 Hyun-Eui Park
Hyun-Eui Park Wonsik Lee
Wonsik Lee Min-Kyoung Shin
Min-Kyoung Shin Sung Jae Shin
Sung Jae Shin- 1Department of Microbiology and Convergence Medical Science, Institute of Health Sciences, College of Medicine, Gyeongsang National University, Jinju, South Korea
- 2School of Pharmacy, Sungkyunkwan University, Suwon, South Korea
- 3Department of Microbiology, Institute for Immunology and Immunological Diseases, Brain Korea 21 Project for Graduate School of Medical Science, Yonsei University College of Medicine, Seoul, South Korea
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb) infection, remains a global health threat despite recent advances and insights into host-pathogen interactions and the identification of diverse pathways that may be novel therapeutic targets for TB treatment. In addition, the emergence and spread of multidrug-resistant Mtb strains led to a low success rate of TB treatments. Thus, novel strategies involving the host immune system that boost the effectiveness of existing antibiotics have been recently suggested to better control TB. However, the lack of comprehensive understanding of the immunomodulatory effects of anti-TB drugs, including first-line drugs and newly introduced antibiotics, on bystander and effector immune cells curtailed the development of effective therapeutic strategies to combat Mtb infection. In this review, we focus on the influence of host immune-mediated stresses, such as lysosomal activation, metabolic changes, oxidative stress, mitochondrial damage, and immune mediators, on the activities of anti-TB drugs. In addition, we discuss how anti-TB drugs facilitate the generation of Mtb populations that are resistant to host immune response or disrupt host immunity. Thus, further understanding the interplay between anti-TB drugs and host immune responses may enhance effective host antimicrobial activities and prevent Mtb tolerance to antibiotic and immune attacks. Finally, this review highlights novel adjunctive therapeutic approaches against Mtb infection for better disease outcomes, shorter treatment duration, and improved treatment efficacy based on reciprocal interactions between current TB antibiotics and host immune cells.
Introduction
Tuberculosis (TB) is a chronic infectious disease caused by an obligate pathogen, Mycobacterium tuberculosis (Mtb), in humans (1). According to the WHO report, in 2020, approximately 10 million people were newly diagnosed, and 1.3 million people died from this notorious disease (2). Moreover, the recent treatment success rate was 82% for drug-sensitive TB and 55% for multidrug-resistant (MDR)-TB (3). There has been a gradual increase in the incidence of MDR-TB, defined as resistance to isoniazid (INH) and rifampicin (RIF), and extensively drug-resistant (XDR)-TB, defined as in vitro drug resistance to not only INH and RIF, but also all fluoroquinolones and at least one injectable aminoglycoside (4).
The presence of a mycobacterial population with more than one bacterial phenotype has been observed in patients with TB, as indicated by bacterial populations with varying growth dynamics in sputum samples (3). TB treatment strategy involves long-term treatment with several drugs for at least six months, which may increase the risk of MDR- and XDR-Mtb emergence (4–6), which is attributed to residual bacteria that are sheltered from or unresponsive to antibiotic treatment in heterogenous mycobacterial populations in patients (3). Thus, enhancing treatment success rate, shortening treatment duration, and preventing MDR Mtb emergence are the most critical factors for successful TB treatment. In this review, we provide an understanding of the mechanism underlying the generation of persistent mycobacteria in heterogeneous mycobacteria populations under immune- or drug-induced stress and discuss the effects of anti-TB drugs on host immune responses as opposed to their effects on Mtb. This review provides insights that may contribute to the development of host immune-mediated therapeutic strategies to eliminate persistent mycobacteria more effectively, thereby enhancing treatment success and preventing the development of MDR-TB.
Mycobacterial Persisters Adapt to Stresses in the Host and Exhibit Antibiotic Tolerance
Antibiotic Tolerance
Host-related stresses, such as hypoxia, acidic conditions, nutrient starvation, oxidative stress, and cytokine responses, alter the metabolic state of pathogens and eventually induces a drug-tolerant phenotype termed “persister” (7–10). These persister cells can maintain an unreplicated status and simultaneously survive antibiotic treatment. After cessation of anti-TB therapy, the surviving persisters revive their metabolism for replication, subsequently causing a relapse. Thus, antibiotic-tolerant persisters are considered surviving bacteria that did not undergo genetic mutations even after long-term antibiotic treatment (11). Although antibiotic tolerance and antibiotic resistance share common characteristics, they differ in several aspects (12, 13). Antibiotic resistance is generally inheritable and occurs in a drug-specific manner, while antibiotic tolerance is not inheritable and functions broadly. Antibiotic resistance is accompanied by an increase in minimum inhibitory concentration (MIC) of drugs, while antibiotic-tolerant and susceptible subpopulations show identical MIC (13). Tolerance refers only to bactericidal antibiotics and not to bacteriostatic antibiotics, unlike resistance (12).
The mechanism of antibiotic tolerance through the formation of persisters in response to a variety of stresses, including nutrient deprivation, oxidative stress, acidic environment, osmotic conditions, and host immune-mediated stresses, has been described in many pathogenic bacteria, including Escherichia coli, Staphylococcus aureus, Pseudomonas aeruginosa, and Mtb (14–18). Several mechanisms underlying the generation of persisters in response to the stresses have been identified; these include metabolic regulation, such as toxin–antitoxin (TA) systems, stringent and SOS responses, and biofilm formation (19–24). Understanding the mechanisms of persister formation under various stresses and developing therapeutic strategies specifically targeting the mechanisms related to antibiotic tolerance are expected to contribute to TB control. Therefore, here, we review the detailed mechanism of persister formation induced by host-mediated stress in Mtb and its effect on antibiotic tolerance.
Mtb Adapts to Host-Mediated Stresses Through Metabolic Regulation
Regulation of Transcription Factors
Mtb encounters various stresses, such as acidic pH, oxidative stress, hypoxia, nutrient deprivation, and cytokine-mediated effectors, during infection. On detecting such a stressful environment, Mtb reprograms its metabolism, at the transcriptional level, to survive in the niche (25, 26). Bacteria combat environmental stress to induce changes in antibiotic resistance and toxicity through two-component systems (TCSs), consisting of a sensor histidine kinase and a modulator of cytoplasmic response integrated into the inner membrane, as a stress recognition and response system (27, 28). To date, 12 complete TCSs have been identified in Mtb, of which PhoPR, PrrAB, MprAB, NarL, and TcrXY are involved in response to stresses, including pH, macrophage infection, detergents, hypoxia, low iron levels, and starvation (27).
The phoPR TCS may be a critical factor for adaptation to a low pH environment (27). When PhoP detects low pH, it activates the transcriptional regulator whiB3, regulates the expression of pH-responsive gene clusters (aprABC, icl, pks2, pks3, pks4, and lipF), and is involved in the survival of Mtb in macrophages (29–33) (Figure 1). Indeed, phoP deletion mutants exhibit growth defects in murine bone marrow-derived macrophages (BMDMs) as well as attenuated virulence with reduced bacterial burden in the lungs, liver, and spleen of a mouse Mtb infection model (34). Interestingly, transcriptional analyses revealed an overlapping of the repressed genes in H37Ra and phoP knockout mutant of H37Rv (35). Moreover, the incorporation of intact phoP into the H37Ra genome increased the bacterial persistence in murine BMDMs (35). In another study, the phoP mutant Mtb strain showed considerable attenuation in severe combined immunodeficient mice compared to the parental and BCG strains (32). Moreover, the Mtb phoP deletion mutant strain conferred protective anti-TB immunity in mouse and guinea pig models, indicating its potential as a live vaccine candidate (32). Liu et al. demonstrated that the expression of five regulons, DosR, MprA, PhoP, Rv1404, and Rv3058c, is responsible for the antibiotic tolerance of Mtb; these five regulons controlled the expression of over 50% of the upregulated genes after treatment with different anti-TB drugs, and their (DosR, PhoP, and MprA) deletion reduced drug tolerance under stress conditions (36).
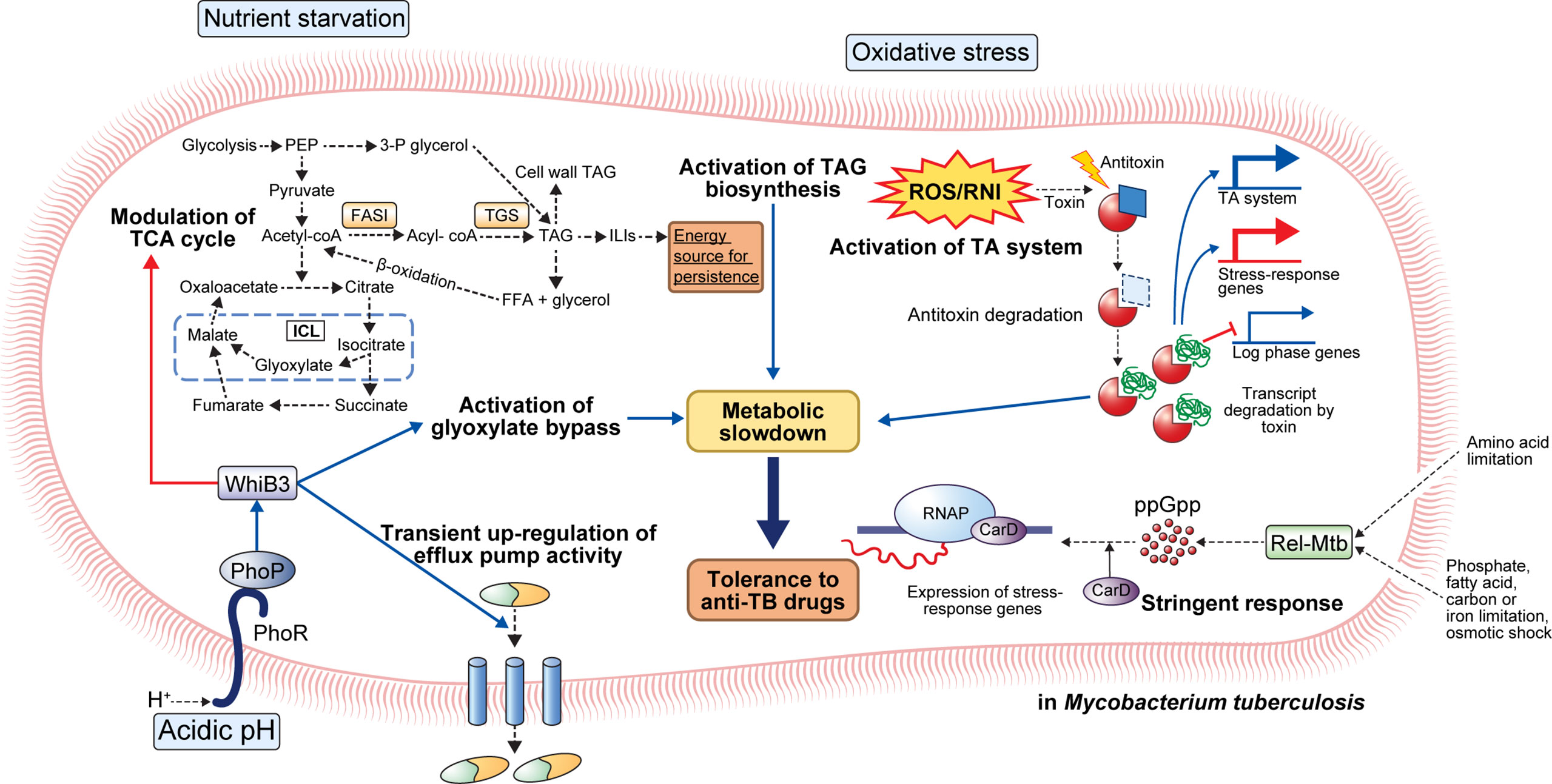
Figure 1 General mechanisms for the establishment of antibiotic tolerance in Mycobacterium tuberculosis. Under host-mediated stresses, M. tuberculosis (Mtb) adapts to stress conditions via several mechanisms. Under acidic pH, the phoPR two-component system activates transcriptional regulator whiB3 that promotes suppression of the TCA cycle, activation of glyoxylate bypass, and transient upregulation of efflux pump activity. Activation of glyoxylate bypass is mediated by isocitrate lyase that converts isocitrate to glyoxylate under stress conditions. Nutrient starvation induces several changes in Mtb metabolism. Nutrient starvation also suppresses the TCA cycle and activates glyoxylate bypass, thereby enhancing the accumulation of triacylglycerol (TAG). The accumulated TAG is stored in the form of intracellular lipophilic inclusions (ILIs). The stored ILIs are used as an energy source in the persistence state. Additionally, the limitation of amino acids, phosphate, fatty acids, carbon, iron, and osmotic shock induces activation of stringent response through the production of ppGpp by Rel-Mtb. Production of ppGpp activates the expression of stress-response genes that causes a metabolic slowdown. Oxidative stress induces the activation of the TA system. Degradation of antitoxin occurs, and toxin degrades the transcript of log-phase genes. Further, upregulation of stress-response genes occurs, facilitating adaptation to stress conditions. Collectively, the adaptation of Mtb to stress conditions leads to metabolic modulation that results in antibiotic tolerance.
Stringent Response
The stringent response is a conserved global signaling system that promotes bacterial survival in various environments, such as nutrient deprivation and other stresses (37). Particularly, stringent responses have been reportedly caused by amino acids, carbon, nitrogen, or phosphorus starvation, as well as UV exposure and fatty acid depletion (37). The stringent response is mediated by the hyperphosphorylated guanine nucleotides ppGpp and pppGpp, collectively referred to as [(p)ppGpp], and inorganic polyphosphate [poly(P)], and the synthesized signaling molecules regulate bacterial transcriptional changes under various stress conditions (38, 39). In Mtb, (p)ppGpp synthesis is induced by nutrient deprivation, long-term culture, and chronic infection in animal models, and it has been reported to be necessary for Mtb survival (37, 40, 41). Two proteins, RelA and SpoT, responsible for the synthesis of (p)ppGpp in gram-negative bacteria, have been identified, but many gram-positive bacteria, including mycobacteria, have only one protein (Rel) homologous to both RelA and SpoT (37). Accumulation of (p)ppGpp synthesized by Rel-Mtb and the transcription factor CarD in hostile environments, such as nutrient deficiency and oxidative stress, leads to transcription and translation of stress-responsive genes in Mtb (37) (Figure 1).
The protein Rel-Mtb modulates the intracellular (p)ppGpp content by regulating its synthesis and hydrolysis via an N-terminal hydrolase and synthetase domain (42). Nutrient starvation induces upregulation of Rv2583c (Rel-Mtb) that subsequently promotes the production of intracellular (p)ppGpp in Mtb (37). Rel-Mtb deletion mutant showed a growth defect in liquid media, and the disrupted growth rate was restored when citrate or phospholipid was used as the sole carbon source in vitro (37). A disrupted growth rate can induce antibiotic tolerance to drugs that kill actively growing cells. Recently, Dutta et al. showed that Rel-Mtb deficiency induces disruption of antibiotic tolerance under stress conditions, increasing susceptibility to INH (43). They reported that the nutrient-starved Rel-Mtb mutants showed similar metabolic activity as wild type bacteria growing in nutrient-rich conditions (43). Disruption of Rel-Mtb induced increased susceptibility to INH in vitro nutrient starvation and BALB/c mouse models (43). Furthermore, they discovered a Rel-Mtb inhibitor through pharmaceutical library screening that showed a direct cytotoxic effect on antibiotic-tolerant Mtb and synergetic effect with INH activity (43). In Mtb, the polyphosphate kinase PPK1 is responsible for poly(P) synthesis, and the exopolyphosphatases, PPX1 and PPX2, and PPK2 are responsible for poly(P) hydrolysis, thereby regulating cellular poly(P) homeostasis (39). The ppx1 or ppk2 deletion mutant strains showed low glycerol-3-phosphate (G3P) and 1-deoxy-xylulose-5-phosphate expression levels in bacterial cells, suggesting downregulated G3P synthesis pathway (39). As a result, the ppk2 and ppx1 deletion mutant increased susceptibility to plumbagin and meropenem, and clofazimine, respectively (39). Similarly, the ppk1 deletion mutants showed increased susceptibility to INH, levofloxacin, and RIF (44). These results suggest that (p)ppGpp and poly(P) synthesis and their modulators play important roles in the development of antibiotic resistance in vivo.
Metabolic Modulation
Numerous acid-inducible genes induce a carbon metabolism shift for microbial persistence in the host macrophages. One such acid-inducible gene encodes isocitrate lyase that converts isocitrate to succinate and glyoxylate (45). Moreover, malate synthase catalyzes malate formation by the addition of acetyl-CoA to glyoxylate (45). Overexpression of isocitrate lyase causes the activation of the glyoxylate shunt, subsequently inducing metabolic shifting; pyruvate, succinate, fumarate, and malate levels were increased while the α-ketoglutarate level was decreased in macrophage infection and low pH culture model (46). The limitation of α-ketoglutarate-derived amino acids and oxaloacetate by glyoxylate shunt activation slows bacterial cell growth and metabolic activity (46). Antibiotics can induce growth and metabolic activity arrest in rapidly growing cells. For example, the antimicrobial effect of INH depends on INH conversion to isonicotinoyl by the catalase-peroxidase katG (3). Converted isonicotinoyl binds NAD+ to make isonicotinoyl-NAD that inhibits mycolic acid synthesis, a bacterial cell wall component, subsequently interfering with mycobacterial cell wall integrity (3). Thus, reduced need for cell wall synthesis due to arrested growth and metabolic activity due to acidic stress induces tolerance to INH (3). RIF kills metabolically active cells by binding to RNA polymerase subunit B and interfering with transcription; RIF resistance is usually acquired through mutation in rpoB that encodes RNA polymerase B protein (47). However, transient antibiotic tolerance has also been reported in previous studies (48–50). In response to environmental stress, Mtb translates a mutated form of RNA polymerase with a lower affinity to RIF, thereby facilitating the acquisition of transient antibiotic tolerance during antibiotic treatment (50). Collectively, growing evidence suggests that metabolically arrested states induce antibiotic tolerance that prevents the complete sterilization of pathogens. Therefore, to eradicate the antibiotic-tolerant bacterial population, a treatment strategy that reactivates the metabolically arrested bacterial population is needed (Figure 1).
Modulation of Lipid Metabolism
Several host immune-mediated stresses induce intracellular triacylglycerol (TAG) droplet accumulation in Mtb by TAG synthase activity. For example, TAG synthase upregulation was confirmed in multiple-stress conditions, such as hypoxia, low pH, and low iron (51–54). Accelerated TAG synthesis induces a reduction in TCA flux and subsequently enhances the survival of Mtb in the presence of antibiotics, such as INH, streptomycin, ciprofloxacin, and ethambutol (EMB) (54). Interestingly, antibiotic tolerance due to TAG accumulation can be reversed by modulating carbon fluxes with complete inhibition of TAG synthase in vitro and in vivo (54). Furthermore, tgs1 deletion mutants continue to grow under stress conditions while wild type strain stops replicating (54). Kapoor et al. developed an in vitro model of human granuloma for pulmonary tuberculosis and discovered unique characteristics of Mtb within the granuloma; Mtb showed dormant phenotypes, including the loss of acid-fastness, accumulation of lipid droplet, transcriptional change of lipid metabolism genes, and tolerance to RIF (51). Moreover, treatment with anti-tumor necrosis factor-alpha (TNF-α) monoclonal antibodies induced resuscitation of Mtb as previously described in human TB (51). Similarly, a multiple-stress model that included low oxygen, high CO2, low nutrient, and acidic pH showed arrested growth, acid-fastness loss, TAG, and wax ester accumulation, along with the rise in antibiotic tolerance to INH and RIF in Mtb (52). Interestingly, antibiotic tolerance was diminished in the tgs1 deletion mutant and restored with the addition of complementation. Furthermore, transcriptome analysis using microarray revealed the achievement of the dormant state showing repression of energy generation, transcription and translation machinery, and induction of stress-responsive genes (52). Recently, Santucci et al. identified the mechanism of TAG accumulation to involve intracytoplasmic lipid inclusions (ILI) induced by carbon excess and nitrogen starvation in M. smegmatis and M. abscessus (53). They also identified tgs1-mediated TAG formation and lipolytic enzyme-mediated TAG breakdown mechanisms. Moreover, they discovered that emergence of antibiotic tolerance against RIF and INH induced by low nitrogen and high ILI environment as previously described (53). Taken together, the importance of TAG synthesis in antibiotic tolerance of Mtb suggests the potential of lipid metabolism-related proteins, such as triacylglycerol synthase and fatty-acyl-CoA reductase, as therapeutic targets for abolishing antibiotic tolerance.
Toxin–Antitoxin (TA) System
The TA system comprises a stable toxin that interferes with indispensable cellular metabolism and an unstable antitoxin that blocks the toxin activity during persister formation (55, 56). The TA systems are generally divided into seven classes depending on their mechanism (57). In detail, type I and III antitoxin include an RNA antitoxin that interferes with translocation of the toxin as an antisense RNA (type I) or binding to toxin protein to neutralize the toxin activity (type III). Type II antitoxins are proteins that interfere with the toxins by direct binding to the toxin protein. Type IV antitoxins inhibit toxin activity by attaching to the toxin target, while type V antitoxins degrade the toxin mRNA target directly. Type VI antitoxins bind to the toxin; they do not directly degrade the toxin itself but promote its degradation by ClpXP (56). In the type VII TA system, antitoxin acts as an enzyme for the chemical modification of the toxin and subsequently neutralizes the toxin (57). As a representative example, in the HipBA toxin/antitoxin module, HipA is a toxin that inhibits cell growth and induces persister formation, while HipB is an antitoxin that binds to HipA and acts as a transcription inhibitor of the hipBA operon. In particular, high HipA expression leads to multidrug resistance in E. coli (55). Characteristically, Mtb has many TA system-related loci in its genome, and at least 88 TAs have been identified (58). According to Keren et al., 10 TA modules were overexpressed in Mtb persister cells, suggesting that the TA system not only contributes to Mtb virulence but also the formation of bacterial persister cells (8). Further, Torrey et al. revealed that multiple pathways such as lipid biosynthesis, carbon metabolism, TA systems, and transcriptional regulators are involved in Mtb persister formation using transcriptional analysis and whole-genome sequencing of Mtb hip mutant (59). Notably, most of the identified Mtb TA systems were Type II, and these include VapBC, MazEF, YefM/YoeB, RelBE, HigBA, and ParDE (60).
Notably, the VapBC TA family is the most abundant type of TA system encoded by Mtb (60). Several studies have demonstrated that host-mediated stress, such as hypoxia and activated macrophages, induces transcriptional activation of multiple VapBC TA loci (61–63). Hudock et al. identified the transcriptional profile from granuloma samples of active and latent TB patients; the expression of eight dosR regulon members (Rv0080, Rv0081, Rv1736c, Rv1737c, Rv2032, Rv2625c, and Rv2630) along with the induction of four pairs of toxin/antitoxin (vapBC19, vapBC21, vapBC33, and vapBC34) were observed within the granulomas of active and latent TB patients (61). Sharma et al. demonstrated that VapC21 overexpression hinders mycobacterial growth, and co-expression of antitoxin VapB21 reverses this effect (62). Moreover, VapC21 overexpression mutant and Mtb cultured in stress conditions, such as nutrient deprivation and hypoxia, exhibited similar transcriptional profiles (62). Furthermore, VapC21 overexpression resulted in upregulated WhiB7 regulon, inducing antibiotic tolerance to aminoglycosides and EMB (62). Talwar et al. identified the role of VapBC12 TA in persister formation under cholesterol-rich conditions; VapC12 RNase toxin targets proT transcript that is indispensable for Mtb growth regulation in a cholesterol-rich environment (63). Therefore, the expression of VapC12 RNase toxin induced the generation of a slow-growing population, and this phenotype occurrence was increased in the presence of cholesterol (63). Interestingly, co-expressing of antitoxin vapB12 disrupted the vapC12-induced phenotype, while vapC12 deletion enhanced the immunopathologic severity and lung bacterial burden compared with the wild type strain (63). Recently, Yu et al. demonstrated a phosphorylation-dependent TA system in Mtb (58). Specifically, phosphorylation of TgIT by TakA induces toxicity neutralization and allows bacterial growth (58). In stressful conditions, TgIT activation via dephosphorylation promotes bacterial growth inhibition, leading to a non-replicating but viable state (58).
SOS Response
Various host-mediated stresses, such as reactive oxygen and nitrogen species, result in DNA damage and subsequently induce a DNA repair mechanism called SOS response (64). The SOS response is controlled by two regulator proteins, RecA and LexA. RecA recognizes damaged single-stranded DNA and induces the proteolysis of LexA repressor leading to the activation of SOS genes (64). Völzing and Brynildsen discovered that DNA repair was essential for the survival of ofloxacin-induced persisters and that delayed DNA repair occurred after ofloxacin treatment (65). Another study indicated that the timing of DNA repair was a key factor for the complete recovery of persisters after ofloxacin treatment. Additionally, nutrient starvation increased the survival rate of E. coli to approximately 100%, following ofloxacin treatment (66). These results indicate that changes in post-antibiotic treatment recovery time are critical to the formation of persister and support the notion that interference of DNA damage repair systems could be an effective strategy to eradicate the persister population.
Previous studies showed that stress-response regulons, including SOS response genes, were upregulated in Mtb persisters (8, 67). In mycobacteria, the DNA damage repair system comprises LexA-mediated and ClpR factor-mediated mechanisms (67). Further, DnaE2 polymerase, induced by ROS and NOS produced in the host immune response, contributes to mutations during the DNA repair process (68). Recently, inhibition of DNA gyrase by fluoroquinolone was found to modulate Mtb growth in intracellular and extracellular environments (69). Interestingly, inhibition of DNA gyrase contributes to the drug tolerance via RecA/LexA-mediated SOS response (69). Choudhary et al. demonstrated that DNA gyrase knockdown Mtb mutant showed decreased drug susceptibility to RIF 24 and 48 h post-treatment, and a similar pattern was observed following INH and EMB treatment (69). Taken together, these findings indicate that changes in post-antibiotic treatment recovery time are critical to the formation of persisters and support the notion that interference by DNA damage repair systems could be an effective strategy to eradicate the persister population.
Mtb Biofilm Formation Contributes to Antibiotic Tolerance
Biofilm is a three-dimensionally organized multicellular bacterial community that grows on surfaces in vitro and in vivo (70). Biofilm induces persistent bacterial infection by protecting bacteria from antibiotics (71). Therefore, the formation of biofilms has been closely linked to antibiotic tolerance in various bacterial pathogens, including E. coli, S. aureus, P. aeruginosa, and Mtb (72–76). Host-mediated stress conditions, such as prolonged hypoxia, oxidative stress, and nutrient starvation, induce biofilm formation leading to the development of antibiotic tolerance (77–81).
Ackart et al. showed that human leukocyte lysis enhanced biofilm formation, subsequently inducing antibiotic tolerance to several anti-TB drugs, such as INH, RIF, and pyrazinamide (PZA) (77). Interestingly, treatment with DNase I or tween scattered the established biofilm. It reversed the antibiotic tolerance, indicating that biofilm formation induced by host-mediated stress provides antibiotic tolerance that leads to persistent infection, and targeting the biofilm enhances drug sensitivity in Mtb (77). Another study induced Mtb biofilm formation in vitro through thiol reductive stress (TRS), resulting in drug-tolerant (INH, RIF, and EMB) phenotypes in which metabolic activity was maintained with the same levels of ATP/ADP, NAD+/NADH, and NADP+/NADPH (78). Furthermore, the TRS-induced biofilm formation was not interrupted by cell wall biosynthesis inhibitor antibiotics (INH and ETB), while DNA synthesis (levofloxacin and ofloxacin), RNA transcription (RIF), and protein synthesis (tetracycline) inhibitors disrupted its formation (78). Recently, Richards et al. identified several indispensable genes for Mtb adaptation during biofilm formation induced by host-mediated stresses, but not in dispersed culture using detergent (79). They observed that the formation of biofilm enhances the enrichment of antibiotic-tolerant cells and subsequently inducing RIF tolerance. Importantly, they established that isonitrile lipopeptide is essential for the structural formation of Mtb biofilm under stress conditions (79). In another study, modulation of trehalose metabolism was observed in antibiotic-tolerant Mtb population isolated from a biofilm; antibiotic-tolerant Mtb utilize trehalose to synthesize central carbon metabolism intermediates required to sustain mandatory cellular functions, whereas planktonic cells use cell-surface glycolipids (81). Moreover, drug-susceptible and MDR Mtb showed similar alteration after antibiotic therapy, suggesting the role of trehalose in both transient and permanent antibiotic tolerance (81). Tripathi et al. showed that ClpB is essential for Mtb survival under host-mediated stress conditions; they demonstrated that ClpB is required for bacterial survival during hypoxia and nutrient starvation (80). The clpB deletion mutant showed abnormal cellular morphology, disrupted biofilm formation, and reduced rate of intracellular survival in THP-1 cells (80). In addition, they showed that ClpB induces the secretion of inflammatory cytokines, such as TNF-α and IL-6, controlled by MAPK and NF-κB pathways (80). Taken together, various virulence factors involved in Mtb biofilm formation may be a potential novel drug target for the elimination of drug-tolerant bacteria.
Antibiotics Can Affect Host Immunity and Influence Clinical Outcomes
Host Metabolic Changes Induced by Antibiotics
Antibiotic activities against bacterial pathogens have traditionally been considered only in terms of their direct killing effects (82). However, growing evidence indicates indirect effects of antibiotics through interaction with host innate immunity that can alter clinical outcomes (83–85). Yang et al. identified antibiotic-induced host metabolic changes during infection and found that antibiotic treatment directly induced the host cells to produce metabolites that reduce drug efficacy and amplify phagocytic killing (86). After ciprofloxacin treatment, the systemic alteration of metabolites was confirmed in mouse tissues, including the peritoneum, plasma, and lungs. On the contrary, E. coli infection induced local changes in the peritoneum alone, not in the plasma or lungs (86). Further, most of the antibiotic-induced metabolic changes were not reliant on the intestinal microbiome and were most likely caused by the direct action of antibiotics on local host cells (86).
Above all, advanced host types of machinery protect cells by detecting and preventing damage due to intrinsic and exogenous inferior substances, such as oxidative stress and toxins (87). These apparatuses are partially responsible for microbial pathogenesis by detecting endogenous factors induced by Mtb infection or exogenous Mtb factors; however, they are responsible for detecting and detoxifying anti-TB drugs or drug-induced endogenous factors (88). These apparatuses may belong to the NRF2-KEAP1 and aryl hydrocarbon receptor (AhR) signaling pathways, whose dual action may be a double-edged sword in Mtb infection. This section evaluates the effect of such a system on Mtb infection and anti-TB treatment.
Keap1-Nrf2 Signaling Pathway
The Keap1-Nrf2 regulatory pathway is a key mechanism for preventing cell damage by detecting intrinsic and exogenous stresses, such as oxidative stress, chemotherapy, and radiation, regulating gene expression to modulate various subsequent antioxidant functions (87). Nrf2 is bound to the inhibitory protein Keap1 in the cytoplasm. When a stressor is detected, the Nrf2 protein is separated from Keap1, causing its cytoplasmic accumulation. Thereafter, it translocates to the cell nucleus, where it acts as a transcription factor, binds to the antioxidant reaction factor (ARE), and then binds to the antioxidant-related genes [e.g., hemeoxygenase-1 (HO-1), NAD(P)H:quinone redox enzyme-1 (NQO1), glutathione S-transferase (GST)] to promote their transcriptional expression (Figure 2) (87). Mtb factors, such as ESAT-6, can induce oxidative damage and apoptosis, counteracted by upregulating antioxidant enzymes via activation of the Keap1-Nrf2 signaling cascade (88, 89). Recent studies have shown that the antioxidant factor expressed by activation of the Keap1-Nrf2 system protects cells by removing infection and damage caused by drugs; however, it also inhibits T cell activation and rather hinders the removal of Mtb. Representatively, HO-1 is a cellular antioxidant enzyme expressed in response to various stress conditions, such as exposure to heavy metals, heat shock, hypoxia, starvation, and immune activation (90–95). HO-1 is the rate-limiting enzyme that degrades heme molecules into free iron, biliverdin, and carbon monoxide (CO) (96). Free iron inhibits nitric oxide (NO) production by acting on inducible NO synthase (iNOS) and, thereby, could improve the survival of intracellular Mtb.
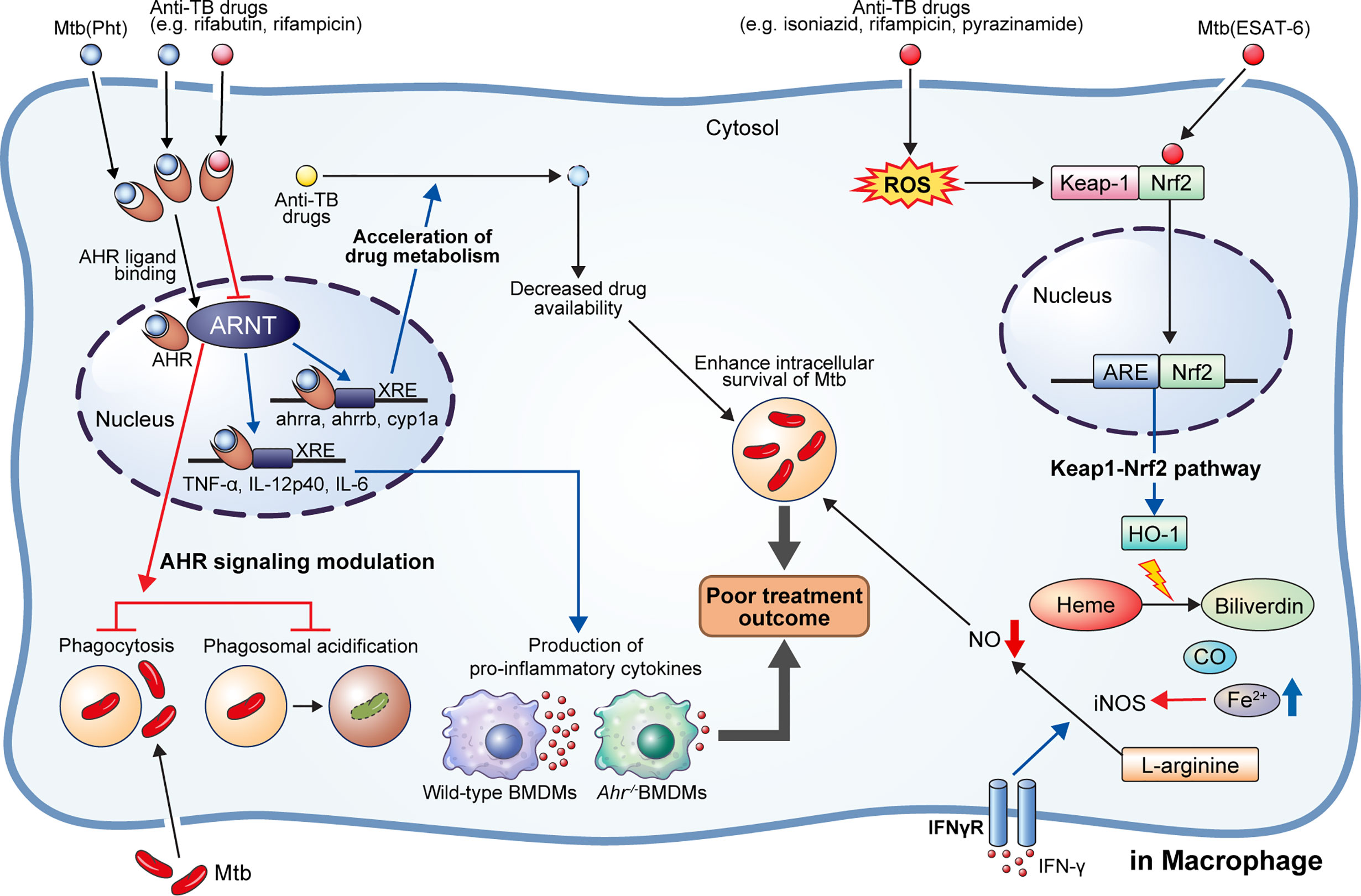
Figure 2 AhR modulation by anti-TB drugs and downstream events. NRF2-KEAP1 signaling and AhR signaling pathways generally protect cells by detecting and preventing damage to endogenous and exogenous substances such as oxidative stress and toxins. They can also detect M. tuberculosis (Mtb) infection or anti-TB drugs and affect host defense and drug metabolism. 1,4-naphthoquinone phthiochol (Pht) produced by Mtb and anti-TB drugs can attach to AhR present in the cytoplasm across the cell membrane. The combined ligand and receptor complex transfers into the nucleus and heterodimerizes with AhR nuclear translocator (ARNT). The ligand, receptor, and ARNT complex binds to xenobiotic response elements (XRE) that are specific DNA sequences found in the target gene promoter region. Activation of the AhR by Pht and anti-TB drugs (e.g., rifabutin, bedaquiline) in macrophages induces impaired phagocytosis of Mtb H37Rv, and phagosome acidification, and production of pro-inflammatory cytokines. Furthermore, activation of AhR facilitates the hepatic metabolism of drugs, ultimately reducing drug availability. Meanwhile, some drugs (e.g., rifampicin) act as AhR inhibitors, inducing impairment of phagocytosis and phagosome acidification, consequently improving the intracellular survival of Mtb in macrophage and zebrafish models. On the other hand, Mtb ESAT-6 and anti-tuberculosis drugs (e.g., isoniazid, rifampicin, pyrazinamide) act on Nrf2-Keap1 signaling to induce the translocation of Nrf2 to the nucleus by degradation of Keap-1. The translocated Nrf2 binds to the antioxidant response element (ARE) and upregulates antioxidant enzymes. Production of hemeoxygenase-1 (HO-1), a representative antioxidant enzyme, is activated, which subsequently induces catabolism of heme to biliverdin, CO, and Fe2+. Elevated Fe2+ inhibits the production of nitric oxide from L-arginine mediated by IFN-γ signaling. Thus, activation of heme catabolism by HO-1 induces the reduction of intracellular bacterial killing. Taken together, activation of AhR signaling and HO-1 production induces a pathogen-beneficial effect that enables persistent infection.
In addition, bactericidal antibiotics cause mitochondrial dysfunction and oxidative damage in mammalian cells (97). A dose- and time-dependent upregulation of intracellular ROS production was confirmed in different human cell lines after treatment with bactericidal antibiotics (ciprofloxacin, ampicillin, and kanamycin) belonging to different classes. Moreover, mitochondrial potential, ATP levels, and metabolic activity were considerably decreased after this treatment, suggesting impairment of mitochondrial function (97). Furthermore, treating human sinonasal epithelial cells with the bactericidal antibiotics, amoxicillin and levofloxacin, leads to increased ROS production, antioxidant gene expression, and cell death (98).
Bactericidal anti-TB drugs, such as RIF, INH, and PZA, can similarly cause mitochondrial dysfunction and oxidative damage in host cells, leading to apoptosis, in addition to their effect on Mtb. Simultaneously, antioxidant mechanisms, such as the Keap1-Nrf2 signaling pathway, may interfere with the removal of Mtb. Interestingly, ROS-mediated damage induced by antibiotics could be rescued by N-acetyl-l-cysteine (NAC) without affecting the antibiotic’s killing ability (97). In addition, an HO-1 inhibitor showed the same effect in the lungs of Mtb-infected mice as anti-TB drugs (99). Thus, the long-term use of bactericidal anti-TB drugs can induce cell death due to ROS production and mitochondrial dysfunction, while simultaneously, the produced ROS act as antioxidants and interfere with the removal of Mtb. These ambivalences need to be more clearly elucidated with respect to the pathogenesis of Mtb. The aforementioned adjuvant treatments are described in detail in section 4.
AhR Signaling Pathway
The AhR is a transcription factor that detects both endogenous and exogenous ligands (100). Initially, AhR function was associated with the detoxification of heterologous ligands, such as benzo[a]pyrene and the highly toxic 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD); subsequently, endogenous molecules, such as tryptophan (Trp), kynurenine, or formindolo [3,2-b] carbazole (FICZ), dietary components, and bacterial-derived ligands, were identified as AhR ligands, broadening the understanding of their function (100).
Particularly, bacterial pigment proteins such as phenazine produced by P. aeruginosa and 1,4-naphthoquinone phthiochol (Pht) produced by Mtb have been identified as bacterial-derived AhR ligands (101). AhR is widely expressed in almost all cell types; in particular, both innate and adaptive immune cells express AhR, suggesting its potential broad-range effects on host immunity (102). AhR is present in the cytoplasm and is activated upon binding to a ligand. Activated AhR binds to the AhR nuclear translocator (ARNT) and regulates the transcription of several target genes, including cytochrome P450 monooxygenases (CYP1A1 and CYP1B1), AhR inhibitor, and pro-inflammatory cytokines (102) (Figure 2).
Ligand-activated AhR translocates to the nucleus from the cytosol and induces immunosuppressive or pro-inflammatory downstream effects depending on the ligand property (102). Further, AhR can modulate macrophage immune response. Shinde et al. showed that phagocytosis of apoptotic cells through toll-like receptor (TLR)9-dependent sensing of the apoptotic cell DNA induces the activation of the AhR pathway (103).
The ligand-activated AhR binds to TB virulence factors and regulates antibacterial responses (104). Puyskens et al. demonstrated that anti-TB drugs, such as RIF and rifabutin bind to AhR and induce modulation of host immune response (104). However, AhR signaling inhibition by a synthetic AhR inhibitor, CH-223191, impairs phagocytosis in THP-1 macrophages. Further, they demonstrated that the rate of internalized zymosan was decreased following RIF treatment, while phagosome acidification was also impaired after RIF as well as CH-223191 treatment (104). In addition to this in vitro study, they confirmed AhR modulation during M. marinum infection in a zebrafish model; a higher bacterial burden was observed in zebrafish embryo following AhR inhibition with CH-223191 than in the untreated control group (104). Similarly, Moura-Alves et al. demonstrated significantly increased bacterial burden in the lungs, liver, and spleen of AhR-/- mice than wild type mice after aerosol Mtb infection (101). Moreover, the production of pro-inflammatory cytokines, such as TNF-α, IL-12p40, and IL-6, were hindered in AhR-/- bone marrow-derived macrophages (101). Furthermore, Memari et al. demonstrated that AhR induced expression of IL-23 and IL-1β, thereby stimulating the production of IL-17 and 22 by specific T cell subsets (Th17, Th22, and ILC3 cells) (105). Upregulation of IL-17 activates parenchymal cells and subsequently induces an influx of polymorphonuclear cells to the infection site mediated by CXCL1, CXCL3, and CXCL5 (106). Further, phagocytosis of apoptotic polymorphonuclear cells by macrophages promotes a phenotypic change of macrophage from M0 to M2c, thereby contributing to inflammation resolution (106).
Modulation of Host Immunity by Anti-TB Drugs
Antibiotics can modulate host immunity either indirectly or directly (107). First, antibiotics alter the host immune system indirectly by affecting the host microbiota composition (108). Second, antibiotics affect the host immune system directly by altering the functions of immune cells (86, 97, 109). Therefore, the interaction between antibiotics and host immunity may influence the clinical outcomes or treatment duration. Several studies have reported the modulation of host immune response by anti-TB drugs. For example, INH induces the apoptosis of activated CD4+ T cells in Mtb-infected mice (110) as well as impairs the production of Mtb-specific interferon (IFN)-γ and anti-CFP10 antibody in household contacts of latent TB patients (111). Similarly, RIF reportedly exerts a mild immunosuppression effect, as indicated by its inhibition of human lymphocytes (112) and significant suppression of T cells compared to that in TB patients without RIF treatment (113). Moreover, RIF partially suppressed the phagocytosis of zymosan by macrophages and moderately suppressed the expression of TNF-α at high doses (114). it was reported to significantly inhibit the secretion of IL-1β and TNF-α while increasing the secretion of IL-6 and IL-10 (115). Furthermore, RIF suppressed LPS-induced production of iNOs, cyclooxygenase-2, IL-1β, TNF-α, and prostaglandin E2 in microglial cells, subsequently improving neuron survival (116). Manca et al. demonstrated that PZA treatment reduces the secretion of pro-inflammatory cytokines and chemokines, such as IL-1β, IL-6, TNF-α, and MCP-1, in Mtb-infected human monocytes and mice (117). Additionally, PZA treatment elevated the expression of adenylate cyclase and peroxisome-proliferator activated receptor in the lungs of Mtb-infected mice (117).
Bedaquiline (BDQ) specifically disrupts intracellular ATP production in bacteria by inhibiting the activity of bacterial ATP synthase, resulting in depleted energy production (118, 119). Recently, a genome-wide transcriptional analysis demonstrated that BDQ promotes the formation of lysosomes, phagocytic vesicle membrane, vacuolar lumen, hydrolase activity, and lipid homeostasis in naïve and Mtb-infected macrophages (120). Moreover, it suppressed basal glycolysis, reduced glycolytic capacity in heat-killed-Mtb-stimulated macrophages, and triggered anti-mycobacterial mechanisms, such as phagosome–lysosome fusion and autophagy (120). Further, BDQ treatment induced the activation of the lysosomal pathway through transcription factor EB and calcium signaling. Interestingly, other classical anti-TB drugs, such as amikacin, EMB, and INH, did not activate the lysosomal pathway. Additionally, BDQ potentiated the anti-mycobacterial activity of PZA but did not show synergistic effects with bactericidal activities of EMB, INH, and RIF (120).
Clofazimine (CFZ) is a riminophenazine compound used for the standard treatment of leprosy (121). In addition to its antimicrobial activity, CFZ has an immune-modulatory activity; CFZ forms biocrystal and modulates innate immune response after phagocytosis as demonstrated by the intracellular CFZ crystal-induced activation of the Akt pathway and enhancement of IL-1RA production in RAW 264.7 cells (122). Moreover, CFZ treatment inhibited TLR2-and TLR4-mediated NF-κB activation and TNF-α production (122). Fukutomi et al. demonstrated that CFZ induces apoptosis of macrophages; representative features of apoptosis, such as decreased metabolic activity, diminished cell size, nuclear condensation, and fragmentation, were observed in CFZ-treated human monocyte-derived macrophages (123). Further, caspase-3 activity was significantly increased in CFZ-treated macrophages (123). Recently, Ahmad et al. showed that BCG revaccination with CFZ treatment induces the differentiation of the stem cell-like memory T (Tsm) cells in mice (124). Differentiation of Tsm cells recovered long-lasting central memory T cells and T effector memory cells to provide enhanced vaccine efficacy in mice (124).
Adjunctive Host-Directed Therapies Improve Anti-TB Drug Activity
Anti-TB therapy involves the combination of several drugs and has a long treatment duration of treatment, resulting in the frequent occurrence of side effects (125, 126). Side effects range from minor ones that disappear spontaneously to serious ones that require treatment (126). The strategies for developing new treatments to control Mtb can be divided into two broad categories: developing novel efficient antibiotics and using existing therapeutic drugs to achieve faster and more effective treatments in a host-specific manner (127, 128). The development of host-directed therapy maximizes treatment efficiency by using the adjunct to elicit a response (127). Essentially, these treatments do not directly target pathogens, thus avoiding the occurrence of drug resistance and reducing drug side effects, thereby making this a promising strategy (127). In this section, we have proposed several such strategies, including various host targets that affect Mtb susceptibility, and discussed the corresponding drugs and their mechanisms of action. A summary of the proposed adjunctive host-directed therapies that improving anti-TB drug activity is presented in Table 1.
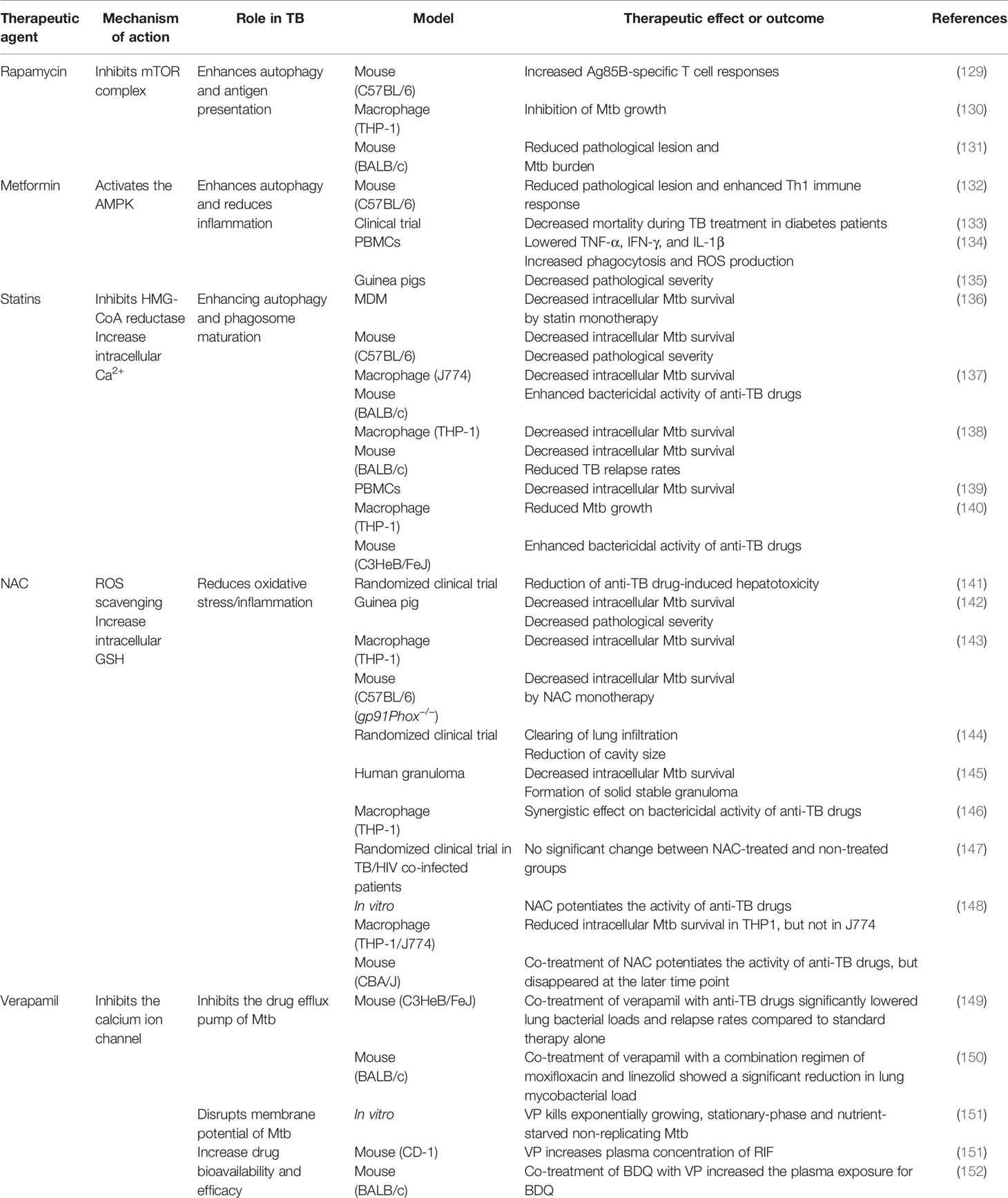
Table 1 Effect of TB representative adjunctive therapeutic agents of anti-TB drugs on host immunity.
Autophagy-Modulating Drugs
Autophagy is an intracellular self-degradation system that transfers cytoplasmic components or specific cytosolic targets to the lysosome for cellular homeostasis maintenance (153). Autophagy is induced by various stress conditions, such as nutrient starvation, hypoxia, and microbial infection (153). Furthermore, diverse pathophysiological conditions, such as aging, autoimmune disease, neurodegeneration, cancer, and inflammation-associated metabolic disorders, are involved in autophagy (154). Autophagy is an essential part of the host immune system against diverse intracellular pathogens, such as Salmonella, Listeria monocytogenes, and Mtb, via the activation of phagolysosome formation (155–157). Therefore, activating autophagy is a promising strategy for eradicating Mtb, especially in the case of MDR-TB.
Rapamycin
Rapamycin (RAP) is a potent antifungal agent produced by Streptomyces hygroscopicus and is used to suppress transplant rejection reactions due to its immunosuppressive property (158, 159). RAP also enhances the T helper 1-driven immune response when co-administered with the BCG vaccine (129). Further, RAP-loaded nanoparticles were efficiently phagocytosed by THP-1 macrophages, significantly reducing the intracellular Mtb load at a concentration of 100 μg/mL (130). In addition to the in vitro results, Gupta et al. showed that inhaled RAP particles reduced pulmonary Mtb loads as well as activated autophagy and phagosome–lysosome fusion in a mouse model (131) (Figure 3). Another study showed that a low dose of RAP (<10 μM) was sufficient to increase autophagy in RAW264.7 cells; however, intracellular mycobacteria killing was only observed at a high concentration of RAP (10 μM) (160). In contrast, Andersson et al. demonstrated that RAP facilitates the increase of Mtb burden in both single and human immunodeficiency virus (HIV) co-infected human monocyte-derived macrophages (161). They suggested that autophagy induction disrupts intracellular killing during a low dose infection. However, they used HIV co-infected human macrophages for their experiment that exhibit impaired immunity and, therefore, are not suitable for investigating the effect of RAP on autophagy.
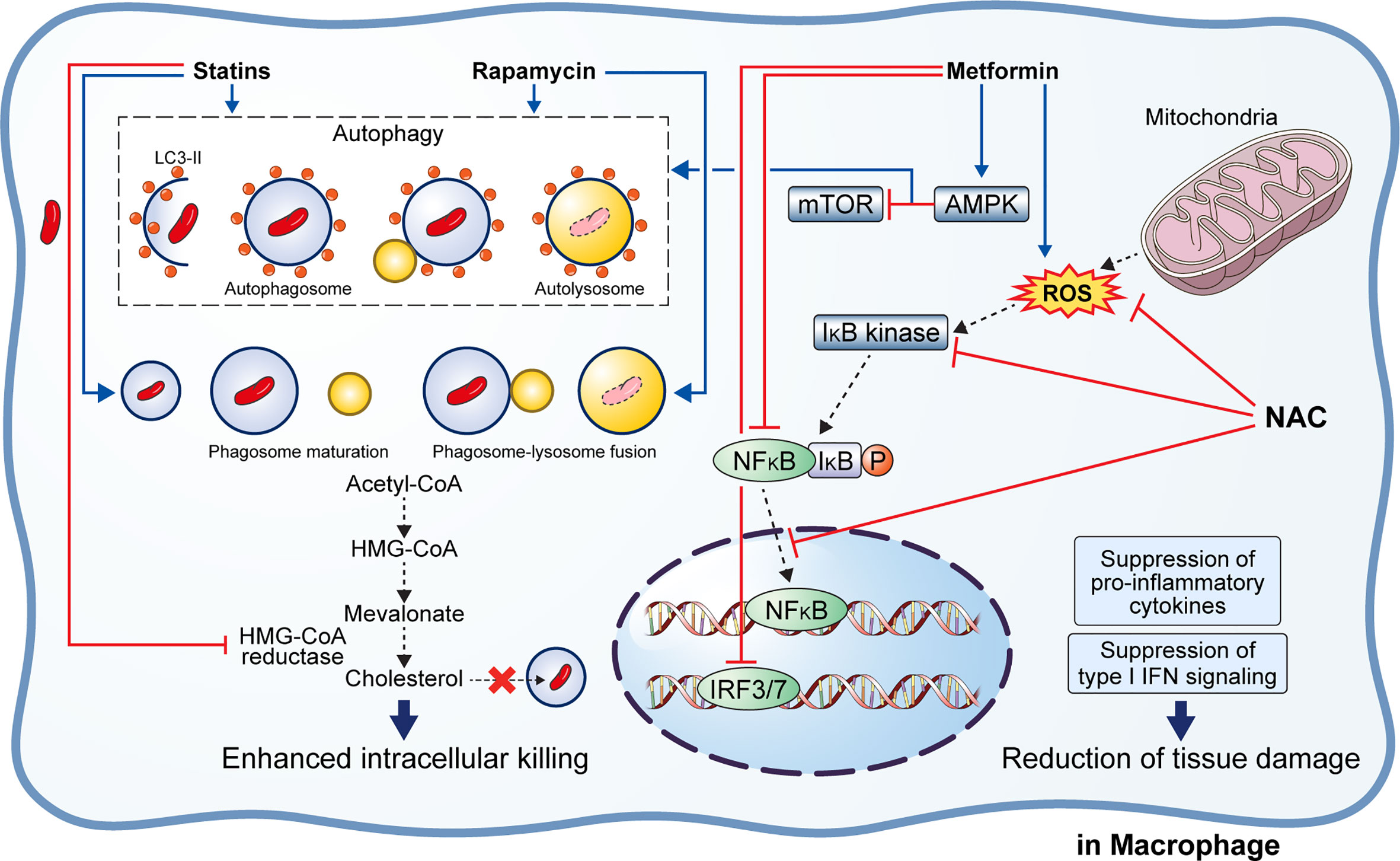
Figure 3 Effect of adjunctive therapeutic agents of anti-TB drugs on host immunity. Various adjunctive drugs aid TB treatment by modulating the host immune response. M. tuberculosis (Mtb) can accumulate cholesterol for use as a source of carbon and energy. Statins bind to the active site of HMG-CoA reductase, thereby inhibiting cholesterol biosynthesis. In addition, statins induce autophagy and phagosome maturation to promote the removal of Mtb. Similarly, rapamycin induces autophagy and phagosome–lysosome fusion to enhance the intracellular killing of Mtb. Metformin inhibits the mTOR complex via AMPK activation in the mitochondria to promote autophagy. Metformin also inhibits ROS production, NFκB signaling, and type I interferon signaling. Similarly, N-acetyl-l-cysteine eliminates the generated ROS and inhibits NFκB signaling. Suppression of pro-inflammatory immune response and type I interferon signaling lead to reduced immunopathological severity that beneficial to the host.
Metformin
Metformin (MET) is the most commonly used medication for diabetes and has been suggested as an adjunctive agent for host-directed TB therapy (132). Singhal et al. discovered that the MET disrupts the intracellular Mtb growth, reduces immunopathological severity, and increases the efficacy of anti-TB drugs (132). In detail, MET treatment reduced the bacterial load (in terms of CFU) 35 days post-infection in Mtb-infected mice (132). The combination of MET and anti-TB drugs, such as INH and ethionamide, significantly decreased lung Mtb load in the mouse model, indicating synergism between MET and anti-TB drugs (132). Additionally, reduction of lung tissue pathology was confirmed in the MET-treated mice, and the number of lung CD8+ IFN-γ+ cells was increased in MET-treated mice in both unstimulated and Mtb-stimulated groups, suggesting an enhanced immune response to TB (132). Similarly, the protective effect of MET was confirmed in a chronically Mtb-infected guinea pig model in which reduced lung legions and Mtb CFU were observed in the MET-treated group compared to those in the untreated group (135). Further, the MET-treated animals showed a higher proportion of lymphocytes in the acute and subacute stages and well-encapsulated granulomas (135). MET reportedly reduces the immunopathological severity by reprogramming T cell metabolism (162, 163). MET-induced oxidative phosphorylation and glycolysis enhanced the host resistance to Mtb infection in the guinea pig model (135). Moreover, Degner et al. demonstrated that MET treatment reduces mortality during TB therapy in a retrospective cohort study in Taiwan (133). Another study reported that MET enhances anti-TB immune responses by altering host responses in humans (134). Additionally, MET induced a significant reduction in TNF-α, IL-1β, IL-6, IFN-γ, and IL-17 release in response to Mtb lysate (134). These studies indicate that MET protects against Mtb infection via modulation of inflammation and metabolism (133–135). Indeed, MET reduces the type I IFN response and pathological severity of TB while enhancing anti-TB immunity, such as autophagy, ROS production, and phagocytosis (132, 164, 165) (Figure 3). In contrast, Dutta et al. showed that MET-treated mice had similar lung Mtb CFU compared to control mice, and the percentage of mice with Mtb culture relapse was similar between the two groups (166). This result was contradictory to a previous in vivo study that used the same MET dose (250 mg/kg) (132). The major difference between the two studies is the co-injection of multiple anti-TB drugs (RIF, INH, PZA, and EMB) in the in vivo model compared with the single anti-TB drug (INH or EMB) used in vitro (132, 166). In addition to its antimicrobial activity, RIF promotes liver metabolism by activating hepatic cytochrome P450 enzymes, such as CYP2D6 and CYP3A4, thereby accelerating the drug metabolism and clearance by the liver (167). However, MET is not metabolized in the liver and is excreted via the urine in its unchanged form (168). Another potential explanation is that MET and RIF compete for the same drug target, namely the AMP-activated protein kinase (AMPK) in the liver (109, 169). Therefore, the combination with RIF reduces the effect of MET in the host.
Statin
Statins are anti-hyperlipidemic drugs that block 3-hydroxy-3-methylglutaryl coenzyme A reductase in the cholesterol synthesis pathway, thereby lowering the risk of stroke and cardiovascular diseases (170) (Figure 3). As cholesterol is an essential intracellular energy source for Mtb, elevated cholesterol level is a risk factor for TB (171). Statins also have immunomodulatory effects, such as the production of natural killer T cells, downregulation of MHC II expression, elevated secretion of IL-1β and IFN-γ, and increased caspase-1 enzyme activity, and thereby promote apoptosis and autophagy (172) (Figure 3). Both retrospective clinical trials and animal model studies have reported that statins are effective in the treatment and prevention of TB (136, 173, 174). Decreased bacterial burden was confirmed in peripheral blood mononuclear cells and monocyte-derived macrophages from patients with familial hypercholesterolemia during statin treatment compared to healthy donors (136). Further, statin treatment reduced the Mtb burden and histopathological severity in the lungs of Mtb-infected mice (136). Lobato et al. evaluated the effects of two statins (atorvastatin and simvastatin) alone and in combination with RIF on M. leprae and Mtb in THP-1 macrophages (175). Both statins showed bactericidal effects on intracellular mycobacteria 72 h post-infection and synergism with RIF at a concentration of 0.2 µM for reducing the viability of intracellular Mtb (175). Skerry et al. investigated the bactericidal activity of simvastatin alone and in combination with anti-TB drugs (RIF, INH, and PZA) in macrophages and a mouse model and found that the addition of 5 mM simvastatin significantly enhanced the bacterial killing of INH in Mtb-infected J774 macrophages (137). In contrast, the addition of 25 mg/kg simvastatin to the standard TB treatment regimen significantly reduced the lung bacterial burden in BALB/c mice (137). Similarly, Dutta et al. found that the addition of simvastatin (60 mg/kg) to the TB treatment regimen (INH/RIF/PZA) shortened the duration required to attain culture-negative results from 4.5 to 3.5 months, i.e., shortened the treatment duration (138). Simvastatin significantly improved the bactericidal activities of anti-TB drugs against intracellular Mtb while having no effect on intracellular RIF concentrations (138). The same research group further showed that various statins, including pravastatin, simvastatin, and Fluvastatin, improved the antimicrobial activity of INH, RIF, and PZA in THP-1 cells and the C3HeB/FeJ mouse model (140). Additionally, pravastatin induced phagosome–lysosome fusion and macrophage activation observed in IFN-γ- and LPS-activated macrophages (140). Interestingly, Guerra-De-Blas et al. demonstrated that simvastatin alone significantly reduced bacterial load in Mtb-infected PBMCs via enhanced production of natural killer T cells, upregulation of co-stimulatory molecules in monocytes, increased the secretion of IL-1β and IL-12p70, and promotion of apoptosis and autophagy in monocytes (139).
ROS Modulating Drugs
ROS and reactive nitrogen species (RNS) are critical host defense mechanisms to eradicate pathogens during infection (176). The ROS and RNS react with the phagosomes and efficiently eliminate intracellular bacteria. However, excessive ROS production may cause mitochondrial damage and cell apoptosis, leading to severe immunopathologic outcomes (177). Therefore, appropriately balanced cellular ROS levels are critical for eliminating intracellular Mtb without causing a detrimental effect in the host.
N-Acetyl-Cysteine (NAC)
Multiple studies have shown that reducing ROS accumulation in the Mtb-infected host by NAC inhibited the Mtb growth and reduced the immunopathological severity despite the contradictory views on the Mtb killing ability of NAC (142, 143, 145, 148, 178–182) (Figure 3). Several studies have demonstrated that NAC ameliorates aminoglycoside-induced ototoxicity (183–185). Venketaraman et al. found that glutathione level was significantly reduced in PBMCs and RBCs isolated from TB patients (181). Further, reduced secretion of IL-10, IL-6, TNF-α, and IL-1 was confirmed in blood cultures of TB patients after NAC treatment (181). Similarly, Palanisamy et al. showed that NAC treatment moderately increased blood glutathione level and the serum antioxidant capacity in Mtb-infected guinea pigs, reduced the bacterial burden in the spleen, and decreased the immunopathological severity in the lungs and spleen of animals (142). Subsequently, Guerra et al. demonstrated that increased glutathione level due to NAC treatment inhibits the intracellular growth of Mtb by causing an increase in the levels of IL-2, IL-12, and IFN-γ secreted by T cells (180). Furthermore, NAC attenuates liver injury induced by anti-TB drugs by promoting free radical scavenging and glutathione synthesis (141).
Similarly, Amaral et al. demonstrated that NAC inhibits the growth of diverse pathogenic mycobacteria such as Mtb, M. bovis, and M. avium (143). The mycobacterial loads in the lungs of NAC-treated animals were significantly reduced compared to that in the untreated animals in both wild type and gp91Phox-/- macrophages, suggesting that the anti-TB activity of NAC is independent of the host NADPH oxidase system (143). Lamprecht et al. showed that NAC potentiates the bactericidal activity of BDQ, Q203, and CFZ in an in vitro macrophage model (179). They found that the addition of NAC significantly improved bactericidal activity of the three anti-TB drugs, leading to complete Mtb sterilization (179). In another study, NAC treatment caused a 50% reduction in bacterial load (in terms of CFU) in THP-1 macrophages and potentiated the bactericidal effect of anti-TB drugs, such as INH, RIF, EMB, or PZA (146). Moreover, NAC treatment can modulate TNF-α levels to maintain granuloma structure without inducing detrimental cell damage to the host (146). Similarly, Teskey et al. showed that incubation of Mtb Erdman strain with NAC significantly inhibited the bacterial growth, while incubation with a combination of NAC and anti-TB drugs (INH, RIF, and EMB) completely sterilized the Mtb culture (145). In addition, NAC treatment significantly increased the IFN-γ level while decreasing that of TNF-α as well as significantly enhanced the phagosome acidification in human granulomas, which indicates improved intracellular killing (145). On the contrary, NAC alone did not kill Mtb in macrophages, whereas INH and NAC combined showed an improved bactericidal activity than INH alone (178). However, Khameneh et al. demonstrated that the combination of anti-TB drugs (RIF and INH) and vitamin C, but not NAC, induced synergistic effects for bacterial killing (182). However, there were a few inconsistencies in their results. For instance, in the presence of 20 μg/mL RIF, treatment with 0.05 mg/mL NAC showed 10% CFU, whereas treatment with 0.1 mg/mL NAC showed 150% CFU compared to untreated controls (182). Recently, Vilchèze et al. showed that NAC improves the sterilizing activity of first and second-line anti-TB drugs in vitro against drug-susceptible and drug-resistant Mtb strains (148). However, a synergistic effect between NAC and anti-TB drugs was not observed in Mtb-infected mice (148). Moreover, although NAC initially inhibited Mtb growth, the NAC-induced growth inhibition was not significant and was lost after the first week of treatment (148). The major difference between the controversial studies is the host species. The direct killing effect of NAC was not seen in studies using in vivo or in vitro mouse models. Similarly, clinical trials on the adjunct effect of NAC on TB therapy in TB patients showed contradictory results (144, 147). Mahakalkar et al. showed that NAC treatment significantly shortened the duration of anti-TB therapy in TB patients (144). On the contrary, a recent clinical trial in Brazil demonstrated that NAC addition to a standard TB regimen did not reduce the duration required to achieve a negative sputum culture, nor did it reduce radiological severity in hospitalized patients with severe TB and HIV co-infection (147) (Table 1). However, these trials did not include a sufficiently large study population. Therefore, a large-scale clinical study is needed to determine the safety and efficacy of NAC treatment in TB.
Vitamin C
Vitamin C (VC) is an essential nutrient for humans that possesses reducing and antioxidant abilities associated with its ability to donate electrons (186). Several studies have investigated the host beneficial or detrimental roles of VC in the pathogenesis of TB (187–192). Vilchèze et al. demonstrated that VC kills drug-susceptible and drug-resistant Mtb via Fenton reaction in a dose-dependent manner in vitro, and 4 mM of VC completely sterilized Mtb culture at three weeks after treatment (187). VC is assumed to kill Mtb by increasing the intracellular ROS level, and this process depends on the intracellular iron concentration (187). The same research group also showed that a combination of VC and anti-TB drugs sterilizes Mtb cultures faster than monotherapy with anti-TB drugs (190). Further, Susanto et al. revealed that administration of VC improves the sputum conversion culture rate in RIF-susceptible Mtb-infected patients (189). On the contrary, Sikri et al. suggested that VC induces Mtb dormancy leading to a viable but non-culturable state (188). VC-treated Mtb showed antibiotic tolerance, thereby exhibiting a higher survival rate than untreated Mtb culture in the presence of anti-TB drugs (188). Similarly, Nandi et al. demonstrated that VC induces the activation of multiple transcriptional regulators for the temporal adaptation to VC, leading to a dormancy response (191). In summary, VC sterilizes Mtb culture by generating ROS via Fenton reaction and promoting oxygen consumption, thus eradicating bacterial persisters. However, several studies report that VC promotes the generation of bacterial persisters in TB. Therefore, further research is needed to elucidate the role of VC in the pathogenesis of TB.
HO-1 Inhibitor
In section 3, we discussed that Mtb and bactericidal anti-TB drugs cause mitochondrial dysfunction and oxidative damage in host cells, consequently ROS-mediated apoptosis as well as simultaneous activation of antioxidant mechanisms, such as the Keap1-Nrf2 signaling pathway, that may interfere with the removal of Mtb (88, 89) A major factor involved in this mechanism is HO-1 (95, 192). However, the detailed role of host HO-1 during the onset and pathogenesis of TB remains controversial and has not been fully elucidated (95, 99, 192–195). HO-1 exerts anti-inflammatory and cytoprotective effects, although the underlying mechanisms are not fully understood (196). Several studies have investigated the host beneficial or detrimental roles of HO-1 during TB infection (95, 99, 192–195). Andrade et al. showed that active TB patients show a negative correlation between plasma levels of HO-1 and MMP-1 (95). Notably, the TB patients with high plasma levels of HO-1 or MMP-1 demonstrated unique clinical presentation and inflammatory cytokine profiles (95). Moreover, a high HO-1 level was induced by the infection of virulent Mtb strain in human or murine macrophages, and MMP-1 expression was inhibited by CO by suppressing c-Jun/AP-1 signaling (95). Costa and colleagues demonstrated that the administration of tin protoporphyrin IX (SnPPIX), an HO-1 enzymatic inhibitor, decreases pulmonary Mtb loads comparable to that accomplished by anti-TB drug therapy as well as improves the bactericidal activity of anti-TB drugs (RIF, INH, and PZA) (192). Interestingly, host T cell immune response was needed to inhibit HO-1 by SnPPIX, and SnPPIX failed to reduce bacterial growth and activity of Mtb HO-1 enzyme in broth culture (192). Rockwood et al. reported HO-1 upregulation in Mtb-infected rabbits, mice, and non-human primates, and anti-TB therapy reduced the HO-1 plasma levels (193). Similar upregulation of HO-1 was observed in the plasma of untreated HIV-1 co-infected TB patients. In these patients, the plasma HO-1 levels positively and negatively correlated with the HIV-1 viral load and CD4+ T cell count, respectively (193). Further, early secreted antigen ESAT-6-mediated nuclear translocation of transcription factor NRF-2 is required for Mtb-induced HO-1 expression (193). Recently, Costa et al. discovered that HO-1 inhibition improves IFN-γ-induced NOS2-dependent bacterial killing by murine macrophages (99). Additionally, HO-1 inhibition induced low intracellular non-protein bound iron in Mtb-infected macrophages and reduced iron deposition in the lungs of Mtb-infected mice (99). Taken together, HO-1 expression inhibits T cell-mediated IFN-γ-induced NOS2-dependent control of Mtb by producing free iron. Therefore, inhibition of HO-1 expression potentiates the anti-TB therapy and improves clinical outcomes.
Calcium Channel Blocker
Verapamil (VP) is a calcium channel blocker used for hypertension treatment that also acts as an inhibitor of drug efflux protein. Several studies have proposed that VP can potentially improve the bactericidal activity of anti-TB drugs, such as RIF, INH, EMB, BDQ, and CFZ (151, 197–199). Machado et al. demonstrated that VP disrupts the heightened antibiotic resistance induced by repetitive exposure to INH (197). Similarly, Gupta et al. showed that VP potentiates the bactericidal activity of BDQ in reference strain H37Rv and eight clinical Mtb isolates (198). Similarly, Li et al. suggested that the addition of VP improves RIF susceptibility in RIF-resistant Mtb isolates (199). Chen et al. suggested that VP monotherapy kills exponentially growing, stationary-phase, nutrient-starved, non-replicating Mtb by disrupting membrane energetics without affecting the physical integrity of the membrane (151). Further, VP potentiates the bactericidal effects of BDQ and CFZ in vitro and in a RIF mouse model without changing the intracellular concentration of the drugs (151). Similarly, Xu et al. demonstrated that VP potentiates the efficacy of BDQ and CFZ against Mtb clinical isolates (152). However, VP increased bioavailability and efficacy of BDQ but not CFZ in Mtb-infected mice (152). Collectively, synergistic activity of VP in vivo may be attributed to improved systemic exposure to co-treated drugs by modulating mammalian transporters without inhibiting bacterial efflux pumps (152). Thus, the combination of VP and anti-TB drugs may be an effective therapy for TB.
Cytokines
Cytokines are small soluble proteins with multifaced aspects of host protective and detrimental effects (200). Excessive TB-induced inflammation interferes with normal lung function and may increase the risk of TB relapse (201). Comprehensive insight into host-pathogen interaction in TB would aid in designing host-directed therapies to shorten antibiotic treatment duration and relieve immunopathology. Immunotherapy with several cytokines protects the host and boosts bacterial clearance (Table 2).
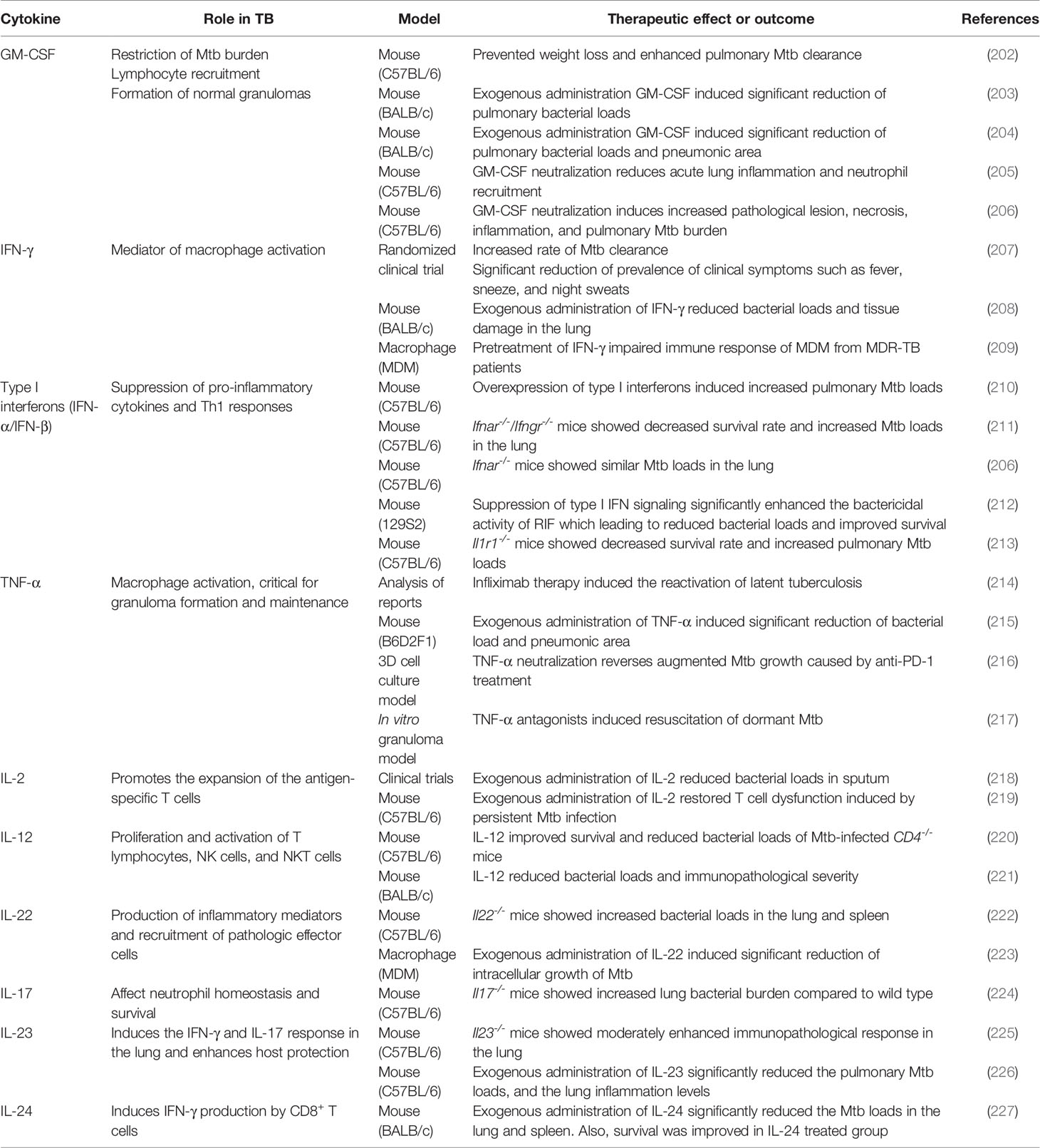
Table 2 Effect of cytokines on host immunity in TB.
IFN-γ and IL-12
IL-12/IFN-γ axis plays a critical role in host immune response for controlling TB, and IFN-γ is the key cytokine in the innate immune response during Mtb infection (219). IFN-γ is responsible for enhancing bactericidal activity through the upregulation of ROS, RNI, and autophagy (155, 228, 229). IFN-γ reverses the blockade of phagosome–lysosome fusion caused by Mtb (230). Moreover, IFN-γ stimulates the production of IL-12 in Mtb-infected macrophages, while IL-12 stimulates the production of IFN-γ by T cells and NK cells, thus activating macrophages that lead to intracellular bacterial killing (231). Dawson et al. reported a significant increase in CD4+ lymphocyte response and significantly reduced Mtb load in the sputum of the recombinant IFN-γ1b-treated group (207). Mata-Espinosa et al. demonstrated that exogenous administration of IFN-γ reduces bacterial loads and tissue damage in the lungs of Mtb-infected mice (208). Along similar lines, Khan et al. showed that treatment with exogenous IFN-γ restored defective immune response of MDMs isolated from MDR-TB patients (209). Exogenous administration of IL-12 improved survival and reduced the bacterial loads of Mtb-infected CD4-/- mice (220). Similarly, treatment with recombinant adenovirus encoding IL-12 (AdIL‐12) significantly reduced the bacterial loads in a progressive pulmonary TB mouse model (221). AdIL-12-treated mice showed significantly higher levels of IFN-γ, TNF-α, and iNOS compared with the untreated group. In addition, AdIL-12-treated mice showed less severe pathological legions than the untreated mice (221).
TNF-α
TNF-α plays an important role in controlling TB in both the initial and late stages (232). TNF-α activates macrophages and contributes to the formation and maintenance of granulomas to suppress the dissemination of Mtb; however, it also induces tissue damage due to the excessive immune responses (233). Keane et al. demonstrated that treatment with TNF-α inhibitor induces the reactivation of latent TB (234). Similarly, treatment with several TNF-α antagonists differentially induced resuscitation of dormant Mtb in a 3D microgranuloma model (214). Adalimumab, a TNF-α antagonist, showed a greater resuscitation rate than etanercept through the TGF-β1-dependent pathway (214). Furthermore, exogenous administration of TNF-α significantly decreased the bacterial load and pneumonic area in Mtb-infected mice (217). In contrast, Tereza et al. demonstrated that excessive TNF-α secretion via PD-1 inhibition facilitated Mtb growth in a micro granuloma model (215).
IL-24
IL-24 plays an immune-regulatory role to induce Th1 cytokines, such as IFN-γ, IL-6, and TNF-α, during TB infection (216). Upregulated IL-24 expression was observed in BCG-vaccinated newborns, suggesting the host protective role of IL-24 during TB infection (235). Wu et al. demonstrated that administration of exogenous IL-24 induces IFN-γ upregulation, whereas IL-24 neutralization causes IFN-γ downregulation (216). Similarly, IL-24 stimulation results in the upregulation of IFN-γ-inducing cytokines, such as IL-12, IL-23, and IL-27 (216). Furthermore, Ma et al. demonstrated that administration of IL-24 induced IFN-γ production and activated CD8+ T cells in mice, indicating its host protective effect in TB (227). Huang et al. found that IL-24 family cytokines, such as IL-19, IL-20, and IL-22, are elevated in BCG-vaccinated non-human primates as well (236). Treatment with exogenous IL-24 significantly increased the survival rate and significantly reduced the bacterial burden compared to the control group (Table 2).
IL-2
IL-2 plays a critical regulatory role during T cell differentiation via induction of the transcription factor eomesodermin and perforin (237). During chronic viral infection, the production of memory T cells and memory T cell-associated molecules, such as CD127 and CD44, was observed after stimulation with low-dose IL-2 (238). IL-2 administration is proven to exert host protective effects in TB infection; it reduced or cleared bacterial burden and increased the number of CD25+ and CD56+ cells (239). A similar protective effect of exogenous IL-2 was reported by Shen et al., who showed that the IL-2-treated group showed decreased sputum smear-positive rates, whereas the control group showed increased sputum smear-positive rates (218). The IL-2-treated group also demonstrated less severe legions than the control group during TB treatment (218). Liu et al. showed that persistent stimulation with Mtb antigen induces disrupted cytokine production by T cells, and IL-2 restores the T cell dysfunction (219). Their findings suggested that administration of exogenous IL-2 leads to maintenance of immune homeostasis in the bone marrow and reactivation of the disrupted hematopoiesis by persistent Mtb infection (219).
Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF)
Previous studies showed that GM-CSF exerts a host protective role during TB infection (202–205, 240–242). It mediates the formation of granuloma and promotes bacterial clearance in the host (241), and induces classical activation of macrophage to M1 polarization state (242).
Treatment with recombinant adenoviruses encoding GM-CSF significantly reduced the bacterial burden in the lungs, increased the number of activated DCs, and elevated the levels of TNF-α, IFN-γ, and iNOS (203). The same group further demonstrated that exogenous administration of GM-CSF significantly reduced the pulmonary Mtb loads and pneumonic area in Mtb-infected mice (202). Similarly, Pasula et al. demonstrated that exogenous keratinocyte growth factor administration protects against Mtb infection through GM-CSF-dependent macrophage activation and phagosome–lysosome fusion (204). Moreover, neutralization of GM-CSF induced higher bacterial burden and increased immunopathologic severity with necrotic granulomatous lesions during INH/RIF treatment in TNF-α-deficient mice (205). Furthermore, GM-CSF blocking by monoclonal antibody enhanced host susceptibility and immunopathological severity in Mtb-infected mice (206). Moreover, the absence of GM-CSF signaling results in type I IFN-induced neutrophil extracellular trap formation that aggravates lung mycobacterial burden and lung pathology (206). In addition, neutrophil extracellular traps were abundant in the lung lesions of Mtb-infected C3HeB/FeJ mice and TB patients who showed impaired response to anti-TB therapy (206).
Type I IFNs
Type I IFNs include IFN-α, IFN-β, and several other IFNs that interact with IFNAR1 and IFNAR2 to activate a range of IFN-stimulated genes via STAT1 and STAT2 signaling (243, 244). Type I IFNs play a complex role in mediating beneficial and detrimental effects in the host during TB infection (244). Whole blood transcriptional profiling of active TB patients revealed that upregulation of type I IFN-αβ-inducible transcripts correlated with radiographic severity of the lungs that was restored to the level of healthy controls after anti-TB therapy (245). Recent studies have demonstrated that type I IFN-inducible blood transcriptional signature, including upregulation of STAT1, IFITs, GBPs, MX1, OAS1, and IRF1, was associated with active TB disease (246–248).
Antonelli et al. demonstrated that overexpression of type I IFNs increased pulmonary bacterial loads and extensively distributed necrotic legions in the poly-l-lysine and carboxymethylcellulose (poly-ICLC)-treated Mtb-infected mice (210). Further, a significant increase of CD11b+F4/80+Gr1int cells that showed diminished MHC II expression was confirmed in their lungs (210). Similarly, Mayer-Barber et al. showed IL-1-dependent host protection by producing eicosanoids that suppress immoderate type I IFN production and control bacterial loads (213). The Il1r1-/- mice showed a significantly decreased survival rate and increased bacterial loads in the lungs compared with the wild type mice (213) (Table 2). Similarly, induction of type I IFN due to loss of GM-CSF signaling or genetic susceptibility facilitated Mtb growth and increased disease severity (206). Interestingly, the same group demonstrated host protective effect of type I IFN in the lungs of Mtb-infected mice lacking IFN-γ signaling; the pulmonary bacterial loads were significantly higher in the Ifnar-/-/Ifngr-/- mice compared with that in the Ifngr−/− and Ifnar−/− mice on post-infection days 24 and 28 (211). Recently, Zhang et al. demonstrated a correlation between type I IFN signaling and cell death of Mtb-infected mouse macrophage (212). Further, suppression of type I IFN signaling significantly enhanced the bactericidal activity of RIF in Mtb-infected mice, leading to reduced bacterial loads and improved survival (212). Collectively, these results provide strong evidence that modulation of type I IFN signaling determines the disease severity and susceptibility of immunopathologic lesions in TB.
Th17 Cytokines
Th17 cytokines secreted by Th17 and Th22 cells may play a regulatory role in the immune response during Mtb infection (249). Th17 cytokines play a protective role in Mtb infection but are also involved in pathology due to excessive immune response (250). IL-22 promotes the production of inflammatory mediators and the recruitment of pathologic effector cells in TB (250). IL-22 also promotes tissue repair and healing in lung epithelial cells (251). Treerat et al. demonstrated a significantly increased bacterial burden in the lungs and spleen of Il22-/- mice (222). Similarly, exogenous administration of IL-22 significantly inhibited intracellular Mtb growth by inducing calgranulin A expression (223). Modulation of Th17 responses is essential to promote anti-TB immunity and block immense immunopathology, leading to a detrimental effect on the host (Table 2). Gopal et al. showed that Il17-/- mice demonstrate increased lung bacterial burden compared to that in wild type mice; however, IL-17 overexpression improved the resistance to TB in Mtb-infected Il17−/− mice (224). IL-23 induces the production of IFN-γ and IL-17 response that promotes anti-TB immunity. Khader et al. observed increased immunopathological severity in the lungs of Il23-/- mice (225). The exogenous administration of IL-23 induced significant reduction of the pulmonary Mtb loads and the immunopathological severity in a mouse model via enhanced local T cell immunity (226). Taken together, appropriate modulation of Th-17 cytokines is critical to control TB with minimal detrimental effects to the host.
Conclusion and Future Perspective
Understanding bacterial adaptation to host-mediated stress contributing to antibiotic tolerance is a critical factor in improving disease outcomes and shortening treatment duration. In addition to eliminating the pathogen, effective TB therapy should relieve the associated clinical symptoms by controlling immune-mediated inflammatory responses and minimize damage to the host, thereby minimizing the sequelae. Within the host, Mtb undergoes metabolic reprogramming by drugs or host-mediated stress, resulting in a drug-tolerant state across drug classes. Representative examples of metabolic reprogramming include metabolic stagnation, activation of metabolic bypass, accumulation of triacylglycerol, biofilm formation, and stringent response. The host metabolism is modulated by Mtb infection or the anti-TB drugs, leading to the critical point that determines the treatment success. Therefore, if the host metabolism can be regulated to induce a host-favorable state, the treatment period can be drastically reduced, and side effects can be minimized, dramatically improving clinical outcomes.
Author Contributions
H-EP, WL, M-KS, and SS wrote the manuscript. M-KS and SS conceived the study, supervised the team, and critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant (NRF-2019R1A2C2003204 and NRF-2021R1C1C2012177) and the Bio & Medical Technology Development Program of NRF (NRF-2020M3A9H5104234) funded by the Korean Government (MSIT), Republic of Korea. The funders had no role in study design, data collection and analysis, decision to publish, or manuscript preparation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank MID (Medical Illustration & Design), a part of the Medical Research Support Services of Yonsei University College of Medicine, for all artistic support related to this work.
Abbreviations
AhR, aryl hydrocarbon receptor; AMPK, AMP-activated kinase; ARE, antioxidant response element; ARNT, AhR nuclear translocator; ESAT-6, early secretory antigenic target-6; HMG-CoA, β-Hydroxy β-methylglutaryl-CoA; NFκB, nuclear factor kappa B; HO-1, hemeoxygenase-1; ICL, isocitrate lyase; ILIs, intracellular lipophilic inclusions; MIC, minimum inhibitory concentration; Mtb, M. tuberculosis; mTOR, mechanistic target of rapamycin; Nrf2-Keap1, NF-E2-related factor 2 (Nrf2)-Kelch-like ECH-associated protein 1; Pht, phthiochol; ppGpp, guanosine pentaphosphate; TA, toxin–antitoxin; TAG, triacylglycerol; TCA, tricarboxylic acid; RNS, reactive nitrogen species; ROS, reactive oxygen species.
References
1. Floyd K, Glaziou P, Zumla A, Raviglione M. The Global Tuberculosis Epidemic and Progress in Care, Prevention, and Research: An Overview in Year 3 of the End TB Era. Lancet Respir Med (2018) 6(4):299–314. doi: 10.1016/S2213-2600(18)30057-2
2. World Health Organization. Global Tuberculosis Report 2020. Geneva:World Health Organization (2020).
3. Goossens SN, Sampson SL, van Rie A. Mechanisms of Drug-Induced Tolerance in Mycobacterium tuberculosis. Clin Microbiol Rev (2021) 34(1):e00141–20. doi: 10.1128/CMR.00141-20
4. Nasiri MJ, Haeili M, Ghazi M, Goudarzi H, Pormohammad A, Fooladi AAI, et al. New Insights in to the Intrinsic and Acquired Drug Resistance Mechanisms in Mycobacteria. Front Microbiol (2017) 8:681. doi: 10.3389/fmicb.2017.00681
5. Mandal S, Njikan S, Kumar A, Early JV, Parish T. The Relevance of Persisters in Tuberculosis Drug Discovery. Microbiology (United Kingdom) (2019) 165(5):492–9. doi: 10.1099/mic.0.000760
6. Zhang Y, Yew WW, Barer MR. Targeting Persisters for Tuberculosis Control. Antimicrob Agents Chemother (2012) 56(5):2223–30. doi: 10.1128/AAC.06288-11
7. Fauvart M, de Groote VN, Michiels J. Role of Persister Cells in Chronic Infections: Clinical Relevance and Perspectives on Anti-Persister Therapies. J Med Microbiol (2011) 60(Pt 6):699–709. doi: 10.1099/jmm.0.030932-0
8. Keren I, Minami S, Rubin E, Lewis K. Characterization and Transcriptome Analysis of Mycobacterium tuberculosis Persisters. mBio (2011) 2(3):e00100–11. doi: 10.1128/mBio.00100-11
9. Sebastian J, Swaminath S, Nair RR, Jakkala K, Pradhan A, Ajitkumar P. De Novo Emergence of Genetically Resistant Mutants of Mycobacterium tuberculosis From the Persistence Phase Cells Formed Against Antituberculosis Drugs In Vitro. Antimicrob Agents Chemother (2017) 61(2):e01343–16. doi: 10.1128/AAC.01343-16
10. Sebastian J, Swaminath S, Ajitkumar P. Reduced Permeability to Rifampicin by Capsular Thickening as a Mechanism of Antibiotic Persistence in Mycobacterium tuberculosis. bioRxiv (2019). doi: 10.1101/624569
11. Balaban NQ, Helaine S, Lewis K, Ackermann M, Aldridge B, Andersson DI, et al. Definitions and Guidelines for Research on Antibiotic Persistence. Nat Rev Microbiol (2019) 17(7):441–8. doi: 10.1038/s41579-019-0196-3
12. Brauner A, Fridman O, Gefen O, Balaban NQ. Distinguishing Between Resistance, Tolerance and Persistence to Antibiotic Treatment. Nat Rev Microbiol (2016) 14(5):320–30. doi: 10.1038/nrmicro.2016.34
13. Levin-Reisman I, Brauner A, Ronin I, Balaban NQ. Epistasis Between Antibiotic Tolerance, Persistence, and Resistance Mutations. Proc Natl Acad Sci USA (2019) 116(29):14734–9. doi: 10.1073/pnas.1906169116
14. Ma D, Mandell JB, Donegan NP, Cheung AL, Ma W, Rothenberger S, et al. The Toxin-Antitoxin mazEF Drives Staphylococcus Aureus Biofilm Formation, Antibiotic Tolerance, and Chronic Infection. mBio (2019) 10(6):e01658–19. doi: 10.1128/mBio.01658-19
15. Rowe SE, Wagner NJ, Li L, Beam JE, Wilkinson AD, Radlinski LC, et al. Reactive Oxygen Species Induce Antibiotic Tolerance During Systemic Staphylococcus Aureus Infection. Nat Microbiol (2020) 5(2):282–90. doi: 10.1038/s41564-020-0679-z
16. Matern WM, Rifat D, Bader JS, Karakousis PC. Gene Enrichment Analysis Reveals Major Regulators of Mycobacterium tuberculosis Gene Expression in Two Models of Antibiotic Tolerance. Front Microbiol (2018) 9:610. doi: 10.3389/fmicb.2018.00610
17. Secor PR, Michaels LA, Ratjen A, Jennings LK, Singh PK. Entropically Driven Aggregation of Bacteria by Host Polymers Promotes Antibiotic Tolerance in Pseudomonas Aeruginosa. Proc Natl Acad Sci USA (2018) 115(42):10780–5. doi: 10.1073/pnas.1806005115
18. Meylan S, Andrews IW, Collins JJ. Targeting Antibiotic Tolerance, Pathogen by Pathogen. Cell (2018) 172(6):1228–38. doi: 10.1016/j.cell.2018.01.037
19. Vega NM, Allison KR, Khalil AS, Collins JJ. Signaling-Mediated Bacterial Persister Formation. Nat Chem Biol (2012) 8(5):431–3. doi: 10.1038/nchembio.915
20. Helaine S, Cheverton AM, Watson KG, Faure LM, Matthews SA, Holden DW. Internalization of Salmonella by Macrophages Induces Formation of Nonreplicating Persisters. Science (2014) 343(6167):204–8. doi: 10.1126/science.1244705
21. Kaiser P, Regoes RR, Dolowschiak T, Wotzka SY, Lengefeld J, Slack E, et al. Cecum Lymph Node Dendritic Cells Harbor Slow-Growing Bacteria Phenotypically Tolerant to Antibiotic Treatment. PloS Biol (2014) 12(2):e1001793. doi: 10.1371/journal.pbio.1001793
22. Manina G, Dhar N, McKinney JD. Stress and Host Immunity Amplify Mycobacterium tuberculosis Phenotypic Heterogeneity and Induce Nongrowing Metabolically Active Forms. Cell Host Microbe (2015) 17(1):32–46. doi: 10.1016/j.chom.2014.11.016
23. Mouton JM, Helaine S, Holden DW, Sampson SL. Elucidating Population-Wide Mycobacterial Replication Dynamics at the Single-Cell Level. Microbiology (United Kingdom) (2016) 162(6):966–78. doi: 10.1099/mic.0.000288
24. Brown DR. Nitrogen Starvation Induces Persister Cell Formation in Escherichia Coli. J Bacteriol (2019) 201(3):e00622–18. doi: 10.1128/JB.00622-18
25. Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, et al. Transcriptional Adaptation of Mycobacterium tuberculosis Within Macrophages: Insights Into the Phagosomal Environment. J Exp Med (2003) 198(5):693–704. doi: 10.1084/jem.20030846
26. Flentie K, Garner AL, Stallings CL. Mycobacterium tuberculosis Transcription Machinery: Ready to Respond to Host Attacks. J Bacteriol (2016) 198(9):1360–73. doi: 10.1128/JB.00935-15
27. Parish T. Two-Component Regulatory Systems of Mycobacteria. Microbiol Spectr (2014) 2(1):MGM2-0010-2013. doi: 10.1128/microbiolspec.MGM2-0010-2013
28. Beceiro A, Tomas M, Bou G. Antimicrobial Resistance and Virulence: A Successful or Deleterious Association in the Bacterial World? Clin Microbiol Rev (2013) 26(2):185–230. doi: 10.1128/CMR.00059-12
29. Galagan JE, Minch K, Peterson M, Lyubetskaya A, Azizi E, Sweet L, et al. The Mycobacterium tuberculosis Regulatory Network and Hypoxia. Nature (2013) 499(7457):178–83. doi: 10.1038/nature12337
30. Walters SB, Dubnau E, Kolesnikova I, Laval F, Daffe M, Smith I. The Mycobacterium tuberculosis PhoPR Two-Component System Regulates Genes Essential for Virulence and Complex Lipid Biosynthesis. Mol Microbiol (2006) 60(2):312–30. doi: 10.1111/j.1365-2958.2006.05102.x
31. Asensio JG, Maia C, Ferrer NL, Barilone N, Laval F, Soto CY, et al. The Virulence-Associated Two-Component PhoP-PhoR System Controls the Biosynthesis of Polyketide-Derived Lipids in Mycobacterium tuberculosis. J Biol Chem (2006) 281(3):1313–6. doi: 10.1074/jbc.C500388200
32. Martin C, Williams A, Hernandez-Pando R, Cardona PJ, Gormley E, Bordat Y, et al. The Live Mycobacterium tuberculosis Phop Mutant Strain Is More Attenuated Than BCG and Confers Protective Immunity Against Tuberculosis in Mice and Guinea Pigs. Vaccine (2006) 24(17):3408–19. doi: 10.1016/j.vaccine.2006.03.017
33. Cimino M, Thomas C, Namouchi A, Dubrac S, Gicquel B, Gopaul DN. Identification of DNA Binding Motifs of the Mycobacterium tuberculosis Phop/Phor Two-Component Signal Transduction System. PloS One (2012) 7(8):e42876. doi: 10.1371/journal.pone.0042876
34. Pérez E, Bordas Y, Guilhot C, Gicquel B, Martı C, Samper S, et al. An Essential Role for Phop in Mycobacterium tuberculosis Virulence. Mol Microbiol (2001) 41(1):179–87. doi: 10.1046/j.1365-2958.2001.02500.x
35. Lee JS, Krause R, Schreiber J, Mollenkopf HJ, Kowall J, Stein R, et al. Mutation in the Transcriptional Regulator PhoP Contributes to Avirulence of Mycobacterium Tuberculosis H37Ra Strain. Cell Host Microbe (2008) 3(2):97–103. doi: 10.1016/j.chom.2008.01.002
36. Liu Y, Tan S, Huang L, Abramovitch RB, Rohde KH, Zimmerman MD, et al. Immune Activation of the Host Cell Induces Drug Tolerance in Mycobacterium Tuberculosis Both In Vitro and In Vivo. J Exp Med (2016) 213(5):809–25. doi: 10.1084/jem.20151248
37. Primm TP, Andersen SJ, Mizrahi V, Avarbock D, Rubin H, Barry CE. The Stringent Response of Mycobacterium tuberculosis Is Required for Long-Term Survival. J Bacteriol (2000) 182(17):4889–98. doi: 10.1128/JB.182.17.4889-4898.2000
38. Avarbock D, Salem J, Li LS, Wang ZM, Rubin H. Cloning and Characterization of a Bifunctional RelA/SpoT Homologue From Mycobacterium tuberculosis. Gene (1999) 233(1–2):261–9. doi: 10.1016/S0378-1119(99)00114-6
39. Chuang YM, Dutta NK, Hung CF, Wu TC, Rubin H, Karakousis PC. Stringent Response Factors PPX1 and PPK2 Play an Important Role in Mycobacterium tuberculosis Metabolism, Biofilm Formation, and Sensitivity to Isoniazid In Vivo. Antimicrob Agents Chemother (2016) 60(11):6460–70. doi: 10.1128/AAC.01139-16
40. Dalebroux ZD, Svensson SL, Gaynor EC, Swanson MS. ppGpp Conjures Bacterial Virulence. Microbiol Mol Biol Rev (2010) 74(2):171–99. doi: 10.1128/MMBR.00046-09
41. Trastoy R, Manso T, Fernández-García L, Blasco L, Ambroa A, Pérez Del Molino ML, et al. Mechanisms of Bacterial Tolerance and Persistence in the Gastrointestinal and Respiratory Environments. Clin Microbiol Rev (2018) 31(4):e00023–18. doi: 10.1128/CMR.00023-18
42. Avarbock A, Avarbock D, Teh JS, Buckstein M, Wang ZM, Rubin H. Functional Regulation of the Opposing (p)ppGpp Synthetase/Hydrolase Activities of RelMtb From Mycobacterium tuberculosis. Biochemistry (2005) 44(29):9913–23. doi: 10.1021/bi0505316
43. Dutta NK, Klinkenberg LG, Vazquez MJ, Segura-Carro D, Colmenarejo G, Ramon F, et al. Inhibiting the Stringent Response Blocks Mycobacterium tuberculosis Entry Into Quiescence and Reduces Persistence. Sci Adv (2019) 5(3):eaav2104. doi: 10.1126/sciadv.aav2104
44. Singh R, Singh M, Arora G, Kumar S, Tiwari P, Kidwai S. Polyphosphate Deficiency in Mycobacterium tuberculosis Is Associated With Enhanced Drug Susceptibility and Impaired Growth in Guinea Pigs. J Bacteriol (2013) 195(12):2839–51. doi: 10.1128/JB.00038-13
45. Sharma V, Sharma S, Hoener Zu Bentrup K, McKinney JD, Russell DG, Jacobs WR, et al. Structure of Isocitrate Lyase, a Persistence Factor of Mycobacterium tuberculosis. Nat Struct Biol (2000) 7(8):663–8. doi: 10.1038/77964
46. Nandakumar M, Nathan C, Rhee KY. Isocitrate Lyase Mediates Broad Antibiotic Tolerance in Mycobacterium tuberculosis. Nat Commun (2014) 5:4306. doi: 10.1038/ncomms5306
47. Nguyen L. Antibiotic Resistance Mechanisms in M. Tuberculosis: An Update. Arch Toxicol (2016) 90(7):1585–604. doi: 10.1007/s00204-016-1727-6
48. Zhu JH, Wang BW, Pan M, Zeng YN, Rego H, Javid B. Rifampicin can Induce Antibiotic Tolerance in Mycobacteria Via Paradoxical Changes in Rpob Transcription. Nat Commun (2018) 9(1):4218. doi: 10.1038/s41467-018-06667-3
49. Hu Y, Mangan JA, Dhillon J, Sole KM, Mitchison DA, Butcher PD, et al. Detection of mRNA Transcripts and Active Transcription in Persistent Mycobacterium tuberculosis Induced by Exposure to Rifampin or Pyrazinamide. J Bacteriol (2000) 182(22):6358–65. doi: 10.1128/JB.182.22.6358-6365.2000
50. Javid B, Sorrentino F, Toosky M, Zheng W, Pinkham JT, Jain N, et al. Mycobacterial Mistranslation Is Necessary and Sufficient for Rifampicin Phenotypic Resistance. Proc Natl Acad Sci USA (2014) 111(3):1132–7. doi: 10.1073/pnas.1317580111
51. Kapoor N, Pawar S, Sirakova TD, Deb C, Warren WL, Kolattukudy PE. Human Granuloma In Vitro Model, for TB Dormancy and Resuscitation. PloS One (2013) 8(1):e53657. doi: 10.1371/journal.pone.0053657
52. Deb C, Lee CM, Dubey VS, Daniel J, Abomoelak B, Sirakova TD, et al. A Novel In Vitro Multiple-Stress Dormancy Model for Mycobacterium tuberculosis Generates a Lipid-Loaded, Drug-Tolerant, Dormant Pathogen. PloS One (2009) 4(6):e6077. doi: 10.1371/journal.pone.0006077
53. Santucci P, Johansen MD, Point V, Poncin I, Viljoen A, Cavalier JF, et al. Nitrogen Deprivation Induces Triacylglycerol Accumulation, Drug Tolerance and Hypervirulence in Mycobacteria. Sci Rep (2019) 9(1):8667. doi: 10.1038/s41598-019-45164-5
54. Baek SH, Li AH, Sassetti CM. Metabolic Regulation of Mycobacterial Growth and Antibiotic Sensitivity. PloS Biol (2011) 9(5):e1001065. doi: 10.1371/journal.pbio.1001065
55. Correia FF, D’Onofrio A, Rejtar T, Li L, Karger BL, Makarova K, et al. Kinase Activity of Overexpressed HipA Is Required for Growth Arrest and Multidrug Tolerance in Escherichia coli. J Bacteriol (2006) 188(24):8360–7. doi: 10.1128/JB.01237-06
56. Page R, Peti W. Toxin-Antitoxin Systems in Bacterial Growth Arrest and Persistence. Nat Chem Biol (2016) 12(4):208–14. doi: 10.1038/nchembio.2044
57. Wang X, Yao J, Sun YC, Wood TK. Type VII Toxin/Antitoxin Classification System for Antitoxins That Enzymatically Neutralize Toxins. Trends Microbiol (2020) 29(5):388–93. doi: 10.1016/j.tim.2020.12.001
58. Yu X, Gao X, Zhu K, Yin H, Mao X, Wojdyla JA, et al. Characterization of a Toxin-Antitoxin System in Mycobacterium tuberculosis Suggests Neutralization by Phosphorylation as the Antitoxicity Mechanism. Commun Biol (2020) 3(1):216. doi: 10.1038/s42003-020-0941-1
59. Torrey HL, Keren I, Via LE, Lee JS, Lewis K. High Persister Mutants in Mycobacterium tuberculosis. PloS One (2016) 11(5):e0155127. doi: 10.1371/journal.pone.0155127
60. Slayden RA, Dawson CC, Cummings JE. Toxin-Antitoxin Systems and Regulatory Mechanisms in Mycobacterium tuberculosis. Pathog Dis (2018) 76(4). doi: 10.1093/femspd/fty039
61. Hudock TA, Foreman TW, Bandyopadhyay N, Gautam US, Veatch AV, LoBato DN, et al. Hypoxia Sensing and Persistence Genes Are Expressed During the Intragranulomatous Survival of Mycobacterium tuberculosis. Am J Respir Cell Mol Biol (2017) 56(5):637–47. doi: 10.1165/rcmb.2016-0239OC
62. Sharma A, Chattopadhyay G, Chopra P, Bhasin M, Thakur C, Agarwal S, et al. VapC21 Toxin Contributes to Drug-Tolerance and Interacts With non-Cognate vapB32 Antitoxin in Mycobacterium tuberculosis. Front Microbiol (2020) 11:2037. doi: 10.3389/fmicb.2020.02037
63. Talwar S, Pandey M, Sharma C, Kutum R, Lum J, Carbajo D, et al. Role of vapBC12 Toxin-Antitoxin Locus in Cholesterol-Induced Mycobacterial Persistence. mSystems (2020) 5(6):e00855–20. doi: 10.1128/mSystems.00855-20
64. Podlesek Z, Zgur Bertok D. The DNA Damage Inducible Sos Response Is a Key Player in the Generation of Bacterial Persister Cells and Population Wide Tolerance. Front Microbiol (2020) 11:1785. doi: 10.3389/fmicb.2020.01785
65. Völzing KG, Brynildsen MP. Stationary-Phase Persisters to Ofloxacin Sustain DNA Damage and Require Repair Systems Only During Recovery. mBio (2015) 6(5):e00731–15. doi: 10.1128/mBio.00731-15
66. Mok WWK, Brynildsen MP. Timing of DNA Damage Responses Impacts Persistence to Fluoroquinolones. Proc Natl Acad Sci USA (2018) 115(27):E6301–9 doi: 10.1073/pnas.1804218115
67. Smollett KL, Smith KM, Kahramanoglou C, Arnvig KB, Buxton RS, Davis EO. Global Analysis of the Regulon of the Transcriptional Repressor LexA, a Key Component of SOS Response in Mycobacterium tuberculosis. J Biol Chem (2012) 287(26):22004–14. doi: 10.1074/jbc.M112.357715
68. Boshoff HIM, Reed MB, Barry CE, Mizrahi V. DnaE2 Polymerase Contributes to In Vivo Survival and the Emergence of Drug Resistance in Mycobacterium tuberculosis. Cell (2003) 113(2):183–93. doi: 10.1016/S0092-8674(03)00270-8
69. Choudhary E, Sharma R, Kumar Y, Agarwal N. Conditional Silencing by CRISPRi Reveals the Role of DNA Gyrase in Formation of Drug-Tolerant Persister Population in Mycobacterium Tuberculosis. Front Cell Infect Microbiol (2019) 9:70. doi: 10.3389/fcimb.2019.00070
70. Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial Biofilms: From the Natural Environment to Infectious Diseases. Nat Rev Microbiol (2004) 2(2):95–108. doi: 10.1038/nrmicro821
71. Stewart PS, Costerton JW. Antibiotic Resistance of Bacteria in Biofilms. Lancet (2001) 358(9276):135–8. doi: 10.1016/S0140-6736(01)05321-1
72. Drenkard E, Ausubel FM. Pseudomonas Biofilm Formation and Antibiotic Resistance Are Linked to Phenotypic Variation. Nature (2002) 416(6882):740–3. doi: 10.1038/416740a
73. Hall CW, Mah TF. Molecular Mechanisms of Biofilm-Based Antibiotic Resistance and Tolerance in Pathogenic Bacteria. FEMS Microbiol Rev (2017) 41(3):276–301. doi: 10.1093/femsre/fux010
74. Yan J, Bassler BL. Surviving as a Community: Antibiotic Tolerance and Persistence in Bacterial Biofilms. Cell Host Microbe (2019) 26(1):15–21. doi: 10.1016/j.chom.2019.06.002
75. Esteban J, García-Coca M. Mycobacterium Biofilms. Front Microbiol (2018) 8:2651. doi: 10.3389/fmicb.2017.02651
76. Basaraba RJ, Ojha AK. Mycobacterial Biofilms: Revisiting Tuberculosis Bacilli in Extracellular Necrotizing Lesions. Microbiol Spectr (2017) 5(3):10.1128/microbiolspec.TBTB2-0024-2016. doi: 10.1128/microbiolspec.TBTB2-0024-2016
77. Ackart DF, Hascall-Dove L, Caceres SM, Kirk NM, Podell BK, Melander C, et al. Expression of Antimicrobial Drug Tolerance by Attached Communities of Mycobacterium tuberculosis. Pathog Dis (2014) 70(3):359–69. doi: 10.1111/2049-632X.12144
78. Trivedi A, Mavi PS, Bhatt D, Kumar A. Thiol Reductive Stress Induces Cellulose-Anchored Biofilm Formation in Mycobacterium tuberculosis. Nat Commun (2016) 7:11392. doi: 10.1038/ncomms11392
79. Richards JP, Cai W, Zill NA, Zhang W, Ojha AK. Adaptation of Mycobacterium tuberculosis to Biofilm Growth Is Genetically Linked to Drug Tolerance. Antimicrob Agents Chemother (2019) 63(11):e01213–19. doi: 10.1128/AAC.01213-19
80. Tripathi P, Singh LK, Kumari S, Hakiem OR, Batra JK. Clpb Is an Essential Stress Regulator of Mycobacterium tuberculosis and Endows Survival Advantage to Dormant Bacilli. Int J Med Microbiol (2020) 310(3):151402. doi: 10.1016/j.ijmm.2020.151402
81. Lee JJ, Lee SK, Song N, Nathan TO, Swarts BM, Eum SY, et al. Transient Drug-Tolerance and Permanent Drug-Resistance Rely on the Trehalose-Catalytic Shift in Mycobacterium tuberculosis. Nat Commun (2019) 10(1):2928. doi: 10.1038/s41467-019-10975-7
82. Aminov RI. A Brief History of the Antibiotic Era: Lessons Learned and Challenges for the Future. Front Microbiol (2010) 1:134. doi: 10.3389/fmicb.2010.00134
83. Bode C, Diedrich B, Muenster S, Hentschel V, Weisheit C, Rommelsheim K, et al. Antibiotics Regulate the Immune Response in Both Presence and Absence of Lipopolysaccharide Through Modulation of Toll-Like Receptors, Cytokine Production and Phagocytosis In Vitro. Int Immunopharmacol (2014) 18(1):27–34. doi: 10.1016/j.intimp.2013.10.025
84. Benoun JM, Labuda JC, McSorley SJ. Collateral Damage: Detrimental Effect of Antibiotics on the Development of Protective Immune Memory. mBio (2016) 7(6):e01520–16. doi: 10.1128/mBio.01520-16
85. Berti A, Rose W, Nizet V, Sakoulas G. Antibiotics and Innate Immunity: A Cooperative Effort Toward the Successful Treatment of Infections. Open Forum Infect Dis (2020) 7(8):ofaa302. doi: 10.1093/ofid/ofaa302
86. Yang JH, Bhargava P, McCloskey D, Mao N, Palsson BO, Collins JJ. Antibiotic-Induced Changes to the Host Metabolic Environment Inhibit Drug Efficacy and Alter Immune Function. Cell Host Microbe (2017) 22(6):757–65.e3. doi: 10.1016/j.chom.2017.10.020
87. Suzuki T, Yamamoto M. Molecular Basis of the Keap1-Nrf2 System. Free Radical Biol Med (2015) 88(Pt B):93–100. doi: 10.1016/j.freeradbiomed.2015.06.006
88. Sun Q, Shen X, Ma J, Lou H, Zhang Q. Activation of Nrf2 Signaling by Oltipraz Inhibits Death of Human Macrophages With Mycobacterium tuberculosis Infection. Biochem Biophys Res Commun (2020) 531(3):312–9. doi: 10.1016/j.bbrc.2020.07.026
89. Rothchild AC, Olson GS, Nemeth J, Amon LM, Mai D, Gold ES, et al. Alveolar Macrophages Generate a Noncanonical NRF2-driven Transcriptional Response to Mycobacterium tuberculosis In Vivo. Sci Immunol (2019) 4(37):eaaw6693. doi: 10.1126/sciimmunol.aaw6693
90. Menzel DB, Rasmussen RE, Lee E, Meacher DM, Said B, Hamadeh H, et al. Human Lymphocyte Home Oxygenase 1 as a Response Biomarker to Inorganic Arsenic. Biochem Biophys Res Commun (1998) 250(3):653–6. doi: 10.1006/bbrc.1998.9363
91. Chang SH, Barbosa-Tessmann I, Chen C, Kilberg MS, Agarwal A. Glucose Deprivation Induces Heme Oxygenase-1 Gene Expression by a Pathway Independent of the Unfolded Protein Response. J Biol Chem (2002) 277(3):1933–40. doi: 10.1074/jbc.M108921200
92. Tsuchihashi S, Zhai Y, Fondevila C, Busuttil RW, Kupiec-Weglinski JW. HO-1 Upregulation Suppresses Type 1 IFN Pathway in Hepatic Ischemia/Reperfusion Injury. Transplant Proc (2005) 37(4):1677–8. doi: 10.1016/j.transproceed.2005.03.080
93. Wang L, Weng CY, Wang YJ, Wu MJ. Lipoic Acid Ameliorates Arsenic Trioxide-Induced HO-1 Expression and Oxidative Stress in THP-1 Monocytes and Macrophages. Chem Biol Interact (2011) 190(2–3):129–38. doi: 10.1016/j.cbi.2011.02.001
94. Neubauer JA, Sunderram J. Heme Oxygenase-1 and Chronic Hypoxia. Respir Physiol Neurobiol (2012) 184(2):178–85. doi: 10.1016/j.resp.2012.06.027
95. Andrade BB, Pavan Kumar N, Amaral EP, Riteau N, Mayer-Barber KD, Tosh KW, et al. Heme Oxygenase-1 Regulation of Matrix Metalloproteinase-1 Expression Underlies Distinct Disease Profiles in Tuberculosis. J Immunol (2015) 195(6):2763–73. doi: 10.4049/jimmunol.1500942
96. Willis D, Moore AR, Frederick R, Willoughby DA. Heme Oxygenase: A Novel Target for the Modulation of the Inflammatory Response. Nat Med (1996) 2(1):87–90. doi: 10.1038/nm0196-87
97. Kalghatgi S, Spina CS, Costello JC, Liesa M, Morones-Ramirez JR, Slomovic S, et al. Bactericidal Antibiotics Induce Mitochondrial Dysfunction and Oxidative Damage in Mammalian Cells. Sci Trans Med (2013) 5(192):192ra85. doi: 10.1126/scitranslmed.3006055
98. Kohanski MA, Tharakan A, Lane AP, Ramanathan M. Bactericidal Antibiotics Promote Reactive Oxygen Species Formation and Inflammation in Human Sinonasal Epithelial Cells. Int Forum Allergy Rhinol (2016) 6(2):191–200. doi: 10.1002/alr.21646
99. Costa DL, Amaral EP, Namasivayam S, Mittereder LR, Fisher L, Bonfim CC, et al. Heme Oxygenase-1 Inhibition Promotes IFNγ- and NOS2-Mediated Control of Mycobacterium tuberculosis Infection. Mucosal Immunol (2021) 14(1):253–66. doi: 10.1038/s41385-020-00342-x
100. Rothhammer V, Quintana FJ. The Aryl Hydrocarbon Receptor: An Environmental Sensor Integrating Immune Responses in Health and Disease. Nat Rev Immunol (2019) 19(3):184–97. doi: 10.1038/s41577-019-0125-8
101. Moura-Alves P, Faé K, Houthuys E, Dorhoi A, Kreuchwig A, Furkert J, et al. AhR Sensing of Bacterial Pigments Regulates Antibacterial Defence. Nature (2014) 512(7515):387–92. doi: 10.1038/nature13684
102. Lamorte S, Shinde R, McGaha TL. Nuclear Receptors, the Aryl Hydrocarbon Receptor, and Macrophage Function. Mol Aspects Med (2021) 78:100942. doi: 10.1016/j.mam.2021.100942
103. Shinde R, Hezaveh K, Halaby MJ, Kloetgen A, Chakravarthy A, Da Silva Medina T, et al. Apoptotic Cell-Induced AhR Activity Is Required for Immunological Tolerance and Suppression of Systemic Lupus Erythematosus in Mice and Humans Article. Nat Immunol (2018) 19(6):571–82. doi: 10.1038/s41590-018-0107-1
104. Puyskens A, Stinn A, van der Vaart M, Kreuchwig A, Protze J, Pei G, et al. Aryl Hydrocarbon Receptor Modulation by Tuberculosis Drugs Impairs Host Defense and Treatment Outcomes. Cell Host Microbe (2020) 27(2):238–48.e7. doi: 10.1016/j.chom.2019.12.005
105. Memari B, Bouttier M, Dimitrov V, Ouellette M, Behr MA, Fritz JH, et al. Engagement of the Aryl Hydrocarbon Receptor in Mycobacterium tuberculosis –Infected Macrophages Has Pleiotropic Effects on Innate Immune Signaling. J Immunol (2015) 195(9):4479–91. doi: 10.4049/jimmunol.1501141
106. Mourik BC, Lubberts E, de Steenwinkel JEM, Ottenhoff THM, Leenen PJM. Interactions Between Type 1 Interferons and the Th17 Response in Tuberculosis: Lessons Learned From Autoimmune Diseases. Front Immunol (2017) 8:294. doi: 10.3389/fimmu.2017.00294
107. Willing BP, Russell SL, Finlay BB. Shifting the Balance: Antibiotic Effects on Host-Microbiota Mutualism. Nat Rev Microbiol (2011) 9(4):233–43. doi: 10.1038/nrmicro2536
108. Ubeda C, Pamer EG. Antibiotics, Microbiota, and Immune Defense. Trends Immunol (2012) 33(9):459–66. doi: 10.1016/j.it.2012.05.003
109. Lee EH, Baek SY, Park JY, Kim YW. Rifampicin Activates AMPK and Alleviates Oxidative Stress in the Liver as Mediated With Nrf2 Signaling. Chem Biol Interact (2020) 315:108889. doi: 10.1016/j.cbi.2019.108889
110. Tousif S, Singh DK, Ahmad S, Moodley P, Bhattacharyya M, Van Kaer L, et al. Isoniazid Induces Apoptosis of Activated CD4+ T Cells: Implications for Post-Therapy Tuberculosis Reactivation and Reinfection. J Biol Chem (2014) 289(44):30190–5. doi: 10.1074/jbc.C114.598946
111. Biraro IA, Egesa M, Kimuda S, Smith SG, Toulza F, Levin J, et al. Effect of Isoniazid Preventive Therapy on Immune Responses to Mycobacterium tuberculosis: An Open Label Randomised, Controlled, Exploratory Study. BMC Infect Dis (2015) 15(1):438. doi: 10.1186/s12879-015-1201-8
112. Nilsson BS. Rifampicin: An Immunosuppressant? Lancet (1971) 298(7720):374. doi: 10.1016/S0140-6736(71)90087-0
113. Gupta S, Grieco MH, Siegel I. Suppression of T Lymphocyte Rosettes by Rifampin. Studies in Normals and Patients With Tuberculosis. Ann Internal Med (1975) 82(4):484–8. doi: 10.7326/0003-4819-82-4-484
114. Mlambo G, Sigola LB. Rifampicin and Dexamethasone Have Similar Effects on Macrophage Phagocytosis of Zymosan, But Differ in Their Effects on Nitrite and TNF-α Production. Int Immunopharmacol (2003) 3(4):513–22. doi: 10.1016/S1567-5769(03)00022-5
115. Ziglam HM, Daniels I, Finch RG. Immunomodulating Activity of Rifampicin. J Chemother (2004) 16(4):357–61. doi: 10.1179/joc.2004.16.4.357
116. Bi W, Zhu L, Wang C, Liang Y, Liu J, Shi Q, et al. Rifampicin Inhibits Microglial Inflammation and Improves Neuron Survival Against Inflammation. Brain Res (2011) 1395:12–20. doi: 10.1016/j.brainres.2011.04.019
117. Manca C, Koo MS, Peixoto B, Fallows D, Kaplan G, Subbian S. Host Targeted Activity of Pyrazinamide in Mycobacterium tuberculosis Infection. PloS One (2013) 8(8):e74082. doi: 10.1371/journal.pone.0074082
118. Koul A, Dendouga N, Vergauwen K, Molenberghs B, Vranckx L, Willebrords R, et al. Diarylquinolines Target Subunit C of Mycobacterial ATP Synthase. Nat Chem Biol (2007) 3(6):323–4. doi: 10.1038/nchembio884
119. Andries K, Verhasselt P, Guillemont J, Göhlmann HWH, Neefs JM, Winkler H, et al. A Diarylquinoline Drug Active on the ATP Synthase of Mycobacterium tuberculosis. Science (2005) 307(5707):223–7. doi: 10.1126/science.1106753
120. Giraud-Gatineau A, Coya JM, Maure A, Biton A, Thomson M, Bernard EM, et al. The Antibiotic Bedaquiline Activates Host Macrophage Innate Immune Resistance to Bacterial Infection. eLife (2020) 9:e55692. doi: 10.7554/eLife.55692
121. Cholo MC, Steel HC, Fourie PB, Germishuizen WA, Anderson R. Clofazimine: Current Status and Future Prospects. J Antimicrob Chemother (2012) 67(2):290–8. doi: 10.1093/jac/dkr444
122. Yoon GS, Sud S, Keswani RK, Baik J, Standiford TJ, Stringer KA, et al. Phagocytosed Clofazimine Biocrystals can Modulate Innate Immune Signaling by Inhibiting TNF-α and Boosting IL-1RA Secretion. Mol Pharm (2015) 12(7):2517–27. doi: 10.1021/acs.molpharmaceut.5b00035
123. Fukutomi Y, Maeda Y, Makino M. Apoptosis-Inducing Activity of Clofazimine in Macrophages. Antimicrob Agents Chemother (2011) 55(9):4000–5. doi: 10.1128/AAC.00434-11
124. Ahmad S, Bhattacharya D, Gupta N, Rawat V, Tousif S, van Kaer L, et al. Clofazimine Enhances the Efficacy of BCG Revaccination Via Stem Cell-Like Memory T Cells. PloS Pathog (2020) 16(5):e1008356. doi: 10.1371/journal.ppat.1008356
125. Yang TW, Park HO, Jang HN, Yang JH, Kim SH, Moon SH, et al. Side Effects Associated With the Treatment of Multidrug-Resistant Tuberculosis at a Tuberculosis Referral Hospital in South Korea. Medicine (United States) (2017) 96(28):e7482. doi: 10.1097/MD.0000000000007482
126. Yee D, Valiquette C, Pelletier M, Parisien I, Rocher I, Menzies D. Incidence of Serious Side Effects From First-Line Antituberculosis Drugs Among Patients Treated for Active Tuberculosis. Am J Respir Crit Care Med (2003) 167(11):1472–7. doi: 10.1164/rccm.200206-626OC
127. Kilinç G, Saris A, Ottenhoff THM, Haks MC. Host-Directed Therapy to Combat Mycobacterial Infections*. Immunol Rev (2021) 301(1):62–83. doi: 10.1111/imr.12951
128. Oh S, Trifonov L, Yadav VD, Barry CE, Boshoff HI. Tuberculosis Drug Discovery: A Decade of Hit Assessment for Defined Targets. Front Cell Infect Microbiol (2021) 11:611304. doi: 10.3389/fcimb.2021.611304
129. Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL, Eissa NT. Autophagy Enhances the Efficacy of BCG Vaccine by Increasing Peptide Presentation in Mouse Dendritic Cells. Nat Med (2009) 15(3):267–76. doi: 10.1038/nm.1928
130. Gupta A, Pant G, Mitra K, Madan J, Chourasia MK, Misra A. Inhalable Particles Containing Rapamycin for Induction of Autophagy in Macrophages Infected With Mycobacterium tuberculosis. Mol Pharm (2014) 11(4):1201–7. doi: 10.1021/mp4006563
131. Gupta A, Sharma D, Meena J, Pandya S, Sachan M, Kumar S, et al. Preparation and Preclinical Evaluation of Inhalable Particles Containing Rapamycin and Anti-Tuberculosis Agents for Induction of Autophagy. Pharm Res (2016) 33(8):1899–912. doi: 10.1007/s11095-016-1926-0
132. Singhal A, Jie L, Kumar P, Hong GS, Leow MKS, Paleja B, et al. Metformin as Adjunct Antituberculosis Therapy. Sci Trans Med (2014) 6(263):263ra159. doi: 10.1126/scitranslmed.3009885
133. Degner NR, Wang JY, Golub JE, Karakousis PC. Metformin Use Reverses the Increased Mortality Associated With Diabetes Mellitus During Tuberculosis Treatment. Clin Infect Dis (2018) 66(2):198–205. doi: 10.1093/cid/cix819
134. Lachmandas E, Eckold C, Böhme J, Koeken VACM, Marzuki MB, Blok B, et al. Metformin Alters Human Host Responses to Mycobacterium tuberculosis in Healthy Subjects. J Infect Dis (2019) 220(1):139–50. doi: 10.1093/infdis/jiz064
135. Frenkel JDH, Ackart DF, Todd AK, DiLisio JE, Hoffman S, Tanner S, et al. Metformin Enhances Protection in Guinea Pigs Chronically Infected With Mycobacterium tuberculosis. Sci Rep (2020) 10(1):16257. doi: 10.1038/s41598-020-73212-y
136. Parihar SP, Guler R, Khutlang R, Lang DM, Hurdayal R, Mhlanga MM, et al. Statin Therapy Reduces the Mycobacterium tuberculosis Burden in Human Macrophages and in Mice by Enhancing Autophagy and Phagosome Maturation. J Infect Dis (2014) 209(5):754–63. doi: 10.1093/infdis/jit550
137. Skerry C, Pinn ML, Bruiners N, Pine R, Gennaro ML, Karakousis PC. Simvastatin Increases the In Vivo Activity of the First-Line Tuberculosis Regimen. J Antimicrob Chemother (2014) 69(9):2453–7. doi: 10.1093/jac/dku166
138. Dutta NK, Bruiners N, Pinn ML, Zimmerman MD, Prideaux B, Dartois V, et al. Statin Adjunctive Therapy Shortens the Duration of TB Treatment in Mice. J Antimicrob Chemother (2016) 71(6):1570–7. doi: 10.1093/jac/dkw014
139. Guerra-De-Blas PDC, Bobadilla-Del-Valle M, Sada-Ovalle I, Estrada-García I, Torres-González P, López-Saavedra A, et al. Simvastatin Enhances the Immune Response Against Mycobacterium tuberculosis. Front Microbiol (2019) 10:2097. doi: 10.3389/fmicb.2019.02097
140. Dutta NK, Bruiners N, Zimmerman MD, Tan S, Dartois V, Gennaro ML, et al. Adjunctive Host-Directed Therapy With Statins Improves Tuberculosis-Related Outcomes in Mice. J Infect Dis (2020) 221(7):1079–87. doi: 10.1093/infdis/jiz517
141. Baniasadi S, Eftekhari P, Tabarsi P, Fahimi F, Raoufy MR, Masjedi MR, et al. Protective Effect of N-Acetylcysteine on Antituberculosis Drug-Induced Hepatotoxicity. Eur J Gastroenterol Hepatol (2010) 22(10):1235–8. doi: 10.1097/MEG.0b013e32833aa11b
142. Palanisamy GS, Kirk NM, Ackart DF, Shanley CA, Orme IM, Basaraba RJ. Evidence for Oxidative Stress and Defective Antioxidant Response in Guinea Pigs With Tuberculosis. PloS One (2011) 6(10):e26254. doi: 10.1371/journal.pone.0026254
143. Amaral EP, Conceição EL, Costa DL, Rocha MS, Marinho JM, Cordeiro-Santos M, et al. N-Acetyl-Cysteine Exhibits Potent Anti-Mycobacterial Activity in Addition to Its Known Anti-Oxidative Functions. BMC Microbiol (2016) 16(1):251. doi: 10.1186/s12866-016-0872-7
144. Mahakalkar S, Nagrale D, Gaur S, Urade C, Murhar B, Turankar A. N-Acetylcysteine as an Add-on to Directly Observed Therapy Short-I Therapy in Fresh Pulmonary Tuberculosis Patients: A Randomized, Placebo-Controlled, Double-Blinded Study. Perspect Clin Res (2017) 8(3):132–6. doi: 10.4103/2229-3485.210450
145. Teskey G, Cao R, Islamoglu H, Medina A, Prasad C, Prasad R, et al. The Synergistic Effects of the Glutathione Precursor, NAC and First-Line Antibiotics in the Granulomatous Response Against Mycobacterium tuberculosis. Front Immunol (2018) 9:2069. doi: 10.3389/fimmu.2018.02069
146. Cao R, Teskey G, Islamoglu H, Abrahem R, Munjal S, Gyurjian K, et al. Characterizing the Effects of Glutathione as an Immunoadjuvant in the Treatment of Tuberculosis. Antimicrob Agents Chemother (2018) 62(11):e01132–18. doi: 10.1128/AAC.01132-18
147. Safe IP, Lacerda MVG, Printes VS, Marins AFP, Rabelo ALR, Costa AA, et al. Safety and Efficacy of N-Acetylcysteine in Hospitalized Patients With HIV-Associated Tuberculosis: An Open-Label, Randomized, Phase II Trial (RIPENACTB Study). PloS One (2020) 15(6):e0235381. doi: 10.1371/journal.pone.0235381
148. Vilchèze C, Jacobs WR. The Promises and Limitations of N-acetylcysteine as a Potentiator of First-Line and Second-Line Tuberculosis Drugs. Antimicrob Agents Chemother (2021) 65(5):e01703–20. doi: 10.1128/AAC.01703-20
149. Gupta S, Tyagi S, Almeida DV, Maiga MC, Ammerman NC, Bishai WR. Acceleration of Tuberculosis Treatment by Adjunctive Therapy With Verapamil as an Efflux Inhibitor. Am J Respir Crit Care Med (2013) 188(5):600–7. doi: 10.1164/rccm.201304-0650OC
150. Pieterman ED, te Brake LHM, de Knegt GJ, van der Meijden A, Alffenaar JWC, Bax HI, et al. Assessment of the Additional Value of Verapamil to a Moxifloxacin and Linezolid Combination Regimen in a Murine Tuberculosis Model. Antimicrob Agents Chemother (2018) 62(9):e01354–18. doi: 10.1128/AAC.01354-18
151. Chen C, Gardete S, Jansen RS, Shetty A, Dick T, Rhee KY, et al. Verapamil Targets Membrane Energetics in Mycobacterium tuberculosis. Antimicrob Agents Chemother (2018) 62(5):e02107–17. doi: 10.1128/AAC.02107-17
152. Xu J, Tasneen R, Peloquin CA, Almeida DV, Li SY, Barnes-Boyle K, et al. Verapamil Increases the Bioavailability and Efficacy of Bedaquiline But Not Clofazimine in a Murine Model of Tuberculosis. Antimicrob Agents Chemother (2018) 62(1):e01692–17. doi: 10.1128/AAC.01692-17
153. Levine B, Mizushima N, Virgin HW. Autophagy in Immunity and Inflammation. Nature (2011) 469(7330):323–35. doi: 10.1038/nature09782
154. Jiang P, Mizushima N. Autophagy and Human Diseases. Cell Res (2014) 24(1):69–79. doi: 10.1038/cr.2013.161
155. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy Is a Defense Mechanism Inhibiting BCG and Mycobacterium tuberculosis Survival in Infected Macrophages. Cell (2004) 119(6):753–66. doi: 10.1016/j.cell.2004.11.038
156. Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, et al. The Ubiquitin Ligase Parkin Mediates Resistance to Intracellular Pathogens. Nature (2013) 501(7468):512–6. doi: 10.1038/nature12566
157. Kim YS, Silwal P, Kim SY, Yoshimori T, Jo EK. Autophagy-Activating Strategies to Promote Innate Defense Against Mycobacteria. Exp Mol Med (2019) 51(12):1–10. doi: 10.1038/s12276-019-0290-7
158. Saunders RN, Metcalfe MS, Nicholson ML. Rapamycin in Transplantation: A Review of the Evidence. Kidney Int (2001) 59(1):3–16. doi: 10.1046/j.1523-1755.2001.00460.x
159. Dumont FJ, Su Q. Mechanism of Action of the Immunosuppressant Rapamycin. Life Sci (1995) 58(5):373–95. doi: 10.1016/0024-3205(95)02233-3
160. Zullo AJ, Jurcic Smith KL, Lee S. Mammalian Target of Rapamycin Inhibition and Mycobacterial Survival Are Uncoupled in Murine Macrophages. BMC Biochem (2014) 15(1):4. doi: 10.1186/1471-2091-15-4
161. Andersson AM, Andersson B, Lorell C, Raffetseder J, Larsson M, Blomgran R. Autophagy Induction Targeting mTORC1 Enhances Mycobacterium tuberculosis Replication in HIV Co-Infected Human Macrophages. Sci Rep (2016) 6:28171. doi: 10.1038/srep28171
162. Son HJ, Lee J, Lee SY, Kim EK, Park MJ, Kim KW, et al. Metformin Attenuates Experimental Autoimmune Arthritis Through Reciprocal Regulation of Th17/Treg Balance and Osteoclastogenesis. Mediators Inflamm (2014) 2014:973986. doi: 10.1155/2014/973986
163. Kang KY, Kim YK, Yi H, Kim J, Jung HR, Kim IJ, et al. Metformin Downregulates Th17 Cells Differentiation and Attenuates Murine Autoimmune Arthritis. Int Immunopharmacol (2013) 16(1):85–92. doi: 10.1016/j.intimp.2013.03.020
164. Titov AA, Baker HV, Brusko TM, Sobel ES, Morel L. Metformin Inhibits the Type 1 IFN Response in Human CD4+ T Cells. J Immunol (2019) 203(2):338–48. doi: 10.4049/jimmunol.1801651
165. Saenwongsa W, Nithichanon A, Chittaganpitch M, Buayai K, Kewcharoenwong C, Thumrongwilainet B, et al. Metformin-Induced Suppression of IFN-α Via mTORC1 Signalling Following Seasonal Vaccination Is Associated With Impaired Antibody Responses in Type 2 Diabetes. Sci Rep (2020) 10(1):3229. doi: 10.1038/s41598-020-60213-0
166. Dutta NK, Pinn ML, Karakousis PC. Metformin Adjunctive Therapy Does Not Improve the Sterilizing Activity of the First-Line Antitubercular Regimen in Mice. Antimicrob Agents Chemother (2017) 61(8):e00652–17. doi: 10.1128/AAC.00652-17
167. Hedrich WD, Hassan HE, Wang H. Insights Into CYP2B6-Mediated Drug-Drug Interactions. Acta Pharm Sin B (2016) 6(5):413–25. doi: 10.1016/j.apsb.2016.07.016
168. Hardie DG. AMP-Activated Protein Kinase as a Drug Target. Annu Rev Pharmacol Toxicol (2007) 47:185–200. doi: 10.1146/annurev.pharmtox.47.120505.105304
169. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-Activated Protein Kinase in Mechanism of Metformin Action. J Clin Invest (2001) 108(8):1167–74. doi: 10.1172/JCI13505
170. Oesterle A, Laufs U, Liao JK. Pleiotropic Effects of Statins on the Cardiovascular System. Circ Res (2017) 120(1):229–43. doi: 10.1161/CIRCRESAHA.116.308537
171. Lee W, VanderVen BC, Fahey RJ, Russell DG. Intracellular Mycobacterium tuberculosis Exploits Host-Derived Fatty Acids to Limit Metabolic Stress. J Biol Chem (2013) 288(10):6788–800. doi: 10.1074/jbc.M112.445056
172. Tahir F, Bin Arif T, Ahmed J, Shah SR, Khalid M. Anti-Tuberculous Effects of Statin Therapy: A Review of Literature. Cureus (2020)12(3):e7404. doi: 10.7759/cureus.7404
173. Liao KF, Lin CL, Lai SW. Population-Based Case-Control Study Assessing the Association Between Statins Use and Pulmonary Tuberculosis in Taiwan. Front Pharmacol (2017) 8:597. doi: 10.3389/fphar.2017.00597
174. Su VYF, Su WJ, Yen YF, Pan SW, Chuang PH, Feng JY, et al. Statin Use Is Associated With a Lower Risk of TB. Chest (2017) 152(3):598–606. doi: 10.1016/j.chest.2017.04.170
175. Lobato LS, Rosa PS, Da Silva Ferreira J, Da Silva Neumann A, Da Silva MG, Nascimento DC, et al. Statins Increase Rifampin Mycobactericidal Effect. Antimicrob Agents Chemother (2014) 58(10):5766–74. doi: 10.1128/AAC.01826-13
176. Vatansever F, de Melo WCMA, Avci P, Vecchio D, Sadasivam M, Gupta A, et al. Antimicrobial Strategies Centered Around Reactive Oxygen Species - Bactericidal Antibiotics, Photodynamic Therapy, and Beyond. FEMS Microbiol Rev (2013) 37(6):955–89. doi: 10.1111/1574-6976.12026
177. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ Res (2018) 122(6):877–902. doi: 10.1161/CIRCRESAHA.117.311401
178. Vilchèze C, Hartman T, Weinrick B, Jain P, Weisbrod TR, Leung LW, et al. Enhanced Respiration Prevents Drug Tolerance and Drug Resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci USA (2017) 114(17):4495–500. doi: 10.1073/pnas.1704376114
179. Lamprecht DA, Finin PM, Rahman MA, Cumming BM, Russell SL, Jonnala SR, et al. Turning the Respiratory Flexibility of Mycobacterium tuberculosis Against Itself. Nat Commun (2016) 7:12393. doi: 10.1038/ncomms12393
180. Guerra C, Morris D, Sipin A, Kung S, Franklin M, Gray D, et al. Glutathione and Adaptive Immune Responses Against Mycobacterium tuberculosis Infection in Healthy and HIV Infected Individuals. PloS One (2011) 6(12):e28378. doi: 10.1371/journal.pone.0028378
181. Venketaraman V, Millman A, Salman M, Swaminathan S, Goetz M, Lardizabal A, et al. Glutathione Levels and Immune Responses in Tuberculosis Patients. Microb Pathog (2008) 44(3):255–61. doi: 10.1016/j.micpath.2007.09.002
182. Khameneh B, Fazly Bazzaz BS, Amani A, Rostami J, Vahdati-Mashhadian N. Combination of Anti-Tuberculosis Drugs With Vitamin C or NAC Against Different Staphylococcus Aureus and Mycobacterium tuberculosis Strains. Microb Pathog (2016) 93:83–7. doi: 10.1016/j.micpath.2015.11.006
183. Ejigu DA, Abay SM. N-Acetyl Cysteine as an Adjunct in the Treatment of Tuberculosis. Tuberc Res Treat (2020) 2020:5907839. doi: 10.1155/2020/5907839
184. Vural A, Koçyiğit İ, Şan F, Eroğlu E, Ketenci İ, Ünal A, et al. Long-Term Protective Effect of N-Acetylcysteine Against Amikacin-Induced Ototoxicity in End-Stage Renal Disease: A Randomized Trial. Perit Dialysis Int (2018) 38(1):57–62. doi: 10.3747/pdi.2017.00133
185. Kocyigit I, Vural A, Unal A, Sipahioglu MH, Yucel HE, Aydemir S, et al. Preventing Amikacin Related Ototoxicity With N-Acetylcysteine in Patients Undergoing Peritoneal Dialysis. Eur Arch Otorhinolaryngol (2015) 272(10):2611–20. doi: 10.1007/s00405-014-3207-z
186. Carr AC, Maggini S. Vitamin C and Immune Function. Nutrients (2017) 9(11):1211. doi: 10.3390/nu9111211
187. Vilchèze C, Hartman T, Weinrick B, Jacobs WR. Mycobacterium tuberculosis Is Extraordinarily Sensitive to Killing by a Vitamin C-induced Fenton Reaction. Nat Commun (2013) 4:1881. doi: 10.1038/ncomms2898
188. Sikri K, Duggal P, Kumar C, Batra SD, Vashist A, Bhaskar A, et al. Multifaceted Remodeling by Vitamin C Boosts Sensitivity of Mycobacterium tuberculosis Subpopulations to Combination Treatment by Anti-Tubercular Drugs. Redox Biol (2018) 15:452–66. doi: 10.1016/j.redox.2017.12.020
189. Susanto L, Siregar Y, Kusumawati L. Vitamin C Supplementation Improve the Sputum Conversion Culture Rate in Pulmonary Tuberculosis Treatment While Rifampicin Susceptible. Iop Conf Ser: Earth Environ Sci (2018) 125:012140. doi: 10.1088/1755-1315/125/1/012140
190. Vilchèze C, Kim J, Jacobs WR. Vitamin C Potentiates the Killing of Mycobacterium tuberculosis by the First-Line Tuberculosis Drugs Isoniazid and Rifampin in Mice. Antimicrob Agents Chemother (2018) 62(3):e02165–17. doi: 10.1128/AAC.02165-17
191. Nandi M, Sikri K, Chaudhary N, Mande SC, Sharma RD, Tyagi JS. Multiple Transcription Factors Co-Regulate the Mycobacterium tuberculosis Adaptation Response to Vitamin C. BMC Genomics (2019) 20(1):887. doi: 10.1186/s12864-019-6190-3
192. Costa DL, Namasivayam S, Amaral EP, Arora K, Chao A, Mittereder LR, et al. Pharmacological Inhibition of Host Heme Oxygenase-1 Suppresses Mycobacterium tuberculosis Infection In Vivo by a Mechanism Dependent on T Lymphocytes. mBio (2016) 7(5):e01675–16. doi: 10.1128/mBio.01675-16
193. Rockwood N, Costa DL, Amaral EP, du Bruyn E, Kubler A, Gil-Santana L, et al. Mycobacterium tuberculosis Induction of Heme Oxygenase-1 Expression Is Dependent on Oxidative Stress and Reflects Treatment Outcomes. Front Immunol (2017) 8:542. doi: 10.3389/fimmu.2017.00542
194. Chinta KC, Rahman MA, Saini V, Glasgow JN, Reddy VP, Lever JM, et al. Microanatomic Distribution of Myeloid Heme Oxygenase-1 Protects Against Free Radical-Mediated Immunopathology in Human Tuberculosis. Cell Rep (2018) 25(7):1938–52.e5. doi: 10.1016/j.celrep.2018.10.073
195. Chinta KC, Pacl HT, Agarwal A, Steyn AJC. Heme Oxygenase-1 as a Pharmacological Target for Host-Directed Therapy to Limit Tuberculosis Associated Immunopathology. Antioxidants (2021) 10(2):177. doi: 10.3390/antiox10020177
196. Canesin G, Hejazi SM, Swanson KD, Wegiel B. Heme-Derived Metabolic Signals Dictate Immune Responses. Front Immunol (2020) 11:66. doi: 10.3389/fimmu.2020.00066
197. Machado D, Couto I, Perdigão J, Rodrigues L, Portugal I, Baptista P, et al. Contribution of Efflux to the Emergence of Isoniazid and Multidrug Resistance in Mycobacterium tuberculosis. PloS One (2012) 7(4):e34538. doi: 10.1371/journal.pone.0034538
198. Gupta S, Cohen KA, Winglee K, Maiga M, Diarra B, Bishai WR. Efflux Inhibition With Verapamil Potentiates Bedaquiline in Mycobacterium tuberculosis. Antimicrob Agents Chemother (2014) 58(1):574–6. doi: 10.1128/AAC.01462-13
199. Li G, Zhang J, Guo Q, Wei J, Jiang Y, Zhao X, et al. Study of Efflux Pump Gene Expression in Rifampicin-Monoresistant Mycobacterium tuberculosis Clinical Isolates. J Antibiot (2015) 68(7):431–5. doi: 10.1038/ja.2015.9
200. Domingo-Gonzalez R, Prince O, Cooper A, Khader SA. Cytokines and Chemokines in Mycobacterium tuberculosis Infection. Microbiol Spectr (2016) 4(5):10.1128/microbiolspec.TBTB2-0018-2016. doi: 10.1128/microbiolspec.TBTB2-0018-2016
201. Verver S, Warren RM, Beyers N, Richardson M, van der Spuy GD, Borgdorff MW, et al. Rate of Reinfection Tuberculosis After Successful Treatment Is Higher Than Rate of New Tuberculosis. Am J Respir Crit Care Med (2005) 171(12):1430–5. doi: 10.1164/rccm.200409-1200OC
202. Francisco-Cruz A, Mata-Espinosa D, Ramos-Espinosa O, Marquina-Castillo B, Estrada-Parra S, Xing Z, et al. Efficacy of Gene-Therapy Based on Adenovirus Encoding Granulocyte-Macrophage Colony-Stimulating Factor in Drug-Sensitive and Drug-Resistant Experimental Pulmonary Tuberculosis. Tuberculosis (2016) 100:5–14. doi: 10.1016/j.tube.2016.05.015
203. Francisco-Cruz A, Mata-Espinosa D, Estrada-Parra S, Xing Z, Hernández-Pando R. Immunotherapeutic Effects of Recombinant Adenovirus Encoding Granulocyte-Macrophage Colony-Stimulating Factor in Experimental Pulmonary Tuberculosis. Clin Exp Immunol (2013) 171(3):283–97. doi: 10.1111/cei.12015
204. Pasula R, Azad AK, Gardner JC, Schlesinger LS, McCormack FX. Keratinocyte Growth Factor Administration Attenuates Murine Pulmonary Mycobacterium tuberculosis Infection Through Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF)-Dependent Macrophage Activation and Phagolysosome Fusion. J Biol Chem (2015) 290(11):7151–9. doi: 10.1074/jbc.M114.591891
205. Benmerzoug S, Marinho FV, Rose S, Mackowiak C, Gosset D, Sedda D, et al. GM-CSF Targeted Immunomodulation Affects Host Response to M. Tuberculosis Infection. Sci Rep (2018) 8(1):8652. doi: 10.1038/s41598-018-26984-3
206. Moreira-Teixeira L, Stimpson PJ, Stavropoulos E, Hadebe S, Chakravarty P, Ioannou M, et al. Type I IFN Exacerbates Disease in Tuberculosis-Susceptible Mice by Inducing Neutrophil-Mediated Lung Inflammation and Netosis. Nat Commun (2020) 11(1):5566. doi: 10.1038/s41467-020-19412-6
207. Dawson R, Condos R, Tse D, Huie ML, Ress S, Tseng CH, et al. Immunomodulation With Recombinant Interferon-γ1b in Pulmonary Tuberculosis. PloS One (2009) 4(9):e6984. doi: 10.1371/journal.pone.0006984
208. Mata-Espinosa DA, Mendoza-Rodríguez V, Aguilar-León D, Rosales R, López-Casillas F, Hernández-Pando R. Therapeutic Effect of Recombinant Adenovirus Encoding Interferon-γ in a Murine Model of Progressive Pulmonary Tuberculosis. Mol Ther (2008) 16(6):1065–72. doi: 10.1038/mt.2008.69
209. Khan TA, Mazhar H, Saleha S, Tipu HN, Muhammad N, Abbas MN. Interferon-Gamma Improves Macrophages Function Against M. Tuberculosis in Multidrug-Resistant Tuberculosis Patients. Chemother Res Pract (2016) 2016:7295390. doi: 10.1155/2016/7295390
210. Antonelli LRV, Rothfuchs AG, Gonçalves R, Roffê E, Cheever AW, Bafica A, et al. Intranasal poly-IC Treatment Exacerbates Tuberculosis in Mice Through the Pulmonary Recruitment of a Pathogen-Permissive Monocyte/Macrophage Population. J Clin Invest (2010) 120(5):1674–82. doi: 10.1172/JCI40817
211. Moreira-Teixeira L, Sousa J, McNab FW, Torrado E, Cardoso F, Machado H, et al. Type I IFN Inhibits Alternative Macrophage Activation During Mycobacterium tuberculosis Infection and Leads to Enhanced Protection in the Absence of IFN-γ Signaling. J Immunol (2016) 197(12):4714–26. doi: 10.4049/jimmunol.1600584
212. Zhang L, Jiang X, Pfau D, Ling Y, Nathan CF. Type I Interferon Signaling Mediates Mycobacterium tuberculosis–Induced Macrophage Death. J Exp Med (2021) 218(2):e20200887. doi: 10.1084/jem.20200887
213. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-Directed Therapy of Tuberculosis Based on Interleukin-1 and Type I Interferon Crosstalk. Nature (2014) 511(7507):99–103. doi: 10.1038/nature13489
214. Arbués A, Brees D, Chibout SD, Fox T, Kammüller M, Portevin D. TNF-α Antagonists Differentially Induce TGFβ1-Dependent Resuscitation of Dormant-Like Mycobacterium tuberculosis. PloS Pathog (2020) 16(2):e1008312. doi: 10.1371/journal.ppat.1008312
215. Tezera LB, Bielecka MK, Ogongo P, Walker NF, Ellis M, Garay-Baquero DJ, et al. Anti-PD-1 Immunotherapy Leads to Tuberculosis Reactivation Via Dysregulation of TNF-α. eLife (2020) 9:e52668. doi: 10.7554/eLife.52668
216. Wu B, Huang C, Kato-Maeda M, Hopewell PC, Daley CL, Krensky AM, et al. IL-24 Modulates IFN-γ Expression in Patients With Tuberculosis. Immunol Lett (2008) 117(1):57–62. doi: 10.1016/j.imlet.2007.11.018
217. Ramos-Espinosa O, Hernández-Bazán S, Francisco-Cruz A, Mata-Espinosa D, Barrios-Payán J, Marquina-Castillo B, et al. Gene Therapy Based in Antimicrobial Peptides and Proinflammatory Cytokine Prevents Reactivation of Experimental Latent Tuberculosis. Pathog Dis (2016) 74(7):ftw075. doi: 10.1093/femspd/ftw075
218. Shen H, Min R, Tan Q, Xie W, Wang H, Pan H, et al. The Beneficial Effects of Adjunctive Recombinant Human Interleukin-2 for Multidrug Resistant Tuberculosis. Arch Med Sci (2015) 11(3):584–90. doi: 10.5114/aoms.2015.52362
219. Liu X, Li F, Niu H, Ma L, Chen J, Zhang Y, et al. IL-2 Restores T-cell Dysfunction Induced by Persistent Mycobacterium tuberculosis Antigen Stimulation. Front Immunol (2019) 10:2350. doi: 10.3389/fimmu.2019.02350
220. Nolt D, Flynn JAL. Interleukin-12 Therapy Reduces the Number of Immune Cells and Pathology in Lungs of Mice Infected With Mycobacterium tuberculosis. Infect Immun (2004) 72(5):2976–88. doi: 10.1128/IAI.72.5.2976-2988.2004
221. Mata-Espinosa DA, Francisco-Cruz A, Marquina-Castillo B, Barrios-Payan J, Ramos-Espinosa O, Bini EI, et al. Immunotherapeutic Effects of Recombinant Adenovirus Encoding Interleukin 12 in Experimental Pulmonary Tuberculosis. Scand J Immunol (2019) 89(3):e12743. doi: 10.1111/sji.12743
222. Treerat P, Prince O, Cruz-Lagunas A, Muñoz-Torrico M, Salazar-Lezama MA, Selman M, et al. Novel Role for IL-22 in Protection During Chronic Mycobacterium tuberculosis HN878 Infection. Mucosal Immunol (2017) 10(4):1069–81. doi: 10.1038/mi.2017.15
223. Dhiman R, Venkatasubramanian S, Paidipally P, Barnes PF, Tvinnereim A, Vankayalapati R. Interleukin 22 Inhibits Intracellular Growth of Mycobacterium tuberculosis by Enhancing Calgranulin a Expression. J Infect Dis (2014) 209(4):578–87. doi: 10.1093/infdis/jit495
224. Gopal R, Monin L, Slight S, Uche U, Blanchard E, A. Fallert Junecko B, et al. Unexpected Role for IL-17 in Protective Immunity Against Hypervirulent Mycobacterium tuberculosis HN878 Infection. PloS Pathog (2014) 10(5):e1004099. doi: 10.1371/journal.ppat.1004099
225. Khader SA, Pearl JE, Sakamoto K, Gilmartin L, Bell GK, Jelley-Gibbs DM, et al. IL-23 Compensates for the Absence of IL-12p70 and Is Essential for the IL-17 Response During Tuberculosis But Is Dispensable for Protection and Antigen-Specific IFN-γ Responses if IL-12p70 Is Available. J Immunol (2005) 175(2):788–95. doi: 10.4049/jimmunol.175.2.788
226. Happel KI, Lockhart EA, Mason CM, Porretta E, Keoshkerian E, Odden AR, et al. Pulmonary Interleukin-23 Gene Delivery Increases Local T-Cell Immunity and Controls Growth of Mycobacterium tuberculosis in the Lungs. Infect Immun (2005) 73(9):5782–8. doi: 10.1128/IAI.73.9.5782-5788.2005
227. Ma Y, Chen HD, Wang Y, Wang Q, Li Y, Zhao Y, et al. Interleukin 24 as a Novel Potential Cytokine Immunotherapy for the Treatment of Mycobacterium tuberculosis Infection. Microbes Infect (2011) 13(12–13):1099–110. doi: 10.1016/j.micinf.2011.06.012
228. Chan J, Xing Y, Magliozzo RS, Bloom BR. Killing of Virulent Mycobacterium tuberculosis by Reactive Nitrogen Intermediates Produced by Activated Murine Macrophages. J Exp Med (1992) 175(4):1111–22. doi: 10.1084/jem.175.4.1111
229. Liu CH, Liu H, Ge B. Innate Immunity in Tuberculosis: Host Defense vs Pathogen Evasion. Cell Mol Immunol (2017) 14(12):963–75. doi: 10.1038/cmi.2017.88
230. MacMicking JD. Cell-Autonomous Effector Mechanisms Against Mycobacterium tuberculosis. Cold Spring Harb Perspect Med (2014) 4(10):a018507. doi: 10.1101/cshperspect.a018507
231. Ramirez-Alejo N, Santos-Argumedo L. Innate Defects of the IL-12/IFN-γ Axis in Susceptibility to Infections by Mycobacteria and Salmonella. J Interferon Cytokine Res (2014) 34(5):307–17. doi: 10.1089/jir.2013.0050
232. Roach DR, Bean AGD, Demangel C, France MP, Briscoe H, Britton WJ. TNF Regulates Chemokine Induction Essential for Cell Recruitment, Granuloma Formation, and Clearance of Mycobacterial Infection. J Immunol (2002) 168(9):4620–7. doi: 10.4049/jimmunol.168.9.4620
233. Pagán AJ, Ramakrishnan L. Immunity and Immunopathology in the Tuberculous Granuloma. Cold Spring Harb Perspect Med (2015) 5(9):a018499. doi: 10.1101/cshperspect.a018499
234. Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis Associated With Infliximab, a Tumor Necrosis Factor α–Neutralizing Agent. N Engl J Med (2001) 345(15):1098–104. doi: 10.1056/NEJMoa011110
235. Wu B, Huang C, Garcia L, De Leon AP, Osornio JS, Bobadilla-del-Valle M, et al. Unique Gene Expression Profiles in Infants Vaccinated With Different Strains of Mycobacterium Bovis Bacille Calmette-Guérin. Infect Immun (2007) 75(7):3658–64. doi: 10.1128/IAI.00244-07
236. Huang D, Qiu L, Wang R, Lai X, Du G, Seghal P, et al. Immune Gene Networks of Mycobacterial Vaccine-Elicited Cellular Responses and Immunity. J Infect Dis (2007) 195(1):55–69. doi: 10.1086/509895
237. Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and Inflammation Induce Distinct Transcriptional Programs That Promote the Differentiation of Effector Cytolytic T Cells. Immunity (2010) 32(1):79–90. doi: 10.1016/j.immuni.2009.11.012
238. West EE, Jin HT, Rasheed AU, Penaloza-MacMaster P, Ha SJ, Tan WG, et al. PD-L1 Blockade Synergizes With IL-2 Therapy in Reinvigorating Exhausted T Cells. J Clin Invest (2013) 123(6):2604–15. doi: 10.1172/JCI67008
239. Johnson B, Bekker LG, Ress S, Kaplan G. Recombinant Interleukin 2 Adjunctive Therapy in Multidrug-Resistant Tuberculosis. Novartis Found Symp (1998) 217:99–106; discussion 106–11. doi: 10.1002/0470846526.ch7
240. Robinson RT. T Cell Production of GM-CSF Protects the Host During Experimental Tuberculosis. mBio (2017) 8(6):e02087–17. doi: 10.1128/mBio.02087-17
241. Szeliga J, Daniel DS, Yang CH, Sever-Chroneos Z, Jagannath C, Chroneos ZC. Granulocyte-Macrophage Colony Stimulating Factor-Mediated Innate Responses in Tuberculosis. Tuberculosis (2008) 88(1):7–20. doi: 10.1016/j.tube.2007.08.009
242. Bhattacharya P, Budnick I, Singh M, Thiruppathi M, Alharshawi K, Elshabrawy H, et al. Dual Role of GM-CSF as a Pro-Inflammatory and a Regulatory Cytokine: Implications for Immune Therapy. J Interferon Cytokine Res (2015) 35(8):585–99. doi: 10.1089/jir.2014.0149
243. Ivashkiv LB, Donlin LT. Regulation of Type I Interferon Responses. Nat Rev Immunol (2014) 14(1):36–49. doi: 10.1038/nri3581
244. Moreira-Teixeira L, Mayer-Barber K, Sher A, O’Garra A. Type I Interferons in Tuberculosis: Foe and Occasionally Friend. J Exp Med (2018) 215(5):1273–85. doi: 10.1084/jem.20180325
245. Berry MPR, Graham CM, McNab FW, Xu Z, Bloch SAA, Oni T, et al. An Interferon-Inducible Neutrophil-Driven Blood Transcriptional Signature in Human Tuberculosis. Nature (2010) 466(7309):973–7. doi: 10.1038/nature09247
246. Zak DE, Penn-Nicholson A, Scriba TJ, Thompson E, Suliman S, Amon LM, et al. A Blood RNA Signature for Tuberculosis Disease Risk: A Prospective Cohort Study. Lancet (2016) 387(10035):2312–22. doi: 10.1016/S0140-6736(15)01316-1
247. Sambarey A, Devaprasad A, Mohan A, Ahmed A, Nayak S, Swaminathan S, et al. Unbiased Identification of Blood-Based Biomarkers for Pulmonary Tuberculosis by Modeling and Mining Molecular Interaction Networks. EBioMedicine (2017) 15:112–26. doi: 10.1016/j.ebiom.2016.12.009
248. Esmail H, Lai RP, Lesosky M, Wilkinson KA, Graham CM, Horswell S, et al. Complement Pathway Gene Activation and Rising Circulating Immune Complexes Characterize Early Disease in HIV-associated Tuberculosis. Proc Natl Acad Sci USA (2018) 115(5):E964–73. doi: 10.1073/pnas.1711853115
249. Papotto PH, Ribot JC, Silva-Santos B. IL-17+γδ T Cells as Kick-Starters of Inflammation. Nat Immunol (2017) 18(6):604–11. doi: 10.1038/ni.3726
250. Shen H, Chen ZW. The Crucial Roles of Th17-Related Cytokines/Signal Pathways in M. Tuberculosis Infection. Cell Mol Immunol (2018) 15(3):216–25. doi: 10.1038/cmi.2017.128
Keywords: mycobacteria, tuberculosis, anti-TB drug, immune response, Mtb response
Citation: Park H-E, Lee W, Shin M-K and Shin SJ (2021) Understanding the Reciprocal Interplay Between Antibiotics and Host Immune System: How Can We Improve the Anti-Mycobacterial Activity of Current Drugs to Better Control Tuberculosis? Front. Immunol. 12:703060. doi: 10.3389/fimmu.2021.703060
Received: 30 April 2021; Accepted: 11 June 2021;
Published: 28 June 2021.
Edited by:
Veronica Schmitz, Oswaldo Cruz Foundation (Fiocruz), BrazilReviewed by:
Dwayne R. Roach, San Diego State University, United StatesC Gopi Mohan, Amrita Vishwa Vidyapeetham University, India
Copyright © 2021 Park, Lee, Shin and Shin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sung Jae Shin, c2pzaGluQHl1aHMuYWM=; Min-Kyoung Shin, bWtzaGluQGdudS5hYy5rcg==