 Sara Alonso-Álvarez1,2,3
Sara Alonso-Álvarez1,2,3 Enrique Colado
Enrique Colado Marco A. Moro-García
Marco A. Moro-García Rebeca Alonso-Arias
Rebeca Alonso-Arias- 1Haematology and Haemotherapy Department, Hospital Universitario Central de Asturias, Oviedo, Spain
- 2Laboratory Medicine Department, Hospital Universitario Central de Asturias, Oviedo, Spain
- 3Department of Hematologic Malignancies, Health Research Institute of the Principality of Asturias (ISPA), Oviedo, Spain
- 4Department of Cardiac Pathology, Health Research Institute of the Principality of Asturias (ISPA), Oviedo, Spain
- 5Immunology Department, Hospital Universitario Central de Asturias, Oviedo, Spain
The exquisite coupling between herpesvirus and human beings is the result of millions of years of relationship, coexistence, adaptation, and divergence. It is probably based on the ability to generate a latency that keeps viral activity at a very low level, thereby apparently minimising harm to its host. However, this evolutionary success disappears in immunosuppressed patients, especially in haematological patients. The relevance of infection and reactivation in haematological patients has been a matter of interest, although one fundamentally focused on reactivation in the post-allogeneic stem cell transplant (SCT) patient cohort. Newer transplant modalities have been progressively introduced in clinical settings, with successively more drugs being used to manipulate graft composition and functionality. In addition, new antiviral drugs are available to treat CMV infection. We review the immunological architecture that is key to a favourable outcome in this subset of patients. Less is known about the effects of herpesvirus in terms of mortality or disease progression in patients with other malignant haematological diseases who are treated with immuno-chemotherapy or new molecules, or in patients who receive autologous SCT. The absence of serious consequences in these groups has probably limited the motivation to deepen our knowledge of this aspect. However, the introduction of new therapeutic agents for haematological malignancies has led to a better understanding of how natural killer (NK) cells, CD4+ and CD8+ T lymphocytes, and B lymphocytes interact, and of the role of CMV infection in the context of recently introduced drugs such as Bruton tyrosine kinase (BTK) inhibitors, phosphoinosytol-3-kinase inhibitors, anti-BCL2 drugs, and even CAR-T cells. We analyse the immunological basis and recommendations regarding these scenarios.
Introduction
Human cytomegalovirus (CMV) is a DNA virus belonging to the herpesvirus family. Its transmission, through saliva, sexual contact, blood and breast milk, makes it highly prevalent, and the seroprevalence increases with age (1). The various studies carried out so far estimate a seroprevalence between 30% and more than 90%, depending on the population under study. This variation may be largely ascribed to age and socio-economic characteristics.
The implications of acute infection are of little relevance in terms of severity of infection and complications, as described below. However, the interest in healthy populations lies in its chronification and latency, and thereby in the development of an immune response that accompanies the host throughout its life, modulating its immune system through mechanisms that are not yet fully understood.
CMV infection plays a very important role in some population groups, such as immunosuppressed patients, and especially haematological patients, since acute infection causes significant morbidity and mortality in such patients.
The objective is to review the knowledge of CMV infection, and to understand its immunological effects in healthy individuals, in general, and in haematological patients.
CMV Infection
Primoinfection and Latent Phase
The infection is usually asymptomatic in adults, although sometimes it occurs in the setting of a mononucleosis-like syndrome. CMV is able to induce a persistent infection throughout the host lifetime. This is due to its ability to remain latent in some cells. It appears that CD34+ cells and CD14+ monocytes and macrophages as well as dendritic cells may constitute the fundamental reservoir (2–4). However, viral DNA has also been found in other cells of the immune system, as well as in epithelial and endothelial cells (3, 5–7) as they get also infected and which would also explain the mechanisms by which it is transmitted (Figure 1).
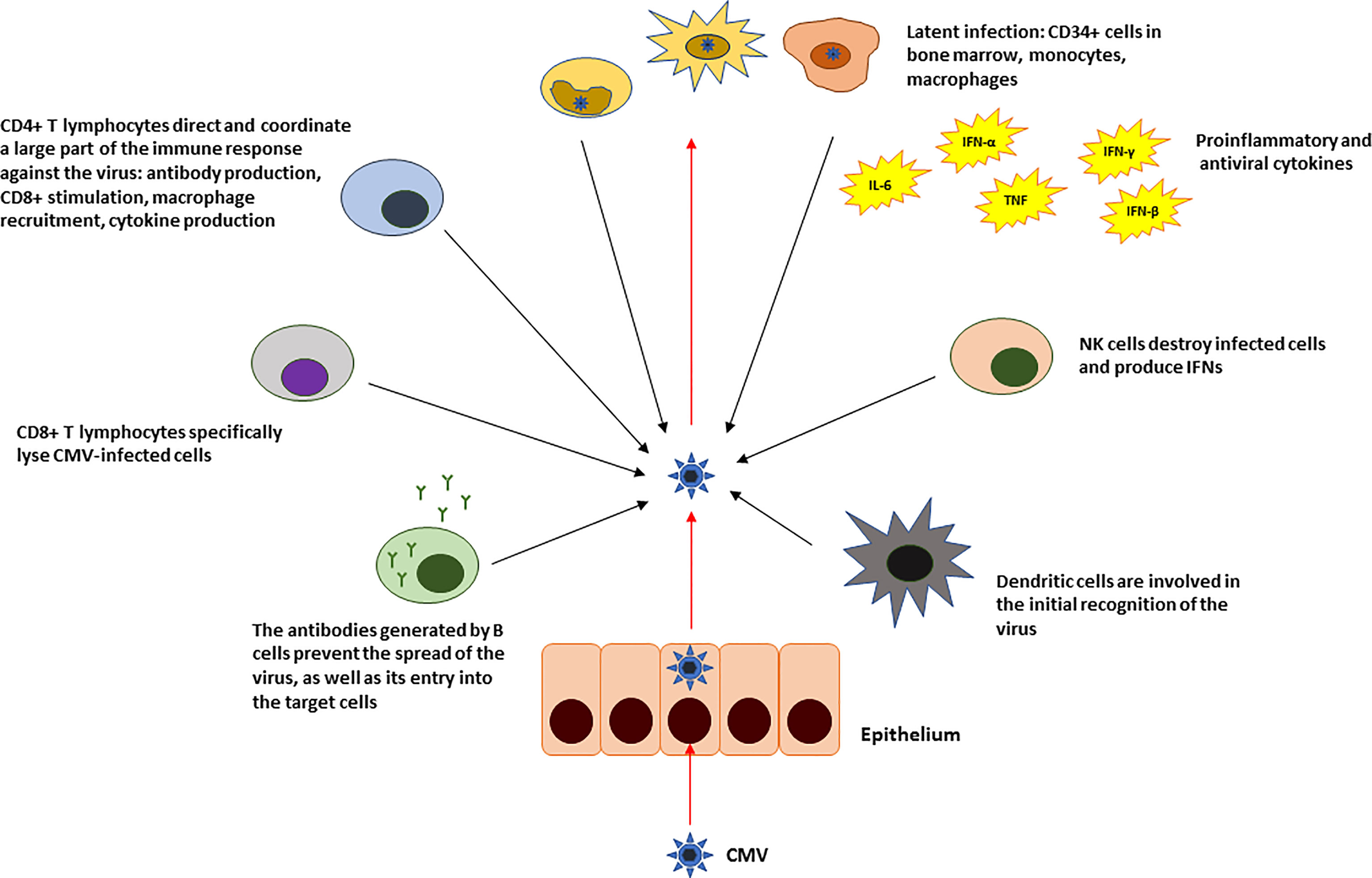
Figure 1 Summary of the innate and adaptive immune response against CMV in immunocompetent individuals.
In the infectious phase (and endogenous reactivation), the initial participation of proteins encoded by immediate-early (IE) genes is key, since they strongly activate the expression of the genes that consolidate the infectious stage (7, 8). These IE genes are expressed in cells that allow it (permissive), which are, apparently, differentiated cells (9). This expression is subject to the activation of the major immediate-early promoter (MIEP) protein. There are a variety of cellular factors that repress the activity of the MIEP in non-permissive cells (2, 10, 11). The terminal differentiation of these cells, which initially does not allow the reactivation of CMV, could decrease the expression of these factors, leaving the promoter complex active, thereby permitting viral reactivation (12, 13).
CMV infection induces specific IgA, IgG, and IgM production. Neutralizing IgGs appear early after infection and are permanently detectable thereafter. IgA can be detected for several months and even years after primoinfection (14). Cellular response is crucial in CMV infection control, CD4+ and CD8+ T lymphocytes are directed to pp65 protein and IE1 protein. When this control is well set, infection usually follows an indolent course (15).
Chronic Infection and Possible Oncogenic Role
Long-term effects on immune system have been described despite the indolent course of CMV infection in the immunocompetent population. CMV has an impact on the T cell pool (and leaves a fingerprint) by large expansions of the CMV-specific memory pool and expansion of terminally differentiated T cells/effector T cells. This might impair the immunological response to neoantigens as well as the number of IL-2 and IL-4-producing CD8+ memory T lymphocytes in the elderly. CMV-specific CD8+ T lymphocytes producing IFN-γ, might contribute to a proinflammatory status, but this is probably less clear. Therefore, some aging-associated processes might be accelerated due to latent CMV infection (15, 16). However, the role of CMV on aging is, in any case, a current matter of debate. And a recent extensive review of this matter by Jackson et al. has revealed that, according to existing data, there is only limited evidence supportive of the formation and maintenance of a large population of CMV specific CD8+ T cells, known as “memory inflation”, as a mechanism of immunosenescence (17).
Potential oncogenicity has also been studied, yielding controversial results in solid and haematological tumours. This may be because the multiple causes, over and above many of the fundamental processes responsible for the development of solid and haematological tumours, make it difficult to assess the specific value of the individual primary causes.
The oncogenic role of other viruses and pathogens (hepatitis C and B, papilloma, and the bacterium Helicobacter pylori) is better established in the context of some solid neoplasms, such as the liver in the case of the first two, the cervix and larynx in the second, and stomach cancer in the case of H. pylori.
The association between CMV infection and numerous solid tumours in relation to its oncogenic and immunomodulatory roles has also been sought and found. In the case of colon cancer, the association between CMV and the development of cancer varies from study to study (18–21), whereby some find an association but others confirm the absence of one. A recent and very detailed review of US28 potential roles in aherosclerotic disease and cancer demonstrates the difficulty of attributing the causation of US28 in carcinogenesis and its role in atherosclerotic disease (22). In this field, the results of the investigations are now very numerous and not always concordant, so they must be integrated in order to establish consistent concepts regarding the functions of this protein. This could later relate it to CMV infection, and this molecule might then be used as a therapeutic target or disease marker and targeting US28 might prevent CMV disease and could benefit immunosuppressed individuals, including transplant patients (23).
CMV is expressed in most human glioma samples (24). However, correlation with peripheral blood CMV detection in glioblastoma patients is variable (25). The low incidence of glioblastoma cases compared with the high prevalence of CMV infection makes it difficult to explain the initiating role of CMV in the development of this neoplasm. In addition, anti-CMV treatment with antiviral drugs such as foscarnet or valganciclovir has not definitively been shown to improve survival in patients with glioblastoma multiforme (26–28). The efficacy of anti-CMV immunotherapy (29) may be due to targeting of CMV-expressing cells that drive tumour growth, activation of other immune cells that cause additional killing of CMV-negative cells, or cross-priming after killing of CMV-positive tumour cells. Therefore, a proposed role for CMV in gliomagenesis is most likely to be associated with an as yet undefined event (30), although it seems that it might be supported by CMV’s oncoimmunomodulatory role.
Although CMV is the virus whose impact seems to be the most significant with respect to T lymphocyte deregulation, it has not been given so much importance from the haematological point of view, and very little is known about its oncogenic role. In fact, very few studies have analysed the influence of CMV on haematological pathology. In contrast, the association of the Epstein–Barr virus with the development of lymphomas is well established (31–34). The fact that the EBV tropism occurs in the B lymphocytes, where it remains latent (35–37), may make the relationship much more direct for the etiological study. In the case of CMV, no indirect association has been sought, perhaps because, although not directly, the effect on, or damage caused to the functionality of B lymphocytes and, especially T lymphocytes, by chronic infection and successive reactivations indirectly affects degree of predisposition to lymphomagenesis.
Although no prospective studies have been carried out, at least three retrospective studies have analysed the influence of the virus in populations that develop lymphomas. Two of the studies did not find a higher seroprevalence in patients with T cell lymphomas compared to the age-controlled population (38, 39). However, another study (40) found a very high seroprevalence in patients with mycosis fungoides and Sézary syndrome (Table 1).
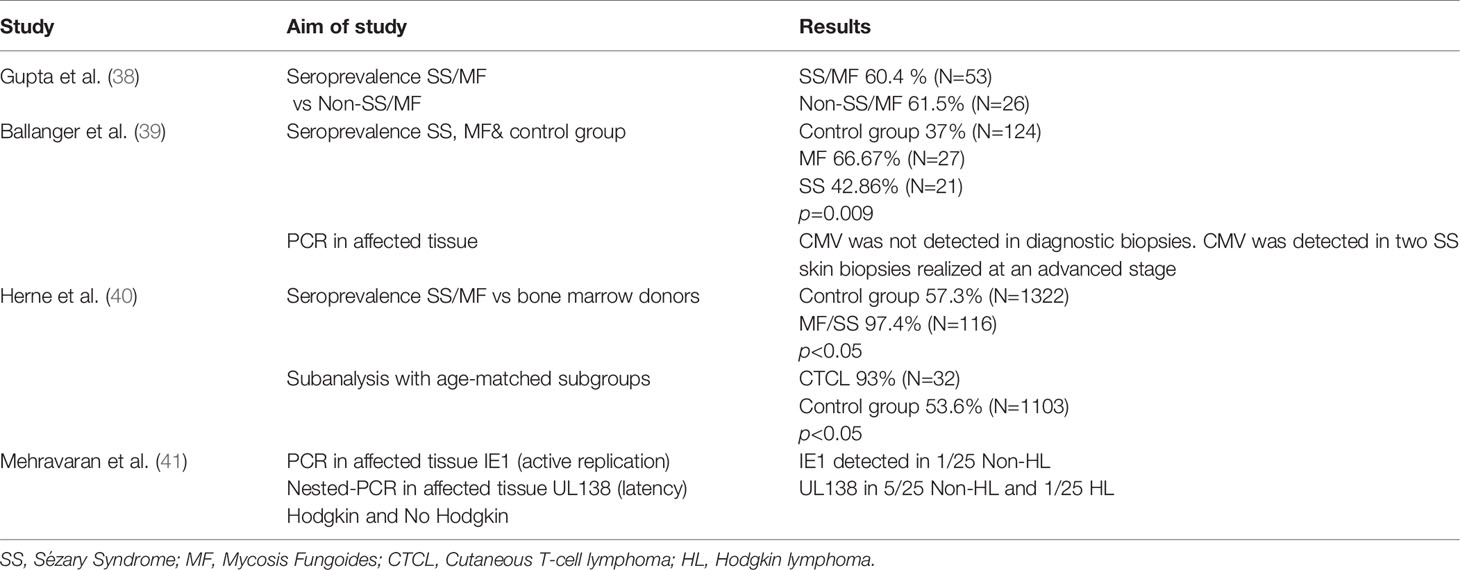
Table 1 CMV role in lymphoma development.
The expression of proteins and transcription factors of the virus has also been observed in some series of patients. Specifically, in a series of Iranian patients, the expression of UL138 mRNAs (as latent infection markers) and IE1 proteins (as reactivation markers) was studied by RT-PCR in patients with Hodgkin and non-Hodgkin lymphomas. The expression of UL138 mRNAs was found to be expressed in 20% of the T cell lymphomas in the series (41). Of note, this observation remains to be explained since T cells are not infected by CMV; casualty seems to be difficult to demonstrate here due to a possible indirect effect; indeed we are yet to know if a potential immune dysregulation motivated by a chronic exposure to the antigen might be responsible of this association.
CMV in the Haematological Setting
CMV in Allogeneic Stem Cell Transplantation
Definitions Regarding CMV Infection
Definitions of CMV infection and disease were initially developed and published as part of the proceedings of the 4th International CMV Conference in Paris in 1993 and have been progressively updated, most recently in 2020 (42–45).
Infection involves the detection of CMV in biological samples. When monitoring patients after transplant, it is usually determined in blood. In these cases, it is worthwhile differentiating whether the infection is detected by finding the antigen (antigenemia), growth in cell culture (viremia), or detecting DNA (DNAemia).
Primary infection takes place in seronegative patients, while reactivation refers to virus detection in previously seropositive patients. Recurrent infection refers to the detection of CMV in a patient with evidence of infection, but when there has been a 4-week infection-free gap between the two determinations. Reinfection refers to a new infection by a different viral strain, while reactivation is established when the same viral strain, of endogenous origin is involved.
CMV disease involves the conjunction of signs and/or symptoms that indicate organ involvement (pulmonary, gastrointestinal, hepatic, retinal, renal, myocardial, encephalic, pancreatic, etc.) together with the detection of CMV (using one or more of the validated techniques) in the affected organ or tissue.
T Lymphocyte Reconstitution and CMV: Cause and Consequence
T lymphocyte reconstitution has an initial thymus-independent phase, during which we observe the antigen-driven expansion of T lymphocytes infused with the graft (Figure 2). The second phase is thymus-dependent. Naïve T lymphocytes derived from the donor with a diverse T lymphocyte receptor (TCR) repertoire expand, although very slowly, so it takes years to complete the reconstitutions of this subset (46). However, the process cannot always be completed in this way because thymic function is conditioned in many allogeneic SCT recipients, thymic involutes in old patients and graft-versus-host disease (GVHD) damages epithelial thymus cells (47). When this occurs, the thymic-independent pathway rapidly generates CD8+T lymphocytes (48), resulting in an inversion of the CD4:CD8 ratio that can persist for years (49). It also leads to peripheral expansion of memory T lymphocytes (CD45RO+CD27+/CD45RO+CD27-) since generation of naïve T lymphocytes (CD45RA+/CD28+) from prethymic progenitors depends on a functional thymus (50). The differences between precursor sources, conditioning regimens and donor features are a consequence of the absolute numbers of CD4+ and CD8+ T lymphocytes infused. In any case, the absolute number of CD4+ T lymphocytes (i.e., regulatory T lymphocytes (Tregs) and conventional CD4+ T lymphocytes), remain at unrecovered levels up to 2 years after hematopoietic stem-cell transplantation (HCT). CD8+ T lymphocytes can recover faster, but also depend on the conditioning regimen and immunosuppressors used. The TCR repertory is considered to drive and be the result of disease control and GVHD activity. With regard to disease control, a study including umbilical cord blood donors (UCDs) and matched related or unrelated donors using in vivo T cell depletion (TCD) showed that patients who remained in remission had greater TCR diversity. A broader TCR spectrum could have an antitumoral role. A narrower TCR spectrum is in turn observed in those with GVHD, which would presumably be related to preferential expansion of particular T lymphocyte clones (51). In contrast, another study reported that grade 2–3 acute GVHD is associated with greater TCR diversity (52). These differences might be due to the high variability among the conditioning regimens, immunosuppressive therapy, and donor source (53).
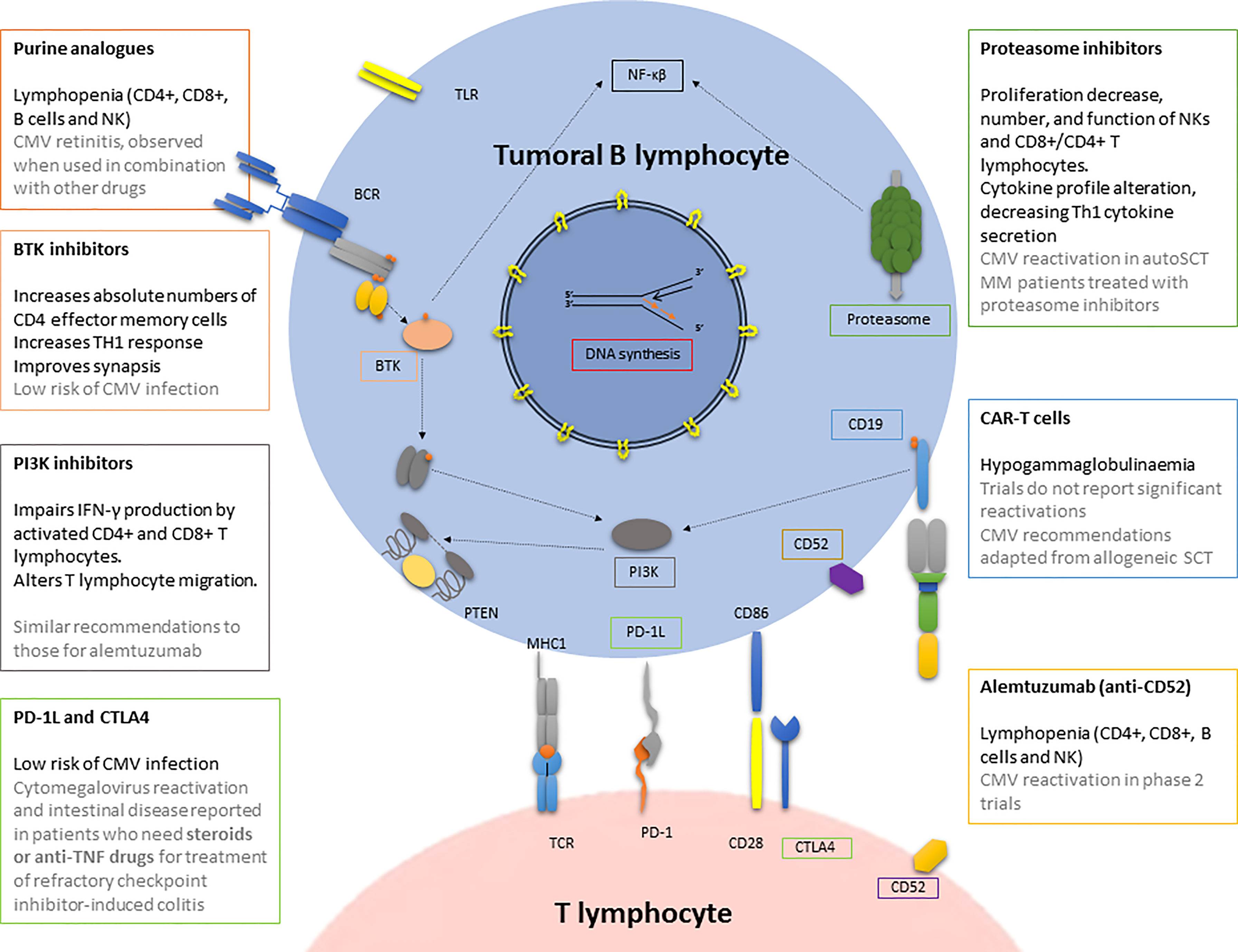
Figure 2 CMV infection/reactivation in the context of antitumoral drugs used in haematological patients. BCR, B cell receptor; BTK, Bruton tyrosine kinase; CAR-T cells, chimeric antigen receptor T cell; CMV, cytomegalovirus; CTLA4, cytotoxic T-lymphocyte antigen 4; MHC, major histocompatibility complex; NK, natural killer; PI3K, phosphatidyl inositol 3 kinase; PD, programmed death; PD-1L, programmed death-ligand 1; PTEN, phosphatase and tensin homologue; SCT, stem cell transplantation; TLR, toll-like receptor.
Activity of T lymphocytes (CD4+ and CD8+) is essential for the control of CMV infection (15, 53, 54). The correct reconstitution of CD8+ and CD4+ T lymphocytes is associated with the control of CMV infection (55). CMV-specific CD8+ T lymphocyte reconstitution is usually delayed by around 3 months after transplantation. There have been reports of prompt recovery, which is thought to be based on the receptor’s ability to bring about T lymphocyte lymphopoiesis (56). Knowing the HLA-typing of the donor, the source of the graft and the type of conditioning are essential for this rapid recovery and therefore the early control of the infection: HLA-typing facilitates the reestablishment, which is faster in identical donors without mismatch, but slower the more T depletion there is. Receptor immunity also influences infection control until immune reconstitution is established, especially in patients on reduced intensity regimens (57).
The relation between T lymphocyte reconstitution and CMV infection is bidirectional, and while delayed T lymphocyte reestablishment compromises anti-CMV protection, prompt reactivation of this virus conditions the characteristics of the recovery (58). CMV expands effector memory T lymphocytes, causing a linked contraction of all naïve T lymphocytes, including putative CD31+ thymic emigrants (59).
Donor Source Role
With respect to the source of the graft, patients who receive an allogeneic umbilical cord blood (UCB) transplant are at the highest risk of CMV infection because of the type of cells present in UCB. In this type of transplant, T lymphocytes are immature, which lower the risk of developing GVHD, but could increase the risk of severe infections (60). UCB contains Tregs, which are more potent suppressors than adult Treg (61). On the other hand, this is consistent with the high dose of anti-thymocyte globulin employed in the conditioning regimen, which is needed to prevent mainly graft rejection but also the effects of GVHD. In summary, T lymphocyte reconstitution is delayed, and this is considered the main cause of the increased risk of infection, particularly by CMV, in this cohort (62).
Patients who receive CD34+ (positively selected) allogeneic SCT or T lymphocyte-depleted allogeneic progenitors belong to the high-risk group for CMV infection because they lack the mature and naïve T lymphocytes that should make cellular immunity reconstitution possible.
The faster engraftment of peripheral blood progenitors compared with bone marrow and cord blood progenitors, and the concordant faster immune reconstitution demonstrated in vitro (63) and in vivo (64), lead us to expect that better immune reconstitution against CMV would reduce the incidence of CMV disease in this group. Several studies have compared the sources and found mixed results for the risk of CMV reactivation in peripheral blood and bone marrow. For example, a non-randomized study of 158 patients showed that the incidence of CMV reactivation (monitoring antigenemia) and CMV interstitial pneumonia were lower in the peripheral blood than in the bone marrow group (65). Another randomized study (n = 172) showed the opposite relationship, with a higher incidence of CMV infection in unmodified peripheral blood SCT recipients (66). This latter theory of a higher risk of CMV infection in peripheral blood than in UCB and bone marrow has been described (63). However, another study found no such differences (64).
This risk of CMV infection drops to an intermediate level as time goes by, as long as anti-T drugs or high-dose steroids are not added.
The influence of the HLA discrepancy between donor and recipient is highly nuanced or dependent on the type of conditioning and immunosuppressive treatment used since these are adjusted based on these discrepancies. The greater the discrepancy, the greater the risk of viral reactivation, but as explained above, this could be a response to the confluence of a more powerful immunosuppressor therapy and the anti-T-lymphocyte agents used.
Patients undergoing haploidentical allogeneic hematopoietic SCT (Haplo-HSCT) have been considered to be at higher risk of CMV reactivation than those receiving HLA-matched allografts due to impaired CMV-specific T lymphocyte reconstitution. Analysis of this and monitoring the CMV DNA load in parallel with CMV-specific IFN-γ-producing CD8+ and CD4+ T lymphocytes revealed that CMV was reactivated approximately as often in PTCy-haplo and HLA-matched recipients, and that CMV-specific T lymphocyte counts were similar in the two groups at most of the times examined. These findings suggest that the two groups reconstitute CMV-specific T lymphocyte immunity in a similar fashion (67).
Immunosuppressors and Anti-T Lymphocyte Agents
Steroids and anti-T lymphocyte agents are the cornerstone of the prevention and treatment of graft rejection and, fundamentally, of the development of graft-versus-recipient disease, the appearance of which considerably increases transplant-related mortality (not associated with relapse) (TRM). These data are well established, and by way of example, in one of the most recent series of patients treated with novel immunosuppressive treatment options, an increase in mortality of 5 to 16% has been described in patients with or without GVHD (68).
Patients who receive an allogeneic transplant from any source and who are treated with high-dose steroids or who receive anti-T lymphocyte agents, such as the aforementioned anti-thymocyte globulin, but also high doses of cyclophosphamide are also at high risk. This drug is necessarily employed in haploidentical transplant after progenitor infusion to avoid the GVHD that would accompany a half-discrepant haplotype. However, T lymphocyte in vivo depletion with cyclofosfamide is being used increasingly often in patients at high risk of GVHD when they undergo non-haploidentical transplants.
Cyclophosphamide
The effects of cyclophosphamide as a T lymphocyte depletion regimen were first studied in the haploidentical transplant setting (69). Its benefits were then extended to other mismatched, or even matched, donors (70). Cyclophosphamide is used as an agent that depletes donor T lymphocytes in vivo. For this purpose, it is administered during the first days of infusing the progenitor cells – the post-transplant-cyclophosphamide (PTCy) strategy. The mechanism by which cyclophosphamide modifies the T lymphocyte response and thereby reduces GVHD has been studied and extrapolated based on murine models of skin graft rejections. However, there has been some disagreement about whether the mechanisms involved are similar.
Nunes et al. developed a murine major histocompatibility complex (MHC)-haploidentical HCT model (B6C3F1 → B6D2F1) that is equivalent to the clinical HCT setting. They described how PTCy, a non-T lymphocyte-cycle-dependent alkylator, affects both highly and lowly proliferative host-alloreactive donor T lymphocytes. After infusion of progenitors, host-alloreactive donor T lymphocytes become activated, proliferative, and give rise to an inflammatory environment. Between post-transplant days +3 and +7, there is continued high-level proliferation of host-alloreactive donor CD8+ effector T lymphocytes and reduced, but continued proliferation of the surviving host-alloreactive donor CD4+ T lymphocytes, both effector and Tregs (70). This time schedule and pattern of proliferation is important for determining when to apply PTCy, because the decrease in host-alloreactive donor CD4+ effector T lymphocyte proliferation is needed to prevent GVHD (71). Around day +5, the functionality of surviving host-alloreactive donor effector T lymphocytes becomes impaired. The severity of this increases over time, and the apparently rapid effect of PTCy is enhanced by preferential reconstitution of donor CD4+ Tregs between days +7 and +21, which suppresses the host-alloreactive donor effector T lymphocytes. Meanwhile, host-non-alloreactive donor T lymphocytes maintain the slow proliferation, so the relative proportion of alloreactive donor T lymphocytes ends up increasing. The dynamics after these first stages might change over time and due to antigenic stimulation.
It has been observed that the regulatory lymphocytes of patients who receive PTCy recover quickly during the post-transplant period; as little as 1 month after the transplant, they are already at levels similar to the baseline of the donor, even when the transplant patient still exhibits lymphopenia (72). In patients receiving PTCy as the sole prophylaxis of GVHD in identical transplants, it has been observed that recipients’ TCR level after infusion of the progenitors is lower than that of the donor in the first moments. However, beyond the first 3 months, it begins to resemble the donor’s repertoire more closely, and in CMV-positive cases, the number and repertoire increasingly resemble those of the donor (73).
PTCy continues to prove to be one of the most beneficial agents for the control of GVHD and even of relapse. In a prospective multi-centre, randomized phase II clinical trial, regimens of (i) tacrolimus, mycophenolate mofetil, and cyclophosphamide, (ii) tacrolimus, methotrexate, and bortezomib, and (iii) tacrolimus, methotrexate, and maraviroc were compared against standard tacrolimus and methotrexate (74). Only the PTCy-containing regimen resulted in superior GVHD-free (severe acute and chronic), relapse-free survival. However, this benefit might alter when CMV infection appears. As previously stated, an increased CMV infection is associated with Haplo-HSCT receiving PTCy (HaploCy). However, the specific roles of the allograft source and the use of PTCy in CMV infection and disease are unresolved. A recent analysis of patients reported to the Center for International Blood and Marrow Transplant Research (CIBMTR) has addressed this aspect by comparing the cumulative incidence of CMV infection at day 180 in three cohorts: one that had received HaploCy (42%), a second group of sibling SCTs with PTCy (37%) and a third cohort of sibling SCTs with calcineurin inhibitor-based (23%) prophylaxis for AML/ALL/MDS. PTCy, regardless of donor, was associated with a higher incidence of CMV infection. The study also concluded that CMV infection could negate the cGVHD protective benefit of PTCy (75).
Methotrexate
Methotrexate is an antitumor and immunosuppressive drug. It is a structural analogue of folic acid; it blocks purine synthesis by inhibiting numerous regulatory enzymes. It does not have protumoral activity, unlike alkylating drugs, so it is of particular interest in the context of patients undergoing multiple therapies with potential induction of secondary tumours. It targets the S phase of the cell cycle, which determines that its action is largely confined to highly proliferative cells. In the early post-transplant period, it predominantly acts on highly proliferative alloreactive lymphocytes. Its use in the context of marrow transplantation dates back to 1970, when Donald Thomas described its role in controlling GVHD in dogs (76). Since then, its use has been maintained with dose modifications and optimizations in its combinations. In combination with a calcineurin inhibitor, it has been the standard of care for immunosuppression in myeloablative matched hematopoietic cell transplants.
Calcineurin Inhibitors
Calcineurin inhibitors stop downstream signalling of the T cell receptor (TCR) of naïve and memory T lymphocytes. This makes them highly effective at suppressing alloimmunity after SCT (77). They have undesirable collateral effects on anti-infectious and tumour-protective immunity, and reactivation of latent herpes viruses including CMV is frequent (78).
Anti-Thymocyte Globulin (ATG)
Low-dose ATG in transplants from high-risk alternative donors reduces GVHD and transplant-related death. All four randomized ATG trials undertaken demonstrated protection against GVHD, and three of them found no detrimental effect on survival (79–81). Two ATG formulations, derived from horse and rabbit, have different mechanisms of action, effects on Tregs, and depths of induced lymphopenia.
A direct association has consistently been found in both formulations between the use of ATG and the occurrence of viral infections, particularly CMV. This association has recently been validated (72).
Steroids
Patients being treated with high-dose steroids in the setting of GVHD, had significantly fewer activated CMV-specific T lymphocytes, both CD8+/IFN-γ+ and CD4+/IFN-γ+ at all developmental stages after allo-SCT. Reconstitution of CMV-specific CD4+ and CD8+ activated lymphocytes was observed at +180 days post-transplant, which was 80 days later than what was observed in the non-steroid counterpart (83). This work explains how and why steroid treatment increases the risk of CMV infection in patients who, because of their serological status and graft source, would otherwise not be at high risk of reactivation.
Some other groups have analysed how GVHD treatment affects T lymphocyte functionality. Young patients with active GVHD (based mainly on steroids) do not have adequate levels of activated CMV-specific CD4+and CD8+T lymphocytes, and do not produce IFN-γ and IL-2 (51). The lack of control of CMV reactivation after allogeneic SCT has been shown to respond to an impaired function of antigen-specific CD8+ T lymphocytes, whereas the amount of CMV-specific T lymphocytes does not have such a marked impact (84).
Monitoring CMV Infection
Serological determination of CMV-specific antibodies is important for determining a patient’s risk of CMV infection after transplantation (85). However, it is worth noting that in polytransfused patients (as exemplified by many haematological patients awaiting an allogeneic transplant), the serological status may be an artefact of a passive immunization mechanism. Discrepancies in CMV have been observed in up to 29%-33% of patients when this has been analysed (86, 87). Routinely determined serostatus is still currently used as a criterion for estimating CMV reactivation risk before transplant.
Without prophylaxis, the disease of 80% of patients who are serologically positive for CMV would reactivate after allogeneic transplantation. Strategies to prevent the development of CMV disease have emerged in recent decades, based on antiviral prophylaxis and CMV viremia monitoring (before developing the disease), and treatment before the disease causes organ damage (pre-emptive therapy). Serological status is not appropriate for the purpose of detecting CMV infection. In turn, detection of CMV in blood is the recommended strategy for preventing CMV disease (88). It can be detected with pp65 antigen in leukocytes, or by the polymerase chain reaction (PCR) technique, which is more sensitive (89) and therefore the most frequently used. The presence of antigen in peripheral blood or high loads of DNA are both predictive of CMV disease in these patients (90, 91). Likewise, patients who are to receive SCT must receive leukodepleted, filtered and irradiated products.
Treatment Options for CMV Infection: Improving Anti-CMV Reconstitution
To decide the best therapy for each patient, we must consider whether the patient has received antiviral prophylaxis, the risk profile for CMV disease, T lymphocyte reconstitution (both general and CMV-specific), viral load and potential drug resistance (92).
Effective agents to control CMV infection have been notably toxic. The three main drugs used in recent years are ganciclovir/valganciclovir, foscarnet and cidofovir. Ganciclovir is an analogue of nucleoside 2’-deoxyguanosine, which functions as a competitive inhibitor with deoxyguanosine triphosphate (dGTP) used by DNA polymerase of viruses for its replication. Foscarnet reversibly blocks the pyrophosphate-binding site of viral polymerase in a non-competitive manner and inhibits the separation of pyrophosphate from deoxynucleotide triphosphates, 100 times more strongly than its action against cellular DNA polymerase α. Cidofovir, in its active form of cidofovir diphosphate, prevents viral replication by selectively inhibiting viral DNA polymerases. It also inhibits human DNA polymerases, but up to 600 times weaklier. Ganciclovir induces haematological toxicity, in which neutropenia contributes to the development of opportunistic bacterial and fungal infections (93, 94). Foscarnet and cidofovir cause kidney damage and require strict analytical control that usually requires the patient’s admission to hospital to receive the treatment (95, 96). They are often used in the context of pre-emptive therapy, and, in fact, when oral agents such as valganciclovir are used as primary prophylaxis, there is no significant benefit (97). The strategy used to date has therefore been pre-emptive therapy, except in patients with a high risk of CMV disease, for whom alternative strategies are warranted. This scenario might soon change with the introduction of letermovir, which is known to reduce CMV reactivation and decrease all-cause mortality, but without being significantly toxic. In fact, it has performed similarly to the placebo in the phase 3 trials (98).
Letermovir inhibits the CMV DNA terminase complex, which is required for cleavage and encapsidation of the resulting viral DNA, thereby interfering with virion formation and without significant toxicities compared to placebo (98). This is extremely important since both ganciclovir, cidofovir and foscarnet have well known hematologic and renal toxicities that frequently limitate their use. The rationale for its potential benefit in comparison with other drugs is a different therapeutic target, that could overcome the resistance observed in the clinic (92, 99). There is an urgent need of drugs that effectively treat CMV reactivation, both in patients who are refractory to ganciclovir and in those who do not admit additional toxicities induced by the antivirals used. For this reason its use has been approved by some regulatory agencies and in the coming years we will verify the real impact on the clinic of transplant patients, fundamentally.
To improve T lymphocyte recovery and CMV control, strategies that aim to improve thymus function could be key. These include protection of thymic epithelium, thymopoiesis enhancement and increasing the number of T lymphoid precursors (47). These in vivo strategies have been highly diverse, including the use of specific lymphokines, growth factors and hormones, among others. On the other hand, cellular therapies have also been developed. Among these, infusion of ex vivo-expanded virus-specific cytotoxic T lymphocytes (CMV-VSTs) has been of particular note (100). These specific cytotoxic lymphocytes have been used either with the original donor source or with partially matched donors. In general, this strategy has proved efficacious in post-transplant CMV reactivation and disease (101, 102). There appear to be correlations (based on retrospective studies) between the baseline CD4+ level (the recipient’s previous immunity) and the rate and duration of engraftment and treatment success, probably because the CD4+ component is essential for mediating the engraftment and activity of the effector cells (103).
Attempts have been made to vaccinate against CMV, the most recent using techniques being based on DNA vaccines as well as peptide-based CMV conjugated with TLR agonists (104). The most advanced vaccines are those combined from CMV glycoprotein B (gB) with the adjuvant MF59. Its use has been tested in transplant patients to prevent post-transplant CMV disease, and in seronegative pregnant women or adolescents. In the first cohort (phase 2 study) of vaccinated vs placebo (105), a decrease in viremia was demonstrated in vaccinated patients proportional to the antibody load generated, although we know that the humoral response constitutes only one line of defence against the CMV, the results were not negligible, and confirmation of efficacy is awaited in phase 3 studies. In the cohort of young women, the vaccine was safe and immunogenic, although with an efficacy of 45% after 2 doses, therefore which was considered insufficient to continue the line of research (105). It is highly likely that the advances in DNA and RNA vaccine technology during 2020 and 2021 will change the spectrum of vaccines, and that the landscape will change in the years to come.
CMV in Other Haematological Settings: CMV Reactivation and Drugs in Haematological Malignancies
CMV and Autologous Stem Cell Transplantation
The role of CMV reactivation or infection has been much less extensively studied in patients receiving an autologous transplant than in those receiving an allogeneic transplant. From the perspective of the treatment of haematological disease, which usually have high short-term mortality rates, CMV reactivations have been studied to determine whether or not the disease will develop. The implications of these reactivations for the immune system in the medium and long term have been considered less important. In fact, the available series confirm that reactivation is a relatively frequent event in patients receiving autologous parental transplants. CMV reactivates in up to 41% of patients during the post-transplant period. Anyway, the rate of CMV disease remains low (106). Those patients receiving high doses of steroids, irradiation, purine analogues or alemtuzumab would require more attention (107).
CMV and Lymphoproliferative Disorders
The immunosuppression observed in many haematological tumours is conditioned by the underlying disease itself, but also fundamentally by the type of treatment used. Lymphoid pathology (acute and chronic) reveals a fundamental involvement of the B and T lymphoid compartments. This creates a tendency to develop viral and fungal infections, as well as infections borne by some intracellular parasites, such as Pneumocystis jirovecii. The cellular immunosuppression observed in this group of patients determines many of the prophylaxis strategies, which are sometimes conditioned to the treatment, but in others are more typical of the immune defect that generates the disease.
Thus, trimethoprim/sulfamethoxazole prophylaxis and the use of acyclovir in lymphoproliferative syndromes are quite widespread.
Acute and chronic myeloid pathologies, such as acute leukaemias or myelodysplastic syndromes, involve the myeloid compartment. Neutropenia mainly conditions bacterial and fungal infections. Their long evolution, the use of purine analogues in treatment regimens, or the frequent consolidation with allogeneic transplantation in this group causes CMV infection to be a concern in more advanced stages of the disease. CMV serostatus and non-relapse mortality rate after transplant are quite well established in acute myeloid leukaemias (108). In turn, recent explorations of series featuring other pathologies such as diffuse large B-cell lymphoma (DLBCL) have not been able to demonstrate such an association (109).
B-cell chronic lymphocytic leukaemias (B-CLL) is the most frequent chronic leukaemia in western countries. It has been suggested that this disease features a CD8+ T lymphocyte expansion that increases as the disease advances. Analysis of specific immune responses with tetrameric CMV-peptide complexes showed that patients exhibiting such an expansion, actually have an increase of CMV-specific CD8+CD45RA+CD27- T lymphocytes. This change is actually specific to CMV-seropositive patients and might reflect the greater effort required to control CMV reactivations (110). Similarly, another analysis of CMV in CLL patients has revealed that the expanded CMV-specific response observed in CLL patients apparently arises with the onset of chemotherapy and stabilizes thereafter (111). Some researchers have called attention to the possible consequence of redirecting autologous CMV-specific cytotoxic T lymphocytes (CTLs) towards B-CLL cells for cancer immunotherapy (112). CMV infection in patients with CLL usually occurs in the context of treatment with purine inhibitors, alemtuzumab, or even alkylating agents such as chlorambucil, and the disease itself. Of all the mechanisms involved in CLL immunosuppression, which falls beyond the scope of this review, hypogammaglobulinemia seems to be of great importance. It has been thoroughly described in CLL and has recently been associated with a shorter time until the next treatment (113).
CMV and Purine Analogues
All purine analogues mimic metabolic purines. Of them, fludarabine is the most extensively used in hematologic cancer. Fludarabine inhibits multiple DNA polymerases, DNA primase, and DNA ligase I, and is S phase-specific (since these enzymes are highly active during DNA replication). Fludarabine acts particularly on T cells, and is therefore very toxic to this compartment.
Normal T lymphocyte function is essential to the control of CMV reinfection. The use of agents such as purine analogues, that have a very potent T lymphocyte suppressor profile while being highly effective for treating chronic lymphoproliferative disorders (114), are highly relevant to the development of CMV reactivations.
Cases of CMV retinitis have been documented in patients with low-grade lymphomas treated with rituximab, fludarabine and steroids (115). Previous serological status was not available for most patients, probably because there is not a specific approach with CMV seropositivity outside transplant scenarios. Despite being a very rare event even in the seropositive population, the sequelae were adverse (blindness) in 86% of patients. CMV monitoring, and clinical observation of possible infection and reactivation, must be considered when these regimens are used.
CMV and Alemtuzumab
Alemtuzumab is a monoclonal antibody which acts on the CD52 protein found on the surface of peripheral blood and lymph node lymphocytes.
CMV reactivations in patients receiving alemtuzumab treatment for CLL are common, with a rate of around 20% in the phase II study with the drug that was evaluated in 78 patients (116). However, viral load monitoring and pre-emptive treatment manages to prevent the disease in most cases (117).
Some studies have even proposed the use of primary prophylaxis with valganciclovir in patients receiving alemtuzumab in whom the drug has a high efficacy (118). However, the main concern with using prophylactic valganciclovir is the additional myelosuppression beyond the already significant amount produced by the alemtuzumab-based regimens themselves. An attempt to reduce cost and toxicity by using lower weekly doses (119) produced lower toxicity and acceptable efficacy. However, due to the aforementioned concerns, primary prophylaxis is not common practice in this group of patients.
CMV and New Agents in the Treatment of Lymphoproliferative Syndromes
Many drugs have been introduced into the therapeutic arsenal to treat lymphoproliferative syndromes, in particular, CLL. However, CMV seropositivity in the era of new therapies has not led to reduced survival or relapse in CLL patients when prospectively compared with seronegative patients (120).
Bruton Tyrosine Kinase Inhibitors
CMV infection is not common in the case of a BTK inhibitor such as ibrutinib, although some cases have been reported (121). Although these are infrequent, we should always be aware of this possibility among immunocompromised patients, particularly those who have been treated with new agents, because this is a curable condition and early therapy seems to be critical if a good outcome is to be achieved.
PI3K Inhibitors
Recommendations for the management and prophylaxis of CMV reactivation have been established in patients treated with idelalisib. These patients can develop a serious infectious disease, with a high risk of CMV reactivation and the involvement of other opportunistic germs (122). It has been shown how idelalisib impairs IFN-γ production by activating T lymphocytes in CLL-treated patients, highlighting the importance of PI3Kδ in this process. Idelalisib inhibits T lymphocytes in relation to generic TCR stimulation and in response to virus-specific stimulation. The CD4+ and CD8+ T lymphocyte subsets both seem to be affected. This might explain the higher rate of CMV reactivations in those CLL patients who are treated with it. Finally, idelalisib has also been shown to alter T lymphocyte migration in vitro (123).
The high risk of CMV reactivation in this therapy group has meant that the guidelines have been adapted to those for the use of alemtuzumab (124). CMV-seronegative patients should receive CMV-negative or filtered blood products (this is blood, platelet, and plasma transfusions. Progenitor cell transplantation products are not irradiated due to the need to maintain the viability of the infused cells). Seropositive patients should be periodically tested for CMV. Idelalisib should be discontinued and ganciclovir or valganciclovir pre-emptively initiated in patients with positive CMV PCR/antigen and symptoms consistent with CMV infection, as well as in patients with fever and no clear source, and for whom quantitative CMV testing is unavailable, and in asymptomatic patients with a rising viral load (125).
Proteasome Inhibitors
Bortezomib and the next-generation proteasome inhibitors have been cornerstones of multiple myeloma (MM) treatment for several years. Although sufficiently relevant to be considered one of the causes of the increase in survival in this group of patients, they have also entailed infectious risks that should be highlighted. Bortezomib was reported to increase the risk of varicella-zoster virus (VZV) reactivation by up to four times (126), and a high incidence of CMV reactivation in fit patients receiving autologous transplantation due MM after treatment with bortezomib-based regimens was recently reported. Of 80 patients who underwent autologous SCT after bortezomib-based therapies, seven (4.1%) received an antiviral treatment for a symptomatic CMV reactivation and one died (127). Although no specific cause has yet been determined, in vitro studies have demonstrated that bortezomib can reduce proliferation, number, and function of natural killer cells and CD8+/CD4+ T and alter the cytokine profile, in particular decreasing Th1 cytokine secretion (128).
In the setting of allogeneic transplantation, bortezomib induces preferential apoptosis among alloreactive T lymphocytes (decreasing Th1 response) by inhibiting nuclear factor-κB (NF-κB) activation, whilst unstimulated T lymphocytes are barely affected. This might explain its potential use as a drug for GVHD treatment.
All these mechanisms could help explain the increased incidence of Herpesviridae family viruses (of which VZV is the best known) in MM patients and might contribute to the increased susceptibility to CMV reactivation in MM patients treated with bortezomib-based regimens followed by ASCT (129).
CMV and CAR-T-Cells
One of the most significant innovations in recent years has been the treatment with the CD19-directed chimerical antigen receptor T lymphocyte. Currently, data regarding the rate and type of viral infections in patients receiving this treatment are scarce. However, there are already some data on the incidence of CMV infections and reactivation in real clinical practice. In a retrospective series of 60 patients with aggressive lymphoma treated with CAR-T, who received antiviral prophylaxis for herpes simplex virus (HSV) according to the recommendations of each hospital, 10 viral infections were documented in the first 30 days, of which two corresponded to CMV reactivations, without organ affectation. During days 31-180, another reactivation by CMV occurred, again without organ disease. None of them, therefore, compromised the life of the patient (130).
Considering now the patients with ALL, data were collected from 83 patients up to 21 years of age who had received CAR-T therapy for this reason (131). Sixteen of them developed viral infections within the first 28 days; these were caused mainly by respiratory viruses, without specifying infection by CMV in these reported cases. Between days 29 and 90, seven patients got viral infections, all of which were due to respiratory viruses.
There are documented cases of CMV reactivation in the first month and during the first three months. Previous therapies, disease stage and patient basal characteristics seem to be crucial.
Regarding prophylaxis against viral infections, there are no unique international recommendations, and these recommendations are heterogeneous (132). The European recommendations are based on data from allogeneic transplant recipients (133). In general, antiviral prophylaxis is established with acyclovir or valacyclovir at least up to one year after CAR-T infusion, or until a CD4+ T lymphocyte level greater than 0.2 x 109/L is documented (134). In paediatrics, nonspecific immunoglobulins are also frequently used to maintain total IgG levels above 0.4 g/L (135). With respect to monitoring, the EBMT guidelines recommend that PCR be performed when clinically indicated (133).
Conclusions
The relationships between CMV infection and haematological pathologies are well known. Fundamentally, as a result of the important repercussions from the management of the infection and reactivation of the virus in the post-allogeneic transplant. However, there are many other situations that give rise to severe immunosuppression, either due to the haematological pathology itself or to the treatments used, which should increase vigilance concerning the complications derived from infection by this virus. Thus, it is necessary to study the effect of new drugs on the immune system and so adapt CMV prophylaxis and infection monitoring to different treatment schemes and situations, now that new anti-CMV drugs with fewer secondary effects are available for this purpose.
In contrast, knowledge about the role of this virus in the development of haematological diseases, in other words, its oncogenic or oncomodulatory potential, is much more limited. The difficulty in finding it may be because CMV infection is associated with age and the fact that its main effect is to bring about the dysfunction of T lymphocytes. T cell lymphomas are quite rare. The search for causality in the more commonly occurring counterpart B-cell lymphomas involves looking for indirect causes arising from the basic relationship between T and B lymphocytes.
Living with this herpesvirus is a situation that has arisen from years of evolution, which has apparently produced a balanced and tolerable relationship. This would imply that its effect, does not directly or indirectly limit survival or favour oncogenesis. In the forthcoming era of medicine we are approaching, there will be infinite possibilities for producing antiviral agents with very low toxicity and for immunizing against the most prevalent microorganisms. These promising possibilities should inspire an exhaustive study of the real effects of CMV and other microorganisms on the oncogenesis and mortality of individuals and populations. It seems clear that the ultimate answer about the oncogenic role of the virus will come from a prospective design approach that will allow us to determine whether human beings who are not infected have a lower risk of developing haematological neoplasms or other conditions than those who are infected.
Author Contributions
The authors’ contributions were as follows: SA-Á and EC designed the study, SA-Á and EC wrote the manuscript, and MM-G and RA-A reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grant PI17/00714 from the Spanish I+D+i 2013–2016 State Program, which was jointly funded by the Instituto de Salud Carlos III and the European Regional Development Fund (ERDF).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Montserrat Mera Torneiro and María Delia Alonso Rodríguez for their excellent technical assistance.
References
1. Staras SAS, Dollard SC, Radford KW, Flanders WD, Pass RF, Cannon MJ. Seroprevalence of Cytomegalovirus Infection in the United States, 1988-1994. Clin Infect Dis (2006) 43(9):1143–51. doi: 10.1086/508173
2. Sinclair J, Sissons P. Latency and Reactivation of Human Cytomegalovirus. J Gen Virol (2006) 87(7):1763–79. doi: 10.1099/vir.0.81891-0
3. Mendelson M, Monard S, Sissons P, Sinclair J. Detection of Endogenous Human Cytomegalovirus in CD34+ Bone Marrow Progenitors. J Gen Virol (1996) 77(12):3099–102. doi: 10.1099/0022-1317-77-12-3099
4. Hahn G, Revello MG, Patrone M, Percivalle E, Campanini G, Sarasini A, et al. Human Cytomegalovirus UL131-128 Genes Are Indispensable for Virus Growth in Endothelial Cells and Virus Transfer to Leukocytes. J Virol (2004) 78(18):10023–33. doi: 10.1128/jvi.78.18.10023-10033.2004
5. Maidji E, Somsouk M, Rivera JM, Hunt PW, Stoddart CA. Replication of CMV in the Gut of HIV-Infected Individuals and Epithelial Barrier Dysfunction. PloS Pathog (2017) 13(2). doi: 10.1371/journal.ppat.1006202
6. Jarvis MA, Nelson JA. Human Cytomegalovirus Persistence and Latency in Endothelial Cells and Macrophages. Curr Opin Microbiol (2002) 5(4):403–7. doi: 10.1016/S1369-5274(02)00334-X
7. Fortunato EA, Sanchez V, Yen JY, Spector DH. Infection of Cells with Human Cytomegalovirus during S Phase Results in a Blockade to Immediate-Early Gene Expression That Can Be Overcome by Inhibition of the Proteasome. J Virol (2002) 76(11):5369–79. doi: 10.1128/jvi.76.11.5369-5379.2002
8. Stenberg RM, Fortney J, Barlow SW, Magrane BP, Nelson JA, Ghazal P. Promoter-Specific Trans Activation and Repression by Human Cytomegalovirus Immediate-Early Proteins Involves Common and Unique Protein Domains. J Virol (1990) 64(4):1556–65. doi: 10.1128/jvi.64.4.1556-1565.1990
9. Sissons JGP, Bain M, Wills MR. Latency and Reactivation of Human Cytomegalovirus. J Infect (2002) 44(2):73–7. doi: 10.1053/jinf.2001.0948
10. Sinclair J, Sissons P. Latent and Persistent Infections of Monocytes and Macrophages. Intervirology (1996) 39(5-6):293–301. doi: 10.1159/000150501
11. Sinclair J, Baillie J, Bryant L, Caswell R. Human Cytomegalovirus Mediates Cell Cycle Progression Through G1 Into Early S Phase in Terminally Differentiated Cells. J Gen Virol (2000) 81(Pt 6):1553–65. doi: 10.1099/0022-1317-81-6-1553
12. Baskar JF, Smith PP, Nilaver G, Jupp RA, Hoffmann S, Peffer NJ, et al. The Enhancer Domain of the Human Cytomegalovirus Major Immediate-Early Promoter Determines Cell Type-Specific Expression in Transgenic Mice. J Virol (1996) 70(5):3207–14. doi: 10.1128/jvi.70.5.3207-3214.1996
13. Meier JL, Keller MJ, McCoy JJ. Requirement of Multiple cis-Acting Elements in the Human Cytomegalovirus Major Immediate-Early Distal Enhancer for Viral Gene Expression and Replication. J Virol (2002) 76(1):313–26. doi: 10.1128/jvi.76.1.313-326.2002
14. Nielsen SL, Sorensen I, Andersen HK. Kinetics of Specific Immunoglobulins M, E, A, and G in Congenital, Primary, and Secondary Cytomegalovirus Infection Studied by Antibody-Capture Enzyme-Linked Immunosorbent Assay. J Clin Microbiol (1988) 26(4):654–61. doi: 10.1128/jcm.26.4.654-661.1988
15. Terrazzini N, Kern F. Cell-Mediated Immunity to Human CMV Infection: A Brief Overview. F1000Prime Rep (2014) 6:28. doi: 10.12703/P6-28
16. Almanzar G, Schwaiger S, Jenewein B, Keller M, Herndler-Brandstetter D, Würzner R, et al. Long-Term Cytomegalovirus Infection Leads to Significant Changes in the Composition of the CD8+ T-Cell Repertoire, Which May Be the Basis for an Imbalance in the Cytokine Production Profile in Elderly Persons. J Virol (2005) 79(6):3675–83. doi: 10.1128/jvi.79.6.3675-3683.2005
17. Moro-García MA, López-Iglesias F, Marcos-Fernández R, Bueno-García E, Díaz-Molina B, Lambert JL, et al. More Intensive CMV-Infection in Chronic Heart Failure Patients Contributes to Higher T-Lymphocyte Differentiation Degree. Clin Immunol (2018) 192:20–9. doi: 10.1016/j.clim.2018.03.015
18. Hart H, Neill WA, Norval M. Lack of Association of Cytomegalovirus With Adenocarcinoma of the Colon. Gut (1982) 23(1):21–30. doi: 10.1136/gut.23.1.21
19. Boguszaková L, Hirsch I, Brichácek B, Faltýn J, Fric P, Dvoráková H, et al. Absence of Cytomegalovirus, Epstein-Barr Virus, and Papillomavirus DNA From Adenoma and Adenocarcinoma of the Colon. Acta Virol (1988) 32(4):303–8.
20. Bricháček B, Hirsch I, Závadová H, Procházka M, Faltýn J, Vonka V. Absence of Cytomegalovirus DNA from Adenocarcinoma of the Colon. Intervirology (1980) 14:223–7. doi: 10.1159/000149187
21. Huang ES, Roche JK. Cytomegalovirus DNA and Adenocarcinoma of the Colon: Evidence for Latent Viral Infection. Lancet (1978) 311:957–60. doi: 10.1016/S0140-6736(78)90248-9
22. Krishna BA, Miller WE, O'Connor CM. US28: HCMV's Swiss Army Knife. Viruses (2018) 10(8):445. doi: 10.3390/v10080445
23. Maussang D, Verzijl D, Van Walsum M, Leurs R, Holl J, Pleskoff O, et al. Human Cytomegalovirus-Encoded Chemokine Receptor US28 Promotes Tumorigenesis. Proc Natl Acad Sci USA (2006) 103:13068–73. doi: 10.1073/pnas.0604433103
24. Mitchell DA, Xie W, Schmittling R, Learn C, Friedman A, McLendon RE, et al. Sensitive Detection of Human Cytomegalovirus in Tumors and Peripheral Blood of Patients Diagnosed With Glioblastoma. Neuro Oncol (2008) 10:10–8. doi: 10.1215/15228517-2007-035
25. Dziurzynski K, Chang SM, Heimberger AB, Kalejta RF, Dallas SRMG, Smit M, et al. Consensus on the Role of Human Cytomegalovirus in Glioblastoma. Neuro Oncol (2012) 14:246–55. doi: 10.1093/neuonc/nor227
26. Stragliotto G, Rahbar A, Solberg NW, Lilja A, Taher C, Orrego A, et al. Effects of Valganciclovir as an add-on Therapy in Patients with Cytomegalovirus-Positive Glioblastoma: A Randomized, Double-Blind, Hypothesis-Generating Study. Int J Cancer (2013) 133:1204–13. doi: 10.1002/ijc.28111
27. Söderberg-Naucler C, Peredo I, Rahbar A, Hansson F, Nordlund A, Stragliotto G. Use of Cox Regression With Treatment Status as a Time-Dependent Covariate to Re-Analyze Survival Benefit Excludes Immortal Time Bias Effect in Patients With Glioblastoma Who Received Prolonged Adjuvant Treatment With Valganciclovir. Int J Cancer (2014) 135:248–9. doi: 10.1002/ijc.28663
28. Hadaczek P, Ozawa T, Soroceanu L, Yoshida Y, Matlaf L, Singer E, et al. Cidofovir: A Novel Antitumor Agent for Glioblastoma. Clin Cancer Res (2013) 19:6473–83. doi: 10.1158/1078-0432.CCR-13-1121
29. Prins RM, Cloughesy TF, Liau LM. Cytomegalovirus Immunity after Vaccination with Autologous Glioblastoma Lysate. N Engl J Med (2008) 359:539–41. doi: 10.1056/nejmc0804818
30. Rahman M, Dastmalchi F, Karachi A, Mitchell D. The role of CMV in Glioblastoma And Implications for Immunotherapeutic Strategies. Oncoimmunology (2019) 8(1):e1514921. doi: 10.1080/2162402X.2018.1514921
31. Deacon EM, Pallesen G, Niedobitek G, Crocker J, Brooks L, Rickinson AB, et al. Epstein-Barr Virus and Hodgkin’s Disease: Transcriptional Analysis of Virus Latency in the Malignant Cells. J Exp Med (1993) 177:339–49. doi: 10.1084/jem.177.2.339
32. Vockerodt M, Yap LF, Shannon-Lowe C, Curley H, Wei W, Vrzalikova K, et al. The Epstein-Barr virus and the Pathogenesis of Lymphoma. J Pathol (2015) 235:312–22. doi: 10.1002/path.4459
33. Shannon-Lowe C, Rickinson AB, Bell AI. Epstein-Barr Virus-Associated Lymphomas. Philos Trans R Soc B Biol Sci (2017) 372(1732):20160271. doi: 10.1098/rstb.2016.0271
34. Murray PG, Young LS. An Etiological Role for the Epstein-Barr Virus in the Pathogenesis of Classical Hodgkin Lymphoma. Blood (2019) 134:591–6. doi: 10.1182/blood.2019000568
35. Qu L, Rowe DT. Epstein-Barr Virus Latent Gene Expression in Uncultured Peripheral Blood Lymphocytes. J Virol (1992) 66:3715–24. doi: 10.1128/jvi.66.6.3715-3724.1992
36. Tierney RJ, Steven N, Young LS, Rickinson AB. Epstein-Barr Virus Latency in Blood Mononuclear Cells: Analysis of Viral Gene Transcription During Primary Infection and in the Carrier State. J Virol (1994) 68:7374–85. doi: 10.1128/jvi.68.11.7374-7385.1994
37. Chen H, Smith P, Ambinder RF, Hayward SD. Expression of Epstein-Barr virus BamHI-A Rightward Transcripts in Latently Infected B cells From Peripheral Blood. Blood (1999) 93(9):3026–32.
38. Gupta RK, Ramble J, Tong CY, Whittaker S, MacMahon E. Cytomegalovirus Seroprevalence is Not Higher in Patients With Mycosis Fungoides/Sezary Syndrome. Blood (2006) 107(3):1241–2.
39. Ballanger F, Bressollette C, Volteau C, Planche L, Dreno B. Cytomegalovirus: Its Potential Role in the Development of Cutaneous T-cell Lymphoma. Exp Dermatol (2009) 18:574–6. doi: 10.1111/j.1600-0625.2008.00817.x
40. Herne KL, Talpur R, Breuer-McHam J, Champlin R, Duvic M. Cytomegalovirus Seropositivity is Significantly Associated With Mycosis Fungoides and Sézary Syndrome. Blood (2003) 101:2132–6. doi: 10.1182/blood-2002-07-2247
41. Mehravaran H, Makvandi M, Zade AS, Neisi N, Kiani H, Radmehr H, et al. Association of Human Cytomegalovirus With Hodgkin’s Disease and Non-Hodgkin’s Lymphomas. Asian Pacific J Cancer Prev (2017) 18:593–7. doi: 10.22034/APJCP.2017.18.3.593
42. Ljungman P, De Bock R, Cordonnier C, Einsele H, Engelhard D, Grundy J, et al. Practices for Cytomegalovirus Diagnosis, Prophylaxis and Treatment In Allogeneic Bone Marrow Transplant Recipients: A Report From the Working Party for Infectious Diseases of the EBMT. Bone Marrow Transplant (1993) 12(4):399–403.
43. Reusser P, Cordonnier C, Einsele H, Engelhard D, Link D, Locasciulli A, et al. European Survey of Herpesvirus Resistance to Antiviral Drugs in Bone Marrow Transplant Recipients. Infectious Diseases Working Party of the European Group for Blood and Marrow Transplantation (EBMT). Bone Marrow Transplant (1996) 17(5):813–7.
44. Styczyński J, Tridello G, Koster L, Iacobelli S, van Biezen A, van der Werf S, et al. Death After Hematopoietic Stem Cell Transplantation: Changes Over Calendar Year Time, Infections and Associated Factors. Bone Marrow Transplant (2020) 55(1):126–36. doi: 10.1038/s41409-019-0624-z
45. Nava T, Ansari M, Dalle JH, de Heredia CD, Güngör T, Trigoso E, et al. Supportive Care During Pediatric Hematopoietic Stem Cell Transplantation: Beyond Infectious Diseases. A Report From Workshops on Supportive Care of the Pediatric Diseases Working Party (PDWP) of the European Society for Blood and Marrow Transplantation (EBMT). Bone Marrow Transplant (2020) 55(6):1126–36. doi: 10.1038/s41409-020-0818-4
46. Ringhoffer S, Rojewski M, Döhner H, Bunjes D, Ringhoffer M. T-Cell Reconstitution After Allogeneic Stem Cell Transplantation: Assessment by Measurement of the sjTREC/βTREC Ratio and Thymic Naïve T Cells. Haematologica (2013) 98:1600–8. doi: 10.3324/haematol.2012.072264
47. Clave E, Busson M, Douay C, De Latour RP, Berrou J, Rabian C, et al. Acute Graft-Versus-Host Disease Transiently Impairs Thymic Output in Young Patients After Allogeneic Hematopoietic Stem Cell Transplantation. Blood (2009) 113:6477–84. doi: 10.1182/blood-2008-09-176594
48. Mackall CL, Fleisher TA, Brown MR, Andrich MP, Chen CC, Feuerstein IM, et al. Distinctions Between CD8+ and CD4+ T-Cell Regenerative Pathways Result In Prolonged T-Cell Subset Imbalance After Intensive Chemotherapy. Blood (1997) 89(10):3700–7.
49. Forman SJ, Nöcker P, Gallagher M, Zaia J, Wright C, Bolen J, et al. Pattern of T Cell Reconstitution Following Allogeneic Bone Marrow Transplantation For Acute Hematological Malignancy. Transplantation (1982) 34:96–8. doi: 10.1097/00007890-198208000-00007
50. Mackall CL, Granger L, Sheard MA, Cepeda R, Gress RE. T-Cell Regeneration After Bone Marrow Transplantation: Differential CD45 Isoform Expression on Thymic-Derived Versus Thymic-Independent Progeny. Blood (1993) 82(8):2585–94.
51. Yew PY, Alachkar H, Yamaguchi R, Kiyotani K, Fang H, Yap KL, et al. Quantitative Characterization of T-Cell Repertoire in Allogeneic Hematopoietic Stem Cell Transplant Recipients. Bone Marrow Transplant (2015) 50:1227–34. doi: 10.1038/bmt.2015.133
52. Van Heijst JWJ, Ceberio I, Lipuma LB, Samilo DW, Wasilewski GD, Gonzales AMR, et al. Quantitative Assessment of T Cell Repertoire Recovery After Hematopoietic Stem Cell Transplantation. Nat Med (2013) 19:372–7. doi: 10.1038/nm.3100
53. Mehta RS, Rezvani K. Immune reconstitution post allogeneic transplant and the impact of immune recovery on the risk of infection. Virulence (2016) 7:901–16. doi: 10.1080/21505594.2016.1208866
54. Pera A, Vasudev A, Tan C, Kared H, Solana R, Larbi A. CMV Induces Expansion of Highly Polyfunctional CD4+ T cell Subset Coexpressing CD57 and CD154. J Leukoc Biol (2017) 101(2):555–66. doi: 10.1189/jlb.4A0316-112R
55. Jeitziner SM, Walton SM, Torti N, Oxenius A. Adoptive Transfer of Cytomegalovirus-Specific Effector CD4+ T Cells Provides Antiviral Protection From Murine CMV Infection. Eur J Immunol (2013) 43(11):2886–95. doi: 10.1002/eji.201343690
56. Lilleri D, Gerna G, Zelini P, Chiesa A, Rognoni V, Mastronuzzi A, et al. Monitoring of Human Cytomegalovirus and Virus-Specific T-Cell Response In Young Patients Receiving Allogeneic Hematopoietic Stem Cell Transplantation. PloS One (2012) 7(7):e41648. doi: 10.1371/journal.pone.0041648
57. Abate D, Cesaro S, Cofano S, Fiscon M, Saldan A, Varotto S, et al. Diagnostic Utility Of Human Cytomegalovirus-Specific T-Cell Response Monitoring in Predicting Viremia in Pediatric Allogeneic Stem-Cell Transplant Patients. Transplantation (2012) 93:536–42. doi: 10.1097/TP.0b013e31824215db
58. Sellar RS, Vargas FA, Henry JY, Verfuerth S, Charrot S, Beaton B, et al. CMV Promotes Recipient T-Cell Immunity Following Reduced-Intensity T-Cell-Depleted HSCT, Significantly Modulating Chimerism Status. Blood (2015) 125(4):731–9. doi: 10.1182/blood-2014-07-589150
59. Suessmuth Y, Mukherjee R, Watkins B, Koura DT, Finstermeier K, Desmarais C, et al. CMV Reactivation Drives Posttransplant T-Cell Reconstitution and Results in Defects in the Underlying TCRβ Repertoire. Blood (2015) 125(25):3835–50. doi: 10.1182/blood-2015-03-631853
60. Crisinel PA, Duval M, Crisinel DT, Mallette B, Bellier N, Vachon MF, et al. Risk of Cytomegalovirus Infection and Disease After Umbilical Cord Blood Transplantation in Children. Can J Infect Dis Med Microbiol (2013) 24(1):e11–5. doi: 10.1155/2013/159691
61. Godfrey WR, Spoden DJ, Ge YG, Baker SR, Liu B, Levine BL, et al. Cord blood CD4+CD25+-Derived T Regulatory Cell Lines Express FoxP3 Protein and Manifest Potent Suppressor Function. Blood (2005) 105:750–8. doi: 10.1182/blood-2004-06-2467
62. Cornetta K, Laughlin M, Carter S, Wall D, Weinthal J, Delaney C, et al. Umbilical Cord Blood Transplantation in Adults: Results of the Prospective Cord Blood Transplantation (COBLT). Biol Blood Marrow Transplant (2005) 11:149–60. doi: 10.1016/j.bbmt.2004.11.020
63. Ottinger HD, Beelen DW, Scheulen B, Schaefer UW, Grosse-Wilde H. Improved Immune Reconstitution After Allotransplantation of Peripheral Blood Stem Cells Instead of Bone Marrow. Blood (1996) 88(7):2775–9.
64. Roberts MM, To LB, Gillis D, Mundy J, Rawling C, Ng K, et al. Immune Reconstitution Following Peripheral Blood Stem Cell Transplantation, Autologous Bone Marrow Transplantation and Allogeneic Bone Marrow Transplantation. Bone Marrow Transplant (1993) 12(5):469–75.
65. Trenschel R, Ross S, Hüsing J, Ottinger H, Elmaagacli A, Roggendorf M, et al. Reduced Risk of Persisting Cytomegalovirus pp65 Antigenemia and Cytomegalovirus Interstitial Pneumonia Following Allogeneic PBSCT. Bone Marrow Transplant (2000) 25:665–72. doi: 10.1038/sj.bmt.1702216
66. Tong J, Sun Z, Liu H, Geng L, Zheng C, Tang B, et al. Risk Factors of CMV Infection in Patients After Umbilical Cord Blood Transplantation: A Multicenter Study in China. Chin J Cancer Res (2013) 25:695–703. doi: 10.3978/j.issn.1000-9604.2013.11.08
67. Huntley D, Giménez E, Pascual MJ, Remigia MJ, Amat P, Vázquez L, et al. Reconstitution of Cytomegalovirus-Specific T-Cell Immunity Following Unmanipulated Haploidentical Allogeneic Hematopoietic Stem Cell Transplantation With Posttransplant Cyclophosphamide. Bone Marrow Transplant (2020) 55(7):1347–56. doi: 10.1038/s41409-020-0865-x
68. Yu J, Parasuraman S, Shah A, Weisdorf D. Mortality, Length of Stay and Costs Associated With Acute Graft-Versus-Host Disease During Hospitalization For Allogeneic Hematopoietic Stem Cell Transplantation. Curr Med Res Opin (2019) 35(6):983–8. doi: 10.1080/03007995.2018.1551193
69. Luznik L, O’Donnell PV, Symons HJ, Chen AR, Leffell MS, Zahurak M, et al. HLA-Haploidentical Bone Marrow Transplantation for Hematologic Malignancies Using Nonmyeloablative Conditioning and High-Dose, Posttransplantation Cyclophosphamide. Biol Blood Marrow Transplant (2008) 14:641–50. doi: 10.1016/j.bbmt.2008.03.005
70. Nunes NS, Kanakry CG. Mechanisms of Graft-Versus-Host Disease Prevention by Post-Transplantation Cyclophosphamide: An Evolving Understanding. Front Immunol (2019) 10:2668. doi: 10.3389/fimmu.2019.02668
71. Wachsmuth LP, Patterson MT, Eckhaus MA, Venzon DJ, Kanakry CG. Optimized Timing of Post-Transplantation Cyclophosphamide in MHC-Haploidentical Murine Hematopoietic Cell Transplantation. Biol Blood Marrow Transplant (2020) 26:230–41. doi: 10.1016/j.bbmt.2019.09.030
72. Kanakry CG, Ganguly S, Zahurak M, Bolaños-Meade J, Thoburn C, Perkins B, et al. Aldehyde Dehydrogenase Expression Drives Human Regulatory T Cell Resistance to Posttransplantation Cyclophosphamide. Sci Transl Med (2013) 5(211):211ra157. doi: 10.1126/scitranslmed.3006960
73. Kanakry CG, Coffey DG, Towlerton AM, Vulic A, Storer BE, Chou J, et al. Origin and Evolution of the T Cell Repertoire After Posttransplantation Cyclophosphamide. JCI Insight (2016) 1(5):e86252. doi: 10.1172/jci.insight.86252
74. Bolaños-Meade J, Reshef R, Fraser R, Fei M, Abhyankar S, Al-Kadhimi Z, et al. Three Prophylaxis Regimens (Tacrolimus, Mycophenolate Mofetil, and Cyclophosphamide; Tacrolimus, Methotrexate, and Bortezomib; or Tacrolimus, Methotrexate, and Maraviroc) Versus Tacrolimus and Methotrexate For Prevention of Graft-Versus-Host Disease With Haemopoietic Cell Transplantation With Reduced-Intensity Conditioning: A Randomised Phase 2 Trial With a Non-Randomised Contemporaneous Control Group (BMT CTN 1203). Lancet Haematol (2019) 6:e132–43. doi: 10.1016/S2352-3026(18)30221-7
75. Goldsmith SR, Abid MB, Auletta JJ, Bashey A, Beitinjaneh A, Castillo P, et al. Posttransplant Cyclophosphamide is Associated With Increased Cytomegalovirus Infection: A CIBMTR Analysis. Blood (2021) 137(23):3291–305. doi: 10.1182/blood.2020009362
76. Storb R, Epstein RB, Graham TC, Thomas ED. Methotrexate Regimens For Control of Graft-Versus-Host Disease in Dogs With Allogeneic Marrow Grafts. Transplantation (1970) 9(3):240–6. doi: 10.1097/00007890-197003000-00007
77. Ekberg H, Bernasconi C, Tedesco-Silva H, Vítko S, Hugo C, Demirbas A, et al. Calcineurin Inhibitor Minimization in The Symphony Study: Observational Results 3 Years After Transplantation. Am J Transplant (2009) 9:1876–85. doi: 10.1111/j.1600-6143.2009.02726.x
78. Fisher RA. Cytomegalovirus Infection and Disease in the New Era of Immunosuppression Following Solid Organ Transplantation. Transpl Infect Dis (2009) 11:195–202. doi: 10.1111/j.1399-3062.2009.00372.x
79. Bacigalupo A, Lamparelli T, Barisione G, Bruzzi P, Guidi S, Alessandrino PE, et al. Thymoglobulin Prevents Chronic Graft-versus-Host Disease, Chronic Lung Dysfunction, and Late Transplant-Related Mortality: Long-Term Follow-Up of a Randomized Trial in Patients Undergoing Unrelated Donor Transplantation. Biol Blood Marrow Transplant (2006) 12:560–5. doi: 10.1016/j.bbmt.2005.12.034
80. Socié G, Schmoor C, Bethge WA, Ottinger HD, Stelljes M, Zander AR, et al. Chronic Graft-Versus-Host Disease: Long-Term Results From A Randomized Trial On Graft-Versus-Host Disease Prophylaxis With or Without Anti-T-cell Globulin ATG-Fresenius. Blood (2011) 117:6375–82. doi: 10.1182/blood-2011-01-329821
81. Kröger N, Solano C, Wolschke C, Bandini G, Patriarca F, Pini M, et al. Antilymphocyte Globulin for Prevention of Chronic Graft-Versus-Host Disease. N Engl J Med (2016) 374:43–53. doi: 10.1056/nejmoa1506002
82. Figgins B, Hammerstrom A, Ariza-Heredia E, Oran B, Milton DR, Yeh J. Characterization of Viral Infections after Antithymocyte Globulin–Based Conditioning in Adults Undergoing Allogeneic Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant (2019) 25:1837–43. doi: 10.1016/j.bbmt.2019.05.020
83. Suárez-Lledó M, Martínez-Cibrián N, Gutiérrez-García G, Dimova-Svetoslavova V, Marcos MA, Martín-Antonio B, et al. Deleterious Effect of Steroids on Cytomegalovirus Infection Rate after Allogeneic Stem Cell Transplantation Depends on Pretransplant Cytomegalovirus Serostatus of Donors and Recipients. Biol Blood Marrow Transplant (2018) 24:2088–93. doi: 10.1016/j.bbmt.2018.05.001
84. Özdemir E, St John LS, Gillespie G, Rowland-Jones S, Champlin RE, Molldrem JJ, et al. Cytomegalovirus Reactivation Following Allogeneic Stem Cell Transplantation is Associated With the Presence of Dysfunctional Antigen-Specific CD8+ T Cells. Blood (2002) 100:3690–7. doi: 10.1182/blood-2002-05-1387
85. Ljungman P, Hakki M, Boeckh M. Cytomegalovirus in Hematopoietic Stem Cell Transplant Recipients. Hematol Oncol Clin North Am (2011) 25(1):151–69. doi: 10.1016/j.hoc.2010.11.011
86. Perry DA, Hakki M. Assessment of a Cytomegalovirus Serology Dual-Testing Strategy in Hematopoietic Stem Cell Transplant Recipients. Transpl Infect Dis (2016) 18:809–14. doi: 10.1111/tid.12591
87. Giulieri S, Manuel O. QuantiFERON®-CMV Assay for the Assessment of Cytomegalovirus Cell-Mediated Immunity. Expert Rev Mol Diagn (2011) 11:17–25. doi: 10.1586/erm.10.109
88. Meyers JD, Ljungman P, Fisher LD. Cytomegalovirus Excretion as a Predictor of Cytomegalovirus Disease After Marrow Transplantation: Importance of Cytomegalovirus Viremia. J Infect Dis (1990) 162(2):373–80. doi: 10.1093/infdis/162.2.373
89. Boeckh M, Huang M, Ferrenberg J, Stevens-Ayers T, Stensland L, Nichols WG, et al. Optimization of Quantitative Detection of Cytomegalovirus DNA in Plasma by Real-Time PCR. J Clin Microbiol (2004) 42(3):1142–8. doi: 10.1128/JCM.42.3.1142-1148.2004
90. Boeckh M, Bowden RA, Goodrich JM, Pettinger M, Meyers JD. Cytomegalovirus Antigen Detection in Peripheral Blood Leukocytes After Allogeneic Marrow Transplantation. Blood (1992) 80(5):1358–64.
91. Einsele H, Hebart H, Kauffmann-Schneider C, Sinzger C, Jahn G, Bader P, et al. Risk Factors for Treatment Failures in Patients Receiving PCR-Based Preemptive Therapy for CMV Infection. Bone Marrow Transplant (2000) 25(7):757–63. doi: 10.1038/sj.bmt.1702226
92. Einsele H, Ljungman P, Boeckh M. How I Treat CMV Reactivation After Allogeneic Hematopoietic Stem Cell Transplantation. Blood (2020) 135(19):1619–29. doi: 10.1182/blood.2019000956
93. Reusser P, Einsele H, Lee J, Volin L, Rovira M, Engelhard D, et al. Infectious Diseases Working Party of the European Group for Blood and Marrow Transplantation. Randomized Multicenter Trial of Foscarnet Versus Ganciclovir For Preemptive Therapy of Cytomegalovirus Infection After Allogeneic Stem Cell Transplantation. Blood (2002) 99(4):1159–64. doi: 10.1182/blood.v99.4.1159
94. Robin M, Porcher R, De Castro Araujo R, de Latour RP, Devergie A, Rocha V, et al. Risk Factors For Late Infections After Allogeneic Hematopoietic Stem Cell Transplantation From a Matched Related Donor. Biol Blood Marrow Transplant (2007) 13(11):1304–12. doi: 10.1016/j.bbmt.2007.07.007
95. Mattes FM, Hainsworth EG, Geretti AM, Nebbia G, Prentice G, Potter M, et al. A Randomized, Controlled Trial Comparing Ganciclovir to Ganciclovir Plus Foscarnet (Each at Half Dose) for Preemptive Therapy of Cytomegalovirus Infection in Transplant Recipients. J Infect Dis (2004) 189(8):1355–61. doi: 10.1086/383040
96. Reusser P. Challenges and Options in the Management of Viral Infections After Stem Cell Transplantation. Support Care Cancer (2002) 10(3):197–203. doi: 10.1007/s005200100299
97. Boeckh M, Nichols WG, Chemaly RF, Papanicolaou GA, Wingard JR, Xie H, et al. Valganciclovir for the Prevention of Complications of Late Cytomegalovirus Infection After Allogeneic Hematopoietic Cell Transplantation: A Randomized Trial. Ann Intern Med (2015) 162(1):1–10. doi: 10.7326/M13-2729
98. Marty FM, Ljungman P, Chemaly RF, Maertens J, Dadwal SS, Duarte RF, et al. Letermovir Prophylaxis for Cytomegalovirus in Hematopoietic-Cell Transplantation. N Engl J Med (2017) 377(25):2433–44. doi: 10.1056/NEJMoa1706640
99. Kilgore JT, Becken B, Varga MG, Parikh S, Prasad V, Lugo D, et al. Use of Letermovir for Salvage Therapy for Resistant Cytomegalovirus in a Pediatric Hematopoietic Stem Cell Transplant Recipient. J Pediatr Infect Dis Soc (2020) 9(4):486–9. doi: 10.1093/jpids/piz050
100. Barrett AJ, Bollard CM. The Coming of Age of Adoptive T-Cell Therapy For Viral Infection After Stem Cell Transplantation. Ann Transl Med (2015) 3(5):62. doi: 10.3978/j.issn.2305-5839.2015.01.18
101. O'Reilly RJ, Prockop S, Hasan AN, Koehne G, Doubrovina E. Virus-Specific T-Cell Banks for 'off the Shelf' Adoptive Therapy of Refractory Infections. Bone Marrow Transplant (2016) 51(9):1163–72. doi: 10.1038/bmt.2016.17
102. Prockop SE, Hasan A, Koehne G, Doubrovina E, Sauter CS, Barker JN, et al. Third Party Donor Derived CMV Specific T Cells for the Treatment of Refractory CMV Viremia and Disease after Hematopoietic Stem Cell Transplant. Blood (2014) 124:184–4. doi: 10.1182/blood.v124.21.184.184
103. Fabrizio VA, Irene Rodriguez-Sanchez M, Mauguen A, Dahi PB, Doubrovina E, O’Reilly RJ, et al. Adoptive Therapy with CMV-Specific Cytotoxic T Lymphocytes Depends on Baseline CD41 Immunity to Mediate Durable Responses. Blood Adv (2021) 5:496–503. doi: 10.1182/bloodadvances.2020002735
104. Kharfan-Dabaja MA, Boeckh M, Wilck MB, Langston AA, Chu AH, Wloch MK, et al. A Novel Therapeutic Cytomegalovirus DNA Vaccine in Allogeneic Haemopoietic Stem-Cell Transplantation: A Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet Infect Dis (2012) 12:290 299. doi: 10.1016/S1473-3099(11)70344-9
105. Nakamura R, La Rosa C, Longmate J, Drake J, Slape C, Zhou Q, et al. Viraemia, Immunogenicity, and Survival Outcomes of Cytomegalovirus Chimeric Epitope Vaccine Supplemented With PF03512676 (CMVPepVax) in Allogeneic Haemopoietic Stem-Cell Transplantation: Randomised Phase 1b Trial. Lancet Haematol (2016) 3:e87–98. doi: 10.1016/S2352-3026(15)00246-X
106. Marchesi F, Pimpinelli F, Ensoli F, Mengarelli A. Cytomegalovirus Infection in Hematologic Malignancy Settings Other Than The Allogeneic Transplant. Hematol Oncol (2018) 36(2):381–91. doi: 10.1002/hon.2453
107. Hebart H, Schröder A, Löffler J, Klingebiel T, Martin H, Wassmann B, et al. Cytomegalovirus Monitoring By Polymerase Chain Reaction of Whole Blood Samples From Patients Undergoing Autologous Bone Marrow or Peripheral Blood Progenitor Cell Transplantation. J Infect Dis (1997) 175:1490–3. doi: 10.1086/516484
108. Zhang YL, Zhu Y, Xiao Q, Wang L, Liu L, Luo XH. Cytomegalovirus Infection is Associated With AML Relapse After Allo-HSCT: A Meta-Analysis of Observational Studies. Ann Hematol (2019) 98(4):1009–20. doi: 10.1007/s00277-018-3585-1
109. Mariotti J, Maura F, Spina F, Roncari L, Dodero A, Farina L, et al. Impact of Cytomegalovirus Replication and Cytomegalovirus Serostatus on the Outcome of Patients With B Cell Lymphoma After Allogeneic Stem Cell Transplantation. Biol Blood Marrow Transplant (2014) 20(6):885–90. doi: 10.1016/j.bbmt.2014.02.015
110. Mackus WJM, Frakking FNJ, Grummels A, Gamadia LE, De Bree GJ, Hamann D, et al. Expansion of CMV-Specific CD8+CD45RA+CD27- T Cells in B-Cell Chronic Lymphocytic Leukemia. Blood (2003) 102:1057–63. doi: 10.1182/blood-2003-01-0182
111. Pourgheysari B, Bruton R, Parry H, Billingham L, Fegan C, Murray J, et al. The Number of Cytomegalovirus-Specific CD4+ T Cells is Markedly Expanded in Patients With B-Cell Chronic Lymphocytic Leukemia and Determines the Total CD4+ T-Cell Repertoire. Blood (2010) 116:2968–74. doi: 10.1182/blood-2009-12-257147
112. Mous R, Savage P, Remmerswaal EBM, van Lier RAW, Eldering E, van Oers MHJ. Redirection of CMV-Specific CTL Towards B-CLL via CD20-Targeted HLA/CMV Complexes. Leukemia (2006) 20:1096–102. doi: 10.1038/sj.leu.2404185
113. Parikh SA, Leis JF, Chaffee KG, Call TG, Hanson CA, Ding W, et al. Hypogammaglobulinemia in Newly Diagnosed Chronic Lymphocytic Leukemia: Natural History, Clinical Correlates, and Outcomes. Cancer (2015) 121:2883–91. doi: 10.1002/cncr.29438
114. Morra E, Nosari A, Montillo M. Infectious Complications in Chronic Lymphocytic Leukaemia. Hematol Cell Ther (1999) 41(4):145–51. doi: 10.1007/s00282-999-0145-0
115. Chan TSY, Cheung CYM, Yeung IYL, Hwang YY, Gill H, Wong IY, et al. Cytomegalovirus Retinitis Complicating Combination Therapy With Rituximab and Fludarabine. Ann Hematol (2015) 94:1043–7. doi: 10.1007/s00277-014-2296-5
116. Ferrajoli A, O’Brien SM, Cortes JE, Giles FJ, Thomas DA, Faderl S, et al. Phase II Study of Alemtuzumab in Chronic Lymphoproliferative Disorders. Cancer (2003) 98:773–8. doi: 10.1002/cncr.11551
117. Laurenti L, Piccioni P, Cattani P, Cingolani A, Efremov D, Chiusolo P, et al. Cytomegalovirus Reactivation During Alemtuzumab Therapy for Chronic Lymphocytic Leukemia: Incidence and Treatment With Oral Ganciclovir. Haematologica (2004) 89(10):1248–52.
118. O’Brien S, Ravandi F, Riehl T, Wierda W, Huang X, Tarrand J, et al. Valganciclovir Prevents Cytomegalovirus Reactivation in Patients Receiving Alemtuzumab-Based Therapy. Blood (2008) 111:1816–9. doi: 10.1182/blood-2007-03-080010
119. Hwang YY, Cheung WWW, Leung AYH, Tse E, Au WY, Kwong YL. Valganciclovir thrice Weekly for Prophylaxis Against Cytomegalovirus Reactivation During Alemtuzumab Therapy. Leukemia (2009) 23:800–1. doi: 10.1038/leu.2008.282
120. Parry HM, Damery S, Hudson C, Maurer MJ, Cerhan JR, Pachnio A, et al. Cytomegalovirus Infection Does Not Impact on Survival or Time to First Treatment in Patients With Chronic Lymphocytic Leukemia. Am J Hematol (2016) 91:776–81. doi: 10.1002/ajh.24403
121. Reddy Y, Baig M, Kalva N, Puli S, Dhillon S. Cytomegalovirus Proctitis in a Patient with Chronic Lymphocytic Leukemia on Ibrutinib Therapy: A Case Report. Cureus (2020) 12(4):e7837. doi: 10.7759/cureus.7837
122. Greenwell IB, Ip A, Cohen JB. PI3K Inhibitors: Understanding Toxicity Mechanisms and Management. Oncol (Williston Park) (2017) 31(11):821–8.
123. Martinelli S, Maffei R, Fiorcari S, Quadrelli C, Zucchini P, Benatti S, et al. Idelalisib impairs T-Cell-Mediated Immunity in Chronic Lymphocytic Leukemia. Haematologica (2018) 103:e598–601. doi: 10.3324/haematol.2017.187070
124. Cuneo A, Barosi G, Danesi R, Fagiuoli S, Ghia P, Marzano A, et al. Management of Adverse Events Associated With Idelalisib Treatment in Chronic Lymphocytic Leukemia And Follicular Lymphoma: A Multidisciplinary Position Paper. Hematol Oncol (2019) 37:3–14. doi: 10.1002/hon.2540
125. Cheah CY, Fowler NH. Idelalisib in the Management of Lymphoma. Blood (2016) 128:331–6. doi: 10.1182/blood-2016-02-702761
126. Chanan-Khan A, Sonneveld P, Schuster MW, Stadtmauer EA, Facon T, Harousseau JL, et al. Analysis of Herpes Zoster Events Among Bortezomib-Treated Patients in the Phase III APEX Study. J Clin Oncol (2008) 26:4784–90. doi: 10.1200/JCO.2007.14.9641
127. Marchesi F, Mengarelli A, Giannotti F, Tendas A, Anaclerico B, Porrini R, et al. High Incidence of Post-Transplant Cytomegalovirus Reactivations in Myeloma Patients Undergoing Autologous Stem Cell Transplantation After Treatment With Bortezomib-Based Regimens: A Survey From the Rome Transplant Network. Transpl Infect Dis (2014) 16:158–64. doi: 10.1111/tid.12162
128. Blanco B, Pérez-Simón JA, Sánchez-Abarca LI, Carvajal-Vergara X, Mateos J, Vidriales B, et al. Bortezomib Induces Selective Depletion of Alloreactive T Lymphocytes and Decreases the Production of Th1 Cytokines. Blood (2006) 107:3575–83. doi: 10.1182/blood-2005-05-2118
129. Basler M, Lauer C, Beck U, Groettrup M. The Proteasome Inhibitor Bortezomib Enhances the Susceptibility to Viral Infection. J Immunol (2009) 183:6145–50. doi: 10.4049/jimmunol.0901596
130. Wudhikarn K, Palomba ML, Pennisi M, Garcia-Recio M, Flynn JR, Devlin SM, et al. Infection During the First Year in Patients Treated With CD19 CAR T Cells for Diffuse Large B Cell Lymphoma. Blood Cancer J (2020) 10(8):79. doi: 10.1038/s41408-020-00346-7
131. Vora SB, Waghmare A, Englund JA, Qu P, Gardner RA, Hill JA. Infectious Complications Following CD19 Chimeric Antigen Receptor T-cell Therapy for Children, Adolescents, and Young Adults. Open Forum Infect Dis (2020) 7(5):ofaa121. doi: 10.1093/ofid/ofaa121
132. Mahmoudjafari Z, Alexander M, Roddy J, Shaw R, Shigle TL, Timlin C, et al. American Society for Transplantation and Cellular Therapy Pharmacy Special Interest Group Position Statement on Pharmacy Practice Management and Clinical Management for COVID-19 in Hematopoietic Cell Transplantation and Cellular Therapy Patients in the United States. Biol Blood Marrow Transplant (2020) 26:1043–9. doi: 10.1016/j.bbmt.2020.04.005
133. Yakoub-Agha I, Chabannon C, Bader P, Basak GW, Bonig H, Ciceri F, et al. Management of Adults and Children Undergoing Chimeric Antigen Receptor T-Cell Therapy: Best Practice Recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Haematologica (2020) 105:297–316. doi: 10.3324/haematol.2019.229781
134. Styczyński J. Infections Following Car-T Cells Therapy: Current State-Of-The-Art Review and Recommendations. Acta Haematol Pol (2020) 51:11–6. doi: 10.2478/ahp-2020-0004
Keywords: CMV, inflammation, lymphoma, transplantation, immunotherapy, CAR-T-cells
Citation: Alonso-Álvarez S, Colado E, Moro-García MA and Alonso-Arias R (2021) Cytomegalovirus in Haematological Tumours. Front. Immunol. 12:703256. doi: 10.3389/fimmu.2021.703256
Received: 30 April 2021; Accepted: 14 September 2021;
Published: 18 October 2021.
Edited by:
Masakazu Kamata, University of Alabama at Birmingham, United StatesReviewed by:
Debbie Van Baarle, National Institute for Public Health and the Environment, NetherlandsBenjamin Anthony Krishna, Cleveland Clinic, United States
Copyright © 2021 Alonso-Álvarez, Colado, Moro-García and Alonso-Arias. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rebeca Alonso-Arias, cmFsb25zb2FyaWFzQGhvdG1haWwuZXM=; Marco A. Moro-García, bWFyY29tb3JvZ0Bob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work and share last authorship