 Kirill V. Ovchinnikov1
Kirill V. Ovchinnikov1 Christian Kranjec1
Christian Kranjec1 Amar Telke1
Amar Telke1 Morten Kjos1
Morten Kjos1 Tage Thorstensen2
Tage Thorstensen2 Siegfried Scherer3
Siegfried Scherer3 Harald Carlsen1
Harald Carlsen1 Dzung B. Diep1*
Dzung B. Diep1*- 1Faculty of Chemistry, Biotechnology and Food Science, Norwegian University of Life Sciences, Ås, Norway
- 2ARD Innovation AS, Ås, Norway
- 3TUM School of Life Sciences, Technical University of Munich, Munich, Germany
Antibiotic-resistant bacterial pathogens have become a serious threat worldwide. One of these pathogens is methicillin-resistant Staphylococcus aureus (MRSA), a major cause of skin and soft tissue infections. In this study we identified a strain of Staphylococcus equorum producing a substance with high antimicrobial activity against many Gram-positive bacteria, including MRSA. By mass spectrometry and whole genome sequencing the antimicrobial substance was identified as the thiopeptide bacteriocin micrococcin P1 (MP1). Based on its properties we developed a one-step purification protocol resulting in high yield (15 mg/L) and high purity (98%) of MP1. For shorter incubation times (5-7 h) MP1 was very potent against MRSA but the inhibitory effect was overshadowed by resistance development during longer incubation time (24h or more). To overcome this problem a synergy study was performed with a number of commercially available antibiotics. Among the antibiotics tested, the combination of MP1 and rifampicin gave the best synergistic effect, with MIC values 25 and 60 times lower than for the individual drugs, respectively. To assess the therapeutic potential of the MP1-rifampicin combination, we used a murine skin infection model based on the use of the multidrug-resistant luciferase-tagged MRSA strain Xen31. As expected, neither of the single antimicrobials (MP1 or rifampicin) could eradicate Xen31 from the wounds. By contrary, the MP1-rifampicin combination was efficient not only to eradicate but also to prevent the recurrence of Xen31 infection. Furthermore, compared to fucidin cream, which is commonly used in skin infection treatments, MP1-rifampicin combination was superior in terms of preventing resistance development. Our results show that combining MP1, and probably other thiopeptides, with antibiotics can be a promising strategy to treat SSTIs caused by MRSA and likely many other Gram-positive bacteria.
Introduction
Skin and soft tissue infections (SSTIs) are among the most common infections in the world and the majority of them is caused by Staphylococcus aureus – a major bacterial human pathogen known for its antibiotic resistance and virulence (1, 2). Methicillin-resistant S. aureus (MRSA) is of particular concern, since patients with SSTIs caused by MRSA have higher risk of bacteremia, hospital re-admission and death, and often require longer and more expensive periods of hospitalization compared to patients infected with non-MRSA (3, 4). European guidelines recommend vancomycin, teicoplanin, linezolid, daptomycin, tigecycline or ceftaroline for the treatment of MRSA infections (5), however resistance development to these antibiotics has already been reported (6–11). Consequently, there is an urgent need for novel antimicrobial agents and strategies to overcome MRSA in SSTIs.
Thiopeptides are sulfur-containing, ribosomally-produced and highly posttranslationally modified bacteriocins – antimicrobial peptides produced by bacteria to inhibit other bacteria in competition for nutrients and habitats (12, 13). These peptides represent a promising class of natural antibacterial molecules, being active against many Gram-positive pathogens, including antibiotic resistant derivatives such as MRSA, vancomycin-resistant enterococci (VRE) and penicillin-resistant Streptococcus pneumoniae (14, 15). Thiopeptides inhibit protein synthesis in sensitive bacteria by binding to a cleft between the ribosomal protein L11 and the 23S rRNA, known as the GTPase-associated center, or by binding to and inactivating the elongation factor Tu (16–18). Besides their antimicrobial properties, some thiopeptides have demonstrated antiplasmodial, antifungal and anticancer activities (19–21). In contrast to non-ribosomally synthesized peptides, thiopeptides are encoded by classical structural genes and synthesized ribosomally, which renders the generation of new analogs by genetic engineering relatively straight-forward (22). These facts, combined with low cytotoxicity of thiopeptides (23) make this class of molecules very appealing for clinical use.
More than one hundred thiopeptides have been discovered so far with most of these molecules being produced by soil bacteria, including Bacillus spp, Streptomyces spp. and Nocardiopsis spp (22). However, despite the great therapeutic potential, their low aqueous solubility and the fact that sensitive bacteria can easily develop resistance to these antimicrobials are major drawbacks that have hindered their introduction to clinical practice (16, 18).
Micrococcin P1 (MP1), which was the first discovered thiopeptide, is a hydrophobic and heat-stable molecule with high activity against a wide range of Gram-positive bacteria as well as Mycobacterium tuberculosis (24). Interestingly MP1 has been shown to be produced by bacteria from different genera, including Micrococcus, Staphylococcus and Bacillus spp. These bacteria are mostly isolated from soil (22), but also from other sources, e.g., French Raclette cheese (25).
In this study we describe a new producer of MP1 with a novel gene cluster. To increase its antimicrobial activity and to circumvent the problem of bacterial resistance development, we explored the synergy of MP1 with several antibiotics and found that it had indeed a strong synergy with some commonly used antibiotics in vitro. Furthermore, we validated this synergistic effect in a murine model of MRSA skin infection.
Materials and Methods
Bacterial Strains and Growth Conditions
All bacterial strains used in this study are listed in Table 1 and Supplemental Table 1. S. equorum KAVA and S. equorum WS 2733 were MP1 producers; the former obtained from this study while the latter from a previous study (25). For in vivo imaging of bacterial infection in mice and antimicrobial synergy study, S. aureus Xen31 (Perkin Elmer, Waltham, MA) was used. The strain was derived from the parental strain S. aureus ATCC 33591, a clinical MRSA isolated from Elmhurst Hospital in New York (26). S. aureus Xen31 possesses a stable copy of the modified Photorhabdus luminescens luxABCDE operon at a single integration site on the bacterial chromosome. To define the inhibition spectrum of MP1, a panel of bacteria from different genera and species were used (see Supplemental Table 1). All bacterial strains were grown in brain heart infusion (BHI) broth (Oxoid, United Kingdom) at 30°C overnight without shaking unless stated otherwise.
Table 1 Strains used in the study.
Sample Collection and Screening for Antimicrobial Activity Against S. aureus
Biological samples used for antimicrobial screening were obtained from fermented fruits and vegetables. Twenty-five different fruits and vegetables were purchased at a local shop (Oslo, Norway). Each sample (20-30 g) was cut into small pieces and left for three weeks in about equal volume of water with or without NaCl (1-2% final concentration) at outdoor temperature (between 15 and 25 °C). After the incubation, the liquid fraction of each sample was mixed with glycerol (final concentration 20%) and stored at -80 °C until use.
To screen for microorganisms with antimicrobial activity, a small volume (50 µl) of each sample was first 10-fold-serially diluted in sterile saline, then 50 µl of each dilution was transferred to 5 ml of BHI soft-agar (0.7% w/v agar at 50 °C) and the mixture was plated on a BHI agar plate, obtaining a plating density of 10-1000 CFUs per plate. The plates were then incubated overnight at 30°C before covering the lawn with 5 ml of BHI soft-agar containing ca 106 CFU/mL of the indicator strain S. aureus LMGT 3258, a methicillin-susceptible S. aureus (MSSA). After a further overnight incubation at 30°C, the colonies surrounded with inhibition zones were selected and streaked on fresh BHI agar plates to obtain single colonies. Antimicrobial producing candidates were reconfirmed by having inhibitory activity toward the indicator strain on agar plate assays. Liquid cultures of the candidate antimicrobial producer strains were mixed with glycerol (20%) and stored at -80 °C until use.
MP1 Purification and Production
The antimicrobial-producing strain S. equorum KAVA was grown for 24 h in 1 L BHI broth at 37°C without shaking. The cells were removed by centrifugation at 10,000 × g for 15 min at room temperature. The supernatant was applied to a Resource reverse-phase chromatography (RPC) column (1 ml) (GE Healthcare Biosciences) connected to an ÄKTA purifier system (Amersham Pharmacia Biotech). A linear gradient of isopropanol (Merck) with 0.1% (vol/vol) trifluoroacetic acid (TFA) (buffer B) at a flow rate of 1.0 ml min−1 was used for elution. RPC fractions were then tested for antimicrobial activity against S. aureus LMGT 3258 before selected fractions were further analyzed by mass spectroscopy (MS).
For comparative analysis of MP1 production, S. equorum KAVA and S. equorum WS 2733 were grown in 5 ml of BHI medium without shaking at 23°C, 30°C and 37°C for 4 days in order to accumulate the bacteriocin. Since MP1 is known to aggregate on the producer cells (27), the cell pellets obtained after centrifugation were treated with 1 ml of 2-propanol to extract MP1. Filter-sterilized supernatants and cell-extracts were analyzed for antimicrobial activity using a microtiter plate assay as previously described (28). The antimicrobial activity of the samples was expressed in bacteriocin units (BU), defined as the minimum amount of bacteriocin that inhibited at least 50% of growth of the indicator strain (S. aureus LMGT 3258) in a 200 μl culture volume.
For large scale purification (the optimized protocol), the selected strain was grown in 2 L of BHI broth at 37°C for 4 days. After centrifugation the supernatant was discarded, the cell pellet was washed with saline and the MP1 extraction was performed with 100 ml of isopropanol (Merck). The extract was diluted 5 times with MiliQ water and applied to a Resource reverse-phase chromatography (RPC) column (3 ml) (GE Healthcare Biosciences) connected to an ÄKTA purifier system (Amersham Pharmacia Biotech). A linear gradient of isopropanol with 0.1% (vol/vol) TFA (buffer B) at a flow rate of 1.0 ml min−1 was used for elution. The MP1 concentration and purity were determined by HPLC using a Phenomenex Axia Luna C8 100A column (Phenomenex, Norway). Commercial MP1 (Cayman Chemical) with ≥ 95% purity was used as a HPLC standard. After purification, the MP1 solution was dried at 55°C in a SpeedVac concentrator (SPD2010 Integrated SpeedVac, ThermoFisher Scientific, USA). The MP1 pellet was resuspended in DMSO (Sigma-Aldrich) to 1 - 10 mg/ml concentrations and stored at -20 °C before use.
MS Analysis
MS data were acquired on an Ultraflex MALDI-TOF/TOF (Bruker Daltonics, Bremen, Germany) operated in reflection mode with delayed extraction. Ions of positive charge in the m/z range of 200 to 6,000 were analyzed using 25 kV acceleration voltage. The sample spectra were calibrated externally with a calibration standard covering the m/z range from 700 to 3,100 (Bruker Daltonics, Bremen, Germany).
DNA Sequencing
For 16S rRNA gene sequencing, DNA from the isolates with antibacterial activity was isolated by using FastPrep Bio101 (Savant Instruments, USA) and DNA minikit (Omega Bio-Tek Inc., GA), according to the manufacturer instructions. Amplification of the 16S rRNA gene by PCR was carried out using the primers 5F (5′-GGTTACCTTGTTACGACTT-3′) and 11R (5′-TAACACATGCAAGTCGAACG-3′) as previously described (29). PCR products was purified with NucleoSpin Extract II (Macherey-Nagel, Düren, Germany) according to the manufacturer instructions and sent to GATC Biotech, Germany, for sequencing.
Whole genome sequencing (WGS) was performed as described previously described (30). Briefly, genomic DNA was extracted from 1 mL of overnight culture using Qiagen DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). DNA libraries were made using the Nextera XT DNA Sample Prep kit (Illumina, San Diego, California, USA) according to the manufacturer instructions. The library was sequenced on Illumina MiSeq platform (Illumina, San Diego, California, USA). Raw Illumina reads were trimmed with Trimmomatic v0.39 (31) to remove the sequencing adapters, quality filtered (Q>20) and de novo assembled using SPAdes (v3.7.1) (32). Contigs shorter than 1000 bp or with < 5 times coverage were removed from each assembly prior to gene annotation. The genomes were annotated using the Prokka pipeline (33). WGS data are publicly available at NCBI (GenBank submission ID 2428870). The gene cluster features were edited in a genome browser Artemis (v18.0.0) (34). The linear comparison of gene cluster was created in a Python application, Easyfig (v2.2.2) (35).
Synergy Assessment
For the assessment of synergistic interactions with MP1, antibiotics with different modes of action and high purity (≥ 97%) were purchased from Sigma-Aldrich. The selected antibiotics were gentamicin, streptomycin, kanamycin, erythromycin, chloramphenicol, tetracycline, penicillin G, fusidic acid and rifampicin. Synergy testing was done with a microtiter plate checkerboard assay as previously described (36). Briefly, equal amount of MP1 was applied on microtiter plate 1 in wells A1-H1 and then diluted two-fold to wells 2-11. Similarly, equal amount of antimicrobial B was applied on microtiter plate 2 in wells A1-A12 and diluted two-fold in wells B-G. Volumes of 50 µl of MP1 from each well of microtiter plate 1 were transferred into microtiter plate 3, except for wells A1-H1. Similarly, the same amounts of antimicrobial B were transferred from microtiter plate 2 into plate 3, except for wells H1-H12 (Supplemental Figure 1). Subsequently, an overnight culture of S. aureus Xen31 was diluted 25 times prior transferring 100 µl aliquots of the bacterial suspension into each well of plate 3. Wells H2-H12 and A1-G1 were used to estimate MIC values of each antimicrobial alone. The fractional inhibition concentration, was used to define the synergy between antimicrobial A (MP1) and B. FIC values were calculated as follows: FIC = FICa + FICb, where the FICa is the MIC of A in combination/MIC of A alone and FICb is the MIC of B in combination/MIC of B alone. Effects were considered as synergistic if FIC was ≤0.5 (37). MIC values were determined in accordance with CLSI/EUCAST recommendations (https://eucast.org/ast_of_bacteria/guidance_documents/).
Selection of Suitable Antimicrobial Vehicles
Due to their poor solubility, we performed a search for a suitable vehicle to deliver rifampicin at a concentration of 0.15 mg/ml and MP1 at 0.01 mg/ml; these concentrations were the final concentrations used in the combinatorial topical treatment in mice. Stock DMSO solutions of rifampicin (30 mg/ml) and MP1 (1 mg/ml) were tested for their solubility against a panel of commercially available skin creams with different fat concentrations (22%, 30%, 47%, 60% and 70%), by diluting the stock solutions 1:65 into each cream. The mixture was heated to 50°C to reduce viscosity, mixed vigorously on a vortex for 5 min and then centrifuged for 15 min at 15000 xg at room temperature. High solubility was reached when no visible pellet was seen at the bottom of the tubes. Based on the levels of antimicrobial solubility, APO base 30% cream (Teva, Finland) was found the most suitable and was selected as the antimicrobial vehicle for all in vivo experiments in this study. The mixture containing 0.15 mg/ml rifampicin and 0.01 mg/ml MP1 in APO base 30% cream did not lose its antimicrobial activity after a two-week storing at 5°C and was chosen for in vivo experiment.
Murine Experiments
Experiments on mice were approved by the Norwegian Food Safety Authority (Oslo, Norway), application no. 20/10793. In total, 39 female BALB/cJRj mice of four weeks of age were purchased from Janvier (Le Genest-Saint-Isle, France). Three to four mice were housed per cage during the whole experiment and maintained on a 12-hour light/12-hour dark cycle with ad libitum access to water and a regular chow diet (RM1; SDS Diet, Essex, UK). Mice were acclimatized in our mouse facilities for two weeks before the start of the experiments; hence the age of mice at the start of the experiments was six weeks.
Before infection and treatment, the mice were shaved as follows: mice were anesthetized with Zoletyl Forte, Rompun, Fentadon (ZRF) cocktail (containing 3.3 mg Zoletil forte, 0.5 mg Rompun and 2.6 µg Fentanyl per 1 ml 0.9% NaCl) by intraperitoneal injection (0.1 ml ZRF/10 g body weight) and shaved on the back and flanks with an electric razor. The remaining hair was removed by hair removal cream (Veet, Reckitt Benckiser, Slough, UK) according to the manufacturer’s instructions. The next day the mice were again anesthetized with ZRF cocktail (0.1 ml/10 g body weight) and two skin wounds were made on the back of every mouse with a sterile biopsy punch 6 mm in diameter (Dermal Biopsy Punch, Miltex Inc, Bethpage, NY). Prior to infection, overnight-grown S. aureus Xen31 cells were washed twice in sterile saline and then suspended in ice-cold PBS buffer. Each wound was inoculated with 10 μl of PBS containing ca 2x107 CFU of S. aureus Xen31 cells using a pipette tip. After bacterial application the mice were kept on a warm pad for 10-15 min to dry the inoculum and the wounds were then covered with a 4×5 cm Tegaderm film (3M Medical Products, St. Paul, MN, USA). Mice were then left for 24h for the infection to establish. The day after (24 h post infection; PI) the mice were anesthetized with 2% isoflurane and the luminescent signal was measured by IVIS Lumina II, Perkin Elmer (2 min exposure time). The luminescent signal was quantified by the software Living Image (Perkin Elmer) from regions of interest (ROIs) around the wound and expressed as photons/second/cm2/steradian.
From this point, the mice were divided into 5 groups and subjected to 5 different treatments: one treated with MP1 (10 µg/ml) in APO base 30% cream (n=8), one treated with rifampicin (0.15 mg/ml) in APO base 30% cream (n=8), one treated with the mixture of MP1 and rifampicin (0.15 mg/ml rifampicin and 10 µg/ml MP1, in APO base 30% cream, n=8), one treated with the vehicle (APO base 30% cream) as a negative control (n=8), and one treated with fucidin cream (2% fusidic acid in a cream base; LEO Pharma, Denmark) as a positive control (n=7). All treatments were performed once a day. To assess if the treatment had a long-lasting effect, 4 mice from each group received the treatment for 4 days and were left untreated in a separate cage until the end of experiment while the remaining 4 mice (3 mice for the fucidin group) continued to receive treatments once a day until the end of the experiment (9 days). In all treatment groups, 50 µl of either antibacterial solution or control substance was injected into each wound under the Tegaderm using an insulin syringe (BD SafetyGlide™; 29G needle). The bioluminescent signal, produced by S. aureus Xen31 luciferase was recorded once per day before each treatment, during the entire course of the experiments. At the end of each experiment mice were euthanized by cervical dislocation.
Statistical Analysis
All in vitro assays were performed three times. For statistical analyses and graphs, R Studio (version 1.0.15; https://rstudio.com/products/rstudio/download/) was used.
Results
Screening for Bacteriocin Producers
Twenty-five fruit and vegetable samples were used as source for screening of bacteria with antimicrobial activity against S. aureus LMG3258. Since nisin producers are frequently found in such samples (38) and we wanted to exclude these from the current screen, the isolates with activity against S. aureus LMG3258 were re-tested against the nisin-immune strain LMGT 2122 (a known nisin producer). Most of the isolates with activity against LMG3258 were indeed nisin producers (data not shown). However, one isolate, called KAVA, inhibited both LMG3258 and LMGT 2122, and was therefore chosen for further analysis. By 16S rRNA genotyping, the isolate KAVA was found to be Staphylococcus equorum (hereafter called S. equorum KAVA).
S. equorum KAVA Produces Micrococcin P1
To define the nature of the active substance, it was purified from the culture supernatant of S. equorum KAVA. The substance was eluted with 45% 2-propanol/TFA during PRC, indicating that the antimicrobial molecule was relatively hydrophobic (Figure 1A). MS analysis showed that the antimicrobial substance had a mass of 1144 Da (Figure 1B), a size corresponding to the known antimicrobial peptide micrococcin P1 (MP1). To prove the identity of the active substance further, we tested the purified fractions of S. equorum KAVA against a panel of 30 different bacterial species. As expected, the substance produced by S. equorum KAVA was active only against Gram-positive bacteria, including Listeria spp, enterococci, staphylococci, but not against Gram-negative bacteria such as Escherichia coli, Acinetobacter baumanii and Pseudomonas aeruginosa (Supplemental Table 1). Such activity spectrum is in line with published results for MP1 (22).
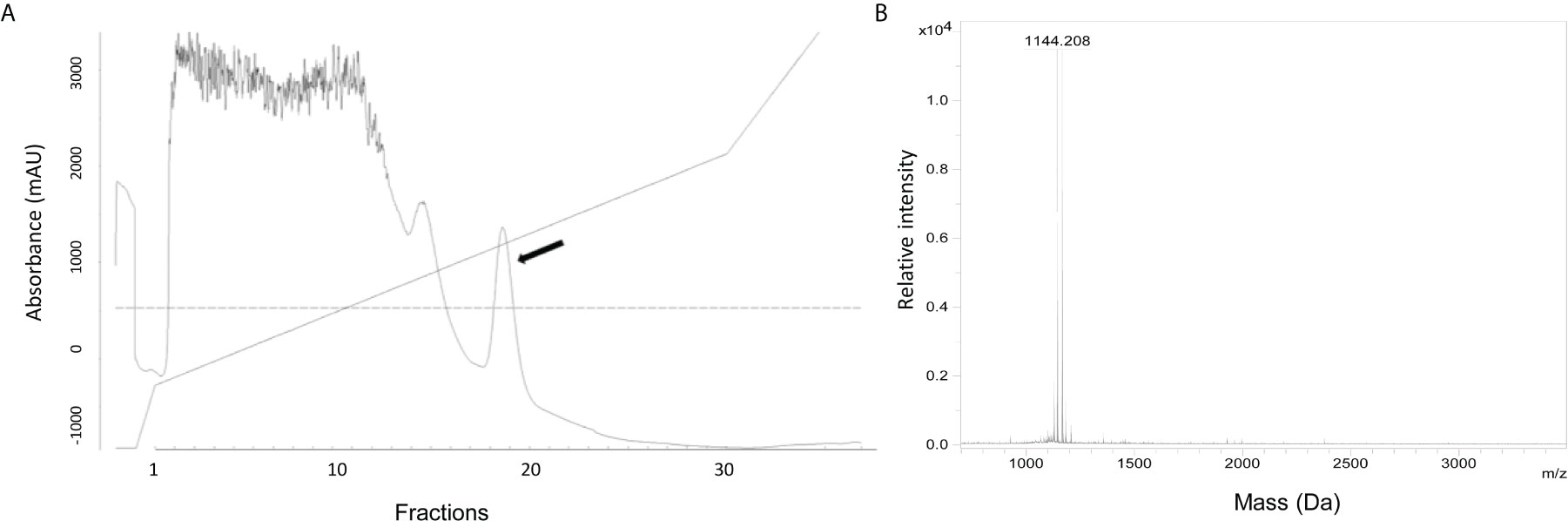
Figure 1 (A) RPC elution profile of MP1 from the cell-free supernatant of S. equorum KAVA. MP1 elution peak indicated with an arrow. (B) Mass spectrometry analysis of the active fraction of MP1 after the RPC. The inset is amplification of the MP1 peak area.
Whole Genome Sequencing of S. equorum Confirms the Presence of Novel MP1 Gene Clusters
To corroborate our finding further, whole genome sequencing of S. equorum KAVA and the strain S. equorum WS 2733, a known MP1 producer isolated from cheese (25), were performed, and indeed two very similar MP1 gene clusters were found (Figure 2). Interestingly, beside these two genomes, a database search led to the identification of similar MP1 gene clusters in other staphylococcal and non-staphylococcal genomes: one in a SSTI-associated S. aureus isolate (accession number VUGU01000042.1), one in a S. felis strain isolated from an otitis infection in a cat (accession number QKYH01000057.1), one in a Bacillus cereus strain (accession number NZ_CP034551.1), and one on the plasmid pBac115 (accession number: KM613043) from Macrococcus caseolyticus.
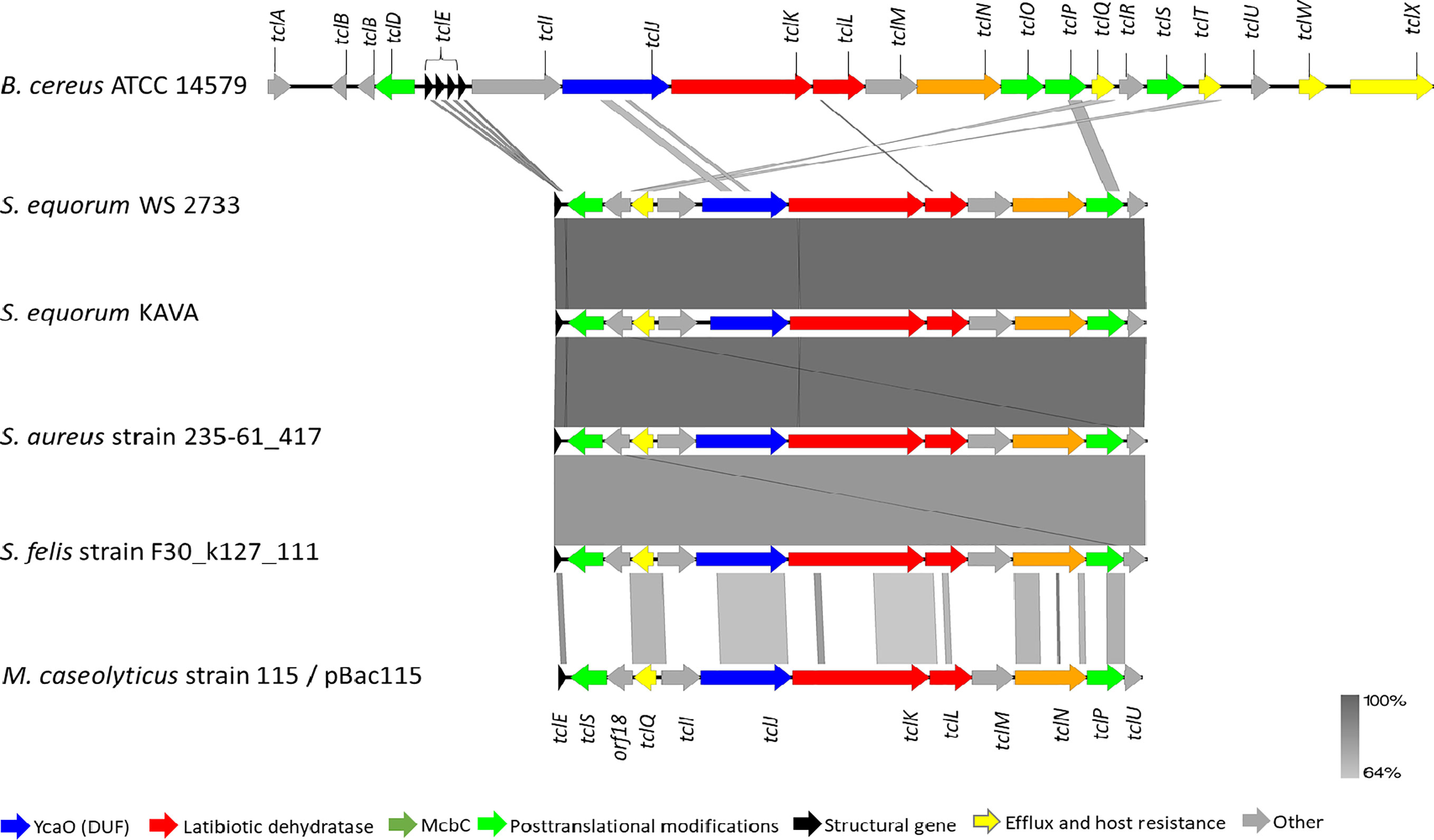
Figure 2 Genetic organization of micrococcin P1 gene cluster of S. equorum WS 2733 and S. equorum KAVA in comparison with reference gene cluster in the plasmid pBac115 of M. caseolyticus (accession number: KM613043), S. aureus strain UP 1591 plasmid unnamed (GenBank accession: CP047810), S. aureus strain 235-61_417 plasmid unnamed (GenBank accession: VUGU01000042.1) and S. felis strain F30_k127_111 (GenBank accession:QKYH01000057). The open reading frames that are involved in micrococcin P1 production are shown in different colors. Areas shaded in grey indicate homologous regions with 69-100% nucleotide identity.
MP1 gene clusters from S. equorum KAVA and S. equorum WS 2733 turn out to be almost identical to each other and both were similar to the pBac115 cluster (39) and other MP1 clusters from staphylococci (Figure 2). All staphylococcal MP1 gene clusters consisted of 12 genes and possessed only one copy of the MP1 structural gene (tclE) and a single immunity gene (tclQ), while MP1 gene cluster of B. cereus ATCC 14579 contained four copies of tclE and two copies of immunity genes tclT and tclQ (Figure 2). Using EMBOSS Needle pairwise sequence alignment (40) we found that the most conserved proteins across the clusters were the MP1 structural peptide, and its immunity protein (Table 2). Other proteins with high similarity were TclJ and TclN (which together catalyze the conversion of all six cysteines in the MP1 backbone to thiazole rings), TclK, TclL (Ser/Thr dehydration), TclM and TclP (unknown function) (41). The genes tclA, tclB, tclD, tclO, tclX were present only in B. cereus ATCC 14579 gene cluster. The staphylococcal MP1 clusters had only one gene with no homology in B. cereus ATCC 14579 gene cluster, namely orf18, encoding a 160 amino acid residue protein with unknown function (42) (Figure 2).
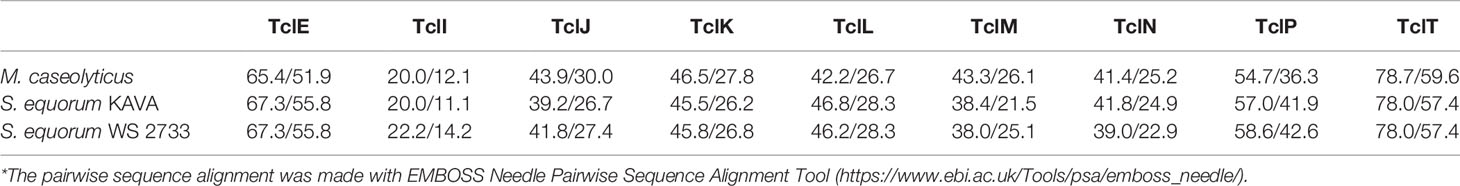
Table 2 Similarity/identity score of core proteins involved in the MP1 production in different strains in comparison with reference MP1 cluster in B. cereus ATCC14579 (*).
Comparison of MP1 Production by S. equorum Strains
Since S. equorum KAVA and S. equorum WS 2733 were readily available, it was of interest to investigate which one had the highest MP1 production ability. To address this point, we compared their growth and bacteriocin production profiles in BHI medium at different temperatures (23°C, 30°C and 37°C). Although both strains grew equally at the three temperatures (data not shown), their bacteriocin production was different. As can be seen in Table 3, S. equorum KAVA produced 80 BU/ml at the two lower temperatures, 23°C and 30°C, and 160 BU/ml at 37°C, while S. equorum WS 2733 produced 40-80 BU/ml at the two lower temperatures but, surprisingly, no or poor production was detected at 37°C (Table 3 and Supplemental Figure 2).

Table 3 Comparison of MP1 production (in BU/ml) by S. equorum KAVA and S. equorum WS 2733 at different temperatures.
As bacteriocins of the thiopeptide family are often adsorbed on the producer cells due to their high hydrophobicity (43), cell pellets from the two producers were also obtained and treated with equal volumes of 2-propanol to extract the bacteriocin into the organic phase. As expected, the organic fractions displayed the highest activity; the increase was 4-30-fold for S. equorum WS 2733 and 15-30-fold for S. equorum KAVA, compared to their respective water-soluble fractions depending on the growth temperature conditions (Table 3). Notably, extracts from S. equorum WS 2733 grown at 37 °C retained no bacteriocin activity.
Given that the extraction of the bacteriocin from S. equorum KAVA cells gave the highest yields, we used this strain as a main source of MP1 for further studies. By modulating the growth conditions (BHI broth, 37°C, four-day incubation), we optimized the protocol (see Materials and Methods) and were able to purify MP1 at a concentration of 15 mg/L of broth with a 98% purity as estimated by RPC HPLC (Supplemental Figure 3).
Search for Synergistic Antimicrobial Activities
MP1 is a peptide with high antimicrobial activity against many Gram-positive bacteria. However, sensitive bacteria can easily become resistant to MP1 by single-point mutations within the gene encoding the L11 ribosomal protein (44), making this antimicrobial less viable in therapeutics. This was also confirmed in recent works against MRSA (45, 46) where we observed numerous MP1-resistant mutants in our antimicrobial sensitivity assays. To avoid this resistance problem, we searched for antimicrobials which could act synergistically with MP1. Nine antibiotics of different classes and with different modes of action were chosen for the synergy experiment (Table 4). Indeed, by using the checkerboard assay, synergistic effects against the strain MRSA Xen31 were found between MP1 and the following antibiotics: tetracycline, penicillin G, chloramphenicol, fusidic acid and especially with rifampicin (Table 4). Fractional inhibitory concentration (FICs) values for tetracycline, penicillin G, chloramphenicol and fusidic acid in combination with MP1 were between 0.13 to 0.18. Most notably, the combination with rifampicin reduced MIC values from >100 µg/ml to 1.5 µg/ml for the antibiotic and from 2.5 µg/ml to 0.1 µg/ml for MP1, resulting in a FIC value equal to 0.05 (FIC values ≤0.5 are considered synergistic between two components) (37). Based on these results we sought to further explore the therapeutic potential of the combinatory effect of MP1 and rifampicin in a murine infection model (see below).

Table 4 Synergy assessment between MP1 and a panel of antibiotics against MRSA Xen31.
Choosing the Vehicle for the Antimicrobials
MP1 and rifampicin were dissolved in the hand cream Apo Base with 30% fat (Teva, Finland), which was the most suitable vehicle in terms of solubility and appropriate viscosity (see Materials and Methods). For topical use in mice the final mixture in APO Base 30 cream contained 10 µg/ml MP1 and 150 µg/ml rifampicin which were about 100 times higher than their MIC values recorded in the checkerboard assay, since MRSA Xen31 parental strain S. aureus ATCC 33591 is known to produce biofilms on surfaces within 24h (47) and staphylococcal biofilms are 10- to 1000-fold more resistant to antimicrobials compared to planktonic cells (48).
The cream by itself did not inhibit MRSA while the cream containing the antimicrobials, hereafter referred to as the MP1-rifampicin mixture, displayed strong antimicrobial activity as expected (Figure 3).

Figure 3 Assessment of the antimicrobial activity of MP1(10 µg/ml), rifampicin (150 µg/ml) and the combination (MP1, 10µg/ml + rifampicin, 150µg/ml) in APO base cream 30%. Cream with not addition was included as negative control. The activity was tested with softagar overlay assay using MRSA Xen31 as indicator strain.
The MP1-Rifampicin Mixture Is Effective Against MRSA in a Murine Skin Wound Infection Model
In order to validate the therapeutic value of the MP1-rifampicin mixture in vivo, we used a recently established murine skin wound infection model (45). This model applies the luciferase-expressing S. aureus Xen31, a derivative of MRSA ATCC33591 (PerkinElmer). This strain allows us to monitor the bacterial growth-dependent luminescence intensity which is proportional to the growth of S. aureus Xen31 (49) during the entire course of the experiment. Mice were divided into five groups (n=8 per group except in one group where n=7), for different treatments: MP1 (group 1), rifampicin (group 2), vehicle (Apo Base 30 cream) alone as negative control (group 3), MP1-rifampicin mixture (group 4) and fucidin cream (group 5) as positive control. Fucidin cream contains 20 mg/ml of fusidic acid and commonly used against MRSA skin infections (50). Wounds on the back of each animal were infected with S. aureus Xen31 (approximately 2x107 CFU/wound), covered with Tegaderm (a transparent wound dressing) and the infection was allowed to establish for 24 h prior beginning each treatment regimen. In addition, each group was divided into two subgroups where one subgroup received daily treatments from day 2 until the end of the experiment (Figures 4A and 5A) while the other subgroup was treated only four times during first four days and after that left untreated until the end of the experiment, in order to examine the long-term effect of the treatments (Figures 4B and 5B).
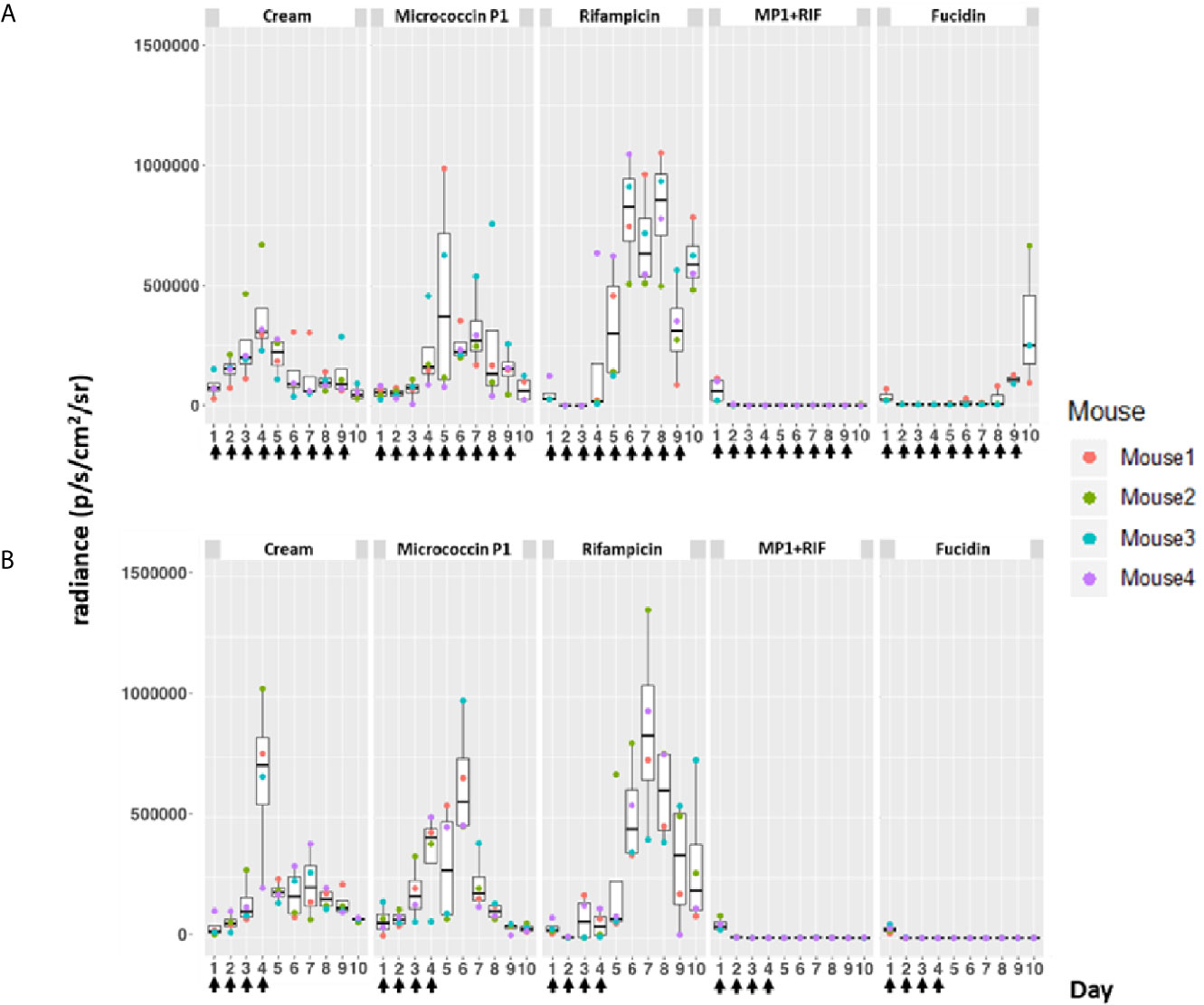
Figure 4 Bioluminescence from mice skin infections during different treatments. Box plots of bioluminescent signals produced by MRSA Xen31 (in photons per second per square centimeter per steradian) in differently treated mouse groups. The days of treatment are indicated with arrows. (A) mice received daily treatments for nine days. (B) mice received daily treatments for four days before they were left untreated for the rest of the experiment. The area within each box represents the interquartile region (IQR), which comprises the second and third quartiles and describes the interval of values where the middle 50% of the observed data are distributed. The horizontal black line within each box represents the median value. The extent of the IQR (box height) express the degree of variability measured within the middle 50% of the observed data, with whiskers extending out at either side of the boxes marking the minimum and maximum observed values, as well as the variability outside the middle 50% of values (whisker length). Outliers are displayed as data that extend out of the whisker limit (1.5 times the IQR).
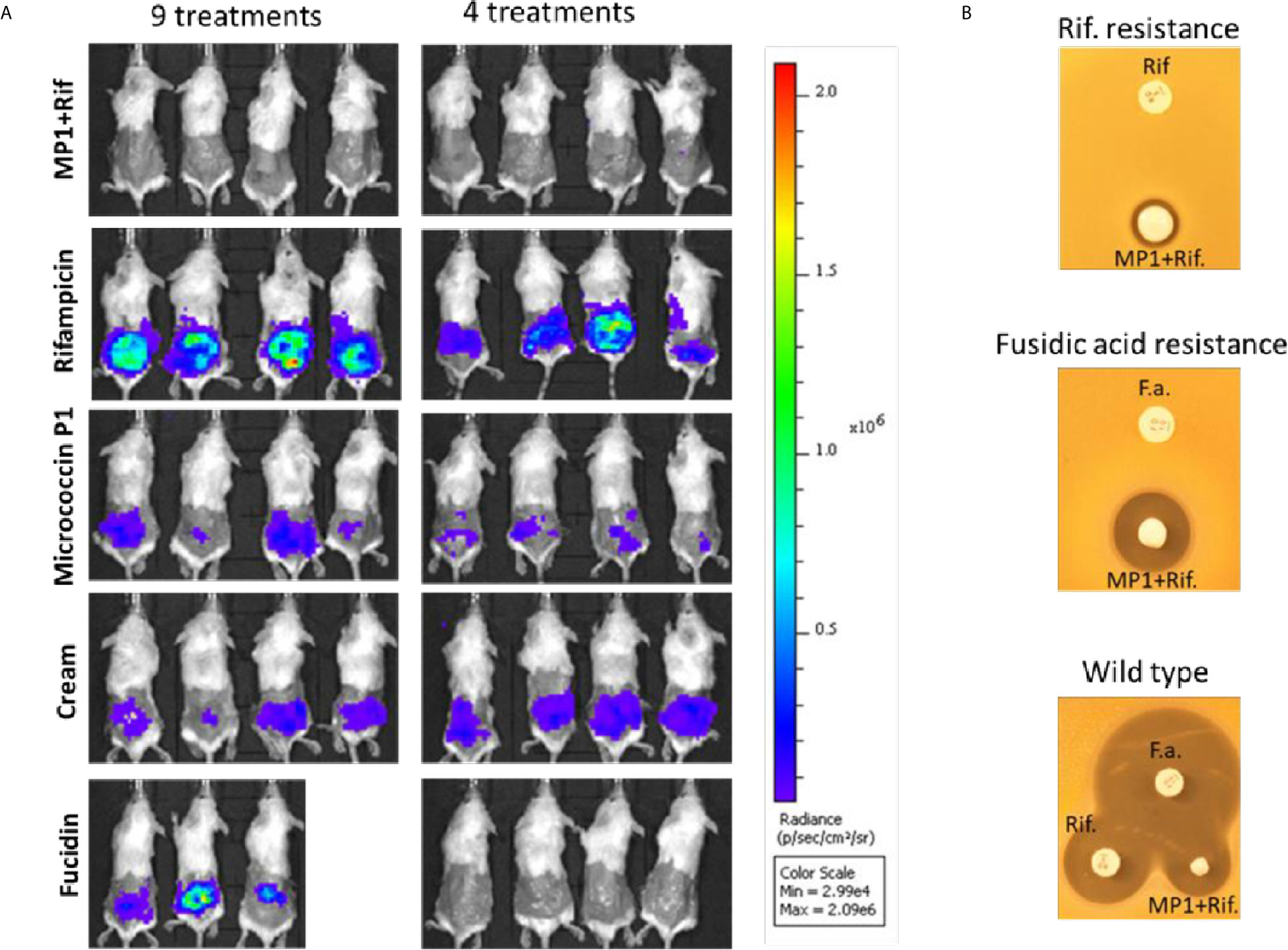
Figure 5 (A) In vivo images of bioluminescent signals produced by MRSA Xen31(in photons/second/cm2/steradian) from the different mouse groups on the last day of the experiment; left column: with nine daily treatments; right column with four daily treatments. (B) Rifampicin and fucidin resistance develops during the treatment of mice. Rifampicin resistant cells (Rif-resistance) and fucidic acid resistant cells (Fucidic acid resistance) isolated from the wounds with strong bioluminescent signals were rechallenged and shown to be resistant to rifampicin (Rif) and fusidic acid (F.a.) but not to the MP1-rifampicin mixture. Wildtype MRSA Xen31 cells exposed to the MP1-ripamicin mixture, rifampicin and fusidic acid were sensitive to all three antimicrobials.
As can be seen in Figure 4, the bioluminescent signals from all mice wounds were clearly visible prior to all treatments (day 1 post infection; PI). Consistent with the in vitro results, the bioluminescent signals generated in the wounds treated with the vehicle (pure APO-base cream) displayed a steady increase from day 2 PI and peaked between day 4 and 7 PI in all mice. As expected, the application of MP1 alone (10 µg/ml) was not able to stop the MRSA wound infection in all mice throughout the experiment. In rifampicin-treated mice (150 µg/ml) the bioluminescent signals sharply declined the first day after treatment in all mice (day 2 PI), however, the next day (day 3 PI), two mice displayed high luminescence, and by day 5 PI this was the case for all rifampicin-treated mice, likely due to the selection of rifampicin-resistant MRSA cells within the wounds (Figure 4B). In contrast, in mice treated with the MP1-rifampicin mixture, the luminescent signals not only declined abruptly within 24 hours after the first treatment but stayed below the detection limit over the entire period of the experiment (10 days). Notably, for the four mice which received only four treatments of MP1-rifampicin mixture, no re-infection or recurrence appeared during the rest of the experiment, indicating that the antimicrobial effect of the MP1-rifampicin combination lasted for at least 5 days after four treatments (Figure 4B). Fucidin was used as a positive control, and, as expected, caused a sharp reduction of luminescent signals within 24 hours after the first treatment. However, on day 6 PI one mouse from the group which received the treatment every day had a slight increase of the luminescent signals (Figure 4A). Next day (Day 7 PI) the signal could not be detected but appeared again on day 8 PI. On day 10, all three mice had high luminescent signals despite being treated with fucidin every day (Figures 4A and 5A). Interestingly, for mice received only 4 fucidin treatments and then left untreated for 5 days, no signs of re-infection were detected after the fourth treatment (Figures 4B and 5A).
The results from the rifampicin and fucidin treatments suggested resistance development in the MRSA strain against these antibiotics. In order to confirm this, on the last day of the experiment, luminescent bacteria were isolated from fucidin-treated and rifampicin-treated mice and rechallenged with the antibiotics. As expected, all isolates were indeed resistant to fucidin and rifampicin, respectively, but not to the MP1-rifampicin mixture (Figure 5B).
Furthermore, no mice showed any obvious signs of abnormal behavior, neither in the non-treated group nor in the treated groups, indicating that the different treatments had no obvious toxic effects.
Discussion
The emergence of multidrug-resistant bacteria has been recognized as a major public health problem. One strategy to combat such bacteria is revitalizing old antimicrobials which were discovered in the past but are not much used in today’s medicine because of different reasons, e.g., low or expensive production, lack of delivery means, or a consequence of resistance development (51, 52). Thiopeptides represent a very promising class of neglected antimicrobials. Despite their potent antimicrobial activity thiopeptides have been poorly exploited in therapeutic treatments so far due to the high rate of resistance development, challenging synthesis, poor aqueous solubility and associated low bioavailability (53). In this work we describe a cost-efficient production of the thiopeptide MP1 and show that MP1 in combination with rifampicin had very good synergistic effects on MRSA. Its therapeutic and synergistic properties were successfully validated in a murine skin infection model.
Several attempts have been made to synthesize MP1 chemically to reduce its cost and possibly modify its structure to make it more water-soluble, but so far these synthetic approaches are not scalable and cost-effective (54, 55). To improve the effectiveness of MP1 production, fermentation can be an alternative to synthetic approaches. In this work we isolated a new S. equorum strain from a sample of fermented vegetable which displayed high and stable MP1 production. We then propose a very simple and cost-effective method for purification of MP1 from S. equorum by extracting MP1 from the producer cells with 2-propanol with subsequent one-step RPC purification of the water-diluted extract. Using this method, we were able to obtain 15 mg of 98% pure MP1 from 1L of BHI medium (Supplemental Figure 3).
The difference in temperature dependent MP1 production between the two S. equorum strains, suggests that the production is somehow regulated differently. However, the two strains have almost identical MP1 gene clusters (Figure 2 and Table 2), indicating that either a subtle difference within the loci or a difference outside the loci could be the cause for this phenotypic difference. Interestingly, there are major genetic differences between the staphylococcal strains and other MP1 producers. For instance, the staphylococcal MP1 gene cluster comprises 12 genes while it has 24 genes in B. cereus ATCC 14579. Furthermore, the MP1 locus contains only one structural gene in the former while four consecutive structural genes are found in the latter. The differences also extend to their final product(s). The staphylococcal strains appear to produce only one product, namely MP1 (39), while B. cereus ATCC 14579 produces a mixture of similar thiopeptides with different posttranslational modifications [thiocillin I, II, III, MP1 and micrococcin P2 (MP2)] (41). In terms of purification of MP1 the staphylococcal strains will be preferable because the purified MP1 will not be contaminated with physico-chemically similar thiopeptide species as it would with B. cereus ATCC 14579.
No cross-resistance has been reported between common antibiotics and thiopeptides (44), suggesting a possible combinatory approach for therapeutic use. Indeed, using MRSA as a target pathogen, MP1 was found to have synergistic effect with several antibiotics, especially with rifampicin which gave the best synergy (Table 4). Rifampicin, also known as rifampin, is a broad-spectrum lipophilic antimicrobial agent that inhibits bacterial RNA polymerase (56). Recently rifampicin has gained much attention due to its bactericidal activity against S. aureus, including MRSA (57). Besides its high cellular permeability, rifampicin is one of the few antimicrobial agents that can penetrate biofilms and kill organisms in the sessile phase of growth (58).
Our results, both in vitro and in vivo showed a clear synergistic effect between MP1 and rifampicin against MRSA Xen31 (Table 4 and Figure 3). While neither MP1 nor rifampicin had any long-lasting therapeutic effect on MRSA Xen31 in the murine model (Figure 4), the mixture of MP1 and rifampicin efficiently removed the pathogen from infection sites and prevented its recurrence and resistance development. The combinatory mixture had a long-lasting effect as no obvious sign of the pathogen was seen at least 5 days after the daily 4-day scheme of treatment (Figure 4). This is not the case for fusidic acid which is a commonly used antibiotic in treatment of skin infections. Our present study and others’ (59) demonstrate that fusidic acid monotherapy is inefficient in the treatment of staphylococcal infections due to rapid resistance development.
It is still debatable whether the combination of rifampicin with other antimicrobials truly confers additional effectiveness over rifampicin monotherapy in human and animal infections, since animal models show contrasting results (58). For example, in a rat model of chronic subcutaneous staphylococcal foreign-body infection rifampicin was an important adjuvant to vancomycin and fleroxacin (60). The synergy with vancomycin was also demonstrated on human patients against MRSA septicemia in burns (61), in a knee prosthetic infection model (62) and in the treatment of nosocomial MRSA induced pneumonia (63). In addition to these examples, several other studies using different animal models and/or other types of infection have also shown good synergistic effects of rifampicin with other antibiotics such as linezolid (64), β-lactams or glycopeptides (65), and some topical antimicrobials (66).Yet, other results provided no overall benefit of the adjunctive effect of rifampicin over standard antibiotic therapy against S. aureus; neither in vitro (67, 68). In addition, the use of rifampicin adjunctive therapy for the treatment of SSTI is not recommended by the Infectious Diseases Society of America (69). These studies with contrasting outcomes highlight a complexity of the types of diseases, the hosts and the combinatory antibiotics in question. Thus, the synergistic properties of rifampicin as therapeutic option should be evaluated with great care to avoid inefficiency or other potential collateral effects before being used in the intended hosts.
Topical treatment often allows the use of relatively high concentrations of antimicrobials at the wound sites compared to systemic treatment (70). However, too high antimicrobial concentrations may cause cytotoxic effects on skin cells and prevent rapid wound healing (71). One possible solution to avoid this is to use synergistic antimicrobial combinations as shown here for MP1-rifampicin. Such an approach also provides an effective mean to prevent resistance development. Nevertheless, further research is needed to unravel the molecular mechanisms underlying the synergistic effects, including the influence on mutation rates, how bacterial cells respond to sublethal antimicrobial concentrations and the mechanism behind the bactericidal effect. Such knowledge will help design safer and more efficient drugs before testing them in clinical settings.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, submission ID 2428870.
Ethics Statement
The animal study was reviewed and approved by Norwegian Food Safety Authority (Oslo, Norway), application no. 20/10793.
Author Contributions
KO: manuscript writing, in vitro and in vivo synergy experiments, MP1 purification. CK: manuscript writing, statistical analysis. AT: WGS, in silico DNA analysis. MK: manuscript writing. TT: funding, conceptualization. SS: manuscript writing, bacterial strain providing. HC: mouse model supervision. DD: project supervision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the research council of Norway, from the program Forny (project no. 296220), and the program Better Health and Quality of Life (project No. 273646).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Linda Godager for technical help during the screening, and Karmen Yade and Vera Vyanni for assistance during the processing of fermentation cultures.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.676534/full#supplementary-material
Supplementary Figure 1 | Microtiter plate checkerboard assay scheme. See description in Materials and Methods.
Supplementary Figure 2 | MP1 production by S. equorum WS 2733 and S. equorum KAVA at different temperatures.
Supplementary Figure 3 | RPC elution profile of MP1 from diluted 2-propanol cell-extract of S. equorum KAVA. B. RPC HPLC purity analysis of the active fractions from the purification. MP1 elution peaks are indicated with arrows.
Supplementary Table 1 | The inhibitory spectrum of the purified fractions of S. equorum KAVA.
References
1. Dryden MS. Complicated Skin and Soft Tissue Infection. J Antimicrob Chemother (2010) 65(Suppl 3):iii35–44. doi: 10.1093/jac/dkq302
2. Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG Jr. Staphylococcus Aureus Infections: Epidemiology, Pathophysiology, Clinical Manifestations, and Management. Clin Microbiol Rev (2015) 28(3):603–61. doi: 10.1128/cmr.00134-14
3. Bassetti M, Carnelutti A, Righi E. The Role of Methicillin-Resistant Staphylococcus Aureus in Skin and Soft Tissue Infections. Curr Opin Infect Dis (2017) 30(2):150–7. doi: 10.1097/qco.0000000000000353
4. Shorr AF. Epidemiology and Economic Impact of Meticillin-Resistant Staphylococcus Aureus: Review and Analysis of the Literature. PharmacoEconomics (2007) 25(9):751–68. doi: 10.2165/00019053-200725090-00004
5. Bassetti M, Baguneid M, Bouza E, Dryden M, Nathwani D, Wilcox M. European Perspective and Update on the Management of Complicated Skin and Soft Tissue Infections Due to Methicillin-Resistant Staphylococcus Aureus After More Than 10 Years of Experience With Linezolid. Clin Microbiol Infect (2014) 20(Suppl 4):3–18. doi: 10.1111/1469-0691.12463
6. Tsiodras S, Gold HS, Sakoulas G, Eliopoulos GM, Wennersten C, Venkataraman L, et al. Linezolid Resistance in a Clinical Isolate of Staphylococcus Aureus. Lancet (Lond Engl) (2001) 358(9277):207–8. doi: 10.1016/s0140-6736(01)05410-1
7. Hayden MK, Rezai K, Hayes RA, Lolans K, Quinn JP, Weinstein RA. Development of Daptomycin Resistance In Vivo in Methicillin-Resistant Staphylococcus Aureus. J Clin Microbiol (2005) 43(10):5285–7. doi: 10.1128/jcm.43.10.5285-5287.2005
8. Chan LC, Basuino L, Diep B, Hamilton S, Chatterjee SS, Chambers HF. Ceftobiprole- and Ceftaroline-Resistant Methicillin-Resistant Staphylococcus Aureus. Antimicrob Agents Chemother (2015) 59(5):2960–3. doi: 10.1128/aac.05004-14
9. Yousefi M, Fallah F, Arshadi M, Pourmand MR, Hashemi A, Pourmand G. Identification of Tigecycline- and Vancomycin-Resistant Staphylococcus Aureus Strains Among Patients With Urinary Tract Infection in Iran. New Microbes New Infect (2017) 19:8–12. doi: 10.1016/j.nmni.2017.05.009
10. Shlaes DM, Shlaes JH, Vincent S, Etter L, Fey PD, Goering RV. Teicoplanin-Resistant Staphylococcus Aureus Expresses a Novel Membrane Protein and Increases Expression of Penicillin-Binding Protein 2 Complex. Antimicrob Agents Chemother (1993) 37(11):2432–7. doi: 10.1128/aac.37.11.2432
11. Bertrand X, Hocquet D, Thouverez M, Plésiat P, Talon D. Characterisation of Methicillin-Resistant Staphylococcus Aureus With Reduced Susceptibility to Teicoplanin in Eastern France. Eur J Clin Microbiol Infect Dis (2003) 22(8):504–6. doi: 10.1007/s10096-003-0966-7
12. Acedo JZ, Chiorean S, Vederas JC, van Belkum MJ. The Expanding Structural Variety Among Bacteriocins From Gram-Positive Bacteria. FEMS Microbiol Rev (2018) 42(6):805–28. doi: 10.1093/femsre/fuy033
13. Eijsink VG, Axelsson L, Diep DB, Håvarstein LS, Holo H, Nes IF. Production of Class II Bacteriocins by Lactic Acid Bacteria; An Example of Biological Warfare and Communication. Antonie Van Leeuwenhoek (2002) 81(1-4):639–54. doi: 10.1023/a:1020582211262
14. Liao R, Duan L, Lei C, Pan H, Ding Y, Zhang Q, et al. Thiopeptide Biosynthesis Featuring Ribosomally Synthesized Precursor Peptides and Conserved Posttranslational Modifications. Chem Biol (2009) 16(2):141–7. doi: 10.1016/j.chembiol.2009.01.007
15. Shen X, Mustafa M, Chen Y, Cao Y, Gao J. Natural Thiopeptides as a Privileged Scaffold for Drug Discovery and Therapeutic Development. Med Chem Res (2019) 28(8):1063–98. doi: 10.1007/s00044-019-02361-1
16. Porse BT, Cundliffe E, Garrett RA. The Antibiotic Micrococcin Acts on Protein L11 at the Ribosomal GTPase Centre. J Mol Biol (1999) 287(1):33–45. doi: 10.1006/jmbi.1999.2600
17. Rosendahl G, Douthwaite S. The Antibiotics Micrococcin and Thiostrepton Interact Directly With 23S rRNA Nucleotides 1067A and 1095A. Nucleic Acids Res (1994) 22(3):357–63. doi: 10.1093/nar/22.3.357
18. Bagley MC, Dale JW, Merritt EA, Xiong X. Thiopeptide Antibiotics. Chem Rev (2005) 105(2):685–714. doi: 10.1021/cr0300441
19. Bhat UG, Halasi M, Gartel AL. Thiazole Antibiotics Target FoxM1 and Induce Apoptosis in Human Cancer Cells. PLoS One (2009) 4(5):e5592. doi: 10.1371/journal.pone.0005592
20. Radhakrishnan SK, Bhat UG, Hughes DE, Wang IC, Costa RH, Gartel AL. Identification of a Chemical Inhibitor of the Oncogenic Transcription Factor Forkhead Box M1. Cancer Res (2006) 66(19):9731–5. doi: 10.1158/0008-5472.Can-06-1576
21. Aminake MN, Schoof S, Sologub L, Leubner M, Kirschner M, Arndt HD, et al. Thiostrepton and Derivatives Exhibit Antimalarial and Gametocytocidal Activity by Dually Targeting Parasite Proteasome and Apicoplast. Antimicrob Agents Chemother (2011) 55(4):1338–48. doi: 10.1128/aac.01096-10
22. Just-Baringo X, Albericio F, Álvarez M. Thiopeptide Antibiotics: Retrospective and Recent Advances. Marine Drugs (2014) 12(1):317–51. doi: 10.3390/md12010317
23. Degiacomi G, Personne Y, Mondésert G, Ge X, Mandava CS, Hartkoorn RC, et al. Micrococcin P1 - A Bactericidal Thiopeptide Active Against Mycobacterium Tuberculosis. Tuberculosis (Edinb) (2016) 100:95–101. doi: 10.1016/j.tube.2016.07.011
24. Su TL. Micrococcin, an Antibacterial Substance Formed by a Strain of Micrococcus. Br J Exp Pathol (1948) 29(5):473–81. doi: 10.1007/978-3-642-46304-4_31
25. Carnio MC, Höltzel A, Rudolf M, Henle T, Jung GN, Scherer S. The Macrocyclic Peptide Antibiotic Micrococcin P1 Is Secreted by the Food-Borne Bacterium Staphylococcus equorum WS 2733 and Inhibits Listeria Monocytogenes on Soft Cheese. Appl Environ Microbiol (2000) 66(6):2378–84. doi: 10.1128/aem.66.6.2378-2384.2000
26. Schaefler S, Perry W, Jones D. Methicillin-Resistant Strains of Staphylococcus Aureus Phage Type 92. Antimicrob Agents Chemother (1979) 15(1):74–80. doi: 10.1128/aac.15.1.74
27. Suri B, Georges C, Peel JE. Method for the Prevention and Treatment of Mastitis. Google Patents (2001).
28. Holo H, Nilssen O, Nes IF. Lactococcin A, A New Bacteriocin From Lactococcus Lactis subsp. Cremoris: Isolation and Characterization of the Protein and its Gene. J Bacteriol (1991) 173(12):3879–87. doi: 10.1128/jb.173.12.3879-3887.1991
29. Birri DJ, Brede DA, Tessema GT, Nes IF. Bacteriocin Production, Antibiotic Susceptibility and Prevalence of Haemolytic and Gelatinase Activity in Faecal Lactic Acid Bacteria Isolated From Healthy Ethiopian Infants. Microb Ecol (2013) 65(2):504–16. doi: 10.1007/s00248-012-0134-7
30. Frantzen CA, Kot W, Pedersen TB, Ardö YM, Broadbent JR, Neve H, et al. Genomic Characterization of Dairy Associated Leuconostoc Species and Diversity of Leuconostocs in Undefined Mixed Mesophilic Starter Cultures. Front Microbiol (2017) 8:132. doi: 10.3389/fmicb.2017.00132
31. Bolger AM, Lohse M, Usadel B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics (2014) 30(15):2114–20. doi: 10.1093/bioinformatics/btu170
32. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: A New Genome Assembly Algorithm and its Applications to Single-Cell Sequencing. J Comput Biol (2012) 19(5):455–77. doi: 10.1089/cmb.2012.0021
33. Seemann T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics (2014) 30(14):2068–9. doi: 10.1093/bioinformatics/btu153
34. Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. ACT: The Artemis Comparison Tool. Bioinformatics (2005) 21(16):3422–3. doi: 10.1093/bioinformatics/bti553
35. Sullivan MJ, Petty NK, Beatson SA. Easyfig: A Genome Comparison Visualizer. Bioinformatics (2011) 27(7):1009–10. doi: 10.1093/bioinformatics/btr039
36. Orhan G, Bayram A, Zer Y, Balci I. Synergy Tests by E Test and Checkerboard Methods of Antimicrobial Combinations Against Brucella Melitensis. J Clin Microbiol (2005) 43(1):140–3. doi: 10.1128/jcm.43.1.140-143.2005
37. Hall MJ, Middleton RF, Westmacott D. The Fractional Inhibitory Concentration (FIC) Index as a Measure of Synergy. J Antimicrob Chemother (1983) 11(5):427–33. doi: 10.1093/jac/11.5.427
38. Cleveland J, Montville TJ, Nes IF, Chikindas ML. Bacteriocins: Safe, Natural Antimicrobials for Food Preservation. Int J Food Microbiol (2001) 71(1):1–20. doi: 10.1016/s0168-1605(01)00560-8
39. Bennallack PR, Burt SR, Heder MJ, Robison RA, Griffitts JS. Characterization of a Novel Plasmid-Borne Thiopeptide Gene Cluster in Staphylococcus Epidermidis Strain 115. J Bacteriol (2014) 196(24):4344–50. doi: 10.1128/jb.02243-14
40. Rice P, Longden I, Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet (2000) 16(6):276–7. doi: 10.1016/s0168-9525(00)02024-2
41. Wieland Brown LC, Acker MG, Clardy J, Walsh CT, Fischbach MA. Thirteen Posttranslational Modifications Convert a 14-Residue Peptide Into the Antibiotic Thiocillin. Proc Natl Acad Sci USA (2009) 106(8):2549–53. doi: 10.1073/pnas.0900008106
42. Bennallack PR, Bewley KD, Burlingame MA, Robison RA, Miller SM, Griffitts JS, et al. Reconstitution and Minimization of a Micrococcin Biosynthetic Pathway in Bacillus subtilis. J Bacteriol (2016) 198(18):2431–8. doi: 10.1128/jb.00396-16
43. Engelhardt K, Degnes KF, Kemmler M, Bredholt H, Fjaervik E, Klinkenberg G, et al. Production of a New Thiopeptide Antibiotic, TP-1161, by a Marine Nocardiopsis Species. Appl Environ Microbiol (2010) 76(15):4969–76. doi: 10.1128/aem.00741-10
44. Baumann S, Schoof S, Bolten M, Haering C, Takagi M, Shin-ya K, et al. Molecular Determinants of Microbial Resistance to Thiopeptide Antibiotics. J Am Chem Soc (2010) 132(20):6973–81. doi: 10.1021/ja909317n
45. Ovchinnikov KV, Kranjec C, Thorstensen T, Carlsen H, Diep DB. Successful Development of Bacteriocins Into Therapeutic Formulation for Treatment of MRSA Skin Infection in a Murine Model. Antimicrob Agents Chemother (2020) 64(12). doi: 10.1128/aac.00829-20
46. Kranjec C, Ovchinnikov KV, Grønseth T, Ebineshan K, Srikantam A, Diep DB. A Bacteriocin-Based Antimicrobial Formulation to Effectively Disrupt the Cell Viability of Methicillin-Resistant Staphylococcus Aureus (MRSA) Biofilms. NPJ Biofilms Microbiomes (2020) 6(1):58. doi: 10.1038/s41522-020-00166-4
47. Bakkiyaraj D, Pandian SK. In Vitro and In Vivo Antibiofilm Activity of a Coral Associated Actinomycete Against Drug Resistant Staphylococcus Aureus Biofilms. Biofouling (2010) 26(6):711–7. doi: 10.1080/08927014.2010.511200
48. Kranjec C, Morales Angeles D, Torrissen Mårli M, Fernández L, García P, Kjos M, et al. Staphylococcal Biofilms: Challenges and Novel Therapeutic Perspectives. Antibiot (Basel) (2021) 10(2). doi: 10.3390/antibiotics10020131
49. Wang X, Chi H, Zhou B, Li W, Li Z, Xia Z. Bacterial Luciferase Gene Cassette as a Real-Time Bioreporter for Infection Model and Drug Evaluation. Curr Pharm Des (2018) 24(8):952–8. doi: 10.2174/1381612824666180213121724
50. Dobie D, Gray J. Fusidic Acid Resistance in Staphylococcus Aureus. Arch Dis Child (2004) 89(1):74–7. doi: 10.1136/adc.2003.019695
51. Tan YT, Tillett DJ, McKay IA. Molecular Strategies for Overcoming Antibiotic Resistance in Bacteria. Mol Med Today (2000) 6(8):309–14. doi: 10.1016/s1357-4310(00)01739-1
52. Phoenix DA, Harris F, Dennison SR. Novel Antimicrobial Agents and Strategies. Weinheim: John Wiley & Sons (2014). doi: 10.1002/cmdc.201500290
53. Chan DCK, Burrows LL. Thiopeptides: Antibiotics With Unique Chemical Structures and Diverse Biological Activities. J Antibiot (2021) 74(3):161–75. doi: 10.1038/s41429-020-00387-x
54. Akasapu S, Hinds AB, Powell WC, Walczak MA. Total Synthesis of Micrococcin P1 and Thiocillin I Enabled by Mo(vi) Catalyst. Chem Sci (2019) 10(7):1971–5. doi: 10.1039/c8sc04885a
55. Aulakh VS, Ciufolini MA. Total Synthesis and Complete Structural Assignment of Thiocillin I. J Am Chem Soc (2011) 133(15):5900–4. doi: 10.1021/ja110166x
56. Goldstein BP. Resistance to Rifampicin: A Review. J Antibiot (2014) 67(9):625–30. doi: 10.1038/ja.2014.107
57. Papich MG. Selection of Antibiotics for Meticillin-Resistant Staphylococcus Pseudintermedius: Time to Revisit Some Old Drugs? Vet Dermatol (2012) 23(4):352–60. doi: 10.1111/j.1365-3164.2011.01030.x
58. Perlroth J, Kuo M, Tan J, Bayer AS, Miller LG. Adjunctive Use of Rifampin for the Treatment of Staphylococcus Aureus Infections: A Systematic Review of the Literature. Arch Intern Med (2008) 168(8):805–19. doi: 10.1001/archinte.168.8.805
59. Howden BP, Grayson ML. Dumb and Dumber–The Potential Waste of a Useful Antistaphylococcal Agent: Emerging Fusidic Acid Resistance in Staphylococcus Aureus. Clin Infect Dis (2006) 42(3):394–400. doi: 10.1086/499365
60. Chuard C, Herrmann M, Vaudaux P, Waldvogel FA, Lew DP. Successful Therapy of Experimental Chronic Foreign-Body Infection Due to Methicillin-Resistant Staphylococcus Aureus by Antimicrobial Combinations. Antimicrob Agents Chemother (1991) 35(12):2611–6. doi: 10.1128/aac.35.12.2611
61. Gang RK, Sanyal SC, Mokaddas E, Lari AR. Rifampicin as an Adjunct to Vancomycin Therapy in MRSA Septicaemia in Burns. Burns (1999) 25(7):640–4. doi: 10.1016/s0305-4179(99)00045-5
62. Saleh-Mghir A, Ameur N, Muller-Serieys C, Ismael F, Lemaitre F, Massias L, et al. Combination of Quinupristin-Dalfopristin (Synercid) and Rifampin is Highly Synergistic in Experimental Staphylococcus Aureus Joint Prosthesis Infection. Antimicrob Agents Chemother (2002) 46(4):1122–4. doi: 10.1128/aac.46.4.1122-1124.2002
63. Niska JA, Shahbazian JH, Ramos RI, Francis KP, Bernthal NM, Miller LS. Vancomycin-Rifampin Combination Therapy has Enhanced Efficacy Against an Experimental Staphylococcus Aureus Prosthetic Joint Infection. Antimicrob Agents Chemother (2013) 57(10):5080–6. doi: 10.1128/aac.00702-13
64. Baldoni D, Haschke M, Rajacic Z, Zimmerli W, Trampuz A. Linezolid Alone or Combined With Rifampin Against Methicillin-Resistant Staphylococcus Aureus in Experimental Foreign-Body Infection. Antimicrob Agents Chemother (2009) 53(3):1142–8. doi: 10.1128/aac.00775-08
65. Russell CD, Lawson McLean A, Saunders C, Laurenson IF. Adjunctive Rifampicin May Improve Outcomes in Staphylococcus Aureus Bacteraemia: A Systematic Review. J Med Microbiol (2014). doi: 10.1099/jmm.0.072280-0
66. De Lucia M, Bardagi M, Fabbri E, Ferreira D, Ferrer L, Scarampella F, et al. Rifampicin Treatment of Canine Pyoderma Due to Multidrug-Resistant Meticillin-Resistant Staphylococci: A Retrospective Study of 32 Cases. Vet Dermatol (2017) 28(2):171–e36. doi: 10.1111/vde.12404
67. Thwaites GE, Scarborough M, Szubert A, Nsutebu E, Tilley R, Greig J, et al. Adjunctive Rifampicin for Staphylococcus Aureus Bacteraemia (ARREST): A Multicentre, Randomised, Double-Blind, Placebo-Controlled Trial. Lancet (Lond Engl) (2018) 391(10121):668–78. doi: 10.1016/s0140-6736(17)32456-x
68. Stein C, Makarewicz O, Forstner C, Weis S, Hagel S, Löffler B, et al. Should Daptomycin-Rifampin Combinations for MSSA/MRSA Isolates be Avoided Because of Antagonism? Infection (2016) 44(4):499–504. doi: 10.1007/s15010-016-0874-2
69. Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, et al. Clinical Practice Guidelines by the Infectious Diseases Society of America for the Treatment of Methicillin-Resistant Staphylococcus Aureus Infections in Adults and Children. Clin Infect Dis (2011) 52(71):e18–55. doi: 10.1093/cid/ciq146
70. Punjataewakupt A, Napavichayanun S, Aramwit P. The Downside of Antimicrobial Agents for Wound Healing. Eur J Clin Microbiol Infect Dis (2019) 38(1):39–54. doi: 10.1007/s10096-018-3393-5
Keywords: MRSA, bacteriocin, skin infection bacteria, micrococcin P1, murine model, Rifampi(ci)n
Citation: Ovchinnikov KV, Kranjec C, Telke A, Kjos M, Thorstensen T, Scherer S, Carlsen H and Diep DB (2021) A Strong Synergy Between the Thiopeptide Bacteriocin Micrococcin P1 and Rifampicin Against MRSA in a Murine Skin Infection Model. Front. Immunol. 12:676534. doi: 10.3389/fimmu.2021.676534
Received: 05 March 2021; Accepted: 11 June 2021;
Published: 02 July 2021.
Edited by:
Fabio Bagnoli, GlaxoSmithKline, ItalyReviewed by:
Lucinda Janete Bessa, LAQV Network of Chemistry and Technology, PortugalDafne Bongiorno, University of Catania, Italy
Copyright © 2021 Ovchinnikov, Kranjec, Telke, Kjos, Thorstensen, Scherer, Carlsen and Diep. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dzung B. Diep, ZHp1bmcuZGllcEBubWJ1Lm5v