 Shreya M. Kanth
Shreya M. Kanth Salina Gairhe
Salina Gairhe- Critical Care Medicine Department, Clinical Center, National Institutes of Health, Bethesda, MD, United States
Semaphorins are a group of proteins that have been studied extensively for their critical function in neuronal development. They have been shown to regulate airway development, tumorigenesis, autoimmune diseases, and the adaptive immune response. Notably, emerging literature describes the role of immunoregulatory semaphorins and their receptors, plexins and neuropilins, as modulators of innate immunity and diseases defined by acute injury to the kidneys, abdomen, heart and lungs. In this review we discuss the pathogenic functions of semaphorins in clinical conditions of acute inflammation, including sepsis and acute lung injury, with a focus on regulation of the innate immune response as well as potential future therapeutic targeting.
Introduction
The semaphorins (SEMA) are a large family of both membrane-bound and secreted proteins defined by the presence of an approximately 500-amino acid extracellular Sema domain located at the N- terminal region (1). Although semaphorins contain the Sema domain, other human proteins also can contain this domain (2). Members of the semaphorin family are subdivided into eight classes, classes 1-7 and V, based on specific domains in their carboxy-terminal regions and phylogenetic tree analysis. Classes 1 and 2 are only found in invertebrate species, classes 3-7 in vertebrates, and class V members are found in DNA viruses, including the viral semaphorin A39R (3) (Supplementary Table 1). Differing semaphorin classes vary in their membrane anchorage, including secreted and membrane-associated forms. Membrane-associated semaphorins exist as transmembrane semaphorins or linked to glycosylphosphatidylinositol (GPI). Class 2, 3, and V semaphorin members are all secreted, whereas classes 4–6 are all transmembrane proteins, and SEMA7A is linked to the plasma membrane via a GPI anchor (4–7). Notably, some of the primarily membrane-associated semaphorins may be cleaved to produce soluble proteins. Proteolytic cleavage of semaphorins to a soluble form allows for diffusion locally and systemically to various effector cells, including immune cells. More recently, a soluble form of semaphorin SEMA5A was described in the context of both pancreatic cancer and rheumatoid arthritis (8, 9). Additionally, SEMA4A and SEMA4D have been found to exist as soluble proteins after proteolytic cleavage mediated by matrix metalloproteinases (10–14).
Activation of downstream signaling pathways involving semaphorins is dependent on the binding of semaphorins to their receptors, resulting in “forward signaling”. Plexins (PLXN) and neuropilins (NRP) are the predominant receptors for semaphorins (15–17), and nine plexins (PLXNA1–PLXNA4, PLXNB1–PLXNB3, PLXNC1 and PLXND1) and two neuropilins (NRP1, NRP2) have been identified in vertebrates. Several other proteins have also been identified as participating in semaphorin signaling, including integrins, T cell immunoglobulin and mucin domain-containing protein 2 (TIM2), and CD72. Furthermore, additional co-receptors have been found to be in close association with plexins and neuropilins on the cell surface. For example, association of adhesion molecule L1-CAM with NRP1 functions as a receptor for SEMA3A and semaphorins can transactivate plexin-associated receptor tyrosine kinases OTK/PTK7, MET, RON, VEGFR2, and ERBB2 (18–23). Although the molecular mechanisms that are responsible for the varying effects of the semaphorin-plexin complex are not fully understood, early studies suggest that activation of Ras and Rho GTPases by plexin GAP has been shown to play a crucial role in cellular proliferation, adhesion and migration (4). Interestingly, transmembrane semaphorins can also function as receptors on cells expressing them to initiate “reverse signaling”. SEMA6D can act as a receptor and upon interaction with a tyrosine kinase, c-Alb, it triggers anti-inflammatory macrophage polarization by controlling fatty-acid metabolism (24). Similarly, binding of PLXNB1 to SEMA4A, which is present as a receptor on dendritic cells, has been shown to regulate migration (25). Beside this, recent studies suggest that semaphorins can act as a co-stimulatory molecule and aid in immune cell activation and regulation of the immune response. For example, dendritic cells (DC) express SEMA3A and inhibit T cell activation whereas SEMA4A binds to Tim-2 on T cells resulting in activation and proliferation. SEMA4D additionally indirectly co-stimulates T cell activity (26). Human monocyte-derived DCs express high levels of SEMA4A which through its newly identified receptor immunoglobulin-like transcript 4 (ILT-4) on CD4+ T cell was shown to stimulate T cell differentiation (10). Additionally, the high evolutionary conservation of semaphorins and plexins among different species suggests that they have fundamental roles in the regulation of multiple cellular functions (27).Taken together, these data suggest that semaphorins can trigger multiple signaling pathways in a wide range of cellular contexts to carry out distinct biological activities, including activation and regulation of immune responses.
Although semaphorins were originally identified for their involvement in neural guidance and development, they have since been found to have important roles in many pathophysiological processes. Work over the past 20 years has demonstrated the function of semaphorins and their receptors in numerous diseases including cancer (28–31), disorders of bone turnover (32–37), angiogenesis (38–42), autoimmune and rheumatologic diseases (43–50), and inflammatory conditions. In this Review, we summarize the rapidly evolving literature on the functional roles of immune semaphorins at a cellular and intercellular level, and discuss their pathogenic roles and therapeutic implications in acute inflammatory diseases and in the innate immune response.
Cellular Function of Semaphorins and Their Receptors in Acute Inflammation
Over the past decade there has been significant evolution in our understanding of immune semaphorins, with a focus on their involvement in the adaptive immune response (51). However, more recent studies have investigated their role in innate immunity including the recruitment and migration of immune cells, modulation of the function and phenotype of myeloid cells, regulation of proinflammatory cytokines, and immune cell survival (Figure 1).
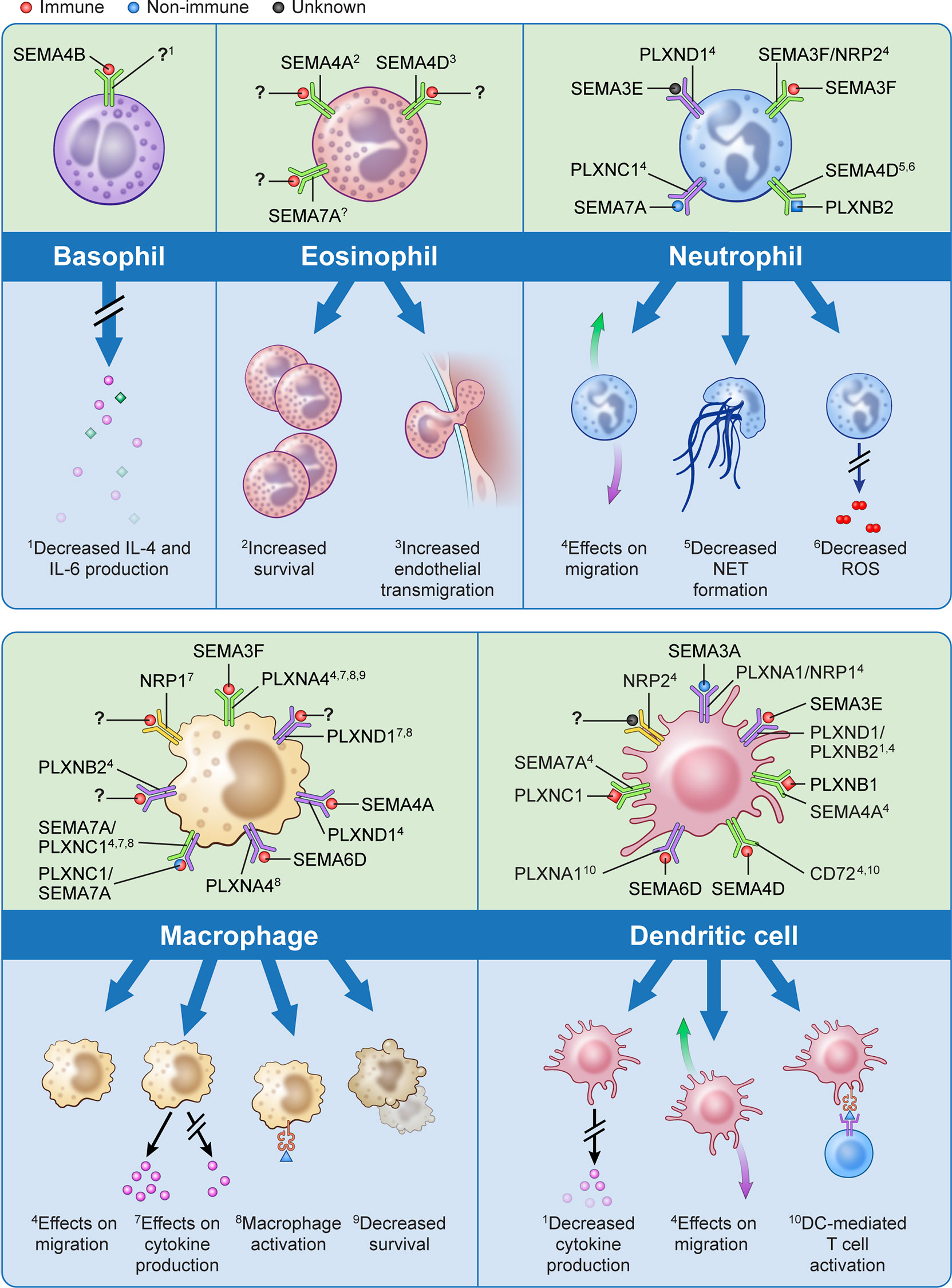
Figure 1 The effects of semaphorins and their receptors on cells of the innate immune system. Semaphorins and their binding partners in relation to innate immune cells are shown here. Ligands depicted in red have been shown to originate from immune cells, those in blue from non-immune cells, and for those depicted in black the cell of origin is unknown.? indicates unknown ligand for the depicted receptor. Superscripts are related to the processes described in the lower part of the panels for each cell type;? indicates that a specific function has not been associated with the interaction shown.
Cell migration is a fundamental process in the immune response, and extracellular guidance cues play a key role in directing this movement in an orchestrated fashion. Semaphorins have been shown to act either as attractive or repulsive cues for migration of neutrophils, macrophages, dendritic cells and eosinophils (13, 39, 44, 52–66). Structurally, both semaphorins and plexins are characterized by the presence of plexin-semaphorin-integrin (PSI) domains adjacent to the N-terminus Sema domain, and plexins also contain a GTPase-activating protein (GAP) domain and a Rho GTPase-binding domain. As such, it is thought that the binding of semaphorins to plexins activates GTPases which modulates integrin-mediated cell attachment, actomyosin contraction, and microtubule destabilization which all may influence cellular migratory abilities (22, 67, 68). For example, treatment of human peripheral blood neutrophils with exogenous SEMA3F has been shown to inhibit neutrophils from migrating towards a chemoattractant in Transwell assays. This slowing of neutrophil migration as well as increased neutrophil rounding is mediated by F-actin reorganization in a zebrafish model of inflammation (57). Conversely, exogenous SEMA3A markedly reduced neutrophil recruitment into the lungs in mouse models of airway allergen exposure (55). This highlights class and subclass differences in semaphorins may have varying and opposing effects on immune cell migration.
Macrophages are key immune effector cells in the innate immune response. They have a remarkable ability to change their phenotype following exposure to pro- or anti-inflammatory stimuli. The semaphorin-plexin axes in the presence or absence of these stimuli have shown to be potent monocyte and macrophage activators with different semaphorin classes giving rise to specific macrophage populations (13, 24, 39, 69–72). For example, while SEMA3A promotes the transition of classically activated (M1) macrophages towards a resolution phenotype, SEMA3E positively regulates macrophage activation towards M1 phenotype (71, 72).
In addition to their aforementioned functions, the expression of semaphorins and their receptors on various innate immune cells plays a role in the release of pro-inflammatory cytokines as well as in innate immune cell survival (44, 50, 73–80). This suggests potential targeted therapies involving semaphorins, plexins, and neuropilins may promote the resolution phase of acute inflammation and mitigate tissue and end-organ damage from inflammatory conditions, such as sepsis and ARDS.
Role of Semaphorins and Their Receptors in Clinical Syndromes
Translational studies and investigations using animal disease models have demonstrated the involvement of various classes of semaphorins, plexins, and neuropilins as both pro- and anti-inflammatory mediators in the pathogenesis of acute inflammation. Here we discuss the role of semaphorins in acute inflammatory clinical syndromes including sepsis, as well as diseases defined by injury to the kidneys, abdomen, heart and lungs (Figure 2).
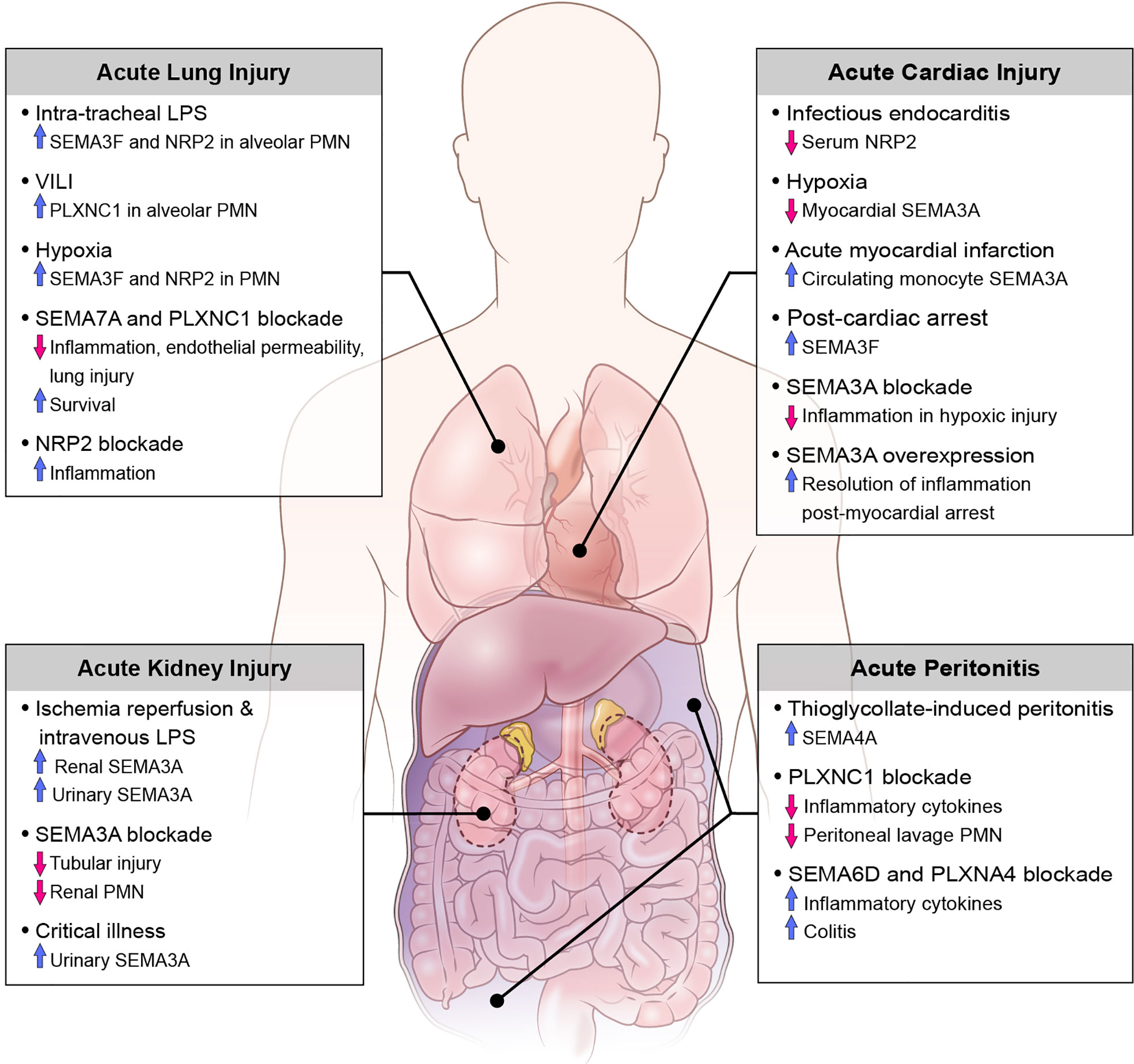
Figure 2 Role of semaphorins and their receptors in clinical conditions of acute inflammation.
Sepsis and Endotoxin-Induced Inflammation
Sepsis is a complex inflammatory syndrome characterized by a dysregulated host response to infection resulting in life-threatening organ dysfunction (81). The initial activation of the innate immune response occurs via binding of pathogen- associated molecular patterns (PAMPs), such as lipopolysaccharide (LPS) or bacterial endotoxin, to specific receptors resulting in activation of downstream signal transduction pathways, and ultimately to both pathogen and host cell and tissue damage (82).
Mouse models of sepsis by means of intraperitoneal injection of LPS reveal the role of SEMA3A, SEMA3E and their receptors PLXNA4 and PLXND1, respectively, in propagating the inflammatory cascade. Congruously, inhibition of these semaphorins and their receptors is associated with an attenuated septic response including reduced levels of proinflammatory cytokines, lower clinical scores of sepsis, and improved survival rates (71, 79, 83). For example, Wen et al. (79) demonstrate a critical role for SEMA3A and its ligand PLXNA4 in the activation and downstream signaling of the Toll-like receptor (TLR) pathways in clinical sepsis. By studying PLXNA4- deficient mice, they show that TLR4 signaling is attenuated by means of decreased inflammatory cytokine production, including TNF-α and IL-6, through intersection of PLXNA4 and TLR4 with the activation of Rac1 and JUN N-terminal kinase (JNK)-mediated nuclear factor- κB (NF-κB) activation. Furthermore, Yamashita et al. (83) explore the application of an anti-SEMA3A neutralizing antibody in mouse models of sepsis which results in increased survival after four days. Although there are no translational studies investigating the use of semaphorins to treat sepsis in humans, these studies suggest an avenue for further research and ultimately a clinical application.
On the contrary, NRP1, which has been identified as a co-receptor for SEMA3A, is shown to down-regulate the systemic inflammatory response of sepsis. In fact, in cecal ligation and puncture as well as intraperitoneal LPS models of sepsis using myeloid cell-specific NRP1-knockout mice there is increased production of pro-inflammatory cytokines (iNOS, TNF-α, IL-6) in peritoneal lavage and serum and higher mortality compared with wild-type mice (73). This suggests that there are differing roles of semaphorins and their receptors within the same class in the regulation of septic inflammation.
Acute Kidney Injury
Previous investigations have demonstrated that SEMA3A is expressed within the developing and mature kidney, specifically in the distal tubules and collecting tubules (84). More recently, reports of acute kidney injury (AKI) in mouse models of sepsis and ischemia-reperfusion describe the pathogenic role of semaphorins in the promotion of tubular epithelial cell injury and restoration of renal function. In mice treated with intra-peritoneal LPS for up to 24 hours, renal histopathology shows increased neutrophil and macrophage infiltration with apoptosis of tubular epithelial cells (TECs) and increased SEMA3A expression in proximal TECs; this injury is attenuated when SEMA3A is inhibited by (−)-epigallocatechin-3-gallate (EGCG) (85). After ischemia-reperfusion injury of the kidneys, a large increase in urinary and renal tissue SEMA3A is detected. Notably, genetic inactivation or functional inhibition of SEMA3A is renoprotective against ischemia-reperfusion renal injury in animal models revealing blunted expression of inflammatory cytokines, reduced renal neutrophil infiltration, and decreased tubular cell apoptosis (86). In a translational single-center prospective cohort study of critically ill patients admitted to an intensive care unit, levels of urinary SEMA3A on admission were significantly higher in patients with AKI compared to non-AKI patients. Additionally, urinary SEMA3A levels were significantly higher in patients with late-onset AKI and AKI progression as compared to established AKI groups and non-AKI progressors. However, urinary SEMA3A was not reflective of AKI severity and there was no difference in levels of SEMA3A in septic versus non-septic patients (87). Although there are many limitations to this study, it is important to recognize the potential use of semaphorins as biomarkers for various disease processes associated with critically ill patients, and further studies to investigate their role in septic shock-related end organ dysfunction are warranted.
Acute Peritonitis
In investigations of animal models with localized acute inflammation, as in with acute peritonitis, SEMA7A and its receptor PLXNC1, as well as SEMA4A are shown to enhance the inflammatory response. Murine models of peritonitis induced by thioglycolate demonstrate the upregulation of SEMA4A on peritoneal myeloid precursors (CD11b+ and CD68+ cells), and SEMA4A is significantly upregulated specifically in inflammatory subsets of circulating monocytes (Ly6Chigh) whose function is to selectively traffic to sites of tissue damage (39). Similarly, in zymosan A (ZyA)- induced peritonitis in mice, genetic inactivation and functional inhibition of PLXNC1 results in reduced peritoneal lavage granulocyte counts, protein content, MPO activity, and cytokine levels (TNF-a, IL-6, MIP-1-a, MIP-2). Furthermore, inactivation of PLXNC1 demonstrates increased leukocyte rolling, reduced adhesion and reduced infiltration into the parietal peritoneum perivascular tissue compared to wild-type mice, resulting in the dampened acute inflammatory response (88). Conversely, SEMA6D and its receptor PLXNA4 have been shown to play a critical role in the polarization of macrophages into anti-inflammatory states, and deficiency of SEMA6D in mice results in increased inflammatory cytokine production by peritoneal macrophages as well as predisposition to colitis after intraperitoneal administration of LPS and dextran sodium sulfate (DSS) (24). Overall, these findings demonstrate class specific responses of various semaphorins in the pathogenesis of acute peritonitis and may reveal opportunities for novel therapeutics.
Acute Cardiac Dysfunction
Semaphorins have previously been shown to play critical functions in cardiogenesis and blood vessel development (89), however more recently they have been explored as biomarkers for various inflammatory cardiovascular conditions in critically ill patients. In a study of patients with clinical suspicion of infectious endocarditis (IE) the investigators performed proteomic analysis of serum to identify putative biomarkers of IE by means of gel electrophoresis and mass spectrometry. Overall, 7 proteins present in at least 70% of all samples were differentially regulated in those with confirmed IE, with neuropilin 2 (NRP) significantly downregulated in IE patients. In models of hypoxia-induced myocardial inflammation SEMA3A levels are decreased in myocardiocytes, and further silencing of the SEMA3A gene is associated with reduced myocardial cell injury after hypoxia and increased resolution of inflammation by means of decreased inflammatory cytokines, decreased cardiomyocyte apoptosis and increased cell viability (90). On the contrary, after myocardial infarction there is increased expression of SEMA3A on circulating monocytes, and stimulation of classically activated monocytes with recombinant SEMA3A promotes their transition towards a resolution phenotype. Additionally, SEMA3A heterozygous mice showed worse cardiac function with decreased fractional shortening of the heart compared to wild type littermates (72). A study of patients who survived out-of-hospital cardiac arrest revealed higher serum SEMA3F levels compared to stable patients with coronary artery disease and healthy volunteers, as well as significantly elevated SEMA3F levels in patients with severe myocardial dysfunction (left ventricular ejection fraction less than 40%) compared to those with normal or mildly reduced ejection fraction. Subgroup analysis noted that elevated SEMA3F levels after return of spontaneous circulation is associated with decreased survival, myocardial dysfunction, as well as prolonged vasopressor therapy (91). More recently, Köhler et al. (92) explored the novel expression and function of SEMA7A in myocardial ischemia-reperfusion injury (MIRI). They demonstrate the increased expression of soluble SEMA7A in patients with acute myocardial infarction associated with a rise in platelet-neutrophil complexes (PNC). Given the hypothesis that SEMA7A plays a pivotal role in the progression of MIRI, the investigators demonstrated that administration of SEMA7A results in increased MIRI and PNC formation and this pathology is due to an interaction of SEMA7A with platelet glycoprotein Ib in animal models. Importantly, they also show that administration of a function-blocking anti-SEMA7A antibody before the start of reperfusion resulted in decreased infarct size and reduced troponin level. These findings have important implications towards the development of treatments that target thrombo-inflammatory MIRI, and further studies to validate these findings are clinically valuable.
Acute Lung Injury
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) describe syndromes of high morbidity and mortality in critically ill patients. The clinical hallmarks include acute onset of respiratory failure with varying degrees of hypoxemia, bilateral infiltrates as seen on chest radiograph, and the absence of cardiogenic pulmonary edema. From a more conceptual standpoint, a plethora of predisposing insults, such as sepsis, trauma, and inhalation of noxious stimuli, leads to inflammation in the lungs with resulting increased pulmonary vascular permeability and alveolar collapse. Pathological hallmarks of ALI are characterized by injury to the alveolar epithelium and endothelium, coagulopathy and activation of the innate immune response, which further amplifies lung damage (93–96). Semaphorins have emerged as key mediators of multiple mechanisms contributing to ALI including neutrophil migration, vascular permeability, cytoskeletal remodeling and the initiation of inflammatory signaling cascades, which we review here.
Neutrophils play a major role in ALI progression in both human and animal studies, and activation and transmigration of neutrophils can lead to tissue damage by release of cytotoxic and immune-cell activating mediators (97–100). SEMA3F and SEMA7A have emerged as regulators of neutrophil migration in models of acute lung injury. In response to intratracheal LPS challenge in a murine model, SEMA3F is released into the airways, with neutrophils from bronchoalveolar lavage expressing SEMA3F and its receptor NRP2 at both the mRNA and protein levels. Neutrophil-specific loss of SEMA3F results in more rapid neutrophil recruitment to and clearance from the lungs in this model, without evidence of increased neutrophil apoptosis. Concurrently, exogenous intratracheal administration of SEMA3F results in increased recovery of neutrophils in BAL and alveolar space. Evaluation of live lung slices from mice stimulated with SEMA3F demonstrates reduced neutrophil speed, lower F-actin content, and increased rounding of neutrophils, all of which likely contribute to retention of neutrophils at sites of inflammation (57). Additionally, SEMA7A induces transendothelial migration of neutrophils into lung tissue in conditions of hypoxia and after LPS inhalation (54, 60), and blockade of SEMA7A in both in vivo and in vitro models reveals attenuated injury-induced influx of neutrophils correlating with dampened lung injury (54, 60, 101). This action is dependent on binding of SEMA7A to the PLXNC1 receptor (53, 54), and animal models of ventilator-induced lung injury (VILI) demonstrate robust induction of PLXNC1 on neutrophils. Furthermore, in vivo inhibition of SEMA7A and PLXNC1 by use of neutralizing antibodies in models of VILI and inhaled LPS models results in improved survival, reduced neutrophil migration and overall reduced cytokine response and lung injury (53, 101), suggesting their use a novel therapeutic agent for the management of ALI.
The pulmonary endothelium serves as a semipermeable barrier between the pulmonary circulation and the interstitium, and disruption of the integrity of this barrier by means of cytoskeletal remodeling leads to worsening pulmonary edema, impaired gas exchange, and release of pro-inflammatory markers and coagulation factors (102). In models of ALI from seawater aspiration, inhibition of SEMA7A and its receptor PLXNC1 results in a significant reduction in actin-dependent cytoskeletal remodeling and decreased monolayer permeability in rat pulmonary microvascular endothelial cells (80, 103).
Lastly, semaphorins and their receptors have been implicated in the regulation of inflammatory signaling cascades and the release of immune cytokines and chemokines in settings of ALI. Cell surface and soluble NRP2 expression in murine alveolar macrophages is dramatically increased following LPS inhalation, and myeloid-specific ablation of NRP2 results in prolonged accumulation of airway neutrophils and macrophages, as well as increased expression of chemokine (C-C motif) ligand 2 (Ccl2) expression in the lungs with ALI, suggesting NRP2 expression suppressed inflammatory responses to inhaled LPS (104). On the contrary, SEMA7A and its receptor PLXNC1 both induce the release of inflammatory cytokines in various models (53, 60, 80, 103). Taken together, this demonstrates that differing classes of semaphorins and their receptors play roles in both the induction and suppression of the innate immune response and inflammatory cascade in ALI.
Future Prospects
Semaphorins and their receptors have an important role in the pathophysiology of acute inflammation, and they serve as potential therapeutic targets in several clinical diseases, as summarized above. To date, preclinical animal models have demonstrated the targeting of semaphorins and plexins in cancer, bone diseases, autoimmune diseases, microvascular diseases, allergic airway disease, and in central nervous system regeneration (7). While there are a currently a number of clinical trials underway targeting semaphorins in the context of cancer (clinicaltrials.gov, NCT 03690986, NCT 03373188, NCT 01313065, NCT 03268057), multiple sclerosis (clinicaltrials.gov, NCT 01764737), and Alzheimer’s Disease (clinicaltrials.gov, NCT 04381468), there is only one observational clinical trial investigating peripheral blood semaphorin levels in patients with early septic shock (clinicaltrials.gov, NCT 02692118), and there are no clinical trials investigating targeting semaphorins in conditions of acute inflammation. As highlighted in this review, the most promising areas for the development of therapeutics directed at semaphorins and their receptors in this field of study are sepsis (79), myocardial ischemia (92), and acute lung injury (53–55, 57, 60). Further elucidation of the expression and function of semaphorins and their receptors on innate immune cells will be important in determining additional prospective use as diagnostic or therapeutic targets.
Author Contributions
SK, SG, and PT-P all contributed to the conception and design of this manuscript. SK contributed to drafting this manuscript. SK, SG, and PT-P all contributed to revisions of this manuscript. All authors contributed to the article and approved the submitted version.
Funding
Intramural funding from the National Institutes of Health supported this work.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Alan Hoofring from the National Institutes of Health Medical Arts Department for illustrating the original figures included in this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.672441/full#supplementary-material
Abbreviations
SEMA, semaphorin; PLXN, plexin; NRP, neuropilin.
References
1. Goodman CS, Kolodkin AL, Luo Y, Püschel AQ, Raper JA. Unified nomenclature for the semaphorins/collapsins. Semaphorin Nomenclature Committee. Cell (1999) 97(5):551–2. doi: 10.1016/S0092-8674(00)80766-7
2. Tamagnone L, Comoglio PM. To Move or Not to Move? Semaphorin signalling in cell migration. EMBO Rep (2004) 5(4):356–61. doi: 10.1038/sj.embor.7400114
4. Alto LT, Terman JR. Semaphorins and Their Signaling Mechanisms. Methods Mol Biol (2017) 1493:1–25. doi: 10.1007/978-1-4939-6448-2_1
5. Messina A, Giacobini P. Semaphorin Signaling in the Development and Function of the Gonadotropin Hormone-Releasing Hormone System. Front Endocrinol (Lausanne) (2013) 4:133. doi: 10.3389/fendo.2013.00133
6. Suzuki K, Kumanogoh A, Kikutani H. Semaphorins and Their Receptors in Immune Cell Interactions. Nat Immunol (2008) 9(1):17–23. doi: 10.1038/ni1553
7. Worzfeld T, Offermanns S. Semaphorins and Plexins as Therapeutic Targets. Nat Rev Drug Discovery (2014) 13(8):603–21. doi: 10.1038/nrd4337
8. Gras C, Eiz-Vesper B, Jaimes Y, Immenschuh S, Jacobs R, Witte T, et al. Secreted Semaphorin 5A Activates Immune Effector Cells and is a Biomarker for Rheumatoid Arthritis. Arthritis Rheumatol (2014) 66(6):1461–71. doi: 10.1002/art.38425
9. Sadanandam A, Sidhu SS, Wullschleger S, Singh S, Varney ML, Yang CS, et al. Secreted Semaphorin 5A Suppressed Pancreatic Tumour Burden But Increased Metastasis and Endothelial Cell Proliferation. Br J Cancer (2012) 107(3):501–7. doi: 10.1038/bjc.2012.298
10. Lu N, Li Y, Zhang Z, Xing J, Sun Y, Yao S, et al. Human Semaphorin-4A Drives Th2 Responses by Binding to Receptor ILT-4. Nat Commun (2018) 9(1):742. doi: 10.1038/s41467-018-03128-9
11. Lu Q, Dong N, Wang Q, Yi W, Wang Y, Zhang S, et al. Increased Levels of Plasma Soluble Sema4D in Patients With Heart Failure. PloS One (2013) 8(5):e64265. doi: 10.1371/journal.pone.0064265
12. Maleki KT, Cornillet M, Björkström NK. Soluble SEMA4D/CD100: A Novel Immunoregulator in Infectious and Inflammatory Diseases. Clin Immunol (2016) 163:52–9. doi: 10.1016/j.clim.2015.12.012
13. Nishide M, Kumanogoh A. The Role of Semaphorins in Immune Responses and Autoimmune Rheumatic Diseases. Nat Rev Rheumatol (2018) 14(1):19–31. doi: 10.1038/nrrheum.2017.201
14. Wang L, Song G, Zheng Y, Tan W, Pan J, Zhao Y, et al. Expression of Semaphorin 4A and its Potential Role in Rheumatoid Arthritis. Arthritis Res Ther (2015) 17(1):227. doi: 10.1186/s13075-015-0734-y
15. Nakamura F, Tanaka M, Takahashi T, Kalb RG, Strittmatter SM. Neuropilin-1 Extracellular Domains Mediate Semaphorin D/III-induced Growth Cone Collapse. Neuron (1998) 21(5):1093–100. doi: 10.1016/S0896-6273(00)80626-1
16. Tamagnone L, Artigiani S, Chen H, He Z, Ming GI, Song H, et al. Plexins are a Large Family of Receptors for Transmembrane, Secreted, and GPI-anchored Semaphorins in Vertebrates. Cell (1999) 99(1):71–80. doi: 10.1016/S0092-8674(00)80063-X
17. Tamagnone L, Comoglio PM. Signalling by Semaphorin Receptors: Cell Guidance and Beyond. Trends Cell Biol (2000) 10(9):377–83. doi: 10.1016/S0962-8924(00)01816-X
18. Castellani V, De Angelis E, Kenwrick S, Rougon G. Cis and Trans Interactions of L1 With Neuropilin-1 Control Axonal Responses to Semaphorin 3A. EMBO J (2002) 21(23):6348–57. doi: 10.1093/emboj/cdf645
19. Franco M, Tamagnone L. Tyrosine Phosphorylation in Semaphorin Signalling: Shifting Into Overdrive. EMBO Rep (2008) 9(9):865–71. doi: 10.1038/embor.2008.139
20. Kumanogoh A, Kikutani H. Immunological Functions of the Neuropilins and Plexins as Receptors for Semaphorins. Nat Rev Immunol (2013) 13(11):802–14. doi: 10.1038/nri3545
21. Pasterkamp RJ. Getting Neural Circuits Into Shape With Semaphorins. Nat Rev Neurosci (2012) 13(9):605–18. doi: 10.1038/nrn3302
22. Takamatsu H, Kumanogoh A. Diverse Roles for Semaphorin-Plexin Signaling in the Immune System. Trends Immunol (2012) 33(3):127–35. doi: 10.1016/j.it.2012.01.008
23. Tamagnone L. Emerging Role of Semaphorins as Major Regulatory Signals and Potential Therapeutic Targets in Cancer. Cancer Cell (2012) 22(2):145–52. doi: 10.1016/j.ccr.2012.06.031
24. Kang S, Nakanishi Y, Kioi Y, Okuzaki D, Kimura T, Takamatsu H, et al. Semaphorin 6D Reverse Signaling Controls Macrophage Lipid Metabolism and Anti-Inflammatory Polarization. Nat Immunol (2018) 19(6):561–70. doi: 10.1038/s41590-018-0108-0
25. Sun T, Yang L, Kaur H, Pestel J, Looso M, Nolte H, et al. A Reverse Signaling Pathway Downstream of Sema4A Controls Cell Migration Via Scrib. J Cell Biol (2017) 216(1):199–215. doi: 10.1083/jcb.201602002
26. Chapoval SP. Neuroimmune Semaphorins as Costimulatory Molecules and Beyond. Mol Med (2018) 24(1):13. doi: 10.1186/s10020-018-0014-9
27. Junqueira Alves C, Yotoko K, Zou H, Friedel RH. Origin and Evolution of Plexins, Semaphorins, and Met Receptor Tyrosine Kinases. Sci Rep (2019) 9(1):1970. doi: 10.1038/s41598-019-38512-y
28. Cagnoni G, Tamagnone L. Semaphorin Receptors Meet Receptor Tyrosine Kinases on the Way of Tumor Progression. Oncogene (2014) 33(40):4795–802. doi: 10.1038/onc.2013.474
29. Maione F, Capano S, Regano D, Zentilin L, Giacca M, Casanovas O, et al. Semaphorin 3A Overcomes Cancer Hypoxia and Metastatic Dissemination Induced by Antiangiogenic Treatment in Mice. J Clin Invest (2012) 122(5):1832–48. doi: 10.1172/JCI58976
30. Wei L, Li H, Tamagnone L, You H. Semaphorins and Their Receptors in Hematological Malignancies. Front Oncol (2019) 9:382. doi: 10.3389/fonc.2019.00382
31. Wu M, Li J, Gao Q, Ye F. The Role of Sema4D/CD100 as a Therapeutic Target for Tumor Microenvironments and for Autoimmune, Neuroimmune and Bone Diseases. Expert Opin Ther Targets (2016) 20(7):885–901. doi: 10.1517/14728222.2016.1139083
32. Bao L, Zhang X, Xu Y, Wang M, Song Y, Gu Y, et al. Dysfunction of MiR-148a-NRP1 Functional Axis Suppresses Osteogenic Differentiation of Periodontal Ligament Stem Cells Under Inflammatory Microenvironment. Cell Reprog (2019) 21(6):314–22. doi: 10.1089/cell.2019.0026
33. Chen TH, Arra M, Mbalaviele G, Swarnkar G, Abu-Amer Y. Inflammatory Responses Reprogram T(REGS) Through Impairment of Neuropilin-1. Sci Rep (2019) 9(1):10429. doi: 10.1038/s41598-019-46934-x
34. Hughes A, Kleine-Albers J, Helfrich MH, Ralston SH, Rogers MJ. A Class III Semaphorin (Sema3e) Inhibits Mouse Osteoblast Migration and Decreases Osteoclast Formation In Vitro. Calcif Tissue Int (2012) 90(2):151–62. doi: 10.1007/s00223-011-9560-7
35. Kenan S, Onur ÖD, Solakoğlu S, Kotil T, Ramazanoğlu M, Çelik HH, et al. Investigation of the Effects of Semaphorin 3A on New Bone Formation in a Rat Calvarial Defect Model. J Craniomaxillofac Surg (2019) 47(3):473–83. doi: 10.1016/j.jcms.2018.12.010
36. Liu L, Wang J, Song X, Zhu Q, Shen S, Zhang W. Semaphorin 3A Promotes Osteogenic Differentiation in Human Alveolar Bone Marrow Mesenchymal Stem Cells. Exp Ther Med (2018) 15(4):3489–94. doi: 10.3892/etm.2018.5813
37. Sun J, Wei X, Wang Z, Liu Y, Lu J, Lu Y, et al. Inflammatory Milieu Cultivated Sema3A Signaling Promotes Chondrocyte Apoptosis in Knee Osteoarthritis. J Cell Biochem (2018) 119(3):2891–9. doi: 10.1002/jcb.26470
38. Joza S, Wang J, Fox E, Hillman V, Ackerley C, Post M. Loss of Semaphorin-Neuropilin-1 Signaling Causes Dysmorphic Vascularization Reminiscent of Alveolar Capillary Dysplasia. Am J Pathol (2012) 181(6):2003–17. doi: 10.1016/j.ajpath.2012.08.037
39. Meda C, Molla F, De Pizzol M, Regano D, Maione F, Capano S, et al. Semaphorin 4A Exerts a Proangiogenic Effect by Enhancing Vascular Endothelial Growth Factor-a Expression in Macrophages. J Immunol (2012) 188(8):4081–92. doi: 10.4049/jimmunol.1101435
40. Mucka P, Levonyak N, Geretti E, Zwaans BMM, Li X, Adini I, et al. Inflammation and Lymphedema are Exacerbated and Prolonged by Neuropilin 2 Deficiency. Am J Pathol (2016) 186(11):2803–12. doi: 10.1016/j.ajpath.2016.07.022
41. Suidan GL, Dickerson JW, Johnson HL, Chan TW, Pavelko KD, Pirko I, et al. Preserved Vascular Integrity and Enhanced Survival Following Neuropilin-1 Inhibition in a Mouse Model of CD8 T Cell-Initiated CNS Vascular Permeability. J Neuroinflamm (2012) 9:218. doi: 10.1186/1742-2094-9-218
42. Torres-Vázquez J, Gitler AD, Fraser SD, Berk JD, Van NP, Fishman MC, et al. Semaphorin-Plexin Signaling Guides Patterning of the Developing Vasculature. Dev Cell (2004) 7(1):117–23. doi: 10.1016/j.devcel.2004.06.008
43. Liu LN, Li XM, Ye DQ, Pan HF. Emerging Role of semaphorin-3A in Autoimmune Diseases. Inflammopharmacology (2018) 26(3):655–65. doi: 10.1007/s10787-018-0484-y
44. Nishide M, Nojima S, Ito D, Takamatsu H, Koyama S, Kang S, et al. Semaphorin 4D Inhibits Neutrophil Activation and is Involved in the Pathogenesis of Neutrophil-Mediated Autoimmune Vasculitis. Ann Rheum Dis (2017) 76(8):1440–8. doi: 10.1136/annrheumdis-2016-210706
45. Rajabinejad M, Asadi G, Ranjbar S, Afshar Hezarkhani L, Salari F, Gorgin Karaji A, et al. Semaphorin 4A, 4C, and 4D: Function Comparison in the Autoimmunity, Allergy, and Cancer. Gene (2020) 746:144637. doi: 10.1016/j.gene.2020.144637
46. Rimar D, Nov Y, Rosner I, Slobodin G, Rozenbaum M, Halasz K, et al. Semaphorin 3A: An Immunoregulator in Systemic Sclerosis. Rheumatol Int (2015) 35(10):1625–30. doi: 10.1007/s00296-015-3269-2
47. Sabag AD, Dias-Polak D, Bejar J, Sheffer H, Bergman R, Vadasz Z. Altered Expression of Regulatory Molecules in the Skin of Psoriasis. Immunol Res (2018) 66(6):649–54. doi: 10.1007/s12026-018-9057-9
48. Tang MW, Malvar Fernández B, Newsom SP, van Buul JD, Radstake T, Baeten DL, et al. Class 3 Semaphorins Modulate the Invasive Capacity of Rheumatoid Arthritis Fibroblast-Like Synoviocytes. Rheumatol (Oxford) (2018) 57(5):909–20. doi: 10.1093/rheumatology/kex511
49. Vadasz Z, Toubi E. Semaphorin3A: A Potential Therapeutic Tool in Immune-Mediated Diseases. Eur J Rheumatol (2018) 5(1):58–61. doi: 10.5152/eurjrheum.2017.17076
50. Xie J, Wang H. Semaphorin 7A as a Potential Immune Regulator and Promising Therapeutic Target in Rheumatoid Arthritis. Arthritis Res Ther (2017) 19(1):10. doi: 10.1186/s13075-016-1217-5
51. Kumanogoh A, Kikutani H. Roles of the Semaphorin Family in Immune Regulation. Adv Immunol (2003) 81:173–98. doi: 10.1016/S0065-2776(03)81005-2
52. Chabbert-de Ponnat I, Marie-Cardine A, Pasterkamp RJ, Schiavon V, Tamagnone L, Thomasset N, et al. Soluble CD100 Functions on Human Monocytes and Immature Dendritic Cells Require Plexin C1 and Plexin B1, Respectively. Int Immunol (2005) 17(4):439–47. doi: 10.1093/intimm/dxh224
53. Granja T, Köhler D, Mirakaj V, Nelson E, König K, Rosenberger P. Crucial Role of Plexin C1 for Pulmonary Inflammation and Survival During Lung Injury. Mucosal Immunol (2014) 7(4):879–91. doi: 10.1038/mi.2013.104
54. Morote-Garcia JC, Napiwotzky D, Köhler D, Rosenberger P. Endothelial Semaphorin 7A Promotes Neutrophil Migration During Hypoxia. Proc Natl Acad Sci U S A (2012) 109(35):14146–51. doi: 10.1073/pnas.1202165109
55. Movassagh H, Saati A, Nandagopal S, Mohammed A, Tatari N, Shan L, et al. Chemorepellent Semaphorin 3e Negatively Regulates Neutrophil Migration In Vitro and In Vivo. J Immunol (2017) 198(3):1023–33. doi: 10.4049/jimmunol.1601093
56. Peng X, Moore M, Mathur A, Zhou Y, Sun H, Gan Y, et al. Plexin C1 Deficiency Permits Synaptotagmin 7-Mediated Macrophage Migration and Enhances Mammalian Lung Fibrosis. FASEB J (2016) 30(12):4056–70. doi: 10.1096/fj.201600373R
57. Plant T, Eamsamarng S, Sanchez-Garcia MA, Reyes L, Renshaw SA, Coelho P, et al. Semaphorin 3F Signaling Actively Retains Neutrophils At Sites of Inflammation. J Clin Invest (2020) 130(6):3221–37. doi: 10.1172/JCI130834
58. Rey-Gallardo A, Delgado-Martín C, Gerardy-Schahn R, Rodríguez-Fernández JL, Vega MA. Polysialic Acid is Required for neuropilin-2a/b-mediated Control of CCL21-driven Chemotaxis of Mature Dendritic Cells and for Their Migration In Vivo. Glycobiology (2011) 21(5):655–62. doi: 10.1093/glycob/cwq216
59. Roney KE, O’Connor BP, Wen H, Holl EK, Guthrie EH, Davis BK, et al. Plexin-B2 Negatively Regulates Macrophage Motility, Rac, and Cdc42 Activation. PloS One (2011) 6(9):e24795. doi: 10.1371/journal.pone.0024795
60. Roth JM, Köhler D, Schneider M, Granja TF, Rosenberger P. Semaphorin 7A Aggravates Pulmonary Inflammation During Lung Injury. PloS One (2016) 11(1):e0146930. doi: 10.1371/journal.pone.0146930
61. Takamatsu H, Takegahara N, Nakagawa Y, Tomura M, Taniguchi M, Friedel RH, et al. Semaphorins Guide the Entry of Dendritic Cells Into the Lymphatics by Activating Myosin II. Nat Immunol (2010) 11(7):594–600. doi: 10.1038/ni.1885
62. Tsuda T, Nishide M, Maeda Y, Hayama Y, Koyama S, Nojima S, et al. Pathological and Therapeutic Implications of Eosinophil-Derived Semaphorin 4D in Eosinophilic Chronic Rhinosinusitis. J Allergy Clin Immunol (2020) 145(3):843–54.e4. doi: 10.1016/j.jaci.2019.12.893
63. van Rijn A, Paulis L, te Riet J, Vasaturo A, Reinieren-Beeren I, van der Schaaf A, et al. Semaphorin 7A Promotes Chemokine-Driven Dendritic Cell Migration. J Immunol (2016) 196(1):459–68. doi: 10.4049/jimmunol.1403096
64. Walzer T, Galibert L, Comeau MR, De Smedt T. Plexin C1 Engagement on Mouse Dendritic Cells by Viral Semaphorin A39R Induces Actin Cytoskeleton Rearrangement and Inhibits Integrin-Mediated Adhesion and Chemokine-Induced Migration. J Immunol (2005) 174(1):51–9. doi: 10.4049/jimmunol.174.1.51
65. Wanschel A, Seibert T, Hewing B, Ramkhelawon B, Ray TD, van Gils JM, et al. Neuroimmune Guidance Cue Semaphorin 3E is Expressed in Atherosclerotic Plaques and Regulates Macrophage Retention. Arterioscler Thromb Vasc Biol (2013) 33(5):886–93. doi: 10.1161/ATVBAHA.112.300941
66. Zhou X, Wahane S, Friedl MS, Kluge M, Friedel CC, Avrampou K, et al. Microglia and Macrophages Promote Corralling, Wound Compaction and Recovery After Spinal Cord Injury Via Plexin-B2. Nat Neurosci (2020) 23(3):337–50. doi: 10.1038/s41593-020-0597-7
67. Casazza A, Fazzari P, Tamagnone L. Semaphorin Signals in Cell Adhesion and Cell Migration: Functional Role and Molecular Mechanisms. Adv Exp Med Biol (2007) 600:90–108. doi: 10.1007/978-0-387-70956-7_8
68. Ford JW, McVicar DW. TREM and TREM-like Receptors in Inflammation and Disease. Curr Opin Immunol (2009) 21(1):38–46. doi: 10.1016/j.coi.2009.01.009
69. Eun SY, O’Connor BP, Wong AW, van Deventer HW, Taxman DJ, Reed W, et al. Cutting Edge: Rho Activation and Actin Polarization are Dependent on plexin-A1 in Dendritic Cells. J Immunol (2006) 177(7):4271–5. doi: 10.4049/jimmunol.177.7.4271
70. Holmes S, Downs AM, Fosberry A, Hayes PD, Michalovich D, Murdoch P, et al. Sema7A is a Potent Monocyte Stimulator. Scand J Immunol (2002) 56(3):270–5. doi: 10.1046/j.1365-3083.2002.01129.x
71. Mohammed A, Okwor I, Shan L, Onyilagha C, Uzonna JE, Gounni AS. Semaphorin 3E Regulates the Response of Macrophages to Lipopolysaccharide-Induced Systemic Inflammation. J Immunol (2020) 204(1):128–36. doi: 10.4049/jimmunol.1801514
72. Rienks M, Carai P, Bitsch N, Schellings M, Vanhaverbeke M, Verjans J, et al. Sema3A Promotes the Resolution of Cardiac Inflammation After Myocardial Infarction. Basic Res Cardiol (2017) 112(4):42. doi: 10.1007/s00395-017-0630-5
73. Dai X, Okon I, Liu Z, Wu Y, Zhu H, Song P, et al. A Novel Role for Myeloid Cell-Specific Neuropilin 1 in Mitigating Sepsis. FASEB J (2017) 31(7):2881–92. doi: 10.1096/fj.201601238R
74. Holl EK, Roney KE, Allen IC, Steinbach E, Arthur JC, Buntzman A, et al. Plexin-B2 and Plexin-D1 in Dendritic Cells: Expression and IL-12/IL-23p40 Production. PloS One (2012) 7(8):e43333. doi: 10.1371/journal.pone.0043333
75. Ji JD, Park-Min KH, Ivashkiv LB. Expression and Function of Semaphorin 3A and its Receptors in Human Monocyte-Derived Macrophages. Hum Immunol (2009) 70(4):211–7. doi: 10.1016/j.humimm.2009.01.026
76. Maeda Y, Tsuda T, Takeda Y, Koyama S, Hayama Y, Nojima S, et al. SEMA4A Promotes Eosinophil Survival and Contributes to Eosinophil-Mediated Allergic Diseases. Allergol Int (2019) 68(2):274–6. doi: 10.1016/j.alit.2018.10.001
77. Movassagh H, Shan L, Mohammed A, Halayko AJ, Gounni AS. Semaphorin 3E Deficiency Exacerbates Airway Inflammation, Hyperresponsiveness, and Remodeling in a Mouse Model of Allergic Asthma. J Immunol (2017) 198(5):1805–14. doi: 10.4049/jimmunol.1601514
78. Nakagawa Y, Takamatsu H, Okuno T, Kang S, Nojima S, Kimura T, et al. Identification of Semaphorin 4B as a Negative Regulator of Basophil-Mediated Immune Responses. J Immunol (2011) 186(5):2881–8. doi: 10.4049/jimmunol.1003485
79. Wen H, Lei Y, Eun SY, Ting JP. Plexin-A4-semaphorin 3A Signaling is Required for Toll-like Receptor- and Sepsis-Induced Cytokine Storm. J Exp Med (2010) 207(13):2943–57. doi: 10.1084/jem.20101138
80. Zhang M, Yan X, Liu W, Sun R, Xie Y, Jin F. Endothelial Semaphorin 7A Promotes Seawater Aspiration-Induced Acute Lung Injury Through Plexin C1 and β1 Integrin. Mol Med Rep (2017) 16(4):4215–21. doi: 10.3892/mmr.2017.7097
81. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). Jama (2016) 315(8):801–10. doi: 10.1001/jama.2016.0287
82. Yu L, Wang L, Chen S. Endogenous Toll-Like Receptor Ligands and Their Biological Significance. J Cell Mol Med (2010) 14(11):2592–603. doi: 10.1111/j.1582-4934.2010.01127.x
83. Yamashita N, Jitsuki-Takahashi A, Ogawara M, Ohkubo W, Araki T, Hotta C, et al. Anti-Semaphorin 3A Neutralization Monoclonal Antibody Prevents Sepsis Development in Lipopolysaccharide-Treated Mice. Int Immunol (2015) 27(9):459–66. doi: 10.1093/intimm/dxv014
84. Reidy K, Tufro A. Semaphorins in Kidney Development and Disease: Modulators of Ureteric Bud Branching, Vascular Morphogenesis, and Podocyte-Endothelial Crosstalk. Pediatr Nephrol (2011) 26(9):1407–12. doi: 10.1007/s00467-011-1769-1
85. Tian X, Gan H, Zeng Y, Zhao H, Tang R, Xia Y. Inhibition of semaphorin-3a Suppresses Lipopolysaccharide-Induced Acute Kidney Injury. J Mol Med (Berl) (2018) 96(7):713–24. doi: 10.1007/s00109-018-1653-6
86. Ranganathan P, Jayakumar C, Mohamed R, Weintraub NL, Ramesh G. Semaphorin 3A Inactivation Suppresses Ischemia-Reperfusion-Induced Inflammation and Acute Kidney Injury. Am J Physiol Renal Physiol (2014) 307(2):F183–94. doi: 10.1152/ajprenal.00177.2014
87. Doi K, Noiri E, Nangaku M, Yahagi N, Jayakumar C, Ramesh G. Repulsive Guidance Cue Semaphorin 3A in Urine Predicts the Progression of Acute Kidney Injury in Adult Patients From a Mixed Intensive Care Unit. Nephrol Dial Transplant (2014) 29(1):73–80. doi: 10.1093/ndt/gft414
88. König K, Marth L, Roissant J, Granja T, Jennewein C, Devanathan V, et al. The Plexin C1 Receptor Promotes Acute Inflammation. Eur J Immunol (2014) 44(9):2648–58. doi: 10.1002/eji.201343968
89. Sun Q, Liu S, Liu K, Jiao K. Role of Semaphorin Signaling During Cardiovascular Development. J Am Heart Assoc (2018) 7(11):1–8. doi: 10.1161/JAHA.118.008853
90. Zhao C, Liu J, Zhang M, Wu Y. Semaphorin 3A Deficiency Improves Hypoxia-Induced Myocardial Injury Via Resisting Inflammation and Cardiomyocytes Apoptosis. Cell Mol Biol (Noisy-le-grand) (2016) 62(2):8–14. doi: 10.14715/cmb/2016.62.2.2
91. Reichert S, Scheid S, Roth T, Herkel M, Petrova D, Linden A, et al. Semaphorin 3F Promotes Transendothelial Migration of Leukocytes in the Inflammatory Response After Survived Cardiac Arrest. Inflammation (2019) 42(4):1252–64. doi: 10.1007/s10753-019-00985-4
92. Köhler D, Granja T, Volz J, Koeppen M, Langer HF, Hansmann G, et al. Red Blood Cell-Derived Semaphorin 7A Promotes Thrombo-Inflammation in Myocardial Ischemia-Reperfusion Injury Through Platelet Gpib. Nat Commun (2020) 11(1):1315. doi: 10.1038/s41467-020-14958-x
93. Butt Y, Kurdowska A, Allen TC. Acute Lung Injury: A Clinical and Molecular Review. Arch Pathol Lab Med (2016) 140(4):345–50. doi: 10.5858/arpa.2015-0519-RA
94. Fanelli V, Ranieri VM. Mechanisms and Clinical Consequences of Acute Lung Injury. Ann Am Thorac Soc (2015) 12 Suppl 1:S3–8. doi: 10.1513/AnnalsATS.201407-340MG
95. Johnson ER, Matthay MA. Acute Lung Injury: Epidemiology, Pathogenesis, and Treatment. J Aerosol Med Pulm Drug Delivery (2010) 23(4):243–52. doi: 10.1089/jamp.2009.0775
96. Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, et al. Acute Respiratory Distress Syndrome: The Berlin Definition. Jama (2012) 307(23):2526–33. doi: 10.1001/jama.2012.5669
97. Abraham E. Neutrophils and Acute Lung Injury. Crit Care Med (2003) 31(4 Suppl):S195–9. doi: 10.1097/01.CCM.0000057843.47705.E8
98. Grommes J, Soehnlein O. Contribution of Neutrophils to Acute Lung Injury. Mol Med (2011) 17(3-4):293–307. doi: 10.2119/molmed.2010.00138
99. Williams AE, Chambers RC. The mercurial nature of neutrophils: still an enigma in ARDS? Am J Physiol Lung Cell Mol Physiol (2014) 306(3):L217–30. doi: 10.1152/ajplung.00311.2013
100. Zemans RL, Colgan SP, Downey GP. Transepithelial Migration of Neutrophils: Mechanisms and Implications for Acute Lung Injury. Am J Respir Cell Mol Biol (2009) 40(5):519–35. doi: 10.1165/rcmb.2008-0348TR
101. Chen X, Wang H, Jia K, Wang H, Ren T. Anti-Semaphorin-7A Single Chain Antibody Demonstrates Beneficial Effects on Pulmonary Inflammation During Acute Lung Injury. Exp Ther Med (2018) 15(3):2356–64. doi: 10.3892/etm.2018.5724
102. Millar FR, Summers C, Griffiths MJ, Toshner MR, Proudfoot AG. The Pulmonary Endothelium in Acute Respiratory Distress Syndrome: Insights and Therapeutic Opportunities. Thorax (2016) 71(5):462–73. doi: 10.1136/thoraxjnl-2015-207461
103. Zhang M, Wang L, Dong M, Li Z, Jin F. Endothelial Semaphorin 7A Promotes Inflammation in Seawater Aspiration-Induced Acute Lung Injury. Int J Mol Sci (2014) 15(11):19650–61. doi: 10.3390/ijms151119650
Keywords: semaphorin, plexin, neuropilin, innate immunity, sepsis, acute inflammation
Citation: Kanth SM, Gairhe S and Torabi-Parizi P (2021) The Role of Semaphorins and Their Receptors in Innate Immune Responses and Clinical Diseases of Acute Inflammation. Front. Immunol. 12:672441. doi: 10.3389/fimmu.2021.672441
Received: 25 February 2021; Accepted: 20 April 2021;
Published: 03 May 2021.
Edited by:
Massimo Collino, University of Turin, ItalyReviewed by:
Roland Horst Friedel, Icahn School of Medicine at Mount Sinai, United StatesDavid Koehler, Tübingen University Hospital, Germany
Svetlana P. Chapoval, University of Maryland, Baltimore, United States
Copyright © 2021 Kanth, Gairhe and Torabi-Parizi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Parizad Torabi-Parizi, dG9yYWJpcGFyaXppcEBjYy5uaWguZ292