 Jun Miyata
Jun Miyata Yoshiyuki Yokokura4
Yoshiyuki Yokokura4 Makoto Arita
Makoto Arita- 1Laboratory of Metabolomics, RIKEN Center for Integrative Medical Sciences, Yokohama, Japan
- 2Division of Pulmonary Medicine, Department of Medicine, Keio University School of Medicine, Tokyo, Japan
- 3Division of Infectious Diseases and Respiratory Medicine, Department of Internal Medicine, National Defense Medical College, Tokorozawa, Japan
- 4Graduate School of Pharmaceutical Sciences, The University of Tokyo, Tokyo, Japan
- 5Laboratory for Innate Immune Systems, RIKEN Center for Integrative Medical Sciences (IMS), Yokohama, Japan
- 6Laboratory for Innate Immune Systems, Department of Microbiology and Immunology, Graduate School of Medicine, Osaka University, Osaka, Japan
- 7Laboratory for Innate Immune Systems, Immunology Frontier Research Center, Osaka University, Osaka, Japan
- 8Division of Physiological Chemistry and Metabolism, Keio University Faculty of Pharmacy, Tokyo, Japan
- 9Cellular and Molecular Epigenetics Laboratory, Graduate School of Medical Life Science, Yokohama City University, Yokohama, Japan
Dysregulated fatty acid metabolism is clinically associated with eosinophilic allergic diseases, including severe asthma and chronic rhinosinusitis. This study aimed to demonstrate the role of 12/15-lipoxygenase (12/15-LOX) in interleukin (IL)-33-induced eosinophilic airway inflammation; to this end, we used 12/15-LOX-deficient mice, which displayed augmented IL-33-induced lung inflammation, characterized by an increased number of infiltrated eosinophils and group 2 innate lymphoid cells (ILC2s) in the airway. Liquid chromatography-tandem mass spectrometry (LC-MS/MS)-based lipidomics revealed that the levels of a series of 12/15-LOX-derived metabolites were significantly decreased, and application of 14(S)-hydroxy docosahexaenoic acid (HDoHE), a major 12/15-LOX-derived product, suppressed IL-33-mediated eosinophilic inflammation in 12/15-LOX-deficient mice. Using bioactive lipid screening, we found that 14(S)-HDoHE and 10(S),17(S)-diHDoHE markedly attenuated ILC2 proliferation and cytokine production at micromolar concentration in vitro. In addition, maresin 1 (MaR1) and resolvin D1 (RvD1), 12/15-LOX-derived specialized proresolving mediators (SPMs), inhibited cytokine production of ILC2s at nanomolar concentration. These findings demonstrate the protective role of endogenous 12/15-LOX-derived lipid mediators in controlling ILC2-mediated eosinophilic airway inflammation and related diseases. Thus, 12/15-LOX-derived lipid mediators may represent a potential therapeutic strategy for ameliorating airway inflammation-associated conditions.
Introduction
Asthma is a common disease affecting more than 300 million people worldwide, and the number of patients with asthma is rapidly increasing (1, 2). Genetic and environmental factors induce diverse immune responses that are classified into atopic and non-atopic phenotypes, mainly characterized by eosinophilic airway inflammation. Severe asthma is characterized by resistance to standardized treatments, including corticosteroids, and frequent exacerbation, which could worsen the quality of life in these patients. However, the exact mechanism underlying severe asthma has not been fully elucidated. Thus, it is necessary to elucidate its pathophysiological process.
Dysregulated metabolism of polyunsaturated fatty acids [arachidonic acid (AA), eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA)] is observed in allergic diseases, including severe asthma and its related diseases, such as aspirin-exacerbated respiratory disease and eosinophilic chronic rhinosinusitis (3, 4). This abnormality is partly characterized by impaired synthesis of specialized proresolving mediators (SPMs; lipoxins (LXs; LXA4 and LXB4), protectins (PD1 and PDX), resolvin D series (RvDs; RvD1-6), and maresins (MaRs; MaR1-2), which promote the resolution of inflammation. SPMs inhibit the migration of polymorphonuclear cells to inflammatory sites and enhance the phagocytic activity of apoptotic cells via macrophages (5, 6). These mediators are mainly biosynthesized via 15-lipoxygenase (LOX) in humans and 12/15-LOX, an ortholog of 15-LOX in mice, that are highly expressed in eosinophils and specific types of macrophages (7–10). Previously, we observed downregulated biosynthesis of 15-LOX-derived SPMs in eosinophils isolated from patients with severe asthma and eosinophilic chronic rhinosinusitis (7, 11–13). Systemic administration of SPMs suppressed pulmonary eosinophilic inflammation in murine models of asthma (14–18). These findings suggest the regulatory roles of 15-LOX and 12/15-LOX in eosinophilic inflammation in the lungs, although the causal relationships between these enzymes and severe allergic diseases remain unclear.
Interleukin (IL)-33 is an IL-1 family cytokine with potent inflammatory properties (19). The gene encoding ST2, an IL-33 receptor, is closely related to asthma susceptibility (20, 21). Group 2 innate lymphoid cells (ILC2s) are potent producers of type 2 cytokines, including IL-5 and IL-13, in response to IL-33 (22–24). Other cell types, including eosinophils, basophils, mast cells, and dendritic cells also respond to IL-33, showing proinflammatory reactions (25–35). IL-33 is highly expressed in airway mucosa or nasal polyps isolated from patients with asthma, and its expression level is well correlated with disease severity (36–39). Therefore, IL-33 is considered a pivotal regulator and potential therapeutic target in respiratory diseases with type-2 airway inflammation, including severe asthma.
In the present study, we investigated the regulatory roles of 12/15-LOX-derived lipid mediators in IL-33-induced eosinophilic airway inflammation in mice. Lipidomic analysis of inflamed lung tissue and in vitro lipid screening analysis using ILC2s were performed to demonstrate the roles of 12/15-LOX.
Materials and Methods
Mouse Experiments
Specific pathogen- and virus-antibody-free, 6-8-week-old, male C57BL/6J (C57BL/6) mice, weighing 25-30 g, were purchased from Charles River Laboratories, Japan. 12/15-LOX-deficient mice were obtained from the Jackson Laboratory (002778, Bar Harbor, ME, USA). All animals were housed at the facility in bubble barrier units (bioBubble, Fort Collins, Colo., USA) under positive pressure. The experimental protocol was reviewed and approved by the Laboratory Animal Care and Use Committee of Keio University of Medicine, the Animal Committee of the University of Tokyo, and the Animal Care and Use Committee of the RIKEN.
Administration of Reagents In Vivo
We administered 40 μL of PBS or IL-33 (R&D, 500 ng per mouse) via intranasal administration under anesthesia with intraperitoneal administration of ketamine (100 mg/kg) and xylazine (10 mg/kg). In some experiments, we simultaneously administered 5 μg/day of 14(S)-HDoHE (Cayman Chemical, Ann Arbor, MI, USA) via intraperitoneal injection.
Establishment of IL-33-Induced Airway Inflammation
We administered IL-33 (R&D Systems, Minneapolis, MN, 500 ng per mouse) for 3 consecutive days and analyzed them 1 or 4 days after the last challenge as previously described (40) with technical modification. Bronchoalveolar lavage fluid (BALF) was collected for cell counts and flow cytometric analysis, and lung tissue for the measurement of mRNA expression and histopathologic analysis. Formalin-fixed paraffin-embedded lung slides were stained with hematoxylin and eosin (HE) or periodic acid Schiff (PAS).
Collection of BALF
The mice were sacrificed by an overdose of intravenous pentobarbital at the indicated times after the last challenge. The trachea was cannulated, and the lungs were lavaged by washing twice with 0.7 mL of ice-cold PBS with EDTA (0.6 mM). The total number of cells in BALF was counted using a hemocytometer, and a differential cell count of 200 cells was determined on Diff-Quik-stained cytospin slides (Baxter Scientific Products, McGraw Park, Ill., USA) prepared with Auto Smear CF12D (Sakura Finetek, Tokyo, Japan). Flow cytometric analysis was performed for cell counts of specific types of lymphocytes (ILC2s and Th2 cells).
Targeted Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS)-Based Lipidomics
LC-MS/MS-based mediator lipidomics was performed as previously described (41). Lung tissues were homogenized in ice-cold methanol and kept in -20°C overnight. The methanolic extract was then diluted with water, acidified with HCl to a pH of 3.5, and applied to Sep-Pak C18 cartridges (Waters) for solid phase extraction. Deuterated internal standards (1 ng of leukotriene (LT) B4-d4, LTD4-d5, prostaglandin (PG) E2-d4, and 15-HETE-d8 (Cayman Chemical, Ann Arbor, MI, USA) were added to the supernatants prior to extraction. For LC-MS/MS analysis, a triple quadrupole linear ion trap mass spectrometer (QTRAP 5500; AB Sciex, Foster City, CA) equipped with a 1.7-μm, 1.0 × 150 mm Acquity UPLC™ BEH C18 column (Waters Corp., Milford, MA) was used. MS/MS analyses were conducted in negative ion mode, and the eicosanoids and docosanoids were identified and quantified by multiple reaction monitoring. Calibration curves were obtained over a range of 1–1,000 pg. The LC retention times for each compound were determined using the corresponding synthetic standards. PD1 and PDX, stereoisomers, were not separable under this LC-MS/MS setting.
Functional Assays of ILC2s In Vitro
Mouse ILC2s were isolated from mesentery using a previously reported method (42). Purified ILC2s were cultured in 96-well round-bottom plates in 200 μL RPMI-1640 media containing 10% FCS, HEPES buffer, non-essential amino acids, penicillin, streptomycin, and 2-mercaptoethanol in the presence of IL-2 (10 ng/mL) at 37°C. To comprehensively evaluate the effect of lipid metabolites, cultured ILC2s were seeded at a density of 10,000 cells per well into 96-well round-bottom plates in the absence of IL-2, and IL-33 was added to the culture medium at a final concentration of 10 ng/mL after pretreatment for 30 min with lipid metabolites (10-11 M – 10-5 M). One hundred microliters of supernatant were collected for the cytokine assay, and ILC2s were counted by flow cytometry on day 4. For apoptosis analysis, ILC2s were stained with Annexin V and propidium iodide (PI) according to the manufacturer’s protocols (Apoptosis Detection Kit, BD Pharmingen), and then analyzed by flow cytometry on day 1 and day 4. All data were analyzed using FlowJo software (TreeStar, Ashland, OR, USA).
Statistical Analysis
Data are presented as the mean ± SEM. Dose-response relationships of lipid metabolites on ILC2 activities were analyzed with repeated measures of analysis of variance, followed by the Bonferroni/Dunn procedure as a post hoc test. Data were analyzed using GraphPad Prism version 4.0c (GraphPad Software, San Diego, CA). Statistical significance was set at P < 0.05.
Results
12/15-LOX Deficiency Augmented IL-33-Induced Airway Eosinophilic Inflammation
To determine whether 12/15-LOX affects the disease onset and/or progression of airway eosinophilic inflammation, we used mice deficient in the gene encoding 12/15-LOX (alox15) in a murine model of IL-33-induced innate airway eosinophilic inflammation. 12/15-LOX-deficient (12/15-LOX-/-) mice developed more severe airway inflammation associated with an increased number of eosinophils and lymphocytes in BALF compared to controls (Figure 1A). Lymphocyte subset analysis revealed a significant increase in the number of ILC2s and Th2 cells (CD4+ST2+ cells) in BALF from IL-33-challenged 12/15-LOX-/- mice compared to wild-type mice (Figure 1B). Importantly, the number of ILC2s was greater than that of Th2 cells (Figure 1B). Expression of mRNA levels of type 2 cytokines (il5, il13) and chemokines for eosinophils (ccl11, ccl24, and ccl26), which are critical inducers of eosinophilic inflammation, in the lungs was significantly higher in IL-33-treated 12/15-LOX-/- mice than in wild-type mice (Figure 1C). Histological analysis demonstrated prominent accumulation of inflammatory cells around the bronchus and increased mucus production in the lung tissue stained with HE and PAS, respectively (Figures 1D, E). In addition, the absence of 12/15-LOX had no impact on the number of cells and lymphocyte subset including Th2 cells and ILC2s in BALF at the steady state (Supplementary Figures 1A, B).
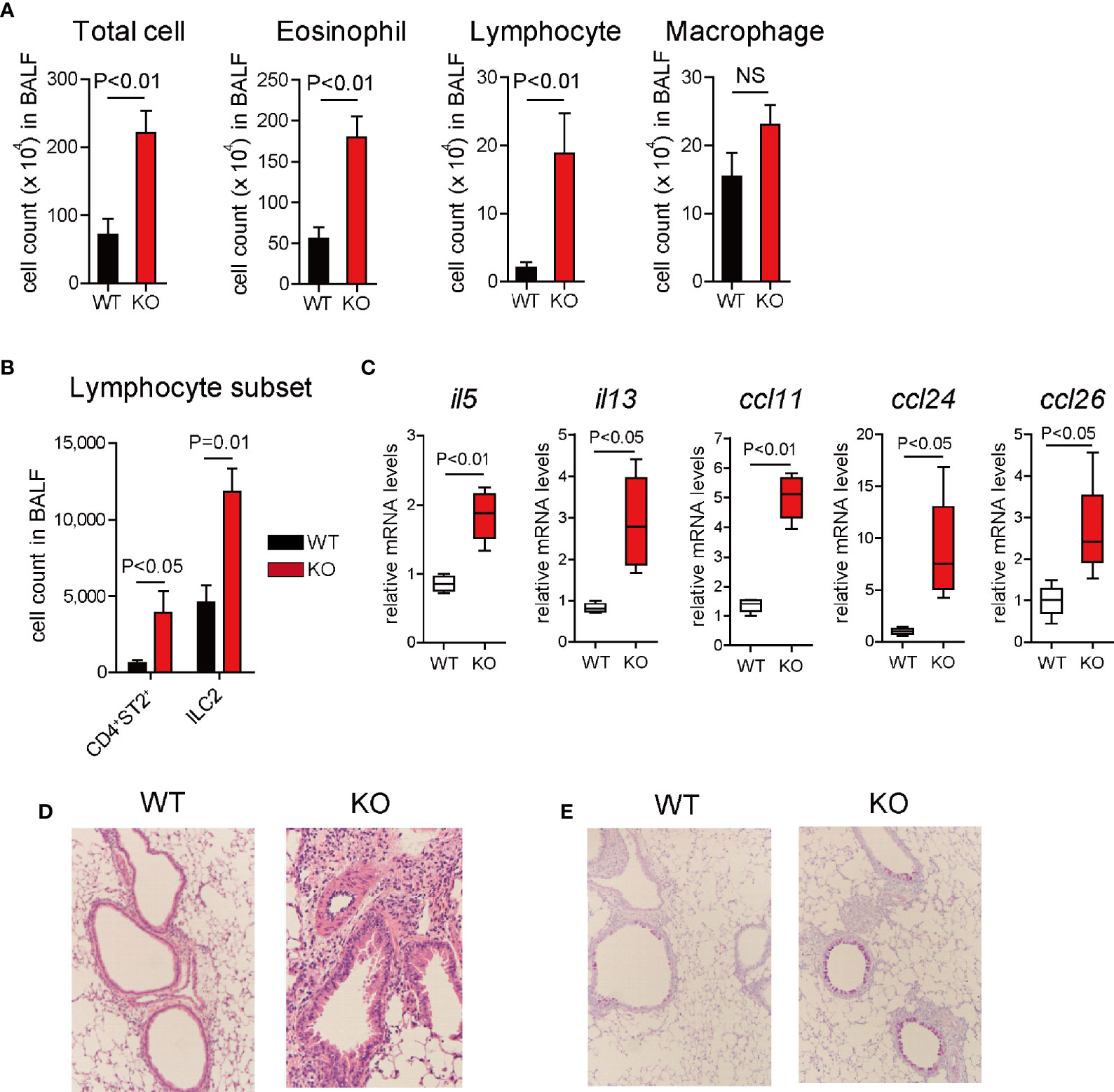
Figure 1 12/15-lipoxygenase deficiency augments IL-33-induced airway eosinophilic inflammation. Airway inflammation was induced by administration of IL-33 (500 ng per mouse, intranasal), for 3 consecutive days, in C57BL/6 and 12/15-lipoxygenase deficient mice. Analysis was performed 4 days after the final administration of IL-33. (A) Number of total cells, eosinophils, lymphocytes, and macrophages in BALF. (B) Number of ILC2 and Th2 cells in BALF by flow cytometric analysis. (C) Relative mRNA expression of cytokines (il5, il13, ccl11, ccl24, and ccl26) was determined by RT-PCR and quantitative real-time PCR analysis. Airway histology was assessed by (D) hematoxylin and eosin and (E) periodic acid–Schiff (PAS)-Alcan blue staining. The data are representative of three independent experiments. Mean ± SEM, n=4 for each group.
Lipidomic Profile of Lung Tissue During IL-33-Induced Airway Eosinophilic Inflammation
To investigate the lipid mediator profiles in the lungs during IL-33-induced eosinophilic pulmonary inflammation, LC-MS/MS-based mediator lipidomics was performed. The amounts of lipid mediators on days 1 and 4 following the last challenge are summarized in Supplementary Table 1. COX-derived products such as PGE2, PGD2, and PGF2α, thromboxane B2, and 12-HHT were abundantly produced in the inflamed lung tissue on day 1, and these amounts were almost the same between wild-type and 12/15-LOX-/- mice (Figure 2A). Among the LOX-derived products, 14-HDoHE, 17-HDoHE, and 12-hydroxyeicosatetraenoic acid (HETE) were generated at substantial levels on day 1. Additionally, their biosynthesis was dependent on 12/15-LOX (Figure 2A). Among the monohydroxylated forms of DHA (4-, 7-, 10-, 13-, 14-, 17-, 20-, and 21-HDoHE), the amount of 14-HDoHE was the highest on day 1 (Figure 2B). The amounts of fatty acids, precursors of lipid mediators (AA, EPA, and DHA), in the lungs did not differ between wild-type and 12/15-LOX-/- mice on day 1 (Figure 2C). Among the downstream lipid mediators (AA-, EPA-, and DHA-derived dihydroxy- or trihydroxy-fatty acids), PD1/PDX, RvD2, RvD5, and RvE3 were also present in a 12/15-LOX-dependent manner on day 1 (Figure 2D). PD1/PDX was relatively abundant compared to other mediators.
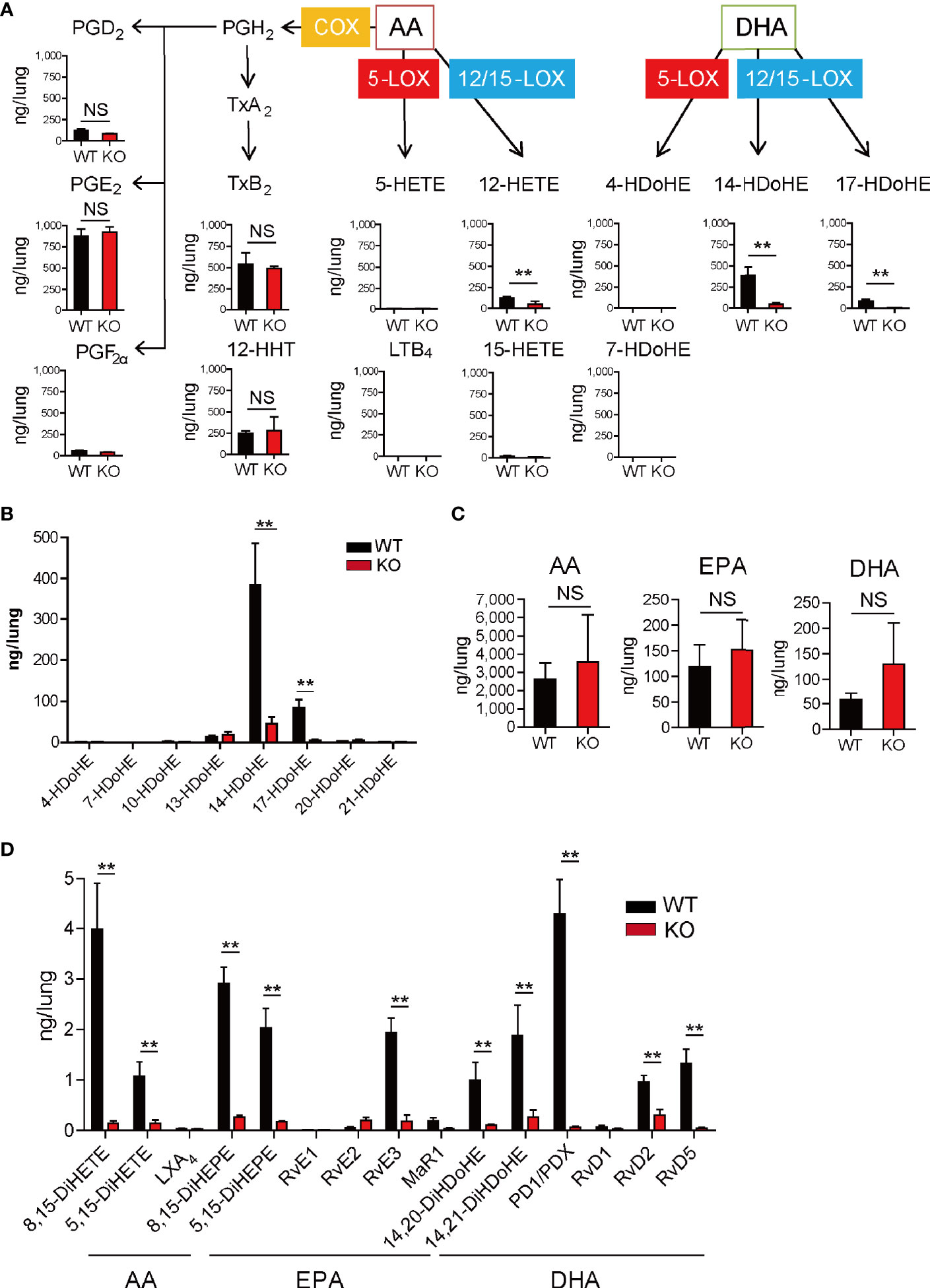
Figure 2 Lipidomic profiles of lungs during IL-33-induced airway eosinophilic inflammation. Airway inflammation was induced by administration of IL-33 (500 ng per mouse, intranasal), for 3 consecutive days, in C57BL/6 and 12/15-lipoxygenase (LOX)-deficient mice. Analysis was performed 1 day after the final administration of IL-33. (A) Lipidomic analysis showed quantitative alterations of arachidonic acid (AA) and docosahexaenoic acid (DHA)-derived metabolites via cyclooxygenase (COX), 5-LOX, and 12/15-LOX, including prostaglandins (PG), thromboxanes (Tx), 12- hydroxyheptadecatrienoic acid (HHT), hydroxyeicosatetraenoic acid (HETE), leukotriene B4 (LTB4), and hydroxy docosahexaenoic acid (HDoHE). (B) Comparative analysis of DHA-derived monohydroxy metabolites in wild-type or 12/15-LOX deficient mice. (C) Comparative analysis of the amounts of polyunsaturated fatty acids [AA, DHA, and eicosapentaenoic acid (EPA)] in wild-type or 12/15-LOX deficient mice. (D) Comparative analysis of 12/15-LOX-derived dihydroxy- or trihydroxy-lipid metabolites, including lipoxin (LX), Rv, MR, and protectin (PD). Mean ± SEM, n=3 for each group. **P < 0.01 (Student’s t-test). NS, not significant.
14(S)-HDoHE and 10(S),17(S)-diHDoHE Suppress ILC2 Cell Activation In Vitro
To further understand the molecular mechanism of 12/15-LOX in the regulation of innate eosinophilic pulmonary inflammation, we performed lipid screening using AA-, EPA-, and DHA-derived metabolites. Their suppressive effects on the proliferation of ILC2 cells in vitro in response to IL-33 were investigated by flow cytometry. The lipid screening assay demonstrated that 14(S)-HDoHE was a potent suppressor of cellular proliferation of ILC2s among DHA-derived monohydroxy metabolites (4-, 7-, 10-, 13-, 14-, 17-, 20-, and 21-HDoHE) (Figure 3A). In addition, among DHA-derived SPMs, including RvDs, PDs, MaRs, and other AA- or EPA-derived dihydroxy or trihydroxy metabolites, 10(S),17(S)-diHDoHE (PDX) most potently suppressed the proliferation of ILC2 cells (Figure 4A).
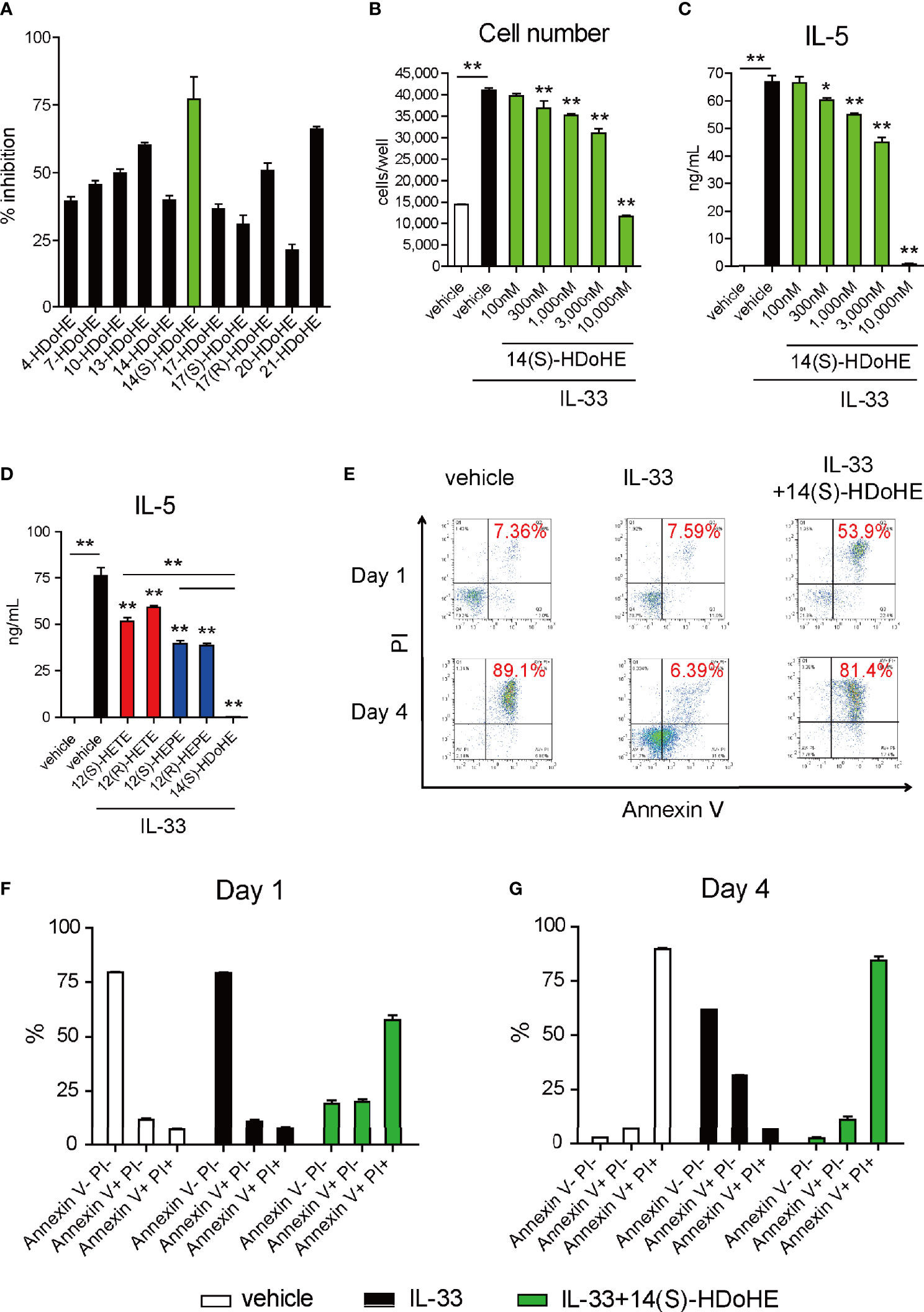
Figure 3 14(S)-HDoHE inhibit proliferation and cytokine production of ILC2s, with pro-apoptotic effects. ILC2 cells (10,000 cells per well) were cultured with IL-33 (10 ng/mL) for 1 or 4 days in the presence or absence of (A) DHA-derived monohydroxy-lipid metabolites (HDoHE, hydroxy docosahexaenoic acid: 10-5 M) or (B–G) in the presence or absence of 14(S)-HDoHE (3 × 10-7 – 10-5 M: (B, C); or 10-5 M: (D–G), (D) 12-hydroxyeicosatetraenoic acid (HETE) and 12-hydroxyeicosapentaenoic acid (HEPE). The total cell count (A, B) and concentrations of IL-5 (C, D) in the culture supernatant were measured. (E–G) Flow cytometric analysis of Annexin V- and PI-stained ILC2 cells cultured with or without IL-33 (10 ng/mL) in the presence or absence of 14(S)-HDoHE (10-5 M) for 1 or 4 days. The data are representative of three independent experiments. Mean ± SEM, n=3-8 for each group. *P < 0.05, **P < 0.01 (Student’s t-test).
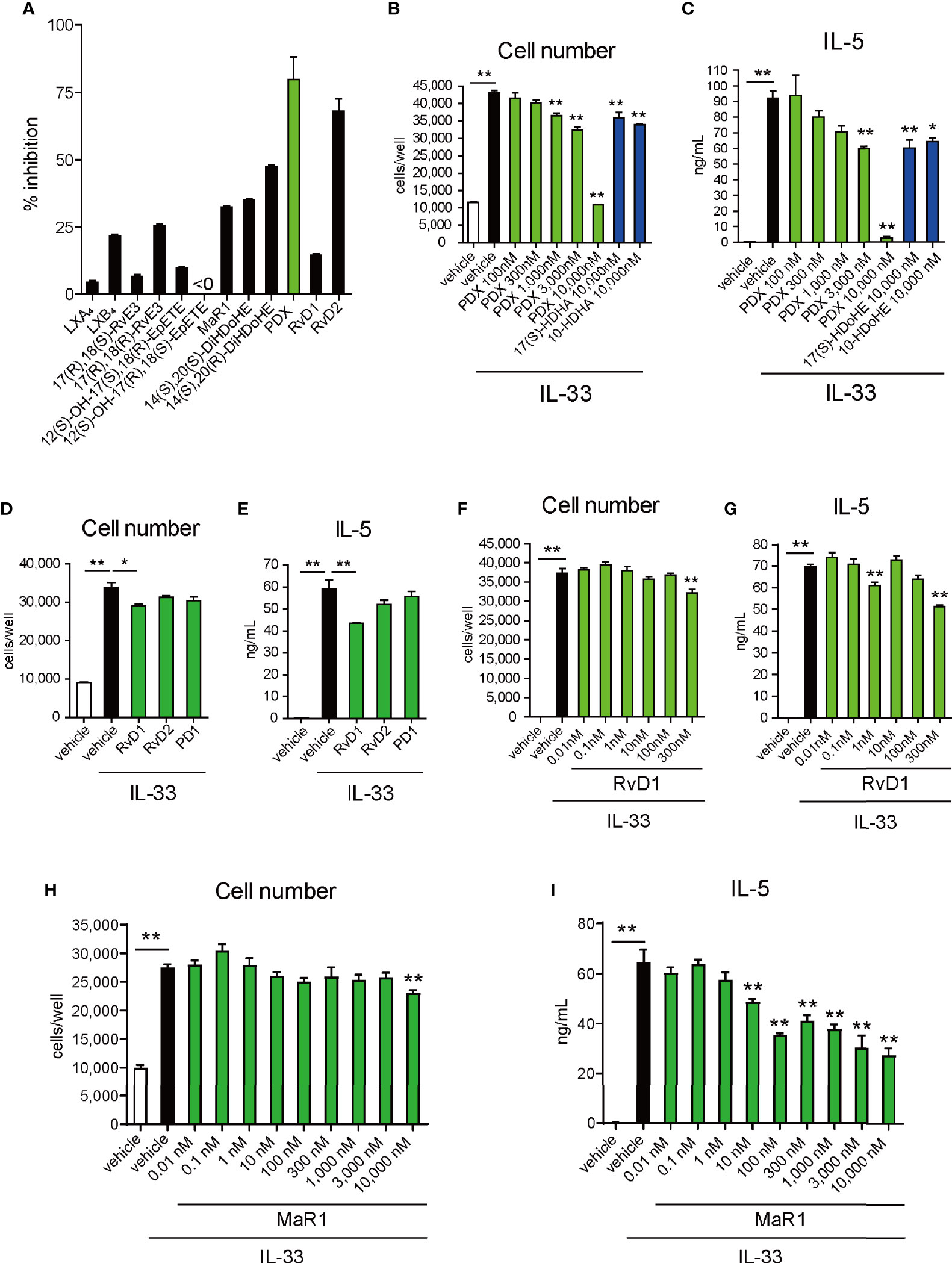
Figure 4 Effects of SPMs on proliferation and cytokine production of ILC2s. (A) ILC2 cells (10,000 cells per well) were cultured with IL-33 (10 ng/mL) for 4 days in the presence or absence of (A) dihydroxy- or trihydroxy-lipid metabolites (LX, lipoxin; Rv, resolvin; MR, maresin; PD, protectin D: 10-5 M) or in the presence or absence of (B, C) 10(S),17(S)-diHDoHE (PDX, 3 × 10-7 – 10-5 M), (D, E) 17(S)-HDoHE-derived SPMs (RvD1, resolvin D1; RvD2, resolvin D2; PD1, protectin D1: 3 x 10-7 nM), (F, G) RvD1 (1 × 10-11 – 3 x 10-7 M: (D, E), or (H, I) MaR1 (MaR1; maresin 1: 10-11 – 10-5 M). The total cell count (A, B, D, F, H) and concentrations of IL-5 (C, E, G, I) in the culture supernatant were measured. Data are shown as Mean ± SEM, n = 3 for each group. *P < 0.05, **P < 0.01 (Student’s t-test).
14(S)-HDoHE suppressed ILC2 cell proliferation in a concentration-dependent manner (Figure 3B). In addition, cytokine release (IL-5) from ILC2 cells was suppressed by 14(S)-HDoHE in a concentration-dependent manner (Figure 3C). The inhibitory effect of 14(S)-HDoHE on IL-5 production was more potent than other related structural isomers such as 12(S)-HETE, 12(R)-HETE, 12(S)-HEPE, and 12(R)-HEPE (Figure 3D). Flow cytometric analysis of Annexin V- and PI-stained ILC2 cells upon stimulation with IL-33 revealed the pro-apoptotic effects of 14(S)-HDoHE on ILC2s after stimulation with IL-33 for 1 or 4 days (Figures 3E–G). PDX similarly inhibited ILC2 proliferation and IL-5 production in a concentration-dependent manner. PDX displayed more potent suppressive effect than its related metabolites including 17(S)-HDoHE and 10(S/R)-HDoHE (Figures 4B, C). The suppressive effects of PD1, RvD1, and RvD2, were also evaluated at nanomolar concentrations. Unlike the others, RvD1 slightly inhibited the proliferation and IL-5 production of ILC2s (Figures 4D–G). In addition, the suppressive effects of MaR1 were evaluated at nanomolar to micromolar concentrations. MaR1 inhibited IL-5 production, not cell proliferation, of ILC2s at nanomolar concentrations (Figures 4H, I). These results suggest that the DHA-derived 12/15-LOX metabolites collectively regulates ILC2 functions to control innate type-2 airway inflammation.
14(S)-HDoHE Suppressed IL-33-Induced Airway Eosinophilic Inflammation In Vivo
Next, we determined the potential preventive effect of 14(S)-HDoHE, a major product of 12/15-LOX in the lung during inflammation, in vivo on IL-33-induced airway eosinophilic inflammation. Intraperitoneal administration of 14(S)-HDoHE reduced the number of eosinophils and macrophages in BALF compared with that in vehicle-treated mice in 12/15-LOX-/- mice (Figure 5A). 14(S)-HDoHE administration also decreased the number of ILC2 and CD4+ST2+ Th2 cells present in BALF in 12/15-LOX-/- mice (Figure 5B). Also, 14(S)-HDoHE reduced the number of ILC2 and Th2 cells in BALF of WT mice (Figure 5B).
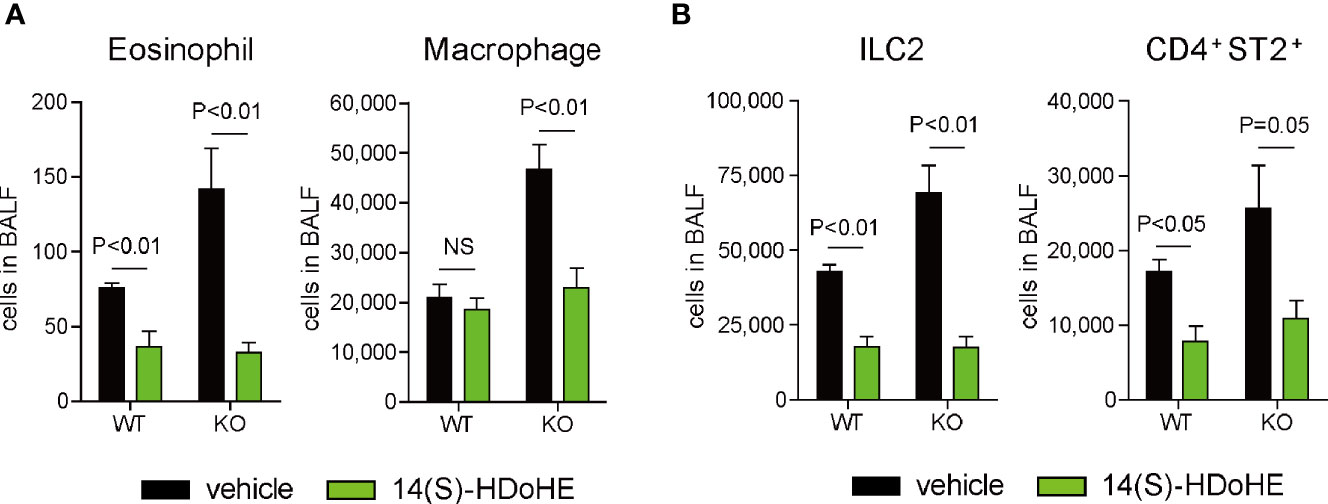
Figure 5 14(S)-HDoHE suppresses IL-33-induced airway eosinophilic inflammation. Airway inflammation was induced by administration of IL-33 (500 ng per mouse, intranasal), for 3 consecutive days, in C57BL/6 and 12/15-lipoxygenase deficient mice. 14(S)-hydroxy docosahexaenoic acid (HDoHE) was administered via intraperitoneal injection prior to IL-33 administration. Flow cytometric analysis was performed 4 days after the final administration of IL-33. (A) Number of eosinophils and macrophages in the BALF. (B) Number of lymphocyte subsets, including ILC2 and Th2 cells, in the BALF. Mean ± SEM, n=4 for each group. NS, not significant.
Discussion
In the present study, we demonstrated that 12/15-LOX, a key enzyme for the biosynthesis of specialized pro-resolving lipid mediators (SPMs), conferred a protective effect on innate pulmonary eosinophilic inflammation in vivo. Lipidomic analysis revealed a series of 12/15-LOX-derived mediators present at substantial levels in the inflamed lung. Using bioactive lipid screening, the potent effect of 14(S)-HDoHE and 10(S),17(S)-diHDoHE in suppressing ILC2 function was observed in vitro. These findings demonstrate the direct effect of DHA-derived pro-resolving mediators in suppressing ILC2 activation. 14(S)-HDoHE also displayed potent anti-inflammatory effects on IL-33-induced eosinophilic airway inflammation when administered in vivo. These results provide a new therapeutic option for 12/15-LOX-derived pro-resolving mediators in ILC2-mediated allergic diseases.
Previous studies have shown the regulatory effects of 14(S)-HDoHE on murine platelets and human alveolar macrophages in vitro (43, 44). PD1/PDX also possesses potent anti-inflammatory functions in regulating neutrophil activation in vitro (45, 46) and ameliorates pulmonary inflammation and fibrosis in vivo (47, 48). Interestingly, we previously reported that peripheral blood eosinophils isolated from patients with severe asthma had a defective biosynthetic capacity of 14(S)-HDoHE, 17(S)-HDoHE, and PD1/PDX (7). Similarly, their biosynthetic capacities have been reported to be impaired in obese mice, and systemic administration of these metabolites ameliorated obesity-induced inflammatory states (49). These findings highlight the therapeutic potential of 14(S)-HDoHE and related SPMs in the regulation of chronic inflammation through DHA metabolism. However, the precise mechanism underlying the 14(S)-HDoHE-mediated effects thorough specific receptors and/or downstream metabolites remains undetermined.
IL-33 induces ILC2 proliferation and activation, which orchestrate innate type-2 inflammation as the dominant IL-5-producing cell in vitro and in vivo (22, 23, 50–55). IL-5 plays a central role in prolonging eosinophils survival and activating eosinophils to elicit degranulation, superoxide generation, cytokine release, and cysteinyl leukotriene synthesis (12, 56). In this study, 14(S)-HDoHE, 17(S)-HDoHE, and other DHA-derived SPMs, such as RvDs, PDs, and MaRs, differentially exerted regulatory effects on cellular functions of ILC2 cells at micromolar or nanomolar concentrations. In humans, LXA4, an AA-derived SPM, decreased IL-13 release from ILC2s stimulated with IL2/IL25/IL-33/PGD2 at nanomolar concentrations (57). In addition, MaR1, a DHA-derived SPM, regulated IL-13 release from ILC2 in vitro at nanomolar concentration and in vivo at ng/mouse administration in a murine model of asthma (58). Also, regulatory T cells synergistically exerted inhibitory effects on ILC2 in the presence of MaR1 (58). Other cell types, including regulatory T cells, may enhance the suppressive effects of SPMs on ILC2s. Additionally, our study suggests that RvD1 and MaR1 can inhibit ILC2 function at nanomolar concentration. Thus, we speculate that these bioactive SPMs collectively, not individually, orchestrate the regulatory circuit for ILC2-mediated eosinophilic inflammation.
The cellular sources of 12/15-LOX-derived mediators during allergic inflammation are of particular interest. 12/15-LOX-expressing eosinophils play pro-resolving functions by enhancing the resolution of neutrophilic inflammation in acute peritonitis (8, 9) and by promoting corneal wound healing in the eye (59). In addition, 12/15-LOX-expressing resident macrophages play an important role in the efferocytosis of apoptotic neutrophils in acute peritonitis (10). Further studies are required to identify the cell types that locally produce 12/15-LOX-derived SPMs to suppress ILC-2-mediated eosinophilic inflammation.
In conclusion, our results demonstrate that 12/15-LOX-derived mediators regulate IL-33-induced eosinophilic airway inflammation. Bioactive lipid screening identified 14(S)-HDoHE and 10(S),17(S)-diHDoHE as potent endogenous suppressors of ILC2 activation. These findings contribute to a better understanding of the cellular and molecular mechanisms underlying the resolution of eosinophilic airway inflammation. Thus, 12/15-LOX-derived lipid mediators and/or 12/15-LOX-mediated lipid metabolism may represent a potential therapeutic strategy for ameliorating airway inflammation-associated conditions.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by the Animal Care and Use Committee of the RIKEN, the Animal Committee of the University of Tokyo, the Laboratory Animal Care and Use Committee of Keio University of Medicine.
Author Contributions
JM and MA designed the experiments. JM and YY performed most of the experiments and compiled the data. KM provided murine group 2 innate lymphoid cells. MA, KF, and HA supervised the project. JM and MA wrote the manuscript, and all authors provided feedback on the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the JSPS Grant-in-Aid for Scientific Research on Innovative Areas (15H05897, 15H05898 to MA), the RIKEN Special Postdoctoral Researchers Program (to JM), and the Grant-in-Aid for research of ONO Medical Research Foundation (to JM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Yosuke Isobe (RIKEN-IMS) for advice and critical input regarding this study; Mie Honda, Kanako Igarashi, and Kazutaka Ikeda (RIKEN-IMS) for technical assistance with LC-MS-based lipidomics.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.687192/full#supplementary-material
Abbreviations
Alox15, 12/15-lipoxygenase; AA, arachidonic acid; BALF, bronchial alveolar lavage fluid; DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid; HDoHE, hydroxy docosahexaenoic acid; HE, hematoxylin eosin; HETE, hydroxyeicosatetraenoic acid; IL, interleukin; LC, liquid chromatography; LOX, lipoxygenase; LT, leukotriene; MaRs, maresins; MS/MS, tandem mass spectrometry; PAS, periodic acid Schiff; PD1, protectin D1; PDX, protectin DX; PG, prostaglandin; RT-PCR, reverse transcription-PCR; Rv, resolvin; SPM, specialized proresolving mediator.
References
1. Bousquet J, Clark TJ, Hurd S, Khaltaev N, Lenfant C, O’Byrne P, et al. GINA Guidelines on Asthma and Beyond. Allergy (2007) 62(2):102–12. doi: 10.1111/j.1398-9995.2006.01305.x
2. Masoli M, Fabian D, Holt S, Beasley R. The Global Burden of Asthma: Executive Summary of the GINA Dissemination Committee Report. Allergy (2004) 59(5):469–78. doi: 10.1111/j.1398-9995.2004.00526.x
3. Yamamoto T, Miyata J, Arita M, Fukunaga K, Kawana A. Current State and Future Prospect of the Therapeutic Strategy Targeting Cysteinyl Leukotriene Metabolism in Asthma. Respir Invest (2019) 57(6):534–43. doi: 10.1016/j.resinv.2019.08.003
4. Miyata J, Fukunaga K, Kawashima Y, Ohara O, Kawana A, Asano K, et al. Dysregulated Metabolism of Polyunsaturated Fatty Acids in Eosinophilic Allergic Diseases. Prostaglandins Other Lipid Mediat (2020) 150:106477. doi: 10.1016/j.prostaglandins.2020.106477
5. Serhan CN. Pro-Resolving Lipid Mediators are Leads for Resolution Physiology. Nature (2014) 510(7503):92–101. doi: 10.1038/nature13479
6. Serhan CN, Levy BD. Resolvins in Inflammation: Emergence of the Pro-Resolving Superfamily of Mediators. J Clin Invest (2018) 128(7):2657–69. doi: 10.1172/jci97943
7. Miyata J, Fukunaga K, Iwamoto R, Isobe Y, Niimi K, Takamiya R, et al. Dysregulated Synthesis of Protectin D1 in Eosinophils From Patients With Severe Asthma. J Allergy Clin Immunol (2013) 131(2):353–60 e1-2. doi: 10.1016/j.jaci.2012.07.048
8. Tani Y, Isobe Y, Imoto Y, Segi-Nishida E, Sugimoto Y, Arai H, et al. Eosinophils Control the Resolution of Inflammation and Draining Lymph Node Hypertrophy Through the Proresolving Mediators and CXCL13 Pathway in Mice. FASEB J (2014) 28(9):4036–43. doi: 10.1096/fj.14-251132
9. Yamada T, Tani Y, Nakanishi H, Taguchi R, Arita M, Arai H. Eosinophils Promote Resolution of Acute Peritonitis by Producing Proresolving Mediators in Mice. FASEB J (2011) 25(2):561–8. doi: 10.1096/fj.10-170027
10. Lim CS, Porter DW, Orandle MS, Green BJ, Barnes MA, Croston TL, et al. Resolution of Pulmonary Inflammation Induced by Carbon Nanotubes and Fullerenes in Mice: Role of Macrophage Polarization. Front Immunol (2020) 11:1186. doi: 10.3389/fimmu.2020.01186
11. Miyata J, Fukunaga K, Kawashima Y, Watanabe T, Saitoh A, Hirosaki T, et al. Dysregulated Fatty Acid Metabolism in Nasal Polyp-Derived Eosinophils From Patients With Chronic Rhinosinusitis. Allergy (2019) 74(6):1113–24. doi: 10.1111/all.13726
12. Miyata J, Fukunaga K, Kawashima Y, Ohara O, Arita M. Cysteinyl Leukotriene Metabolism of Human Eosinophils in Allergic Disease. Allergol Int Off J Japanese Soc Allergol (2020) 69(1):28–34. doi: 10.1016/j.alit.2019.06.002
13. Miyata J, Arita M. Role of Omega-3 Fatty Acids and Their Metabolites in Asthma and Allergic Diseases. Allergol Int Off J Japanese Soc Allergol (2015) 64(1):27–34. doi: 10.1016/j.alit.2014.08.003
14. Levy BD, Lukacs NW, Berlin AA, Schmidt B, Guilford WJ, Serhan CN, et al. Lipoxin A4 Stable Analogs Reduce Allergic Airway Responses Via Mechanisms Distinct From CysLT1 Receptor Antagonism. FASEB J (2007) 21(14):3877–84. doi: 10.1096/fj.07-8653com
15. Levy BD, Kohli P, Gotlinger K, Haworth O, Hong S, Kazani S, et al. Protectin D1 Is Generated in Asthma and Dampens Airway Inflammation and Hyperresponsiveness. J Immunol (2007) 178(1):496–502. doi: 10.4049/jimmunol.178.1.496
16. Rogerio AP, Haworth O, Croze R, Oh SF, Uddin M, Carlo T, et al. Resolvin D1 and Aspirin-Triggered Resolvin D1 Promote Resolution of Allergic Airways Responses. J Immunol (2012) 189(4):1983–91. doi: 10.4049/jimmunol.1101665
17. Levy BD, De Sanctis GT, Devchand PR, Kim E, Ackerman K, Schmidt BA, et al. Multi-Pronged Inhibition of Airway Hyper-Responsiveness and Inflammation by Lipoxin a(4). Nat Med (2002) 8(9):1018–23. doi: 10.1038/nm748
18. Mochimaru T, Fukunaga K, Miyata J, Matsusaka M, Masaki K, Kabata H, et al. 12-OH-17,18-Epoxyeicosatetraenoic Acid Alleviates Eosinophilic Airway Inflammation in Murine Lungs. Allergy (2018) 73(2):369–78. doi: 10.1111/all.13297
19. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. Il-33, an Interleukin-1-Like Cytokine That Signals Via the IL-1 Receptor-Related Protein ST2 and Induces T Helper Type 2-Associated Cytokines. Immunity (2005) 23(5):479–90. doi: 10.1016/j.immuni.2005.09.015
20. Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A Large-Scale, Consortium-Based Genomewide Association Study of Asthma. N Engl J Med (2010) 363(13):1211–21. doi: 10.1056/NEJMoa0906312
21. Gudbjartsson DF, Bjornsdottir US, Halapi E, Helgadottir A, Sulem P, Jonsdottir GM, et al. Sequence Variants Affecting Eosinophil Numbers Associate With Asthma and Myocardial Infarction. Nat Genet (2009) 41(3):342–7. doi: 10.1038/ng.323
22. Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. Il-33-responsive Lineage- CD25+ CD44(Hi) Lymphoid Cells Mediate Innate Type 2 Immunity and Allergic Inflammation in the Lungs. J Immunol (2012) 188(3):1503–13. doi: 10.4049/jimmunol.1102832
23. Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate Production of T(H)2 Cytokines by Adipose Tissue-Associated c-Kit(+)Sca-1(+) Lymphoid Cells. Nature (2010) 463(7280):540–4. doi: 10.1038/nature08636
24. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes Represent a New Innate Effector Leukocyte That Mediates Type-2 Immunity. Nature (2010) 464(7293):1367–70. doi: 10.1038/nature08900
25. Besnard AG, Togbe D, Guillou N, Erard F, Quesniaux V, Ryffel B. IL-33-Activated Dendritic Cells Are Critical for Allergic Airway Inflammation. Eur J Immunol (2011) 41(6):1675–86. doi: 10.1002/eji.201041033
26. Eiwegger T, Akdis CA. Il-33 Links Tissue Cells, Dendritic Cells and Th2 Cell Development in a Mouse Model of Asthma. Eur J Immunol (2011) 41(6):1535–8. doi: 10.1002/eji.201141668
27. Kurokawa M, Matsukura S, Kawaguchi M, Ieki K, Suzuki S, Odaka M, et al. Expression and Effects of IL-33 and ST2 in Allergic Bronchial Asthma: IL-33 Induces Eotaxin Production in Lung Fibroblasts. Int Arch Allergy Immunol (2011) 155 Suppl 1:12–20. doi: 10.1159/000327259
28. Cherry WB, Yoon J, Bartemes KR, Iijima K, Kita H. A Novel IL-1 Family Cytokine, IL-33, Potently Activates Human Eosinophils. J Allergy Clin Immunol (2008) 121(6):1484–90. doi: 10.1016/j.jaci.2008.04.005
29. Rank MA, Kobayashi T, Kozaki H, Bartemes KR, Squillace DL, Kita H. Il-33-activated Dendritic Cells Induce an Atypical TH2-type Response. J Allergy Clin Immunol (2009) 123(5):1047–54. doi: 10.1016/j.jaci.2009.02.026
30. Saglani S, Lui S, Ullmann N, Campbell GA, Sherburn RT, Mathie SA, et al. Il-33 Promotes Airway Remodeling in Pediatric Patients With Severe Steroid-Resistant Asthma. J Allergy Clin Immunol (2013) 132(3):676–85 e13. doi: 10.1016/j.jaci.2013.04.012
31. Bouffi C, Rochman M, Zust CB, Stucke EM, Kartashov A, Fulkerson PC, et al. IL-33 Markedly Activates Murine Eosinophils by an NF-kappaB-Dependent Mechanism Differentially Dependent Upon an IL-4-driven Autoinflammatory Loop. J Immunol (2013) 191(8):4317–25. doi: 10.4049/jimmunol.1301465
32. Komai-Koma M, Gilchrist DS, McKenzie AN, Goodyear CS, Xu D, Liew FY. Il-33 Activates B1 Cells and Exacerbates Contact Sensitivity. J Immunol (2011) 186(4):2584–91. doi: 10.4049/jimmunol.1002103
33. Kurowska-Stolarska M, Kewin P, Murphy G, Russo RC, Stolarski B, Garcia CC, et al. Il-33 Induces Antigen-Specific IL-5+ T Cells and Promotes Allergic-Induced Airway Inflammation Independent of IL-4. J Immunol (2008) 181(7):4780–90. doi: 10.4049/jimmunol.181.7.4780
34. Yagami A, Orihara K, Morita H, Futamura K, Hashimoto N, Matsumoto K, et al. Il-33 Mediates Inflammatory Responses in Human Lung Tissue Cells. J Immunol (2010) 185(10):5743–50. doi: 10.4049/jimmunol.0903818
35. Suzukawa M, Koketsu R, Iikura M, Nakae S, Matsumoto K, Nagase H, et al. Interleukin-33 Enhances Adhesion, CD11b Expression and Survival in Human Eosinophils. Lab Invest (2008) 88(11):1245–53. doi: 10.1038/labinvest.2008.82
36. Fux M, Pecaric-Petkovic T, Odermatt A, Hausmann OV, Lorentz A, Bischoff SC, et al. Il-33 is a Mediator Rather Than a Trigger of the Acute Allergic Response in Humans. Allergy (2014) 69(2):216–22. doi: 10.1111/all.12309
37. Hardman CS, Panova V, McKenzie AN. Il-33 Citrine Reporter Mice Reveal the Temporal and Spatial Expression of IL-33 During Allergic Lung Inflammation. Eur J Immunol (2013) 43(2):488–98. doi: 10.1002/eji.201242863
38. Prefontaine D, Nadigel J, Chouiali F, Audusseau S, Semlali A, Chakir J, et al. Increased IL-33 Expression by Epithelial Cells in Bronchial Asthma. J Allergy Clin Immunol (2010) 125(3):752–4. doi: 10.1016/j.jaci.2009.12.935
39. Prefontaine D, Lajoie-Kadoch S, Foley S, Audusseau S, Olivenstein R, Halayko AJ, et al. Increased Expression of IL-33 in Severe Asthma: Evidence of Expression by Airway Smooth Muscle Cells. J Immunol (2009) 183(8):5094–103. doi: 10.4049/jimmunol.0802387
40. Kabata H, Moro K, Fukunaga K, Suzuki Y, Miyata J, Masaki K, et al. Thymic Stromal Lymphopoietin Induces Corticosteroid Resistance in Natural Helper Cells During Airway Inflammation. Nat Commun (2013) 4:2675. doi: 10.1038/ncomms3675
41. Arita M. Mediator Lipidomics in Acute Inflammation and Resolution. J Biochem (2012) 152(4):313–9. doi: 10.1093/jb/mvs092
42. Moro K, Ealey KN, Kabata H, Koyasu S. Isolation and Analysis of Group 2 Innate Lymphoid Cells in Mice. Nat Protoc (2015) 10(5):792–806. doi: 10.1038/nprot.2015.047
43. Yamaguchi A, Stanger L, Freedman CJ, Standley M, Hoang T, Adili R, et al. Dha 12-LOX-derived Oxylipins Regulate Platelet Activation and Thrombus Formation Through a PKA-dependent Signaling Pathway. J Thromb Haemost (2021) 19(3):839–51. doi: 10.1111/jth.15184
44. Wendell SG, Golin-Bisello F, Wenzel S, Sobol RW, Holguin F, Freeman BA. 15-Hydroxyprostaglandin Dehydrogenase Generation of Electrophilic Lipid Signaling Mediators From Hydroxy ω-3 Fatty Acids. J Biol Chem (2015) 290(9):5868–80. doi: 10.1074/jbc.M114.635151
45. Serhan CN, Gotlinger K, Hong S, Lu Y, Siegelman J, Baer T, et al. Anti-Inflammatory Actions of Neuroprotectin D1/protectin D1 and Its Natural Stereoisomers: Assignments of Dihydroxy-Containing Docosatrienes. J Immunol (2006) 176(3):1848–59. doi: 10.4049/jimmunol.176.3.1848
46. Stein K, Stoffels M, Lysson M, Schneiker B, Dewald O, Krönke G, et al. A Role for 12/15-Lipoxygenase-Derived Proresolving Mediators in Postoperative Ileus: Protectin DX-Regulated Neutrophil Extravasation. J leukocyte Biol (2016) 99(2):231–9. doi: 10.1189/jlb.3HI0515-189R
47. Li H, Hao Y, Zhang H, Ying W, Li D, Ge Y, et al. Posttreatment With Protectin DX Ameliorates Bleomycin-Induced Pulmonary Fibrosis and Lung Dysfunction in Mice. Sci Rep (2017) 7:46754. doi: 10.1038/srep46754
48. Xia H, Ge Y, Wang F, Ming Y, Wu Z, Wang J, et al. Protectin DX Ameliorates Inflammation in Sepsis-Induced Acute Lung Injury Through Mediating Pparγ/Nf-κb Pathway. Immunologic Res (2020) 68(5):280–8. doi: 10.1007/s12026-020-09151-7
49. Crouch MJ, Kosaraju R, Guesdon W, Armstrong M, Reisdorph N, Jain R, et al. Frontline Science: A Reduction in DHA-Derived Mediators in Male Obesity Contributes Toward Defects in Select B Cell Subsets and Circulating Antibody. J leukocyte Biol (2019) 106(2):241–57. doi: 10.1002/jlb.3hi1017-405rr
50. Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, et al. Administration of IL-33 Induces Airway Hyperresponsiveness and Goblet Cell Hyperplasia in the Lungs in the Absence of Adaptive Immune System. Int Immunol (2008) 20(6):791–800. doi: 10.1093/intimm/dxn037
51. Snelgrove RJ, Gregory LG, Peiro T, Akthar S, Campbell GA, Walker SA, et al. Alternaria-Derived Serine Protease Activity Drives IL-33 Mediated Asthma Exacerbations. J Allergy Clin Immunol (2014) 134(3):583–92.e6. doi: 10.1016/j.jaci.2014.02.002
52. Ikutani M, Yanagibashi T, Ogasawara M, Tsuneyama K, Yamamoto S, Hattori Y, et al. Identification of Innate IL-5-producing Cells and Their Role in Lung Eosinophil Regulation and Antitumor Immunity. J Immunol (2012) 188(2):703–13. doi: 10.4049/jimmunol.1101270
53. Salmond RJ, Mirchandani AS, Besnard AG, Bain CC, Thomson NC, Liew FY. Il-33 Induces Innate Lymphoid Cell-Mediated Airway Inflammation by Activating Mammalian Target of Rapamycin. J Allergy Clin Immunol (2012) 130(5):1159–66.e6. doi: 10.1016/j.jaci.2012.05.018
54. Barlow JL, Peel S, Fox J, Panova V, Hardman CS, Camelo A, et al. IL-33 Is More Potent Than IL-25 in Provoking IL-13-producing Nuocytes (Type 2 Innate Lymphoid Cells) and Airway Contraction. J Allergy Clin Immunol (2013) 132(4):933–41. doi: 10.1016/j.jaci.2013.05.012
55. Kim HY, Chang YJ, Subramanian S, Lee HH, Albacker LA, Matangkasombut P, et al. Innate Lymphoid Cells Responding to IL-33 Mediate Airway Hyperreactivity Independently of Adaptive Immunity. J Allergy Clin Immunol (2012) 129(1):216–27.e1-6. doi: 10.1016/j.jaci.2011.10.036
56. Pelaia C, Paoletti G, Puggioni F, Racca F, Pelaia G, Canonica GW, et al. Interleukin-5 in the Pathophysiology of Severe Asthma. Front Physiol (2019) 10:1514. doi: 10.3389/fphys.2019.01514
57. Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, et al. Lipoxin A4 Regulates Natural Killer Cell and Type 2 Innate Lymphoid Cell Activation in Asthma. Sci Transl Med (2013) 5(174):174ra26. doi: 10.1126/scitranslmed.3004812
58. Krishnamoorthy N, Burkett PR, Dalli J, Abdulnour RE, Colas R, Ramon S, et al. Cutting Edge: Maresin-1 Engages Regulatory T Cells to Limit Type 2 Innate Lymphoid Cell Activation and Promote Resolution of Lung Inflammation. J Immunol (2015) 194(3):863–7. doi: 10.4049/jimmunol.1402534
Keywords: 12/15-lipoxygenase, IL-33, docosahexaenoic acid, 14(S)-HDoHE, group 2 innate lymphoid cell, lipidomics, maresin, specialized proresolving mediator
Citation: Miyata J, Yokokura Y, Moro K, Arai H, Fukunaga K and Arita M (2021) 12/15-Lipoxygenase Regulates IL-33-Induced Eosinophilic Airway Inflammation in Mice. Front. Immunol. 12:687192. doi: 10.3389/fimmu.2021.687192
Received: 29 March 2021; Accepted: 04 May 2021;
Published: 19 May 2021.
Edited by:
Amiram Ariel, University of Haifa, IsraelReviewed by:
Nan Chiang, Brigham and Women’s Hospital and Harvard Medical School, United StatesMichelle Amantea Sugimoto, University College London, United Kingdom
Copyright © 2021 Miyata, Yokokura, Moro, Arai, Fukunaga and Arita. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Makoto Arita, bWFrb3RvLmFyaXRhQHJpa2VuLmpw