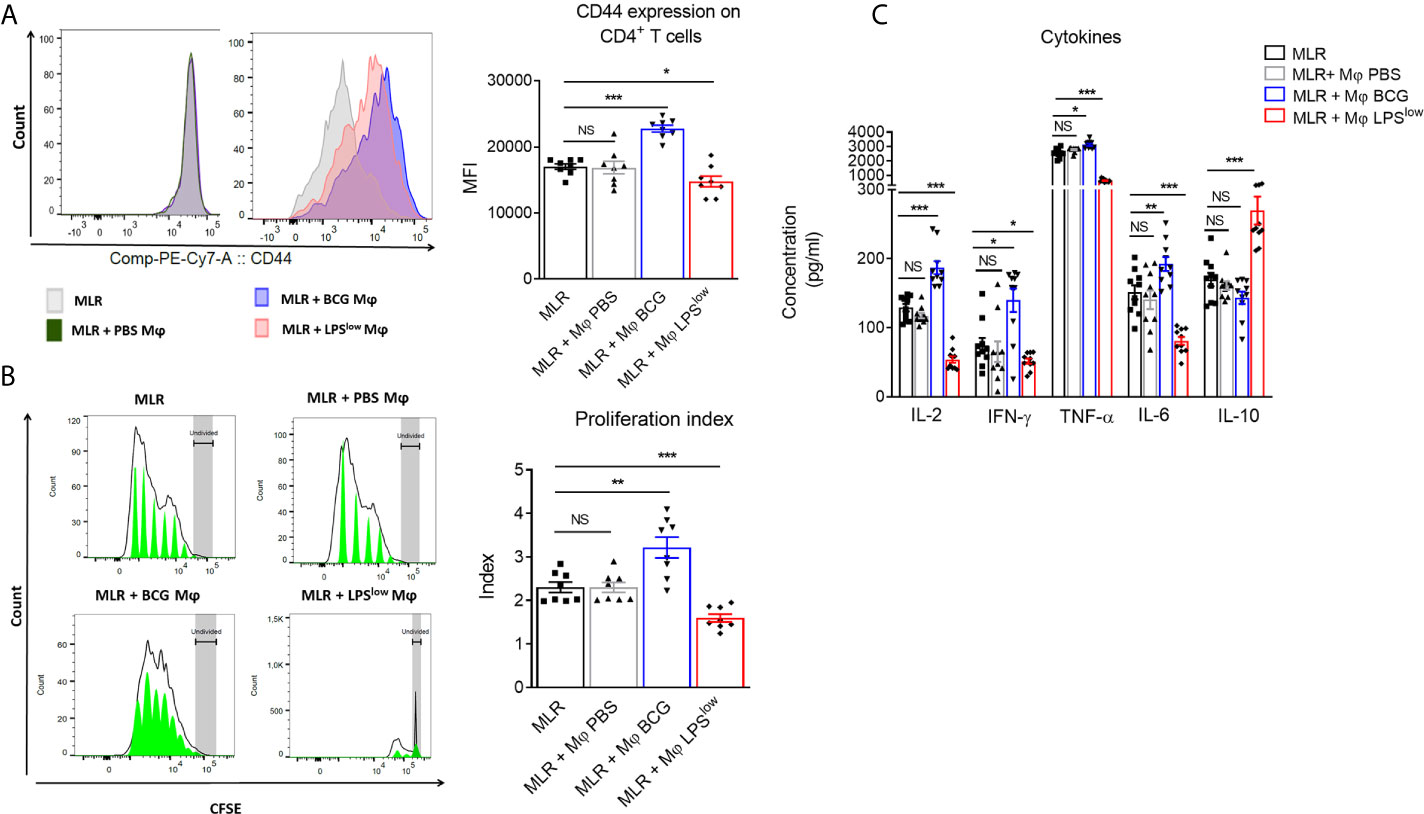
Figure 1 Effects of trained-macrophages adding on CD4+ T cell proliferation, activation, and cytokine secretion in a mixed lymphocyte reaction. (A) Flow cytometry analysis of CD4+ T-cell activation after in vivo training during mixed lymphocyte reaction. Responder cells were constituted by purified CD4+ T cells derived from B10.D2 mice. Irradiated splenic cells from Balb/c mice constituted the stimulator cells. Cells were cultured in different conditions. Control groups were: CD4+ T cells alone in medium, irradiated Balb/c splenic cells alone in medium, CD4+ T cells with anti-CD3/CD28 antibody. Tested groups were: CD4+ T cells with irradiated Balb/c (MLR), MLR with PBS-trained Balb/c Mφ (bone marrow derived macrophages), MLR with LPSlow-trained Mφ, MLR with BCG-trained Mφ. Each condition of co-culture was performed in a 10-plicate, and plates were incubated for 4 days at 37°C in 5% CO2. Overlaid histograms indicate a representative CD44 expression on gated CD4+ T cells of each condition. The column graph represents the mean fluorescence intensity of CD44 expression ± SEM obtained from the 10-plicate of three independent experiments. (B) Flow cytometry assessment of CFSE-stained B10.D2 CD4+ T-cell proliferation cultured for 96 h in different conditions. CFSE graphs are representative of each co-culture condition. The first peak represents the undivided cells. The black line represents the different cell populations that underwent division, and the green area shows the peaks of different generations of proliferating cells. The column graph represents the mean of the proliferative index of each condition ± SEM obtained from the 10-plicate of three independent experiments. (C) Levels of IL-2, IFN-γ, TNF-α, IL-6, and IL-10 cytokines in the supernatant of the different MLR-trained Mφ conditions. Concentrations were assessed by ELISA and bar graphs represent the mean (pg/ml) ± SEM of 10-plicate of three independent experiments. The ANOVA test with Bonferroni correction was used to detect significant differences between the groups. Statistics are shown between the MLR group and the MLR + trained macrophages conditions. NS, non-significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Adoptive Transfer of Trained Macrophages Modulates cGvHD in Mice
We next investigated the in vivo effects of the adoptive transfer of trained macrophages on the course of cGvHD. We used the well-established animal model of sclerodermatous cGvHD triggered upon transplantation of B10.D2 (H-2d) BM and spleen cells across minor histocompatibility loci into lethally irradiated Balb/c (H-2d) recipients (Figure 2A). Clinical score assessment started 10 days after transplantation. At day 40, cGvHD mice had a score of 4.60 ± 0.36 while syngeneic remained null (p < 0.0001). Weight loss was marked in cGvHD mice compared to syngeneic animals (p = 0.005, Figure 2B). Importantly, infusion of LPSlow-macrophages significantly attenuated the severity of the clinical score and the weight loss compared to the untreated mice (p = 0.005 and p = 0.006, respectively, Figures 2B, C). Adoptive transfer of PBS-macrophages did not affect the clinical score nor the weight loss, whereas BCG-macrophage infusion tended to aggravate them though not significantly (p = 0.15 and p = 0.23, respectively). Representative photographs of mice at day 40 after BMT showed spiky coat and alopecia in the cGvHD animals with a favorable resolution in the cGvHD-LPSlow group (Figure 2D). Histological hematoxylin–eosin staining showed mixed lymphomonocytic infiltrations around the portal vein, which was significantly suppressed in the cGvHD-LPSlow group (Figure 2E). While mice receiving PBS or BCG-macrophages did not significantly affect cGVHD-induced increase in serum ALT, a marker of GvHD-associated liver injury, a virtually complete inhibition was observed in cGvHD-LPSlow group compared to the cGVHD group (p < 0.0001) (Figure 2F).
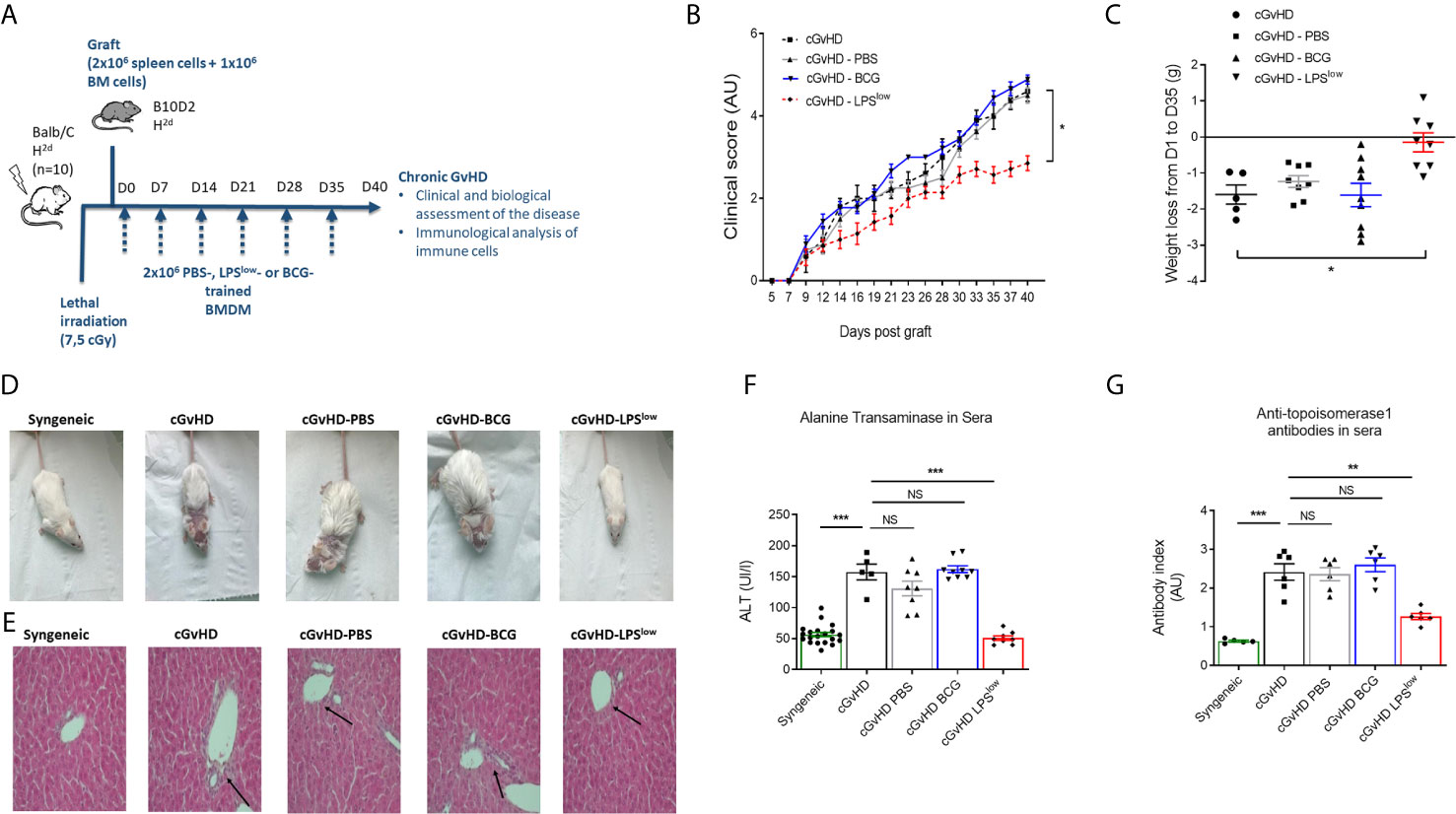
Figure 2 Adoptive transfer of trained macrophages modulates cGvHD in mice. (A) Schematic representation of the experimental induction of sclerodermatous chronic graft-versus-host disease in mice and scheduling of trained macrophages infusion. Mice (n = 7 at least per group) were monitored for survival, weight, clinical signs of chronic GvHD for 40 days in one experiment. (B) Clinical score assessment of the different groups of cGvHD mice. The scoring system included five clinical criteria (maximum index of 5) representing the sum of the recorded values and expressed in arbitrary unit (AU). (C) Weight loss assessment of the different groups of cGvHD mice. Mice were weighted at day 0 of the experiment and equitably distributed into the different groups according to their initial weight. At day of the sacrifice, each identified mouse was weighted to determine the weight loss delta. Scatter plots represent individual data with mean ± SEM obtained from at least n = 5 biologically independent mice: syngeneic (n = 5); cGvHD (n =5); cGvHD-PBS (n = 8); cGvHD-BCG (n = 9); cGvHD-LPSlow (n = 8). Statistical differences between the groups were detected by the ANOVA test with Bonferroni correction. (D) Representative photographs of mice at day 40 after BMT showing hair aspect (spiky coat and alopecia in the cGvHD animals compared to the syngeneic group). (E) Representative liver section of 5 μm stained with hematoxylin and eosin showing increased periportal mononuclear cell infiltration in cGhVD mice compared to syngeneic group. Photographs were taken with a Nikon Eclipse 80i microscope. Original magnification ×40. (F) Levels of serum alanine aminotransferase (ALT) in the different experimental groups. Each box represents the mean of the concentration expressed in international unit per liter ± SEM. (G) Anti-topoisomerase 1 antibody levels in the different experimental groups measured by ELISA. Each box represents the mean of the antibody index expressed in arbitrary unit ± SEM. The ANOVA test with Bonferroni correction was used to detect significant differences between the groups (unless stated). NS, non-significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Chronic GvHD is characterized by dysregulated adaptive immune response with an inappropriate activation of allogeneic CD4+ T cells that leads to an alteration of B and CD8+ T-cell responses, responsible for the autoimmune features and tissue damage (33, 34). Anti-DNA topoisomerase I autoantibody production, indicative of this immune dysregulation, was increased in cGvHD mice compared to the syngeneic mice (p < 0.001, Figure 2G). Importantly, LPSlow-macrophages induced a significant reduction in serum anti-topoisomerase 1 antibody index compared to the untreated cGvHD mice (p = 0.002), while the adoptive transfer of PBS- and BCG-macrophages had no affect (Figure 2G). We then assessed the immunological effects of the infused macrophages on the course of the cGvHD. Activation of T and B cells as well as a shift toward a predominant memory phenotype of CD4+ T cells is a hallmark of established cGvHD (35) (Supplementary Figures S2A–C). As shown in Figures 3A–C, adoptive transfer of LPSlow-macrophages significantly reduced CD69 expression on CD4+ T and B cells (p = 0.02 and p = 0.005) but not on CD8+ T cells and was associated with a reduced frequency of memory CD4+ CD62LLow CD44High T cells compared to untreated cGvHD mice (p = 0.006). By contrast, BCG-macrophage treatment increased the activation of CD4+ T cells (p = 0.004) and significantly augmented the proportion of memory over naïve CD4+ T cell subpopulation (p = 0.01). The inducible costimulatory molecule ICOS (CD278), which participates in the augmented T-cell response during cGvHD (36) was downregulated in cGvHD-LPSlow mice (p = 0.001) but enhanced in cGvHD-BCG group (p = 0.04, Figure 3C).
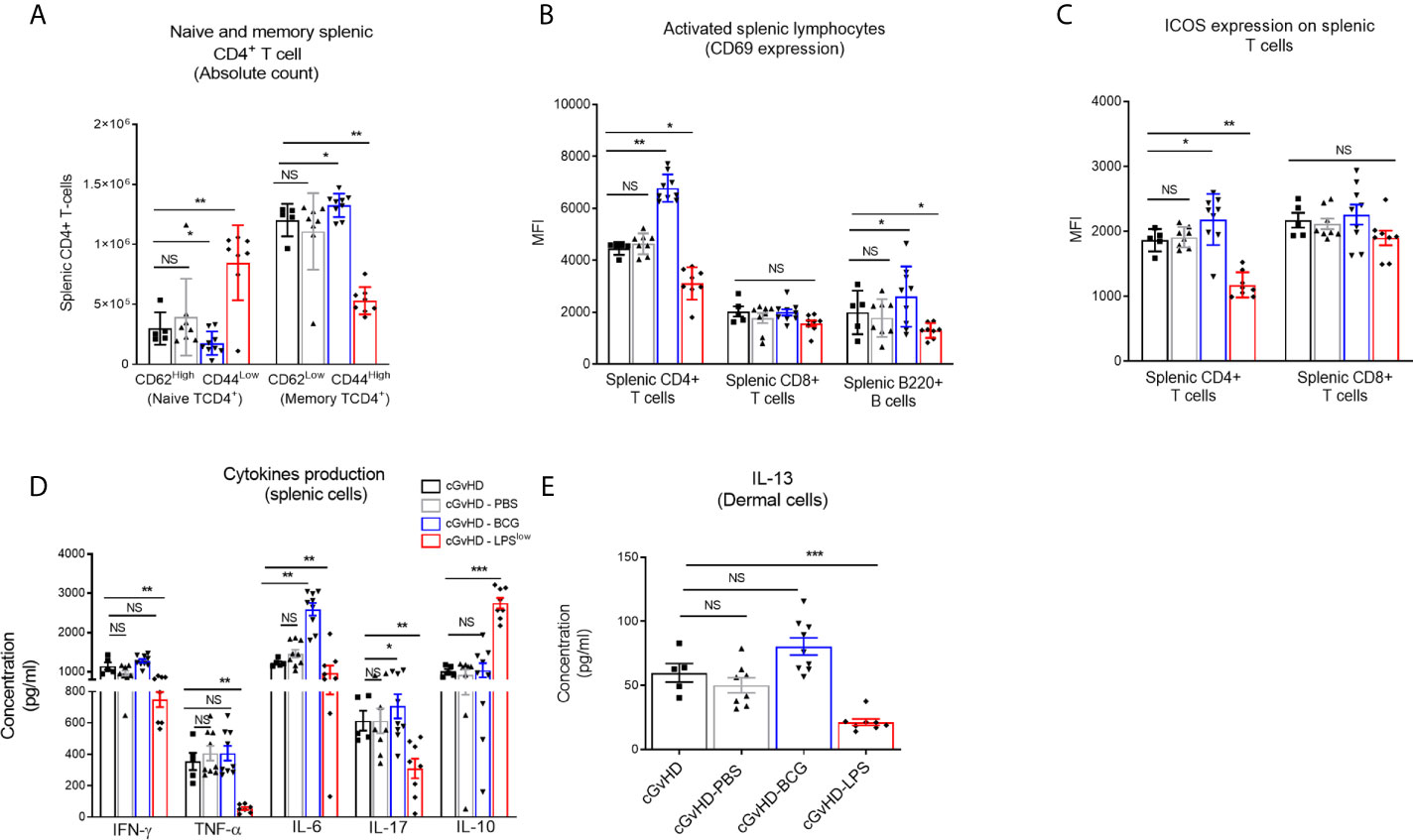
Figure 3 Trained macrophage infusion affects the activation and the cytokine production of splenic immune cells in cGvHD mice. (A) Frequency of naive and memory CD4+ T-cell subpopulations in the spleen of the different experimental groups. Data represent absolute count of naive (CD62LHigh CD44Low) and memory (CD62LLow and CD44High) CD4+ T cells among the total splenic CD4+ T-cell population ± SEM obtained from at least n = 7 biologically independent mice. (B, C) Activation of splenic CD4+, CD8+ T cells and B220+ B cells assessed by flow cytometric measurement of the CD69 expression (B) and ICOS expression for the T-cell subpopulations (C). Data represent the MFI ± SEM from at least n = 7 biologically independent mice. (D) Cytokine production in the supernatant of stimulated splenic cells. Spleen cell suspensions were stimulated with 5 μg ml−1 of Concanavalin A and incubated for 48 h at 37 °C with 5% CO2. Levels of IFN-γ, TNF-α, IL-6, IL-17, and IL-10 were assessed by ELISA, and concentrations are expressed in pg/ml. Data represent the mean ± SEM from the duplicate of at least n = 7 biologically independent samples. (E) ELISA assessment of IL-13 production (pg/ml) by skin-derived immune cells. Each box represents mean ± SEM of triplicate obtained with cell culture from at least n = 7 biologically independent mice. NS, Non-significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
cGvHD-Associated Dysregulation of Cytokine and Chemokine Production Is Alleviated Upon Adoptive Transfer of LPSlow-Macrophages
Cytokine network in the cGvHD is adversely altered, with an early increase in Th1 and Th17 pro-inflammatory cytokine production, but a predominance of the Th2 subset at the chronic stage (37) (Supplementary Figures S2D, E). Spleen cells from cGvHD-LPSlow had a significant reduction in TNF-α (p = 0.003), IL-6 (p = 0.006), IFN-γ (p = 0.009), and IL-17 (p = 0.007) production but a concomitant increase in IL-10 secretion (p = 0.0005, Figure 3D). In contrast, splenic cells from cGvHD mice treated with BCG-macrophages (cGvHD-BCG) displayed a heightened capacity to produce the pro-inflammatory cytokine IL-6 (p = 0.005) and IL-17 (p = 0.03) compared to the untreated cGvHD mice (Figure 3D). Furthermore, dermal cells from cGvHD-LPSlow animals showed a significantly reduced capacity to secrete IL-13 compared to the cGvHD mice (p = 0.0008), which was not affected by infusion with BCG-macrophages (Figure 3E).
Next, we set out to decipher whether the trained-macrophage infusion could affect the expression of chemokine receptors on allogeneic T cells that play an important role in lymphocyte migration to the sites of allo-recognition and priming (38). Chronic GvHD development was accompanied by an upregulation of CCR5 and CXCR3 on splenic T CD4+ and CD8+ cells and increased frequency and expression of CCR10 and CCR4 on dermal CD4+ T cells (Figures 4A–D). Of note, infusion of LPSlow-macrophages significantly downregulated the expression of CXCR3 (p = 0.001 and p = 0.03) and CCR5 (p = 0.005 and p = 0.03) on splenic CD4+ and CD8+ T cells; the expression of these chemokine receptors was strongly induced by adoptive transfer of BCG-macrophages (Figures 4A, B). Similarly, at the dermal level (Figures 4C, D), LPSlow-macrophage transfer significantly diminished CCR4 expression on CD4+ and CD8+ T cells (p = 0.02 and p = 0.03), whereas CCR10 was only decreased on CD4+ T cells (p = 0.001). cGvHD-BCG mice had an increased expression of only CCR10 on their dermal CD4+ T cells (p = 0.03). In parallel, we evaluated the dermal mRNA expression of the skin homing chemokines ccl17 and ccl27 and observed that they were increased in cGvHD group compared to the syngeneic mice (p < 0.0001). As shown in Figure 4E, BCG-macrophage infusion only upregulated the expression of ccl27 (p = 0.04), while LPSlow-macrophages abolished both the overexpression of ccl17 (p = 0.005) and ccl27 (p = 0.003). Finally, we analyzed the mRNA expression of two chemokines that promote the migration of the leukocyte subsets to the liver, ccl2, and cxcl2 (39, 40). Untreated cGVHD mice exhibited an increase of hepatic ccl2 and cxcl2 compared to syngeneic grafted mice (p < 0.0001). A downregulation of ccl2 (p = 0.004) and trend towards a decrease of cxcl2 expression (p = 0.18) were observed in the liver of cGvHD-LPSlow mice. Neither PBS- nor BCG-trained macrophage injections have a significant impact on the ccl2 and cxcl2 expression in the hepatic tissue (Figure 4F).
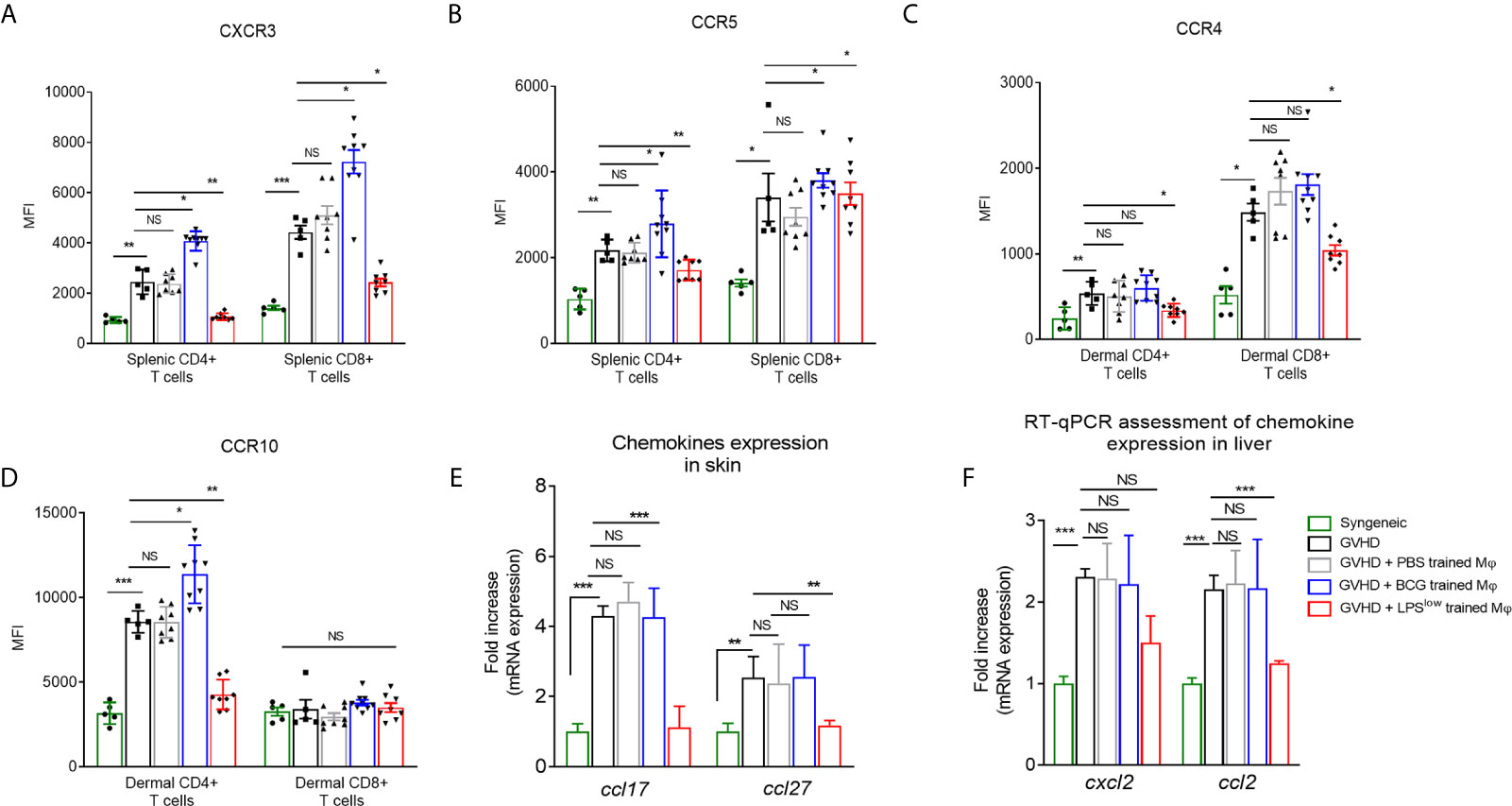
Figure 4 LPSlow-macrophage injections influence the chemokine network in cGvHD mice. (A, B) Flow cytometric analysis of splenic CD4+ and CD8+ T-cells CXCR3 and CCR5 expression at day 40. (C, D) Flow cytometric analysis of dermal CD4+ and CD8+ T-cells CCR4 and CCR10 expression at day 40. Gating strategy is represented in Supplementary Figure S4. Data represent the mean fluorescence intensity of the chemokine receptors expression on T cells obtained from at least n = 7 biologically independent mice. The ANOVA test with Bonferroni correction was used to detect significant differences between the groups. NS, non-significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. (E, F) RT-qPCR assessment of ccl17 and ccl27 mRNA levels in the skin (E) and cxcl2 and ccl2 mRNA levels in the liver (F) of cGvHD mice at day 40 of the experiment. Results were expressed as fold increase versus the syngeneic group, derived from at least n = 7 biologically independent mice. Unpaired T-test was used to detect the significance between the groups. NS, non-significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Altogether, these results indicate that infusion of LPSlow-macrophages exerts a suppressive effect on allogeneic responses during cGvHD with a drop in inflammatory cytokines and chemokine expression in target organs, but increased IL-10 production and a downregulation of chemokine and activation receptors on allogeneic T cells.
LPSlow-Macrophages Serve as Protective Barrier Against cGvHD-Induced Tissue/Organ Injury
Chronic GvHD is accompanied by serious skin and visceral injuries including fibrotic lesions of the lung and severe intestinal crypt damage (41). As a next step to evaluate the in vivo effects of infusion of trained macrophages on the visceral involvement of cGvHD, we quantified the mRNA expression of the fibrosis-associated type I collagen Col1a1 and α-smooth muscle actin genes in skin and lungs. As expected, we observed an upregulation of mRNA expression of Col1a1 and α-sma in skin and lungs in the cGvHD group compared to the syngeneic mice (p < 0.0001). BCG-macrophage infusion increased the expression of Col1a1 in skin (p = 0.03) while cGvHD-LPSlow mice showed a significant reduction of Col1a1 (p = 0.02 and p = 0.0008) and α-sma mRNA expression in skin and lungs (p = 0.0007 and p = 0.0006, respectively, Figures 5A, B). Moreover, cGvHD was accompanied by an upregulation of il13 mRNA expression in lungs compared to the syngeneic group (p < 0.0001). LPSlow-macrophages attenuated il13 expression (p = 0.0009) while the BCG-macrophages did not have any effect compared to the cGVHD group (Figure 5B). Sirius red-dyed staining of dermal and pulmonary sections showed clear evidence of collagen accumulation driven by the cGvHD. Infusion with LPSlow-macrophages resulted in alleviated fibrosis in skin and lungs (Figure 5C). We then analyzed the expression of ccn2, a matricellular protein that is upregulated in fibrotic hepatic disorders (42) and sox9/krt19 expression, markers associated with chronic liver damage (43). We observed an increased expression of ccn2, sox9, and krt19 in cGvHD mice compared to syngeneic (p = 0.03, p = 0.002, and p = 0.007 respectively). LPSlow-macrophage treatment diminished the expression of the assessed markers, while BCG-macrophage injection worsened the expression of ccn2 and sox9 compared to the cGVHD mice (Figure 5D). Lastly, the intestinal manifestation of cGvHD was assessed by mRNA quantification of il22 and il17 for the barrier integrity and mpo expression for quantification of the inflammatory infiltrate. As expected, cGvHD mice showed a significant upregulation of il22 (p = 0.0008), il17 (p = 0.007), and mpo (p = 0.0007) genes compared to the syngeneic group. Results showed that infusion with BCG-macrophages exacerbated the expression of il22 (p = 0.0008), whereas the adoptive transfer of LPSlow-macrophages significantly reduced mRNA levels of il22, il17, and mpo (p = 0.0007, p = 0.03, and p = 0.009, respectively) (Figure 5E).
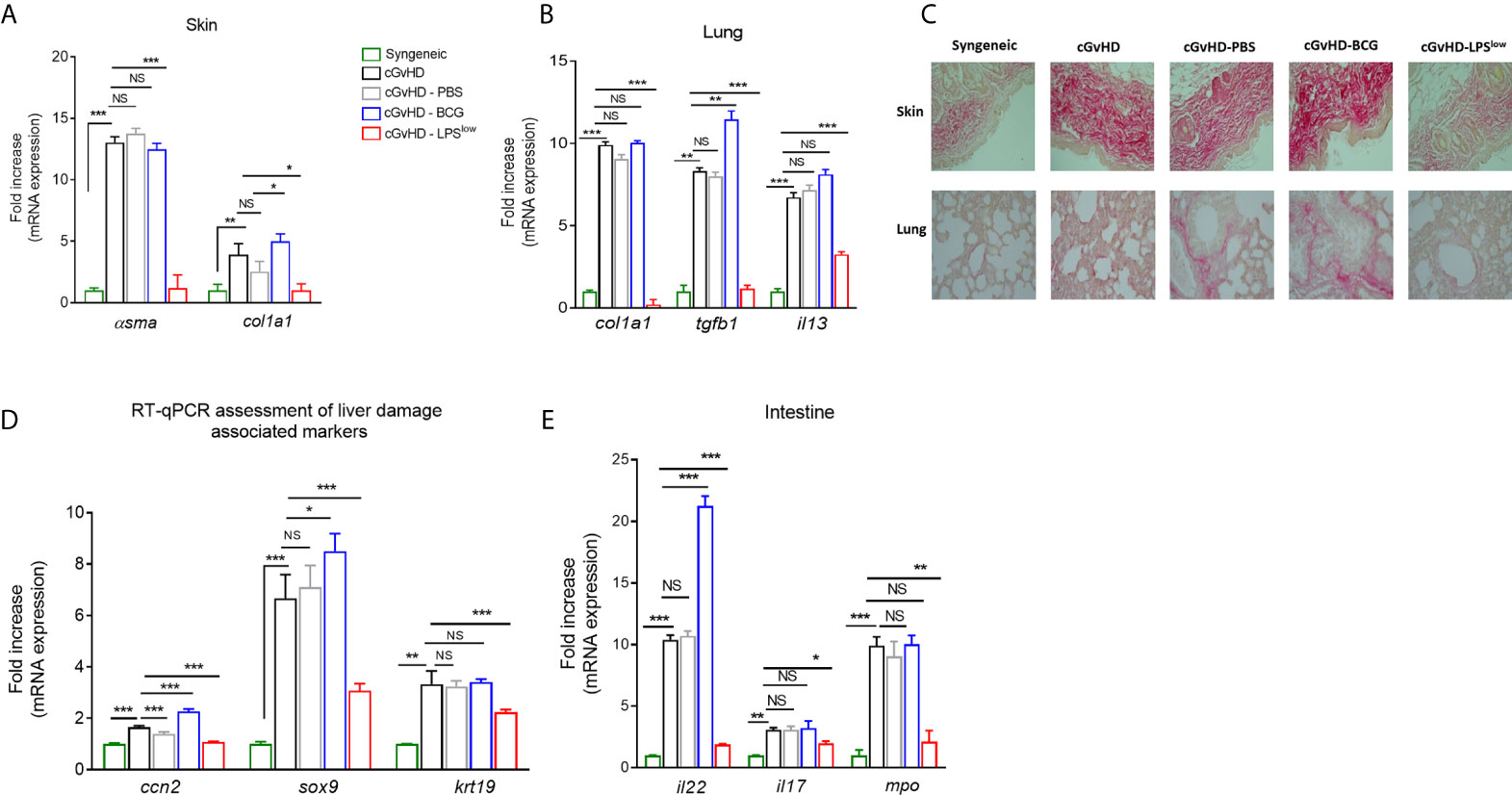
Figure 5 LPSlow-macrophages serve as protective barrier against cGvHD-induced tissue/organ injury. (A, B) RT-qPCR assessment of α-sma and col1a1 mRNA levels in the skin (A) and of col1a1, tgfb1 and il13 mRNA levels in the lungs (B) of cGvHD mice at sacrifice. Results were expressed as fold increase versus the syngeneic group, derived from at least n = 7 biologically independent mice. (C) Representative Sirius red-dyed skin and lung sections of 5 μm, showing enhanced fibrosis with accumulation of collagen in the dermis and thickening of inter-alveolar septa reflecting the obliterative bronchiolitis of cGvHD mice compared to syngeneic mice. Photographs were taken with a Nikon Eclipse 80i microscope. Original magnification ×40. (D, E) RT-qPCR assessment of il22, il17, and mpo mRNA levels in the intestines (D) and of ccn2, sox9, and krt19 in the liver tissue (E) of cGvHD mice at day 40 of the experiment. Results were expressed as fold increase versus the syngeneic group, derived from at least n = 7 biologically independent mice. Unpaired T-test was used to detect the significance. NS, non-significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Collectively, these findings show the beneficial effects of LPSlow-macrophages on the onset and clinical features of cGvHD and highlight the correlation between the specific immunological modifications and the impact on the visceral manifestation of the disease.
Weekly Infusion of LPSlow-Trained Macrophages Alleviates Acute GvHD in Mice
Acute GvHD was induced using the fully mismatched MHC mouse model of hematopoietic stem cell transplantation of lethally irradiated Balb/c recipient mice with splenocytes and bone marrow cells from C57BL/6 mice (Figure 6A). Survival rate was monitored daily. As shown in Figure 6B, lethally irradiated but non-transplanted mice had a median survival of 8 days, while all mice from the syngeneic group remained alive at the end of the experiment (day 30). Mice started to lose weight after the irradiation, but all transplanted animals regained weight after bone marrow transplantation (BMT). Allogeneic-transplanted mice started to lose weight around day 10 and reached the nadir at day 25. Clinical signs of acute GvHD started at day 11 of the experiment with decreased activity, hunched posture, and progressive alopecia (Figure 6C). Daily monitoring showed that aGvHD mice infused with LPSlow-macrophages had a trend towards an improved survival rate compared to the untreated group. PBS and BCG-macrophage treatment did not influence the survival rate of the aGvHD mice (Figure 6C). Thus, adoptive transfer of LPSlow-macrophages may exert a protective effect against acute GvHD, similar to what was observed with the cGvHD model.
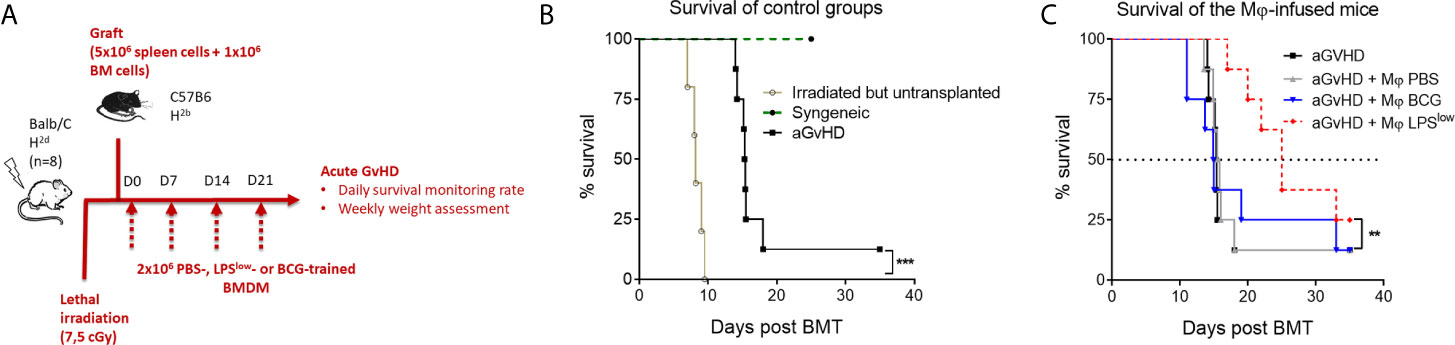
Figure 6 Weekly infusion of LPSlow-trained macrophages alleviates acute murine GvHD. (A) Schematic representation of the in vivo induction of MHC-mismatched acute GvHD and scheduling of trained macrophages infusion. Mice (n = 8 per group) from one experiment were monitored for survival. (B) Kaplan–Meier survival curve of the control groups of the acute GvHD experiment showing the survival rate of syngeneic, irradiated, but untransplanted and aGvHD mice. (C) Survival rate of the different groups of the experiment. Each group: aGvHD, aGvHD + Mφ PBS, aGvHD + Mφ BCG, aGvHD + Mφ LPSlow contained (n = 8) mice. Statistics are shown between the non-treated aGvHD and the aGvHD + Mφ LPSlow group. The log-rank (Mantel–Cox) and the Gehan–Breslow–Wilcoxon statistic tests were used to detect the differences between the groups. NS, non-significant; **p ≤ 0.01; ***p ≤ 0.001.
LPSlow-Trained Macrophages Hamper Allogeneic T-Cell Activity in an IL-10-Dependent Manner
We have shown in the first experiment that LPSlow-macrophage addition to the MLR system was associated with a marked increase of IL-10 level in the supernatant. Thus, we focused on that cytokine to get further into the mechanistic features of the protective effect of LPSlow-macrophages in the aGvHD and cGvHD models. We showed that adding recombinant mouse IL-10 (rm-IL-10) cytokine in the MLR system upregulated the phosphorylation of STAT3 and the mRNA expression of the IL-10 target gene arnt2 (44) but decreased the phosphorylation of T-cell specific protein kinase ZAP-70 (45) and the mRNA expression of the T-cell activation marker cd40l (46) (Figures 7A, B). Similarly, co-culture of LPSlow-macrophages with allogeneic CD4+ T cells increased arnt2 expression but was associated with a decrease of cd40l mRNA level (Figure 7C), a downregulation of the proliferation and cell-cycle associated proteins CDK4 and Cyclin A (Figure 7D), and a drop of IL-2, IL-6, and IFN-γ levels in the supernatant of the MLR milieu (Figure 7E). Notably, the inhibition of the STAT3 signaling pathway (Figures 7A, B), as well as the mRNA IL-10 silencing of the LPSlow-macrophages co-cultured with the allogeneic CD4+ T cells abrogated the effects observed with wild-type LPSlow-macrophages (Figures 7C–E). Altogether, these results highlight the involvement of IL10 in the modulation of the allogeneic response of CD4+ T cells and deliver further mechanistic insights into the immunomodulatory effect of LPSlow-macrophages in the aGvHD and CGvHD models.
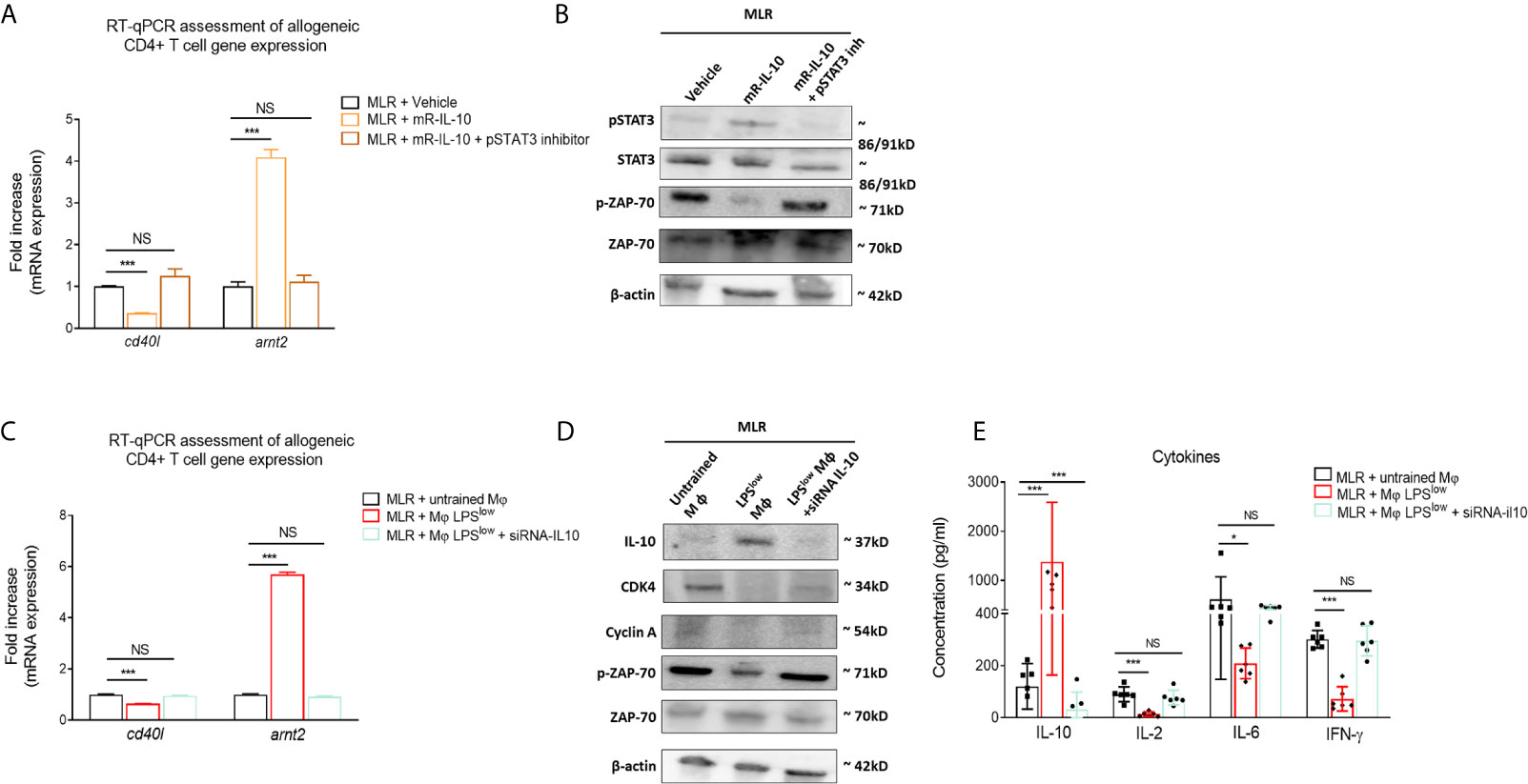
Figure 7 LPSlow-trained macrophages hamper allogeneic T-cell activity in an IL-10-dependent manner. (A, B) Purified CD4+ T cells derived from B10.D2 mice co-cultured with irradiated splenic cells from Balb/c mice were treated with 10 µM of STAT3 inhibitor V or vehicle (PBS + 1%DMSO) for about 30 min before mouse recombinant IL-10 stimulation for 24 h. (A) RT-qPCR assessment of arnt2 and cd40l mRNA expression. (B) Western blot analysis of pSTAT3, STAT3, pZAP70, ZAP-70, and β-actin expression. Representative results of two experiments performed. mR-IL-10, mouse recombinant IL-10. (C, D) Bone marrow derived macrophages obtained from Balb/c mice were either stimulated by PBS (untrained) or trained with LPSlow, and subjected to IL-10 mRNA invalidation with small interfering RNA. Cells were then co-cultured with allogeneic CD4+ T cells from the MLR for 72 h in six-plicate. Cells were then analyzed by RT-qPCR for cd40l and arnt2 mRNA expression (C) and by Western blot for IL-10, CDK4, Cyclin A, pZAP70, ZAP-70, and β-actin expression (D). Representative results of two experiments performed. (E) ELISA measurement of IL-10, IL-2, IL-6, and IFN-γ levels in the supernatant of the allogeneic CD4+ T cells co-cultured with wild-type untrained macrophages, LPSlow -macrophages, or IL-10 KO LPSlow-macrophages. Data represent the mean (pg/ml) ± SEM of six-plicate of two independent experiments. Unpaired T-test and the ANOVA test with Bonferroni correction were used to detect the significance. NS, non-significant; *p ≤ 0.05; ***p ≤ 0.001.
Adoptive Transfer of LPSlow-Trained Macrophages Does Not Exacerbate B Lymphoma in Mice
It is well established that therapeutic strategies used for the prophylaxis and treatment of GvHD can diminish the beneficial effect of graft-versus-leukemia effects (4). Hence, we evaluated whether the protective/inhibitory effects of LPSlow-macrophages on the course of acute or chronic GvHD observed in our study could influence tumor progression in a B lymphoma (A20) murine model. Mice were injected subcutaneously with 106 A20 cells and subjected to weekly infusion of trained macrophages (Figure 8A). Tumors were detected as early as day 10 of the injections. During the experiment (34 days), only three animals died spontaneously as a result of rapid tumor progression, and for ethical reasons, two were humanly euthanized at day 28 when the tumor reached a pre-defined volume size of 3,000 mm3. As shown in Figure 8B, tumors expanded rapidly in the different groups, though less importantly in the A20-BCG group. At the end of the experiment (day 34), no difference in tumor volume was detected between the A20-CTRL, the A20-PBS, and the A20-LPSlow groups. Interestingly, A20-BCG mice had significantly reduced tumor volume at day 34 compared to the other groups (p = 0.03). As a next step to decipher the effects of the infusion of trained macrophages observed in this experiment, we assessed the activation status and cytokine production by splenic and tumor infiltrating T cells. BCG-macrophage treatment resulted in an increase in TNF-α and IL-6 production by splenic cells compared to untreated A20 mice (p = 0.03 and p = 0.04, respectively), while PBS and LPSlow-macrophage infusions had no effects. Splenic IL-10 production capacity remained unchanged in the four groups (Figure 8C). Activation of CD4+ and CD8+ T cells was assessed by CD69 expression at both the systemic level on the spleen and in situ on tumors. BCG-macrophage treatment enhanced the activation of both CD4+ and CD8+ in situ (p = 0.0006 and p = 0.0008), while it only activated CD4+ T cells in the spleen (p = 0.006, Figure 8D). The mean fluorescence intensity of CD69 on CD4+ and CD8+ T cells in spleen and tumors did not differ in untreated A20 mice or in mice infused with PBS or LPSlow-macrophages (Figure 8E). Finally, to get further into the molecular changes underlying the effects of the infused macrophages on the tumor growth, we analyzed the expression of several genes involved in both the anti-tumoral and the allogenic immune responses (Figures 8F, G). PBS-macrophage infusion did not induce any change in gene expression in the isolated A20 tumors or in the spleen tissues compared to the untreated A20-CTRL group. A20 mice infused with LPSlow-macrophages upregulated the expression of fizz1 (found in inflammatory zone-1) gene which is usually associated with the alternatively activated macrophages phenotype (M2 macrophages), in the spleen (p = 0.0005) and in the tumor (p = 0.03) and il10 gene only in the spleen (p = 0.02) compared to untreated A20 mice. Moreover, LPSlow-macrophages downregulated the expression of inos in spleen (p = 0.03) and il6 both in spleen (p = 0.005) and the tumor (p = 0.02). All the other genes assessed remained unchanged. BCG-macrophage infusion was associated with an upregulation of the splenic and tumoral expression of inos (p = 0.03), il6 (p = 0.0005), and infg (p = 0.03), markers that are associated with classically activated macrophage phenotype (M1 macrophages). By contrast, we observed a decrease in the expression of fizz1 (p = 0.01) and the T regulator cell transcription factor foxp3 (p = 0.03) in both the spleen and the tumor tissues. Furthermore, A20-BCG mice had a diminished il4 (p = 0.03) expression in spleen and a drop in il10 and tgfb expression in tumors (p = 0.03 and p = 0.02, respectively).
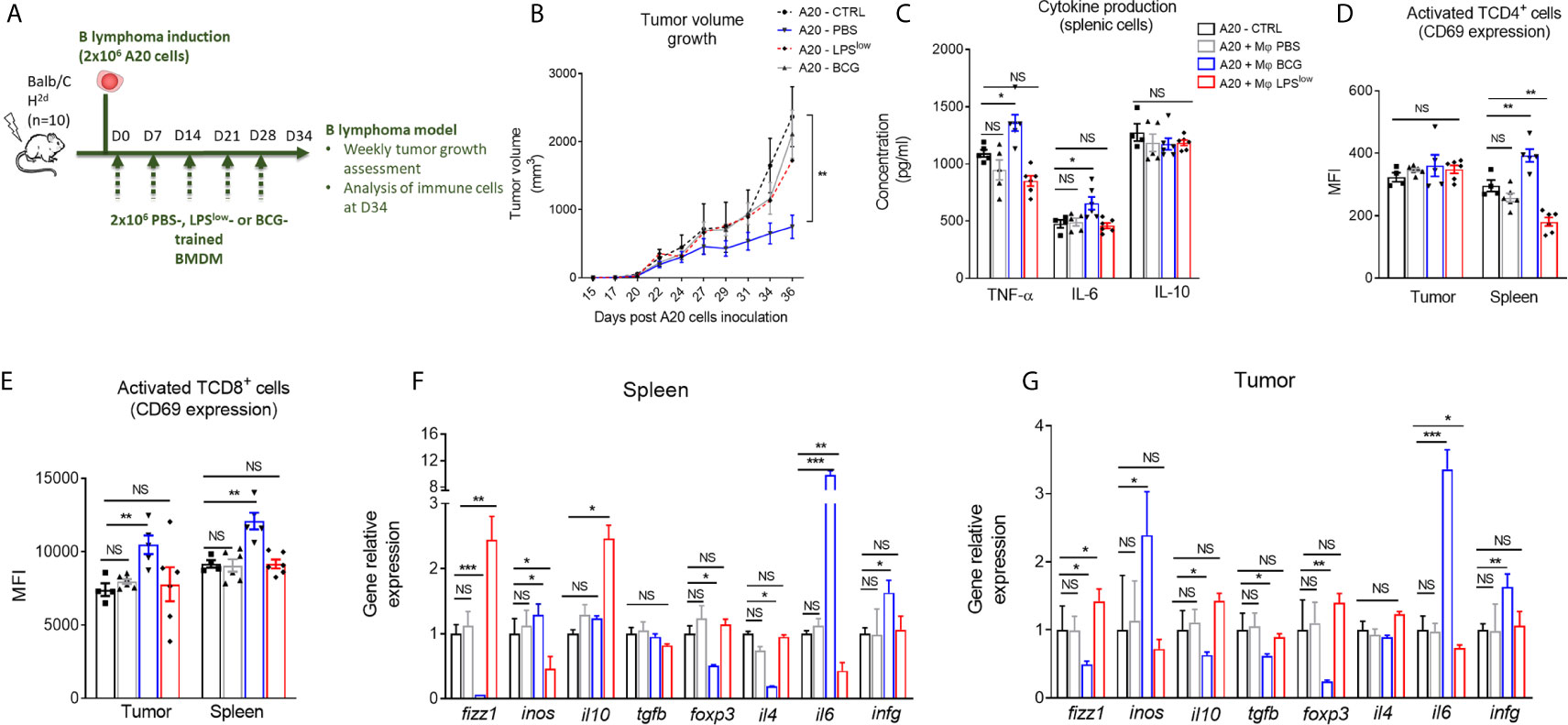
Figure 8 Clinical and immunological impact of the adoptive transfer of trained-macrophages on A20 cells B lymphoma tumor growth in mice. (A) Schematic representation of the in vivo induction of B lymphoma in mice and scheduling of trained macrophage infusion. Mice (n = 7 in each group) were subcutaneously injected with 2 × 106 A20 cells and sacrificed at day 34 in one experiment. (B) Tumor volume evolution in the different groups of A20-challenged mice (n = 7). Tumor growth was monitored three times a week with a microcaliper, and tumor volumes were calculated as length × width2 × 0.5236 and expressed in mm3. Statistics are shown between the BCG-macrophages infused (A20-BCG) and the untreated A20 groups. Differences were detected at sacrifice using the Mann–Whitney test. **p ≤ 0.01. (C) Cytokine production in the supernatant of splenic cells harvested at sacrifice, stimulated with 5 μg/ml of Concanavalin A and incubated for 48 h at 37 °C with 5% CO2. Levels of TNF-α, IL-6, and IL-10 were assessed by ELISA and concentrations are expressed in pg/ml. Data represent the mean ± SEM from the duplicate of at least n = 5 biologically independent samples. (D, E) Flow cytometric analysis of splenic and tumoral CD4+ (C) and CD8+ (D) T-cell phenotype at day 34 of the experiment. Tumor infiltrating T cells were gated on CD3 and CD4 or CD8 positive cells among CD45 positive population. Data represent the mean fluorescence intensity of CD69 ± SEM, reflecting the activation status of the assessed cells, obtained from at least n = 5 biologically independent samples. (F, G) RT-qPCR assessment of fizz1, inos, il10, tgfb, foxp3, il4, il6, and infg mRNA levels in the spleen and tumor lesions of the different groups of A20-challenged mice at day 36 of the experiment. Results were expressed as fold increase versus the untreated A20-mice group, derived from at least n=5 biologically independent samples. Unpaired T-test was used to detect the differences between the groups. NS, non-significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Cumulatively, these results show that LPSlow-macrophages do not adversely affect tumor growth and are not accompanied by major modifications of T cell activation, cytokine production, or gene expression. Interestingly, infusion with BCG-macrophages exhibit an anti-tumoral activity with an upregulation of the M1-associated genes and an increase of T-cell activation and pro-inflammatory cytokine secretion.
Discussion
Immunological memory is traditionally associated with the adaptive immune system. Yet, this dogma has been recently challenged as innate immune cells like macrophages can be trained to mount a heightened inflammatory response upon non-specific antigenic challenge, or on the contrary can be rendered hypo-responsive with immunosuppressive effects (11, 47). In this study, we show that LPSlow-macrophages are efficient in targeting inflammation-mediated allo-immune responses associated with acute and chronic GvHD, resulting in significant disease alleviation. On the contrary, BCG-macrophages tend to aggravate the clinical and biological features of cGvHD. The pathophysiology of GvHD is complex, highly dependent on major histocompatibility complex (MHC) antigenic disparity between donor and recipient, and involves a variety of cell types in both its generation and subsequent pathologic manifestations (48). The release of pro-inflammatory cytokines from activated immune cells including macrophages and donor T allo-reactive cells lies at the center of GvHD pathogenesis (41). This process enhances the interaction between antigen presenting cells and donor-derived lymphocyte populations, including CD4+ T cells, resulting in the generation of effector populations (6). As a first step to decipher the immunomodulatory effects of trained macrophages in our in vivo GvHD model, we reproduced in vitro the onset of the GvHD allo-immune reaction using a minor MHC mismatched MLR. LPSlow-macrophage addition limited the activation and proliferation of CD4+ T cells, along with a decrease of IL-2, IFN-γ, TNF-α and IL-6 production but an increase of IL-10 release, while BCG-macrophages had an opposite effect. T-cell receptor (TCR) recognition of the MHC-allo antigen complex and costimulatory molecule engagement results in IL-2 release by CD4+ T cells that exerts a potent proliferative and growth autocrine effect (49). These activated allogeneic CD4+ T cells upregulate CD44 expression that binds to the lymphocyte-specific protein kinase (Lck) and thereby enhances T-cell receptor signaling (50). During the allo-immune response, activated T cells undergo various polarization status influenced by the exogenous cytokine milieu (51), which generates heterogeneous cell populations predominated by the IFN-γ Th1 releasing subtype that triggers the initial effector mechanisms (52). Studies on innate immune memory demonstrated that BCG-training of macrophages results in upregulation of pattern recognition receptors and costimulatory molecules with heightened non-specific production of the pro-inflammatory cytokines, IL-6, and TNF-α (13, 23). IL-6 has anti-apoptotic properties as it prolongs CD4+ T cell survival in vitro by inducing Bcl-2 expression in the T cells (53) and downregulating FasL expression (54). Furthermore, TNF-α has the capacity to promote TCR-dependent T-cell activation (55) via the activation of NFκB pathway (56). Conversely, the immunoregulatory IL-10 cytokine, the expression of which is upregulated in LPSlow-macrophages following inflammatory stimulation (10, 23, 57), reduces MHC class II expression on monocytes/macrophages, inhibits T-cell proliferation, and abrogates Th1, Th2, and Th17 cytokine production (58). In addition, LPSlow-trained macrophages have been shown to adopt a hypo-responsive phenotype with a downregulation of the co-stimulatory molecules such as CD80, inflammatory markers (Ly6C) but an upregulation of the mannose receptor CD206 and the DC-SIGN (CD209). These phenotypes persist in vivo after peritoneal injections and can be also transferred to splenic and peritoneal macrophages (23, 59).
Our findings indicate that macrophages acquire a modified phenotype according to the nature of immune training and thus react differently once in contact with the allogeneic T cells, either to promote or to inhibit the allo-immune response.
Chronic inflammation, fibrosis, and autoimmune disorders are the pathologic hallmark of cGvHD (60). We therefore used the murine sclerodermatous cGvHD model that mimics clinical and biological features of the human chronic form, involving both inflammatory and pro-fibrotic components (5). In the first stage of the disease, tissue damage through irradiation results in the upregulation in TNF-α, IL-1β, and IL-6 inflammatory cytokines that favors activation of host dendritic cells and monocytes/macrophages. In addition, recognition of the host minor antigens by donor CD4+ T cells leads to an activation of a Th1 response that drives the initial inflammatory reaction with an early activation of the Th17 lymphocyte subset, known to exert potent chemoattractant effects and promote the recruitment of T lymphocytes in target tissues (61). This initial inflammatory reaction is followed by a progressive shift toward the Th2 response with IL-13 and IL-4 production that drives the activation of profibrotic macrophages through TGF-β signaling (62, 63) and induces hyperactivation of the autoreactive B cells responsible for the autoimmune features of the disease (64). The major finding of this study was that LPSlow-macrophage infusion attenuated the development of sclerodermatous cGvHD, while BCG-macrophages tended to aggravate it. Consistent with our in vitro results, we observed a drop in splenic TNF-α, IL-6, IFN-γ, and IL-17, and dermal IL-13 production in cGvHD-LPSlow mice, while their capacity for systemic IL-10 secretion was enhanced. Concomitantly, LPSlow-macrophage infusion diminished CD69 expression on T and B cells and ICOS expression on CD4+ T cells. Current evidence suggests that dysregulated cytokine production occurs as a cascade during sequential macrophage and T-cell activation and is responsible for the clinical manifestation of the cGvHD (6). Also, IL-6 overproduction mediated by the translocation of microbiome-derived PAMPs perpetuates the inflammation in cGvHD (65), and its blockade alleviates the clinical severity (66). High amounts of TNF-α release following the conditioning regimen drive the initiating events of the GvHD and amplify the disease process once established through T-cell activation and apoptosis induction on target tissues (67). It has been shown that high levels of IFN-γ detected in a mixed lymphocyte culture in allogeneic bone marrow transplant recipient correlated with GVHD development in the corresponding patients (68). ICOS (CD278) is a costimulatory molecule expressed on activated T cells that favors Th17 and Th2 lymphocyte differentiation, and drives B cell interaction with germinal centers for antibody production (69). Its role in cGvHD is highly documented as recipients of icos(−/−) CD4+ T cells exhibited less GVHD morbidity (70) with a significant decrease in donor T follicular helper population, Th17 skewing, and lower antibody production (71).
Chemokines act on many aspects of the cGvHD pathophysiology including HSC homing and mobilization, T-cell activation, and immune cell recruitment to GvHD target organs (40). Adoptive transfer of LPSlow-macrophages was accompanied with a drop in CCR4 and CCR10 expression on dermal CD4+ T cells as well as a decrease in CXCR3 expression on splenic T cells, along with a reduction in the skin ccl17 and ccl27 mRNA content. The CCR10–CCL27 and CCR4–CCL17 chemokine axes play a critical role in CD4+ recruitment of T cells to inflamed skin (72, 73). Peripheral blood T cells from GvHD patients exhibit a high proportion of CD4+ CCR10+ T cells that disappear after GvHD resolution (74). CXCR3 is highly expressed on effector T cells upon activation and plays an important role in T-cell trafficking into inflamed tissues (75). Studies revealed a central role of CXCR3 in the pathogenesis of dermal damage during cGVHD (76), and its blockade inhibited allo-reactive CD8+ T cell and alleviated GvHD in mice (77). Here, the impairment of these chemokine axes may explain the attenuated skin damage and fibrosis observed in LPSlow-cGvHD mice. After transplantation, naïve allogeneic CD4+ T cells were primed in the lymph nodes, proliferated and activated CD8+ T and B cells. This process induced effector T-cell populations and a progressive shift towards a predominant memory T-cell phenotype which was responsible for persistent host tissue injury (35, 78). CCR5 expression is essential to the recruitment of memory and effector T cells to the inflamed tissue (79), and this receptor has been linked to the pathogenesis of liver damage in GvHD (80). The blockade of CCR5 has also been described to inhibit leukocyte trafficking and reduce mucosal inflammation in murine colitis (81). Our findings indicate that the attenuation of the allo-immune response intensity and the lack of CCR5 expression on T cells provoked by LPSlow-macrophages blocked the aberrant differentiation into memory phenotype and prevented cellular infiltrates into the liver and the intestine. Reduced levels of serum ALT as well as reduction in mpo, il22, and il17 expression in the gut of LPSlow-cGvHD mice, compared to the cGvHD controls, reflect the attenuated organ damage associated with the disease.
In sclerodermatous GvHD, visceral fibrosis is a hallmark of the advanced stage of disease and responsible for the devastating clinical outcome (82). Expression of α-sma and col1a, indicative of the activation of extracellular matrix secreting myofibroblasts, as well as the pro-fibrotic cytokines il13 and tgfb1 was notably reduced upon infusion of LPSlow-macrophages. We have previously shown that the in vivo LPSlow training and adoptive transfer of LPSlow-macrophages were effective in alleviating fibrotic manifestations in the skin and lungs in a murine model of systemic sclerosis (23). Fibrosis is a common pathological consequence of chronic inflammation and alteration of the immune response regulation (83). Here, the ability of LPSlow-macrophages to dampen the inflammatory process by downregulating the T and B cell activation and reorganizing the networks of cytokines and chemokines counterbalanced the disproportionate immune response and prevented excessive fibrotic pathway activation and tissue damage in target organs.
Despite the differences in clinical phenotype and time of onset, acute and chronic murine GvHD share multiple immunopathological characteristics in which initial inflammation that drives and amplifies the allogeneic response remains the main actor (84). We showed that aGvHD mice infused with LPSlow-macrophages had a marked improvement in overall survival and displayed attenuated clinical features. Numerous studies have proposed cellular therapy to hamper GvHD. So far, infusions of regulatory T-cells (Tregs), mesenchymal stromal cells (MSCs), and the Myeloid-Derived Suppressor Cells (MDSCs) have been the most studied, with promising results (9, 85), while the exact mechanism by which these cells improve GvHD outcome remains incompletely elucidated; multiple immunosuppressive properties such as IL-10, indoleamine 2-3 dioxygenase, ectonucleotidase, and arginase-1 production, and inhibitory T cell co-receptor expression have been suggested as mechanisms to suppress T cell function (86–88). As in cGvHD, the important inflammatory milieu associated with aGvHD acts as a trigger for the infused trained macrophages. In turn, those cells react with either an overproduction of inflammatory cytokines (BCG-macrophages) or an IL-10 release (LPSlow-macrophages) that shuts down the inflammatory process. As GvHD is characterized by a very early overproduction of inflammatory cytokines and a short median survival, infusion of BCG-macrophages failed to mount an important synergistic effect that could translate clinically, while LPSlow-macrophages treatment profoundly ameliorated the two clinical forms of the disease.
Successful treatment of GvHD is limited due to possible loss of the graft-versus-leukemia effect that leads to an increased relapse rate of a malignant hematological disease (89). The relapse usually results from immunosuppressive drugs that target host T cells and weaken their ability to recognize and control residual tumoral cells. In our A20 mice model of B lymphoma, infusion of LPSlow-macrophages did not aggravate mortality rate nor accelerated tumor growth. Tumor growth is typically associated with the establishment of an immunosuppressive milieu that favors tumoral escape (90). Bascuas et al. reported a drop of a large array of chemokines and cytokines responsible for T-cell recruitment and activation within the tumor microenvironment using the A20 lymphoma cell murine model (91). Also, it has been shown that A20 tumor cells show altered expression of the costimulatory CD80 and are a potent source of IL-10 secretion, suggesting an important immunosuppressive status that accompanies A20 cell establishment (92). The lack of an inflammatory trigger in the context of a significant immunosuppression driven by the A20 cells may explain the absence of an added effect of the LPSlow-macrophages to the tumoral growth. Notably, we observed that BCG-macrophage infusion attenuated tumor growth and was associated with an upregulation of the M1 macrophages associated genes including ifng, il6, and inos as well as with T cell activation and pro-inflammatory cytokine secretion. We and others have reported that BCG-trained macrophages acquire an M1-like phenotype with an enhanced capacity for TNF-α and IL-6 secretion and an overexpression of pattern recognition and co-stimulatory receptors (21, 23, 93). Macrophages are the most abundant cells within the tumor stroma, displaying a high plasticity with either beneficial or deleterious effects on tumor lesions (94). A shift toward the inflammatory M1 phenotype that orchestrates Th1 responses is classically associated with anti-tumoral effects and an improved prognosis for the underlying tumor (95). Although BCG immunotherapy is widely used in clinics to treat bladder cancer and has been tested successfully in malignant melanoma (96), the exact mechanism of its action is still debated (97). Our findings suggest that the anti-tumoral activity of BCG may act through macrophage training by conferring them a pro-inflammatory status that enhance their capacity to drive an efficient local immune response. Recent experiments have shown that the immunosuppressive status of myeloid cell can be successfully reversed by exposure to β-glucan, known to exert potent training effects on macrophages, resulting in a decreased frequency of MDSCs associated with lung carcinoma (98).
However, additional investigations are needed to assess the immunomodulatory impact of LPSlow-macrophages on the separation of anti-leukemia effect from GvHD after BMT in an established GVL model.
This work is the first to highlight the immunomodulatory effects of trained macrophages on the course of two GvHD models, showing that cellular therapy may efficiently affect the immunological and fibrotic hallmark of this disease. Here, LPSlow-macrophages strongly reduce the proliferation and the activation of allogeneic CD4+ T cells in an IL-10-dependent manner, early at the pathogenesis of the GvHD, slowing down the overwhelming inflammatory process that characterizes this disease. Survival time of injected macrophages has already been shown in previous studies to persist from 2 weeks to up to 1 year in some models and according to the macrophage tissue origin (99, 100). However, further experiments are needed to trace precisely the migration and the lifespan of the injected trained-macrophages. Importantly, our study opens the way to get further into the immune mechanisms of trained immunity by analyzing the effects of trained macrophages on the phenotype and function of T and B cells, and thus better understand how they shape the adaptive immune response. Furthermore, the novel immunotherapeutic approach addressed in this study is a promising approach to target either excessive immune response that drives graft rejection or defective immunity associated with malignant disease progression.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by the Ethics Committee from Paris Descartes University (N° CEEA34.CN.017.12).
Author Contributions
Conceptualization, Methodology, MJ, FB and CN. Animal experiments MJ, BS, MT and CN. Investigation, MJ, BS, CN and CC. Formal analysis, MJ. Writing –Original Draft, MJ, CN and FB. Writing – Review, MJ, CN and LD. Editing, MJ and FB. Funding Acquisition, CN and FB. Supervision, CN and FB. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from University Paris Descartes INSERM.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank the Plateform of Cytometry and Immuno-biology CYBIO of Cochin Institute, Paris, for flow cytometry and data analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.670776/full#supplementary-material
Supplementary Figure 1 | (A) Flow cytometric analysis of CD44 expression of CD4+ T cells in the MLR condition (B10D2 CD4+ T cells with irradiated splenic Balb/c cells), B10D2 CD4+ T cells incubated with anti CD3 and CD28 antibodies (Positive control), B10D2 CD4+ T cells cultured alone and irradiated Balb/c splenocytes (Negative controls). Each bar represents the mean fluorescence intensity of CD44 expression ± SEM obtained from the 10-plicate of three independent experiments. (B) Proliferation index of CFSE-stained cells in the MLR condition (B10D2 CD4+ T cells cultured with irradiated splenic Balb/c cells), B10D2 CD4+ T cells incubated with anti CD3 and CD28 antibodies (Positive control), B10D2 CD4+ T cells cultured alone and irradiated Balb/c splenocytes (Negative controls). Each bar represents the mean proliferation index of ± SEM obtained from the 10-plicates of three independent experiments. (C) ELISA assessment of IL-2, IFN-γ, TNF-α, IL-6, and IL-10 cytokine concentrations in the supernatant of the different MLR control conditions. Bar graphs represent the mean (pg/ml) ± SEM of 10-plicate of three independent experiments. The ANOVA test with Bonferroni correction was used to detect significant differences between the groups. Statistics are shown between the MLR group and the different control conditions. NS, non-significant, *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Supplementary Figure 2 | (A) Frequency of naive and memory CD4+ T-cell subpopulations in the spleen of the syngeneic versus the cGvHD groups. Data represent the absolute count of naive (CD62LHigh CD44Low) and memory (CD62LLow and CD44High) CD4+ T cells among the total splenic CD4+ T-cell population ± SEM obtained from at least n = 5 biologically independent mice. (B) Flow cytometric measurement of the CD69 expression on splenic CD4+ and CD8+ T cells and B220+ B cells. (C) Flow cytometric analysis of ICOS expression on splenic CD4+ and CD8+ T cells. (D) Cytokine production in the supernatant of stimulated splenic cells from the syngeneic and the cGvHD groups. Levels of IFN-γ, TNF-α, IL-17, and IL-10 were assessed by ELISA and concentrations are expressed in pg/ml. Data represent the mean ± SEM from the duplicate of at least n = 5 biologically independent samples. (E) ELISA assessment of IL-13 production (pg/ml) by skin-derived immune cells from the syngeneic and the cGvHD mice. Each box represents mean ± SEM of triplicate obtained with cell culture from at least n = 5 biologically independent samples. The ANOVA test with Bonferroni correction was used to detect significant differences between the groups. NS, non-significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Supplementary Figure 3 | (A, B) Histograms showing the absolute count of dermal CD4+ T and CD8+ T-cell populations. Cell suspensions were obtained from the lysis of dermal punches of tissue collected from the shaved back of each mouse. Statistics are shown between the cGvHD group and the cGvHD + trained macrophages groups. The ANOVA test with Bonferroni correction was used to detect significant differences between the groups. NS: non-significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. (C) Representative skin and lung sections of 5 μm stained with hematoxylin and eosin in the different groups. Overall, cGvHD mice have an increased dermis or inter-alveolar mononuclear cell infiltration compared to syngeneic group. Photographs were taken with a Nikon Eclipse 80i microscope. Original magnification ×40.
Supplementary Figure 4 | (A) Flow cytometric gating strategy to detect dermal CD4+ and CD8+ T cells from Balb/c mice for further analysis of surface marker expression. CD4+ T cells were identified as CD3+ CD4+ double positive cells and CD8+ T cells were identified as CD3+ CD8+ double positive cells. (B) Flow cytometric gating strategy to detect splenic CD4+ and CD8+ T cells from Balb/c mice for further analysis of surface markers expression. CD4+ T cells were identified as CD3+ CD4+ double positive cells and CD8+ T cells were identified as CD3+ CD8+ double positive cells. B cells were identified as CD3- B220+. (C) Flow cytometric gating strategy to identify splenic macrophages from Balb/c mice for further surface marker analysis. Total splenic cells were gated on CD11b+ and F4/80+ to identify macrophage population.
References
1. Flowers MED, Martin PJ. How We Treat Chronic Graft-Versus-Host Disease. Blood (2015) 125:606–15. doi: 10.1182/blood-2014-08-551994
2. Wingard JR, Majhail NS, Brazauskas R, Wang Z, Sobocinski KA, Jacobsohn D, et al. Long-Term Survival and Late Deaths After Allogeneic Hematopoietic Cell Transplantation. J Clin Oncol (2011) 29:2230–9. doi: 10.1200/JCO.2010.33.7212
3. Zheng C-C, Zhu X-Y, Tang B-L, Zhang X-H, Zhang L, Geng L-Q, et al. Clinical Separation of Cgvhd and GvL and Better GvHD-Free/Relapse-Free Survival (GRFS) After Unrelated Cord Blood Transplantation for AML. Bone Marrow Transplant (2017) 52:88–94. doi: 10.1038/bmt.2016.182
4. Chang Y-J, Zhao X-Y, Huang X-J. Strategies for Enhancing and Preserving Anti-Leukemia Effects Without Aggravating Graft-Versus-Host Disease. Front Immunol (2018) 9:3041. doi: 10.3389/fimmu.2018.03041
5. MacDonald KPA, Hill GR, Blazar BR. Chronic Graft-Versus-Host Disease: Biological Insights From Preclinical and Clinical Studies. Blood (2017) 129:13–21. doi: 10.1182/blood-2016-06-686618
6. Cooke KR, Luznik L, Sarantopoulos S, Hakim FT, Jagasia M, Fowler DH, et al. The Biology of Chronic Graft-Versus-Host Disease: A Task Force Report From the National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-Versus-Host Disease. Biol Blood Marrow Transplant (2017) 23:211–34. doi: 10.1016/j.bbmt.2016.09.023
7. Magenau J, Runaas L, Reddy P. Advances in Understanding the Pathogenesis of Graft-Versus-Host Disease. Br J Haematol (2016) 173:190–205. doi: 10.1111/bjh.13959
8. Toubai T, Mathewson ND, Magenau J, Reddy P. Danger Signals and Graft-Versus-Host Disease: Current Understanding and Future Perspectives. Front Immunol (2016) 7:539. doi: 10.3389/fimmu.2016.00539
9. Blazar BR, Murphy WJ, Abedi M. Advances in Graft-Versus-Host Disease Biology and Therapy. Nat Rev Immunol (2012) 12:443–58. doi: 10.1038/nri3212
10. Gardiner CM, Mills KHG. The Cells That Mediate Innate Immune Memory and Their Functional Significance in Inflammatory and Infectious Diseases. Semin Immunol (2016) 28:343–50. doi: 10.1016/j.smim.2016.03.001
11. Netea MG, Joosten LAB, Latz E, Mills KHG, Natoli G, Stunnenberg HG, et al. Trained Immunity: A Program of Innate Immune Memory in Health and Disease. Science (2016) 352:aaf1098. doi: 10.1126/science.aaf1098
12. Netea MG. Training Innate Immunity: The Changing Concept of Immunological Memory in Innate Host Defence. Eur J Clin Invest (2013) 43:881–4. doi: 10.1111/eci.12132
13. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LAB, Ifrim DC, Saeed S, et al. Bacille Calmette-Guerin Induces NOD2-Dependent Nonspecific Protection From Reinfection via Epigenetic Reprogramming of Monocytes. Proc Natl Acad Sci USA (2012) 109:17537–42. doi: 10.1073/pnas.1202870109
14. Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida Albicans Infection Affords Protection Against Reinfection via Functional Reprogramming of Monocytes. Cell Host Microbe (2012) 12:223–32. doi: 10.1016/j.chom.2012.06.006
15. Kleinnijenhuis J, Quintin J, Preijers F, Benn CS, Joosten LAB, Jacobs C, et al. Long-Lasting Effects of BCG Vaccination on Both Heterologous Th1/Th17 Responses and Innate Trained Immunity. J Innate Immun (2014) 6:152–8. doi: 10.1159/000355628
16. Biswas SK, Lopez-Collazo E. Endotoxin Tolerance: New Mechanisms, Molecules and Clinical Significance. Trends Immunol (2009) 30:475–87. doi: 10.1016/j.it.2009.07.009
17. Hotchkiss RS, Monneret G, Payen D. Sepsis-Induced Immunosuppression: From Cellular Dysfunctions to Immunotherapy. Nat Rev Immunol (2013) 13:862–74. doi: 10.1038/nri3552
18. Zhang S, Yang N, Ni S, Li W, Xu L, Dong P, et al. Pretreatment of Lipopolysaccharide (LPS) Ameliorates D-GalN/LPS Induced Acute Liver Failure Through TLR4 Signaling Pathway. Int J Clin Exp Pathol (2014) 7:6626–34.
19. Quinn SM, Cunningham K, Raverdeau M, Walsh RJ, Curham L, Malara A, et al. Anti-Inflammatory Trained Immunity Mediated by Helminth Products Attenuates the Induction of T Cell-Mediated Autoimmune Disease. Front Immunol (2019) 10:1109. doi: 10.3389/fimmu.2019.01109
20. Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining Trained Immunity and Its Role in Health and Disease. Nat Rev Immunol (2020) 20(6):375–88. doi: 10.1038/s41577-020-0285-6
21. Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonça LE, Pacis A, et al. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity Against Tuberculosis. Cell (2018) 172:176–90.e19. doi: 10.1016/j.cell.2017.12.031
22. Libraty DH, Zhang L, Woda M, Acosta LP, Obcena A, Brion JD, et al. Neonatal BCG Vaccination Is Associated With Enhanced T-Helper 1 Immune Responses to Heterologous Infant Vaccines. Trials Vaccinol (2014) 3:1–5. doi: 10.1016/j.trivac.2013.11.004
23. Jeljeli M, Riccio LGC, Doridot L, Chêne C, Nicco C, Chouzenoux S, et al. Trained Immunity Modulates Inflammation-Induced Fibrosis. Nat Commun (2019) 10:5670. doi: 10.1038/s41467-019-13636-x
24. Garnett C, Apperley JF, Pavlů J. Treatment and Management of Graft-Versus-Host Disease: Improving Response and Survival. Ther Adv Hematol (2013) 4:366–78. doi: 10.1177/2040620713489842
25. Chávez-Galán L, Vesin D, Martinvalet D, Garcia I. Low Dose BCG Infection as a Model for Macrophage Activation Maintaining Cell Viability. J Immunol Res (2016) 2016:4048235. doi: 10.1155/2016/4048235
26. Cevey ÁC, Penas FN, Alba Soto CD, Mirkin GA, Goren NB. IL-10/STAT3/SOCS3 Axis Is Involved in the Anti-Inflammatory Effect of Benznidazole. Front Immunol (2019) 10:1267. doi: 10.3389/fimmu.2019.01267
27. Anderson BE, McNiff JM, Matte C, Athanasiadis I, Shlomchik WD, Shlomchik MJ. Recipient CD4+ T Cells That Survive Irradiation Regulate Chronic Graft-Versus-Host Disease. Blood (2004) 104(5):1565–73. doi: 10.1182/blood-2004-01-0328
28. Brubaker AL, Schneider DF, Palmer JL, Faunce DE, Kovacs EJ. An Improved Cell Isolation Method for Flow Cytometric and Functional Analyses of Cutaneous Wound Leukocytes. J Immunol Methods (2011) 373:161–6. doi: 10.1016/j.jim.2011.08.013
29. Kavian N, Marut W, Servettaz A, Laude H, Nicco C, Chéreau C, et al. Arsenic Trioxide Prevents Murine Sclerodermatous Graft-Versus-Host Disease. J Immunol (2012) 188:5142–9. doi: 10.4049/jimmunol.1103538
30. Cetkovic-Cvrlje M, Roers BA, Waurzyniak B, Liu XP, Uckun FM. Targeting Janus Kinase 3 to Attenuate the Severity of Acute Graft-Versus-Host Disease Across the Major Histocompatibility Barrier in Mice. Blood (2001) 98:1607–13. doi: 10.1182/blood.v98.5.1607
31. Kim KJ, Kanellopoulos-Langevin C, Merwin RM, Sachs DH, Asofsky REstablishment and Characterization of BALB/c Lymphoma Lines With B Cell Properties. J Immun (1979) 122(2):549–54.
32. Bascuas T, Moreno M, Mónaco A, Reyes L, Paolino A, Oliver P, et alA Novel Non-Hodgkin Lymphoma Murine Model Closer to the Standard Clinical Scenario. J Transl Med (2016) 14:323.
33. Coghill JM, Sarantopoulos S, Moran TP, Murphy WJ, Blazar BR, Serody JS. Effector CD4+ T Cells, the Cytokines They Generate, and GVHD: Something Old and Something New. Blood (2011) 117:3268–76. doi: 10.1182/blood-2010-12-290403
34. Paz Morante M, Briones J, Canto E, Sabzevari H, Martino R, Sierra J, et al. Activation-Associated Phenotype of CD3+ T Cells in Acute Graft-Versus-Host Disease. Clin Exp Immunol (2006) 145:36–43. doi: 10.1111/j.1365-2249.2006.03104.x
35. Zhang Y, Joe G, Hexner E, Zhu J, Emerson SG. Alloreactive Memory T Cells Are Responsible for the Persistence of Graft-Versus-Host Disease. J Immunol (Baltimore Md : 1950) (2005) 174(5):3051–8. doi: 10.4049/jimmunol.174.5.3051
36. Ogawa S, Nagamatsu G, Watanabe M, Watanabe S, Hayashi T, Horita S, et al. Opposing Effects of Anti-Activation-Inducible Lymphocyte-Immunomodulatory Molecule/Inducible Costimulator Antibody on the Development of Acute Versus Chronic Graft-Versus-Host Disease. J Immunol (2001) 167:5741–8. doi: 10.4049/jimmunol.167.10.5741
37. MacDonald KP, Blazar BR, Hill GR. Cytokine Mediators of Chronic Graft-Versus-Host Disease. J Clin Invest (2017) 127(7):2452–63. doi: 10.1172/JCI90593
38. Cooke KR, Coghill JM, Hildebrandt GC, Serody JS. Chemokines and Graft-Versus-Host Disease. Johns Hopkins Univ (2013) 18:393–424. doi: 10.1016/B978-0-12-416004-0.00017-3
39. Bouazzaoui A, Spacenko E, Mueller G, Miklos S, Huber E, Holler E, et al. Chemokine and Chemokine Receptor Expression Analysis in Target Organs of Acute Graft-Versus-Host Disease. Genes Immun (2009) 10:687–701. doi: 10.1038/gene.2009.49
40. Castor MGM, Pinho V, Teixeira MM. The Role of Chemokines in Mediating Graft Versus Host Disease: Opportunities for Novel Therapeutics. Front Pharmacol (2012) 3:23. doi: 10.3389/fphar.2012.00023
41. Ghimire S, Weber D, Mavin E, Nong Wang X, Dickinson AM, Holler E, et al. Pathophysiology of GvHD and Other HSCT-Related Major Complications. Front Immunol (2017) 8:79. doi: 10.3389/fimmu.2017.00079
42. Aw R, Dr B. Connective Tissue Growth Factor (CTGF/CCN2) in Hepatic Fibrosis. Hepatol research: Off J Japan Soc Hepatol (2003) 26(1):1–9. doi: 10.1016/s1386-6346(03)00115-3
43. Tarlow BD, Pelz C, Naugler WE, Wakefield L, Wilson EM, Finegold MJ, et al. Bipotential Adult Liver Progenitors are Derived From Chronically Injured Mature Hepatocytes. Cell Stem Cell (2014) 15(5):605–18. doi: 10.1016/j.stem.2014.09.008
44. Tt A, Lb I. Interleukin-10-Induced Gene Expression and Suppressive Function Are Selectively Modulated by the PI3K-Akt-GSK3 Pathway. Immunology (2011) 132(4):567–77. doi: 10.1111/j.1365-2567.2010.03402.x
45. Kadlecek TA, van ONSC, Lefrancois L, Olson S, Finlay D, DH C, et al. Differential Requirements for ZAP-70 in TCR Signaling and T Cell Development. J Immunol (1998) 161:4688–94.
46. Ford GS, Barnhart B, Shone S, Covey LR. Regulation of CD154 (CD40 Ligand) mRNA Stability During T Cell Activation. J Immunol (1999) 162:4037–44.
47. Arts RJW, Netea MG. Adaptive Characteristics of Innate Immune Responses in Macrophages. Microbiol Spectr (2016) 4(4):4. doi: 10.1128/microbiolspec.MCHD-0023-2015
48. Choi SW, Levine JE, Ferrara JLM. Pathogenesis and Management of Graft Versus Host Disease. Immunol Allergy Clin North Am (2010) 30:75–101. doi: 10.1016/j.iac.2009.10.001
49. Ross SH, Cantrell DA. Signaling and Function of Interleukin-2 in T Lymphocytes. Annu Rev Immunol (2018) 36:411–33. doi: 10.1146/annurev-immunol-042617-053352
50. Lefebvre DC, Lai JCY, Maeshima N, Ford JL, Wong ASL, Cross JL, et al. CD44 Interacts Directly With Lck in a Zinc-Dependent Manner. Mol Immunol (2010) 47(10):1882–9. doi: 10.1016/j.molimm.2010.03.018
51. Silva R, Morgado JM, Freitas A, Couceiro A, Orfao A, Regateiro F, et al. Influence of Pro- and Anti-Inflammatory Cytokines in Th1 Polarization After Allogeneic Stimulation. Int J BioMed Sci (2005) 1:46–52.
52. Krenger W, Ferrara JLM. Graft-Versus-Host Disease and the Th1/Th2 Paradigm. Immunol Res (1996) 15(1):50–73. doi: 10.1007/bf02918284
53. Teague TK, Marrack P, Kappler JW, Vella AT. IL-6 Rescues Resting Mouse T Cells From Apoptosis. J Immunol (1997) 158:5791–6.
54. Ayroldi E, Zollo O, Cannarile L, D’ Adamio F, Grohmann U, Delfino DV, et al. Interleukin-6 (IL-6) Prevents Activation-Induced Cell Death: IL-2-Independent Inhibition of Fas/fasL Expression and Cell Death. Blood (1998) 92:4212–9. doi: 10.1182/blood.V92.11.4212.423k42_4212_4219
55. Yokota S, Geppert TD, Lipsky PE. Enhancement of Antigen- And Mitogen-Induced Human T Lymphocyte Proliferation by Tumor Necrosis Factor-Alpha. J Immunol (1988) 140(2):531–6.
56. Banerjee D, Liou H-C, Sen R. C-Rel-Dependent Priming of Naive T Cells by Inflammatory Cytokines. Immunity (2005) 24(4):445–58. doi: 10.1016/j.immuni.2005.09.012
57. Seeley JJ, Ghosh S. Molecular Mechanisms of Innate Memory and Tolerance to LPS. J Leukoc Biol (2017) 101:107–19. doi: 10.1189/jlb.3MR0316-118RR
58. Couper KN, Blount DG, Riley EM. IL-10: The Master Regulator of Immunity to Infection. J Immunol (2008) 180:5771–7. doi: 10.4049/jimmunol.180.9.5771
59. Jeljeli M, Riccio LGC, Chouzenoux S, Moresi F, Toullec L, Doridot L, et al. Macrophage Immune Memory Controls Endometriosis in Mice and Humans. Cell Rep (2020) 33(5):108325. doi: 10.1016/j.celrep.2020.108325
60. Baird K, Pavletic SZ. Chronic Graft Versus Host Disease. Curr Opin Hematol (2006) 13:426–35. doi: 10.1097/01.moh.0000245689.47333.ff
61. Serody JS, Hill GR. The IL-17 Differentiation Pathway and Its Role in Transplant Outcome. Biol Blood Marrow Transplant (2012) 18:S56–61. doi: 10.1016/j.bbmt.2011.10.001
62. McCormick LL, McCormick LL, Zhang Y, Tootell E, Gilliam ACAnti-TGF-β Treatment Prevents Skin and Lung Fibrosis in Murine Sclerodermatous Graft-Versus-Host Disease: A Model for Human Scleroderma. J Immunol (1999) 163(10):5693–9.
63. Zhang Y, McCormick LL, Desai SR, Wu C, Gilliam AC. Murine Sclerodermatous Graft-Versus-Host Disease, a Model for Human Scleroderma: Cutaneous Cytokines, Chemokines, and Immune Cell Activation. J Immunol (2002) 168:3088–98. doi: 10.4049/jimmunol.168.6.3088
64. Zeiser R, Sarantopoulos S, Blazar BR. B-Cell Targeting in Chronic Graft-Versus-Host Disease. Blood (2018) 131:1399–405. doi: 10.1182/blood-2017-11-784017
65. Piper C, Drobyski WR. Inflammatory Cytokine Networks in Gastrointestinal Tract Graft vs. Host Disease. Front Immunol (2019) 10:163. doi: 10.3389/fimmu.2019.00163
66. Le Huu D, Matsushita T, Jin G, Hamaguchi Y, Hasegawa M, Takehara K, et al. IL-6 Blockade Attenuates the Development of Murine Sclerodermatous Chronic Graft-Versus-Host Disease. J Invest Dermatol (2012) 132:2752–61. doi: 10.1038/jid.2012.226
67. Levine JE. Implications of TNF-α in the Pathogenesis and Management of GVHD. Int J Hematol (2011) 93:571–7. doi: 10.1007/s12185-011-0803-1
68. Tanaka J, Imamura M, Kasai M, Kobayashi S, Hashino S, Kobayashi H, et al. Cytokine Gene Expression in the Mixed Lymphocyte Culture in Allogeneic Bone Marrow Transplants as a Predictive Method for Transplantation-Related Complications. Br J Haematol (1994) 87(2):415–8. doi: 10.1111/j.1365-2141.1994.tb04935.x
69. Wikenheiser DJ, Stumhofer JS. ICOS Co-Stimulation: Friend or Foe? Front Immunol (2016) 7:304. doi: 10.3389/fimmu.2016.00304
70. Yu X-Z, Liang Y, Nurieva RI, Guo F, Anasetti C, Dong C. Opposing Effects of ICOS on Graft-Versus-Host Disease Mediated by CD4 and CD8 T Cells. J Immunol (2006) 176:7394–401. doi: 10.4049/jimmunol.176.12.7394
71. Zhang M, Wu Y, Bastian D, Iamsawat S, Chang J, Daenthanasanmak A, et al. Inducible T-Cell Co-Stimulator Impacts Chronic Graft-Versus-Host Disease by Regulating Both Pathogenic and Regulatory T Cells. Front Immunol (2018) 9:1461. doi: 10.3389/fimmu.2018.01461
72. Soler D, Humphreys TL, Spinola SM, Campbell JJ. CCR4 Versus CCR10 in Human Cutaneous TH Lymphocyte Trafficking. Blood (2003) 101:1677–82. doi: 10.1182/blood-2002-07-2348
73. Gilet J, Chang Y, Chenivesse C, Legendre B, Vorng H, Duez C, et al. Role of CCL17 in the Generation of Cutaneous Inflammatory Reactions in Hu-PBMC-SCID Mice Grafted With Human Skin. J Invest Dermatol (2009) 129:(4)879–90. doi: 10.1038/jid.2008.333
74. Faaij CMJM, Lankester AC, Spierings E, Hoogeboom M, Bowman EP, Bierings M, et al. A Possible Role for CCL27/CTACK-CCR10 Interaction in Recruiting CD4 T Cells to Skin in Human Graft-Versus-Host Disease. Br J Haematol (2006) 133:538–49. doi: 10.1111/j.1365-2141.2006.06058.x
75. Groom JR, Luster AD. CXCR3 in T Cell Function. Exp Cell Res (2011) 317:620–31. doi: 10.1016/j.yexcr.2010.12.017
76. Croudace JE, Inman CF, Abbotts BE, Nagra S, Nunnick J, Mahendra P, et al. Chemokine-Mediated Tissue Recruitment of CXCR3+ CD4+ T Cells Plays a Major Role in the Pathogenesis of Chronic GVHD. Blood (2012) 120(20):4246–55. doi: 10.1182/blood-2012-02-413260
77. He S, Cao Q, Qiu Y, Mi J, Zhang JZ, Jin M, et al. A New Approach to the Blocking of Alloreactive T Cell-Mediated Graft-Versus-Host Disease by In Vivo Administration of Anti-CXCR3 Neutralizing Antibody. J Immunol (2008) 181:7581–92. doi: 10.4049/jimmunol.181.11.7581
78. Loschi M, Porcher R, Peffault de Latour R, Vanneaux V, Robin M, Xhaard A, et al. High Number of Memory T Cells Is Associated With Higher Risk of Acute Graft-Versus-Host Disease After Allogeneic Stem Cell Transplantation. Biol Blood marrow transplantation : J Am Soc Blood Marrow Transplant (2015) 21(3):569–74. doi: 10.1016/j.bbmt.2014.12.009
79. Crawford A, Angelosanto JM, Nadwodny KL, Blackburn SD, Wherry EJ. A Role for the Chemokine RANTES in Regulating CD8 T Cell Responses During Chronic Viral Infection. PloS Pathog (2011) 7:e1002098. doi: 10.1371/journal.ppat.1002098
80. Murai M, Yoneyama H, Harada A, Yi Z, Vestergaard C, Guo B, et al. Active Participation of CCR5(+)CD8(+) T Lymphocytes in the Pathogenesis of Liver Injury in Graft-Versus-Host Disease. J Clin Invest (1999) 104(1):49–57. 10.1172/JCI6642
81. Mencarelli A, Cipriani S, Francisci D, Santucci L, Baldelli F, Distrutti E, et al. Highly Specific Blockade of CCR5 Inhibits Leukocyte Trafficking and Reduces Mucosal Inflammation in Murine Colitis. Sci Rep (2016) 6:30802. doi: 10.1038/srep30802
82. Kitko CL, White ES, Baird K. FIBROTIC AND SCLEROTIC MANIFESTATIONS OF CHRONIC GRAFT VERSUS HOST DISEASE. Biol Blood Marrow Transplant (2012) 18:S46–52. doi: 10.1016/j.bbmt.2011.10.021
83. Wynn TA, Ramalingam TR. Mechanisms of Fibrosis: Therapeutic Translation for Fibrotic Disease. Nat Med (2012) 18:1028–40. doi: 10.1038/nm.2807
84. Schroeder MA, DiPersio JF. Mouse Models of Graft-Versus-Host Disease: Advances and Limitations. Dis Model Mech (2011) 4:318–33. doi: 10.1242/dmm.006668
85. Hashimoto D, Chow A, Greter M, Saenger Y, Kwan W-H, Leboeuf M, et al. Pretransplant CSF-1 Therapy Expands Recipient Macrophages and Ameliorates GVHD After Allogeneic Hematopoietic Cell Transplantation. J Exp Med (2011) 208:1069–82. doi: 10.1084/jem.20101709
86. Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, et al. Bone Marrow Myeloid-Derived Suppressor Cells (MDSCs) Inhibit Graft-Versus-Host Disease (GVHD) via an Arginase-1-Dependent Mechanism That Is Up-Regulated by Interleukin-13. Blood (2010) 116(25):5738–47. doi: 10.1182/blood-2010-06-287839
87. Dunavin N, Dias A, Li M, McGuirk J. Mesenchymal Stromal Cells: What Is the Mechanism in Acute Graft-Versus-Host Disease? Biomedicines (2017) 5(3):39. doi: 10.3390/biomedicines5030039
88. Riegel C, Boeld TJ, Doser K, Huber E, Hoffmann P, Edinger M. Efficient Treatment of Murine Acute GvHD by In Vitro Expanded Donor Regulatory T Cells. Leukemia (2019) 34(3):895–908. doi: 10.1038/s41375-019-0625-3
89. Weisdorf D, Zhang M-J, Arora M, Horowitz MM, Rizzo JD, Eapen M. Graft-Versus-Host Disease Induced Graft-Versus-Leukemia Effect: Greater Impact on Relapse and Disease-Free Survival After Reduced Intensity Conditioning. Biol Blood Marrow Transplant (2012) 18:1727–33. doi: 10.1016/j.bbmt.2012.06.014
90. Cao J, Yan Q. Cancer Epigenetics, Tumor Immunity, and Immunotherapy. Trends Cancer (2020) 6(7):580–92. doi: 10.1016/j.trecan.2020.02.003
91. Bascuas T, Moreno M, Mónaco A, Reyes L, Paolino A, Oliver P, et al. A Novel non-Hodgkin Lymphoma Murine Model Closer to the Standard Clinical Scenario. J Trans Med (2016) 14:323. doi: 10.1186/s12967-016-1073-8
92. Elpek KG, Lacelle C, Singh NP, Yolcu ES, Shirwan H. CD4+CD25+ T Regulatory Cells Dominate Multiple Immune Evasion Mechanisms in Early But Not Late Phases of Tumor Development in a B Cell Lymphoma Model. J Immunol (Baltimore Md : 1950) (2007) 178(11):6840–8. doi: 10.4049/jimmunol.178.11.6840
93. Liu Q, Tian Y, Zhao X, Jing H, Xie Q, Li P, et al. NMAAP1 Expressed in BCG-Activated Macrophage Promotes M1 Macrophage Polarization. Mol Cells (2015) 38:886–94. doi: 10.14348/molcells.2015.0125
94. Najafi M, Hashemi Goradel N, Farhood B, Salehi E, Nashtaei MS, Khanlarkhani N, et al. Macrophage Polarity in Cancer: A Review. J Cell Biochem (2019) 120(3):2756–65. doi: 10.1002/jcb.27646
95. Noy R, Pollard JW. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity (2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
96. Morton DL, Eilber FR, Holmes EC, Hunt JS, Ketcham AS, Silverstein MJ, et al. BCG Immunotherapy of Malignant Melanoma: Summary of a Seven-Year Experience. Ann Surg (1974) 180:635–41. doi: 10.1097/00000658-197410000-00029
97. Pettenati C, Ingersoll MA. Mechanisms of BCG Immunotherapy and its Outlook for Bladder Cancer. Nat Rev Urol (2018) 15(10):615–25. doi: 10.1038/s41585-018-0055-4
98. Albeituni SH, Ding C, Liu M, Hu X, Luo F, Kloecker G, et al. Yeast-Derived Particulate β-Glucan Treatment Subverts the Suppression of Myeloid-Derived Suppressor Cells (MDSC) by Inducing Polymorphonuclear MDSC Apoptosis and Monocytic MDSC Differentiation to APC in Cancer. J Immunol (Baltimore Md : 1950) (2016) 196(5):2167–80. doi: 10.4049/jimmunol.1501853
99. Suzuki T, Arumugam P, Sakagami T, Lachmann N, Chalk C, Sallese A, et al. Pulmonary Macrophage Transplantation Therapy. Nature (2014) 514(7523):450–4. doi: 10.1038/nature13807
Keywords: macrophages, trained immunity, graft-versus-host disease, alloimmunity, inflammation, anti-tumoral action
Citation: Jeljeli M, Chêne C, Chouzenoux S, Thomas M, Segain B, Doridot L, Nicco C and Batteux F (2021) LPSlow-Macrophages Alleviate the Outcome of Graft-Versus-Host Disease Without Aggravating Lymphoma Growth in Mice. Front. Immunol. 12:670776. doi: 10.3389/fimmu.2021.670776
Received: 22 February 2021; Accepted: 07 July 2021;
Published: 03 August 2021.
Edited by:
Aurore Saudemont, GlaxoSmithKline, United KingdomReviewed by:
Lanping Xu, Peking University People’s Hospital, ChinaSebastiaan Meijsing, Max-Planck-Gesellschaft (MPG), Germany
Depei Wu, The First Affiliated Hospital of Soochow University, China
Copyright © 2021 Jeljeli, Chêne, Chouzenoux, Thomas, Segain, Doridot, Nicco and Batteux. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frédéric Batteux, ZnJlZGVyaWMuYmF0dGV1eEBhcGhwLmZy
†These authors have contributed equally to this work and share last authorship