 Markus Zeisbrich
Markus Zeisbrich Nina Chevalier
Nina Chevalier Bettina Sehnert
Bettina Sehnert Marta Rizzi
Marta Rizzi Nils Venhoff
Nils Venhoff Jens Thiel
Jens Thiel Reinhard E. Voll
Reinhard E. Voll- Department of Rheumatology and Clinical Immunology, Medical Center – University of Freiburg, University of Freiburg, Freiburg, Germany
Objectives: ANCA-associated vasculitides (AAV) affect small- and medium-sized blood vessels. In active disease, vessel wall infiltrates are mainly composed of monocytes and macrophages. Immune checkpoint molecules are crucial for the maintenance of self-tolerance and the prevention of autoimmune diseases. After checkpoint inhibitor therapy, the development of autoimmune vasculitis has been observed. However, defects of immune checkpoint molecules in AAV patients have not been identified yet.
Methods: Monocytes and monocyte-derived macrophages from AAV patients and healthy age-matched controls were tested for surface expression of immunoinhibitory checkpoint programmed cell death ligand-1 (PD-L1). Using in vitro co-culture approaches, the effect of monocyte PD-L1 expression on CD4+ T cell activation and proliferation was tested.
Results: Monocytes from AAV patients displayed lower PD-L1 expression and a defective PD-L1 presentation upon activation, an effect that was correlated with disease activity. Lower PD-L1 expression was due to increased lysosomal degradation of PD-L1 in AAV monocytes. We identified a reduced expression of CMTM6, a protein protecting PD-L1 from lysosomal breakdown, as the underlying molecular defect. PD-L1low AAV monocytes showed increased stimulatory capacity and induced T cell activation and proliferation. Inhibiting lysosomal function corrected this phenotype by increasing PD-L1, thus normalizing the pro-stimulatory behavior of AAV monocytes.
Conclusions: This study identifies a defect of the immunoinhibitory checkpoint PD-L1 in monocytes from patients with AAV. Low expression of CMTM6 results in enhanced lysosomal degradation of PD-L1, thus providing insufficient negative signaling to T cells. Correcting this defect by targeting lysosomal function may represent a novel strategy to treat AAV.
Introduction
Antineutrophil cytoplasmic antibody (ANCA)–associated vasculitides (AAV) are characterized by necrotizing inflammation of small blood vessels and the presence of ANCA with specificity for proteinase-3 (PR3) or myeloperoxidase (MPO). Frequent target tissues are the respiratory tract, kidneys, skin, and peripheral nerves. Immunopathologically, AAV present with an absence or paucity of immunoglobulin and complement deposition in affected vessels. Early vascular lesions of AAV consist of neutrophils with admixed monocytes. Within days, the initial inflammatory lesion is replaced by inflammation with a predominance of monocytes and macrophages (1). Also in renal biopsies, monocytes and macrophages are the predominant cells in glomeruli of patients with AAV (2, 3).
In AAV, systemic monocyte activation has been observed and their activation state persisted during remission (4–6). Transmembrane receptors that facilitate extracellular matrix adhesion are upregulated on AAV monocytes indicating an increased ability to interact with vascular endothelium (7). Monocytes also express antigens recognized by ANCA, and stimulation in vitro with ANCA promoted cytokine and reactive oxygen species (ROS) production (8, 9). Moreover, AAV monocytes recognize neutrophil extracellular traps (NETs) to upregulate the alarmin S100A9, which results in the induction of metalloproteinase-9 and enables monocytes to invade into the extracellular matrix (10).
Immune checkpoint molecules are crucial for the maintenance of self-tolerance. Programmed death ligand-1 (PD-L1) is an inhibitory checkpoint molecule that counterbalances the overwhelming activation of the immune system. PD-L1 is expressed on antigen-presenting cells (monocytes, macrophages, and dendritic cells) and endothelial cells (11). By engaging its ligand programmed death-1 (PD-1) on T cells, TCR- and CD28-mediated activation cascades are down-regulated. In animal studies, disruption of PD-L1 signals has been associated with inflammatory disease (12–14). In line with this, a defect in the PD-1/PD-L1 axis was reported in large vessel vasculitis (15, 16). In cancer immunotherapy, the use of checkpoint inhibitors that block the PD-1/PD-L1 pathway has proven to be very effective (17–19). In parallel, increasing reports of immune-related adverse events were noted (irAEs) (20). In fact, several forms of vasculitis have been reported to develop or re-activate after checkpoint inhibitor therapy, including AAV (21–24).
Here, we report that monocytes from AAV patients express low levels of the inhibitory checkpoint PD-L1. Patient-derived cells showed a defect in presenting PD-L1 in response to inflammatory stimuli, thus leading to increased activation of T cells in co-culture experiments. Mechanistically, inhibiting lysosome function in monocytes from AAV patients corrected their hyperstimulatory phenotype by upregulating PD-L1, indicating that increased lysosomal degradation of PD-L1 occurs in AAV monocytes. In line with this, AAV monocytes display a lower expression of chemokine-like factor-like MARVEL transmembrane domain containing family member 6 (CMTM6), a protein protecting PD-L1 from lysosomal breakdown (25, 26).
This study establishes a link between autoimmune small-vessel vasculitis and immunoinhibitory checkpoint deficiency, highlighting the potential role of uncontrolled innate immunity in promoting the development of autoimmune disease.
Materials and Methods
Patients and Controls
The study population included 26 AAV patients classified as granulomatosis with polyangiitis (GPA, n=21) or microscopic polyangiitis (MPA, n=5) as defined by the Chapel Hill Consensus Conference nomenclature (27) and 29 age-matched healthy controls. All AAV patients tested positive for ANCA by indirect immunofluorescence and PR3 or MPO-ELISA and were enrolled between July 2019 and July 2020. Besides, seven RA patients fulfilling the 2010 EULAR/ACR diagnostic criteria were enrolled. Patient blood samples were provided by the Imm-Rheum Biobank of the Department of Rheumatology and Clinical Immunology, University Medical Center Freiburg. Clinical characteristics are given in Table 1. Demographically matched healthy individuals were obtained from the Institute for Cell and Gene Therapy at the University Medical Center Freiburg. They had no history of autoimmune disease, cancer, chronic viral infection, or any other inflammatory syndrome. The study was approved by the Institutional Review Board (Ek 218/20, Ek 383/19). Active disease was defined as new onset or recurrence of symptoms that are associated with AAV combined with an increase in C-reactive protein (CRP), which was not explained by an infection, and a Birmingham Vasculitis Activity Score (BVAS) ≥ 1 (28).
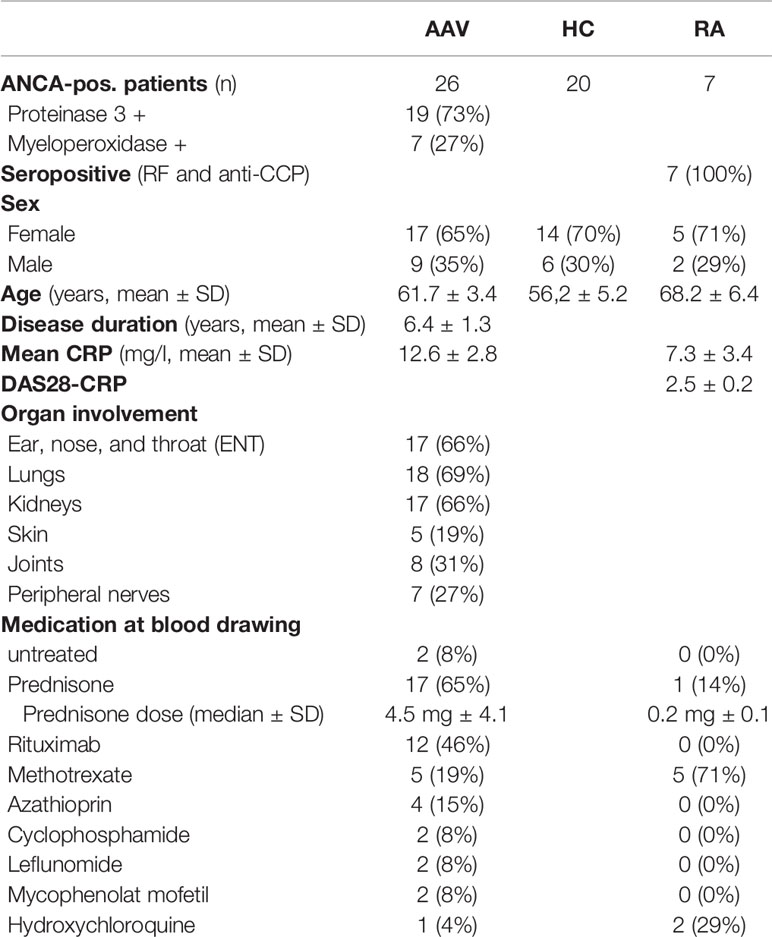
Table 1 Clinical characteristics of patients and controls.
PR3- and MPO-ELISA
The ANCA staining pattern (cytoplasmatic or perinuclear) was assessed by indirect immunofluorescence. ANCA specificity for proteinase 3 (PR3, Organtec) or myeloperoxidase (MPO, Euroimmun) was measured by enzyme-linked immunosorbent assay (ELISA) and interpreted according to the manufacturers’ reference ranges with the upper limit of the normal of <10 U/ml for PR3 and <20 U/ml for MPO.
Cells and Culture
Peripheral blood mononuclear cells (PBMCs) were isolated from the peripheral blood by density gradient centrifugation with Lymphoprep (anprotec). Monocytes were isolated using EasySep Human Monocyte Enrichment Kit without CD16 Depletion (Stemcell Technologies) and stimulated with IFNγ (100 IU/ml; PeproTech) or TNFα (5 ng/ml; BioLegend) for 24 hours. To inhibit lysosomal function, cells were treated with Bafilomycin A1 (20 nM; Cayman Chemicals) or chloroquine (10 μM; Sigma-Aldrich). For additional experiments, hydrocortisone (Sigma-Aldrich) was added or monocytes were pre-treated with TNFα (5ng/ml) for 30 minutes and stimulated with 5 μg/ml anti-PR3 (Santa Cruz Biotechnology) or anti-MPO (Miltenyi Biotec).
In co-culture experiments, monocytes were pretreated with IFNγ (100 IU/ml; PeproTech) for 24 hours and then co-cultured with CD4+ T cells in a ratio of 1:3 (400,000 monocytes to 1.2 million T cells). CD4+ T cells were derived from different HLA-mismatched healthy donors and were distributed equally throughout experiments in order to assess the T cell response based on the difference of the monocyte population. To assess early T cell activation, CD4+CD25+ T cells were quantified by flow cytometry after 48 hours of co-culture. To assess T cell proliferation, CD4+ T cells were labeled with carboxyfluorescein succinimidyl ester (CFSE) and co-cultured with monocytes for 5 days. Proliferation rates were analyzed by CFSE dilution. Monocyte-derived macrophages were differentiated in RPMI 1640 medium (Life technologies) supplemented with 10% FBS (Gibco) and 20 ng/ml M-CSF (BioLegend) or 20 ng/ml GM-CSF (BioLegend) for 6 days as reported previously (29). Cells were detached using StemPro Accutase Cell Dissociation (Life Technologies, Thermo Fisher).
Quantitative RT-PCR
Total RNA was extracted with Quick-RNA Microprep Kit (Zymo Research), cDNA was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Gene expression was determined using SYBR Select Master Mix (Applied Biosystems) on an OneStepPlus RT-PCR machine (Applied Biosystems). Used primers are listed below (Table 2). Gene transcript numbers were adjusted relative to β-actin transcripts.

Table 2 Primer list.
Flow Cytometry
For intracellular staining, cells were permeabilized using the Cytofix/Cytoperm Kit (BD Biosciences). Surface staining and intracellular protein evaluation were done using BD LSR Fortessa. Data were analyzed with FlowJo software (Tree Star). The following antibodies or stainings were used: CMTM6 (biorbyt), PD-L1, PD-L2, CD80, CD86, CD4, CD25 (all BioLegend), and CFSE (Molecular Probes).
Statistical Analysis
Statistical significance was assessed after data sets were tested for normal Gaussian distribution by D’Agostino normality testing. For data sets with normal distribution, parametric testing was applied (unpaired t-test and paired t-test). For other data sets, non-parametric testing was applied (Mann-Whitney test, Wilcoxon test, Kruskal-Wallis test with Dunn’s multiple comparisons test, and Spearman correlation). All data were analyzed by Prism V.9.1.0 (GraphPad).
Results
Reduced Frequency of PD-L1+ Monocytes in Peripheral Blood of AAV Patients
Monocytes from patients with PR3- or MPO-ANCA-positive AAV were tested for surface expression of the immune checkpoint molecule PD-L1. The frequency of PD-L1+ monocytes in AAV patients was reduced compared to cells from healthy control (HC) donors (Figure 1A). A reduction of PD-L1 expression on AAV monocytes was observed across all monocyte subsets (Figure 1B; for subset identification see Supplementary Figure 1). To evaluate whether this reduction in PD-L1 expression is associated with defects in the expression of other inhibitory or stimulatory molecules, we tested the surface expression of PD-L2, CD80, and CD86. While the inhibitory PD-L2 and the stimulatory CD80 were expressed only weakly, almost all monocytes expressed CD86. Overall, no differences between HC and AAV patients were detected (Figures 1C–E). Together, these data demonstrate that patient-derived monocytes have a defect in the expression of PD-L1.
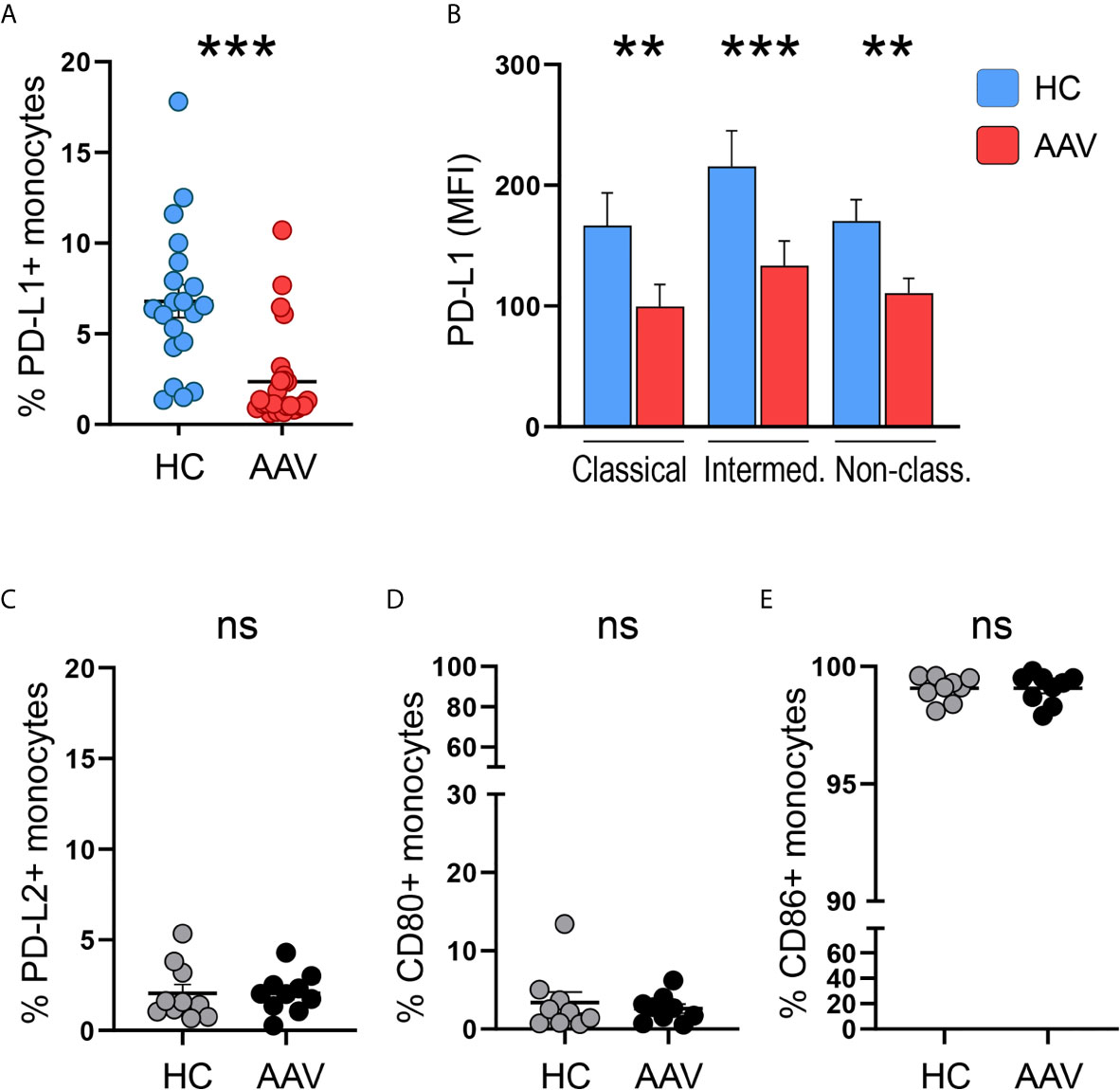
Figure 1 Less PD-L1+ monocytes in patients with AAV. (A) Percentages of PD-L1+ monocytes in HC (n=20) and AAV patients (n=26). (B) Surface expression of PD-L1 (MFI) on classical (CD14+CD16-), intermediate (CD14+CD16+), and non-classical monocytes (CD14dim CD16++) in healthy control donors (n=20) and AAV patients (n=26). (C–E) Percentages of PD-L2+, CD80+, and CD86+ monocytes in HC and AAV patients (n=9-10 each cohort). Mann-Whitney (A, B, D) test or unpaired t-test (C, E) were applied. **P<0.01; ***P<0.001. Bar graph shows mean ± SEM. AAV, ANCA-associated vasculitis; HC, healthy control donors; MFI, mean fluorescence intensity; PD-L1, Programmed death-ligand 1; ns, statistically not significant.
AAV Monocytes Display a Defect in PD-L1 Presentation Upon Activation
Monocytes acquire PD-L1 expression through extracellular stimuli, e.g. when entering tissue sites and through cytokine stimulation. Analog to tissue infiltration, the culture of monocytes on tissue culture plastic alone is enough to induce PD-L1 expression (30). Still, the strongest known inducer of PD-L1 is IFNγ (31, 32). We tested for PD-L1 induction on monocytes from HC and AAV patients after 24 hours of in vitro culture alone or with additional stimulation by IFNγ. As a control, we used monocytes from patients with another chronic inflammatory disease, rheumatoid arthritis (RA), to assess the impact of chronic inflammation on PD-L1 expression.
When left untreated for 24 hours, induction of PD-L1 was weaker in monocytes from AAV patients compared to monocytes from HC and RA patients. After 24 hours of stimulation, IFNγ potently induced PD-L1 expression. Again, monocytes from AAV patients displayed less PD-L1 than HC and RA monocytes. In general, control monocytes from RA patients did not differ from HC monocytes in their PD-L1 expression (Figures 2A, B). We further tested TNFα as this cytokine was reported to have at least some effect on PD-L1 expression (31, 32). Overall, its effect was rather weak compared to IFNγ, and also in this set of experiments AAV monocytes failed to properly upregulate PD-L1 (Supplementary Figure 2).
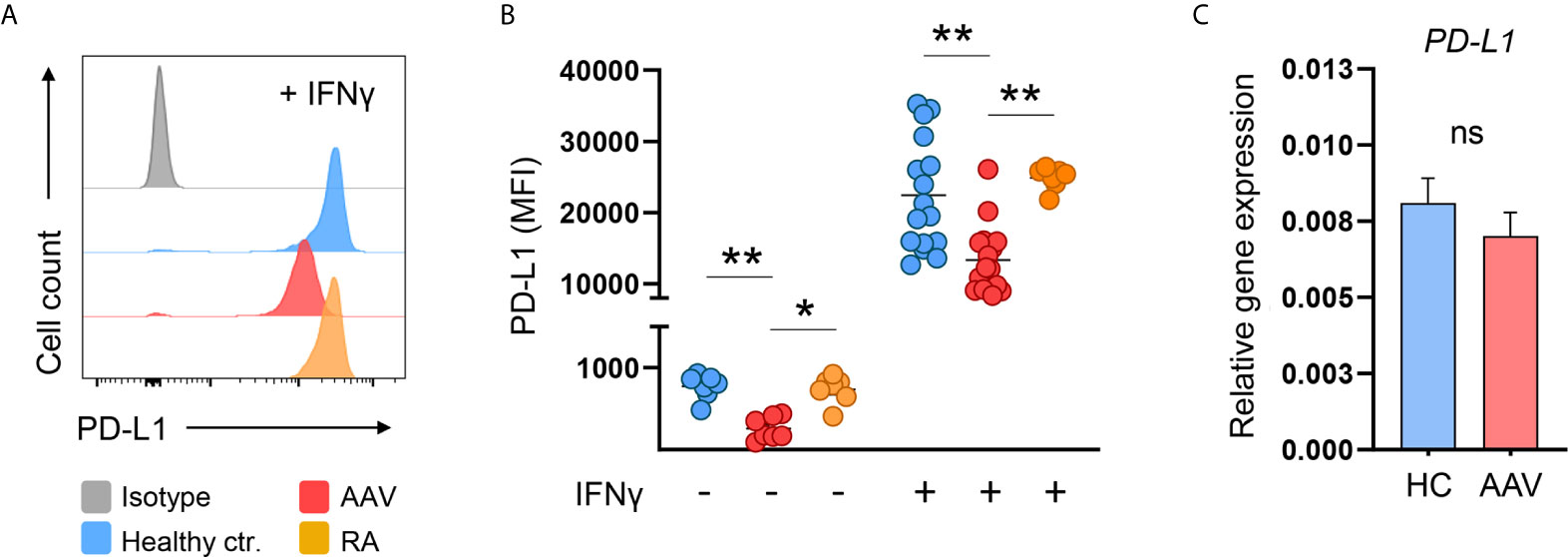
Figure 2 AAV monocytes fail to upregulate PD-L1. (A) Representative histograms and (B) summarizing scatter plot of PD-L1 surface expression of monocytes left untreated for 24 hours (n=7 each group) or after stimulation with IFNγ for 24 hours (100 IU/ml; HC n=21, AAV n=26, and RA n=7). (C) Gene expression of PD-L1 in monocytes from HC and AAV patients (n=14 each group) after stimulation with IFNγ for 24 hours measured by RT-PCR, relative to housekeeping gene β-actin. Kruskal-Wallis test with Dunn’s multiple comparisons test (B) and Mann-Whitney test (C) were applied. *P<0.05; **P<0.01. Bar graphs show mean ± SEM. AAV, ANCA-associated vasculitis; HC, healthy control donors; MFI, mean fluorescence intensity; PD-L1, Programmed death-ligand 1; RA, Rheumatoid arthritis; ANCA, Anti-neutrophil cytoplasmic antibodies; ns, statistically not significant.
The defect in PD-L1 protein presentation of AAV monocytes was not accompanied by a reduction of PD-L1 mRNA on the transcriptional level (Figure 2C). Thus, a global defect in enhancing PD-L1 expression upon activation was detected in AAV monocytes that appeared to be regulated at the protein level.
Current Medication of AAV Patients Is Not Associated With PD-L1 Expression
As the majority of AAV patients received medical treatment (Table 1), we performed a subgroup analysis and examined PD-L1 expression of AAV patients depending on their medication.
PD-L1 was low on monocytes from untreated patients on circulating cells (Figure 3A) and after activation with IFNγ (Figure 3D) but due to the low sample size (n=2) no statistical conclusion can be drawn. We further analyzed subgroups based on cortisone intake (Figures 3B, E) and rituximab infusion (Figures 3C, F) and observed no relevant effect.
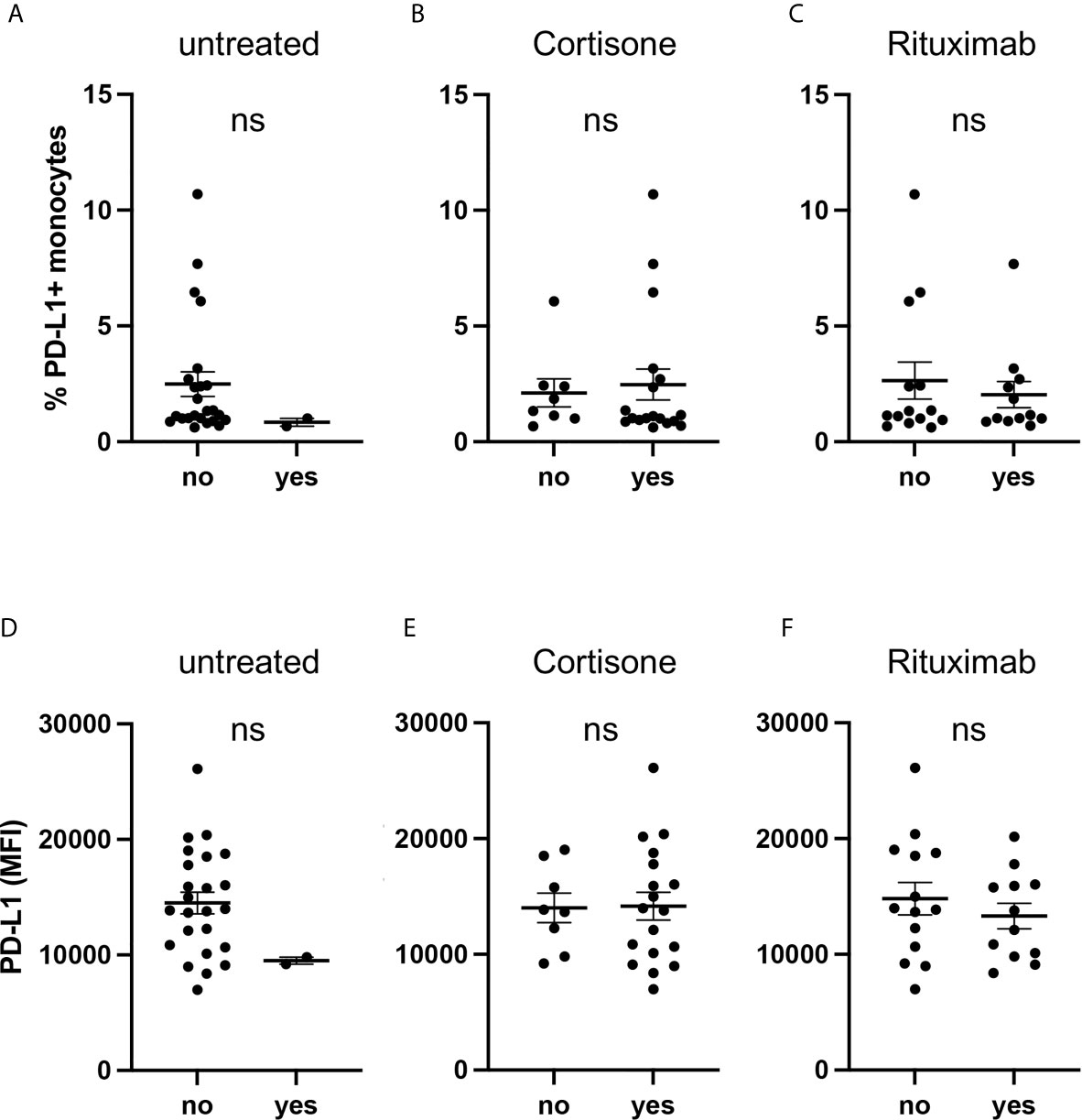
Figure 3 Current medication of AAV patients is not associated with PD-L1 expression. Percentages of PD-L1+ monocytes (A–C) and PD-L1 expression of monocytes stimulated with IFNγ for 24 hours (D–F) from AAV patients subgrouped depending on their medication. Mann-Whitney test (A–D) and unpaired t-test (E, F) were applied. MFI, mean fluorescence intensity; PD- L1, Programmed death-ligand 1; ns, statistically not significant.
Glucocorticoids (GC) have pleiotropic effects and can act directly on monocytes. We, therefore, treated GC-naïve monocytes with hydrocortisone in different concentrations, either in the presence or absence of additional IFNγ. In summary, hydrocortisone did not alter PD-L1 expression (Supplementary Figure 3).
These data indicate that the medication of AAV patients did not bias their monocytes towards low PD-L1 expression.
PD-L1 Expression on Monocytes Correlates With Disease Activity Markers
We hypothesized that the reduced frequency of PD-L1+ monocytes in the peripheral blood of AAV patients and the induced PD-L1 expression after cell activation are correlated with disease activity.
The low frequency of circulating PD-L1+ monocytes was correlated with high ANCA titers in AAV patients (Supplementary Figure 4A), while no correlation was observed between C-reactive protein (CRP) serum concentrations, blood monocyte counts, and renal organ involvement of AAV with the frequency of PD-L1+ monocytes (Supplementary Figures 4B, C, E). Although there was a trend for patients with active disease to show lower frequencies of PD-L1+ monocytes, it did not reach statistical significance (Supplementary Figure 4D).
IFNγ-induced PD-L1 expression correlated inversely with ANCA titers as well as CRP serum concentrations of AAV patients (Figures 4A, B), while blood monocyte counts did not correlate with PD-L1 (Figure 4C). Moreover, PD-L1 expression after stimulation with IFNγ was lower in patients with active disease (Figure 4D) but similar between patients with or without kidney involvement of AAV (Figure 4E).
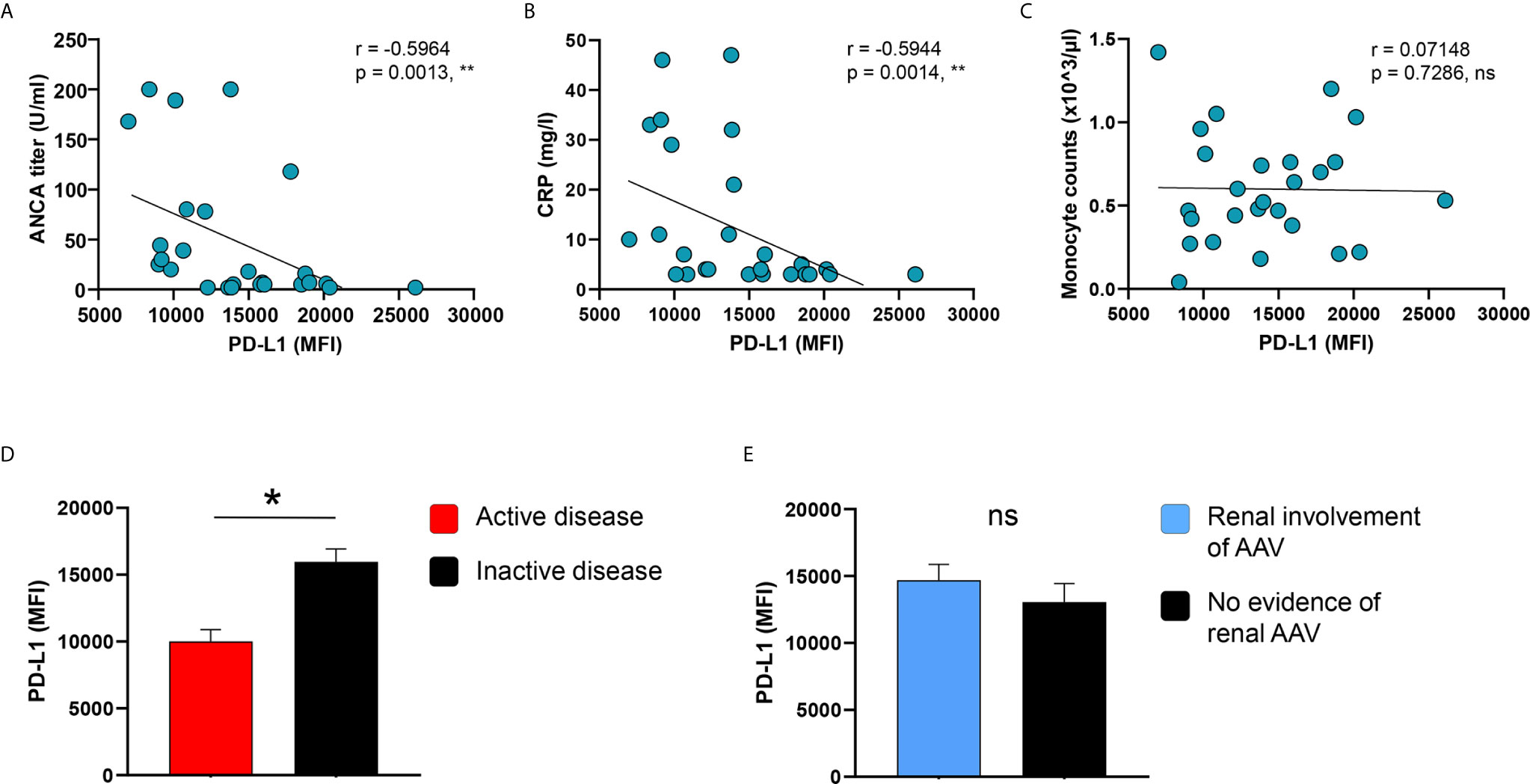
Figure 4 PD-L1 induction on monocytes correlates with disease markers and activity. (A–C) Correlation of IFNγ-induced PD-L1 expression with ANCA titers, serum levels of CRP, and blood monocyte counts in patients with AAV (n=26). IFNγ-induced PD-L1 expression on monocytes compared between AAV patients with (D) either active or inactive disease (active n=8, inactive n=18) and (E) with or without renal AAV (renal AAV n=17, without renal AAV n=9). Spearman correlation (A–C) and unpaired t-test (D, E) were applied. *P<0.05; **P<0.01. Bar graph shows mean ± SEM. AAV, ANCA-associated vasculitis; HC, healthy control donors; MFI, mean fluorescence intensity; PD-L1, Programmed death-ligand 1; ANCA, Anti-neutrophil cytoplasmic antibodies; CRP, C-reactive protein; ns, statistically not significant.
As high ANCA titers correlated with low PD-L1 expression, we tested whether direct stimulation of monocytes with anti-PR3 or anti-MPO antibodies could downregulate PD-L1. Short-term treatment (4 hours) did not alter PD-L1 expression in healthy or AAV monocytes (Supplementary Figures 5A, B). After 24 hours, anti-MPO antibodies did not affect PD-L1 expression, while there was a trend for anti-PR3 antibodies to upregulated PD-L1 at least in some of the healthy and patient samples tested (Supplementary Figures 5C, D). Also, adding anti-PR3 or anti-MPO antibodies when cells are stimulated with IFNγ did not change PD-L1 levels (Supplementary Figure 6).
These data indicate that lower surface presentation of PD-L1 on monocytes is associated with high ANCA titers and disease activity. This effect is not attributed to direct effects of ANCA antibodies on PD-L1.
Blocking Lysosomal Function Restores PD-L1 Expression in AAV Monocytes That Have Low Expression of CMTM6
PD-L1 mRNA was not downregulated in AAV monocytes, indicating a regulation on the post-transcriptional level. Breakdown and degradation of surface PD-L1 have been reported to occur in lysosomes (33). Thus, we hypothesized that lysosomal degradation contributes to PD-L1 deficiency in AAV monocytes. Indeed, Bafilomycin A1 treatment that inhibits lysosomal function restored PD-L1 expression on AAV monocytes without affecting PD-L1 expression on HC monocytes (Figures 5A, B). A similar effect was observed when AAV monocytes were treated with chloroquine (Supplementary Figure 7B). One of the functions of chloroquine is the inhibition of lysosomes.
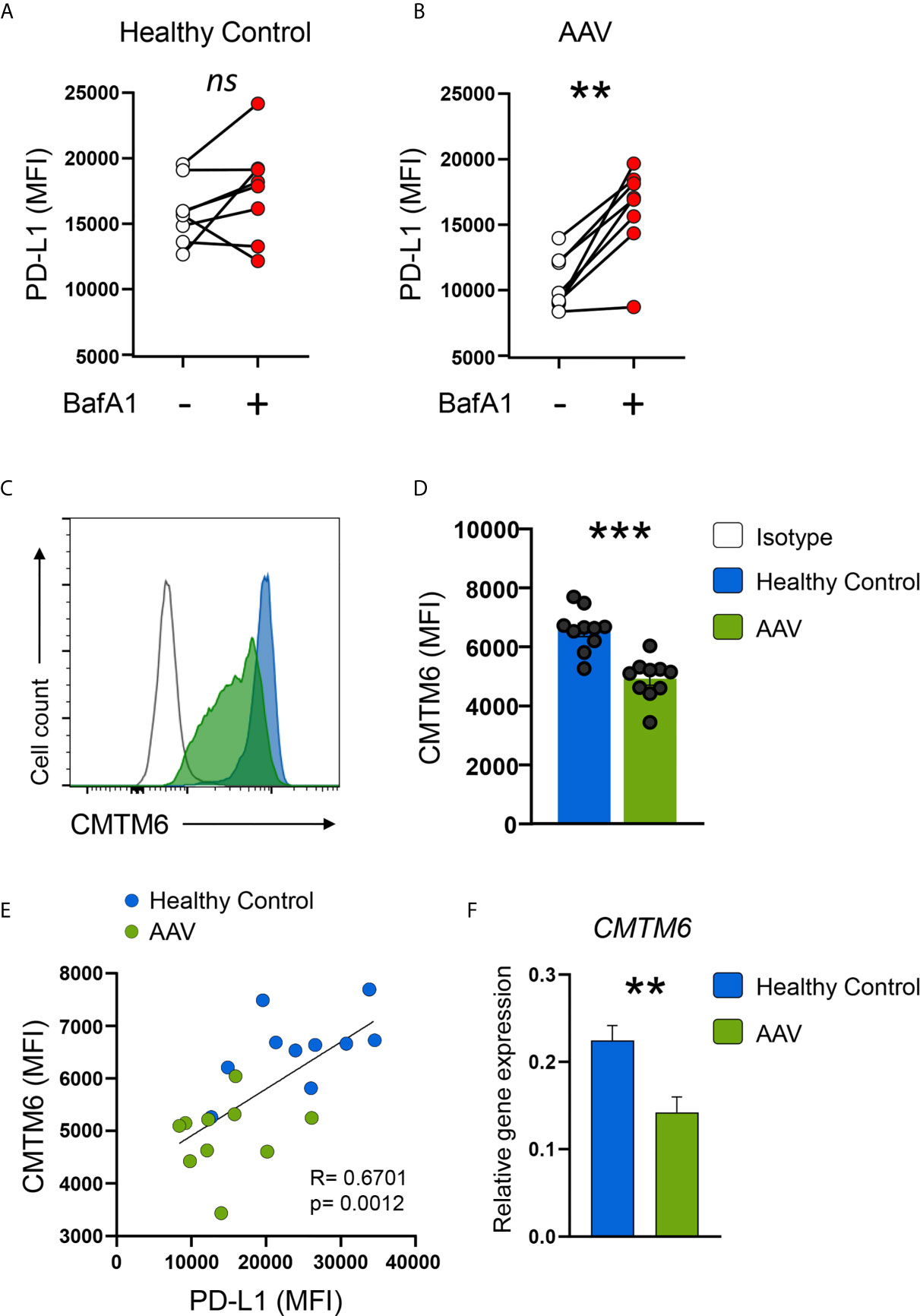
Figure 5 AAV monocytes are deficient for CMTM6, blocking lysosomal function restores capacity to upregulate PD-L1. (A, B) Monocytes from either HC (E) or AAV patients (F) were stimulated with IFNγ for 24 hours in the presence or absence of the lysosomal inhibitor BafA1 (20nM). Monocytes not treated with BafA1 inhibitor were treated with the corresponding vehicle. (C) Representative histograms of CMTM6 protein expression in IFNγ-stimulated (24 hours) monocytes from HC and AAV patients. (D) Summarizing scatter dot plot showing results from 10 experiments. (E) Correlation of PD-L1 expression with CMTM6 in IFNγ-stimulated monocytes in HC and AAV patients (each group n=10). (F) Gene expression of CMTM6 in monocytes from HC and AAV patients after stimulation with IFNγ for 24 hours measured by RT-PCR, relative to housekeeping gene β-actin. Paired t-test (A), Wilcoxon test (B), unpaired t-test (D), Spearman correlation (E), and Mann-Whitney test (F) were applied. ***P<0.01; ***P<0.001. Bar graph shows mean ± SEM. AAV, ANCA-associated vasculitis; HC, healthy control donors; MFI, mean fluorescence intensity; PD-L1, Programmed death-ligand 1; CMTM6, CKLF-like MARVEL transmembrane domain containing 6; BafA1, Bafilomycin A1; ns, statistically not significant.
The protein chemokine-like factor-like MARVEL transmembrane domain containing family member 6 (CMTM6) emerged as a master regulator of the PD-L1 protein pool by preventing PD-L1 from being targeted for lysosome-mediated degradation (25, 26). CMTM6 protein levels were reduced in circulating AAV monocytes (Supplementary Figure 5B) as wells as after activation with IFNγ (Figures 5C, D). CMTM6 protein levels correlated with PD-L1 expression (Figure 5E). Lower expression of CMTM6 in AAV monocytes corresponded to a reduction in CMTM6 mRNA (Figure 5F).
Collectively, these data demonstrated that CMTM6 is reduced in AAV monocytes, thus facilitating lysosomal degradation of PD-L1. Blocking lysosomal function restored their capacity to upregulate PD-L1.
PD-L1/CMTM6-Defect Is Preserved in Monocyte-Derived Macrophages
Monocytes can alter their phenotype based on environmental signals, e.g. they can differentiate into monocyte-derived macrophages. To study whether the defect in PD-L1 and CMTM6 expression was present also in macrophages, we differentiated monocytes with M-CSF or GM-CSF as previously described (29). After 6 days of differentiation, GM-CSF was a more potent inducer of PD-L1 than M-CSF. Monocyte-derived macrophages from AAV patients had lower expression of PD-L1 after differentiation with GM-CSF; after M-CSF differentiation there was a trend for lower PD-L1 expression in patients (Figures 6A, B) and CMTM6 (Figure 6C) compared to macrophages from HC donors. This suggests that the underlying defect for PD-L1 deficiency is imprinted to monocytes from AAV patients and carried on to corresponding cells once they differentiate.
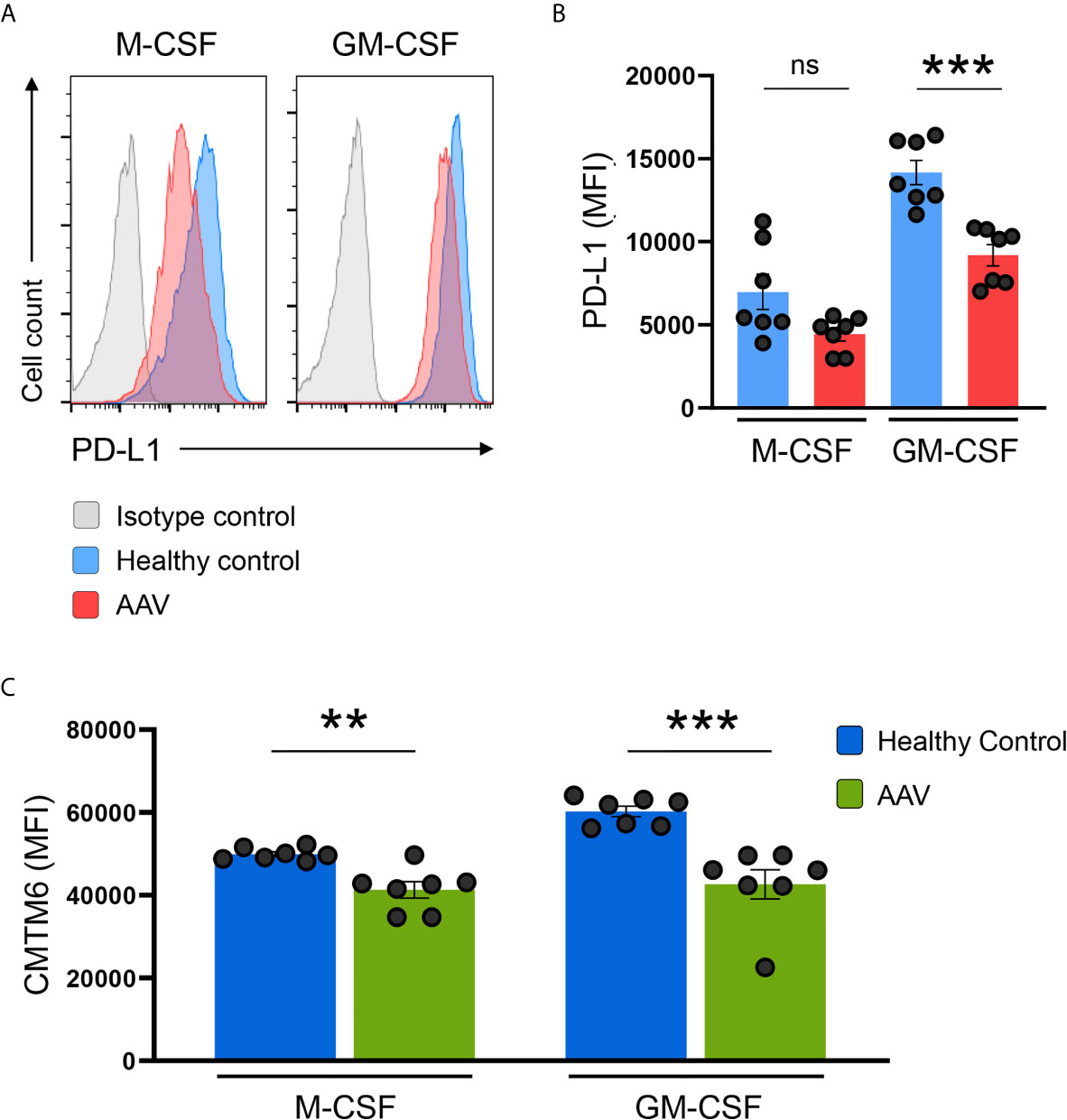
Figure 6 PD-L1/CMTM6-defect is passed to monocyte-derived macrophages. (A) Representative histograms of PD-L1 expression (MFI) on monocyte-derived macrophages after differentiation with M-CSF or GM-CSF for 6 days. (B) Summarizing scatter dot plot presenting results from experiments with cells from HC or AAV patients (n=7 each group). (C) Expression of CMTM6 protein in monocyte-derived macrophages from HC or AAV patients (n=7 each group). Mann-Whitney test (B, C) was applied. **P<0.01; ***P<0.001. Bar graph shows mean ± SEM. Bar graph shows mean ± SEM. AAV, ANCA-associated vasculitis; HC, healthy control donors; MFI, mean fluorescence intensity; PD-L1, Programmed death-ligand 1; M-CSF, macrophage colony-stimulating factor; GM-CSF, Granulocyte-macrophage colony-stimulating factor; ns, statistically not significant.
Monocytes From AAV Patients Show an Enhanced Stimulatory Capacity
To examine the ability of PD-L1lo AAV monocytes to stimulate T cells, we measured monocyte-induced T cell activation and expansion (in HC-derived T cells) by adapting a previously published co-culture model (16). AAV monocytes and HC monocytes were cultured with CFSE-labeled CD4+ T cells, after 5 days frequencies of dividing CD4+ T cells were measured. Frequencies of proliferating CD4+ T cells were higher in co-cultures with AAV monocytes (Figures 7A, B). Additionally, AAV monocytes enhanced early T cell activation as measured by the frequency of CD4+ CD25+ T cells after 48 hours (Figures 7C, D).
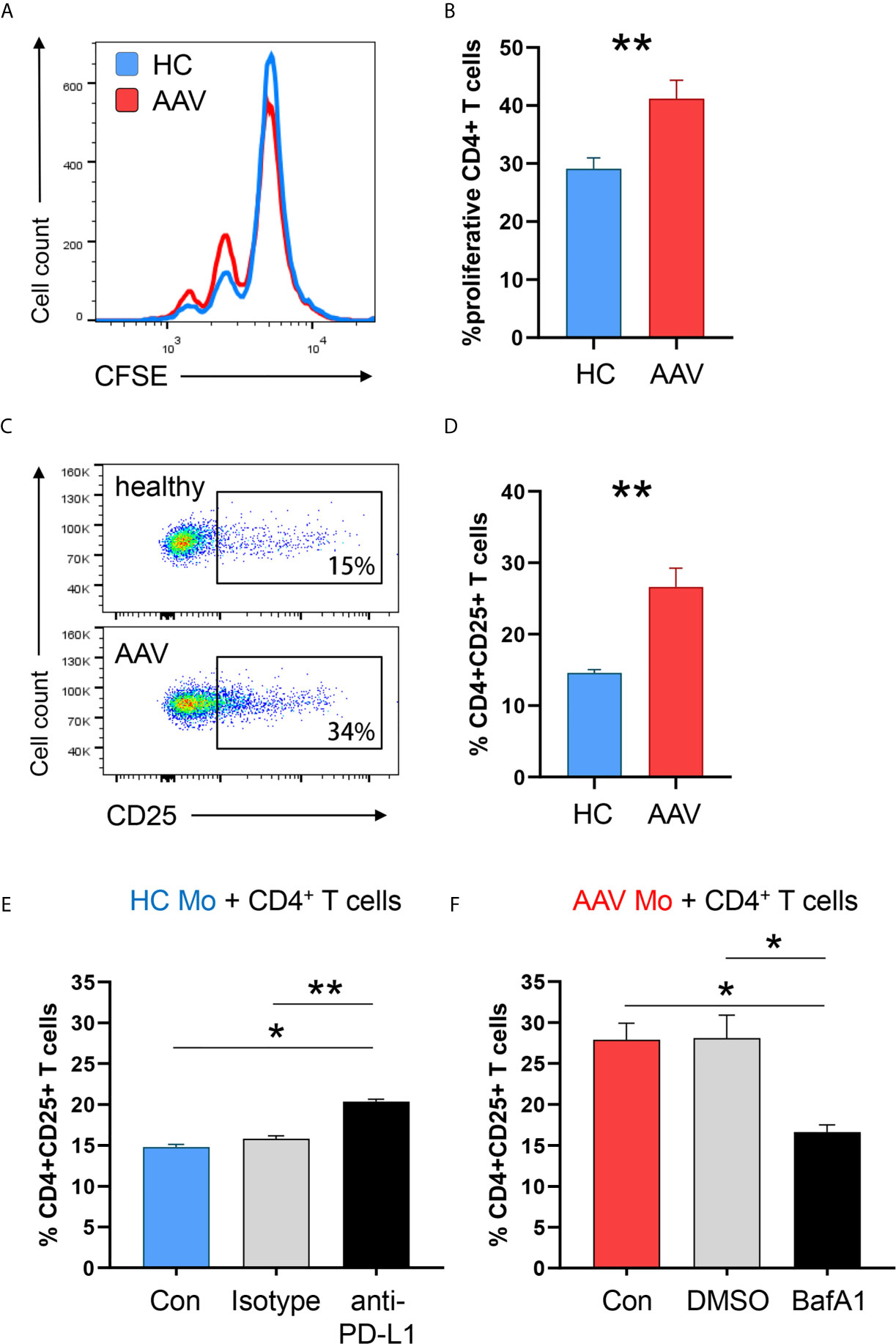
Figure 7 PD-L1lo monocytes from AAV patients show enhanced stimulatory capacity. Monocytes from healthy or AAV donors were pretreated with IFNγ for 24h. Then, their capacity to stimulate T cells was probed by coculturing them with CD4+ T cells from healthy donors (ratio monocytes/T cells 1:3). T-cell proliferation was determined through CFSE dilution and T-cell activation was quantified by the frequency of CD4+CD25+ T cells. (A) Proliferation of CD4+ T cells was measured by flow cytometry after 5 days of co-culture. Representative histograms of CSFE expression. (B) Frequencies of proliferating CD4+ T cells when cocultured with either HC or AAV patient-derived monocytes (n=8 each group). (C) Activated CD4+CD25+ T cells quantified by flow cytometry after 48 h. (D) Percentage of activated CD4+ T cells after coculture (n=6 each group). (E) Co-culture with monocytes from healthy donors was performed in the presence of anti-PD-L1 antibodies or isotype control. Frequencies of activated CD4+CD25+ T cells from 6 independent experiments (isotype n=3) were measured after 48 h by flow cytometry. (F) Co-culture with monocytes from AAV patients was performed after monocytes were pre-treated with the lysosomal inhibitor BafA1 (20nM) or DMSO vector control. Frequencies of activated CD4+CD25+ T cells from 6 independent experiments (DMSO n=3) were measured after 48 h by flow cytometry. Mann-Whitney test (B), unpaired t-test (D) and Kruskal-Wallis test with Dunn’s multiple comparisons test (E, F) were applied. *P<0.05; **P<0.01. Bar graph shows mean ± SEM. Bar graph shows mean ± SEM. AAV, ANCA-associated vasculitis; HC, healthy control donors; MFI, mean fluorescence intensity; PD-L1, Programmed death-ligand 1; M-CSF, macrophage colony-stimulating factor; GM-CSF, Granulocyte-macrophage colony-stimulating factor.
To understand whether the enhanced activation of T cells primed by monocytes is directly attributed to PD-L1 expression, anti–PD-L1 antibodies were added to co-cultures with monocytes from HC donors. Blocking PD-L1 on HC monocytes simulated PD-L1 deficiency and increased CD4+ T-cell activation (Figure 7E), confirming published data (16). To further elucidate whether blockade of PD-L1 degradation in lysosomes normalizes the hyperstimulatory behavior of patient-derived cells, AAV monocytes were pre-treated with the lysosomal inhibitor Bafilomycin A1. Inhibiting lysosome function in AAV monocytes restored their ability to balance immune cell interaction with T cells, resulting in less CD4+ T cell activation (Figure 7F).
In essence, PD-L1lo monocytes from AAV patients cause enhanced stimulation of CD4+ T cells. Targeting lysosomal degradation of PD-L1 in AAV monocytes corrected this phenotype.
Discussion
The development of autoimmune small-vessel vasculitis has been observed after checkpoint inhibitor therapy (21–24), indicating a relevant role of immune checkpoint molecules in the disease process. However, no molecular alterations of such molecules in the immune system of patients with AAV have been reported so far. In this study, we found that monocytes from AAV patients show a defect in presenting the immunoinhibitory checkpoint PD-L1 leading to enhanced stimulation of T cells.
Physiologically, the co-inhibitory ligand PD-L1 shows limited expression on circulating monocytes (30) and in normal tissues (11). Induction of PD-L1 on antigen-presenting cells occurs rapidly after cell activation to instigate a negative feedback loop thereby preventing overactivation of the adaptive immune system, e.g. of CD4+ T cells. Failure of PD-L1 induction promotes autoimmunity as reported for giant cell arteritis, an immune-mediated large vessel vasculitis, in which PD-L1-deficient dendritic cells facilitate inflammatory vascular damage (16). In AAV lesions, monocyte infiltration is a characteristic hallmark and vascular infiltrates show a predominance of monocytes and monocyte-derived macrophages. When entering tissue sites, monocytes encounter a multitude of pro- and anti-inflammatory stimuli and their response determines whether inflammation resolves or amplifies. In the case of AAV, PD-L1lo monocytes may disturb the immunomodulatory PD-L1/PD-1 axis, thereby contributing to enhanced activation of CD4+ T cells and, thus, consolidating the chronic inflammatory process.
PD-L1 protein deficiency was not accompanied by decreased PD-L1 mRNA transcripts in AAV monocytes, which indicated a regulation on the post-transcriptional level. Cleavage of PD-L1 by metalloproteinases (MMP) has been reported (34, 35) with tumor-derived MMP-13 potently degrading PD-L1 (36). In our experiments, inhibition of MMP-13 did not affect PD-L1 expression of monocytes (data not shown). Lysosomes are the cell’s degradation center and are responsible for the breakdown of proteins, polysaccharides, and complex lipids (37). Recently, two independent reports identified CMTM6, a protein of previously unknown function, as a major regulator of the PD-L1 protein pool. CMTM6 co-localizes with PD-L1 and prevents PD-L1 from being targeted for lysosome-mediated degradation (25, 26). Blocking lysosomal function with a specific inhibitor corrected PD-L1 deficiency in AAV monocytes indicating increased lysosomal breakdown of PD-L1 due to low levels of CMTM6. A similar effect was observed when AAV monocytes were treated with chloroquine. Chloroquine and hydroxychloroquine (HCQ) also impair lysosomal function but are less specific as they interfere with toll-like receptors and intracellular nucleic acid sensors (38). Interestingly, some groups report the successful use of HCQ in patients with AAV (39), and currently, a phase-II study is ongoing that evaluates HCQ in the treatment of AAV (HAVEN: Hydroxychloroquine in ANCA Vasculitis Evaluation, NCT04316494).
Emphasizing the role of monocyte PD-L1 expression in the disease course of AAV, higher ANCA titers correlated with lower numbers of circulating PD-L1+ monocytes in vasculitis patients. Moreover, their diminished capacity to present PD-L1 upon cell activation predicted higher ANCA titers, higher CRP serum concentrations, and active disease.
An unexpected finding of this study was the influence of colony-stimulating factors (CSFs) on PD-L1 expression. Higher PD-L1 surface expression was observed when monocytes were differentiated to macrophages with GM-CSF instead of M-CSF. In the literature, only one report describes the effect of GM-CSF on PD-L1, showing that tumor-derived GM-CSF induced PD-L1 expression in neutrophils (40). PD-L1 induction by GM-CSF could be part of a negative feedback loop to prevent uncontrolled inflammation sparked by more pro-inflammatory GM-CSF-differentiated macrophages. Alternatively, lower PD-L1 expression by M-CSF may reflect the greater lysosomal activity of M-CSF-derived macrophages (41). Serum levels of M-CSF are increased in AAV patients with active nephritis (42) and renal M-CSF production is upregulated in vasculitic glomeruli where it associates with local macrophage proliferation (43), suggesting an M-CSF-skewed macrophage phenotype in renal disease in AAV.
The design of this study bears limitations. The majority of AAV patients receive immunosuppressive therapy, which could alter any kind of read-out. AAV is a rare disease and often presents with severe symptoms making an immediate start of therapy inevitable. Our subgroup analysis as well as experiments testing the direct effects of cortisone on PD-L1 expression did not indicate evidence for a medication bias towards lower PD-L1 expression. Although we cannot exclude other factors in the natural course of the disease, the correlation of PD-L1 with markers of inflammation and disease activity suggests that low PD-L1 expression on monocytes is associated with disease activity.
Another limitation is the use of RA monocytes as disease control. To exclude that chronic inflammation lowers PD-L1 expression, we tested monocytes from RA patients, a typical chronic-inflammatory disease. It could be argued that inflammation in those patients is localized mainly in the joints and not in the vessel wall. Still, RA patients do have a component of systemic inflammation, which results in their increased cardiovascular risk (44) - as such a vascular inflammatory process. Further studies in other autoimmune disease would be needed to clarify whether low PD-L1 expression is specific for vasculitis. In connective tissue disease, in SLE for example, contradictory results have been reported with one group finding low PD-L1 expression on monocytes in a pediatric cohort (45) and another study reporting upregulation of PD-L1 on SLE monocytes (46).
In summary, this study identified a defective immunoinhibitory PD-L1 checkpoint on monocytes and monocyte-derived macrophages from patients with AAV. CMTM6-deficient vasculitic monocytes degrade PD-L1 in lysosomes, thus providing insufficient negative signaling to CD4+ cells, fostering the development of highly activated T cells in patients with autoimmune small-vessel vasculitis. Correcting this defect in monocytes by targeting lysosomal function may be a promising novel strategy to treat AAV, especially to maintain remission.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional Review Board (Ek 218/20, Ek 383/19) of the University of Freiburg. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
MZ, JT, and RV conceived the study. MZ performed experiments. MZ and NV analyzed data. BS contributed technical expertise. NC and MR enrolled patients and oversaw patient recruitment. MZ, NV, JT, and RV wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the “Else-Kröner-Fresenius-Stiftung” (NAKSYS– 016_Kolleg.03) providing a fellowship to MZ, and the European Regional Development Fund of the European Union (INTERREG V programs RARENET and PERSONALIS to RV and MR). MZ was funded by the Research Commission of the Faculty of Medicine, University of Freiburg. JT was supported by the Berta-Ottenstein-Programme for Advanced Clinician Scientists, Faculty of Medicine, University of Freiburg. NC was supported by the Ministry of Science, Research, and Arts Baden-Wurttemberg (Margarete von Wrangell Programme).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Theresa Schleyer from the Rheum-Imm Biobank for assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.673912/full#supplementary-material
References
1. Jennette JC, Falk RJ. Pathogenesis of Antineutrophil Cytoplasmic Autoantibody-Mediated Disease. Nat Rev Rheumatol (2014) 10(8):463–73. doi: 10.1038/nrrheum.2014.103
2. Weidner S, Carl M, Riess R, Rupprecht HD. Histologic Analysis of Renal Leukocyte Infiltration in Antineutrophil Cytoplasmic Antibody-Associated Vasculitis: Importance of Monocyte and Neutrophil Infiltration in Tissue Damage. Arthritis Rheum (2004) 50(11):3651–7. doi: 10.1002/art.20607
3. Zhao L, David MZ, Hyjek E, Chang A, Meehan SM. M2 Macrophage Infiltrates in the Early Stages of ANCA-associated Pauci-Immune Necrotizing GN. Clin J Am Soc Nephrol (2015) 10(1):54–62. doi: 10.2215/CJN.03230314
4. Muller Kobold AC, Kallenberg CG, Tervaert JW. Monocyte Activation in Patients With Wegener’s Granulomatosis. Ann Rheum Dis (1999) 58(4):237–45. doi: 10.1136/ard.58.4.237
5. Tarzi RM, Liu J, Schneiter S, Hill NR, Page TH, Cook HT, et al. CD14 Expression is Increased on Monocytes in Patients With Anti-Neutrophil Cytoplasm Antibody (ANCA)-Associated Vasculitis and Correlates With the Expression of ANCA Autoantigens. Clin Exp Immunol (2015) 181(1):65–75. doi: 10.1111/cei.12625
6. Wikman A, Lundahl J, Jacobson SH. Sustained Monocyte Activation in Clinical Remission of Systemic Vasculitis. Inflammation (2008) 31(6):384–90. doi: 10.1007/s10753-008-9089-8
7. Haller H, Eichhorn J, Pieper K, Gobel U, Luft FC. Circulating Leukocyte Integrin Expression in Wegener’s Granulomatosis. J Am Soc Nephrol (1996) 7(1):40–8. doi: 10.1681/ASN.V7140
8. Hattar K, van Burck S, Bickenbach A, Grandel U, Maus U, Lohmeyer J, et al. Anti-Proteinase 3 Antibodies (c-ANCA) Prime CD14-dependent Leukocyte Activation. J Leukoc Biol (2005) 78(4):992–1000. doi: 10.1189/jlb.0902442
9. Weidner S, Neupert W, Goppelt-Struebe M, Rupprecht HD. Antineutrophil Cytoplasmic Antibodies Induce Human Monocytes to Produce Oxygen Radicals In Vitro. Arthritis Rheum (2001) 44(7):1698–706. doi: 10.1002/1529-0131(200107)44:7<1698::AID-ART294>3.0.CO;2-J
10. Akiyama M, Zeisbrich M, Ibrahim N, Ohtsuki S, Berry GJ, Hwang PH, et al. Neutrophil Extracellular Traps Induce Tissue-Invasive Monocytes in Granulomatosis With Polyangiitis. Front Immunol (2019) 10:2617. doi: 10.3389/fimmu.2019.02617
11. Weyand CM, Berry GJ, Goronzy JJ. The Immunoinhibitory PD-1/PD-L1 Pathway in Inflammatory Blood Vessel Disease. J Leukoc Biol (2018) 103(3):565–75. doi: 10.1189/jlb.3MA0717-283
12. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J Exp Med (2000) 192(7):1027–34. doi: 10.1084/jem.192.7.1027
13. Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, et al. Tissue Expression of PD-L1 Mediates Peripheral T Cell Tolerance. J Exp Med (2006) 203(4):883–95. doi: 10.1084/jem.20051776
14. Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, et al. PD-L1-deficient Mice Show That PD-L1 on T Cells, Antigen-Presenting Cells, and Host Tissues Negatively Regulates T Cells. Proc Natl Acad Sci USA (2004) 101(29):10691–6. doi: 10.1073/pnas.0307252101
15. Watanabe R, Hilhorst M, Zhang H, Zeisbrich M, Berry GJ, Wallis BB, et al. Glucose Metabolism Controls Disease-Specific Signatures of Macrophage Effector Functions. JCI Insight (2018) 3(20):e123047. doi: 10.1172/jci.insight.123047
16. Zhang H, Watanabe R, Berry GJ, Vaglio A, Liao YJ, Warrington KJ, et al. Immunoinhibitory Checkpoint Deficiency in Medium and Large Vessel Vasculitis. Proc Natl Acad Sci USA (2017) 114(6):E970–E9. doi: 10.1073/pnas.1616848114
17. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and Activity of anti-PD-L1 Antibody in Patients With Advanced Cancer. N Engl J Med (2012) 366(26):2455–65. doi: 10.1056/NEJMoa1200694
18. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and Tumor Responses With Lambrolizumab (anti-PD-1) in Melanoma. N Engl J Med (2013) 369(2):134–44. doi: 10.1056/NEJMoa1305133
19. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, Activity, and Immune Correlates of anti-PD-1 Antibody in Cancer. N Engl J Med (2012) 366(26):2443–54. doi: 10.1056/NEJMoa1200690
20. Suarez-Almazor ME, Kim ST, Abdel-Wahab N, Diab A. Review: Immune-Related Adverse Events With Use of Checkpoint Inhibitors for Immunotherapy of Cancer. Arthritis Rheumatol (2017) 69(4):687–99. doi: 10.1002/art.40043
21. Sibille A, Alfieri R, Malaise O, Detrembleur N, Pirotte M, Louis R, et al. Granulomatosis With Polyangiitis in a Patient on Programmed Death-1 Inhibitor for Advanced non-Small-Cell Lung Cancer. Front Oncol (2019) 9:478. doi: 10.3389/fonc.2019.00478
22. van den Brom RR, Abdulahad WH, Rutgers A, Kroesen BJ, Roozendaal C, de Groot DJ, et al. Rapid Granulomatosis With Polyangiitis Induced by Immune Checkpoint Inhibition. Rheumatol (Oxford) (2016) 55(6):1143–5. doi: 10.1093/rheumatology/kew063
23. Mamlouk O, Lin JS, Abdelrahim M, Tchakarov AS, Glass WF, Selamet U, et al. Checkpoint Inhibitor-Related Renal Vasculitis and Use of Rituximab. J Immunother Cancer (2020) 8(2):e000750. doi: 10.1136/jitc-2020-000750
24. Nabel CS, Severgnini M, Hung YP, Cunningham-Bussel A, Gjini E, Kleinsteuber K, et al. Anti-PD-1 Immunotherapy-Induced Flare of a Known Underlying Relapsing Vasculitis Mimicking Recurrent Cancer. Oncologist (2019) 24(8):1013–21. doi: 10.1634/theoncologist.2018-0633
25. Burr ML, Sparbier CE, Chan YC, Williamson JC, Woods K, Beavis PA, et al. CMTM6 Maintains the Expression of PD-L1 and Regulates Anti-Tumour Immunity. Nature (2017) 549(7670):101–5. doi: 10.1038/nature23643
26. Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, de Vries E, Wu W, et al. Identification of CMTM6 and CMTM4 as PD-L1 Protein Regulators. Nature (2017) 549(7670):106–10. doi: 10.1038/nature23669
27. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum (2013) 65(1):1–11. doi: 10.1002/art.37715
28. Luqmani RA, Bacon PA, Moots RJ, Janssen BA, Pall A, Emery P, et al. Birmingham Vasculitis Activity Score (BVAS) in Systemic Necrotizing Vasculitis. QJM (1994) 87(11):671–8. doi: 10.1093/oxfordjournals.qjmed.a068882
29. Zeisbrich M, Yanes RE, Zhang H, Watanabe R, Li Y, Brosig L, et al. Hypermetabolic Macrophages in Rheumatoid Arthritis and Coronary Artery Disease Due to Glycogen Synthase Kinase 3b Inactivation. Ann Rheum Dis (2018) 77(7):1053–62. doi: 10.1136/annrheumdis-2017-212647
30. Brown JA, Dorfman DM, Ma FR, Sullivan EL, Munoz O, Wood CR, et al. Blockade of Programmed Death-1 Ligands on Dendritic Cells Enhances T Cell Activation and Cytokine Production. J Immunol (2003) 170(3):1257–66. doi: 10.4049/jimmunol.170.3.1257
31. Chen S, Crabill GA, Pritchard TS, McMiller TL, Wei P, Pardoll DM, et al. Mechanisms Regulating PD-L1 Expression on Tumor and Immune Cells. J Immunother Cancer (2019) 7(1):305. doi: 10.1186/s40425-019-0770-2
32. Hartley G, Regan D, Guth A, Dow S. Regulation of PD-L1 Expression on Murine Tumor-Associated Monocytes and Macrophages by Locally Produced TNF-Alpha. Cancer Immunol Immunother (2017) 66(4):523–35. doi: 10.1007/s00262-017-1955-5
33. Gou Q, Dong C, Xu H, Khan B, Jin J, Liu Q, et al. Pd-L1 Degradation Pathway and Immunotherapy for Cancer. Cell Death Dis (2020) 11(11):955. doi: 10.1038/s41419-020-03140-2
34. Aguirre JE, Beswick EJ, Grim C, Uribe G, Tafoya M, Chacon Palma G, et al. Matrix Metalloproteinases Cleave Membrane-Bound PD-L1 on CD90+ (Myo-)Fibroblasts in Crohn’s Disease and Regulate Th1/Th17 Cell Responses. Int Immunol (2020) 32(1):57–68. doi: 10.1093/intimm/dxz060
35. Romero Y, Wise R, Zolkiewska A. Proteolytic Processing of PD-L1 by ADAM Proteases in Breast Cancer Cells. Cancer Immunol Immunother (2020) 69(1):43–55. doi: 10.1007/s00262-019-02437-2
36. Hira-Miyazawa M, Nakamura H, Hirai M, Kobayashi Y, Kitahara H, Bou-Gharios G, et al. Regulation of Programmed-Death Ligand in the Human Head and Neck Squamous Cell Carcinoma Microenvironment is Mediated Through Matrix Metalloproteinase-Mediated Proteolytic Cleavage. Int J Oncol (2018) 52(2):379–88. doi: 10.3892/ijo.2017.4221
37. Xu H, Ren D. Lysosomal Physiology. Annu Rev Physiol (2015) 77:57–80. doi: 10.1146/annurev-physiol-021014-071649
38. Gies V, Bekaddour N, Dieudonne Y, Guffroy A, Frenger Q, Gros F, et al. Beyond Anti-viral Effects of Chloroquine/Hydroxychloroquine. Front Immunol (2020) 11:1409. doi: 10.3389/fimmu.2020.01409
39. Casian A, Sangle SR, D’Cruz DP. New Use for an Old Treatment: Hydroxychloroquine as a Potential Treatment for Systemic Vasculitis. Autoimmun Rev (2018) 17(7):660–4. doi: 10.1016/j.autrev.2018.01.016
40. Wang TT, Zhao YL, Peng LS, Chen N, Chen W, Lv YP, et al. Tumour-Activated Neutrophils in Gastric Cancer Foster Immune Suppression and Disease Progression Through GM-CSF-PD-L1 Pathway. Gut (2017) 66(11):1900–11. doi: 10.1136/gutjnl-2016-313075
41. Akagawa KS. Functional Heterogeneity of Colony-Stimulating Factor-Induced Human Monocyte-Derived Macrophages. Int J Hematol (2002) 76(1):27–34. doi: 10.1007/BF02982715
42. Ramirez GA, Blasi M, Sciorati C, Rovere-Querini P, Manfredi AA. Plasma Levels of M-CSF are Increased in ANCA-associated Vasculitides With Active Nephritis. Results Immunol (2015) 5:33–6. doi: 10.1016/j.rinim.2015.10.002
43. Isbel NM, Nikolic-Paterson DJ, Hill PA, Dowling J, Atkins RC. Local Macrophage Proliferation Correlates With Increased Renal M-CSF Expression in Human Glomerulonephritis. Nephrol Dial Transplant (2001) 16(8):1638–47. doi: 10.1093/ndt/16.8.1638
44. Hansildaar R, Vedder D, Baniaamam M, Tausche A-K, Gerritsen M, Nurmohamed MT, et al. Cardiovascular Risk in Inflammatory Arthritis: Rheumatoid Arthritis and Gout. Lancet Rheumatol (2021) 3(1):e58–70. doi: 10.1016/S2665-9913(20)30221-6
45. Mozaffarian N, Wiedeman AE, Stevens AM. Active Systemic Lupus Erythematosus is Associated With Failure of Antigen-Presenting Cells to Express Programmed Death Ligand-1. Rheumatol (Oxford) (2008) 47(9):1335–41. doi: 10.1093/rheumatology/ken256
Keywords: ANCA vasculitis, PD-L1, macrophages, lysosomes, immune checkpoint, vasculitis < rheumatic diseases, monocytes
Citation: Zeisbrich M, Chevalier N, Sehnert B, Rizzi M, Venhoff N, Thiel J and Voll RE (2021) CMTM6-Deficient Monocytes in ANCA-Associated Vasculitis Fail to Present the Immune Checkpoint PD-L1. Front. Immunol. 12:673912. doi: 10.3389/fimmu.2021.673912
Received: 28 February 2021; Accepted: 14 April 2021;
Published: 24 May 2021.
Edited by:
Alexandre Wagner Silva De Souza, Federal University of São Paulo, BrazilReviewed by:
Yannick van Sleen, University Medical Center Groningen, NetherlandsCarlo Chizzolini, Université de Genève, Switzerland
Copyright © 2021 Zeisbrich, Chevalier, Sehnert, Rizzi, Venhoff, Thiel and Voll. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus Zeisbrich, bWFya3VzLnplaXNicmljaEB1bmlrbGluaWstZnJlaWJ1cmcuZGU=
†These authors have contributed equally to this work and share last authorship