 Yves Carlier
Yves Carlier Carine Truyens
Carine Truyens Eric Muraille
Eric Muraille- 1Laboratoire de Parasitologie, Faculté de Médecine, Université Libre de Bruxelles (ULB), Bruxelles, Belgium
- 2Department of Tropical Medicine, School of Public Health and Tropical Medicine, Tulane University, New Orleans, LA, United States
The newborns of women infected with the parasite Trypanosoma cruzi (the agent of Chagas disease) can be infected either before birth (congenitally), or after birth (as e.g., by vector route). Congenital Chagas disease can induce high levels of neonatal morbidity and mortality. Parasite-infected pregnant women transmit antibodies to their fetus. Antibodies, by opsonizing parasites, can promote phagocytosis and killing of T. cruzi by cells expressing FcγR, on the mandatory condition that such cells are sufficiently activated in an inflammatory context. Antibody-dependent enhancement (ADE) is a mechanism well described in viral infections, by which antibodies enhance entry of infectious agents into host cells by exploiting the phagocytic FcγR pathway. Previously reported Chagas disease studies highlighted a severe reduction of the maternal-fetal/neonatal inflammatory context in parasite-transmitting pregnant women and their congenitally infected newborns. Otherwise, experimental observations brought to light ADE of T. cruzi infection (involving FcγR) in mouse pups displaying maternally transferred antibodies, out of an inflammatory context. Herein, based on such data, we discuss the previously unconsidered possibility of a role of ADE in the trans-placental parasite transmission, and/or the development of severe and mortal clinical forms of congenital/neonatal Chagas disease in newborns of T. cruzi-infected mothers.
Introduction
Antibody-dependent enhancement (ADE) is a mechanism by which certain viral infections are enhanced in the presence of antibodies mediating entry of infectious agents into host cells by exploiting the phagocytic FcγR pathway. A lot of studies emphasized the role of ADE in infections with dengue-, zika-, influenza-, Ebola-viruses, HIV, RSV and more recently coronaviruses (reviewed in 1). ADE can occur when non-neutralizing antibodies or antibodies at sub-neutralizing levels bind to viral antigens without blocking or clearing infection. It is particularly relevant in the context of pre-existing immunity, when viruses are in the presence of circulating antibodies resulting from either a previous infection or maternal transmission to neonates. Interestingly, ADE was initially mentioned to explain an increased incidence of dengue hemorrhagic fever in Thai infected infants born to immune mothers that ostensibly had pre-existing antibodies to dengue virus (2, 3).
The ADE concept can be extended to infections with intracellular protozoan parasites and particularly to the infection with the hemoflagellate Trypanosoma cruzi. This parasite comes in two forms: the trypomastigote (the free circulating parasite form) and the amastigote (the intracellular multiplication form) (4). It is the agent of the tropical Chagas disease (CD), transmitted in humans by insect vectors, blood transfusion, organ transplantation, orally (by ingestion of food or liquid contaminated with T. cruzi), or by maternal-fetal route (5). Congenital CD (CCD) remains an important global and neglected public health problem. In the currently 21 endemic Latin American countries, 1,125,000 women in fertile age are estimated infected with T. cruzi (mainly since childhood through insect vector transmission), with an incidence of congenital infection of 8668 cases/year (6). About 5% of pregnant women chronically infected with T. cruzi (the most frequent clinical phase of CD) give birth to infected newborns (7). Through migrations of Latin American people, cases of CCD are also reported in North America, Europe, Australia and Japan, where vector transmission does not occur (or is extremely rare) (8). Beside asymptomatic cases, T. cruzi-infected newborns can exhibit fever, low birth weight, prematurity, hepatosplenomegaly, pneumonitis, premature rupture of membranes, until presenting pejorative outcomes with high levels of neonatal morbidity (meningoencephalitis, myocarditis) and mortality. Left untreated, the neonatal infection can progress to chronic CD later in life (susceptible to induce myocardiopathy or digestive megaviscera) (reviewed in 9, 10).
Here, based on reported human and experimental data from our team and others, we aim to discuss the previously unconsidered possibility of a role of ADE in the development and/or worsening of CD in neonates born to T. cruzi-infected mothers. Such newborns display maternally derived antibodies and can be infected either congenitally (as in endemic and non-endemic areas) or after birth, by another transmission route (as e.g. by vector route as occurring in LA endemic countries).
Antibody responses in T. cruzi infection
Humoral immune response in humans as well as experimental T. cruzi infection in mice is diverse and complex, including both parasite-specific and unspecific antibodies (Ab) arising from polyclonal B cell activation. Parasitemia (blood amount of trypomastigotes) starts to decrease when T. cruzi-specific Ab reach their highest levels (in the transition from acute to chronic phase of infection), suggesting their protective role (reviewed in 11).
Some Ab have neutralizing functions, e.g. against the glycoproteins protecting the trypomastigotes from the direct action of complement, likewise allowing parasite lysis (12). Others Ab inhibit the activity of major virulence factors of T. cruzi, such as the proteolytic enzyme cruzipain. Still other Ab, directed against the parasite trans-sialidases (13), allow the action of anti-α-Gal Ab, another class of lytic Ab, abundantly produced during human infection (14).
Ab, by opsonizing trypomastigotes, can also mediate parasite clearance by promoting their phagocytosis and killing by activated granulocytes, monocytes and macrophages expressing FcγR (15, 16). Such cell activation, resulting from intracellular signals transduced upon FcγR crosslinking and interactions between cytokine(s) and their membrane receptor(s), promotes the generation of reactive nitrogen species able to kill extracellular T. cruzi (17).
IgG Ab appear in acute infection and persist life-long during the chronic infection. IgG1-, followed by IgG3-Ab isotypes are mainly produced in human infection, whereas IgG2a-, IgG2b- and IgG1-Ab are more frequently observed in mouse infection, all isotypes being able to bind cell FcγR (18, 19).
Vertical Transmission of T. cruzi-specific Ab
There is a fundamental difference between man and mouse regarding the transfer of maternal IgG to offspring. It occurs through the placenta (from the 13-14th week of pregnancy to delivery) in humans, and by breast milk in mice (from birth to the weaning on day 21 after birth). Such transfer is carried out by transcytosis through a FcRn localized on the membrane of trophoblastic cells in humans or digestive tube cells in mice (20). Most IgG, whatever their Fab specificities are transferred, though some isotypes are preferentially transmitted, due to higher affinities between their Fc portion and the FcRn (21).
In human infection with T. cruzi, the trans-placental transmission of Ab occurs as in other infections (22–25). Although few studies have compared the features of transferred antibodies in infected and uninfected neonates from infected mothers, their amounts and repertoires appear similar in infected mothers and their uninfected neonates at birth (23), whereas congenitally infected newborns seem display lower Ab levels than their mothers (26). Such transferred Ab persist up to 8-9 months after birth before becoming undetectable (10, 27).
In murine infection with T. cruzi, maternal-offspring transfer of Ab occurs through lactation (28–30), but direct kinetic studies of such transfer are lacking. Our report performed with uninfected mice having received purified T. cruzi-specific Ab during gestation and lactation period, showed that Ab levels were reduced by 4 times in offspring at the weaning time (i.e. 3 weeks after birth), compared to the maternal Ab level (30). Other infection models indicate that such transferred Ab can persist more than 6 weeks after birth, i.e. more than 3 weeks after weaning in mouse pups (31).
Experimental Evidences of Ade of T. cruzi infection in Mouse Pups Displaying Maternally Transferred Ab
As shown in various experiments, whatever the used T. cruzi genotype/strain, acute infection in mice either prevents gestation or, in case of gestation development, induces high pup mortality (probably related to the inflammatory storm produced by the parasite inoculation) with rare congenital infection in the few surviving pups. When gestation occurs during chronic infection, congenital transmission is not observed, even after looking for cryptic infection in pups immunosuppressed by cyclophosphamide (32, 33).
The mouse model being not suitable for studying T. cruzi congenital infection, studies were performed using offspring (uninfected) born to chronically infected or uninfected dams, and experimentally infected after weaning (on two months after birth). Higher parasitemia and mortality rates were observed in offspring of infected mice compared with control offspring. The most severe infections were noted when offspring were born and suckled by their mothers, but no more when offspring was infected 5 (instead of 2) months after birth. Such effect was not seen if offspring was infected with Plasmodium chabaudi or Schistosoma mansoni. Such results highlight a maternally induced T. cruzi-specific enhancement of infection in offspring of infected dams (34). However, the mechanism of such enhancement and the nature of what was transferred from dams to pups to induce it remained unclear.
In order to study more specifically the role of Ab in the previously demonstrated enhancement of post-natal infection in progeny of infected mice, another experiment was performed by injecting either serum from chronically infected animals or purified T. cruzi-specific Ab into uninfected mice (born to uninfected dams) during gestation and lactation periods. It was verified that injected Ab were transferred to offspring. When infected two months after birth (as in the experiments mentioned above), offspring of mice treated with chronic serum or purified Ab displayed significantly higher parasitemia and mortality rates than offspring from mothers receiving control serum or immunoglobulins unrelated to T. cruzi (30). These results indicate an ADE-like phenomenon in experimental T. cruzi infection, and particularly in mice receiving maternally derived Ab.
Experimental Evidences of a Fcγr-Involvement in Ade of T. cruzi Infection in Mice
Flow cytometry studies were performed in mice using the 2.4G2 monoclonal Ab (MoAb), specific to the extracellular domains of low-affinity Fc receptors for IgG (FcγRII/III, CD31 and CD16 respectively). Membrane of splenic and mesenteric lymph nodes cells of T. cruzi-infected mice displayed higher expression and absolute number of FcγR in the early and late parasitemic phase (before and after the rise of Ab), compared to uninfected mice (35).
In order to investigate the role of such FcγR, the 2.4G2 MoAb was injected into mice (2 months old). Repeated injections every 3-4 days decreased the availability of FcγR on peritoneal, lymph node, and spleen cells in control uninfected mice. Injections of 2.4G2 MoAb just before parasite inoculation and during the acute phase of T. cruzi infection strongly reduced mortality and parasitemia in comparison to control animals receiving an unrelated MoAb, whereas the levels of immunoglobulins and/or T. cruzi-specific Ab remained similar in infected and control mice (36).
These results, associated with those mentioned above, indicate that FcγR play a role in the ADE-like phenomenon observed in mice infected with T. cruzi. They confirm previous in vitro experiments showing the role of parasite-opsonizing Ab in the enhancement of cell parasitic infection (37), besides the various other mechanisms allowing invasion of phagocytic and non-phagocytic host cells by the infective trypomastigote form of T. cruzi (38).
Pregnancy/Gestation, Placental and Neonatal Inflammasomes During T. cruzi infection
It is important to note that in the experiments mentioned above (30), apart the Ab, no inflammatory- or other immunological-components were transferred to the offspring. Moreover, there was neither placental inflammation, nor other disturbances likely to be a source of interference in offspring, since dams were uninfected. This suggests that the absence or, at least, a severe reduction of the maternally-derived inflammatory environment might be a key factor promoting ADE by limiting in offspring the cell activation necessary to the protective role of parasite opsonizing Ab (see above). This raises the question of the maternal/fetal/neonatal inflammatory status in case of T. cruzi infection.
Although few studies were performed in experimental models, it was shown that gestation associated with acute or chronic T. cruzi infection in mice promotes a strong inflammatory response (32, 39, 40), and that an inflammatory status induced in mother rat contributes to the control of T. cruzi infection in offspring (41).
The first and third trimesters of human pregnancy are known to induce a strong physiological inflammation status (42). Recent studies showed that the second trimester of pregnancy also induces inflammasome signaling in placental trophoblasts to promote fetal and maternal antimicrobial defenses (43).
Infection during pregnancy increases still more such inflammatory context. Indeed, T. cruzi-infected pregnant women display a hyperactivation of blood cells releasing various pro-inflammatory cytokines such as IL-1β, IL-6, TNF-α and IFN-γ, in response to T. cruzi or LPS/PHA, compared to non-infected pregnant women (44, 45). IFN-γ is the key cytokine controlling T. cruzi infection, as other intracellular pathogens. It activates monocytes/macrophages and stimulates, in synergy with TNF-α, the generation of NO which kills parasites (11, 17). However, there is an important difference in the inflammatory status, between pregnant women having given birth to either infected or uninfected neonates. Cells from transmitting pregnant women are phenotypically less activated, and produce less inflammatory cytokines compared to mothers of uninfected newborns (44, 46, 47). Indeed, cells of such mothers produce 3 times less IFN-γ when stimulated with T. cruzi, and such defective response persist after delivery. The observation that mothers of infected newborns harbor higher parasitemia than those delivering uninfected babies (9, 47, 48), argues still more for the lower inflammatory capacity of these firsts.
Interestingly, the inflammatory status observed in infected pregnant women is also observed in their neonates (maternal imprinting). Uninfected neonates of infected mothers display leukocytes releasing more TNF-α and higher circulating levels of TNF-α, IFN-γ and IL-18 compared to infected neonates (49, 50). They also present a higher proportion of CD56bright NK cells producing IFN-γ (44, 51). Such Th1 pro-inflammatory status persisted in infant life, with a promoting effect on responses to vaccines (52).
Altogether, such results indicate that: i) a stronger inflammatory state than in normal gestation is induced on both sides of placenta when gestation is associated to acute or chronic infection with T. cruzi (in mouse and human infection); ii) such inflammatory context is strongly reduced in case of congenital parasite transmission (in human CCD).
Might Maternal Ab Enhance Trans-Placental Transmission of Parasites in Humans?
CCD results from a two step process: i) the parasite transmission from mother to fetus through the placenta, and, ii) the development of parasitic infection in the fetus/neonate (multiplication of parasites resulting in CCD more or less severe, see above). Both steps depend on specific mechanisms detailed elsewhere (9, 53, 54). However, as far as we know, the possible enhancing role of Ab present in maternal blood, in the trans-placental transmission of parasites through the trophoblastic FcRn, has not been studied. Interestingly, it has been reported that human cytomegalovirus co-opt FcRn-mediated transcytosis and are transported across syncytiotrophoblasts in immune complexes that infect underlying cytotrophoblasts and are captured by macrophages in the villus core (55, 56). If this is a plausible mechanism also for T. cruzi is worth questioning.
This might occur only if parasites are observed in trophoblast and placental tissues (histopathological studies). Such cases are encountered only when very high maternal parasitemia have overwhelmed the placental capacity of defenses, resulting in abortions or delivery of neonates with severe CCD. By contrast, this is unlikely in pregnant women displaying moderated blood parasite amounts (the most frequent case nowadays), since the activation of placental innate defenses (and particularly the trophoblast turnover) is sufficient enough to prevent the trophoblast crossing. In such cases, the parasite population remaining present into the intervillous space, infects the epithelial cells of the marginal zone deprived of trophoblast to be transferred into the fetal blood vessels (9, 53, 54).
The fact that congenital infection is almost absent in mice in which there is neither trophoblastic FcRn nor subsequent trans-placental Ab transfer (see above), might argue for a possible role of the trophoblastic FcRn in the transmission of Ab-opsonized parasites in human infection,
Might ADE Contribute to the Severity of Human Neonatal CD?
Clinical surveys in endemic areas have clearly shown that severe and lethal CCD are related to high parasitemia in newborns (53, 57, 58).
Indeed, data mentioned above indicate that:
i) Besides their capacity to neutralize essential parasite molecules, Ab, by opsonizing parasites, can promote their phagocytosis and killing by cells expressing FcγR on the mandatory condition that such cells are sufficiently activated in an inflammatory context;
ii) The maternal-fetal/neonatal inflammatory context is seriously reduced in the 5% of infected and parasite-transmitting pregnant women, and their congenitally infected newborns;
iii) The levels of transmitted Ab seems lower in congenitally infected neonates than in uninfected newborns delivered by infected mothers;
iv) Experiments in mice clearly indicate the occurrence of ADE of T. cruzi infection, particularly when parasites are in presence of low levels of transmitted maternal Ab, outside of an inflammatory context.
Would be the observations in mice extrapolable to human? The question raised here concerns the potential role of ADE in neonates infected either before- (congenitally) or after-birth (by another transmission route), both having previously received maternally transmitted Ab. In other words, might low amounts of Ab (likely insufficient to sustain a frank protective activity) opsonize circulating parasites and favor their entry into FcγR expressing cells not sufficiently activated to trigger their protective killing burst? Might such enhancement of their intracellular multiplication (leading to high parasite load), worsen infection and lead to more severe and lethal clinical forms of CCD, in some infected newborns (Figure 1)? Indeed, such severe forms, though less frequent nowadays, are always observed in endemic as well as non-endemic areas of CD [see below; (9)].
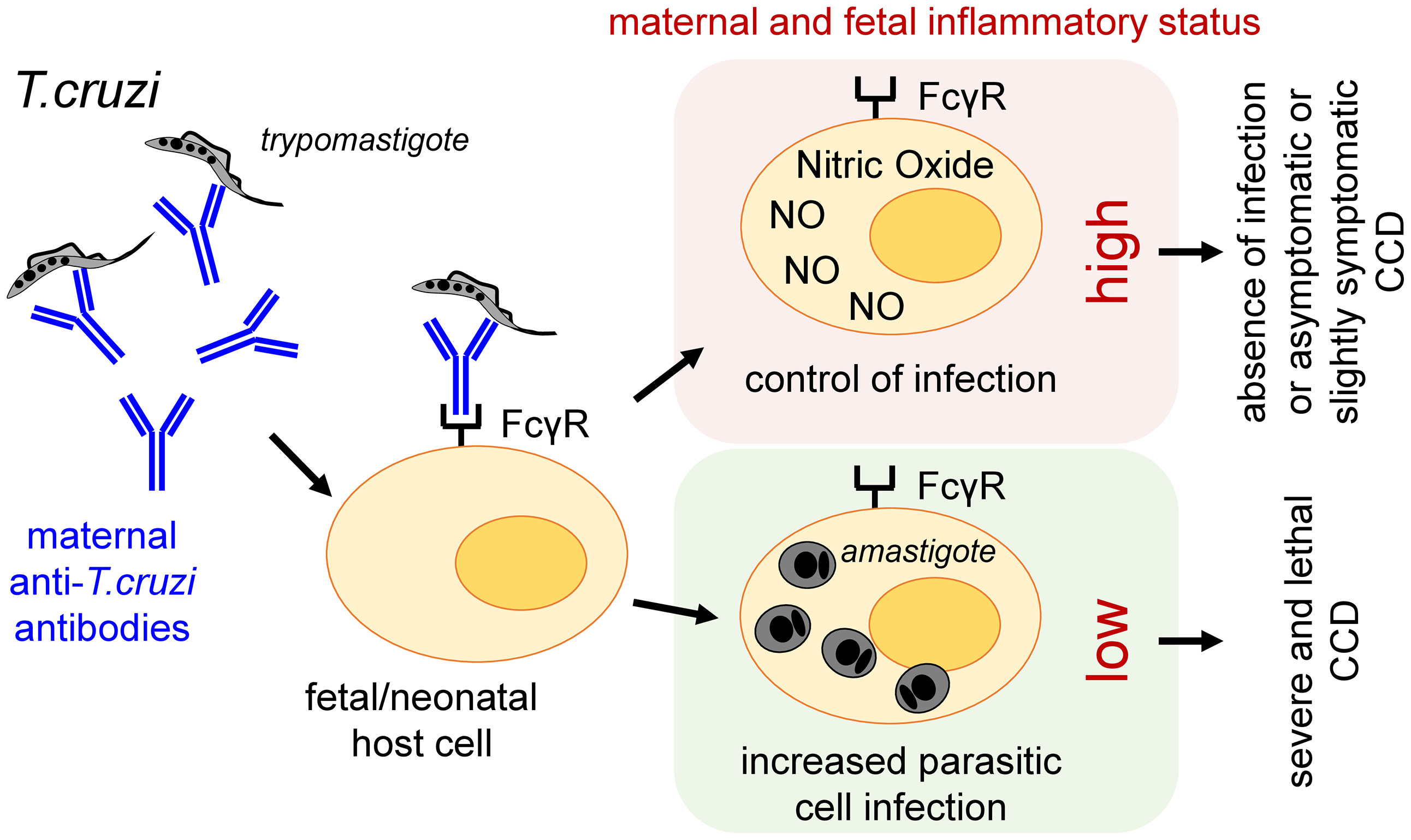
Figure 1 Proposed role of ADE in the development and/or worsening of CCD in humans.
Possible Factors Biasing Host Towards ADE
Internal as well as external factors might drive towards ADE.
Beside the weakness of the inflammatory context and the global availability of Ab, other intrinsic individual factors might contribute to ADE, such as the distribution of IgG isotypes (presenting different affinities for FcγR) among available Ab, or the allelic distribution of FcγR on host cells (1). Information is lacking on qualitative or quantitative differences in maternal Ab isotype population transmitted in uninfected vs infected babies and between the severity gradients of CCD among infected ones. The allelic distribution of FcγR in human population is not homogenous and some variants do not allow cell activation. Whether such a possibility would condition ADE in T. cruzi infection remains to be investigated by characterizing the Fc domain structure and allelic distribution of FcγR genetic variants in neonates of infected mothers. Indeed, epidemiological elements suggest that genetic traits might be, at least partially, related to CCD, such as the familial clustering of congenital cases and the repetition of congenital transmission in successive pregnancies in the same women (59, 60), as well the persistent lower capacity of cells of such women to produce IFN-γ (47).
In addition, other external factors, such as malnutrition and poverty, by affecting the features of innate immune response with cytokine patterns skewed towards a Th2-response, and reducing the Ab production (61), might contribute to favor ADE of T. cruzi infection. In line with this possibility, are the reported observations on the progressive decrease of severity of CCD overtime, which was much more severe and deadly in the past when malnutrition and poverty was particularly important in endemic LA countries (9). For instance, we reported mortality rates up to 13% in a Bolivian cohort of congenitally infected newborns studied between 1992 and 1994, while such rate dropped to 2% in another study in 1999-2001, when Bolivia benefited from improved socio-economic conditions and better maternal care (62).
Conclusions And Perspectives
Although the ADE concept was initially described in viral infections, it has been also shown in bacterial infections and diseases (likely with distinct underlying mechanisms; reviewed in 63). Its extension to protozoan parasite would open new perspectives on the balance between protective and enhancing/facilitating Ab in the immunological homeostasis in response to infections.
Data from clinical surveys, combined with experimental observations, allow us to propose the working hypothesis of a role of ADE in the trans-placental parasite transmission, and/or the development of particularly severe and mortal clinical forms of congenital/neonatal CD. Such amplification of fetal or neonatal infection, through the phagocytic FcγR pathway, in fetuses/newborns having previously received antibodies from their mothers, might contribute to explain the more detrimental pole of the large clinical spectrum of CCD, as well as its historical evolution.
The balance between ADE and Ab-protective responses might depend on the maternal-fetal/neonatal relationship, and particularly on the features of the Ab populations and inflammatory context (with a possible inverse correlation between ADE and the maternal/fetal/neonatal inflammatory context), likely associated to other external or genetic factors. This working hypothesis should stimulate further research works exploring the potential role of FcRn and ADE in the trans-placental transmission of parasites in humans, using ex vivo infection of placental explants or trophoblastic cell lines with opsonized T. cruzi parasites, or, perhaps also, humanized mice expressing placental FcRn. Other investigations would be particularly welcome on Ab isotype concentrations, inflammation markers, and allelic distribution of cellular FcγR in T. cruzi-infected mothers and their neonates, in order to determine the conditions favoring ADE. A better knowledge of conditions contributing to the severity of human neonatal CD might lead to improvements of our diagnosis tools to establish a prognosis of more severe CCD, and focus more attention on such risky group of pregnant women (9).
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary files. Further inquiries can be directed to the corresponding author.
Author Contributions
YC sustained the working hypothesis which was discussed with EM and CT. YC wrote the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bournazos S, Gupta A, Ravetch JV. The Role of IgG Fc Receptors in Antibody-Dependent Enhancement. Nat Rev Immunol (2020) 20(10):633–43. doi: 10.1038/s41577-020-00410-0
2. Halstead SB, Nimmannitya S, Cohen SN. Observations Related to Pathogenesis of Dengue Hemorrhagic Fever. IV. Relation of Disease Severity to Antibody Response and Virus Recovered. Yale J Biol Med (1970) 42(5):311–28.
3. Katzelnick LC, Gresh L, Halloran ME, Mercado JC, Kuan G, Gordon A, et al. Antibody-Dependent Enhancement of Severe Dengue Disease in Humans. Science (2017) 358(6365):929–32. doi: 10.1126/science.aan6836
4. Batista MF, Nájera CA, Meneghelli I, Bahia D. The Parasitic Intracellular Lifestyle of Trypanosomatids: Parasitophorous Vacuole Development and Survival. Front Cell Dev Biol (2020) 8:.396. doi: 10.3389/fcell.2020.00.396
5. Pérez-Molina JA, Molina I. Chagas Disease. Lancet (2018) 391(10115):82–94. doi: 10.1016/S0140-6736(17)31612-4
6. WHO. Chagas Disease in Latin America: An Epidemiological Update Based on 2010 Estimates. Relev Epidemiol Hebd (2015) 90:33–43.
7. Howard E, Xiong X, Carlier Y, Sosa-Estani S, Buekens P. Frequency of the Congenital Transmission of Trypanosoma Cruzi : A Systematic Review and Meta-Analysis. BJOG (2014) 121:22–33. doi: 10.1111/1471-0528.12396
8. Gascon J, Bern C, Pinazo M-J. Chagas Disease in Spain, the United States and Other non-Endemic Countries. Acta Trop (2010) 115(1-2):22–7. doi: 10.1016/j.actatropica.2009.07.019
9. Carlier Y, Truyens C. Maternal-Fetal Transmission of Trypanosoma Cruzi. In: Telleria J, Tibayrenc M, editors. American Trypanosomiasis-Chagas Disease. One Hundred Years of Research, 2d edition. UK, USA: Elsevier (2017), ISBN: ISBN 978-0-12-801029-7. p. 517–59. chap 23.
10. Carlier Y, Altcheh J, Angheben A, Freilij H, Luquetti AO, Schijman AG, et al. Congenital Chagas Disease: Updated Recommendations for Prevention, Diagnosis, Treatment, and Follow-Up of Newborns and Siblings, Girls, Women of Childbearing Age, and Pregnant Women. PloS Negl Trop Dis (2019) 13(10):e0007694. doi: 10.1371/journal.pntd.0007694
11. Truyens C, Carlier Y. Protective Host Response to Trypanosoma Cruzi and its Limitations. In: Telleria J, Tibayrenc M, editors. American Trypanosomiasis-Chagas Disease. One Hundred Years of Research, 2d edition. UK, USA: Elsevier (2017). p. 579–604. chap 25.
12. Krautz GM, Kissinger JC, Krettli AU. The Targets of the Lytic Antibody Response Against Trypanosoma Cruzi. Parasitol Today (2000) 16(1):31–4. doi: 10.1016/s0169-4758(99)01581-1
13. Leguizamón MS, Campetella O, Russomando G, Almiron M, Guillen I, Gonzalez Cappa SM, et al. Antibodies Inhibiting Trypanosoma Cruzi Trans-Sialidase Activity in Sera From Human Infections. J Infect Dis (1994) 170(6):1570–4. doi: 10.1093/infdis/170.6.1570
14. Gazzinelli RT, Pereira ME, Romanha A, Gazzinelli G, Brener Z. Direct Lysis of Trypanosoma Cruzi: A Novel Effector Mechanism of Protection Mediated by Human Anti-Gal Antibodies. Parasite Immunol (1991) 13(4):345–56. doi: 10.1111/j.1365-3024.1991.tb00288.x
15. Scott MT, Moyes L. 75Se-Methionine Labelled Trypanosoma Cruzi Blood Trypomastigotes: Opsonization by Chronic Infection Serum Facilitates Killing in Spleen and Liver. Clin Exp Immunol (1982) 48(3):754–7.
16. Umekita LF, Takehara HA, Mota I. Role of the Antibody Fc in the Immune Clearance of Trypanosoma Cruzi. Immunol Lett (1988) 17(1):85–9. doi: 10.1016/0165-2478(88)90106-x
17. Metz G, Carlier Y, Vray B. Trypanosoma Cruzi Upregulates Nitric Oxide Release by IFN-Gamma-Preactivated Macrophages, Limiting Cell Infection Independently of the Respiratory Burst. Parasite Immunol (1993) 15(12):693–9. doi: 10.1111/j.1365-3024.1993.tb00584.x
18. Brodskyn CI, Silva AM, Takehara HA, Mota I. IgG Subclasses Responsible for Immune Clearance in Mice Infected With Trypanosoma Cruzi. Immunol Cell Biol (1989) 67(Pt 6):343–8. doi: 10.1038/icb.1989.50
19. el Bouhdidi A, Truyens C, Rivera M-T, Bazin H, Carlier Y. Trypanosoma Cruzi Infection in Mice Induces a Polyisotypic Hypergammaglobulinaemia and Parasite-Specific Response Involving High IgG2a Concentrations and Highly Avid IgG1 Antibodies. Parasite Immunol (1994) 16(2):69–76. doi: 10.1111/j.1365-3024.1994.tb00325.x
20. Simister NE. Placental Transport of Immunoglobulin G. Vaccine (2003) 21(24):3365–9. doi: 10.1016/s0264-410x(03)00334-7
21. Clements T, Rice TF, Vamvakas G, Barnett S, Barnes M, Donaldson B, et al. Update on Transplacental Transfer of IgG Subclasses: Impact of Maternal and Fetal Factors. Front Immunol (2020) 11:1920. doi: 10.3389/fimmu.2020.01920
22. Miles MA, Castro C, Macedo V, Draper CC. Letter: Trypanosoma Cruzi–Prenatal Transfer of Maternal Antibody in Man. Trans R Soc Trop Med Hyg (1975) 69(2):286. doi: 10.1016/0035-9203(75)90171-6
23. Breniere SF, Bailly M, Carrasco R, Carlier Y. Transmission Transplacentaire Des Anticorps Anti-Trypanosoma Cruzi. Cah. O.R.S.T.O.M., sér. Ent. méd. et Parasitol (1983) 21:139–40.
24. Truyens C, Hermann E, Alonso-Vega C, Rodriguez P, Vekemans J, Torrico F, et al. Immune Responses of non-Infected Neonates of Mothers Infected With Trypanosoma Cruzi. Rev Soc Bras Med Trop (2005) 38 Suppl 2:96–100.
25. Sosa-Estani S, Gamboa-León MR, del Cid-Lemus J, Althabe F, Alger J, Almendares, et al. Use of a Rapid Test on Umbilical Cord Blood to Screen for Trypanosoma Cruzi Infection in Pregnant Women in Argentina, Bolivia, Honduras and México. Am J Trop Med Hyg (2008) 79:755–9. doi: 10.4269/ajtmh.2008.79.755
26. Rodriguez P, Truyens C, Alonso-Vega C, Flores A, Cordova M, Suarez E, et al. Serum Levels for IgM and IgA Antibodies to Anti-Trypanosoma Cruzi in Samples of Blood From Newborns From Mothers With Positive Serology for Chagas Disease. Rev Soc Bras Med Trop (2005) 38 Suppl 2:62–4.
27. Chippaux JP, Clavijo AN, Santalla JA, Postigo JR, Schneider D, Brutus L. Antibody Drop in Newborns Congenitally Infected by Trypanosoma Cruzi Treated With Benznidazole. Trop.Med.Int.Health (2010) 15:87–93. doi: 10.1111/j.1365-3156.2009.02431.x
28. Kolodny MH. The Transmission of Immunity in Experimental Trypanosomiasis (Trypanosoma Cruzi) From Mother Rats to Their Offspring. Parasitology (1939) 30:19–39.
29. Miles MA. Trypanosoma Cruzi-Milk Transmission of Infection and Immunity From Mother to Young. Parasitology (1972) 65(1):1–9. doi: 10.1017/s0031182000044188
30. Marques de Araujo S, Rivera M-T, Bouhdidi A, de Maertelaer V, Carlier Y. Maternal Trypanosoma Cruzi-Specific Antibodies and Worsening of Acute Infection in Mouse Offspring. Am J Trop Med Hyg (1996) 54(1):13–7. doi: 10.4269/ajtmh.1996.54.13
31. Richter MY, Jakobsen H, Birgisdottir A, Haeuw J-F, Power UF, Del Giudice G, et al. Immunization of Female Mice With Glycoconjugates Protects Their Offspring Against Encapsulated Bacteria. Infect Immun (2004) 72(1):187–95. doi: 10.1128/iai.72.1.187-195.2004
32. Mjihdi A, Lambot MA, Stewart IJ, Detournay O, Noël JC, Carlier Y, et al. Acute Trypanosoma Cruzi Infection in Mouse Induces Infertility or Placental Parasite Invasion and Ischemic Necrosis Associated With Massive Fetal Loss. Am J Pathol (2002) 161(2):673–80. doi: 10.1016/S0002-9440(10)64223-X
33. Cencig S, Coltel N, Truyens C, Carlier Y. Fertility, Gestation Outcome and Parasite Congenital Transmissibility in Mice Infected With TcI, TcII and TcVI Genotypes of Trypanosoma Cruzi. PloS Negl Trop Dis (2013) 7(6):e2271. doi: 10.1371/journal.pntd.0002271
34. Carlier Y, Rivera MT, Truyens C, Ontivero M, Flament J, Van Marck E, et al. Chagas' Disease: Decreased Resistance to Trypanosoma Cruzi Acquired Infection in Offspring of Infected Mice. Am J Trop Med Hyg (1992) 46(2):116–22. doi: 10.4269/ajtmh.1992.46.116
35. Araujo-Jorge T, el Bouhdidi A, Rivera MT, Daëron M, Carlier Y. Trypanosoma Cruzi Infection in Mice Enhances the Membrane Expression of Low-Affinity Fc Receptors for IgG and the Release of Their Soluble Forms. Parasite Immunol (1993a) 15(9):539–46. doi: 10.1111/j.1365-3024.1993.tb00642
36. Araujo-Jorge T, Rivera MT, Bouhdidi A, Daëron M, Carlier Y. An Fc Gamma RII-, Fc Gamma RIII-Specific Monoclonal Antibody (2.4G2) Decreases Acute Trypanosoma Cruzi Infection in Mice. Infect Immun (1993b) 61(11):4925–8. doi: 10.1128/IAI.61.11.4925-4928.1993
37. Kipnis TL, Calich VL, da Silva WD. Active Entry of Bloodstream Forms of Trypanosoma Cruzi Into Macrophages. Parasitology (1979) 78(1):89–98. doi: 10.1017/s0031182000048617
38. Barrias ES, de Carvalho TM, De Souza W. Trypanosoma Cruzi: Entry Into Mammalian Host Cells and Parasitophorous Vacuole Formation. Front Immunol (2013) 4:186. doi: 10.3389/fimmu.2013.00186
39. Rivera MT, Marques de Araujo S, Lucas R, Deman J, Truyens C, Defresne MP, et al. High Tumor Necrosis Factor Alpha (TNF-Alpha) Production in Trypanosoma Cruzi-Infected Pregnant Mice and Increased TNF-Alpha Gene Transcription in Their Offspring. Infect Immun (1995) 63(2):591–5. doi: 10.1128/IAI.63.2.591-595.1995
40. Mjihdi A, Truyens C, Detournay O, Carlier Y. Systemic and Placental Productions of Tumor Necrosis Factor Contribute to Induce Fetal Mortality in Mice Acutely Infected With Trypanosoma Cruzi. Exp Parasitol (2004) 107(1-2):58–64. doi: 10.1016/j.exppara.2004.03.016
41. Didoli GL, Davila HO, Feldman S, di Masso R, Revelli SS, Bottasso OA. Protected Trypanosoma Cruzi Infection in Rats Born to Mothers Receiving Interferon-Gamma During Gestation is Associated With a Decreased Intramacrophage Parasite Growth and Preferential Synthesis of Specific IgG2b Antibodies. Int J Immunopharmaco (2000) 22(1):45–55. doi: 10.1016/s0192-0561(99)00063-6
42. Dekel N, Gnainsky Y, Granot I, Racicot K, Mor G. The Role of Inflammation for a Successful Implantation. Am J Reprod Immunol (2014) 72(2):141–7. doi: 10.1111/aji.12266
43. Burgener SS, Schroder K. Placental Inflammasome Signaling: Protection for Mother and Baby. .J Exp Med (2021) 218(1):e20201738. doi: 10.1084/jem.20201738
44. Vekemans J, Truyens C, Torrico F, Solano M, Torrico MC, Rodriguez P, et al. Maternal Trypanosoma Cruzi Infection Upregulates Capacity of Uninfected Neonate Cells to Produce Pro- and Anti-Inflammatory Cytokines. Infect Immun (2000) 68(9):5430–4. doi: 10.1128/iai.68.9.5430-5434.2000
45. Alonso-Vega C, Hermann E, Truyens C, Rodriguez P, Torrico MC, Torrico F, et al. Immunological Status of Mothers Infected With Trypanosoma Cruzi. Rev Soc Bras Med Trop (2005) 38(Suppl 2):101–4.
46. Cardoni RL, García MM, De Rissio AM. Proinflammatory and Anti-Inflammatory Cytokines in Pregnant Women Chronically Infected With Trypanosoma Cruzi. Acta Trop (2004) 90(1):65–72. doi: 10.1016/j.actatropica.2003.09.020
47. Hermann E, Truyens C, Alonso-Vega C, Rodriguez P, Berthe A, Torrico F, et al. Congenital Transmission of Trypanosoma Cruzi is Associated With Maternal Enhanced Parasitemia and Decreased Production of Interferon- Gamma in Response to Parasite Antigens. J Infect Dis (2004) 189(7):1274–81. doi: 10.1086/382511
48. Bua J, Volta BJ, Velazquez EB, Ruiz AM, Rissio AM, Cardoni RL. Vertical Transmission of Trypanosoma Cruzi Infection: Quantification of Parasite Burden in Mothers and Their Children by Parasite DNA Amplification. Trans R Soc Trop Med Hyg (2012) 106(10):623–8. doi: 10.1016/j.trstmh.2012.03.015
49. García MM, De Rissio AM, Villalonga X, Mengoni E, Cardoni RL. Soluble Tumor Necrosis Factor (TNF) Receptors (sTNF-R1 and -R2) in Pregnant Women Chronically Infected With Trypanosoma Cruzi and Their Children. Am J Trop Med Hyg (2008) 78(3):499–503. doi: 10.4269/ajtmh.2008.78.499
50. Mayer JP, Biancardi M, Altcheh J, Freilij H, Weinke T, Liesenfeld O. Congenital Infections With Trypanosoma Cruzi or Toxoplasma Gondii are Associated With Decreased Serum Concentrations of Interferon-Gamma and Interleukin-18 But Increased Concentrations of Interleukin-10. Ann Trop Med Parasitol (2010) 104(6):485–92. doi: 10.1179/136485910X12786389891362
51. Hermann E, Alonso-Vega C, Berthe A, Truyens C, Flores A, Cordova M, et al. Human Congenital Infection With Trypanosoma Cruzi Induces Phenotypic and Functional Modifications of Cord Blood NK Cells. Pediatr Res (2006) 60(1):38–43. doi: 10.1203/01.pdr.0000220335.05588.ea
52. Dauby N, Alonso-Vega C, Suarez E, Flores A, Hermann E, Córdova M, et al. Maternal Infection With Trypanosoma Cruzi and Congenital Chagas Disease Induce a Trend to a Type 1 Polarization of Infant Immune Responses to Vaccines. PloS Negl Trop Dis (2009) 3(12):e571. doi: 10.1371/journal.pntd.0000571
53. Carlier Y, Truyens C. Congenital Chagas Disease as an Ecological Model of Interactions Between Trypanosoma Cruzi Parasites, Pregnant Women, Placenta and Fetuses. Acta Trop (2015) 151:103–15. doi: 10.1016/j.actatropica.2015.07.016
54. Carlier Y, Schijman AG, Kemmerling U. Placenta, Trypanosoma Cruzi and Congenital Chagas Disease. Curent Trop Med Rep (2020) 7(4):172–82. doi: 10.1007/s40475-020-00210-5
55. Maidji E, McDonagh S, Genbacev O, Tabata T, Pereira L. Maternal Antibodies Enhance or Prevent Cytomegalovirus Infection in the Placenta by Neonatal Fc Receptor-Mediated Transcytosis. Am J Pathol (2006) 168(4):1210–26. doi: 10.2353/ajpath.2006.050482
56. Pereira L. Congenital Viral Infection: Traversing the Uterine-Placental Interface. Annu Rev Virol (2018) 5(1):273–99. doi: 10.1146/annurev-virology-092917-043236
57. Laucella SA, Postan M, Martin D, Fralish BH, Albareda MC, Alvarez MG, et al. Frequency of Interferon- Gamma -Producing T Cells Specific for Trypanosoma Cruzi Inversely Correlates With Disease Severity in Chronic Human Chagas Disease. J Infect Dis (2004) 189:909–18. doi: 10.1086/381682
58. Torrico MC, Solano M, Guzman JM, Parrado R, Suarez E, Alonzo-Vega C, et al. Estimation of the Parasitemia in Trypanosoma Cruzi Human Infection: High Parasitemias Are Associated With Severe and Fatal Congenital Chagas Disease. Rev Soc Bras Me Trop (2005) 38(Suppl 2):58–61.
59. Sánchez Negrette O, Mora MC, Basombrío MA. High Prevalence of Congenital Trypanosoma Cruzi Infection and Family Clustering in Salta, Argentina. Pediatrics (2005) 115(6):e668–72. doi: 10.1542/peds.2004-1732
60. Danesi E, Fabbro DL, Segura EL, Sosa-Estani S. Higher Congenital Transmission Rate of Trypanosoma Cruzi Associated With Family History of Congenital Transmission. Rev Soc Bras Med Trop (2020) 53:e20190560. doi: 10.1590/0037-8682-0560-2019
61. Rytter MJH, Kolte L, Briend A, Friis H, Christensen VB. The Immune System in Children With Malnutrition–a Systematic Review. PloS One (2014) 9(8):e105017. doi: 10.1371/journal.pone.0105017
62. Torrico F, Alonso-Vega C, Suarez E, Rodriguez P, Torrico MC, Dramaix M, et al. Maternal Trypanosoma Cruzi Infection, Pregnancy Outcome, Morbidity, and Mortality of Congenitally Infected and Non-Infected Newborns in Bolivia. Am J Trop Med Hyg (2004) 70:201–9. doi: 10.4269/ajtmh.2004.70.201
Keywords: Trypanosoma cruzi, chagas disease, congenital/neonatal infection, antibodies, FcR, antibody-dependent enhancement
Citation: Carlier Y, Truyens C and Muraille E (2021) Is Antibody-Dependent Enhancement of Trypanosoma cruzi Infection Contributing to Congenital/Neonatal Chagas Disease? Front. Immunol. 12:723516. doi: 10.3389/fimmu.2021.723516
Received: 10 June 2021; Accepted: 26 August 2021;
Published: 10 September 2021.
Edited by:
Diana Bahia, Federal University of Minas Gerais, BrazilReviewed by:
José Ronnie Carvalho Vasconcelos, Federal University of São Paulo, BrazilMomar Ndao, McGill University, Canada
Copyright © 2021 Carlier, Truyens and Muraille. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yves Carlier, eXZlcy5jYXJsaWVyQHVsYi5iZQ==