 Shuai Liu*
Shuai Liu* Keji Zhao*
Keji Zhao*- Laboratory of Epigenome Biology, Systems Biology Center, NHLBI, NIH, Bethesda, MD, United States
The code of life is not only encrypted in the sequence of DNA but also in the way it is organized into chromosomes. Chromosome architecture is gradually being recognized as an important player in regulating cell activities (e.g., controlling spatiotemporal gene expression). In the past decade, the toolbox for elucidating genome structure has been expanding, providing an opportunity to explore this under charted territory. In this review, we will introduce the recent advancements in approaches for mapping spatial organization of the genome, emphasizing applications of these techniques to immune cells, and trying to bridge chromosome structure with immune cell activities.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Introduction
The spatial-temporal gene expression determines the identity and activity of cells (1, 2). Gene expression is controlled by transcription factors working on the dynamically structured genome (3–6). Elucidating genome architecture is pivotal for understanding the fundamental mechanisms of gene expression regulation. Thanks to technological developments, our understanding of genome structure has been revolutionized in the past decade (7–10).
Eukaryotic genomes are organized into chromatin with nucleosomes as fundamental structural units (11). The spatial organization of chromatin in the nucleus has been investigated at several scales. First, formation of chromatin loops brings distant genomic regions from tens to hundreds of kilobases to spatial proximity, which involves the Cohesin complex through an extrusion process (12–17). The next level of chromatin organization is topologically associating domains (TADs), ranging from hundreds of kilobases to several million bases (18–20). The boundaries of TADs are usually marked by the binding of insulator CCCTC-binding factor (CTCF), which plays a key role in the formation of TADs (20–22). TAD boundaries appear to restrict chromatin loops within TADs and few chromatin loops form across TAD boundaries (20, 23). Chromatin can also be separated into even larger domains, termed as A compartment and B compartment, which are enriched for active and repressive chromatin modification marks, respectively (23, 24). A compartment is composed of active TADs, which often contain expressed genes, while B compartments contain silent genes. Each chromosome occupies a specific location in the 3D nuclear space, which is termed as chromosome territory (24, 25). From nucleosome to chromosome territory, the architecture of chromatin features a hierarchical pattern.
From a methodology perspective, RNA-seq method provides transcriptome information (26), and ChIP-seq are used for investigating transcription factor binding and histone modification profiles on the genome (27, 28). Techniques for mapping the landscape of chromatin accessibility include ATAC-seq and DNase-seq (29). The techniques for cracking the three-dimensional genome architecture have been rapidly evolving during the last decade. In this review, we will introduce current state-of-art techniques for untangling the three-dimensional (3D) organization of chromatin, and then discuss their applications to the immune system. Two classes of approaches are extensively used in exploring the 3D genome organization: microscopy-imaging based techniques and sequencing based methods (Table 1). We will briefly introduce imaging-based methods, and then focus on sequencing-based approaches.
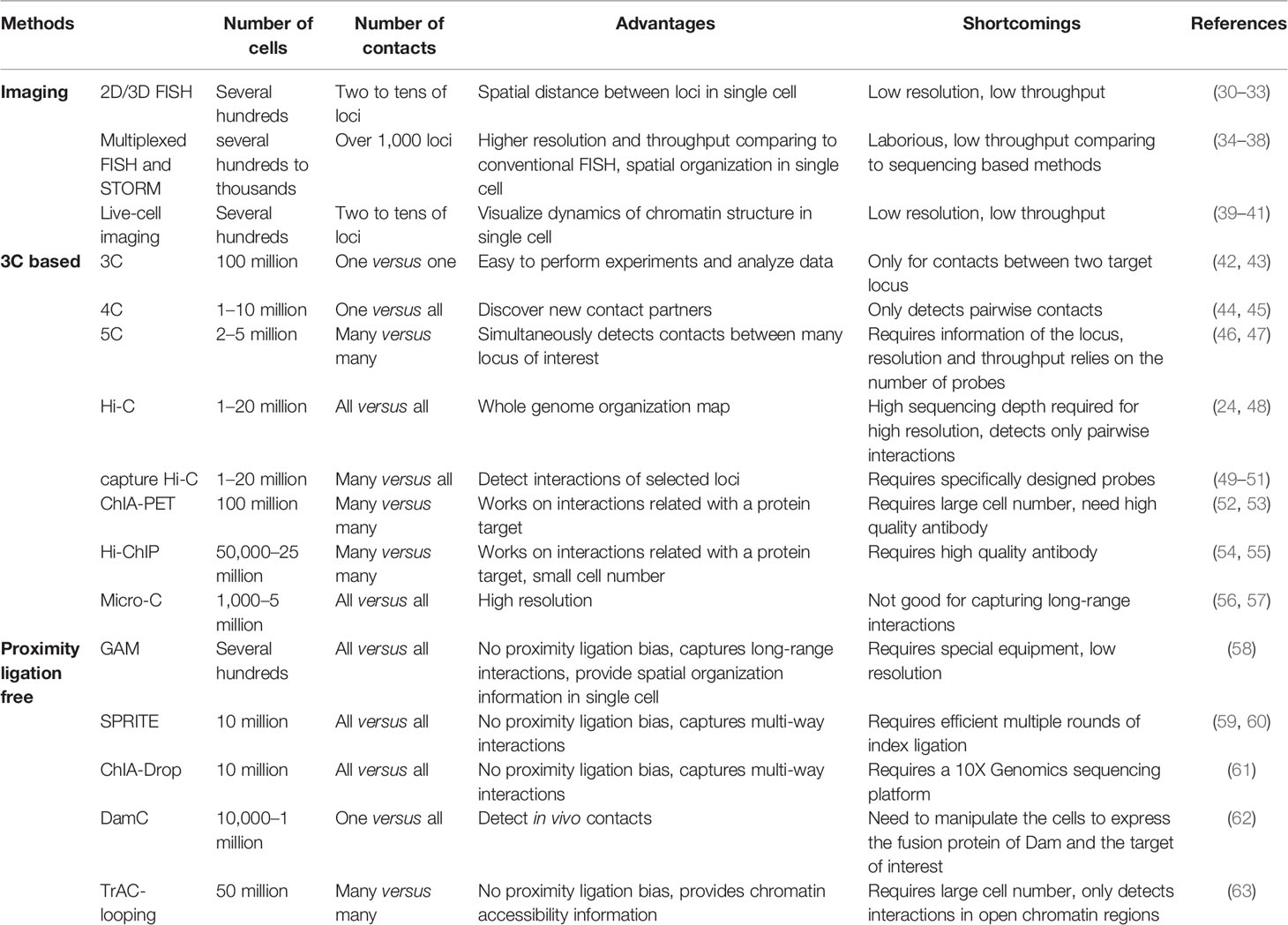
Table 1 Comparison of the features of techniques for elucidating chromatin structures.
Imaging-Based Techniques
Fluorescence in situ hybridization (FISH) was once the dominating method for studying genome structure (30, 31, 64, 65). The spatial distance between chromatin loci is visualized under the microscope by hybridizing fluorescently labeled probes to target regions in fixed cells. DNA-FISH is suitable for detecting the spatial distances between two or a few loci. However, the two major limitations, resolution and throughput, hamper its applications. The resolution of FISH was constrained by microscopy and the probe, making it not suitable of resolving loci within relatively close spatial distance (shorter than several hundred nanometers) or close locations on the genome (less than 100 kb). Recently, there have been significant improvements in the resolution and throughput of FISH microscopy with the development of short multiplexed probes and super-resolution microscopy (32–36, 66). Genome architecture in single cells can now be visualized at spatial resolution of tens of nanometers and genome resolution of tens of kilobase pairs (36, 37, 67, 68). One major advantage of imaging based techniques is that they are capable of monitoring the dynamics of chromatin structure in living cells (38, 69), which was reviewed recently (39). Even though the resolution of these approaches is relatively low, they still provide invaluable knowledge about the dynamics of genome architecture. It would be of interest to observe how the genome architecture of immune cells changes in response to environmental stimulus. Fluorescence and electron imaging techniques are evolving rapidly for elucidating chromatin organization. There are excellent reviews on this topic, which will not be discussed in detail here (40, 41, 70, 71). Although these cutting-edge imaging techniques have not been widely applied to studying the immune system, with the improvement of resolution and throughput, they will be sure to illuminate the connection between genome organization and immune cell activity.
Sequencing-Based Techniques
In our discussion, sequencing based approaches for mapping genome architecture are separated into two categories: proximity ligation-based methods (Figure 1) and proximity ligation-free methods.
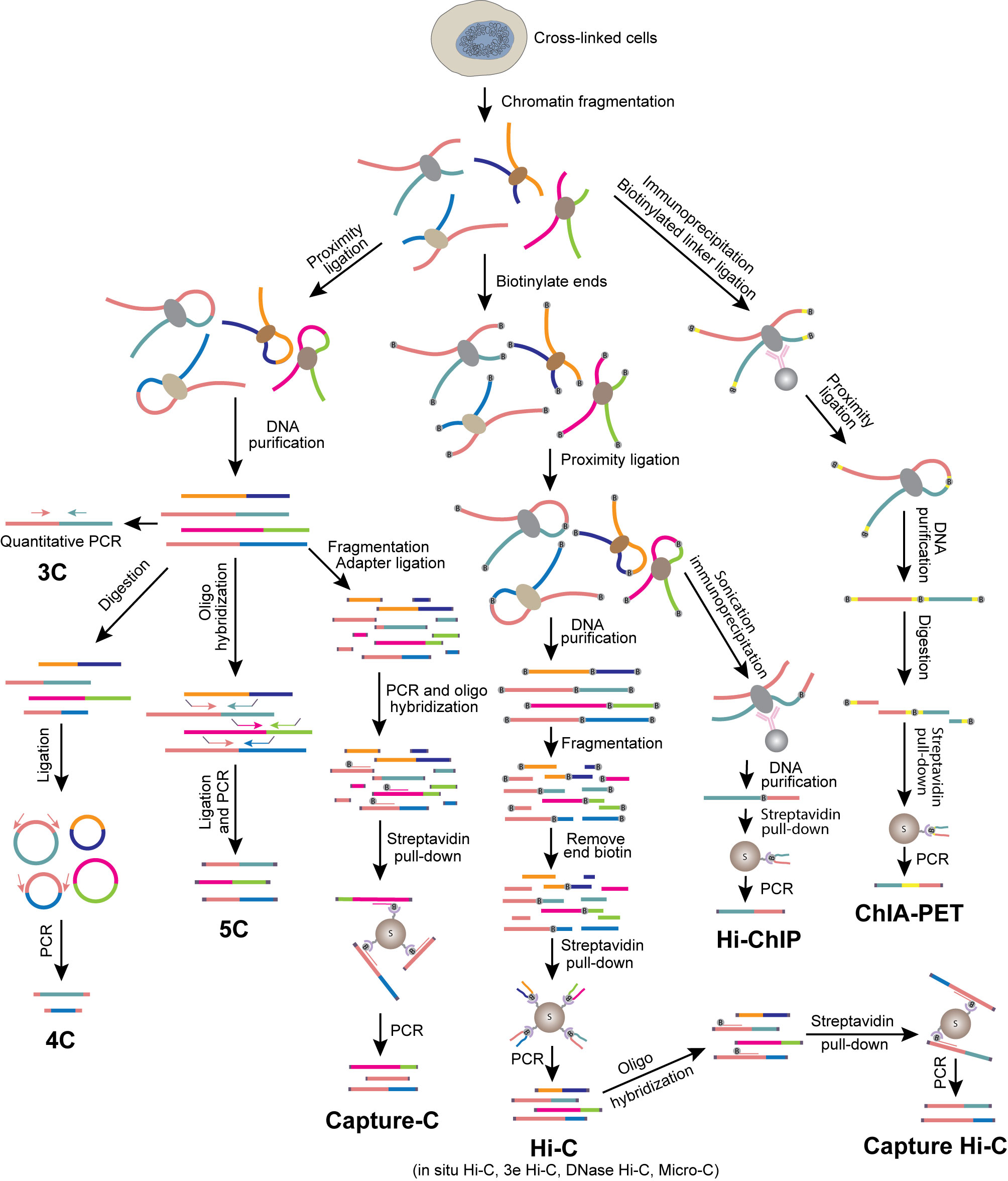
Figure 1 Overview of 3C-based methods. Schematics illustrate experimental procedures of different methods.
Proximity Ligation-Based Methods
(1) Chromosome Conformation Capture (3C)
Unlike FISH directly presenting the physical distance between genomic loci, chromosome conformation capture assay (3C) detects the spatial proximity between two genomic loci by interpreting the efficiency of them being ligated together (72, 73). Cells are fixed with formaldehyde to preserve the spatial conformation of chromatin, and chromatin is then digested with a restriction enzyme to generate free ends of DNA. The DNA ends from chromatin regions in close spatial proximity are prone to be ligated in the presence of DNA ligase. The frequency of ligation between two loci can be examined by gel electrophoresis or qPCR using primers specific to these regions. 3C detects the interaction between two candidate loci (one-to-one). FISH and 3C are frequently used together to cross-validate each other’s findings. Even with various more advanced methods available as discussed below, 3C remains a good choice if one is interested in examining the changes of interaction between two specific genomic regions, especially transcriptional regulatory regions, during cell differentiation or activation.
(2) Circular Chromosome Conformation Capture (4C) and Chromosome Conformation Capture Carbon Copy (5C)
4C was developed to identify genome-wide interacting partners of a target locus (the “viewpoint”) (74, 75). Unlike 3C, which requires knowledge of the interacting locus, 4C is capable of discovering unknown interactions. The 4C protocol requires a DNA circularization step after proximity ligation, followed by reverse PCR with primers designed according to the sequence of the viewpoint. All interacting partners can then be identified by sequencing. Thus, 4C is a method for detecting one-to-all interactions. By comparison, 5C is a method for detecting many-to-many interactions. Since all interacting partners are supposed to be ligated at the proximity ligation step in 3C, multiple interacting pairs may be amplified and detected by using properly designed multiplexed primers in a 5C library. The coverage and resolution of 5C depends on the diversity of the primers (76, 77).
(3) Hi-C and Derivatives
In the past decade, a major breakthrough in the field of elucidating genome architecture was the development of Hi-C, a method for analyzing whole genome organization (all-to-all interactions) (24). Hi-C follows the original 3C protocol with some modifications including addition of a biotinylated nucleotide fill-in step before proximity ligation. To generate Hi-C libraries, biotin-labeled and ligated DNAs are enriched, digested and ligated to Y-shaped adaptors for next generation sequencing (NGS). After sequencing on an NGS platform and mapping the reads to the genome, a genome-wide contact map can be established from the paired-end sequencing reads. Hi-C has provided tremendous information about genome organization, from compartments and TADs to chromatin loops. Efforts have been made to optimize Hi-C to generate higher resolution genome organization map (78). In situ Hi-C decreases random ligation between chromatin fragments due to reduced nucleus disruption (23, 42). The resolution of a genome-wide chromatin contact map generated by Hi-C is limited by the available restriction enzyme cleavage sites in the genome. Thus, digesting the chromatin by using a combination of restriction enzymes (3e Hi-C) (43), DNase I (DNase Hi-C) (44) or micrococcal nuclease (Micro-C) significantly increases the resolution (45, 46). Application of Hi-C to immune cells has revealed cell-specific three-dimensional chromatin interactions and provided insights into the mechanisms of their regulation in immune cells (43, 47).
Although Hi-C has the advantage of being capable of providing genome-wide chromatin contact maps, it suffers from the need for very deep sequencing in order to obtain information on chromatin interaction among transcriptional regulatory elements such as promoters and enhancers. The cost of sequencing to examine the fine chromatin structures is often prohibitory (23, 24, 48). Only a very limited number of interactions between promoters and enhancers are detected even with a sequencing depth of billions of PETs from a Hi-C library. To increase the resolution and efficiency and reduce the sequencing cost of Hi-C, a few derivative approaches were developed. One strategy is the Capture Hi-C method that detects chromatin contacts of selected chromatin regions such as gene promoters (56, 79–81). After proximity ligations, genomic loci of interest are captured and enriched by hybridizing biotinylated oligonucleotide probes. This strategy provides the chromatin interaction information of the selected genomic regions. ChIA-PET (Chromatin Interaction Analysis with Paired-End Tag) and HiChIP are strategies of detecting chromatin interactions of a subset of genomic regions by combining Hi-C and ChIP (chromatin immunoprecipitation) (49, 57, 82–84). ChIA-PET performs proximity ligation after the ChIP step by using a specific antibody against a transcription factor (TF), chromatin modifier or histone modification to enrich target regions, while HiChIP performs ChIP after the proximity ligation step. These two approaches are capable of profiling chromatin interactions genome-wide at locations bound by a specific chromatin protein or carrying a specific histone modification. For example, the promoter-enhancer interaction network explored by RNA Pol II ChIA-PET in GM12878 cells provided comprehensive view of the regulation of B cell transformation triggered by Epstein-Barr Virus (EBV) infection (50, 51).
Hi-C is a powerful technique for detecting chromatin interactions; however, the heterogeneity of chromatin structure in each individual cell is concealed in this population averaged approach. In situ single-cell Hi-C and Dip-C are two methods for exploring the diversity of genome organization at a single cell level (52–55). Nagano et al. applied single-cell Hi-C to tracking the dynamics of chromatin structure in cell cycle at a single cell level (85). Flyamer et al. discovered reorganization of chromatin in the transition of oocyte to zygote (86). By using Dip-C, Tan et al. investigated genome organization in neurons of mouse retina, olfactory epithelium and developing brain (87, 88). By applying these techniques to immune cells, it would reveal interesting diversity features of genome architecture at a single cell level, such as the recombination of antigen receptors.
When performing 3C experiments, chromatin conformation is normally preserved by fixing cells with formaldehyde. However, chemical crosslinking may introduce bias in detecting chromatin interactions. Intrinsic 3C/4C/Hi-C (i3C/i4C/iHi-C) and liquid chromatin Hi-C are developed to study native genome structure (89, 90). i3C captures chromatin structures detected by conventional 3C with lower background. Liquid chromatin Hi-C is capable of tracking dynamics of chromatin interactions. By using i4C, Weiterer et al. studied chromatin structures at CXCL2 and IL8 loci with the stimulation of interleukin (IL)-1α, and found that IL-1α–induced chromatin remodeling depends on TAK1 kinase and NF-κB pathways (91).
(4) Other Proximity Ligation-Based Methods
Conventional 3C techniques reconstruct genome architecture based on averaged pairwise chromatin interactions. It remains to be elucidated how these interaction pairs are synergized into higher order structures. Recently developed chromosomal walks (C-walks), multi-contact 4C (MC-4C), Tri-C, multi-contact 3C (MC-3C) and other methods provide insights into concurrent chromatin interactions at single allele levels (92–97).
Majority of noncoding RNAs (ncRNAs) localize in the nucleus, and have contacts with chromatin. They play important roles in gene regulation and chromatin remodeling (98). Proximity ligation-based methods, such as MARGI (mapping RNA-genome interactions), GRID-seq (global RNA interactions with DNA by deep-sequencing), ChAR-seq (chromatin-associated RNA sequencing) and RADICL-seq (RNA and DNA interacting complexes ligated and sequenced), were developed to detect genome-wide RNA-chromatin contacts (99–102). They share a similar approach by bridging interacting RNA and DNA with a bivalent linker.
Proximity Ligation-Free Approaches
All 3C-derived methods require proximity-based ligation. They identify ligation frequency between loci instead of direct physical contact information. Since the products are generated by pair-wise ligation between different genomic regions, these methods are not effective at capturing multiple contact partners of a locus simultaneously. Furthermore, artifacts could also be generated by the proximity ligation step. To address these caveats, proximity ligation-free techniques have been emerging.
(1) Genome Architecture Mapping (GAM)
GAM is performed by cryo-sectioning fixed and sucrose-embedded nuclei (103). Hundreds of nuclei are sectioned in random orientations. DNA from each slice is extracted, indexed and sequenced. Chromatin contact information can be inferred from the sequencing data by counting the chance of DNA loci co-existing in the same slice. A mathematical model named SLICE (statistical inference of co-segregation) was developed for analyzing GAM data. GAM reveals long-distance chromatin loops and TADs structure similar to that from Hi-C analysis but at the single-cell level. However, due to the limited number of slices that can be generated from each nucleus, the method suffers from low resolution, particularly for studies of promoter-enhancer interactions.
(2) Split-Pool Recognition of Interactions by Tag Extension (SPRITE) and ChIA-Drop
Chromatin complexes are formed by chromatin binding proteins and their target DNA, which could be stabilized by formaldehyde crosslinking. The interacting chromatin regions that remain in the same complex after cleavage of chromatin will be labeled by the same set of barcoding indexes after multiple of rounds of index ligation. Chromatin regions, which are in different chromatin complexes, are labeled by different barcoding indexes in this process. Based on this property, SPRITE and ChIA-drop assign chromatin regions to different complexes, which infers the spatial proximity and thus potential interaction between different regions of chromatin (104–106). Tagging in SPRITE is performed by ligation in multiple rounds of split-pool, whereas, the reaction adding barcoding indexes to each complex in ChIA-drop takes place in a droplet. DNA loci carrying the same barcode are considered to be in the same interacting complex. Multiple chromatin regions could be assigned to the same chromatin complex, which implies multiple ways of interactions in addition to pairwise interactions.
(3) DamC
DamC uses Escherichia coli DNA adenine methyltransferase (Dam) fused with rTetR, which binds to TetOs inserted into the genome in the presence of doxycycline (Dox) (107). The bound rTetR-Dam methylate adenines in GATC sequences near its binding sites or regions in spatial proximity. Methylated locations are identified by DpnI, which specifically cleaves adenine methylated GATC sequences in the genome. The cleavage sites are ligated to sequencing adapters and analyzed by sequencing. This method generates 4C-like data (one-to-all) with a viewpoint of TetOs that are inserted into the genome. Data of many-to-all interactions can be generated using cell lines with multiple sites of TetOs in the genome. DamC provides genome structure information in living cells; however, it requires manipulating the genome to insert “viewpoints” and to express rTetR-Dam protein in the cells of interest.
(4) Transposase-Mediated Analysis of Chromatin Looping (TrAC-Looping)
Our lab developed a method termed as TrAC-looping (transposase-mediated analysis of chromatin looping), which detects chromatin interactions at transposase Tn5 accessible regions (108). This method relies on Tn5’s ability to insert DNA into the genome. Tn5 forms a tetramer complex with a specially designed bivalent mosaic ends (ME) oligonucleotide linker, which inserts one ME end of the bivalent linker to a chromatin region and the other ME end to an interacting chromatin region. By this way, the two interacting regions are directly linked by an oligonucleotide bridge and thus can be amplified by PCR as one DNA template using primers recognizing the ME sequence and oligonucleotide linker. TrAC-looping detects all interactions between transcription regulatory regions, which include all active promoters and enhancers. TrAC-looping data simultaneously inform both chromatin accessibility and chromatin interactions. Only modest sequencing depth is required for measuring genome-wide regulatory chromatin interactions at high resolution.
Profiling Genome Organization in Immune Cell Development and Differentiation
Next, we will discuss recent applications of these various methods to investigate the genome architecture in immune cells during development and differentiation. The development and differentiation of immune cells are accompanied by reorganization of the genome architecture, from compartment switch to the formation of loops between regulatory elements (Figure 2). Tissue microenvironment triggers the transformation of genome structure, producing key transcription factors (TFs), which regulate the expression of lineage specific genes, determining the fate and function of the cells. The genome organization during immune cell development and differentiation, especially during lineage commitment and antigen receptor V(D)J recombination, has been extensively studied by various approaches.
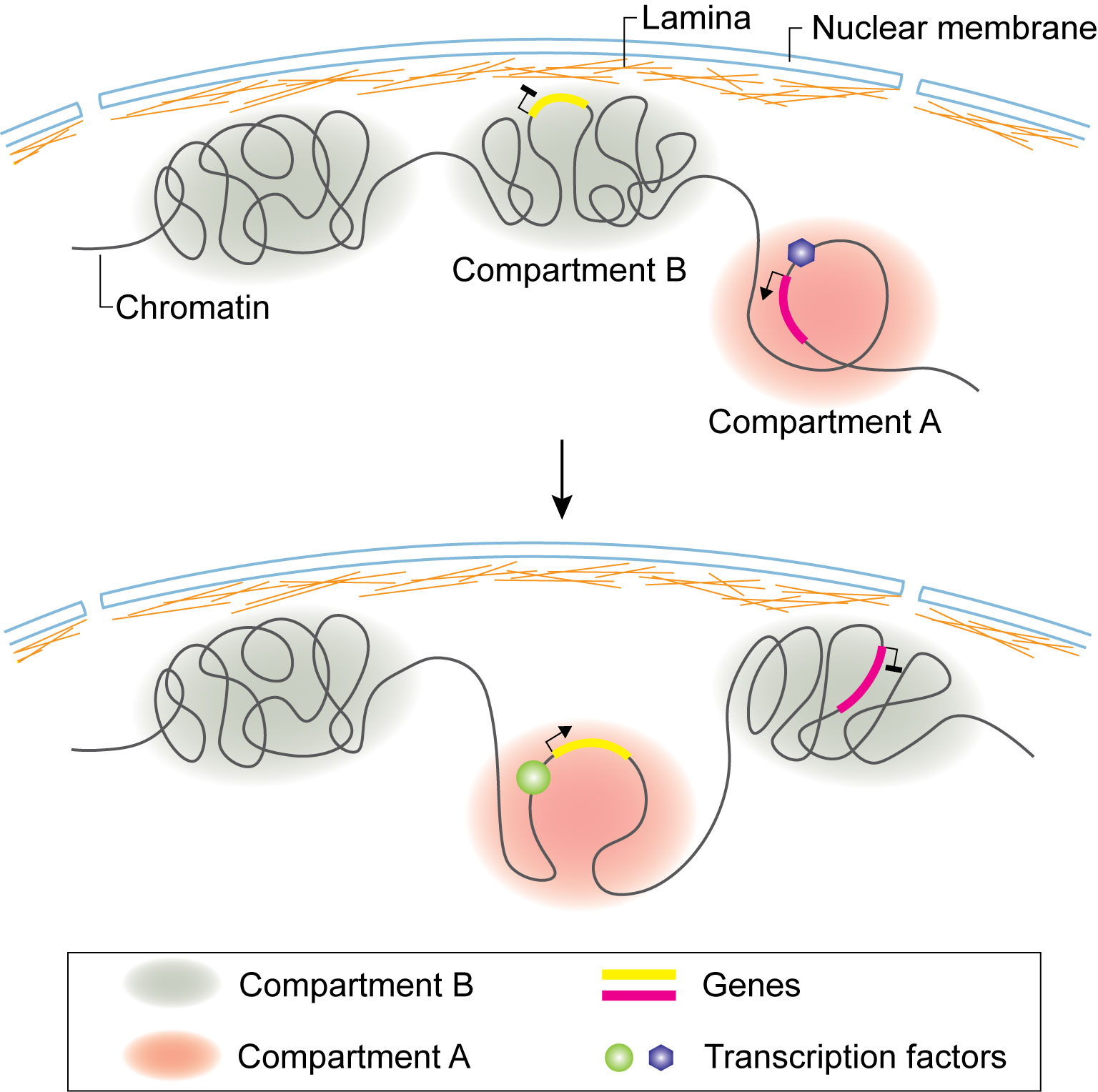
Figure 2 Compartment switch in B cell and T cell lineage commitment. Loci of B or T cell lineage specific genes dissociate from nuclear lamina and are flipped from inactive B compartment to active A compartment. Compartment switch is accompanied by expression of specific transcription factors regulating target gene expression.
Genome Architecture of B Cells
B cell development takes place in bone marrow. Early B cell factor (EBF1) is one of the key transcription factors that control the commitment of progenitor cells to B-cell lineage. By combining Hi-C and 3D-FISH, it was observed that the Ebf1gene locus is sequestered at the nuclear lamina as a repressive B compartment in progenitor cells. When the progenitor cells transitioned to the pro-B stage, the Ebf1 locus switched to an active A compartment, with the establishment of stage specific new interactions (Figure 2) (109). Transcription factor Paired box 5 (PAX5) locks B-cell commitment by regulating the expression of lineage specific genes. By using Hi-C, it was found that PAX5 plays a critical role in establishing and maintaining the unique genome structural features in B cells throughout the B cell differentiation process (58). Ultimately, it regulates gene expression by rewiring the interactions between enhancers and promoters (58).
To fight against various antigens, the adaptive immune system depends on B and T cells producing a diverse pool of antigen specific receptors, immunoglobulins and T cell receptors (TCRs), respectively, through the process of V(D)J gene recombination (Figure 3) (59). The recombination of immunoglobulin heavy chain (Igh) and light chain (Igl) takes place in Pro-B and Small pre-B stages, respectively. The initiation of recombination is carried out by RAG recombinases, which are expressed in developing T and B cells. A Hi-C study revealed that the expression of RAGs is regulated by transcription factor E2A, which facilitates the formation of chromatin loops between their promoters with the T cell-specific enhancer (R-TEn) or B cell-specific elements (R1B and R2B) (60). During the transition from progenitor cells to pro-B cells, by using 3D FISH, it was observed that the Igh locus is released from the nuclear periphery and undergoes chromatin decompaction, suggesting that nuclear periphery localization represses the transcription and recombination of Ig genes during lymphocyte development (61). It was also visualized by FISH that distal Igh V regions are brought into close proximity of the DJ cluster to generate diverse Igh populations (62, 63), which is mediated by the formation of chromatin looping (110). By combining FISH and 3C, Guo et al. discovered that the Igh locus folds into several multi-looped domains, which are subsequently brought into proximity for selective recombination of the enhancer Eµ dependent looping with specific sites within VH region (111).
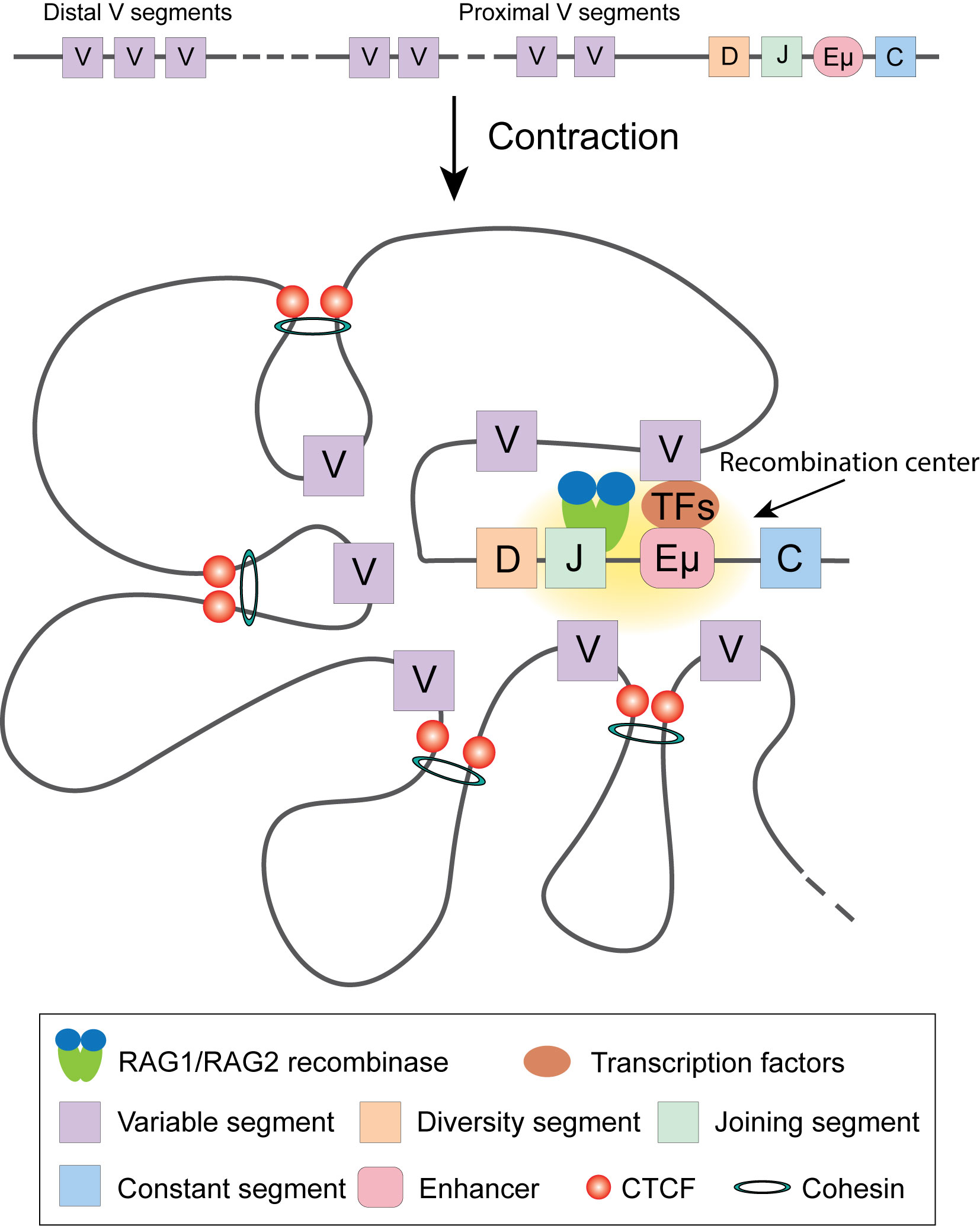
Figure 3 V(D)J recombination of Igh locus. Transcription factors target Enhancer µ and recruit RAG1/RAG2 recombinase to execute recombination. In committed pro-B cells, Cohesin complex and CTCF facilitate Igh locus contraction by looping, ensuring each V gene segment has equal opportunity to target DJ segment.
Close inspection of Igh recombination in live pro-B cells revealed that the contact between VH and DHJH loci happens in minutes and the interaction is controlled by the spatial confinement from the formation of topological domains (112). Using 4C, Medvedovic et al. extensively studied the organization of the Igh locus from different viewpoints (113). They found that distal VH cluster showed more flexibility than the proximal domain, ensuring equal chance of distal VH to recombine with DHJH. The long-range looping requires regulators PAX5, YY1, and CTCF. Convincing data indicated the important roles of CTCF and Cohesin in organizing chromatin loops at the Igh locus and their involvement in Igh recombination (114–117). Cohesin complex facilitates RAG endonuclease scanning along the chromatin fiber by loop extrusion, which initiates immunoglobulin recombination. CTCF bound at its target sites stabilizes the contact between RAG and the distal VH locus, promoting the recombination to happen (17, 110, 118–120). PAX5 specifically represses the expression of Cohesin release factor WAPl to ensure successful loop extrusion across the entire Igh locus (121).
Another major chromatin reorganization event takes place upon the activation of B cells. To elucidate the cis-regulomes in activated B cells, Chaudhri et al. studied the interaction network of cis-regulatory elements by Hi-C. They discovered multiplex enhancer-promoter interaction configurations, including genes regulated by multiple enhancers and enhancers controlling multiple genes, which are engaged in B cell activation, cycling and differentiation (122).
Genome Architecture of T Cells
Multipotent hematopoietic progenitor cells migrate into the thymus, then initiate differentiation from double negative (DN, CD4− CD8−) to double positive (DP, CD4+ CD8+) and finally the CD4 or CD8 single positive T cells. DN cells can be further separated into several stages from ETP, DN2, DN3 to DN4. While ETP and DN2 cells can take alternative cell fates such as dendritic cells, NK cells and mast cells, DN3 cells are already committed to T cell lineage. Thus, the transition from DN2 to DN3 is a key step of T cell fate commitment. Previous studies identified BCL11B, which is upregulated at the DN2 stage, as a key transcription factor required for DN2 to DN3 transition (123–125). Hu et al. used Hi-C to survey the dynamic changes of genome architecture from HSCs to DP cells and observed a striking reorganization of chromatin during the transition from DN2 to DN3 (47). While all other transitions display a relatively smooth change in chromatin interaction, the DN2 to DN3 transition is accompanied by an abrupt global transformation of the 3D genome structure, suggesting that the global changes in chromatin organization may lock the cells in the T cell lineage by creating an energy barrier for reversing cell fate. This observation led to the question of what process causes this global reorganization of chromatin? BCL11B was found to bind to numerous key regulatory regions accompanying T cell commitment (47, 124). Interestingly, BCL11B is a core subunit of the ATP-dependent chromatin remodeling BAF complexes, suggesting that it may be a key player in promoting the global transformation of chromatin reorganization during the DN2 to DN3 transition. Although it has not been demonstrated yet whether BCL11B regulates the changes of chromatin interaction during the early development of T progenitor cells, its deletion in CD4 T cells resulted in decreased chromatin interaction at its bound regions (47). The Bcl11b locus is also associated with a switch from the repressive B compartment to active A compartment when BCL11B expression is upregulated at the DN2 stage. DNA-FISH confirmed the locus moves away from the repressive nuclear periphery during this process. This work demonstrated that, during T cell differentiation, remodeling of chromatin landscape initiates the expression of key transcription factors, which reorganize chromatin structure and alter gene expression to determine the fate of the cells. The activation of Bcl11b is mediated by a non-coding RNA ThymoD (thymocyte differentiation factor). Combining Hi-C and DNA-FISH, Isoda et al. found that ThymoD promotes BCL11B expression by facilitating the formation of chromatin looping between its enhancer and promoter mediated by CTCF and Cohesin (126).
Similar to the recombination of immunoglobulin loci in B cell development, reorganization of T cell antigen receptors also takes place during T cell development. Based on the composition of T cell receptors (TCRs), T cells are separated into Tαβ and Tγδ lineages. The rearrangement of β, γ and δ TCRs takes place in DN cells, whereas TCRα recombination takes place in DP cells. Lineage-specific recombination of TCR is partially determined by the cell specific expression of RAG1 and RAG2. Hi-C analysis revealed that there is a unique organization pattern of cis-regulatory elements in developing T cells (60). Transcription factor E2A binds to a T cell lineage-specific enhancer (R-TEn) to orchestrate the chromatin conformation and expression of the Rag genes in T cells. The chromatin organization landscape of the Tcr locus changes in a spatial and temporal pattern during T cell development. A DamID study identified a lamina-associated domain (LAD), which represses the recombination and expression of Tcrb (127). In DN cells, LAD disassociates from lamina, and Tcrb adopts a compact conformation, enabling the recombination between Vβ and DβJβ segments (128). 3C and 4C analysis indicated that the distal Vβ segments are separated from the recombination center in DP cells (129). Enhancer Eβ is important for the expression of Tcrb. Zhao et al. studied the looping between Eβ and Tcrb promoter by 3C (130). They found transcription factor RUNX1 facilitates the rearrangement of chromatin loops. Different transcription factors may contribute to the rearrangement of chromatin interaction landscape of different Tcr loci. For example, STAT5 binding to Jγ promoter is essential for the change of chromatin conformation at the Tcrg locus (131). A series of work, by combining 3C, 4C, Hi-C and DNA-FISH, have demonstrated that chromatin looping organized by CTCF and Cohesin plays an important role in shaping the diversity of TCR repertoire (117, 132–136).
Mature naïve CD4 or CD8 single positive (SP) cells exit the thymus. They are activated upon encountering antigens and differentiate to effector or memory cells. This process is associated with global epigenomic changes (137). Three-dimensional chromatin organization has been extensively studied in these cells by different approaches, especially for CD4+ T helper (Th) cells. Our lab looked into the genome-wide promoter-enhancer interaction in naïve and activated human CD4+ T cells by TrAC-looping and ChIA-PET (84, 108). It was found that genes with promoters which have interacting enhancers are expressed at higher levels than those without interacting enhancers, and their expression levels are positively correlated with the number of interacting enhancers. A large number of promoters interact with other promoters and they are co-expressed in a tissue-specific manner (84, 108). Activation of naïve CD4+ T cells is associated with significant changes of chromatin interaction at thousands of promoters and enhancers, which are correlated with the changes in gene expression (84, 108). The FOS family motif, TGAGTCA, was significantly enriched in chromatin regions with increased interaction and ChIP-seq assays confirmed that the binding of FOSL1/Fra-1 to these regions after T cell activation. These data suggest that the binding of the FOS family factors may contribute to the increased interactions.
Naïve CD4+ cells differentiate into Th1, Th2, Treg, Th17, or Tfh effector cells by the expression of key transcription factors. T-bet is the key transcription factor for Th1 differentiation and the expression of cytokine interferon-γ (IFNG). DNA-FISH combined with 3C analysis indicated that T-bet collaborates with CTCF and Cohesin to facilitate the formation of chromatin loops between the regulatory elements at Ifng gene locus and promote its expression (138, 139). The transcription factor GATA3 critically regulates Th2 differentiation and the expression of Th2 cytokines, IL4, IL13, and IL5. It was found that the Th2 locus control regions (LCR) contributes to the expression of Th2 cytokines through the formation of long-range chromatin loops with the promoter regions (140). GATA3 collaborates with STAT6 and YY1 to regulate the chromatin interaction of the Th2 cytokine locus (141, 142). Interestingly, interchromosomal interactions between Th2 LCR (on chromosome 11) and the promoter of Ifng (on chromosome 10) or IL-17 (on chromosome 1) were discovered by 3C and FISH analysis (143, 144). These results suggest long-range chromatin interactions between functionally important genes may play a role in coordinating the expression of key cytokines in the T cell fate decision. The basic leucine zipper transcription factor, BATF, is one of the key transcription factors for the differentiation of Th17 and Tfh cells. A recent study using Hi-C and 3C assays demonstrated that BATF, cooperating with ETS1, recruits CTCF to lineage-specific genes and reorganizes chromatin landscapes in developing effector cells (145). By using a combination of ChIP-seq, Hi-C and CRISPR-mediated genomic editing, our lab discovered a novel transcriptional regulatory element about 8.5 kb of upstream of the Foxp3 gene promoter for the expression of FOXP3 in Treg cells (146). The data revealed that this element interacts with the Foxp3 promoter and intronic enhancers and its deletion impaired the expression of FOXP3. Interestingly, the histone methyltransferase MLL4 binds to this upstream regulatory region and remotely regulates the H3K4 methylation at the promoter and intronic enhancer regions via long-distance chromatin looping (Figure 4), which exemplifies that a chromatin modification enzyme can modify a chromatin region from a distant binding site through long-distance chromatin interactions. The differentiation of T cells is regulated by not only transcription factors, but also by cytokines. By applying ChIA-PET, Li et al. found that IL-2 activated STAT5, which then regulates the chromatin looping between super-enhancers and promoter of Il2ra gene (147).
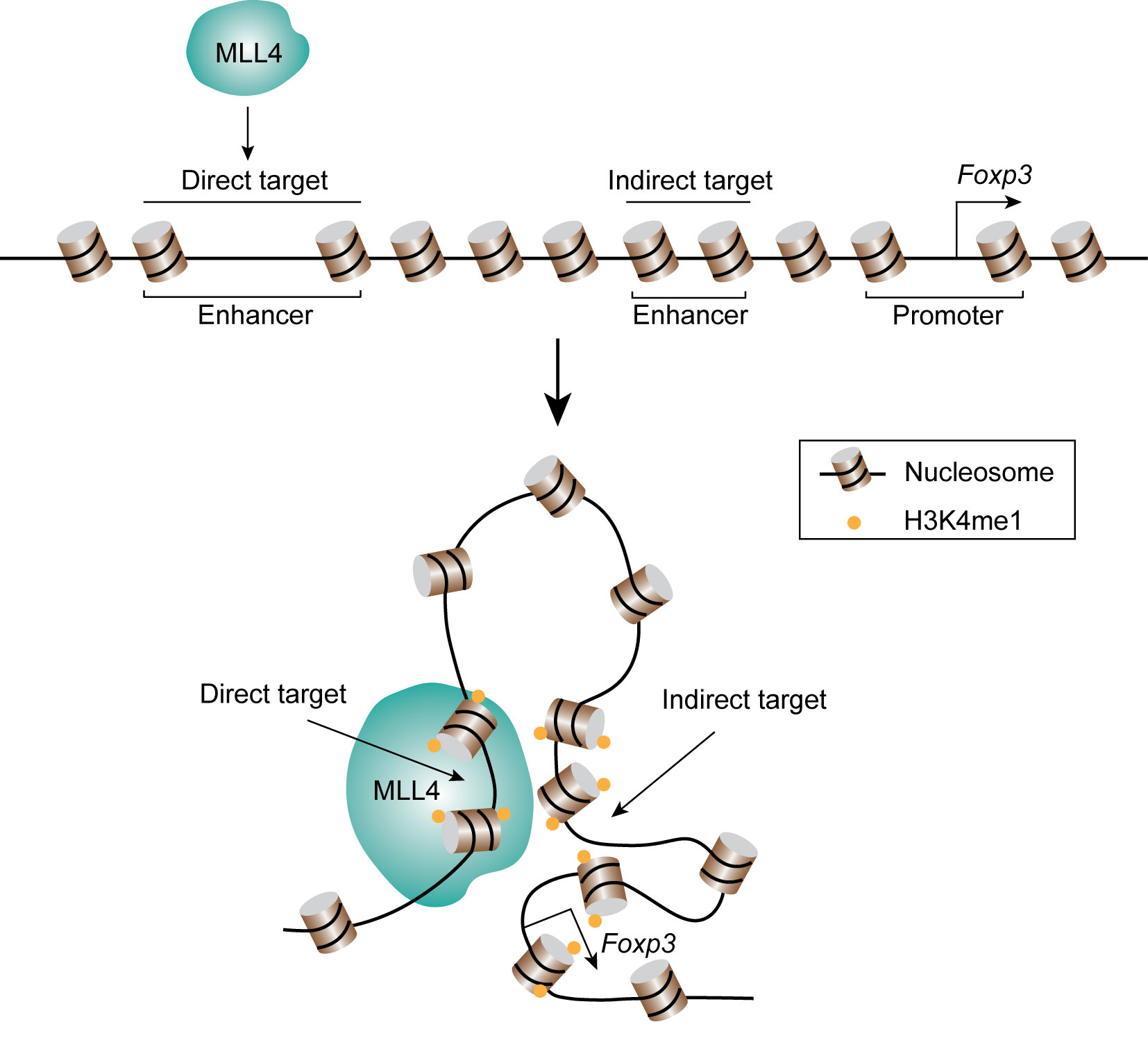
Figure 4 MLL4 catalyzes H3K4 methylation through chromatin looping. MLL4 binds its direct target region, and the lysine 4 of histone H3 at distal indirect target regions are also methylated through chromatin looping.
Mutations in key transcription factors may hamper the differentiation of corresponding T cells, leading to diseases caused by abnormal immune activity. For example, a M370I mutation in FOXP3, identified in an IPEX (immune dysregulation polyendocrinopathy enteropathy X-linked) patient, led to increased chromatin interaction and de-repression of the Th2 cytokine locus in Treg cells in mutation-recapitulated mice as revealed by H3K27ac HiChIP assays (148). These abnormal Treg cells were unable to suppress extrinsic Th2 cells and the mice developed an autoimmune syndrome consistent with an unrestrained T helper type 2 (Th2) immune response. In addition to gene coding regions, mutations in regulatory elements may also alter protein expression and result in the onset of immune diseases. Methods for untangling 3D chromatin structure, such as HiChIP and Hi-C, have been applied to investigate the connection between alternation of the regulome and diseases (49, 79, 149). For example, by applying HiChIP to T cells from type 1 diabetes patients or mouse models, Fasolino et al. discovered the formation of chromatin loops between enhancers and promoters at diabetes risk-conferring loci, indicating that genetic variation leading to the alternation of chromatin structure and gene expression may result in the pathogenesis of autoimmune disorders (150). Widespread alternations in TAD boundary and intra-TAD chromatin interactions were found in T cells of T cell acute lymphoblastic leukemia (T-ALL) patients using Hi-C assays (151). These studies suggest that 3D chromatin structure may critically contribute to the pathogenesis of human diseases and elucidation of the structural changes and their regulation may provide novel insights into the diseases.
Future Perspectives
In this review, we summarized the approaches for elucidating 3D genome organization, with emphasis on their applications in the immune system. Studies using these tools have advanced our understanding of how genome architecture such as TADs and A/B compartments contributes to the development, differentiation and function of various immune cells. However, many techniques need further improvements in sensitivity and resolution and decreasing background noise. Hi-C related assays could provide high resolution data, which may even reveal promoter-enhancer interactions; however, the sequencing cost is prohibitory to reach its full capacity. Since promoter-enhancer interactions are critical to cell-specific gene expression, highly sensitive and less costly methods for this purpose will be instrumental for elucidating this aspect of chromatin looping. In this regard, TrAC-looping is a method of choice, which specifically reveals interactions among chromatin regulatory elements and thus requires only very modest sequencing depth to cover the entire genome. However, the current TrAC-looping protocol requires 50 to 100 million cells for one experiment, which may be prohibitory for many types of primary cells or clinical samples. A more sensitive TrAC-looping protocol requiring fewer cells is highly demanded.
Genome organization data from bulk cells provides average images of genome architecture of many cells. More studies are demanded to address unique features of individual cells and elucidate how the cellular heterogeneity impacts cellular differentiation and function. Current single cell Hi-C and Dip-C assays suffer from limitations including low efficiency and resolution. To study the genome organization at a single cell level, more versatile approaches are required. For imaging-based approaches, improvements in throughput and resolution should be made to visualize the dynamics of chromatin structure in single cell. With new technical improvement and development, genome architecture, particularly promoter-enhancer interactions, can be analyzed using rare primary cells or patient samples, which will provide more insights into the contribution and mechanisms of chromatin looping in the normal development of immune cells and pathogenic immune disorders.
Author Contributions
SL and KZ conceived the review idea and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Acknowledgments
The research in the authors’ laboratory is supported by Division of Intramural Research, National Heart, Lung, and Blood Institute, National Institutes of Health.
References
1. Zheng H, Xie W. The Role of 3D Genome Organization in Development and Cell Differentiation. Nat Rev Mol Cell Biol (2019) 20:535–50. doi: 10.1038/s41580-019-0132-4
2. Stadhouders R, Filion GJ, Graf T. Transcription Factors and 3D Genome Conformation in Cell-Fate Decisions. Nature (2019) 569:345–54. doi: 10.1038/s41586-019-1182-7
3. Zabidi MA, Stark A. Regulatory Enhancer-Core-Promoter Communication Via Transcription Factors and Cofactors. Trends Genet (2016) 32:801–14. doi: 10.1016/j.tig.2016.10.003
4. Schoenfelder S, Fraser P. Long-Range Enhancer-Promoter Contacts in Gene Expression Control. Nat Rev Genet (2019) 20:437–55. doi: 10.1038/s41576-019-0128-0
5. Robson MI, Ringel AR, Mundlos S. Regulatory Landscaping: How Enhancer-Promoter Communication Is Sculpted in 3D. Mol Cell (2019) 74:1110–22. doi: 10.1016/j.molcel.2019.05.032
6. Andersson R, Sandelin A. Determinants of Enhancer and Promoter Activities of Regulatory Elements. Nat Rev Genet (2020) 21:71–87. doi: 10.1038/s41576-019-0173-8
7. Bonev B, Cavalli G. Organization and Function of the 3D Genome. Nat Rev Genet (2016) 17:661–78. doi: 10.1038/nrg.2016.112
8. Rowley MJ, Corces VG. Organizational Principles of 3D Genome Architecture. Nat Rev Genet (2018) 19:789–800. doi: 10.1038/s41576-018-0060-8
9. Gibcus JH, Dekker J. The Hierarchy of the 3D Genome. Mol Cell (2013) 49:773–82. doi: 10.1016/j.molcel.2013.02.011
10. Misteli T. The Self-Organizing Genome: Principles of Genome Architecture and Function. Cell (2020) 183:28–45. doi: 10.1016/j.cell.2020.09.014
11. Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal Structure of the Nucleosome Core Particle At 2.8 A Resolution. Nature (1997) 389:251–60. doi: 10.1038/38444
12. Naumova N, Imakaev M, Fudenberg G, Zhan Y, Lajoie BR, Mirny LA, et al. Organization of the Mitotic Chromosome. Science (2013) 342:948–53. doi: 10.1126/science.1236083
13. Sanborn AL, Rao SS, Huang SC, Durand NC, Huntley MH, Jewett AI, et al. Chromatin Extrusion Explains Key Features of Loop and Domain Formation in Wild-Type and Engineered Genomes. Proc Natl Acad Sci USA (2015) 112:E6456–65. doi: 10.1073/pnas.1518552112
14. Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, Mirny LA. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep (2016) 15:2038–49. doi: 10.1016/j.celrep.2016.04.085
15. Davidson IF, Bauer B, Goetz D, Tang W, Wutz G, Peters JM. DNA Loop Extrusion by Human Cohesin. Science (2019) 366:1338–45. doi: 10.1126/science.aaz3418
16. Ganji M, Shaltiel IA, Bisht S, Kim E, Kalichava A, Haering CH, et al. Real-Time Imaging of DNA Loop Extrusion by Condensin. Science (2018) 360:102–5. doi: 10.1126/science.aar7831
17. Vian L, Pekowska A, Rao SSP, Kieffer-Kwon KR, Jung S, Baranello L, et al. The Energetics and Physiological Impact of Cohesin Extrusion. Cell (2018) 173:1165–78.e20. doi: 10.1016/j.cell.2018.03.072
18. Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, et al. Three-Dimensional Folding and Functional Organization Principles of the Drosophila Genome. Cell (2012) 148:458–72. doi: 10.1016/j.cell.2012.01.010
19. Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, et al. Spatial Partitioning of the Regulatory Landscape of the X-inactivation Centre. Nature (2012) 485:381–5. doi: 10.1038/nature11049
20. Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, et al. Topological Domains in Mammalian Genomes Identified by Analysis of Chromatin Interactions. Nature (2012) 485:376–80. doi: 10.1038/nature11082
21. Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU, et al. Crispr Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. Cell (2015) 162:900–10. doi: 10.1016/j.cell.2015.07.038
22. Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, et al. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains From Genomic Compartmentalization. Cell (2017) 169:930–44.e22. doi: 10.1016/j.cell.2017.05.004
23. Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, et al. A 3D Map of the Human Genome At Kilobase Resolution Reveals Principles of Chromatin Looping. Cell (2014) 159:1665–80. doi: 10.1016/j.cell.2014.11.021
24. Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science (2009) 326:289–93. doi: 10.1126/science.1181369
25. Han Z, Cui K, Placek K, Hong N, Lin C, Chen W, et al. Diploid Genome Architecture Revealed by Multi-Omic Data of Hybrid Mice. Genome Res (2020) 30:1097–106. doi: 10.1101/gr.257568.119
26. Wang Z, Gerstein M, Snyder M. Rna-Seq: A Revolutionary Tool for Transcriptomics. Nat Rev Genet (2009) 10:57–63. doi: 10.1038/nrg2484
27. Park PJ. ChIP-seq: Advantages and Challenges of a Maturing Technology. Nat Rev Genet (2009) 10:669–80. doi: 10.1038/nrg2641
28. Furey TS. ChIP-seq and Beyond: New and Improved Methodologies to Detect and Characterize protein-DNA Interactions. Nat Rev Genet (2012) 13:840–52. doi: 10.1038/nrg3306
29. Klemm SL, Shipony Z, Greenleaf WJ. Chromatin Accessibility and the Regulatory Epigenome. Nat Rev Genet (2019) 20:207–20. doi: 10.1038/s41576-018-0089-8
30. Gall JG, Pardue ML. Formation and Detection of RNA-DNA Hybrid Molecules in Cytological Preparations. Proc Natl Acad Sci USA (1969) 63:378–83. doi: 10.1073/pnas.63.2.378
31. Bayani J, Squire JA. Fluorescence in Situ Hybridization (Fish). Curr Protoc Cell Biol Chapter (2004) 22:Unit 22 4 doi: 10.1002/0471143030.cb2204s23.
32. Beliveau BJ, Joyce EF, Apostolopoulos N, Yilmaz F, Fonseka CY, McCole RB, et al. Versatile Design and Synthesis Platform for Visualizing Genomes With Oligopaint FISH Probes. Proc Natl Acad Sci USA (2012) 109:21301–6. doi: 10.1073/pnas.1213818110
33. Beliveau BJ, Boettiger AN, Avendano MS, Jungmann R, McCole RB, Joyce EF, et al. Single-Molecule Super-Resolution Imaging of Chromosomes and in Situ Haplotype Visualization Using Oligopaint FISH Probes. Nat Commun (2015) 6:7147. doi: 10.1038/ncomms8147
34. Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, et al. Solution Hybrid Selection With Ultra-Long Oligonucleotides for Massively Parallel Targeted Sequencing. Nat Biotechnol (2009) 27:182–9. doi: 10.1038/nbt.1523
35. Huang B, Wang W, Bates M, Zhuang X. Three-Dimensional Super-Resolution Imaging by Stochastic Optical Reconstruction Microscopy. Science (2008) 319:810–3. doi: 10.1126/science.1153529
36. Wang S, Su JH, Beliveau BJ, Bintu B, Moffitt JR, Wu CT, et al. Spatial Organization of Chromatin Domains and Compartments in Single Chromosomes. Science (2016) 353:598–602. doi: 10.1126/science.aaf8084
37. Boettiger AN, Bintu B, Moffitt JR, Wang S, Beliveau BJ, Fudenberg G, et al. Super-Resolution Imaging Reveals Distinct Chromatin Folding for Different Epigenetic States. Nature (2016) 529:418–22. doi: 10.1038/nature16496
38. Ma H, Tu LC, Naseri A, Huisman M, Zhang S, Grunwald D, et al. Multiplexed Labeling of Genomic Loci With dCas9 and Engineered sgRNAs Using Crisprainbow. Nat Biotechnol (2016) 34:528–30. doi: 10.1038/nbt.3526
39. Shaban HA, Seeber A. Monitoring the Spatio-Temporal Organization and Dynamics of the Genome. Nucleic Acids Res (2020) 48:3423–34. doi: 10.1093/nar/gkaa135
40. Shim SH. Super-Resolution Microscopy of Genome Organization. Genes Genomics (2021) 43:281–7. doi: 10.1007/s13258-021-01044-9
41. Botchway SW, Farooq S, Sajid A, Robinson IK, Yusuf M. Contribution of Advanced Fluorescence Nano Microscopy Towards Revealing Mitotic Chromosome Structure. Chromosome Res (2021) 29:19–36. doi: 10.1007/s10577-021-09654-5
42. Nagano T, Varnai C, Schoenfelder S, Javierre BM, Wingett SW, Fraser P. Comparison of Hi-C Results Using in-Solution Versus in-Nucleus Ligation. Genome Biol (2015) 16:175. doi: 10.1186/s13059-015-0753-7
43. Ren G, Jin W, Cui K, Rodrigez J, Hu G, Zhang Z, et al. Ctcf-Mediated Enhancer-Promoter Interaction Is a Critical Regulator of Cell-to-Cell Variation of Gene Expression. Mol Cell (2017) 67:1049–58.e6. doi: 10.1016/j.molcel.2017.08.026
44. Ma W, Ay F, Lee C, Gulsoy G, Deng X, Cook S, et al. Fine-Scale Chromatin Interaction Maps Reveal the Cis-Regulatory Landscape of Human lincRNA Genes. Nat Methods (2015) 12:71–8. doi: 10.1038/nmeth.3205
45. Hsieh TS, Cattoglio C, Slobodyanyuk E, Hansen AS, Rando OJ, Tjian R, et al. Resolving the 3D Landscape of Transcription-Linked Mammalian Chromatin Folding. Mol Cell (2020) 78:539–53.e8. doi: 10.1016/j.molcel.2020.03.002
46. Krietenstein N, Abraham S, Venev SV, Abdennur N, Gibcus J, Hsieh TS, et al. Ultrastructural Details of Mammalian Chromosome Architecture. Mol Cell (2020) 78:554–65.e7. doi: 10.1016/j.molcel.2020.03.003
47. Hu G, Cui K, Fang D, Hirose S, Wang X, Wangsa D, et al. Transformation of Accessible Chromatin and 3D Nucleome Underlies Lineage Commitment of Early T Cells. Immunity (2018) 48:227–42.e8. doi: 10.1016/j.immuni.2018.01.013
48. Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, et al. A High-Resolution Map of the Three-Dimensional Chromatin Interactome in Human Cells. Nature (2013) 503:290–4. doi: 10.1038/nature12644
49. Mumbach MR, Satpathy AT, Boyle EA, Dai C, Gowen BG, Cho SW, et al. Enhancer Connectome in Primary Human Cells Identifies Target Genes of Disease-Associated DNA Elements. Nat Genet (2017) 49:1602–12. doi: 10.1038/ng.3963
50. Tang Z, Luo OJ, Li X, Zheng M, Zhu JJ, Szalaj P, et al. Ctcf-Mediated Human 3d Genome Architecture Reveals Chromatin Topology for Transcription. Cell (2015) 163:1611–27. doi: 10.1016/j.cell.2015.11.024
51. Jiang S, Zhou H, Liang J, Gerdt C, Wang C, Ke L, et al. The Epstein-Barr Virus Regulome in Lymphoblastoid Cells. Cell Host Microbe (2017) 22(4):561–73.e4. doi: 10.1016/j.chom.2017.09.001
52. Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, et al. Single-Cell Hi-C Reveals Cell-to-Cell Variability in Chromosome Structure. Nature (2013) 502:59–64. doi: 10.1038/nature12593
53. Ramani V, Deng X, Qiu R, Gunderson KL, Steemers FJ, Disteche CM, et al. Massively Multiplex Single-Cell Hi-C. Nat Methods (2017) 14:263–6. doi: 10.1038/nmeth.4155
54. Stevens TJ, Lando D, Basu S, Atkinson LP, Cao Y, Lee SF, et al. 3D Structures of Individual Mammalian Genomes Studied by Single-Cell Hi-C. Nature (2017) 544:59–64. doi: 10.1038/nature21429
55. Tan L, Xing D, Chang CH, Li H, Xie XS. Three-Dimensional Genome Structures of Single Diploid Human Cells. Science (2018) 361:924–8. doi: 10.1126/science.aat5641
56. Hughes JR, Roberts N, McGowan S, Hay D, Giannoulatou E, Lynch M, et al. Analysis of Hundreds of Cis-Regulatory Landscapes At High Resolution in a Single, High-Throughput Experiment. Nat Genet (2014) 46:205–12. doi: 10.1038/ng.2871
57. Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, et al. An Oestrogen-Receptor-Alpha-Bound Human Chromatin Interactome. Nature (2009) 462:58–64 doi: 10.1038/nature08497.
58. Johanson TM, Lun ATL, Coughlan HD, Tan T, Smyth GK, Nutt SL, et al. Transcription-Factor-Mediated Supervision of Global Genome Architecture Maintains B Cell Identity. Nat Immunol (2018) 19:1257–64. doi: 10.1038/s41590-018-0234-8
59. Schatz DG, Ji Y. Recombination Centres and the Orchestration of V(D)J Recombination. Nat Rev Immunol (2011) 11:251–63. doi: 10.1038/nri2941
60. Miyazaki K, Watanabe H, Yoshikawa G, Chen K, Hidaka R, Aitani Y, et al. The Transcription Factor E2A Activates Multiple Enhancers That Drive Rag Expression in Developing T and B Cells. Sci Immunol (2020) 5:eabb1455. doi: 10.1126/sciimmunol.abb1455
61. Kosak ST, Skok JA, Medina KL, Riblet R, Le Beau MM, Fisher AG, et al. Subnuclear Compartmentalization of Immunoglobulin Loci During Lymphocyte Development. Science (2002) 296:158–62. doi: 10.1126/science.1068768
62. Sayegh CE, Jhunjhunwala S, Riblet R, Murre C. Visualization of Looping Involving the Immunoglobulin Heavy-Chain Locus in Developing B Cells. Genes Dev (2005) 19:322–7. doi: 10.1101/gad.1254305
63. Jhunjhunwala S, van Zelm MC, Peak MM, Cutchin S, Riblet R, van Dongen JJ, et al. The 3D Structure of the Immunoglobulin Heavy-Chain Locus: Implications for Long-Range Genomic Interactions. Cell (2008) 133:265–79. doi: 10.1016/j.cell.2008.03.024
64. Speicher MR, Gwyn Ballard S, Ward DC. Karyotyping Human Chromosomes by Combinatorial Multi-Fluor FISH. Nat Genet (1996) 12:368–75. doi: 10.1038/ng0496-368
65. Levsky JM, Singer RH. Fluorescence in Situ Hybridization: Past, Present and Future. J Cell Sci (2003) 116:2833–8. doi: 10.1242/jcs.00633
66. Rust MJ, Bates M, Zhuang X. Sub-Diffraction-Limit Imaging by Stochastic Optical Reconstruction Microscopy (STORM). Nat Methods (2006) 3:793–5. doi: 10.1038/nmeth929
67. Bintu B, Mateo LJ, Su JH, Sinnott-Armstrong NA, Parker M, Kinrot S, et al. Super-Resolution Chromatin Tracing Reveals Domains and Cooperative Interactions in Single Cells. Science (2018) 362:eaau1783. doi: 10.1126/science.aau1783
68. Su JH, Zheng P, Kinrot SS, Bintu B, Zhuang X. Genome-Scale Imaging of the 3D Organization and Transcriptional Activity of Chromatin. Cell (2020) 182:1641–59.e26. doi: 10.1016/j.cell.2020.07.032
69. Ma H, Reyes-Gutierrez P, Pederson T. Visualization of Repetitive DNA Sequences in Human Chromosomes With Transcription Activator-Like Effectors. Proc Natl Acad Sci USA (2013) 110:21048–53. doi: 10.1073/pnas.1319097110
70. Kubalova I, Nemeckova A, Weisshart K, Hribova E, Schubert V. Comparing Super-Resolution Microscopy Techniques to Analyze Chromosomes. Int J Mol Sci (2021) 22:1903. doi: 10.3390/ijms22041903
71. Boettiger A, Murphy S. Advances in Chromatin Imaging At Kilobase-Scale Resolution. Trends Genet (2020) 36:273–87. doi: 10.1016/j.tig.2019.12.010
72. Dekker J, Rippe K, Dekker M, Kleckner N. Capturing Chromosome Conformation. Science (2002) 295:1306–11. doi: 10.1126/science.1067799
73. Naumova N, Smith EM, Zhan Y, Dekker J. Analysis of Long-Range Chromatin Interactions Using Chromosome Conformation Capture. Methods (2012) 58:192–203. doi: 10.1016/j.ymeth.2012.07.022
74. Zhao Z, Tavoosidana G, Sjolinder M, Gondor A, Mariano P, Wang S, et al. Circular Chromosome Conformation Capture (4C) Uncovers Extensive Networks of Epigenetically Regulated Intra- and Interchromosomal Interactions. Nat Genet (2006) 38:1341–7. doi: 10.1038/ng1891
75. Simonis M, Klous P, Homminga I, Galjaard RJ, Rijkers EJ, Grosveld F, et al. High-Resolution Identification of Balanced and Complex Chromosomal Rearrangements by 4C Technology. Nat Methods (2009) 6:837–42. doi: 10.1038/nmeth.1391
76. Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, et al. Chromosome Conformation Capture Carbon Copy (5C): A Massively Parallel Solution for Mapping Interactions Between Genomic Elements. Genome Res (2006) 16:1299–309. doi: 10.1101/gr.5571506
77. Dostie J, Zhan Y, Dekker J. Chromosome Conformation Capture Carbon Copy Technology. Curr Protoc Mol Biol Chapter (2007) 21:Unit 21 14. doi: 10.1002/0471142727.mb2114s80
78. Belaghzal H, Dekker J, Gibcus JH. Hi-C 2.0: An Optimized Hi-C Procedure for High-Resolution Genome-Wide Mapping of Chromosome Conformation. Methods (2017) 123:56–65. doi: 10.1016/j.ymeth.2017.04.004
79. Mifsud B, Tavares-Cadete F, Young AN, Sugar R, Schoenfelder S, Ferreira L, et al. Mapping Long-Range Promoter Contacts in Human Cells With High-Resolution Capture Hi-C. Nat Genet (2015) 47:598–606. doi: 10.1038/ng.3286
80. Schoenfelder S, Javierre BM, Furlan-Magaril M, Wingett SW, Fraser P. Promoter Capture Hi-C: High-resolution, Genome-Wide Profiling of Promoter Interactions. J Vis Exp (2018) 136:57320. doi: 10.3791/57320
81. Dryden NH, Broome LR, Dudbridge F, Johnson N, Orr N, Schoenfelder S, et al. Unbiased Analysis of Potential Targets of Breast Cancer Susceptibility Loci by Capture Hi-C. Genome Res (2014) 24:1854–68. doi: 10.1101/gr.175034.114
82. Kieffer-Kwon KR, Tang Z, Mathe E, Qian J, Sung MH, Li G, et al. Interactome Maps of Mouse Gene Regulatory Domains Reveal Basic Principles of Transcriptional Regulation. Cell (2013) 155:1507–20. doi: 10.1016/j.cell.2013.11.039
83. Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, et al. HiChIP: Efficient and Sensitive Analysis of Protein-Directed Genome Architecture. Nat Methods (2016) 13:919–22. doi: 10.1038/nmeth.3999
84. Chepelev I, Wei G, Wangsa D, Tang Q, Zhao K. Characterization of Genome-Wide Enhancer-Promoter Interactions Reveals Co-Expression of Interacting Genes and Modes of Higher Order Chromatin Organization. Cell Res (2012) 22:490–503. doi: 10.1038/cr.2012.15
85. Nagano T, Lubling Y, Varnai C, Dudley C, Leung W, Baran Y, et al. Cell-Cycle Dynamics of Chromosomal Organization At Single-Cell Resolution. Nature (2017) 547:61–7. doi: 10.1038/nature23001
86. Flyamer IM, Gassler J, Imakaev M, Brandao HB, Ulianov SV, Abdennur N, et al. Single-Nucleus Hi-C Reveals Unique Chromatin Reorganization At Oocyte-to-Zygote Transition. Nature (2017) 544:110–4. doi: 10.1038/nature21711
87. Tan L, Xing D, Daley N, Xie XS. Three-Dimensional Genome Structures of Single Sensory Neurons in Mouse Visual and Olfactory Systems. Nat Struct Mol Biol (2019) 26:297–307. doi: 10.1038/s41594-019-0205-2
88. Tan L, Ma W, Wu H, Zheng Y, Xing D, Chen R, et al. Changes in Genome Architecture and Transcriptional Dynamics Progress Independently of Sensory Experience During Post-Natal Brain Development. Cell (2021) 184:741–58.e17. doi: 10.1016/j.cell.2020.12.032
89. Brant L, Georgomanolis T, Nikolic M, Brackley CA, Kolovos P, van Ijcken W, et al. Exploiting Native Forces to Capture Chromosome Conformation in Mammalian Cell Nuclei. Mol Syst Biol (2016) 12:891. doi: 10.15252/msb.20167311
90. Belaghzal H, Borrman T, Stephens AD, Lafontaine DL, Venev SV, Weng Z, et al. Liquid Chromatin Hi-C Characterizes Compartment-Dependent Chromatin Interaction Dynamics. Nat Genet (2021) 53:367–78. doi: 10.1038/s41588-021-00784-4
91. Weiterer SS, Meier-Soelch J, Georgomanolis T, Mizi A, Beyerlein A, Weiser H, et al. Distinct IL-1alpha-responsive Enhancers Promote Acute and Coordinated Changes in Chromatin Topology in a Hierarchical Manner. EMBO J (2020) 39:e101533. doi: 10.15252/embj.2019101533
92. Olivares-Chauvet P, Mukamel Z, Lifshitz A, Schwartzman O, Elkayam NO, Lubling Y, et al. Capturing Pairwise and Multi-Way Chromosomal Conformations Using Chromosomal Walks. Nature (2016) 540:296–300. doi: 10.1038/nature20158
93. Allahyar A, Vermeulen C, Bouwman BAM, Krijger PHL, Verstegen M, Geeven G, et al. Enhancer Hubs and Loop Collisions Identified From Single-Allele Topologies. Nat Genet (2018) 50:1151–60. doi: 10.1038/s41588-018-0161-5
94. Oudelaar AM, Davies JOJ, Hanssen LLP, Telenius JM, Schwessinger R, Liu Y, et al. Single-Allele Chromatin Interactions Identify Regulatory Hubs in Dynamic Compartmentalized Domains. Nat Genet (2018) 50:1744–51. doi: 10.1038/s41588-018-0253-2
95. Tavares-Cadete F, Norouzi D, Dekker B, Liu Y, Dekker J. Multi-Contact 3C Reveals That the Human Genome During Interphase is Largely Not Entangled. Nat Struct Mol Biol (2020) 27:1105–14. doi: 10.1038/s41594-020-0506-5
96. Gavrilov AA, Chetverina HV, Chermnykh ES, Razin SV, Chetverin AB. Quantitative Analysis of Genomic Element Interactions by Molecular Colony Technique. Nucleic Acids Res (2014) 42:e36. doi: 10.1093/nar/gkt1322
97. Ay F, Vu TH, Zeitz MJ, Varoquaux N, Carette JE, Vert JP, et al. Identifying Multi-Locus Chromatin Contacts in Human Cells Using Tethered Multiple 3C. BMC Genomics (2015) 16:121. doi: 10.1186/s12864-015-1236-7
98. Morris KV, Mattick JS. The Rise of Regulatory RNA. Nat Rev Genet (2014) 15:423–37. doi: 10.1038/nrg3722
99. Sridhar B, Rivas-Astroza M, Nguyen TC, Chen W, Yan Z, Cao X, et al. Systematic Mapping of RNA-Chromatin Interactions In Vivo. Curr Biol (2017) 27:602–9. doi: 10.1016/j.cub.2017.01.011
100. Li X, Zhou B, Chen L, Gou LT, Li H, Fu XD. GRID-Seq Reveals the Global RNA-chromatin Interactome. Nat Biotechnol (2017) 35:940–50. doi: 10.1038/nbt.3968
101. Bell JC, Jukam D, Teran NA, Risca VI, Smith OK, Johnson WL, et al. Chromatin-Associated RNA Sequencing (ChAR-seq) Maps Genome-Wide RNA-to-DNA Contacts. Elife (2018) 7:e27024. doi: 10.7554/eLife.27024
102. Bonetti A, Agostini F, Suzuki AM, Hashimoto K, Pascarella G, Gimenez J, et al. RADICL-Seq Identifies General and Cell Type-Specific Principles of Genome-Wide RNA-chromatin Interactions. Nat Commun (2020) 11:1018. doi: 10.1101/681924
103. Beagrie RA, Scialdone A, Schueler M, Kraemer DC, Chotalia M, Xie SQ, et al. Complex Multi-Enhancer Contacts Captured by Genome Architecture Mapping. Nature (2017) 543:519–24. doi: 10.1038/nature21411
104. Quinodoz SA, Ollikainen N, Tabak B, Palla A, Schmidt JM, Detmar E, et al. Higher-Order Inter-chromosomal Hubs Shape 3d Genome Organization in the Nucleus. Cell (2018) 174:744–57.e24. doi: 10.1016/j.cell.2018.05.024
105. Vangala P, Murphy R, Quinodoz SA, Gellatly K, McDonel P, Guttman M, et al. High-Resolution Mapping of Multiway Enhancer-Promoter Interactions Regulating Pathogen Detection. Mol Cell (2020) 80(2):359–73.e8. doi: 10.1016/j.molcel.2020.09.005
106. Zheng M, Tian SZ, Capurso D, Kim M, Maurya R, Lee B, et al. Multiplex Chromatin Interactions With Single-Molecule Precision. Nature (2019) 566:558–62. doi: 10.1038/s41586-019-0949-1
107. Redolfi J, Zhan Y, Valdes-Quezada C, Kryzhanovska M, Guerreiro I, Iesmantavicius V, et al. Damc Reveals Principles of Chromatin Folding In Vivo Without Crosslinking and Ligation. Nat Struct Mol Biol (2019) 26:471–80. doi: 10.1038/s41594-019-0231-0
108. Lai B, Tang Q, Jin W, Hu G, Wangsa D, Cui K, et al. Trac-Looping Measures Genome Structure and Chromatin Accessibility. Nat Methods (2018) 15:741–7. doi: 10.1038/s41592-018-0107-y
109. Lin YC, Benner C, Mansson R, Heinz S, Miyazaki K, Miyazaki M, et al. Global Changes in the Nuclear Positioning of Genes and Intra- and Interdomain Genomic Interactions That Orchestrate B Cell Fate. Nat Immunol (2012) 13:1196–204. doi: 10.1038/ni.2432
110. Zhang Y, Zhang X, Ba Z, Liang Z, Dring EW, Hu H, et al. The Fundamental Role of Chromatin Loop Extrusion in Physiological V(D)J Recombination. Nature (2019) 573:600–4. doi: 10.1038/s41586-019-1547-y
111. Guo C, Gerasimova T, Hao H, Ivanova I, Chakraborty T, Selimyan R, et al. Two Forms of Loops Generate the Chromatin Conformation of the Immunoglobulin Heavy-Chain Gene Locus. Cell (2011) 147:332–43. doi: 10.1016/j.cell.2011.08.049
112. Lucas JS, Zhang Y, Dudko OK, Murre C. 3D Trajectories Adopted by Coding and Regulatory DNA Elements: First-Passage Times for Genomic Interactions. Cell (2014) 158:339–52. doi: 10.1016/j.cell.2014.05.036
113. Medvedovic J, Ebert A, Tagoh H, Tamir IM, Schwickert TA, Novatchkova M, et al. Flexible Long-Range Loops in the VH Gene Region of the Igh Locus Facilitate the Generation of a Diverse Antibody Repertoire. Immunity (2013) 39:229–44. doi: 10.1016/j.immuni.2013.08.011
114. Degner SC, Verma-Gaur J, Wong TP, Bossen C, Iverson GM, Torkamani A, et al. CCCTC-Binding Factor (CTCF) and Cohesin Influence the Genomic Architecture of the Igh Locus and Antisense Transcription in Pro-B Cells. Proc Natl Acad Sci USA (2011) 108:9566–71. doi: 10.1073/pnas.1019391108
115. Degner SC, Wong TP, Jankevicius G, Feeney AJ. Cutting Edge: Developmental Stage-Specific Recruitment of Cohesin to CTCF Sites Throughout Immunoglobulin Loci During B Lymphocyte Development. J Immunol (2009) 182:44–8. doi: 10.4049/jimmunol.182.1.44
116. Guo C, Yoon HS, Franklin A, Jain S, Ebert A, Cheng HL, et al. CTCF-Binding Elements Mediate Control of V(D)J Recombination. Nature (2011) 477:424–30. doi: 10.1038/nature10495
117. Seitan VC, Hao B, Tachibana-Konwalski K, Lavagnolli T, Mira-Bontenbal H, Brown KE, et al. A Role for Cohesin in T-cell-receptor Rearrangement and Thymocyte Differentiation. Nature (2011) 476:467–71. doi: 10.1038/nature10312
118. Jain S, Ba Z, Zhang Y, Dai HQ, Alt FW. Ctcf-Binding Elements Mediate Accessibility of RAG Substrates During Chromatin Scanning. Cell (2018) 174:102–116 e14. doi: 10.1016/j.cell.2018.04.035
119. Zhang X, Zhang Y, Ba Z, Kyritsis N, Casellas R, Alt FW. Fundamental Roles of Chromatin Loop Extrusion in Antibody Class Switching. Nature (2019) 575:385–9. doi: 10.1038/s41586-019-1723-0
120. Ba Z, Lou J, Ye AY, Dai HQ, Dring EW, Lin SG, et al. CTCF Orchestrates Long-Range Cohesin-Driven V(D)J Recombinational Scanning. Nature (2020) 586:305–10. doi: 10.1038/s41586-020-2578-0
121. Hill L, Ebert A, Jaritz M, Wutz G, Nagasaka K, Tagoh H, et al. Wapl Repression by Pax5 Promotes V Gene Recombination by Igh Loop Extrusion. Nature (2020) 584:142–7. doi: 10.1038/s41586-020-2454-y
122. Chaudhri VK, Dienger-Stambaugh K, Wu Z, Shrestha M, Singh H. Charting the Cis-Regulome of Activated B Cells by Coupling Structural and Functional Genomics. Nat Immunol (2020) 21:210–20. doi: 10.1038/s41590-019-0565-0
123. Ikawa T, Hirose S, Masuda K, Kakugawa K, Satoh R, Shibano-Satoh A, et al. An Essential Developmental Checkpoint for Production of the T Cell Lineage. Science (2010) 329:93–6. doi: 10.1126/science.1188995
124. Li L, Leid M, Rothenberg EV. An Early T Cell Lineage Commitment Checkpoint Dependent on the Transcription Factor Bcl11b. Science (2010) 329:89–93. doi: 10.1126/science.1188989
125. Li P, Burke S, Wang J, Chen X, Ortiz M, Lee SC, et al. Reprogramming of T Cells to Natural Killer-Like Cells Upon Bcl11b Deletion. Science (2010) 329:85–9. doi: 10.1126/science.1188063
126. Isoda T, Moore AJ, He Z, Chandra V, Aida M, Denholtz M, et al. Non-Coding Transcription Instructs Chromatin Folding and Compartmentalization to Dictate Enhancer-Promoter Communication and T Cell Fate. Cell (2017) 171:103–119 e18. doi: 10.1016/j.cell.2017.09.001
127. Chen S, Luperchio TR, Wong X, Doan EB, Byrd AT, Roy Choudhury K, et al. A Lamina-Associated Domain Border Governs Nuclear Lamina Interactions, Transcription, and Recombination of the Tcrb Locus. Cell Rep (2018) 25:1729–40.e6. doi: 10.1016/j.celrep.2018.10.052
128. Jiang T, Raviram R, Snetkova V, Rocha PP, Proudhon C, Badri S, et al. Identification of Multi-Loci Hubs From 4C-Seq Demonstrates the Functional Importance of Simultaneous Interactions. Nucleic Acids Res (2016) 44:8714–25. doi: 10.1093/nar/gkw568
129. Majumder K, Rupp LJ, Yang-Iott KS, Koues OI, Kyle KE, Bassing CH, et al. Domain-Specific and Stage-Intrinsic Changes in Tcrb Conformation During Thymocyte Development. J Immunol (2015) 195:1262–72. doi: 10.4049/jimmunol.1500692
130. Zhao JY, Osipovich O, Koues OI, Majumder K, Oltz EM. Activation of Mouse Tcrb: Uncoupling Runx1 Function From Its Cooperative Binding With ETS1. J Immunol (2017) 199:1131–41. doi: 10.4049/jimmunol.1700146
131. Wagatsuma K, Tani-ichi S, Liang B, Shitara S, Ishihara K, Abe M, et al. Stat5 Orchestrates Local Epigenetic Changes for Chromatin Accessibility and Rearrangements by Direct Binding to the TCRgamma Locus. J Immunol (2015) 195:1804–14. doi: 10.4049/jimmunol.1302456
132. Shih HY, Verma-Gaur J, Torkamani A, Feeney AJ, Galjart N, Krangel MS. Tcra Gene Recombination is Supported by a Tcra Enhancer- and CTCF-dependent Chromatin Hub. Proc Natl Acad Sci USA (2012) 109:E3493–502. doi: 10.1073/pnas.1214131109
133. Chaumeil J, Micsinai M, Ntziachristos P, Deriano L, Wang JM, Ji Y, et al. Higher-Order Looping and Nuclear Organization of Tcra Facilitate Targeted Rag Cleavage and Regulated Rearrangement in Recombination Centers. Cell Rep (2013) 3:359–70. doi: 10.1016/j.celrep.2013.01.024
134. Chen L, Carico Z, Shih HY, Krangel MS. A Discrete Chromatin Loop in the Mouse Tcra-Tcrd Locus Shapes the TCRdelta and TCRalpha Repertoires. Nat Immunol (2015) 16:1085–93. doi: 10.1038/ni.3232
135. Carico ZM, Roy Choudhury K, Zhang B, Zhuang Y, Krangel MS. Tcrd Rearrangement Redirects a Processive Tcra Recombination Program to Expand the Tcra Repertoire. Cell Rep (2017) 19:2157–73. doi: 10.1016/j.celrep.2017.05.045
136. Zhao H, Li Z, Zhu Y, Bian S, Zhang Y, Qin L, et al. A Role of the CTCF Binding Site At Enhancer Ealpha in the Dynamic Chromatin Organization of the Tcra-Tcrd Locus. Nucleic Acids Res (2020) 48:9621–36. doi: 10.1093/nar/gkaa711
137. Barski A, Cuddapah S, Kartashov AV, Liu C, Imamichi H, Yang W, et al. Rapid Recall Ability of Memory T Cells is Encoded in Their Epigenome. Sci Rep (2017) 7:39785. doi: 10.1038/srep39785
138. Sekimata M, Perez-Melgosa M, Miller SA, Weinmann AS, Sabo PJ, Sandstrom R, et al. CCCTC-Binding Factor and the Transcription Factor T-bet Orchestrate T Helper 1 Cell-Specific Structure and Function At the Interferon-Gamma Locus. Immunity (2009) 31:551–64. doi: 10.1016/j.immuni.2009.08.021
139. Hadjur S, Williams LM, Ryan NK, Cobb BS, Sexton T, Fraser P, et al. Cohesins Form Chromosomal Cis-Interactions At the Developmentally Regulated IFNG Locus. Nature (2009) 460:410–3. doi: 10.1038/nature08079
140. Lee GR, Spilianakis CG, Flavell RA. Hypersensitive Site 7 of the TH2 Locus Control Region is Essential for Expressing TH2 Cytokine Genes and for Long-Range Intrachromosomal Interactions. Nat Immunol (2005) 6:42–8. doi: 10.1038/ni1148
141. Hwang SS, Kim YU, Lee S, Jang SW, Kim MK, Koh BH, et al. Transcription Factor YY1 is Essential for Regulation of the Th2 Cytokine Locus and for Th2 Cell Differentiation. Proc Natl Acad Sci USA (2013) 110:276–81. doi: 10.1073/pnas.1214682110
142. Spilianakis CG, Flavell RA. Long-Range Intrachromosomal Interactions in the T Helper Type 2 Cytokine Locus. Nat Immunol (2004) 5:1017–27. doi: 10.1038/ni1115
143. Spilianakis CG, Lalioti MD, Town T, Lee GR, Flavell RA. Interchromosomal Associations Between Alternatively Expressed Loci. Nature (2005) 435:637–45. doi: 10.1038/nature03574
144. Kim LK, Esplugues E, Zorca CE, Parisi F, Kluger Y, Kim TH, et al. Oct-1 Regulates IL-17 Expression by Directing Interchromosomal Associations in Conjunction With CTCF in T Cells. Mol Cell (2014) 54:56–66. doi: 10.1016/j.molcel.2014.02.004
145. Pham D, Moseley CE, Gao M, Savic D, Winstead CJ, Sun M, et al. Batf Pioneers the Reorganization of Chromatin in Developing Effector T Cells Via Ets1-Dependent Recruitment of Ctcf. Cell Rep (2019) 29:1203–20.e7. doi: 10.1016/j.celrep.2019.09.064
146. Placek K, Hu G, Cui K, Zhang D, Ding Y, Lee JE, et al. MLL4 Prepares the Enhancer Landscape for Foxp3 Induction Via Chromatin Looping. Nat Immunol (2017) 18:1035–45. doi: 10.1038/ni.3812
147. Li P, Mitra S, Spolski R, Oh J, Liao W, Tang Z, et al. STAT5-Mediated Chromatin Interactions in Superenhancers Activate IL-2 Highly Inducible Genes: Functional Dissection of the Il2ra Gene Locus. Proc Natl Acad Sci USA (2017) 114:12111–9. doi: 10.1073/pnas.1714019114
148. Van Gool F, Nguyen MLT, Mumbach MR, Satpathy AT, Rosenthal WL, Giacometti S, et al. A Mutation in the Transcription Factor Foxp3 Drives T Helper 2 Effector Function in Regulatory T Cells. Immunity (2019) 50:362–77.e6. doi: 10.1016/j.immuni.2018.12.016
149. Jeng MY, Mumbach MR, Granja JM, Satpathy AT, Chang HY, Chang ALS. Enhancer Connectome Nominates Target Genes of Inherited Risk Variants From Inflammatory Skin Disorders. J Invest Dermatol (2019) 139:605–14. doi: 10.1016/j.jid.2018.09.011
150. Fasolino M, Goldman N, Wang W, Cattau B, Zhou Y, Petrovic J, et al. Genetic Variation in Type 1 Diabetes Reconfigures the 3D Chromatin Organization of T Cells and Alters Gene Expression. Immunity (2020) 52:257–74.e11. doi: 10.1016/j.immuni.2020.01.003
Keywords: three-dimensional chromatin organization, enhancer-promoter interactions, transcriptional regulation, epigenome, cell differentiation
Citation: Liu S and Zhao K (2021) The Toolbox for Untangling Chromosome Architecture in Immune Cells. Front. Immunol. 12:670884. doi: 10.3389/fimmu.2021.670884
Received: 22 February 2021; Accepted: 06 April 2021;
Published: 29 April 2021.
Edited by:
Ageliki Tsagaratou, University of North Carolina at Chapel Hill, United StatesReviewed by:
Argyris Papantonis, University Medical Center Göttingen, GermanyCharalampos Spilianakis, University of Crete, Greece
Copyright © 2021 Liu and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Keji Zhao, emhhb2tAbmhsYmkubmloLmdvdg==; Shuai Liu, c2h1YWkubGl1QG5paC5nb3Y=