 Bryan A. Joosse1
Bryan A. Joosse1 James H. Jackson1,2Alberto Cisneros 3rd1
James H. Jackson1,2Alberto Cisneros 3rd1 Austin B. Santhin1Scott A. Smith3,4,5
Austin B. Santhin1Scott A. Smith3,4,5 Daniel J. Moore4,5,6Leslie J. Crofford1,4,5Erin M. Wilfong1,7
Daniel J. Moore4,5,6Leslie J. Crofford1,4,5Erin M. Wilfong1,7 Rachel H. Bonami1,4,5*
Rachel H. Bonami1,4,5*- 1Department of Medicine, Division of Rheumatology and Immunology, Vanderbilt University Medical Center, Nashville, TN, United States
- 2Department of Biomedical Sciences, School of Medicine Greenville, University of South Carolina, Greenville, SC, United States
- 3Department of Medicine, Division of Infectious Diseases, Vanderbilt University Medical Center, Nashville, TN, United States
- 4Department of Pathology, Microbiology and Immunology, Vanderbilt University Medical Center, Nashville, TN, United States
- 5Vanderbilt Institute for Infection, Immunology, and Inflammation (VI4), Nashville, TN, United States
- 6Department of Pediatrics, Division of Endocrinology & Diabetes, Vanderbilt University Medical Center, Nashville, TN, United States
- 7Department of Medicine, Allergy, Pulmonary, and Critical Care, Vanderbilt University Medical Center, Nashville, TN, United States
Antigen-specific B cells (ASBCs) can drive autoimmune disease by presenting autoantigen to cognate T cells to drive their activation, proliferation, and effector cell differentiation and/or by differentiating into autoantibody-secreting cells. Autoantibodies are frequently used to predict risk and diagnose several autoimmune diseases. ASBCs can drive type 1 diabetes even when immune tolerance mechanisms block their differentiation into antibody-secreting cells. Furthermore, anti-histidyl tRNA synthetase syndrome patients have expanded IgM+ Jo-1-binding B cells, which clinically diagnostic IgG Jo-1 autoantibodies may not fully reflect. Given the potential disconnect between the pathologic function of ASBCs and autoantibody secretion, direct study of ASBCs is a necessary step towards developing better therapies for autoimmune diseases, which often have no available cure. We therefore developed a high-throughput screening pipeline to 1) phenotypically identify specific B cell subsets, 2) expand them in vitro, 3) drive them to secrete BCRs as antibody, and 4) identify wells enriched for ASBCs through ELISA detection of antibody. We tested the capacity of several B cell subset(s) to differentiate into antibody-secreting cells following this robust stimulation. IgM+ and/or IgD+, CD27- memory, memory, switched memory, and BND B cells secreted B cell receptor (BCR) as antibody following in vitro stimulation, whereas few plasmablasts responded. Bimodal responses were observed across autoimmune donors for IgM+ CD21lo and IgM- CD21lo B cells, consistent with documented heterogeneity within the CD21lo subset. Using this approach, we detected insulin-binding B cell bias towards CD27- memory and CD27+ memory subsets in pre-symptomatic type 1 diabetes donors. We took advantage of routine detection of Jo-1-binding B cells in Jo-1+ anti-histidyl tRNA synthetase syndrome patients to show that Jo-1-binding B cells and total B cells expanded 20-30-fold using this culture system. Overall, these studies highlight technology that is amenable to small numbers of cryopreserved peripheral blood mononuclear cells that enables interrogation of phenotypic and repertoire attributes of ASBCs derived from autoimmune patients.
Introduction
B lymphocytes contribute to immune responses by presenting antigens to T cells, secreting cytokines, and differentiating into antibody-secreting cells. Autoantibodies are frequently used to predict and diagnose autoimmune diseases, highlighting the important role that B cells play in promoting autoimmunity (1–7). In some diseases, such as Sjögren’s syndrome, systemic lupus erythematosus, and rheumatoid arthritis, autoantibodies have pathologic function via immune complex formation (8). In others, such as type 1 diabetes, autoantibodies are not directly pathogenic (9); rather, it is the antigen-presenting function of the B cell that is essential for disease (9–13). Autoimmune disease treatments such as prednisone, rituximab, or abatacept involve broad immune suppression. For example, rituximab globally depletes B cells which is effective at treating several autoimmune diseases, including rheumatoid arthritis, systemic lupus erythematosus, anti-histidyl tRNA synthetase syndrome, and systemic sclerosis (14–20). Rituximab is well-tolerated in adults, but results in diminution of vaccine responses, a key consideration for treatment of pediatric autoimmune diseases such as type 1 diabetes (21). Therapies that selectively target ASBCs would avoid the problem of broad immune suppression and should thus be safer. Selection elimination of anti-insulin B cells prevents disease in type 1 diabetes-prone mice (22); targeting ASBCs may thus offer an effective alternative to broad immunosuppression for autoimmune disease prevention and treatment. Understanding the mechanisms that govern immune tolerance breach by autoreactive B cells requires identification and study of ASBCs.
B lymphocytes express antigen-specific, membrane-bound B cell receptors but are not a major source of circulating antibody. Rather, B lymphocytes must receive the right stimulation to differentiate into plasmablasts or plasma cells that secrete BCR as circulating antibody (23). Different immune checkpoints govern whether autoreactive B cells 1) expand, 2) undergo mutation and affinity maturation, and 3) differentiate into antibody-secreting cells (23, 24). In Sjögren’s syndrome, sustained Ro60 autoantibody production is due to continual generation of plasmablasts from ASBCs, rather than long-lived plasma cells, suggesting continual autoreactive B cell seeding of the peripheral repertoire is required (25). Studies in mice show that autoantigen-specific B cells (ASBCs) can retain disease-relevant autoantigen-presenting function even when immune tolerance mechanisms block their differentiation into autoantibody-secreting cells (26–28). This points to a need to identify the specific mechanisms by which ASBCs escape immune tolerance to expand and drive pathology, a process which may differ between autoimmune diseases.
Methods have been developed to track ASBCs in the broad repertoire that are as rare as 1 in 20 million cells (29). Many different B cell subsets can potentially contribute to a protective or autoimmune response that may have different responsiveness to specific stimuli. For example, whereas naïve B cells proliferate in response to BCR stimulation, anergic (BND) and CD21lo B cells do not (30, 31). BND and CD21lo subsets may serve as reservoirs for autoreactive B cells in several autoimmune diseases, including type 1 diabetes, Sjögren’s syndrome, anti-histidyl tRNA synthetase syndrome, and systemic sclerosis (32–36). We sought to develop high-throughput stimulation and screening methods to identify ASBCs among total PBMCs using ELISA detection of BCRs secreted as antibody. Given the signaling differences present among autoreactive-prone B cell subsets of interest, we examined the potential of each of these subsets to respond to robust stimulation in vitro that includes BCR, CpG, CD40L, IL-21, and BAFF stimulation (29). We further tested the capacity of several memory B cell subsets and plasmablasts to differentiate/secrete BCR as antibody in this culture system as a mean to identify ASBCs within a polyclonal repertoire.
Materials and Methods
Participant Selection and Clinical Information
Systemic sclerosis and Jo-1+ anti-histidyl tRNA synthetase syndrome patients were selected from enrollees in the Vanderbilt Myositis and Scleroderma Treatment and Investigative Center (MYSTIC) cohort. A subset of systemic sclerosis patients was defined as having interstitial lung disease if a radiologist determined that fibrosis was present on a CT scan. Sjögren’s syndrome patients were selected from enrollees in the B Lymphocytes in Sjögren’s syndrome (BLISS) at Vanderbilt University Medical Center. Patients were eligible for the study if they were 18 years or older and had been diagnosed with systemic sclerosis by a rheumatologist or pulmonologist or Sjögren’s syndrome by a rheumatologist. Clinical data were collected from the electronic medical record. One donor was obtained from the Human Immunology Discovery Initiative (HIDI) cohort. Additional autoimmune diagnoses were obtained via patient-reported questionnaire. These studies were approved by the Vanderbilt Institutional Review Board. Participant characteristics are outlined in Table 1.
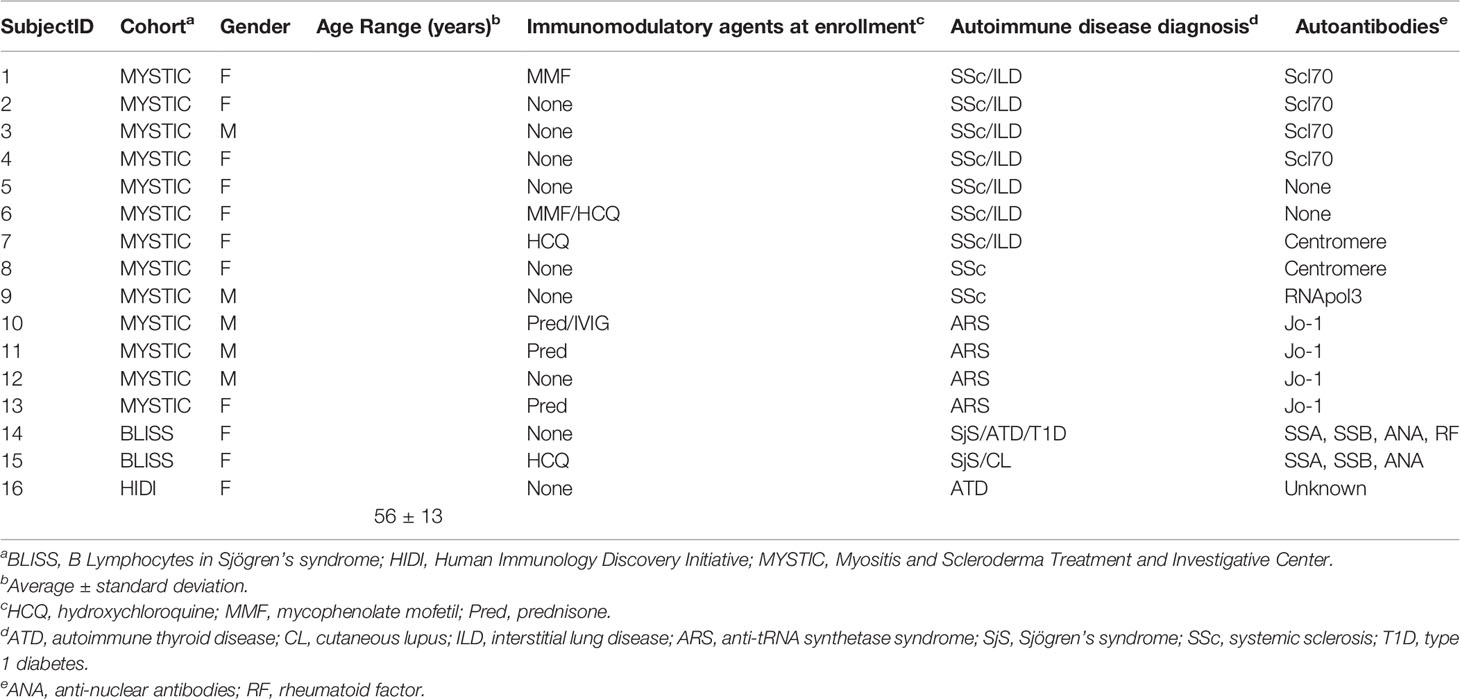
Table 1 Demographics for participants included from MYSTIC, BLISS, and HIDI cohorts.
Donors at risk for type 1 diabetes were recruited at Vanderbilt through their participation in Type 1 Diabetes TrialNet Pathway to Prevention (37). Type 1 Diabetes TrialNet participants were first, second, or third-degree relatives of type 1 diabetes probands that were defined as having pre-symptomatic type 1 diabetes based on a history of positivity for two or more islet autoantibodies (IAA, GAD65, ICA, IA2, or ZNT8, defined in Table 2, one of which was insulin autoantibody (IAA). Participants were excluded from this cohort if they previously received insulin therapy or were diagnosed with diabetes at the time of blood draw. Participants were excluded for any of the following: fever 101°F in preceding 48hrs of clinic visit, receiving antibiotic therapy at clinic visit, weight less than 8 kg, known pregnancy, anemia at time of study initiation (Hgb <12), comorbid conditions which would render giving blood samples a risk, chronic use of immune suppressive or depleting medications, prior use of immune modulating medications, inability to meet study visit requirements, ongoing use of Accutane, age <1 year, > 60 years at the time of baseline visit. Participant characteristics are outlined in Table 2. This study received IRB approval by Vanderbilt and ancillary study approval by Type 1 Diabetes TrialNet. All data were managed using the REDCap software system (38).
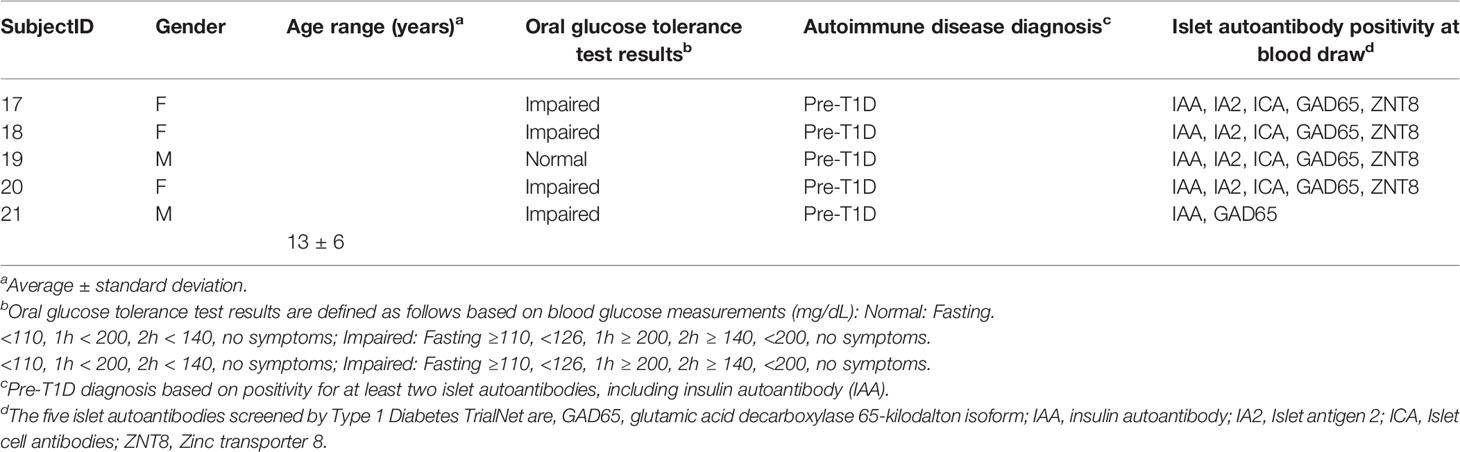
Table 2 Type 1 Diabetes TrialNet participant demographics.
Sample Collection and Processing
Peripheral blood mononuclear cells (PBMCs) were obtained by collecting whole blood into mononuclear cell preparation tubes with sodium heparin or sodium citrate (BD) via peripheral venipuncture. Cells were washed twice in PBS and red blood cells were lysed using Ack Lysis Buffer (Gibco). Cells were again washed in PBS and counted. Cells were cooled in 10% dimethyl sulfoxide (DMSO) in fetal bovine serum (FBS) at a rate of -1°C/minute until they reached a temperature of -80°C for 24-72 hours. Cells were then stored in liquid nitrogen until the time of analysis. Viability was assessed at time of sample thawing with ∼ 80% average viability.
ELISA
384-well ELISA plates (Maxisorp) were coated with either 500 ng/ml goat anti-human Ig (Southern Biotech, 2010-01) in 1X PBS overnight at 4°C or 1 μg/mL recombinant human insulin (Sigma I2643) in borate buffered saline overnight at 37°C to assess total antibody or anti-insulin antibody, respectively. The next day, plates were washed 5X with 0.5X PBS and then blocked with 5% chicken serum in 1X PBS plus 0.5% Tween (1X PBS-T) for 1h at room temperature. Block was decanted from plates, blotted dry, and then stimulated PBMC culture supernatants (diluted 1:2 in 1X PBS-T) were added to the plates. Plates were then incubated for 1.5h at room temperature. Bound antibody was detected either by goat anti-human IgG-HRP (Southern Biotech, 2040-05) or goat anti-human IgM-HRP (Southern Biotech, 2020-05) secondary antibodies diluted in 1X PBS-T. Plates were washed 10X with 0.5% PBS. Finally, TMB Ultra ELISA substrate (Thermo Fisher, PI34029) was added, and plates were read at O.D. 370nm using a microplate reader (SpectraMax M3). Wells with fluorescence values above a cutoff of the mean + 3 standard deviations of a blank well were considered positive. Antibody concentrations were calculated using IgG/IgM standards (Sigma, I4506 and I8260, respectively).
Flow Cytometry Purification of B Cell Subsets and Analysis
Cells were stained in flow cytometry staining buffer (1X PBS containing 5% FBS, 0.02% EDTA, and 0.1% sodium azide) with the following reactive antibodies and reagents: CD19 (SJ25C1), CD21 (Bu32), CD24 (ML5), CD27 (O323), CD38 (HIT2), IgD (IA6-2), IgG (G18-145), IgM (MHM-88) (BioLegend or BD Biosciences). B cell subsets were purified using a FACSAria sorter (BD Biosciences). Data were analyzed using FlowJo software (Tree Star, Inc.). Jo-1-binding B cells were identified by flow cytometry using GST-tagged Jo-1 antigen we previously cloned and recombinantly expressed, followed by detection with anti-GST secondary antibody (Abcam), as previously described (36).
B Cell Stimulation
Cryopreserved PBMCs were rapidly thawed, washed, and resuspended in ClonaCell-HY Medium A (StemCell). PBMCs or sorted B cell subsets plated as indicated in figure legends in Medium A with 0.833 μg/ml of CpG (ZOEZOEZZZZZOEEZOEZZZT, Invitrogen) + 0.133 μg/ml each of mouse-anti-human kappa and lambda antibodies (Southern Biotech), as well as 0.033x106 cells/mL viable gamma-irradiated NIH3T3 fibroblasts that were genetically engineered to express cell-surface human CD40L (CD154), secreted human B cell activating factor (BAFF), and human IL-21 (unpublished line related to (39, 40), originally provided to Dr. Smith by Dr. Deepta Bhattacharya; Washington University in St. Louis, St. Louis, MO). These stimuli drive B cells to secrete BCR as antibody to enable screening for ASBCs (29). The mixture was then plated into 96-well (300 μL total volume) or 384-well (100 μL total volume) flat bottom plates (Corning). Plates were incubated with 5% CO2 at 37°C and screened for antibody production by ELISA after 1 wk, as described above.
Statistical Analysis
Standard statistical tests utilized for each experiment are indicated in the corresponding figure legends and significance values were calculated using Prism (GraphPad).
Results
BCRs Secreted as Antibody Are Captured From Stimulated Mature Naïve and Memory B Cell Subsets
ASBCs are not always readily detectable in autoimmune individuals. A robust stimulation method allows capture of IgE ASBCs as rare as 1 in 20 million as hybridomas (29). This method relies on high-throughput ELISA screening for antigen-specific BCRs secreted as antibody following robust stimulation. One caveat with this method is that it does not provide information about the phenotypic subset from which the ASBC was derived. B cell phenotype correlates with dramatic differences in functional capacity; anergic B cells (e.g. BND cells in humans) are functionally silenced for proliferation and differentiation into antibody-secreting cells, but memory B cells are poised to rapidly contribute to protective immune responses (29). Thus, consideration of which subset(s) ASBCs are expanded in provides important information about how autoreactive B cell clones arise and function in the repertoire to promote autoimmunity. We sought to test whether this stimulation/screening method could be adapted to use flow cytometry-purified B cell subsets from autoimmune disease patient peripheral blood as inputs, as this would enable ASBCs identification coupled with phenotypic information, such as whether the ASBC had a memory phenotype in vivo. Figure 1 provides an overview of the methods applied to each figure. As shown in Figure 2A, specific B cell subsets were purified using flow cytometry sorting. The focus of this manuscript is on B cells derived from autoimmune donors; we presented detailed immunophenotyping to compare B cell subset percentages between patients with several of the autoimmune diseases highlighted in this manuscript and healthy controls in separate manuscripts (36, 41). Purified B cell subsets (e.g. memory) were subsequently divided across wells and stimulated as in Methods to drive B cells to differentiate into antibody-secreting cells (Figure 2B). ELISA detects the presence of BCR secreted as antibody in CD19+ IgM+ and/or IgD+, class-switched CD27+ memory (CD19+ IgM- IgD- CD27+), and CD27- memory (CD19+ IgM- IgD- CD27-) B cell subsets (Figure 2C). BCR was also detected as antibody from CD27+ memory cells which included IgM+ cells (Figure 2F). These data show our in vitro stimulation protocol can be used to screen BCRs expressed by non-class-switched and memory B cell subsets.
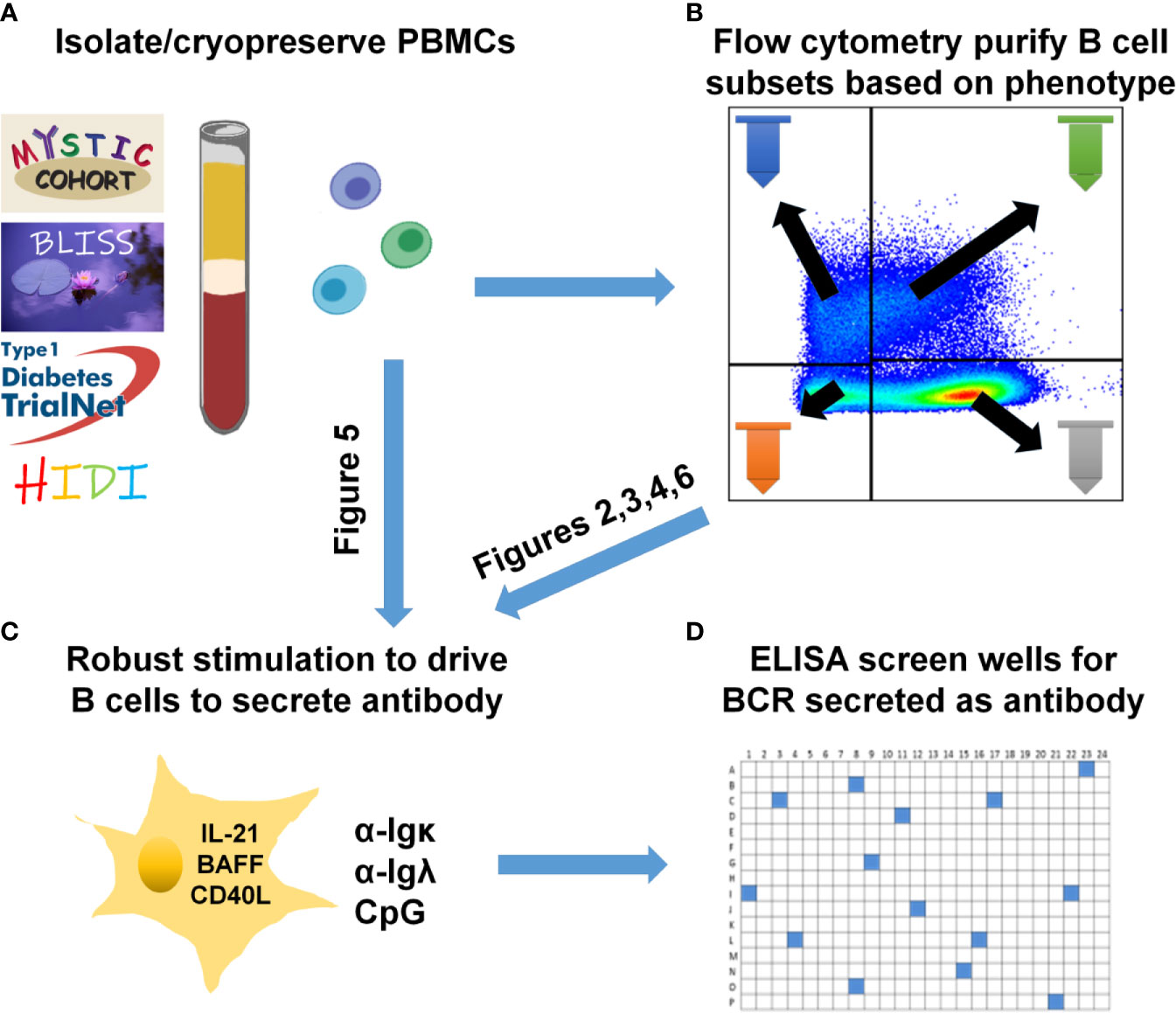
Figure 1 Human B cell stimulation and high-throughput screening methods overview. (A) PBMCs were isolated and cryopreserved from the indicated cohorts. (B) Specific B cell subsets were flow cytometry-purified. (C) As specified for each figure, total PBMCs (A) were plated into 96-well plates or purified B cell subsets (B) were plated into 384-well plates and stimulated as in Methods to drive B cell proliferation and differentiation into antibody-secreting cells. (D) High-throughput ELISA screening of well supernatants identified wells containing ASBCs.
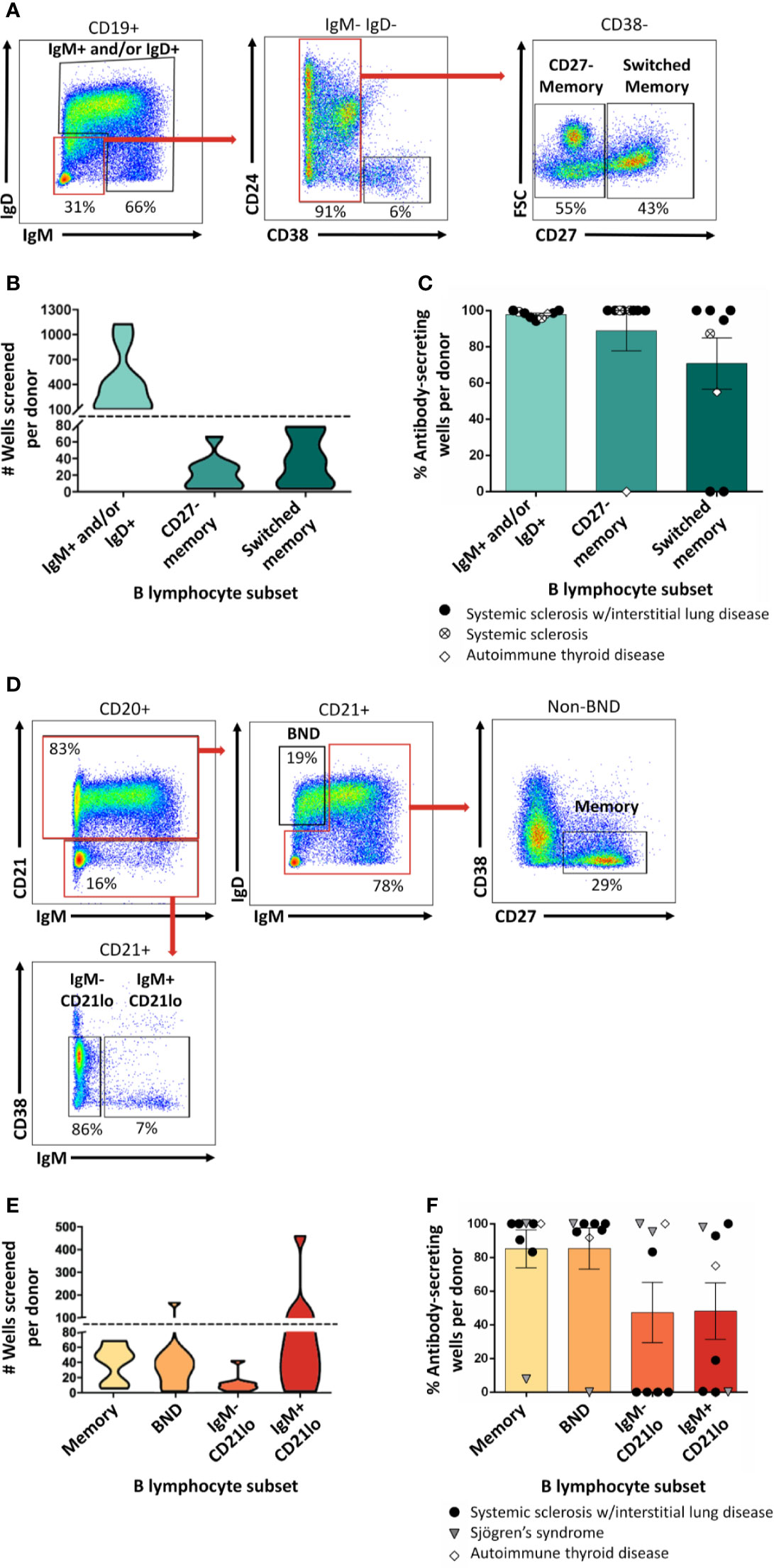
Figure 2 Mature naïve, memory, and autoreactive-prone B cell subsets differentiate into antibody-secreting cells following in vitro stimulation. (A) Representative plots show flow cytometry identification of the indicated B cell subsets (gated on CD19+ live singlet lymphocytes). Subsets were defined as follows: IgM+ and/or IgD+, CD27- memory (IgD+ IgM+ CD27- CD38-), and CD27+ switched memory (IgD- IgM- CD27+ CD38-). (B) Violin plots show the data distribution and density for the number of all wells screened for each B cell subset per donor in (C), (C) PBMCs were isolated from systemic sclerosis/interstitial lung disease (black circle, n=6), systemic sclerosis (open circle/X, n=2), or ATD patients (open diamond, n=1) and B cell subsets were identified as in (A) and flow cytometry purified. Individual B cell subsets were plated in 384-well plates at ~300 B cells/well and stimulated as in Methods for 1 wk. ELISA was used to measure antibody present in culture supernatants. Wells were scored positively for antibody secretion for ELISA OD370nm > 0.5. The frequency of positive wells was calculated per subset, per donor; individual points represent individual donors. (D) Representative plots show flow cytometry identification of the indicated B cell subsets (gated on CD19+ live singlet lymphocytes). Subsets were defined as follows: memory (IgM-/IgD- or IgMmid/high CD21+ CD27+ CD38-), BND (IgMlow/neg/IgD+ CD21+), IgM- CD21lo (IgM- CD27+ CD38mid/neg), or IgM+ CD21lo (IgM+ CD21lo CD38mid/neg). (E) Violin plots show the data distribution and density for the number of all wells screened for each B cell subset per donor in (F), (F) PBMCs were isolated from systemic sclerosis/interstitial lung disease (black circle, n=5), Sjögren’s syndrome (filled triangle, n=2), or ATD patients (open diamond, n=1) and B cell subsets were identified as in (D) and flow cytometry purified. Individual B cell subsets were plated in 384-well plates at ~ 300 B cells/well and stimulated as in Methods for 1 wk. Wells were scored positively for antibody secretion for ELISA OD370nm > 0.5. The frequency of positive wells was calculated per subset, per donor; individual points represent individual donors.
Autoreactive-Prone BND (Anergic) and CD21low B Cells Are Stimulated to Secrete BCRs as Antibody
We previously identified ASBCs using fluorescently-labeled autoantigen (36). This detection method could however miss B cells that downregulate surface BCR because of chronic antigen stimulation and/or immune tolerance mechanisms such as anergy. Anergy is an immune tolerance mechanism by which autoreactive B cells are functionally silenced but retained in the repertoire; they can be phenotypically identified as BND cells (IgMlo/- IgD+ CD27-) (32). CD21lo B cells are an autoreactive-prone subset that is increased in the peripheral blood of patients with several autoimmune diseases (31, 33, 42). CD21lo B cells have been reported to be anergic, yet also to express highly mutated BCRs that are consistent with germinal center origin (31, 33, 43). Given the potential for BND and CD21lo B cell subsets to serve as autoreactive B cell reservoirs, we flow cytometry purified these subsets as shown in Figure 2D and screened wells to determine (Figure 2E) whether our stimulation protocol could drive B cell subsets to differentiate and secrete their BCRs as antibody (44). BND and CD21lo BCRs were detected as antibody following stimulation (Figure 2F), demonstrating that BCRs from these subsets can be evaluated using this protocol.
Plasmablast Secretion of Antibody Is Not Sustained Following In Vitro BCR/CD40/BAFF/Cpg/IL-21 Stimulation
B cells do not secrete antibody; rather, if they receive the right signals, they differentiate into antibody-secreting cells. Whereas plasmablasts are short-lived antibody-secreting cells that can be found in peripheral blood, plasma cells are long-lived antibody-secreting cells that can be found in the bone marrow and other tissues. Plasmablasts are enriched for ASBCs, thus we wanted to test whether this approach captures plasmablast BCRs. Flow cytometry purification identified plasmablasts as in Figure 3A, which were stimulated in vitro as in Methods (Figure 3B). Antibody was not detected by ELISA for plasmablast isolated from the majority of donors following this stimulation protocol (Figure 3C), thus other approaches are required to assay antigen-specific plasmablasts.
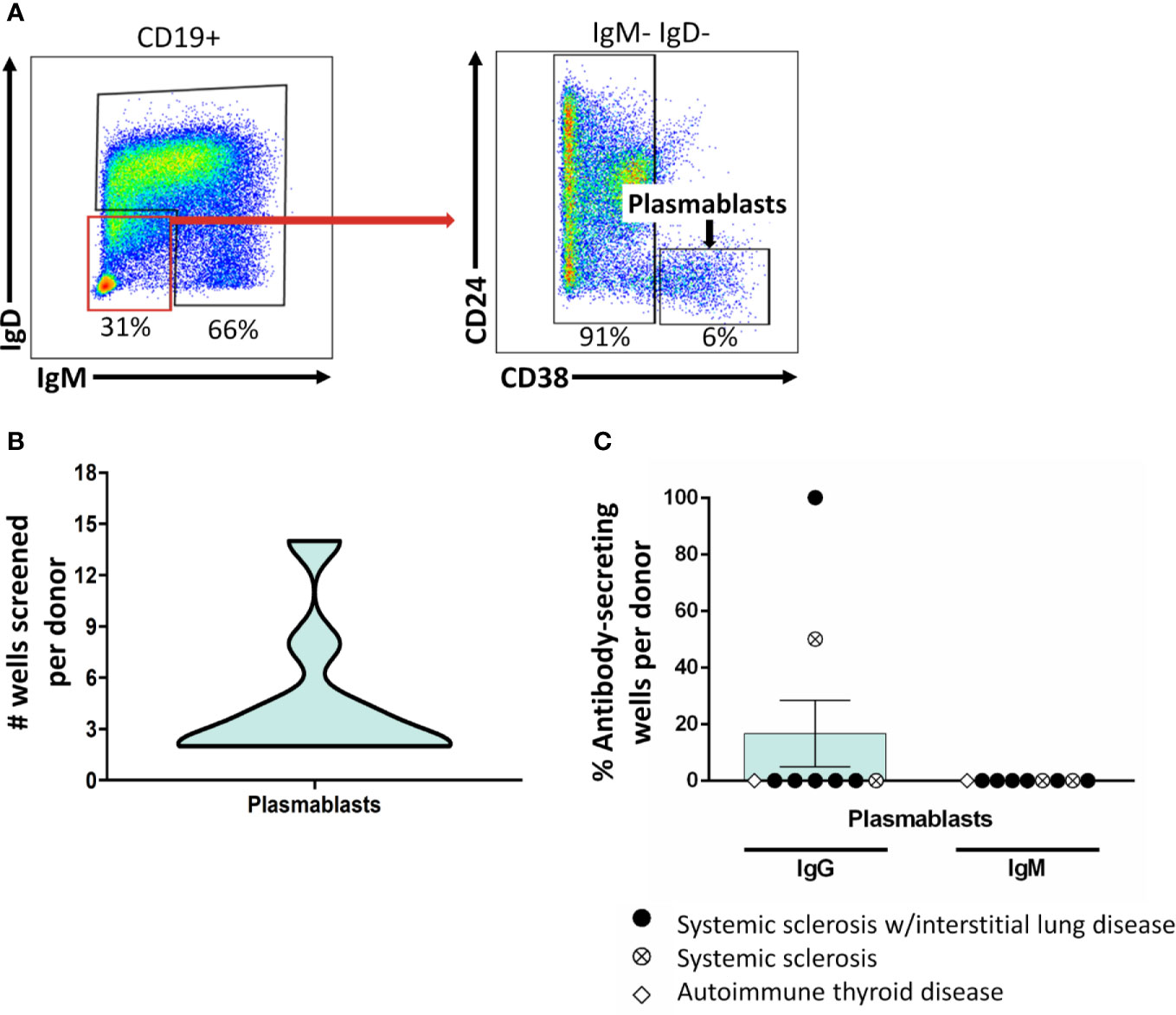
Figure 3 Class-switched plasmablasts do not sustain antibody secretion following BCR/CD40/BAFF/TLR9/IL-21 stimulation. (A) PBMCs were isolated from autoimmune disease patients. Representative plots show flow cytometry identification of plasmablasts (CD19+ IgD- IgM- CD24- CD38- live singlets). (B) B cell subsets were identified as in (A) and flow cytometry purified. Plasmablasts were plated in 384-well plates at ~300 plasmablasts/well and stimulated as in Methods for 1 wk. The data distribution and density of the number of plasmablast wells that were screened per donor is expressed as a violin plot. (C) Culture supernatants from plasmablasts plated as in (B) were screened for antibody production by ELISA. Wells were scored positively for antibody secretion for ELISA OD370nm > 0.5. The frequency of positive wells was calculated per subset, per donor; individual points represent individual donors. Results from systemic sclerosis/interstitial lung disease (black circle, n=6), systemic sclerosis (open circle/X, n=2), or ATD patients (open diamond, n=1) are shown.
Limited Class-Switching Is Observed for IgM+/IgD+ B Cells Following In Vitro Stimulation
Non-class-switched B cell subsets were stimulated for 6d as in Methods to test whether they undergo class-switching to IgG (Figure 4A). Flow cytometry analysis revealed limited class-switching away from the IgM isotype for IgM+ and/or IgD+, BND, or IgM+ CD21lo B cells following stimulation (Figures 4B, F, H). Conversely, the majority of switched memory B cells were identified as IgG+ or IgM- IgG- at the end of culture (Figure 4D), potentially indicating IgA class switch, which was not measured in this assay. The memory B cell subset, which included both IgM+ and IgM- B cells as input, contained both IgM+ and switched B cells following stimulation (Figure 4E), suggesting both IgM+ and class-switched CD27+ B cells survive in this culture. IgG+ and IgM- IgG- B cells were observed among cultured CD27- memory and IgM- CD21lo B cells as expected (Figures 4C, G), however the relatively high proportion of IgM- CD21lo B cells that expressed IgM at the end of culture suggests class-switched B cells in these subsets may not respond as strongly as IgM+ CD21lo B cells to this stimulation.
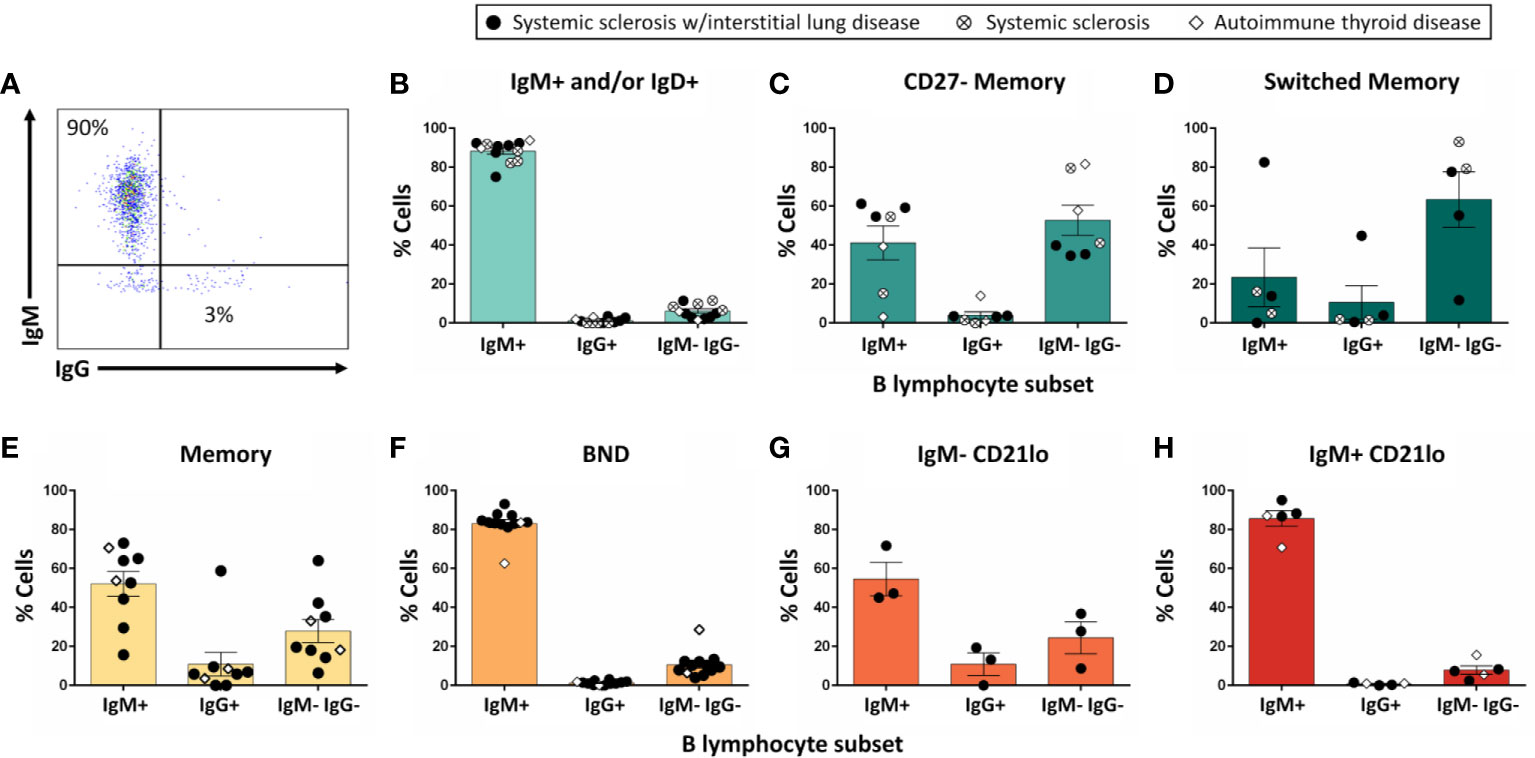
Figure 4 The majority of IgM+ and/or IgD+ B cells do not undergo class-switch to IgG following in vitro stimulation. PBMCs were isolated from systemic sclerosis/interstitial lung disease (black circle, n=6 donors; n=35 wells), systemic sclerosis (open circle/X, n=2; n=8 wells), or ATD patients (open diamond, n=1; n=11 wells). Flow cytometry-purified B cell subsets (identified as in Figure 1) were plated in 384-well plates at ∼300 cells/well and stimulated as in Methods for 1 wk. Flowcytometry was used to measure the frequency of IgM+, IgG+, and IgM- IgG- cells among CD19+ live singlet lymphocytes (A). Flow phenotyping inclusion criteria for this analysis was > 95 lymphocyte events and > 35 CD19+ events. Results for (B) IgM+ and/or IgD+, (C) CD27- memory, (D) CD27+ switched memory, (E) memory, (F) BND, (G) IgM- CD21lo, and (H) IgM+ CD21lo B cell subsets are shown.
Insulin-Binding ASBCs Are Detected Among Memory B Cell Subsets From Donors at High Risk for Type 1 Diabetes
ASBCs are not always readily detectable in autoimmune individuals. For example, ex vivo detection of anti-insulin B cells is challenging in both humans and mice at high risk for type 1 diabetes, despite being seropositive for insulin autoantibody (22, 32). We sought to couple phenotypic information with our ASBC screening pipeline to provide methods to better understand how ASBC functional capacity changes with respect to autoimmune disease progression, or in response to immune therapy using small volumes (2-10mL) of peripheral blood. Whereas CD27+ memory B cells arise through germinal center reactions, CD27- IgD- memory B cells (increased in the autoimmune disease, SLE) do not (45, 46). Insulin is a major autoantigen in type 1 diabetes and insulin autoantibodies are one of five islet autoantibodies used by Type 1 Diabetes TrialNet to identify individuals at high risk for type 1 diabetes (3). Donors at risk for type 1 diabetes were identified as in Methods based on donor positivity for insulin autoantibody (IAA) plus at least one other islet autoantibody and PBMCs were flow cytometry sorted to identify IgM+ and/or IgD+, IgD- CD27- memory, or CD27+ memory B cells and stimulated as in Methods (Figure 5A). Wells that contained insulin-binding B cells were identified by ELISA using a secondary antibody confirmed to bind both IgM and IgG to ensure fair comparison across subsets regardless of isotype (Figure 5B). An increased frequency of CD27- and CD27+ memory subset wells contained insulin-binding ASBCs relative to the IgM+ and/or IgD+ subset, suggesting insulin-binding B cells are enriched among memory B cells (Figure 5C). Thus, these approaches can be used to detect expanded ASBCs within specific B cell subsets.
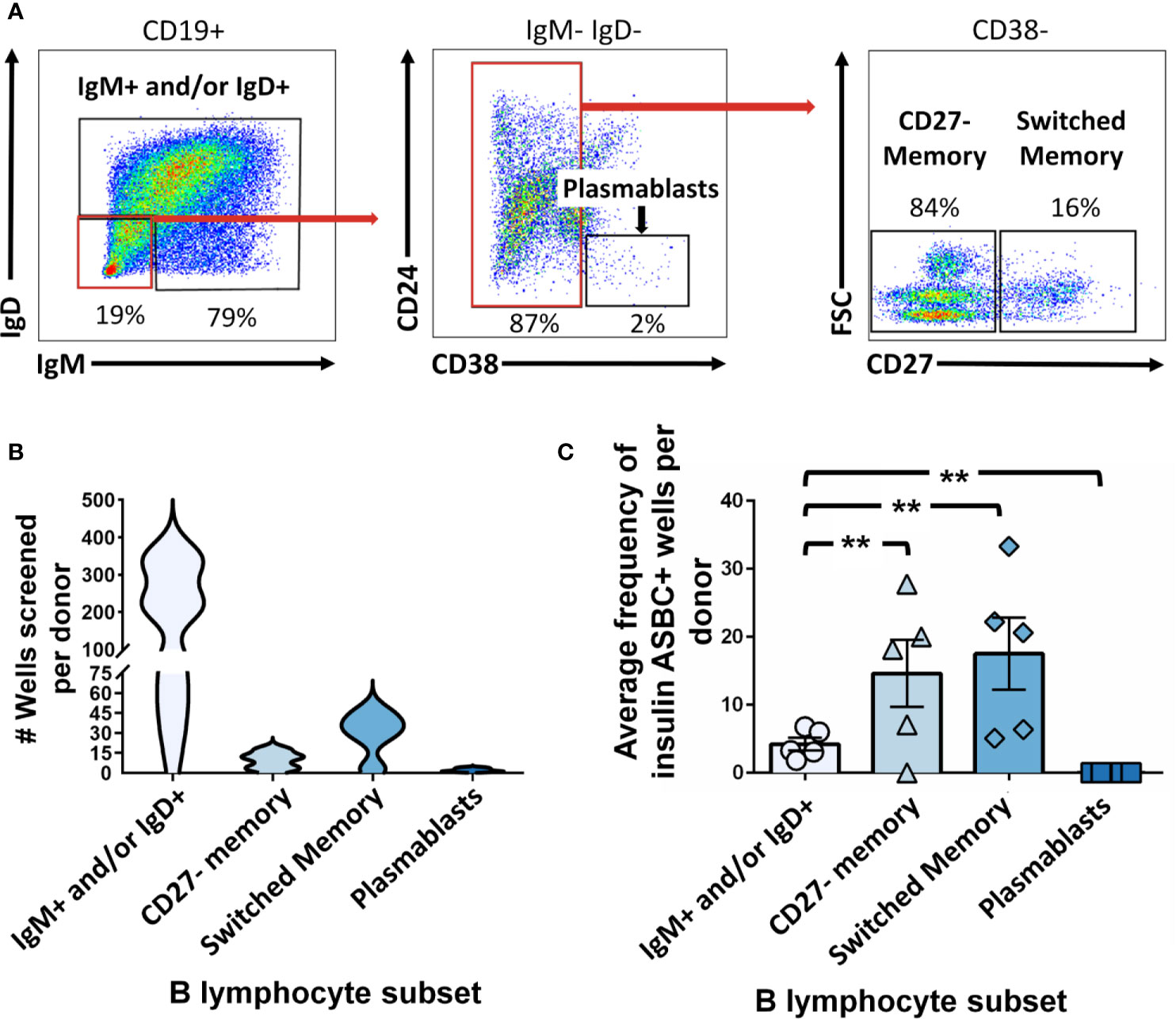
Figure 5 Insulin-binding ASBCs are identified within specific B cell subsets. (A) Representative plots show flow cytometry identification of the indicated B cell subsets (gated on CD19+ live singlet lymphocytes). Subsets were defined as follows: IgM+ and/or IgD+, CD27- memory (IgD+ IgM+ CD27- CD38-), and CD27+ switched memory (IgD- IgM- CD27+ CD38-). (B) Data distribution and density of wells screened per donor are expressed as a violin plot per subset. (C) PBMCs were isolated from n=5 participants at high risk for type 1 diabetes [positive for ≥ two islet autoantibodies, one of which was insulin autoantibody (IAA)]; flow cytometry-purified B cell subsets (identified as in Figure 1) were plated in 384-well plates at ~300 cells/well and stimulated as in Methods for 1 wk. ELISA was used to measure anti-insulin antibody in culture supernatants to identify wells containing insulin-binding ASBCs (positive wells defined as O.D.370nm ≥ 0.85 which was one standard deviation above the mean and which displayed reduced insulin antibody binding when parallel supernatants were incubated with 100-fold excess soluble insulin competitor). Errors bars shown are SEM per subset. Each data point represents the average % insulin-binding ASBCs identified per donor for each B cell subset. **p < 0.01, Mann-Whitney U test.
Jo-1-Binding ASBCs Expand Following In Vitro Stimulation
Flow cytometry detection of ASBCs can be challenging due to low frequency of target cells. However, we recently identified a readily detectable population of Jo-1-binding B cells in the peripheral blood of Jo-1 autoantibody-positive anti-histidyl tRNA synthetase syndrome patients using flow cytometry staining with labeled autoantigen (36). This created an opportunity to directly examine expansion of ASBCs following our in vitro stimulation protocol. The frequency of Jo-1-binding B cells was measured for n=10 Jo-1+ anti-histidyl tRNA synthetase syndrome donors; PBMCs from n=4 of these donors were stimulated for 1 wk as in Methods and cells from individual wells were flow cytometry phenotyped (Figures 6A, E) to determine the frequency of Jo-1-binding B cells (ASBCs), as well as total B cells following stimulation. B cells underwent an average 18-fold expansion (Figure 6B), whereas Jo-1-binding ASBCs expanded 32-fold on average (Figure 6C) following in vitro stimulation. These data confirm that Jo-1-binding ASBCs expand at least as well as total B cells via this stimulation protocol. PBMCs are divided across 384-well plates, enabling bias for particular wells towards a given antigen specificity. This is illustrated in Figure 6D, which shows the relative frequency of ASBCs is increased following in vitro stimulation as compared to the ex vivo frequency of ASBCs. Additionally, we showed a heterogeneity across IgM and IgG isotypes within Jo-1-binding ASBCs across different donors (Figure 6F). A small percentage of Jo-1-binding ASBCs were isotypes other than IgM and IgG (5-30%; IgM-, IgG-).
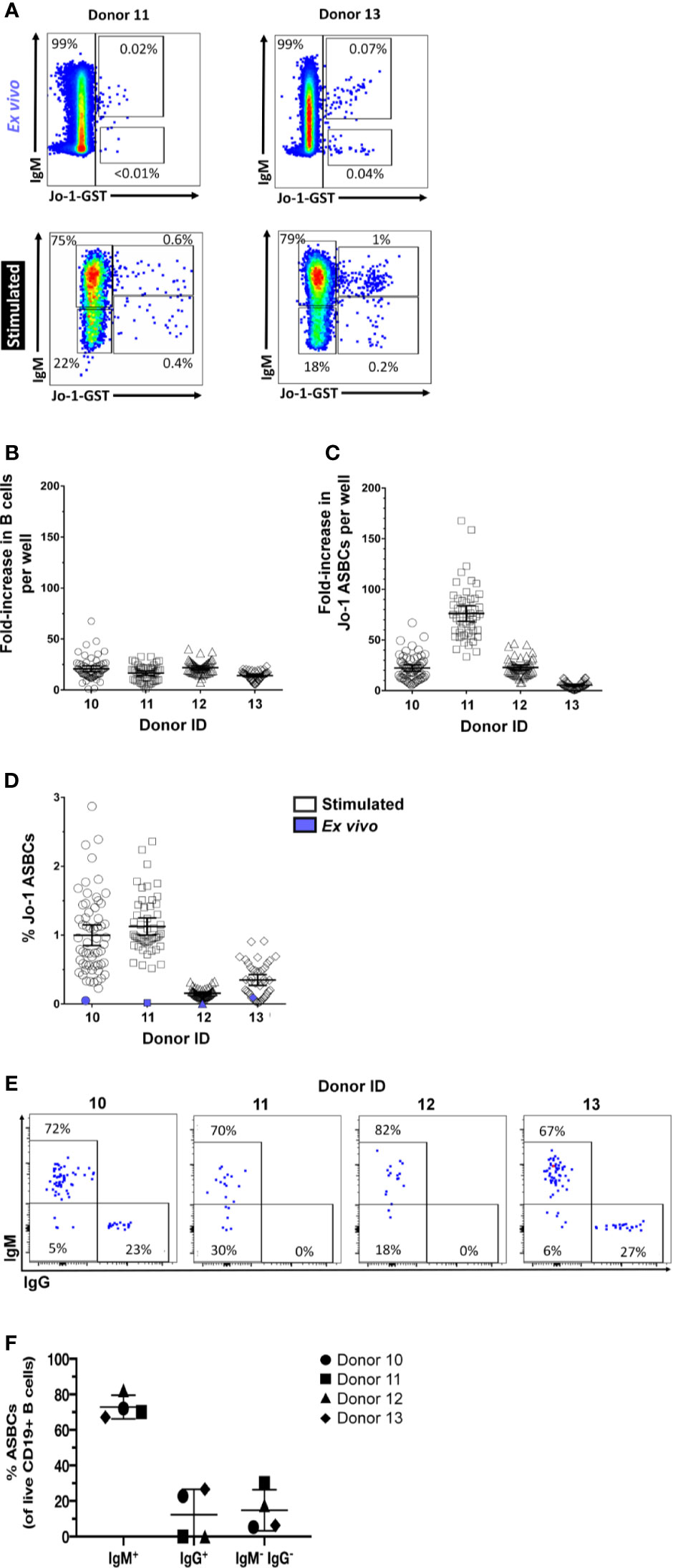
Figure 6 Autoreactive Jo-1-binding ASBCs expand in response to in vitro stimulation. PBMCs were isolated from n = 4 Jo-1 anti-histidyl tRNA synthetase syndrome patients, and (A) representative plots show flow cytometry identification of total B cells as well as Jo-1-binding ASBCs ex vivo and following stimulation in vitro for 1 wk as in Methods (gated on CD19+ live singlet lymphocytes). 3.3x104 PBMCs were stimulated per well in 96 well plates as in Methods. An average input total B cell and Jo-1-binding B cell frequency per well was calculated based on ex vivo flow phenotyping data. After 1 wk of stimulation, individual wells were harvested and flow cytometry was used to determine output total B cell and Jo-1-binding ASBC frequency per well. Output frequency/input frequency was used to calculate estimated fold expansion for (B) total B cells or (C) Jo-1-binding ASBCs in each well. Individual wells are plotted. (D) The frequency of Jo-1-binding ASBCs ex vivo (purple symbol) or following in vitro stimulation (open symbols) is shown for each donor; individual wells are plotted. (E) Representative plots show flow cytometry identification of Jo-1-binding ASBCs by IgM and IgG isotypes (gated on Jo-1-GST+ CD19+ live singlet lymphocytes). (F) The isotype frequency of Jo-1-binding ASBCs following in vitro stimulation is shown for each donor; individual donors are plotted.
This approach could thus be applied to expand the original number of ASBCs to support downstream assays for which ASBC numbers are a limiting factor.
Discussion
Specific B cell subsets function differently to coordinate protective or autoimmune responses. Hybridoma technology advancements enable capture of ASBCs as rare as 1 in 20 million cells (29), but this approach does not capture B cell subset phenotypic data. We present data to show that flow cytometry-purified B cell subsets can be stimulated using this published protocol to additionally discern the phenotypic subset of ASBCs. Knowledge about which subset (e.g. memory) ASBCs derive from and how this might change with disease progression could enhance efforts to track ASBCs as sensitive disease biomarkers to predict increasing disease severity or flares, or alternatively as barometers for immunomodulatory therapy success.
Different autoimmune diseases may arise through distinct immune tolerance checkpoint breaches. B cell differentiation into an antibody-secreting cell may be governed differently for different ASBC specificities and across different diseases; for example, whereas Ro60 autoantibodies are typically sustained and Jo-1 autoantibody levels track with IIM disease severity, insulin autoantibodies are frequently transient (25, 47–49). To encompass this potential heterogeneity, B cell subset responses were surveyed from patients with several autoimmune diseases. We find that most of the B cell subsets tested secrete BCR as antibody using this stimulation protocol. Mature naïve, BND, CD27+ memory, IgM- IgD- CD27+ memory, IgM- IgD- CD27- memory B cell subsets isolated from at least 70% of patients responded to this stimulation protocol by secreting antibody. This study was underpowered to uncover specific functional differences present among B cells isolated from patients/participants with different autoimmune diseases. Additional studies will be required to uncover autoimmune disease-specific differences in B cell subset responses to this stimulation protocol.
Responses were more variable among IgM- CD21lo and IgM+ CD21lo B cell subsets; for each subset, roughly 50% of patients showed a response. Of note, an abnormal CD19+ IgM- IgD- CD21lo subset has been identified in systemic sclerosis and anti-histidyl tRNA synthetase syndrome, which expresses many innate immune cell markers (unpublished observations). If these cells are not of the B lineage, that would explain their inability to secrete antibody. Alternatively, if CD21lo B cells have already upregulated a plasmablast/plasma cell transcriptional program, as has been suggested in the setting of infection (43), they may respond less well to stimulation, as plasmablast responses were largely absent with this stimulation protocol. Given the known functional difference identified among the broad category of CD21lo B cells (31, 33, 43), our data highlight the need to test this method in specific autoimmune diseases in which aberrant CD21lo B cell populations have been identified.
The limited antibody secretion detected from plasmablasts following this in vitro stimulation points to a need to apply other approaches, such as single-cell BCR sequencing, to examine the antigen-specific plasmablast repertoire. Alternatively, this stimulation protocol could be modified to include factors essential for plasmablast/plasma cell survival and differentiation, such as IL-6, IL-10, IFN-α and APRIL (50, 51). Such modifications would need to be separately tested for their impact on other B cell subsets. Innate stimulation, such as through TLR7 and TLR9, is known to drive activation of autoreactive B cells in systemic lupus erythematosus (52, 53). Studies of murine anti-insulin B cells showed that LPS (TLR4), but not ssDNA (TLR7) or CpG (TLR9) stimulation, overrode immune tolerance checkpoints to drive anti-insulin B cell proliferation (54). The innate stimuli that can drive immune tolerance breach by human anti-insulin B cells have not been investigated. The data presented here confirm insulin and Jo-1-binding B cells are captured using this method; we also identify Ro-52 and Ro-60-binding B cells using this method (unpublished observations).
Purified B cells that were IgM+ or IgD+ underwent limited class-switching away from the IgM isotype following in vitro stimulation, suggesting isotype is largely preserved for non-class-switched cells. Co-expression of multiple IgH transcripts has been noted in memory B cells and is enhanced in patients with Sjögren’s syndrome (55). Thus, it is possible that the limited IgG class switching among memory-phenotype B cells may reflect transcriptional programming that occurred in vivo, rather than in response to stimulation in vitro. IgG+ B cells were detected among stimulated IgM-/IgD- B cell subsets, as expected, but many cells surveyed were negative for both IgM and IgG surface expression. IgM-/IgG- cells may be of the IgA isotype, which was not examined in this study. B cells terminally differentiated to the IgE isotype are captured by this stimulation protocol (29), but future studies are warranted to confirm whether IgA+ B cells can be assessed using this method.
The ease with which ASBCs can be studied depends on repertoire frequency, as well as the sensitivity of the technology employed. Jo-1-binding ASBCs were detected in 50% of Jo1+ anti-histidyl tRNA synthetase syndrome patients surveyed (36), whereas magnetic sorting enrichment of insulin-binding ASBCs was used to overcome their low frequencies in at-risk/type 1 diabetic donors (32). We took advantage of the relatively high frequency of Jo-1-binding B cells ex vivo to measure autoreactive ASBC expansion in this culture system. Jo-1-binding ASBCs expanded 32-fold on average, suggesting this approach might be coupled with other downstream assays in which expansion of ASBCs is necessary.
In this study, we identify insulin-binding ASBCs among CD27+ and CD27- memory subsets in donors at high risk for type 1 diabetes, whereas relatively fewer wells derived from IgM+ and/or IgD+ (presumably mature naïve) B cells from the same donors scored positive for the presence of anti-insulin ASBCs. Polyreactive B cells that bind several antigens, including insulin, have been identified in autoimmune donors among recent bone marrow emigrants and mature naïve B cells (56–58). These data suggest that if polyreactive B cells were responsible for the anti-insulin ASBCs identified, they should be skewed away from, not towards class-switched memory subsets. One limitation of this approach is that multiple B cell clones (and thus antibody specificities) are present in each well; this ELISA-based approach limits accurate assessment of polyreactivity of ASBCs, which are not assayed separately from non-ASBCs present in the same well. Parallel ELISA screening using excess unlabeled antigen competitor is thus the most reliable method for determining antigen specificity, which we applied in this study to identify wells containing anti-insulin ASBCs. Individual B cell receptor binding attributes must be assayed directly to confirm autoantigen specificity; as such, efforts are presently underway in our laboratory to interrogate B cell receptors isolated from ASBCs present in specific B cell subsets that were immortalized as hybridomas. We demonstrate this stimulation protocol can expand ASBCs, thus it may also be amenable to combination with downstream single-cell profiling that can combine BCR repertoire (BCRseq), transcriptional profiling (RNAseq), phenotypic profiling (CITEseq), and/or antigen-reactivity profiling (LIBRAseq) (59, 60). This stimulation protocol produces B cells with sufficient viability that they can be fused to myelomas as hybridoma lines (29). We presume it will also be amenable to combination with these single-cell techniques, but this will require confirmation in future studies. Figure 7 shows examples of downstream applications that could be coupled to B cell subset purification, stimulation, and high-throughput screening presented in this report, which include the development of human hybridoma cell lines to interrogate B cell receptor sequence and function.
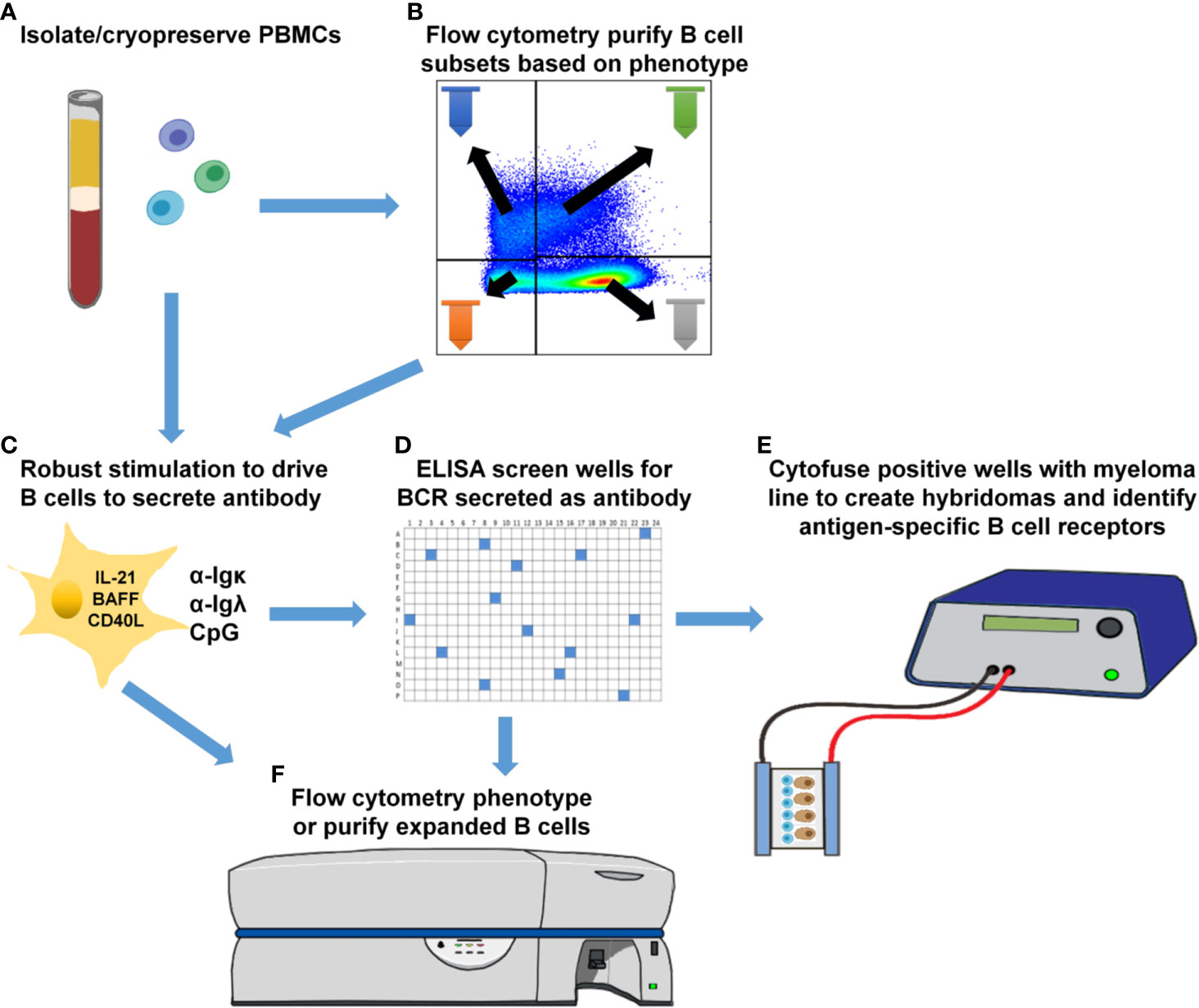
Figure 7 Downstream applications for methods to phenotypically-define, expand, and identify ASBCs. (A) Isolate and cryopreserve PBMCs. (B) Optionally purify specific B cell subsets using flow cytometry cell sorting. (C) Plate total PBMCs (panel A) or purified B cell subsets (panel B) into 384-well plates and stimulate as in Methods to drive B cell proliferation and differentiation into antibody-secreting cells. (D) High-throughput ELISA screening of well supernatants to identify wells containing ASBCs. (E) Cytofuse candidate wells to produce human hybridoma lines or (F) enrich for ASBCs prior to flow cytometry phenotyping.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Vanderbilt Institutional Review Board, Vanderbilt University Medical Center. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
BJ, JJ, AS, AC, and RB designed and performed experiments and analyzed resulting data. SS designed experiments and provided critical feedback. DM, LC, and EW recruited donors for these studies. BJ and RB wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
National Institutes of Health Grants NIH T32AR059039, and K12HD043483, Vanderbilt Institute for Clinical and Translational Research Pilot and Feasibility Grant VR53141 (supported by Vanderbilt NIH/CTSA UL1 RR024975), Vanderbilt NIH/CTSA UL1TR000445, Juvenile Diabetes Research Foundation Strategic Research Agreement 3-SRA-2019-790-S-B, Porter Family Fund for Autoimmunity Research, the Vanderbilt Medical Research Student Research Training Program in Diabetes and Obesity [supported by the Vanderbilt Short Term Research Training Program for Medical Students (NIH grant DK007383), Vanderbilt Research Training in Diabetes and Endocrinology (NIH grant T32DK007061), and the Vanderbilt Diabetes Research and Training Center (NIH grant DK20593)], and the Vanderbilt Human Immunology Discovery Initiative supported this work, along with the Vanderbilt Medical Center Flow Cytometry Shared Resource [supported by Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404)].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge the Vanderbilt Medical Center Flow Cytometry Shared Resource for performing flow cytometry sorting.
Abbreviations
ASBCs, Antigen-specific B cells; BCR, B cell receptor; IAA, insulin autoantibody; PBMCs, peripheral blood mononuclear cells.
References
1. Steck AK, Johnson K, Barriga KJ, Miao D, Yu L, Hutton JC, et al. Age of Islet Autoantibody Appearance and Mean Levels of Insulin, But Not GAD or IA-2 Autoantibodies, Predict Age of Diagnosis of Type 1 Diabetes: Diabetes Autoimmunity Study in the Young. Diabetes Care (2011) 34:1397–9. doi: 10.2337/dc10-2088
2. Yu L, Robles DT, Abiru N, Kaur P, Rewers M, Kelemen K, et al. Early Expression of Antiinsulin Autoantibodies of Humans and the NOD Mouse: Evidence for Early Determination of Subsequent Diabetes. Proc Natl Acad Sci USA (2000) 97:1701–6. doi: 10.1073/pnas.040556697
3. Sosenko JM, Skyler JS, Palmer JP, Krischer JP, Yu L, Mahon J, et al. The Prediction of Type 1 Diabetes by Multiple Autoantibody Levels and Their Incorporation Into an Autoantibody Risk Score in Relatives of Type 1 Diabetic Patients. Diabetes Care (2013) 36:2615–20. doi: 10.2337/dc13-0425
4. Shiboski CH, Shiboski SC, Seror R, Criswell LA, Labetoulle M, Lietman TM, et al. 2016 American College of Rheumatology/European League Against Rheumatism Classification Criteria for Primary Sjogren’s Syndrome: A Consensus and Data-Driven Methodology Involving Three International Patient Cohorts. Arthritis Rheumatol (2017) 69:35–45. doi: 10.1002/art.39859
5. Bohan A, Peter JB. Polymyositis and Dermatomyositis (First of Two Parts). N Engl J Med (1975) 292:344–7. doi: 10.1056/NEJM197502132920706
6. Bohan A, Peter JB. Polymyositis and Dermatomyositis (Second of Two Parts). N Engl J Med (1975) 292:403–7. doi: 10.1056/NEJM197502202920807
7. McHugh NJ, Tansley SL. Autoantibodies in Myositis. Nat Rev Rheumatol (2018) 14:290–302. doi: 10.1038/nrrheum.2018.56
8. Elkon K, Casali P. Nature and Functions of Autoantibodies. Nat Clin Pract Rheumatol (2008) 4:491–8. doi: 10.1038/ncprheum0895
9. Serreze DV, Fleming SA, Chapman HD, Richard SD, Leiter EH, Tisch RM. B Lymphocytes are Critical Antigen-Presenting Cells for the Initiation of T Cell-Mediated Autoimmune Diabetes in Nonobese Diabetic Mice. J Immunol (1998) 161:3912–8.
10. Silveira PA, Johnson E, Chapman HD, Bui T, Tisch RM, Serreze DV. The Preferential Ability of B Lymphocytes to Act as Diabetogenic APC in NOD Mice Depends on Expression of Self-Antigen-Specific Immunoglobulin Receptors. Eur J Immunol (2002) 32:3657–66. doi: 10.1002/1521-4141(200212)32:12<3657::AID-IMMU3657>3.0.CO;2-E
11. Noorchashm H, Lieu YK, Noorchashm N, Rostami SY, Greeley SA, Schlachterman A, et al. I-Ag7-mediated Antigen Presentation by B Lymphocytes is Critical in Overcoming a Checkpoint in T Cell Tolerance to Islet Beta Cells of Nonobese Diabetic Mice. J Immunol (1999) 163:743–50.
12. Serreze DV, Chapman HD, Varnum DS, Hanson MS, Reifsnyder PC, Richard SD, et al. B Lymphocytes are Essential for the Initiation of T Cell-Mediated Autoimmune Diabetes: Analysis of a New “Speed Congenic” Stock of NOD.Ig Mu Null Mice. J Exp Med (1996) 184:2049–53. doi: 10.1084/jem.184.5.2049
13. Marino E, Tan B, Binge L, Mackay CR, Grey ST. B-Cell Cross-Presentation of Autologous Antigen Precipitates Diabetes. Diabetes (2012) 61:2893–905. doi: 10.2337/db12-0006
14. Aggarwal R, Oddis CV, Goudeau D, Koontz D, Qi Z, Reed AM, et al. Autoantibody Levels in Myositis Patients Correlate With Clinical Response During B Cell Depletion With Rituximab. Rheumatol (Oxford) (2016) 55:991–9. doi: 10.1093/rheumatology/kev444
15. Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, et al. Efficacy of B-cell-targeted Therapy With Rituximab in Patients With Rheumatoid Arthritis. N Engl J Med (2004) 350:2572–81. doi: 10.1056/NEJMoa032534
16. Smith KG, Jones RB, Burns SM, Jayne DR. Long-Term Comparison of Rituximab Treatment for Refractory Systemic Lupus Erythematosus and Vasculitis: Remission, Relapse, and Re-Treatment. Arthritis Rheum (2006) 54:2970–82. doi: 10.1002/art.22046
17. Jordan S, Distler JH, Maurer B, Huscher D, van Laar JM, Allanore Y, et al. Effects and Safety of Rituximab in Systemic Sclerosis: An Analysis From the European Scleroderma Trial and Research (EUSTAR) Group. Ann Rheum Dis (2015) 74:1188–94. doi: 10.1136/annrheumdis-2013-204522
18. Franks SE, Getahun A, Hogarth PM, Cambier JC. Targeting B Cells in Treatment of Autoimmunity. Curr Opin Immunol (2016) 43:39–45. doi: 10.1016/j.coi.2016.09.003
19. Vital EM, Dass S, Buch MH, Henshaw K, Pease CT, Martin MF, et al. B Cell Biomarkers of Rituximab Responses in Systemic Lupus Erythematosus. Arthritis Rheum (2011) 63:3038–47. doi: 10.1002/art.30466
20. Fasano S, Gordon P, Hajji R, Loyo E, Isenberg DA. Rituximab in the Treatment of Inflammatory Myopathies: A Review. Rheumatol (Oxford) (2017) 56:26–36. doi: 10.1093/rheumatology/kew146
21. Pescovitz MD, Torgerson TR, Ochs HD, Ocheltree E, McGee P, Krause-Steinrauf H, et al. Effect of Rituximab on Human In Vivo Antibody Immune Responses. J Allergy Clin Immunol (2011) 128:1295–302. doi: 10.1016/j.jaci.2011.08.008
22. Henry RA, Kendall PL, Thomas JW. Autoantigen-Specific B Cell Depletion Overcomes Failed Immune Tolerance in Type 1 Diabetes. Diabetes (2012) 61:2037–44. doi: 10.2337/db11-1746
23. Manjarrez-Orduno N, Quach TD, Sanz I. B Cells and Immunological Tolerance. J Invest Dermatol (2009) 129:278–88. doi: 10.1038/jid.2008.240
24. Meffre E, O’Connor KC. Impaired B-cell Tolerance Checkpoints Promote the Development of Autoimmune Diseases and Pathogenic Autoantibodies. Immunol Rev (2019) 292:90–101. doi: 10.1111/imr.12821
25. Lindop R, Arentz G, Bastian I, Whyte AF, Thurgood LA, Chataway TK, et al. Long-Term Ro60 Humoral Autoimmunity in Primary Sjogren’s Syndrome is Maintained by Rapid Clonal Turnover. Clin Immunol (2013) 148:27–34. doi: 10.1016/j.clim.2013.03.015
26. Kendall PL, Case JB, Sullivan AM, Holderness JS, Wells KS, Liu E, et al. Tolerant Anti-Insulin B Cells are Effective Apcs. J Immunol (2013) 190:2519–26. doi: 10.4049/jimmunol.1202104
27. Acevedo-Suarez CA, Hulbert C, Woodward EJ, Thomas JW. Uncoupling of Anergy From Developmental Arrest in Anti-Insulin B Cells Supports the Development of Autoimmune Diabetes. J Immunol (2005) 174:827–33. doi: 10.4049/jimmunol.174.2.827
28. Felton JL, Maseda D, Bonami RH, Hulbert C, Thomas JW. Anti-Insulin B Cells Are Poised for Antigen Presentation in Type 1 Diabetes. J Immunol (2018) 201:861–73. doi: 10.4049/jimmunol.1701717
29. Wurth MA, Hadadianpour A, Horvath DJ, Daniel J, Bogdan O, Goleniewska K, et al. Human IgE mAbs Define Variability in Commercial Aspergillus Extract Allergen Composition. JCI Insight (2018) 3. doi: 10.1172/jci.insight.123387
30. Duty JA, Szodoray P, Zheng NY, Koelsch KA, Zhang Q, Swiatkowski M, et al. Functional Anergy in a Subpopulation of Naive B Cells From Healthy Humans That Express Autoreactive Immunoglobulin Receptors. J Exp Med (2009) 206:139–51. doi: 10.1084/jem.20080611
31. Isnardi I, Ng YS, Menard L, Meyers G, Saadoun D, Srdanovic I, et al. Complement Receptor 2/CD21- Human Naive B Cells Contain Mostly Autoreactive Unresponsive Clones. Blood (2010) 115:5026–36. doi: 10.1182/blood-2009-09-243071
32. Smith MJ, Packard TA, O’Neill SK, Henry Dunand CJ, Huang M, Fitzgerald-Miller L, et al. Loss of Anergic B Cells in Prediabetic and New-Onset Type 1 Diabetic Patients. Diabetes (2015) 64:1703–12. doi: 10.2337/db13-1798
33. Saadoun D, Terrier B, Bannock J, Vazquez T, Massad C, Kang I, et al. Expansion of Autoreactive Unresponsive CD21-/Low B Cells in Sjogren’s Syndrome-Associated Lymphoproliferation. Arthritis Rheum (2013) 65:1085–96. doi: 10.1002/art.37828
34. Wehr C, Eibel H, Masilamani M, Illges H, Schlesier M, Peter HH, et al. A New CD21low B Cell Population in the Peripheral Blood of Patients With SLE. Clin Immunol (2004) 113:161–71. doi: 10.1016/j.clim.2004.05.010
35. Forestier A, Guerrier T, Jouvray M, Giovannelli J, Lefevre G, Sobanski V, et al. Altered B Lymphocyte Homeostasis and Functions in Systemic Sclerosis. Autoimmun Rev (2018) 17:244–55. doi: 10.1016/j.autrev.2017.10.015
36. Young-Glazer J, Cisneros A 3rd, Wilfong EM, Smith SA, Crofford LJ, Bonami RH. Jo-1 Autoantigen-Specific B Cells are Skewed Towards Distinct Functional B Cell Subsets in Anti-Synthetase Syndrome Patients. Arthritis Res Ther (2021) 23:33. doi: 10.1186/s13075-020-02412-8
37. Type 1 Diabetes Trialnet (2021). Available at: https://www.trialnet.org/.
38. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research Electronic Data Capture (Redcap)–a Metadata-Driven Methodology and Workflow Process for Providing Translational Research Informatics Support. J BioMed Inform (2009) 42:377–81. doi: 10.1016/j.jbi.2008.08.010
39. Chou C, Verbaro DJ, Tonc E, Holmgren M, Cella M, Colonna M, et al. The Transcription Factor AP4 Mediates Resolution of Chronic Viral Infection Through Amplification of Germinal Center B Cell Responses. Immunity (2016) 45:570–82. doi: 10.1016/j.immuni.2016.07.023
40. Nojima T, Haniuda K, Moutai T, Matsudaira M, Mizokawa S, Shiratori I, et al. In-Vitro Derived Germinal Centre B Cells Differentially Generate Memory B or Plasma Cells In Vivo. Nat Commun (2011) 2:465. doi: 10.1038/ncomms1475
41. Wilfong EM, Bartkowiak T, Vowell KN, Westlake CS, Irish JM, Kendall PL, et al. High-Dimensional Analysis Reveals Abnormal B Cell Subsets Associated With Specific Changes to Circulating T and Myeloid Cell Populations in Patients With Idiopathic Inflammatory Myopathies. medRxiv (2021) 2021:03. doi: 10.1101/2021.03.23.21253635
42. Glauzy S, Boccitto M, Bannock JM, Delmotte FR, Saadoun D, Cacoub P, et al. Accumulation of Antigen-Driven Lymphoproliferations in Complement Receptor 2/CD21(-/Low) B Cells From Patients With Sjogren’s Syndrome. Arthritis Rheumatol (2018) 70:298–307. doi: 10.1002/art.40352
43. Lau D, Lan LY, Andrews SF, Henry C, Rojas KT, Neu KE, et al. Low CD21 Expression Defines a Population of Recent Germinal Center Graduates Primed for Plasma Cell Differentiation. Sci Immunol (2017) 2. doi: 10.1126/sciimmunol.aai8153
44. Townsend MJ, Monroe JG, Chan AC. B-Cell Targeted Therapies in Human Autoimmune Diseases: An Updated Perspective. Immunol Rev (2010) 237:264–83. doi: 10.1111/j.1600-065X.2010.00945.x
45. Fecteau JF, Cote G, Neron S. A New Memory CD27-Igg+ B Cell Population in Peripheral Blood Expressing VH Genes With Low Frequency of Somatic Mutation. J Immunol (2006) 177:3728–36. doi: 10.4049/jimmunol.177.6.3728
46. Wei C, Anolik J, Cappione A, Zheng B, Pugh-Bernard A, Brooks J, et al. A New Population of Cells Lacking Expression of CD27 Represents a Notable Component of the B Cell Memory Compartment in Systemic Lupus Erythematosus. J Immunol (2007) 178:6624–33. doi: 10.4049/jimmunol.178.10.6624
47. Krystufkova O, Hulejova H, Mann HF, Pecha O, Putova I, Ekholm L, et al. Serum Levels of B-cell Activating Factor of the TNF Family (BAFF) Correlate With anti-Jo-1 Autoantibodies Levels and Disease Activity in Patients With anti-Jo-1positive Polymyositis and Dermatomyositis. Arthritis Res Ther (2018) 20:158. doi: 10.1186/s13075-018-1650-8
48. Barker JM, Barriga KJ, Yu L, Miao D, Erlich HA, Norris JM, et al. Prediction of Autoantibody Positivity and Progression to Type 1 Diabetes: Diabetes Autoimmunity Study in the Young (Daisy). J Clin Endocrinol Metab (2004) 89:3896–902. doi: 10.1210/jc.2003-031887
49. Kulmala P, Rahko J, Savola K, Vahasalo P, Veijola R, Sjoroos M, et al. Stability of Autoantibodies and Their Relation to Genetic and Metabolic Markers of Type I Diabetes in Initially Unaffected Schoolchildren. Diabetologia (2000) 43:457–64. doi: 10.1007/s001250051329
50. Cocco M, Stephenson S, Care MA, Newton D, Barnes NA, Davison A, et al. In Vitro Generation of Long-Lived Human Plasma Cells. J Immunol (2012) 189:5773–85. doi: 10.4049/jimmunol.1103720
51. Jourdan M, de Boussac H, Viziteu E, Kassambara A, Moreaux J. In Vitro Differentiation Model of Human Normal Memory B Cells to Long-lived Plasma Cells. J Vis Exp (2019) (143). doi: 10.3791/58929
52. Soni C, Wong EB, Domeier PP, Khan TN, Satoh T, Akira S, et al. B Cell-Intrinsic TLR7 Signaling is Essential for the Development of Spontaneous Germinal Centers. J Immunol (2014) 193:4400–14. doi: 10.4049/jimmunol.1401720
53. Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-Like Receptor 9 Controls anti-DNA Autoantibody Production in Murine Lupus. J Exp Med (2005) 202:321–31. doi: 10.1084/jem.20050338
54. Williams JM, Bonami RH, Hulbert C, Thomas JW. Reversing Tolerance in Isotype Switch-Competent Anti-Insulin B Lymphocytes. J Immunol (2015) 195:853–64. doi: 10.4049/jimmunol.1403114
55. Hansen A, Gosemann M, Pruss A, Reiter K, Ruzickova S, Lipsky PE, et al. Abnormalities in Peripheral B Cell Memory of Patients With Primary Sjogren’s Syndrome. Arthritis Rheum (2004) 50:1897–908. doi: 10.1002/art.20276
56. Samuels J, Ng YS, Coupillaud C, Paget D, Meffre E. Impaired Early B Cell Tolerance in Patients With Rheumatoid Arthritis. J Exp Med (2005) 201:1659–67. doi: 10.1084/jem.20042321
57. Meffre E, Schaefer A, Wardemann H, Wilson P, Davis E, Nussenzweig MC. Surrogate Light Chain Expressing Human Peripheral B Cells Produce Self-Reactive Antibodies. J Exp Med (2004) 199:145–50. doi: 10.1084/jem.20031550
58. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant Autoantibody Production by Early Human B Cell Precursors. Science (2003) 301:1374–7. doi: 10.1126/science.1086907
59. Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al. Simultaneous Epitope and Transcriptome Measurement in Single Cells. Nat Methods (2017) 14:865–8. doi: 10.1038/nmeth.4380
Keywords: B cells, B cell receptor (BCR), autoimmune disease, autoantigen, myositis, Sjogren’s syndrome, systemic sclerosis (scleroderma), type 1 diabetes
Citation: Joosse BA, Jackson JH, Cisneros A 3rd, Santhin AB, Smith SA, Moore DJ, Crofford LJ, Wilfong EM and Bonami RH (2021) High-Throughput Detection of Autoantigen-Specific B Cells Among Distinct Functional Subsets in Autoimmune Donors. Front. Immunol. 12:685718. doi: 10.3389/fimmu.2021.685718
Received: 25 March 2021; Accepted: 06 May 2021;
Published: 24 May 2021.
Edited by:
Zhenming Xu, The University of Texas Health Science Center at San Antonio, United StatesReviewed by:
Hong Zan, The University of Texas Health Science Center at San Antonio, United StatesZiaur S. M. Rahman, Penn State Milton S. Hershey Medical Center, United States
Copyright © 2021 Joosse, Jackson, Cisneros, Santhin, Smith, Moore, Crofford, Wilfong and Bonami. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rachel H. Bonami, cmFjaGVsLmguYm9uYW1pQHZ1bWMub3Jn