 Shota Aoyama
Shota Aoyama Ryosuke Nakagawa
Ryosuke Nakagawa James J. Mulé
James J. Mulé Adam W. Mailloux
Adam W. Mailloux- 1Department of Surgery, Institute of Gastroenterology, Tokyo Women’s Medical University, Tokyo, Japan
- 2Immunology Program, Moffitt Cancer Center, Tampa, FL, United States
- 3Cutaneous Oncology Program, Moffitt Cancer Center, Tampa, FL, United States
- 4Department of Microbiology and Immunology, University of Iowa, Iowa City, IA, United States
Tertiary lymphoid structures (TLS) are ectopically formed aggregates of organized lymphocytes and antigen-presenting cells that occur in solid tissues as part of a chronic inflammation response. Sharing structural and functional characteristics with conventional secondary lymphoid organs (SLO) including discrete T cell zones, B cell zones, marginal zones with antigen presenting cells, reticular stromal networks, and high endothelial venues (HEV), TLS are prominent centers of antigen presentation and adaptive immune activation within the periphery. TLS share many signaling axes and leukocyte recruitment schemes with SLO regarding their formation and function. In cancer, their presence confers positive prognostic value across a wide spectrum of indications, spurring interest in their artificial induction as either a new form of immunotherapy, or as a means to augment other cell or immunotherapies. Here, we review approaches for inducible (iTLS) that utilize chemokines, inflammatory factors, or cellular analogues vital to TLS formation and that often mirror conventional SLO organogenesis. This review also addresses biomaterials that have been or might be suitable for iTLS, and discusses remaining challenges facing iTLS manufacturing approaches for clinical translation.
Introduction
The presence of infiltrating immune cell populations is a prominent histological feature of most solid tumors that with some exceptions (1, 2), often confers positive prognostic significance across a wide spectrum of indications (3). This benefit is often contingent on the number and phenotypic makeup of the immune infiltrate, and on the ratios of beneficial effector cells to immune suppressive populations (3, 4). This may entail elevated numbers of activated CD8+ cytotoxic T cells (TC), type-I polarized CD4+ helper T (TH1) cells, and B cells, signifying an adaptive anti-tumor immune response (3, 5, 6). In a similar fashion, infiltrating antigen-presenting cells such as macrophage and dendritic cells (DC) confer positive prognostic value in many tumor types (7, 8), and in particular those antigen presenting cells with type I polarization attributes are especially equipped to support anti-tumor immunity (9, 10). It is an important understated fact that elements associated with antigen presentation and immune polarization are found inside solid tumors and confer prognostic benefit alongside effector lymphocyte populations. This infers that active antigen presentation, and the structural organization needed to support it, must occur at the tumor site; thus, an anti-tumor immune response is not limited to remote activation of effector lymphocytes in draining secondary lymphoid organs (SLO), but also occurs locally within and proximal to the tumor mass (4).
It is now understood that many tumors are associated with the presence of tertiary lymph node structures (TLS) (11). TLS consist of structural features analogous to conventional SLO, including discrete B cell zones, T cell zones, marginal zones with activated macrophage and DC, reticular fibroblast cell (RFC) networks (or RFC-like stromal networks), and vasculature permissive to immune cell extravasation (11–13). In mature TLS, this high level of organization can consist of networks of supportive infrastructure are compartmentalized just as they are in SLO, with activated mature DC supporting TH1 activation in T cell zones (14, 15), and follicular DC localizing to B cell zones in support of humoral immunity (16, 17). TLS form de novo in the microenvironment of solid tissues in response to protracted inflammatory stimuli, and may dissipate upon the resolution of inflammation (18). TLS can additionally foster tumor antigen presentation and T cell activation, including germinal centers (19, 20), B cell class switching (21), activated antigen presenting cells (22), and T cell clonal expansion (23, 24). In human cancers, TLS are associated with better disease outcomes across a broad spectrum of indications including ovarian (25, 26), metastatic melanoma (27, 28), breast (29, 30), colorectal (11, 31), and non-small cell lung cancers (7, 14), and can augment the efficacy of immunotherapies such as imune chackpoint inhibitors (28). In murine models, TLS can reduce orthotopic growth of colon carcinoma (32), melanoma (33), and fibrosarcoma (34). These associations and clear demonstrations of beneficial anti-tumor immunity by TLS embody a majority of scenarios that are overwhelmingly positive in nature and that provide a strong basis for pursuing the artificial induction of TLS as a therapeutic modality. However, there are reports in which TLS are associated with negative prognostication or disease progression. This is best exemplified in hepatocellular carcinoma (HCC) (35), and suggests that while TLS represent an integral part of the anti-tumor immune response, their function is likely influenced by a number of contextual signals, including those afforded by local stroma, secreted inflammatory factors, other resident immune populations, local vasculature, and epithelium (36). This may also indicate that different types of TLS exist that are susceptible to immune polarization or can even serve an immune suppressive role depending on and subsequent to microenvironmental context (37). This review will focus on approaches that can be taken to artificially induce TLS as a novel immunotherapy or as a means of augmenting immunotherapies. The prognostic value of TLS has been well reviewed (38, 39).
The clear benefit of TLS has prompted investigation into their potential therapeutic use, both as a standalone treatment or as an adjuvant to adoptive transfer-based cell therapies (40, 41). As such, artificial or inducible TLS (iTLS) hold great promise as a novel immunotherapy, but significant challenges must first be overcome that preclude their advent. These challenges range from knowledge gaps in basic TLS biology to complexities associated with clinical grade biomaterials and autologous cell processing. This review provides an overview of what strategies have been and might be employed to artificially induce TLS, how iTLS may be employed as a novel therapeutic, and what technical difficulties must be addressed prior to manufacturing iTLS at a clinical level.
Lessons From SLO Organogenesis: Strategies for Therapeutic TLS Induction
TLS formation is a complex process incorporating many processes that overlap conceptually with conventional SLO organogenesis (42), although multiple contextual and spacial constraints add complexity to TLS formation. In addition, not all TLS develop to the same level of structural and functional maturity. The level of TLS organization, what ectopic factors contribute to their function and development and how these factors play into prognostication have been well reviewed (36). In this review, we focus on approaches to artificially induce TLS formation, which have thus far been guided by our understanding of shared pathways between SLO organogenesis and natural TLS formation. Any successfully implemented iTLS will likely be subject to the same functional and organizational variations as seen in natural TLS caused by diverse microenvironmental cues present in different organs and indications. Thus, any clincial translation of iTLS must expect disease-specific challenges and variations regarding efficacy.
SLO initiate during embryogenesis following expression of lymphotoxin alpha-1, beta-2 (LTα1β2) on specialized lymphoid tissue inducer cells (LTi) (43) that binds to lymphotoxin beta receptor (LTBR) expressed on lymphoid tissue organizer cells (LTo), an early mesenchymal-derived fibroblast (42). Engagement of LTBR induces the expression of numerous NF-κB target genes (44, 45) that orchestrate the recruitment of different immune cells. NF-κB signals via two separate pathways, canonical and non-canonical. Canonical signaling leads to the translocation of p50/RelA dimers to the nucleus, where they induce CCL4, CXCL2, and CCL2 among other gene targets, while non-canonical NF-κB signaling leads to the translocation of p52/RelB dimers to the nucleus inducing CXCL13, CCL19, and CCL21 (44). This results in the recruitment of early CD11c+ myeloid populations followed by mass immigration of B and T cells which segregate into discrete T cell zones and B cell follicles (46, 47). This influx of lymphocyte subsets coincides with the LTBR-dependent development of high endothelial venules (HEV), and is followed by the LTBR-dependent appearance of antigen-presenting cells such as follicular DC (FDC) (48–50). As the SLO develops, LTo cells differentiate into RFC through continued LTBR signaling (51). Importantly, SLO formation requires both NF-κB signaling pathways to properly develop, although the non-canonical pathway appears more indispensable (45). In adults, there is no clear evidence that LTi or LTo cells persist, and so for TLS formation it is less clear which cell types fill these roles. However, overexpression of LTα1β2 markedly increase TLS (2), whereas LTBR blockade prevents TLS in murine models (52); this suggests that any cell expressing LTα1β2 has the potential to function as a LTi analogue, and any LTBR+ stromal cell capable of chemokine production has the potential to function as a LTo analogue. Importantly, most mesenchymal-derived stroma throughout the body expresses LTBR (53), including RFC (54). Additionally, LTα1β2 is expressed on activated T cells, B cells, and dendritic cells (55), giving this signaling axis wide-reaching potential for TLS induction if the correct environmental inflammatory cues are met. Importantly, engagement of LTBR on many types of mesenchymal-derived stroma induces analogous expression of both canonical and non-canonical NF-κB target genes as compared to LTo, including the lymphoid tissue homeostatic cytokines CXCL13, CCL19 and CCL21 necessary for SLO development (1). In addition to LTα1β2, another LTBR ligand, homologous to lymphotoxin, exhibits inducible expression and competes with HSV glycoprotein D for binding to herpesvirus entry mediator, a receptor expressed on T lymphocytes (LIGHT) effectively elicits chemokine gene targets on LTBR+ stroma through NF-κB (56). LIGHT signaling can also lead to TLS formation, and while also binding to other receptors such as Herpesvirus entry mediator (HVEM), LIGHT acts analogous to LTα1β2 in its capacity to induce TLS formation (34, 57).
The role of chemokines in both SLO organogenesis and in TLS formation cannot be understated. For SLO, the narrow set of homeostatic chemokines required for organogenesis reflects the chemokine receptor patterns expressed by naïve and resting memory T cells, and coincides with the chemokine receptors expressed by DC and macrophage (58). Activated lymphocytes follow different trafficking patterns throughout the periphery owing to the downregulation of SLO-homing chemokine receptors and the up-regulation of alternative chemokine receptor sets that allow for emigration from SLO and infiltration into inflamed peripheral sites (59). It is thus unsurprising that gene signatures associated with TLS formation in tumors encompass not only SLO-associated homeostatic chemokines, but many other chemokines capable of recruiting lymphocytes in various stages of activation and effector function and that are associated with peripheral lymphocyte trafficking (13). A TLS gene signature which incorporates 12 chemokines (12CK-GES) that was associated with better patient survival independent of tumor staging, was first identified in patients with colorectal carcinoma (11) and was soon after used to predict the presence of TLS in wide range of tumor types including melanoma, lung, breast, and colorectal (13, 29). Importantly, nine of the chemokines identified in the 12CK-GES have reported up-regulation by LTBR signaling in mesenchymal-derived stroma through canonical or non-canonical NF-κB signaling, whereas the remaining three are hallmark products of tumor-associated macrophages (TAM), or type-II polarized macrophages (60–64) which themselves are recruited by multiple members of the 12CK-GES (Table 1). Insights from the 12CK-GES, and the parallels to SLO organogenesis can easily lead one to speculate that TLS form via a sequential or semi-sequential recruitment of immune subsets in response to chronic LTBR stimulation, and that any chemokines in the 12CK-GES not directly produced by LTBR+ stroma might be indirectly accounted for by subsequently recruited immune populations. In addition to TLS-associated chemokines, LTBR signaling also regulates the expression of a number of homeostatic cytokines and growth factors important to SLO organogenesis and to TLS formation, including IL-7, IL-15, and B cell activating factor (BAFF) (44, 72). However, to prove that any components of TLS organization form through sequential recruitment steps requires an experimental model of TLS formation in which temporal data can be acquired. When considering TLS induction as an anti-cancer therapeutic, such models may be necessary to deduce which components of TLS formation are required for anti-tumor activity, and which component might be expendable for anti-tumor effect.

Table 1 12CK-GES and associated NF-κB signaling pathways.
Strategies for iTLS can either utilize methods to initiate sustained LTBR signaling, thereby taking advantage of the same cascade of events that leads to naturally occurring TLS, by introducing cellular components engineered with constitutively active LTBR or with transgenic expression of LTBR gene targets, by some combination of the above approaches, or by complete TLS manufacture ex vivo prior to adoptive transfer/retransfer (Figure 1). iTLS methods that involve the introduction of isolated or cultivated cellular components have an additional appeal beyond the iTLS itself. The central role that antigen presenting cells play in TLS formation and function (73) and the effector cell-recruiting potential TLS create in the tumor microenvironment (57) make iTLS an ideal platform for the delivery of DC-based anti-tumor vaccines or as an adjuvant for chimeric antigen receptor-transduced T cell (CAR-T) or tumor-infiltrating lymphocyte (TIL) adoptive transfer therapies. Given the breadth of possible approaches, novel iTLS-based therapies can be designed with goals ranging from early interventional to multi-modal combination therapies that bridge cellular therapies, immunotherapies, and/or chemotherapeutic and radiation therapies.
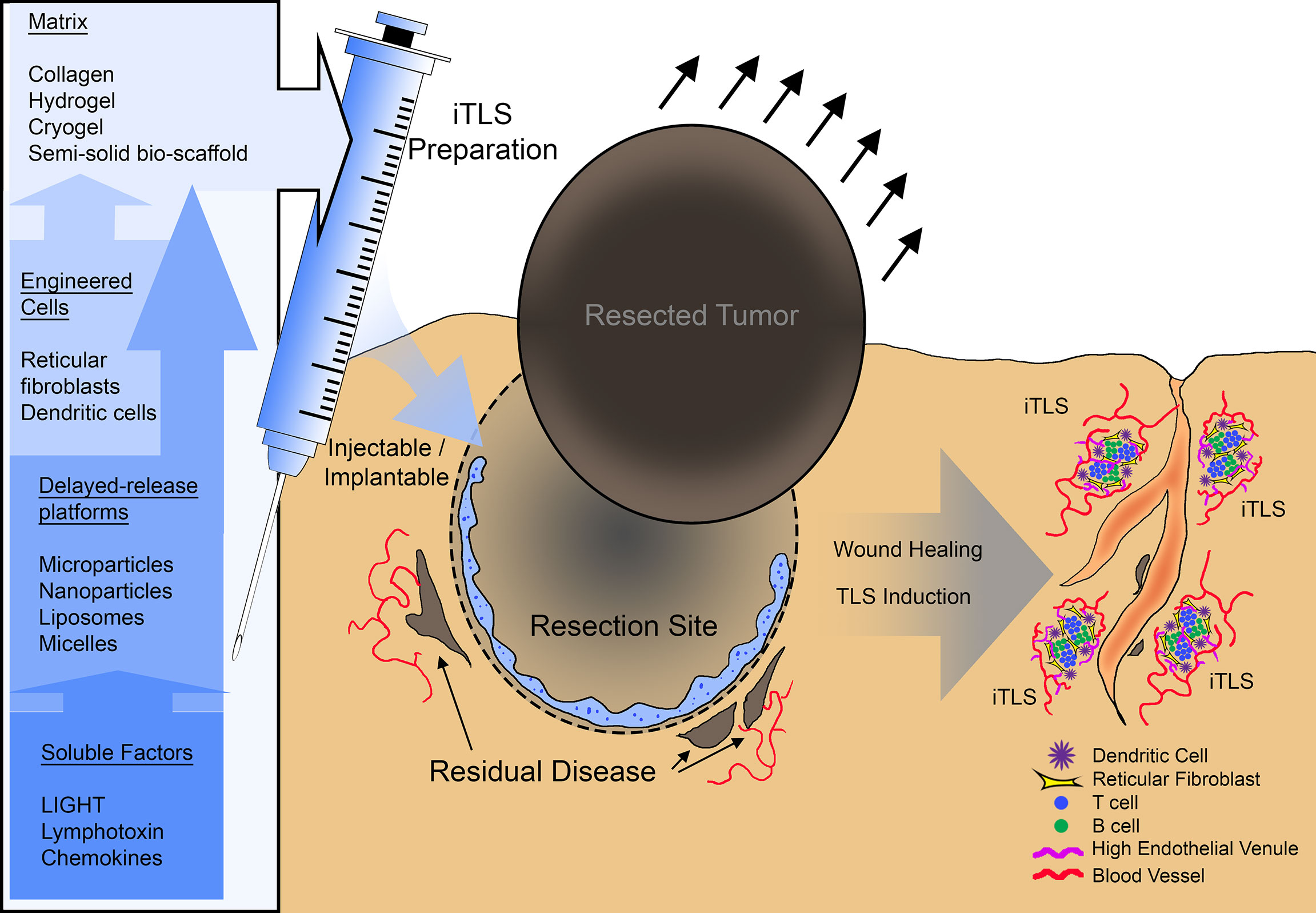
Figure 1 A summary schematic of potential therapeutic use of inducible tertiary lymphoid structures (iTLS). iTLS preparations may include cellular components such as dendritic cells, or reticular fibroblasts modified to engage the LTBR pathway, or co-delivered with soluble LTBR-ligands via a delayed-release platform such as liposomes, nanoparticles, or micelles. Injectable or implantable iTLS preparations could be administered at the site of tumor resection to induce TLS and subsequently control residual disease or counteract reoccurrence.
Early demonstrations that the LTBR-chemokine axis can be utilized for iTLS occurred in transgenic model systems in which mice overexpressing chemokines or LTα developed lymph node-like structures in certain tissues (74). In mice expressing LTα under the rat insulin promoter, a promotor with transgene expression limited to pancreatic beta cells and proximal tubule of the kidney, TLS formation was observed in both the pancreas and kidney, especially in proximity to vasculature where the formation of HEV was evident (74). Under the same promoter, transgenic CCL21 induced HEV-containing TLS in the pancreas as well (75, 76), but using a promoter with skin-specific CCL21 expression did not result in TLS; these data suggest that additional cues or microenvironmental constraints are needed for TLS beyond CCL21 expression alone (75). Other examples include LTBR ligand targeting strategies, such as delivery of recombinant LIGHT tagged to vascular-targeting peptide. This chimeric compound induced TLS in pancreatic neuroendocrine tumors and in glioblastoma in areas surrounding dense vasculature that contain discrete B cell zones, T cell zones, macrophage, HEV, and DC (32, 77). Delivery of LTBR ligands to induce TLS is also possible via adoptive cell transfer of transgenic cells. DC transduced with the type-I polarizing transcription factor T-bet induced the expression of LIGHT and LTα, and subsequently CCL21, when injected into murine colon adenocarcinoma, slowing tumor growth (73, 78). Lastly, delivery of LTBR+ stroma demonstrates functional TLS formation when injected subcutaneously juxtaposed to established MC-38 murine colon carcinoma tumors, slowing tumor growth and actively priming T cell response (79). Despite the precedent that TLS can be induced by taking advantage of the LTBR-chemokine axis, the greatest benefit from iTLS will likely result from injectable or implantable preparations that do not require additional microenvironmental cues from the recipient host. This way, they may be applied to a wider range of tissues and organs as part of a microenvironment reprogramming strategy. Discussed below are biocompatible matrixes and micro/nanoparticles that may be suitable as scaffolding material for iTLS or for the sustained release of biologics aimed at TLS induction respectively.
Biomaterials With Proposed Suitability for iTLS
A variety of three-dimensional materials that are permissible to cellular infiltration and that may allow for cell-scaffold interactions have been used for tissue engineering, regenerative medicine, and ex vivo scientific investigation (80). Some bio-scaffolds derived from animal or cell-based products such as Matrigel® (Corning Life Sciences) have a decades-long precedent. These products isolate entire acid-soluble constituents of tumor cell line-derived extracellular matrix, and thus represent a more physiologically complete microenvironment than synthetic scaffolds. However, such products are not immunologically inert, and contain growth factors and biologically active components that have well described angiogenic, adipogenic, and inflammatory properties (81). This is even true of “growth factor reduced” product versions (albeit improved) (81, 82). As such, use of such cell or animal-derived matrixes often invites scientific scrutiny (83), and thus more advanced synthetic alternatives are typically sought for bioengineering endeavors which contain less lot-to-lot variation, display a more immunologically inert background, and lack any confounding variables caused by biologically active carryover components (81).
Collagen Matrixes
Collagens constitute the major framework of the extracellular matrix (ECM) with distinct primary, secondary, tertiary, and quaternary structures that create a range of ECM scaffold superstructures in different tissues and organs from rope-like fibrils to web-like networks, anchoring structures, and can even complex with transmembrane collagens (84). In vertebrates, at least 29 types of collagen are coded by at least 45 separate genes (85). Given the complexity of collagen, tissues from animals or human-sourced raw materials are often used to make collagen-based scaffolding materials (86). Recombinant sources of collagen monomers or peptides are commercially available (87) but small yields and the lack of the tertiary and quaternary structural complexity that would be afforded by multi-collagen type complexes limit their use as a scaffold for bioengineering (86). Most collagen products are manufactured in one of two ways: from decellularization of existing ECM, resulting in an intact ECM superstructure (88), or through the breakdown, solubilization, extraction, and reformulation of collagen, often with the addition of crosslinking agents such as glycosaminoglycans (89, 90), elastins (91, 92), or chitosans (93, 94). The precise methodologies used to manufacture commercial collagen matrix products should be carefully considered prior to implementation in iTLS approaches since most collagen-based matrixes are derived from animal or human tissues (86). While more purified than bulk ECM products like Matrigel®, biologically active impurities may still carry over from raw materials (95). A major strength of collagen-based matrixes is their versatility in available formats, including sheets, sponges, disks, granulated tablets, or even nano-scale spheres (96).
One of the first successful demonstrations of iTLS using implantable artificial matrix was performed by Suematsu and Wantanabe using a collagen sponge biomatrix impregnated with a thymus-derived stromal cell line modified to constitutively express LTα. Following implantation, functionality of iTLS was demonstrated by vaccination with 4-hydroxy3-nitrophenylacetyl-ovalbumin (NP-OVA). Three weeks after subcutaneous implantation, discrete B cell and T cell zones formed in the stromal cell-impregnated collagen sponge with interacting DC, as well as HEV-like structures. Recipient mice that were vaccinated with NP-OVA produced anti-NP-OVA IgG-producing B cells within iTLS. This effect was bolstered if NP-OVA-pulsed DC were included in preparations. Furthermore, these NP-OVA-primed iTLS could be resected after their formation, and transplanted to syngeneic recipient mice wherein they could mount effective secondary response to NP-OVA (97). These iTLS were later shown to be able to produce a potent secondary immune response when transplanted into severe combined immunodeficiency (SCID) recipient mice, repopulating SLO and bone marrow with anti-NP-OVA IgG-producing B cells (98). An alternative approach using chemokine and VCAM-1-loaded collagen matrix instead of stromal cells soon followed, recapitulating the successes of stromal-cell line-loaded collagen sponges. These chemokine-induced TLS incorporated B cell zones, T cell zones, and supporting DC and were able to prime a similar anti-NP-OVA response. HEV were not interrogated in these iTLS (99).
Hydrogels
Hydrogel refers to a large class of biomaterials that are made of three-dimensional crosslinked polymers with a large capacity for water uptake and retention that are prepared in aqueous solution (100). Depending on the types of polymers and crosslinking reagents used, a wide variety of hydrogels can be prepared that have different properties regarding biologic interactivity or inertness, physical rigidity or elasticity, temperature sensitivity, pH sensitivity, shape memory properties, or capacity to carry and deliver soluble drugs or cellular payloads (101–103). Hydrogels can be classified according to physical structure, charge, size or chemical properties (102). Of relevance to iTLS efforts, most hydrogel preparations can be injectable or transplantable depending on the timing and chemistries of the crosslinking steps, and can be formulated in large injection/implantation volumes or in micro or even nanoscale particle preparations (104). Because of their large capacity to hold aqueous solution, many hydrogel preparation methodologies are widely compatible with cell culture conditions so long as the chemistries required for crosslinking do not stray from physiologic pH, temperature, salinity, and other cell culture ranges (105–107). For this reason, crosslinking chemistries that utilize mild temperature or pH changes such as warming from 4°C to 37°C or changing from a slightly acidic solution to slightly basic are ideal for injectable preparations, as such hydrogels would crosslink after injection into living recipients (105, 107). When used as an in vitro model for iTLS, hydrogel preparations with BAFF-producing stromal cells and IL-4 support the compartmentalization, expansion, and class-switching of primary B cells (108, 109). When used as a model of cell therapy delivery, hydrogels have been successfully deployed to carry CAR-T cells in conjunction with delivering stimulator of IFN genes (STING) agonist cyclic di-GMP. This preparation was then placed in resection sites of murine pancreatic tumors, or alongside pancreatic tumors mimicking non-resectable masses. These implants produced a significant anti-tumor effect compared to intravenously delivered CAR-T, activated host DC, and induced significant infiltration of immune cells at the implantation sites (110).
Cryogels, a subset of hydrogels prepared at sub 0°C, may be of particular interest to iTLS efforts. Their preparation creates a larger pore size than typical hydrogels, which are typically measured in the nanometer range. Such a small pore size requires hydrogel breakdown or active turnover by infiltrating populations to emigrate or infiltrate transferred materials, significantly limiting cellular involvement (111, 112). Cryogels are formed when polymer and cross linkers are displaced by ice crystal formation, causing concentration spikes in localized spaces in between ice crystals. When the cryogels are subsequently thawed, the space occupied by the ice crystals leaves a porous network measured on a micron scale or larger (113, 114). Thus, cryogels provide a more cell-invasive alternative to conventional cryogels, although, their freeze/thaw preparation method requires any would-be cellular components to be loaded after formation and precludes any formation post injection, somewhat limiting their use to implantation. Cryogels have been be used to deliver chemotherapies and cancer vaccines (115), and when incorporated with DC activating components and tumor antigen vaccination strategies, induces the recruitment of DC and lymphocytes (113, 116, 117), although the organization of these infiltrates was not investigated.
Other Solid or Semi-Solid Bio-Scaffolds
A wide array of scaffolding material distinct from hydrogels or tissue-derived collagen matrixes have been derived for use in tissue repair, wound-healing, or other bioengineering endeavors (118). Among these are matrixes made from mesoporous silica rods, which have been used to boost the immunogenicity of tumor antigen peptide-based vaccine approaches in mice bearing B16F10 melanoma or CT26 colon carcinoma tumors. In these models, tumor associated antigen pools were loaded into mesoporous silica rod matrixes and injected subcutaneously. While the organization of infiltrates was not analyzed histologically, vaccination using this matrix approach greatly enhanced lymphocyte infiltration, and activation, as well as mediated anti-tumor effects on lung nodule growth (119). While not evidence of iTLS, this study does provide precedence for silica as a biomaterial supportive of lymphocyte recruitment, DC activation, and antigen priming. Another bio-matrix that may be permissive to iTLS are polyamide fiber preparations. In what represents one of the earliest attempt to manufacture iTLS entirely in situ, non-woven sheets of polyamide fibers were loaded with antigen-primed primed DC (in this case, cytomegalovirus lysate) and sealed inside a closed and chambered bioreactor system in which lymphocytes isolated from peripheral blood mononuclear cells (PBMC) were continuously circulated. After two weeks, this bioreactor approach resulted in TLS-like structures with discrete B cell and T cell clusters around DC inside the polyamide fiber sheets. Cytokine production suggests that these lymphocytes were activated by the primed DC (120).
Micro and Nanoparticles in iTLS
While bio-scaffolds can, in some instances, impart delayed release of soluble factors, another class of biomaterials has been refined over time with delayed-release of soluble factors as one of several defining characteristics. Microparticles or nanoparticles represent entire fields of materials science in their own right (121). Here, microparticles and nanoparticles are proposed as cooperative biomaterial elements that can be used as an incorporated element within bio-scaffolding to mediate controlled release of chemokines, LTBR ligands, or other TLS-inducing factors. Of these, liposomes represent the most studied and characterized class of micro or nanoscale biomaterials that can serve as carriers for a wide range of compounds. Liposomes form when lipid components assemble into spherical bilayers leaving an aqueous compartment that carry a water-soluble payload (122). The physical properties of liposomes can be easily controlled by altering the types of lipids used for incorporation (123–126), and their size can be controlled by deploying different production methods such as sonication, which delivers liposomes in the 20-40 nm range (127), microfluidic mixing in the 20-80 nm range (128, 129), high pressure homogenization in the 20-140 nm range (130), flow focusing in the 50-150 nm range (131), and extrusion in the 70-415 nm range (132). Many liposomes are already incorporated as part of FDA-approved drugs (133, 134) giving precedent to their clinical translatability and patient safety, and there are now multitudes of methodologies that can deliver on a wide range of specifications, cost, uniformity, and bulk manufacturing requirements (135).
Unmodified liposomes were first described 55 years ago (136), and while successful, have limitations due to their unprotected outer lipid surface, making them subject to fusion with other liposomes as natural result of surface tension reduction (137, 138). Unmodified liposomes are also susceptible to opsonization of serum protein following injection which can lead to phagocytic uptake, or clearance in the liver (139, 140). Any such alteration manipulates drug release kinetics or limits payload delivery to intended targets. A second generation of liposomes was created by modifying liposomal surfaces with integrated polymers, providing structural stabilization and interfering with serum protein binding (139, 141), the most successful of which has been polyethylene glycol (PEG) (142, 143). Additional modification strategies have been employed to liposomes within the last 20 years that not only aim to stabilize liposomal formulations, but to also impart drug target selectivity or more precisely control drug release. Examples of such modifications include liposomes with surface-attached bioactive ligands, such as aptamers, peptides, or most commonly immunoglobulin or immunoglobulin fragments (144). By incorporating moieties with known affinities for antigens expressed on target tissues (or tumors), liposomes can then specifically interact with intended targets. Such incorporations can be accomplished by including recombinant protein products in the initial lipid formulations that either naturally contain hydrophobic regions or that are themselves modified to contain hydrophobic regions, or by covalent binding to hydrophilic regions of incorporated lipids (145). Of particular relevance to iTLS, liposomes can offer extended release kinetics of their payloads, or even be designed for content release in response to an external trigger, such as temperature (146), magnetic fields (147), or light (148, 149). In addition, liposomes are compatible with other bio-scaffolding materials such as hydrogels, cryogels, or polymeric matrixes (150), and have a demonstrated ability for delayed release of chemokines such as CXCL13 (151).
Other nanoscale particles capable of delivering or releasing inflammatory mediators include micelles which are created by self-assembling amphiphilic polymers (152, 153). Micelles have proven capacity to deliver cytokines (154), antigens (155), and interfering RNAs (156, 157), and thus represent a plausible alternative to liposomes to deliver factors for iTLS. In addition, nanoparticles made from aliphatic polyesters (PLGA) are also an attractive option to incorporate delayed-release of soluble factors, and are already FDA-approved in many clinical contexts (158–160). PLGA nanoparticles are biodegradable, and like micelles, are capable of tumor antigen delivery (161, 162) and immunotherapeutic biologics (163, 164). In what is perhaps the most robust example of iTLS generated in animal models, Kobayashi and Watanabe combined microscale gelatin-based hydrogels loaded with LTα1β2, CCL19, CCL21, CXCL12, CXCL13, and soluble RANKL inside a macroscale implantable collagen sponge. This preparation was then implanted onto the kidney capsule of recipient mice, and after three weeks produced mature iTLS with discrete T cell zones, B cell zones, RFC networks, FDC in what appears to be a marginal zone, HEV, and lymphatic vasculature. These iTLS were also able to prime primary and secondary IgG responses to NP-OVA (165). Another example of combined biomaterial approaches utilized lipid-coated silica microspheres harboring IL-15/IL-15Ra fusion proteins and anti-CD3, anti-CD18, and anti-CD137 antibodies to act as artificial antigen-presenting cells inserted into alginate hydrogels loaded with NKG2D-CAR-expressing murine T cells. This construct, when implanted next to partially resected established subcutaneous 4T1 breast cancer tumor-bearing mice, elicited significant anti-tumor reactivity and slowed tumor growth (166). While not necessarily iTLS, this study further establishes precedent that combination biomaterials can deliver and expand effector lymphocytes.
Challenges Awaiting iTLS for Clinical Translation
Avoiding Foreign Body Response for Incorporated Biomaterials
One general drawback to the use of biomaterials is the induction of a foreign body response (FBR), an acute inflammatory reaction against the material itself (167). FBR can result in a wide range of unintended consequences including but not limited to vascularization, fibrotic encapsulation, and infiltration of innate immune cells (168, 169). Neutrophils and macrophages are among the earliest effector cells responding to the FBR and can destroy implanted biomaterials through the release of cytotoxic granules, reactive oxygen species, proteolytic enzymes, and phagocytosis (170–173). Of particular importance to iTLS, recruited and activated macrophages and neutrophils produce high levels of chemokines associated with FBR such as CXCL8, CCL2, CCL4 (174, 175). While CCL2 and CCL4 are part of the 12CK-GES associated with TLS presence in human tumors, CXCL8 is not (11), and none of these chemokines encompass the SLO homeostatic chemokines CXCL13, CCL19 and CCL21 previously used for iTLS, as discussed above. In addition, physical macrophage adherence to many biomaterials polarizes them to a M2 phenotype, which may be detrimental to iTLS formation for anti-tumor immunity (176). To avoid a FBR, it is advantageous to select biomaterials with low antigenicity that have little or no carryover of soluble factors from animal sources (167). In addition, biomaterial topography has also been identified as a contributing factor to FBR, and thus biomaterial size, shape, and texture can be modified to minimize FBR (177, 178). However, any such measure would need to be weighed against the need to recruit cellular infiltrate as part of the iTLS.
Generating cGMP Materials for iTLS Manufacture
As ever more complex components and methodologies are used to innovate iTLS as a potential therapy, so too do the challenges associated with translating such approaches to the clinic. To fully qualify for the Food and Drug Administration’s (FDA) approval, components used to make any would-be iTLS therapy need to graduate to clinical-grade materials by the time pivotal trials are conducted, meaning the components themselves must be manufactured under cGMP conditions (179). Not only does this add difficulty to the process, but in almost every scenario, results in elevated manufacturing cost (180, 181). Cell therapies utilizing one cell type with one gene modification can easily exceed $400,000 per dose due in no small part to the elevated cost of manufacturing cell therapies at a cGMP level (182). Given the potentially multimodal processes involved in iTLS development, this new class of immunotherapy may incur clinical-grade manufacturing costs reflective of cell therapies, biomaterials, and biologics combined. In addition to cost, there are also regulatory challenges. Biomaterial products which contain no cellular or bioactive components, such as an inert scaffolding material, might be considered from a regulatory perspective as a “medical device” depending on their mode of action, but should such material be combined with cellular components or biologics, it will most certainly be considered a drug (183). Careful consideration will then need to be taken when defining what components are drug product versus drug substance. Conventionally, the drug substance is whatever components entail the “active ingredient(s),” whereas the drug product is the entirety of the components and compositions used in the manufacturing process. These definitions are critical to the regulatory success of new investigational drugs, but may be less clear for iTLS, which may combine novel biomaterials with varying amounts of bioactivity (184), with active biologics and cell therapies which may contain genetic modifications. Similar to the advent of cell therapies over the past few decades, the clinical translation of bioengineered constructs such as iTLS may be codependent on the FDA’s creation of a new guidelines that are developed in concert with the scientific field (185).
Concluding Remarks
The progress toward utilizing TLS as a therapeutic intervention has made great strides over the last few decades and has come to incorporate many new and technologies, particularly in the biomaterials space. iTLS have to potential to become an entire new class of immunotherapy combining elements of biologic compounds, cellular therapeutics, and biofabrication techniques. However, significant challenges and unanswered questions remain. These include identifying the optimal bio-scaffold/nanomaterial combinations for sustained release of TLS-inducing soluble factors and identifying the minimal combination of chemokines, LTBR ligands, and other soluble factors required for robust iTLS formation. Another prominent point of consideration is to further evaluate if stromal and/or DC components are needed for iTLS approaches. Their involvement has been critical to early iTLS successes, but recent advances demonstrate iTLS can be achieved using cell-free constructs (165). This would have obvious benefits when translating to clinical-grade manufacturing processes, and allow for less costly clinical translation.
Author Contributions
SA and RN performed literature searches, outlined sections, and reviewed and edited the manuscript, JJM reviewed and edited the manuscript, and provided funding, AWM wrote the manuscript, created the figure and table, reviewed and edited the manuscript, and provided funding. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the NCI-NIH (1R01 CA148995, 1R01 CA184845, P30 CA076292, P50 CA168536, and 5R21CA214285), Cindy and Jon Gruden Fund, Chris Sullivan Fund, V Foundation, and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Fernandes MT, Dejardin E, dos Santos NR. Context-Dependent Roles for Lymphotoxin-Beta Receptor Signaling in Cancer Development. Biochim Biophys Acta (2016) 1865(2):204–19. doi: 10.1016/j.bbcan.2016.02.005
2. Wolf MJ, Seleznik GM, Zeller N, Heikenwalder M. The Unexpected Role of Lymphotoxin Beta Receptor Signaling in Carcinogenesis: From Lymphoid Tissue Formation to Liver and Prostate Cancer Development. Oncogene (2010) 29(36):5006–18. doi: 10.1038/onc.2010.260
3. Senovilla L, Vacchelli E, Galon J, Adjemian S, Eggermont A, Fridman WH, et al. Trial Watch: Prognostic and Predictive Value of the Immune Infiltrate in Cancer. Oncoimmunology (2012) 1(8):1323–43. doi: 10.4161/onci.22009
4. Goc J, Fridman WH, Sautes-Fridman C, Dieu-Nosjean MC. Characteristics of Tertiary Lymphoid Structures in Primary Cancers. Oncoimmunology (2013) 2(12):e26836. doi: 10.4161/onci.26836
5. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The Immune Contexture in Human Tumours: Impact on Clinical Outcome. Nat Rev Cancer (2012) 12(4):298–306. doi: 10.1038/nrc3245
6. Nielsen JS, Sahota RA, Milne K, Kost SE, Nesslinger NJ, Watson PH, et al. CD20+ Tumor-Infiltrating Lymphocytes Have an Atypical CD27- Memory Phenotype and Together With CD8+ T Cells Promote Favorable Prognosis in Ovarian Cancer. Clin Cancer Res (2012) 18(12):3281–92. doi: 10.1158/1078-0432.CCR-12-0234
7. Dieu-Nosjean MC, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, et al. Long-Term Survival for Patients With Non-Small-Cell Lung Cancer With Intratumoral Lymphoid Structures. J Clin Oncol (2008) 26(27):4410–7. doi: 10.1200/JCO.2007.15.0284
8. Remark R, Alifano M, Cremer I, Lupo A, Dieu-Nosjean MC, Riquet M, et al. Characteristics and Clinical Impacts of the Immune Environments in Colorectal and Renal Cell Carcinoma Lung Metastases: Influence of Tumor Origin. Clin Cancer Res (2013) 19(15):4079–91. doi: 10.1158/1078-0432.CCR-12-3847
9. Najafi M, Hashemi Goradel N, Farhood B, Salehi E, Nashtaei MS, Khanlarkhani N, et al. Macrophage Polarity in Cancer: A Review. J Cell Biochem (2019) 120(3):2756–65. doi: 10.1002/jcb.27646
10. Jayasingam SD, Citartan M, Thang TH, Mat Zin AA, Ang KC, Ch’ng ES. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front Oncol (2019) 9:1512. doi: 10.3389/fonc.2019.01512
11. Coppola D, Nebozhyn M, Khalil F, Dai H, Yeatman T, Loboda A, et al. Unique Ectopic Lymph Node-Like Structures Present in Human Primary Colorectal Carcinoma are Identified by Immune Gene Array Profiling. Am J Pathol (2011) 179(1):37–45. doi: 10.1016/j.ajpath.2011.03.007
12. Dieu-Nosjean MC, Goc J, Giraldo NA, Sautes-Fridman C, Fridman WH. Tertiary Lymphoid Structures in Cancer and Beyond. Trends Immunol (2014) 35(11):571–80. doi: 10.1016/j.it.2014.09.006
13. Messina JL, Fenstermacher DA, Eschrich S, Qu X, Berglund AE, Lloyd MC, et al. 12-Chemokine Gene Signature Identifies Lymph Node-Like Structures in Melanoma: Potential for Patient Selection for Immunotherapy? Sci Rep (2012) 2:765. doi: 10.1038/srep00765
14. Goc J, Germain C, Vo-Bourgais TK, Lupo A, Klein C, Knockaert S, et al. Dendritic Cells in Tumor-Associated Tertiary Lymphoid Structures Signal a Th1 Cytotoxic Immune Contexture and License the Positive Prognostic Value of Infiltrating CD8+ T Cells. Cancer Res (2014) 74(3):705–15. doi: 10.1158/0008-5472.CAN-13-1342
15. Movassagh M, Spatz A, Davoust J, Lebecque S, Romero P, Pittet M, et al. Selective Accumulation of Mature DC-Lamp+ Dendritic Cells in Tumor Sites is Associated With Efficient T-Cell-Mediated Antitumor Response and Control of Metastatic Dissemination in Melanoma. Cancer Res (2004) 64(6):2192–8. doi: 10.1158/0008-5472.CAN-03-2969
16. Bergomas F, Grizzi F, Doni A, Pesce S, Laghi L, Allavena P, et al. Tertiary Intratumor Lymphoid Tissue in Colo-Rectal Cancer. Cancers (Basel) (2011) 4(1):1–10. doi: 10.3390/cancers4010001
17. Drayton DL, Ying X, Lee J, Lesslauer W, Ruddle NH. Ectopic LT Alpha Beta Directs Lymphoid Organ Neogenesis With Concomitant Expression of Peripheral Node Addressin and a HEV-Restricted Sulfotransferase. J Exp Med (2003) 197(9):1153–63. doi: 10.1084/jem.20021761
18. Moyron-Quiroz JE, Rangel-Moreno J, Kusser K, Hartson L, Sprague F, Goodrich S, et al. Role of Inducible Bronchus Associated Lymphoid Tissue (Ibalt) in Respiratory Immunity. Nat Med (2004) 10(9):927–34. doi: 10.1038/nm1091
19. de Chaisemartin L, Goc J, Damotte D, Validire P, Magdeleinat P, Alifano M, et al. Characterization of Chemokines and Adhesion Molecules Associated With T Cell Presence in Tertiary Lymphoid Structures in Human Lung Cancer. Cancer Res (2011) 71(20):6391–9. doi: 10.1158/0008-5472.CAN-11-0952
20. Silina K, Soltermann A, Attar FM, Casanova R, Uckeley ZM, Thut H, et al. Germinal Centers Determine the Prognostic Relevance of Tertiary Lymphoid Structures and are Impaired by Corticosteroids in Lung Squamous Cell Carcinoma. Cancer Res (2018) 78(5):1308–20. doi: 10.1158/0008-5472.CAN-17-1987
21. Schroder AE, Greiner A, Seyfert C, Berek C. Differentiation of B Cells in the Nonlymphoid Tissue of the Synovial Membrane of Patients With Rheumatoid Arthritis. Proc Natl Acad Sci USA (1996) 93(1):221–5. doi: 10.1073/pnas.93.1.221
22. Hughes CE, Benson RA, Bedaj M, Maffia P. Antigen-Presenting Cells and Antigen Presentation in Tertiary Lymphoid Organs. Front Immunol (2016) 7:481. doi: 10.3389/fimmu.2016.00481
23. Joshi NS, Akama-Garren EH, Lu Y, Lee DY, Chang GP, Li A, et al. Regulatory T Cells in Tumor-Associated Tertiary Lymphoid Structures Suppress Anti-Tumor T Cell Responses. Immunity (2015) 43(3):579–90. doi: 10.1016/j.immuni.2015.08.006
24. Zhu W, Germain C, Liu Z, Sebastian Y, Devi P, Knockaert S, et al. A High Density of Tertiary Lymphoid Structure B Cells in Lung Tumors is Associated With Increased CD4(+) T Cell Receptor Repertoire Clonality. Oncoimmunology (2015) 4(12):e1051922. doi: 10.1080/2162402X.2015.1051922
25. Kroeger DR, Milne K, Nelson BH. Tumor-Infiltrating Plasma Cells are Associated With Tertiary Lymphoid Structures, Cytolytic T-Cell Responses, and Superior Prognosis in Ovarian Cancer. Clin Cancer Res (2016) 22(12):3005–15. doi: 10.1158/1078-0432.CCR-15-2762
26. Truxova I, Kasikova L, Hensler M, Skapa P, Laco J, Pecen L, et al. Mature Dendritic Cells Correlate With Favorable Immune Infiltrate and Improved Prognosis in Ovarian Carcinoma Patients. J Immunother Cancer (2018) 6(1):139. doi: 10.1186/s40425-018-0446-3
27. Mattlage AE, Ashenden AL, Lentz AA, Rippee MA, Billinger SA. Submaximal and Peak Cardiorespiratory Response After Moderate-High Intensity Exercise Training in Subacute Stroke. Cardiopulm Phys Ther J (2013) 24(3):14–20. doi: 10.1097/01823246-201324030-00003
28. Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary Lymphoid Structures Improve Immunotherapy and Survival in Melanoma. Nature (2020) 577(7791):561–5. doi: 10.1038/s41586-019-1914-8
29. Prabhakaran S, Rizk VT, Ma Z, Cheng CH, Berglund AE, Coppola D, et al. Evaluation of Invasive Breast Cancer Samples Using a 12-Chemokine Gene Expression Score: Correlation With Clinical Outcomes. Breast Cancer Res (2017) 19(1):71. doi: 10.1186/s13058-017-0864-z
30. Liu X, Tsang JYS, Hlaing T, Hu J, Ni YB, Chan SK, et al. Distinct Tertiary Lymphoid Structure Associations and Their Prognostic Relevance in HER2 Positive and Negative Breast Cancers. Oncologist (2017) 22(11):1316–24. doi: 10.1634/theoncologist.2017-0029
31. Posch F, Silina K, Leibl S, Mundlein A, Moch H, Siebenhuner A, et al. Maturation of Tertiary Lymphoid Structures and Recurrence of Stage II and III Colorectal Cancer. Oncoimmunology (2018) 7(2):e1378844. doi: 10.1080/2162402X.2017.1378844
32. Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, et al. Facilitating T Cell Infiltration in Tumor Microenvironment Overcomes Resistance to PD-L1 Blockade. Cancer Cell (2016) 29(3):285–96. doi: 10.1016/j.ccell.2016.02.004
33. Schrama D, thor Straten P, Fischer WH, McLellan AD, Brocker EB, Reisfeld RA, et al. Targeting of Lymphotoxin-Alpha to the Tumor Elicits an Efficient Immune Response Associated With Induction of Peripheral Lymphoid-Like Tissue. Immunity (2001) 14(2):111–21. doi: 10.1016/S1074-7613(01)00094-2
34. Yu P, Lee Y, Liu W, Chin RK, Wang J, Wang Y, et al. Priming of Naive T Cells Inside Tumors Leads to Eradication of Established Tumors. Nat Immunol (2004) 5(2):141–9. doi: 10.1038/ni1029
35. Finkin S, Yuan D, Stein I, Taniguchi K, Weber A, Unger K, et al. Ectopic Lymphoid Structures Function as Microniches for Tumor Progenitor Cells in Hepatocellular Carcinoma. Nat Immunol (2015) 16(12):1235–44. doi: 10.1038/ni.3290
36. Rodriguez AB, Engelhard VH. Insights Into Tumor-Associated Tertiary Lymphoid Structures: Novel Targets for Antitumor Immunity and Cancer Immunotherapy. Cancer Immunol Res (2020) 8(11):1338–45. doi: 10.1158/2326-6066.CIR-20-0432
37. Sautes-Fridman C, Verneau J, Sun CM, Moreira M, Chen TW, Meylan M, et al. Tertiary Lymphoid Structures and B Cells: Clinical Impact and Therapeutic Modulation in Cancer. Semin Immunol (2020) 48:101406. doi: 10.1016/j.smim.2020.101406
38. Munoz-Erazo L, Rhodes JL, Marion VC, Kemp RA. Tertiary Lymphoid Structures in Cancer - Considerations for Patient Prognosis. Cell Mol Immunol (2020) 17(6):570–5. doi: 10.1038/s41423-020-0457-0
39. Jacquelot N, Tellier J, Nutt SI, Belz Gt. Tertiary Lymphoid Structures and B Lymphocytes in Cancer Prognosis and Response to Immunotherapies. Oncoimmunology (2021) 10(1):1900508. doi: 10.1080/2162402X.2021.1900508
40. Zhu G, Falahat R, Wang K, Mailloux A, Artzi N, Mule JJ. Tumor-Associated Tertiary Lymphoid Structures: Gene-Expression Profiling and Their Bioengineering. Front Immunol (2017) 8:767. doi: 10.3389/fimmu.2017.00767
41. Sautes-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary Lymphoid Structures in the Era of Cancer Immunotherapy. Nat Rev Cancer (2019) 19(6):307–25. doi: 10.1038/s41568-019-0144-6
42. Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic Lymphoid-Like Structures in Infection, Cancer and Autoimmunity. Nat Rev Immunol (2014) 14(7):447–62. doi: 10.1038/nri3700
43. Yoshida H, Naito A, Inoue J, Satoh M, Santee-Cooper SM, Ware CF, et al. Different Cytokines Induce Surface Lymphotoxin-Alphabeta on IL-7 Receptor-Alpha Cells That Differentially Engender Lymph Nodes and Peyer’s Patches. Immunity (2002) 17(6):823–33. doi: 10.1016/S1074-7613(02)00479-X
44. Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, et al. The Lymphotoxin-Beta Receptor Induces Different Patterns of Gene Expression Via Two NF-Kappab Pathways. Immunity (2002) 17(4):525–35. doi: 10.1016/S1074-7613(02)00423-5
45. Yilmaz ZB, Weih DS, Sivakumar V, Weih F. Relb is Required for Peyer’s Patch Development: Differential Regulation of P52-Relb by Lymphotoxin and TNF. EMBO J (2003) 22(1):121–30. doi: 10.1093/emboj/cdg004
46. Mebius RE, Streeter PR, Michie S, Butcher EC, Weissman IL. A Developmental Switch in Lymphocyte Homing Receptor and Endothelial Vascular Addressin Expression Regulates Lymphocyte Homing and Permits CD4+ CD3- Cells to Colonize Lymph Nodes. Proc Natl Acad Sci USA (1996) 93(20):11019–24. doi: 10.1073/pnas.93.20.11019
47. Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, et al. Human Fetal Lymphoid Tissue-Inducer Cells are Interleukin 17-Producing Precursors to RORC+ CD127+ Natural Killer-Like Cells. Nat Immunol (2009) 10(1):66–74. doi: 10.1038/ni.1668
48. Browning JL, Allaire N, Ngam-Ek A, Notidis E, Hunt J, Perrin S, et al. Lymphotoxin-Beta Receptor Signaling is Required for the Homeostatic Control of HEV Differentiation and Function. Immunity (2005) 23(5):539–50. doi: 10.1016/j.immuni.2005.10.002
49. Krautler NJ, Kana V, Kranich J, Tian Y, Perera D, Lemm D, et al. Follicular Dendritic Cells Emerge From Ubiquitous Perivascular Precursors. Cell (2012) 150(1):194–206. doi: 10.1016/j.cell.2012.05.032
50. Jeucken KCM, Koning JJ, Mebius RE, Tas SW. The Role of Endothelial Cells and TNF-Receptor Superfamily Members in Lymphoid Organogenesis and Function During Health and Inflammation. Front Immunol (2019) 10:2700. doi: 10.3389/fimmu.2019.02700
51. Cheng HW, Onder L, Novkovic M, Soneson C, Lutge M, Pikor N, et al. Origin and Differentiation Trajectories of Fibroblastic Reticular Cells in the Splenic White Pulp. Nat Commun (2019) 10(1):1739. doi: 10.1038/s41467-019-09728-3
52. Grabner R, Lotzer K, Dopping S, Hildner M, Radke D, Beer M, et al. Lymphotoxin Beta Receptor Signaling Promotes Tertiary Lymphoid Organogenesis in the Aorta Adventitia of Aged Apoe-/- Mice. J Exp Med (2009) 206(1):233–48. doi: 10.1084/jem.20080752
53. Degli-Esposti MA, Davis-Smith T, Din WS, Smolak PJ, Goodwin RG, Smith CA. Activation of the Lymphotoxin Beta Receptor by Cross-Linking Induces Chemokine Production and Growth Arrest in A375 Melanoma Cells. J Immunol (1997) 158(4):1756–62.
54. Chai Q, Onder L, Scandella E, Gil-Cruz C, Perez-Shibayama C, Cupovic J, et al. Maturation of Lymph Node Fibroblastic Reticular Cells From Myofibroblastic Precursors is Critical for Antiviral Immunity. Immunity (2013) 38(5):1013–24. doi: 10.1016/j.immuni.2013.03.012
55. Browning JL, Sizing ID, Lawton P, Bourdon PR, Rennert PD, Majeau GR, et al. Characterization of Lymphotoxin-Alpha Beta Complexes on the Surface of Mouse Lymphocytes. J Immunol (1997) 159(7):3288–98.
56. Ware CF. Targeting Lymphocyte Activation Through the Lymphotoxin and LIGHT Pathways. Immunol Rev (2008) 223:186–201. doi: 10.1111/j.1600-065X.2008.00629.x
57. Lee Y, Chin RK, Christiansen P, Sun Y, Tumanov AV, Wang J, et al. Recruitment and Activation of Naive T Cells in the Islets by Lymphotoxin Beta Receptor-Dependent Tertiary Lymphoid Structure. Immunity (2006) 25(3):499–509. doi: 10.1016/j.immuni.2006.06.016
58. Randall TD, Carragher DM, Rangel-Moreno J. Development of Secondary Lymphoid Organs. Annu Rev Immunol (2008) 26:627–50. doi: 10.1146/annurev.immunol.26.021607.090257
59. Hampton HR, Chtanova T. Lymphatic Migration of Immune Cells. Front Immunol (2019) 10:1168. doi: 10.3389/fimmu.2019.01168
60. Zhang X, Chen L, Dang WQ, Cao MF, Xiao JF, Lv SQ, et al. CCL8 Secreted by Tumor-Associated Macrophages Promotes Invasion and Stemness of Glioblastoma Cells Via ERK1/2 Signaling. Lab Invest (2020) 100(4):619–29. doi: 10.1038/s41374-019-0345-3
61. Pascual-Garcia M, Bonfill-Teixidor E, Planas-Rigol E, Rubio-Perez C, Iurlaro R, Arias A, et al. LIF Regulates CXCL9 in Tumor-Associated Macrophages and Prevents CD8(+) T Cell Tumor-Infiltration Impairing Anti-PD1 Therapy. Nat Commun (2019) 10(1):2416. doi: 10.1038/s41467-019-10369-9
62. Qu Y, Wen J, Thomas G, Yang W, Prior W, He W, et al. Baseline Frequency of Inflammatory Cxcl9-Expressing Tumor-Associated Macrophages Predicts Response to Avelumab Treatment. Cell Rep (2020) 32(1):107873. doi: 10.1016/j.celrep.2020.107873
63. Kodelja V, Muller C, Politz O, Hakij N, Orfanos CE, Goerdt S. Alternative Macrophage Activation-Associated CC-Chemokine-1, a Novel Structural Homologue of Macrophage Inflammatory Protein-1 Alpha With a Th2-Associated Expression Pattern. J Immunol (1998) 160(3):1411–8.
64. Chen J, Yao Y, Gong C, Yu F, Su S, Chen J, et al. Ccl18 From Tumor-Associated Macrophages Promotes Breast Cancer Metastasis Via Pitpnm3. Cancer Cell (2011) 19(4):541–55. doi: 10.1016/j.ccr.2011.02.006
65. Bista P, Zeng W, Ryan S, Bailly V, Browning JL, Lukashev ME. TRAF3 Controls Activation of the Canonical and Alternative Nfkappab by the Lymphotoxin Beta Receptor. J Biol Chem (2010) 285(17):12971–8. doi: 10.1074/jbc.M109.076091
66. Lau TS, Chung TK, Cheung TH, Chan LK, Cheung LW, Yim SF, et al. Cancer Cell-Derived Lymphotoxin Mediates Reciprocal Tumour-Stromal Interactions in Human Ovarian Cancer by Inducing CXCL11 in Fibroblasts. J Pathol (2014) 232(1):43–56. doi: 10.1002/path.4258
67. Yeh DY, Wu CC, Chin YP, Lu CJ, Wang YH, Chen MC. Mechanisms of Human Lymphotoxin Beta Receptor Activation on Upregulation of CCL5/RANTES Production. Int Immunopharmacol (2015) 28(1):220–9. doi: 10.1016/j.intimp.2015.06.010
68. Grandoch M, Feldmann K, Gothert JR, Dick LS, Homann S, Klatt C, et al. Deficiency in Lymphotoxin Beta Receptor Protects From Atherosclerosis in Apoe-Deficient Mice. Circ Res (2015) 116(8):e57–68. doi: 10.1161/CIRCRESAHA.116.305723
69. Bonizzi G, Karin M. The Two NF-Kappab Activation Pathways and Their Role in Innate and Adaptive Immunity. Trends Immunol (2004) 25(6):280–8. doi: 10.1016/j.it.2004.03.008
70. Vulcano M, Struyf S, Scapini P, Cassatella M, Bernasconi S, Bonecchi R, et al. Unique Regulation of CCL18 Production by Maturing Dendritic Cells. J Immunol (2003) 170(7):3843–9. doi: 10.4049/jimmunol.170.7.3843
71. Proost P, Verpoest S, Van de Borne K, Schutyser E, Struyf S, Put W, et al. Synergistic Induction of CXCL9 and CXCL11 by Toll-Like Receptor Ligands and Interferon-Gamma in Fibroblasts Correlates With Elevated Levels of CXCR3 Ligands in Septic Arthritis Synovial Fluids. J Leukoc Biol (2004) 75(5):777–84. doi: 10.1189/jlb.1003524
72. Vondenhoff MF, Greuter M, Goverse G, Elewaut D, Dewint P, Ware CF, et al. Ltbetar Signaling Induces Cytokine Expression and Up-Regulates Lymphangiogenic Factors in Lymph Node Anlagen. J Immunol (2009) 182(9):5439–45. doi: 10.4049/jimmunol.0801165
73. Weinstein AM, Chen L, Brzana EA, Patil PR, Taylor JL, Fabian KL, et al. Tbet and IL-36gamma Cooperate in Therapeutic DC-Mediated Promotion of Ectopic Lymphoid Organogenesis in the Tumor Microenvironment. Oncoimmunology (2017) 6(6):e1322238. doi: 10.1080/2162402X.2017.1322238
74. Kratz A, Campos-Neto A, Hanson MS, Ruddle NH. Chronic Inflammation Caused by Lymphotoxin is Lymphoid Neogenesis. J Exp Med (1996) 183(4):1461–72. doi: 10.1084/jem.183.4.1461
75. Chen SC, Vassileva G, Kinsley D, Holzmann S, Manfra D, Wiekowski MT, et al. Ectopic Expression of the Murine Chemokines CCL21a and CCL21b Induces the Formation of Lymph Node-Like Structures in Pancreas, But Not Skin, of Transgenic Mice. J Immunol (2002) 168(3):1001–8. doi: 10.4049/jimmunol.168.3.1001
76. Fan L, Reilly CR, Luo Y, Dorf ME, Lo D. Cutting Edge: Ectopic Expression of the Chemokine TCA4/SLC is Sufficient to Trigger Lymphoid Neogenesis. J Immunol (2000) 164(8):3955–9. doi: 10.4049/jimmunol.164.8.3955
77. Johansson-Percival A, He B, Li ZJ, Kjellen A, Russell K, Li J, et al. De Novo Induction of Intratumoral Lymphoid Structures and Vessel Normalization Enhances Immunotherapy in Resistant Tumors. Nat Immunol (2017) 18(11):1207–17. doi: 10.1038/ni.3836
78. Chen L, Fabian KL, Taylor JL, Storkus WJ. Therapeutic Use of Dendritic Cells to Promote the Extranodal Priming of Anti-Tumor Immunity. Front Immunol (2013) 4:388. doi: 10.3389/fimmu.2013.00388
79. Zhu G, Nemoto S, Mailloux AW, Perez-Villarroel P, Nakagawa R, Falahat R, et al. Induction of Tertiary Lymphoid Structures With Antitumor Function by a Lymph Node-Derived Stromal Cell Line. Front Immunol (2018) 9:1609. doi: 10.3389/fimmu.2018.01609
80. Weiden J, Tel J, Figdor CG. Synthetic Immune Niches for Cancer Immunotherapy. Nat Rev Immunol (2018) 18(3):212–9. doi: 10.1038/nri.2017.89
81. Aisenbrey EA, Murphy WL. Synthetic Alternatives to Matrigel. Nat Rev Mater (2020) 5(7):539–51. doi: 10.1038/s41578-020-0199-8
82. Vukicevic S, Kleinman HK, Luyten FP, Roberts AB, Roche NS, Reddi AH. Identification of Multiple Active Growth Factors in Basement Membrane Matrigel Suggests Caution in Interpretation of Cellular Activity Related to Extracellular Matrix Components. Exp Cell Res (1992) 202(1):1–8. doi: 10.1016/0014-4827(92)90397-Q
83. Polykandriotis E, Arkudas A, Horch RE, Kneser U. To Matrigel or Not to Matrigel. Am J Pathol (2008) 172(5):1441; author reply –2. doi: 10.2353/ajpath.2008.071215
84. Liu X, Zheng C, Luo X, Wang X, Jiang H. Recent Advances of Collagen-Based Biomaterials: Multi-Hierarchical Structure, Modification and Biomedical Applications. Mater Sci Eng C Mater Biol Appl (2019) 99:1509–22. doi: 10.1016/j.msec.2019.02.070
85. Shoulders MD, Raines RT. Collagen Structure and Stability. Annu Rev Biochem (2009) 78:929–58. doi: 10.1146/annurev.biochem.77.032207.120833
86. Meyer M. Processing of Collagen Based Biomaterials and the Resulting Materials Properties. BioMed Eng Online (2019) 18(1):24. doi: 10.1186/s12938-019-0647-0
87. Werkmeister JA, Ramshaw JA. Recombinant Protein Scaffolds for Tissue Engineering. BioMed Mater (2012) 7(1):012002. doi: 10.1088/1748-6041/7/1/012002
88. Gilbert TW, Sellaro TL, Badylak SF. Decellularization of Tissues and Organs. Biomaterials (2006) 27(19):3675–83. doi: 10.1016/j.biomaterials.2006.02.014
89. Chen P, Marsilio E, Goldstein RH, Yannas IV, Spector M. Formation of Lung Alveolar-Like Structures in Collagen-Glycosaminoglycan Scaffolds in Vitro. Tissue Eng (2005) 11(9-10):1436–48. doi: 10.1089/ten.2005.11.1436
90. Boyce ST, Christianson DJ, Hansbrough JF. Structure of a Collagen-GAG Dermal Skin Substitute Optimized for Cultured Human Epidermal Keratinocytes. J BioMed Mater Res (1988) 22(10):939–57. doi: 10.1002/jbm.820221008
91. Gerhold D, Caskey CT. It’s the Genes! EST Access to Hum Genome content. Bioessays (1996) 18(12):973–81. doi: 10.1002/bies.950181207
92. Aprahamian M, Lambert A, Balboni G, Lefebvre F, Schmitthaeusler R, Damge C, et al. A New Reconstituted Connective Tissue Matrix: Preparation, Biochemical, Structural and Mechanical Studies. J BioMed Mater Res (1987) 21(8):965–77. doi: 10.1002/jbm.820210803
93. Wu X, Black L, Santacana-Laffitte G, Patrick CW Jr. Preparation and Assessment of Glutaraldehyde-Crosslinked Collagen-Chitosan Hydrogels for Adipose Tissue Engineering. J BioMed Mater Res A (2007) 81(1):59–65. doi: 10.1002/jbm.a.31003
94. Shahabeddin L, Berthod F, Damour O, Collombel C. Characterization of Skin Reconstructed on a Chitosan-Cross-Linked Collagen-Glycosaminoglycan Matrix. Skin Pharmacol (1990) 3(2):107–14. doi: 10.1159/000210857
95. Irawan V, Sung TC, Higuchi A, Ikoma T. Collagen Scaffolds in Cartilage Tissue Engineering and Relevant Approaches for Future Development. Tissue Eng Regener Med (2018) 15(6):673–97. doi: 10.1007/s13770-018-0135-9
96. Khan R, Khan MH. Use of Collagen as a Biomaterial: An Update. J Indian Soc Periodontol (2013) 17(4):539–42. doi: 10.4103/0972-124X.118333
97. Suematsu S, Watanabe T. Generation of a Synthetic Lymphoid Tissue-Like Organoid in Mice. Nat Biotechnol (2004) 22(12):1539–45. doi: 10.1038/nbt1039
98. Okamoto N, Chihara R, Shimizu C, Nishimoto S, Watanabe T. Artificial Lymph Nodes Induce Potent Secondary Immune Responses in Naive and Immunodeficient Mice. J Clin Invest (2007) 117(4):997–1007. doi: 10.1172/JCI30379
99. Kobayashi Y, Kato K, Watanabe T. Synthesis of Functional Artificial Lymphoid Tissues. Discovery Med (2011) 12(65):351–62.
100. Bashir S, Hina M, Iqbal J, Rajpar AH, Mujtaba MA, Alghamdi NA, et al. Fundamental Concepts of Hydrogels: Synthesis, Properties, and Their Applications. Polymers (Basel) (2020) 12(11). doi: 10.3390/polym12112702
101. Lowenberg C, Balk M, Wischke C, Behl M, Lendlein A. Shape-Memory Hydrogels: Evolution of Structural Principles to Enable Shape Switching of Hydrophilic Polymer Networks. Acc Chem Res (2017) 50(4):723–32. doi: 10.1021/acs.accounts.6b00584
102. Mahinroosta M, Jomeh Farsangi Z, Allahverdi A, Shakoori Z. Hydrogels as Intelligent Materials: A Brief Review of Synthesis, Properties and Applications. Materials Today Chem (2018) 8:42–55. doi: 10.1016/j.mtchem.2018.02.004
103. Wang Q. Smart Materials for Tissue Engineering: Applications: Royal Society of Chemistry. Royal Society of Chemistry (2017). doi: 10.1039/9781788010542
104. Kabanov AV, Vinogradov SV. Nanogels as Pharmaceutical Carriers: Finite Networks of Infinite Capabilities. Angewandte Chemie (2009) 48: (30):5418–29. doi: 10.1002/anie.200900441
105. Dusek K, Duskova-Smrckova M. Volume Phase Transition in Gels: Its Discovery and Development. Gels (2020) 6(3):1–12. doi: 10.3390/gels6030022
106. Grinberg VY, Dubovik AS, Kuznetsov DV, Grinberg NV, Grosberg AY, Tanaka T. Studies of the Thermal Volume Transition of Poly(N-Isopropylacrylamide) Hydrogels by High-Sensitivity Differential Scanning Microcalorimetry. 2. Thermodynamic Functions. Macromolecules (2000) 33(23):8685–92. doi: 10.1021/ma000527w
107. Schild HG. Poly(N-Isopropylacrylamide): Experiment, Theory and Application. Prog Polymer Sci (1992) 17(2):163–249. doi: 10.1016/0079-6700(92)90023-R
108. Purwada A, Singh A. Immuno-Engineered Organoids for Regulating the Kinetics of B-Cell Development and Antibody Production. Nat Protoc (2017) 12(1):168–82. doi: 10.1038/nprot.2016.157
109. Purwada A, Jaiswal MK, Ahn H, Nojima T, Kitamura D, Gaharwar AK, et al. Ex Vivo Engineered Immune Organoids for Controlled Germinal Center Reactions. Biomaterials (2015) 63:24–34. doi: 10.1016/j.biomaterials.2015.06.002
110. Smith TT, Moffett HF, Stephan SB, Opel CF, Dumigan AG, Jiang X, et al. Biopolymers Codelivering Engineered T Cells and STING Agonists Can Eliminate Heterogeneous Tumors. J Clin Invest (2017) 127(6):2176–91. doi: 10.1172/JCI87624
111. Pedde RD, Mirani B, Navaei A, Styan T, Wong S, Mehrali M, et al. Emerging Biofabrication Strategies for Engineering Complex Tissue Constructs. Adv Mater (2017) 29(19):1–27. doi: 10.1002/adma.201606061
112. Wegst UG, Bai H, Saiz E, Tomsia AP, Ritchie RO. Bioinspired Structural Materials. Nat Mater (2015) 14(1):23–36. doi: 10.1038/nmat4089
113. Bencherif SA, Warren Sands R, Ali OA, Li WA, Lewin SA, Braschler TM, et al. Injectable Cryogel-Based Whole-Cell Cancer Vaccines. Nat Commun (2015) 6:7556. doi: 10.1038/ncomms8556
114. Bencherif SA, Sands RW, Bhatta D, Arany P, Verbeke CS, Edwards DA, et al. Injectable Preformed Scaffolds With Shape-Memory Properties. Proc Natl Acad Sci USA (2012) 109(48):19590–5. doi: 10.1073/pnas.1211516109
115. Bauleth-Ramos T, Shih T-Y, Shahbazi M-A, Najibi AJ, Mao AS, Liu D, et al. Acetalated Dextran Nanoparticles Loaded Into an Injectable Alginate Cryogel for Combined Chemotherapy and Cancer Vaccination. Adv Funct Mater (2019) 29(35):1903686. doi: 10.1002/adfm.201903686
116. Singh A, Suri S, Roy K. In-Situ Crosslinking Hydrogels for Combinatorial Delivery of Chemokines and Sirna-DNA Carrying Microparticles to Dendritic Cells. Biomaterials (2009) 30(28):5187–200. doi: 10.1016/j.biomaterials.2009.06.001
117. Singh A, Qin H, Fernandez I, Wei J, Lin J, Kwak LW, et al. An Injectable Synthetic Immune-Priming Center Mediates Efficient T-Cell Class Switching and T-Helper 1 Response Against B Cell Lymphoma. J Control Release (2011) 155(2):184–92. doi: 10.1016/j.jconrel.2011.06.008
118. Najibi AJ, Mooney DJ. Cell and Tissue Engineering in Lymph Nodes for Cancer Immunotherapy. Adv Drug Delivery Rev (2020) 161-162:42–62. doi: 10.1016/j.addr.2020.07.023
119. Li AW, Sobral MC, Badrinath S, Choi Y, Graveline A, Stafford AG, et al. A Facile Approach to Enhance Antigen Response for Personalized Cancer Vaccination. Nat Mater (2018) 17(6):528–34. doi: 10.1038/s41563-018-0028-2
120. Giese C, Demmler CD, Ammer R, Hartmann S, Lubitz A, Miller L, et al. A Human Lymph Node in Vitro–Challenges and Progress. Artif Organs (2006) 30(10):803–8. doi: 10.1111/j.1525-1594.2006.00303.x
121. Lakshmanan VK, Jindal S, Packirisamy G, Ojha S, Lian S, Kaushik A, et al. Nanomedicine-Based Cancer Immunotherapy: Recent Trends and Future Perspectives. Cancer Gene Ther (2021). doi: 10.1038/s41417-021-00299-4
122. Kim EM, Jeong HJ. Liposomes: Biomedical Applications. Chonnam Med J (2021) 57(1):27–35. doi: 10.4068/cmj.2021.57.1.27
123. Plotnick AN. Lipid-Based Formulations of Amphotericin B. J Am Vet Med Assoc (2000) 216(6):838–41. doi: 10.2460/javma.2000.216.838
124. Patel HM. Serum Opsonins and Liposomes: Their Interaction and Opsonophagocytosis. Crit Rev Ther Drug Carrier Syst (1992) 9(1):39–90.
125. Scherphof GL, Dijkstra J, Spanjer HH, Derksen JT, Roerdink FH. Uptake and Intracellular Processing of Targeted and Nontargeted Liposomes by Rat Kupffer Cells in Vivo and in Vitro. Ann N Y Acad Sci (1985) 446:368–84. doi: 10.1111/j.1749-6632.1985.tb18414.x
126. Alving CR, Steck EA, Chapman WL Jr., Waits VB, Hendricks LD, Swartz GM Jr., et al. Therapy of Leishmaniasis: Superior Efficacies of Liposome-Encapsulated Drugs. Proc Natl Acad Sci USA (1978) 75(6):2959–63. doi: 10.1073/pnas.75.6.2959
127. Mayer LD, Hope MJ, Cullis PR. Vesicles of Variable Sizes Produced by a Rapid Extrusion Procedure. Biochim Biophys Acta (1986) 858(1):161–8. doi: 10.1016/0005-2736(86)90302-0
128. Jahn A, Stavis SM, Hong JS, Vreeland WN, DeVoe DL, Gaitan M. Microfluidic Mixing and the Formation of Nanoscale Lipid Vesicles. ACS Nano (2010) 4(4):2077–87. doi: 10.1021/nn901676x
129. Zhigaltsev IV, Belliveau N, Hafez I, Leung AK, Huft J, Hansen C, et al. Bottom-Up Design and Synthesis of Limit Size Lipid Nanoparticle Systems With Aqueous and Triglyceride Cores Using Millisecond Microfluidic Mixing. Langmuir (2012) 28(7):3633–40. doi: 10.1021/la204833h
131. Jahn A, Vreeland WN, DeVoe DL, Locascio LE, Gaitan M. Microfluidic Directed Formation of Liposomes of Controlled Size. Langmuir (2007) 23(11):6289–93. doi: 10.1021/la070051a
132. Hinna A, Steiniger F, Hupfeld S, Stein P, Kuntsche J, Brandl M. Filter-Extruded Liposomes Revisited: A Study Into Size Distributions and Morphologies in Relation to Lipid-Composition and Process Parameters. J Liposome Res (2016) 26(1):11–20. doi: 10.3109/08982104.2015.1022556
133. Panahi Y, Farshbaf M, Mohammadhosseini M, Mirahadi M, Khalilov R, Saghfi S, et al. Recent Advances on Liposomal Nanoparticles: Synthesis, Characterization and Biomedical Applications. Artif Cells Nanomed Biotechnol (2017) 45(4):788–99. doi: 10.1080/21691401.2017.1282496
134. Bulbake U, Doppalapudi S, Kommineni N, Khan W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics (2017) 9(2):1–33. doi: 10.3390/pharmaceutics9020012
135. Has C, Sunthar P. A Comprehensive Review on Recent Preparation Techniques of Liposomes. J Liposome Res (2020) 30(4):336–65. doi: 10.1080/08982104.2019.1668010
136. Bangham AD, Horne RW. Negative Staining of Phospholipids and Their Structural Modification by Surface-Active Agents as Observed in the Electron Microscope. J Mol Biol (1964) 8:660–8. doi: 10.1016/S0022-2836(64)80115-7
137. Haluska CK, Riske KA, Marchi-Artzner V, Lehn JM, Lipowsky R, Dimova R. Time Scales of Membrane Fusion Revealed by Direct Imaging of Vesicle Fusion With High Temporal Resolution. Proc Natl Acad Sci USA (2006) 103(43):15841–6. doi: 10.1073/pnas.0602766103
138. Marrink SJ, Mark AE. The Mechanism of Vesicle Fusion as Revealed by Molecular Dynamics Simulations. J Am Chem Soc (2003) 125(37):11144–5. doi: 10.1021/ja036138+
139. Gabizon A, Papahadjopoulos D. Liposome Formulations With Prolonged Circulation Time in Blood and Enhanced Uptake by Tumors. Proc Natl Acad Sci USA (1988) 85(18):6949–53. doi: 10.1073/pnas.85.18.6949
140. Senior J, Gregoriadis G. Is Half-Life of Circulating Liposomes Determined by Changes in Their Permeability? FEBS Lett (1982) 145(1):109–14. doi: 10.1016/0014-5793(82)81216-7
141. Immordino ML, Dosio F, Cattel L. Stealth Liposomes: Review of the Basic Science, Rationale, and Clinical Applications, Existing and Potential. Int J Nanomedicine (2006) 1(3):297–315.
142. Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Optimizing Liposomes for Delivery of Chemotherapeutic Agents to Solid Tumors. Pharmacol Rev (1999) 51(4):691–743.
143. Allen TM, Hansen C, Martin F, Redemann C, Yau-Young A. Liposomes Containing Synthetic Lipid Derivatives of Poly(Ethylene Glycol) Show Prolonged Circulation Half-Lives in Vivo. Biochim Biophys Acta (1991) 1066(1):29–36. doi: 10.1016/0005-2736(91)90246-5
144. Khan AA, Allemailem KS, Almatroodi SA, Almatroudi A, Rahmani AH. Recent Strategies Towards the Surface Modification of Liposomes: An Innovative Approach for Different Clinical Applications. 3 Biotech (2020) 10(4):163. doi: 10.1007/s13205-020-2144-3
145. Willis M, Forssen E. Ligand-Targeted Liposomes. Adv Drug Delivery Rev (1998) 29(3):249–71. doi: 10.1016/S0169-409X(97)00083-5
146. Ponce AM, Vujaskovic Z, Yuan F, Needham D, Dewhirst MW. Hyperthermia Mediated Liposomal Drug Delivery. Int J Hyperthermia (2006) 22(3):205–13. doi: 10.1080/02656730600582956
147. Kaur R, Morris R, Bencsik M, Vangala A, Rades T, Perrie Y. Development of a Novel Magnetic Resonance Imaging Contrast Agent for Pressure Measurements Using Lipid-Coated Microbubbles. J BioMed Nanotechnol (2009) 5(6):707–15. doi: 10.1166/jbn.2009.1087
148. Shum P, Kim JM, Thompson DH. Phototriggering of Liposomal Drug Delivery Systems. Adv Drug Delivery Rev (2001) 53(3):273–84. doi: 10.1016/S0169-409X(01)00232-0
149. Yavlovich A, Singh A, Blumenthal R, Puri A. A Novel Class of Photo-Triggerable Liposomes Containing DPPC:DC(8,9)PC as Vehicles for Delivery of Doxorubcin to Cells. Biochim Biophys Acta (2011) 1808(1):117–26. doi: 10.1016/j.bbamem.2010.07.030
150. Yu JR, Janssen M, Liang BJ, Huang HC, Fisher JP. A Liposome/Gelatin Methacrylate Nanocomposite Hydrogel System for Delivery of Stromal Cell-Derived Factor-1alpha and Stimulation of Cell Migration. Acta Biomater (2020) 108:67–76. doi: 10.1016/j.actbio.2020.03.015
151. Rubio AJ, Zhong X, Porter TM. In Vitro Characterization of Chemokine-Loaded Liposomes. Cogent Biol (2019) 5(1):1–17. doi: 10.1080/23312025.2019.1662931
152. Liu T, Liu Z, Chen J, Jin R, Bai Y, Zhou Y, et al. Redox-Responsive Supramolecular Micelles for Targeted Imaging and Drug Delivery to Tumor. J BioMed Nanotechnol (2018) 14(6):1107–16. doi: 10.1166/jbn.2018.2573
153. Letchford K, Burt H. A Review of the Formation and Classification of Amphiphilic Block Copolymer Nanoparticulate Structures: Micelles, Nanospheres, Nanocapsules and Polymersomes. Eur J Pharm Biopharm (2007) 65(3):259–69. doi: 10.1016/j.ejpb.2006.11.009
154. Ishii S, Kaneko J, Nagasaki Y. Development of a Long-Acting, Protein-Loaded, Redox-Active, Injectable Gel Formed by a Polyion Complex for Local Protein Therapeutics. Biomaterials (2016) 84:210–8. doi: 10.1016/j.biomaterials.2016.01.029
155. Li H, Li Y, Wang X, Hou Y, Hong X, Gong T, et al. Rational Design of Polymeric Hybrid Micelles to Overcome Lymphatic and Intracellular Delivery Barriers in Cancer Immunotherapy. Theranostics (2017) 7(18):4383–98. doi: 10.7150/thno.20745
156. Liu L, Yi H, Wang C, He H, Li P, Pan H, et al. Integrated Nanovaccine With Microrna-148a Inhibition Reprograms Tumor-Associated Dendritic Cells by Modulating Mir-148a/DNMT1/SOCS1 Axis. J Immunol (2016) 197(4):1231–41. doi: 10.4049/jimmunol.1600182
157. Li C, Zhang X, Chen Q, Zhang J, Li W, Hu H, et al. Synthetic Polymeric Mixed Micelles Targeting Lymph Nodes Trigger Enhanced Cellular and Humoral Immune Responses. ACS Appl Mater Interfaces (2018) 10(3):2874–89. doi: 10.1021/acsami.7b14004
158. Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V. PLGA-Based Nanoparticles: An Overview of Biomedical Applications. J Control Release (2012) 161(2):505–22. doi: 10.1016/j.jconrel.2012.01.043
159. Shive MS, Anderson JM. Biodegradation and Biocompatibility of PLA and PLGA Microspheres. Adv Drug Delivery Rev (1997) 28(1):5–24. doi: 10.1016/S0169-409X(97)00048-3
160. Hamdy S, Haddadi A, Hung RW, Lavasanifar A. Targeting Dendritic Cells With Nano-Particulate Plga Cancer Vaccine Formulations. Adv Drug Delivery Rev (2011) 63(10-11):943–55. doi: 10.1016/j.addr.2011.05.021
161. Guo Y, Wang D, Song Q, Wu T, Zhuang X, Bao Y, et al. Erythrocyte Membrane-Enveloped Polymeric Nanoparticles as Nanovaccine for Induction of Antitumor Immunity Against Melanoma. ACS Nano (2015) 9(7):6918–33. doi: 10.1021/acsnano.5b01042
162. Fang RH, Hu CM, Luk BT, Gao W, Copp JA, Tai Y, et al. Cancer Cell Membrane-Coated Nanoparticles for Anticancer Vaccination and Drug Delivery. Nano Lett (2014) 14(4):2181–8. doi: 10.1021/nl500618u
163. Mi Y, Smith CC, Yang F, Qi Y, Roche KC, Serody JS, et al. A Dual Immunotherapy Nanoparticle Improves T-Cell Activation and Cancer Immunotherapy. Adv Mater (2018) 30(25):e1706098. doi: 10.1002/adma.201706098
164. Rosalia RA, Cruz LJ, van Duikeren S, Tromp AT, Silva AL, Jiskoot W, et al. CD40-Targeted Dendritic Cell Delivery of PLGA-Nanoparticle Vaccines Induce Potent Anti-Tumor Responses. Biomaterials (2015) 40:88–97. doi: 10.1016/j.biomaterials.2014.10.053
165. Kobayashi Y, Watanabe T. Gel-Trapped Lymphorganogenic Chemokines Trigger Artificial Tertiary Lymphoid Organs and Mount Adaptive Immune Responses in Vivo. Front Immunol (2016) 7:316. doi: 10.3389/fimmu.2016.00316
166. Stephan SB, Taber AM, Jileaeva I, Pegues EP, Sentman CL, Stephan MT. Biopolymer Implants Enhance the Efficacy of Adoptive T-Cell Therapy. Nat Biotechnol (2015) 33(1):97–101. doi: 10.1038/nbt.3104
167. Mariani E, Lisignoli G, Borzi RM, Pulsatelli L. Biomaterials: Foreign Bodies or Tuners for the Immune Response? Int J Mol Sci (2019) 20(3):1–42. doi: 10.3390/ijms20030636
168. Chung L, Maestas DR Jr., Housseau F, Elisseeff JH. Key Players in the Immune Response to Biomaterial Scaffolds for Regenerative Medicine. Adv Drug Delivery Rev (2017) 114:184–92. doi: 10.1016/j.addr.2017.07.006
169. Christo SN, Diener KR, Bachhuka A, Vasilev K, Hayball JD. Innate Immunity and Biomaterials At the Nexus: Friends or Foes. BioMed Res Int (2015) 2015:342304. doi: 10.1155/2015/342304
170. Labow RS, Meek E, Santerre JP. Neutrophil-Mediated Biodegradation of Medical Implant Materials. J Cell Physiol (2001) 186(1):95–103. doi: 10.1002/1097-4652(200101)186:1<95::AID-JCP1008>3.0.CO;2-0
171. Nimeri G, Ohman L, Elwing H, Wettero J, Bengtsson T. The Influence of Plasma Proteins and Platelets on Oxygen Radical Production and F-Actin Distribution in Neutrophils Adhering to Polymer Surfaces. Biomaterials (2002) 23(8):1785–95. doi: 10.1016/S0142-9612(01)00305-2
172. Nimeri G, Majeed M, Elwing H, Ohman L, Wettero J, Bengtsson T. Oxygen Radical Production in Neutrophils Interacting With Platelets and Surface-Immobilized Plasma Proteins: Role of Tyrosine Phosphorylation. J BioMed Mater Res A (2003) 67(2):439–47. doi: 10.1002/jbm.a.10081
173. Wettero J, Bengtsson T, Tengvall P. Complement Activation on Immunoglobulin G-Coated Hydrophobic Surfaces Enhances the Release of Oxygen Radicals From Neutrophils Through an Actin-Dependent Mechanism. J BioMed Mater Res (2000) 51(4):742–51. doi: 10.1002/1097-4636(20000915)51:4<742::AID-JBM24>3.0.CO;2-D
174. Jones JA, Chang DT, Meyerson H, Colton E, Kwon IK, Matsuda T, et al. Proteomic Analysis and Quantification of Cytokines and Chemokines From Biomaterial Surface-Adherent Macrophages and Foreign Body Giant Cells. J BioMed Mater Res A (2007) 83(3):585–96. doi: 10.1002/jbm.a.31221
175. Yamashiro S, Kamohara H, Wang JM, Yang D, Gong WH, Yoshimura T. Phenotypic and Functional Change of Cytokine-Activated Neutrophils: Inflammatory Neutrophils are Heterogeneous and Enhance Adaptive Immune Responses. J Leukoc Biol (2001) 69(5):698–704.
176. Garg K, Pullen NA, Oskeritzian CA, Ryan JJ, Bowlin GL. Macrophage Functional Polarization (M1/M2) in Response to Varying Fiber and Pore Dimensions of Electrospun Scaffolds. Biomaterials (2013) 34(18):4439–51. doi: 10.1016/j.biomaterials.2013.02.065
177. Chen S, Jones JA, Xu Y, Low HY, Anderson JM, Leong KW. Characterization of Topographical Effects on Macrophage Behavior in a Foreign Body Response Model. Biomaterials (2010) 31(13):3479–91. doi: 10.1016/j.biomaterials.2010.01.074
178. Fink J, Fuhrmann R, Scharnweber T, Franke RP. Stimulation of Monocytes and Macrophages: Possible Influence of Surface Roughness. Clin Hemorheol Microcirc (2008) 39(1-4):205–12. doi: 10.3233/CH-2008-1090
179. Nutrition CfFSaA, Medicine CfV, Affairs OoR, Research CfDEa, Health CfDaR, Research CfBEa. Part 11, Electronic Records; Electronic Signatures - Scope and Application. U.S. Food and drug Administration (2003).
180. Tinkle S, McNeil SE, Muhlebach S, Bawa R, Borchard G, Barenholz YC, et al. Nanomedicines: Addressing the Scientific and Regulatory Gap. Ann N Y Acad Sci (2014) 1313:35–56. doi: 10.1111/nyas.12403
181. Teli MK, Mutalik S, Rajanikant GK. Nanotechnology and Nanomedicine: Going Small Means Aiming Big. Curr Pharm Des (2010) 16(16):1882–92. doi: 10.2174/138161210791208992
182. Hernandez I, Prasad V, Gellad WF. Total Costs of Chimeric Antigen Receptor T-Cell Immunotherapy. JAMA Oncol (2018) 4(7):994–6. doi: 10.1001/jamaoncol.2018.0977
183. Administration FaD. Definition of Primary Mode of Action of a Combination Product. Federal Register (2004). pp. 25527–33.
184. Boehler RM, Graham JG, Shea LD. Tissue Engineering Tools for Modulation of the Immune Response. Biotechniques (2011) 51(4):239–40, 42, 44 passim. doi: 10.2144/000113754
Keywords: immunotherapy, tertiary lymphoid structure (TLS), cancer, bioengineering, biomaterials
Citation: Aoyama S, Nakagawa R, Mulé JJ and Mailloux AW (2021) Inducible Tertiary Lymphoid Structures: Promise and Challenges for Translating a New Class of Immunotherapy. Front. Immunol. 12:675538. doi: 10.3389/fimmu.2021.675538
Received: 03 March 2021; Accepted: 27 April 2021;
Published: 14 May 2021.
Edited by:
Vivek Verma, Georgetown University, United StatesReviewed by:
Iain Comerford, University of Adelaide, AustraliaSaba Nayar, University of Birmingham, United Kingdom
Copyright © 2021 Aoyama, Nakagawa, Mulé and Mailloux. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adam W. Mailloux, YWRhbS1tYWlsbG91eEB1aW93YS5lZHU=
†These authors have contributed equally to this work and share first authorship