 Sultan Tousif1
Sultan Tousif1 Yong Wang1
Yong Wang1 Joshua Jackson1
Joshua Jackson1 Kenneth P. Hough1John G. Strenkowski1
Kenneth P. Hough1John G. Strenkowski1 Mohammad Athar2
Mohammad Athar2 Victor J. Thannickal1
Victor J. Thannickal1 Robert H. McCusker3
Robert H. McCusker3 Selvarangan Ponnazhagan4
Selvarangan Ponnazhagan4 Jessy S. Deshane1*
Jessy S. Deshane1*- 1Department of Medicine, University of Alabama at Birmingham, Birmingham, AL, United States
- 2Department of Dermatology, University of Alabama at Birmingham, Birmingham, AL, United States
- 3Department of Animal Sciences, University of Illinois at Urbana Champaign, Urbana, IL, United States
- 4Department of Pathology, University of Alabama at Birmingham, Birmingham, AL, United States
Regulatory B cells (Breg) are IL-10 producing subsets of B cells that contribute to immunosuppression in the tumor microenvironment (TME). Breg are elevated in patients with lung cancer; however, the mechanisms underlying Breg development and their function in lung cancer have not been adequately elucidated. Herein, we report a novel role for Indoleamine 2, 3- dioxygenase (IDO), a metabolic enzyme that degrades tryptophan (Trp) and the Trp metabolite L-kynurenine (L-Kyn) in the regulation of Breg differentiation in the lung TME. Using a syngeneic mouse model of lung cancer, we report that Breg frequencies significantly increased during tumor progression in the lung TME and secondary lymphoid organs, while Breg were reduced in tumor-bearing IDO deficient mice (IDO-/-). Trp metabolite L-Kyn promoted Breg differentiation in-vitro in an aryl hydrocarbon receptor (AhR), toll-like receptor-4-myeloid differentiation primary response 88, (TLR4-MyD88) dependent manner. Importantly, using mouse models with conditional deletion of IDO in myeloid-lineage cells, we identified a significant role for immunosuppressive myeloid-derived suppressor cell (MDSC)-associated IDO in modulating in-vivo and ex-vivo differentiation of Breg. Our studies thus identify Trp metabolism as a therapeutic target to modulate regulatory B cell function during lung cancer progression.
Introduction
Lung Cancer is the leading cause of cancer-associated morbidity and mortality among men and women, claiming ~1.3 million deaths annually worldwide (1–3). The predominant form of lung cancer is non-small-cell lung carcinoma (NSCLC), which accounts for about 85 percent of all lung cancers. Immunotherapy strategies have provided only limited improvement in 5-year survival for NSCLC.
The tumor microenvironment (TME) plays a dynamic role in regulating immune responses during the progression of lung cancer (4, 5). Tumors infiltrating immune cells not only inhibit but also promote tumor growth (6–8). Tumor promoting myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Treg) are known contributors of immunosuppression in the TME (9, 10). But recent studies have emphasized a regulatory function also for subsets of B cells called regulatory B cells (Breg) in inhibiting anti-tumor immune responses (11–16). Increased frequency of circulating Breg was recently reported in patients with pathological stage of lung cancer (17). Among the widely described immunosuppressive characteristics of Breg is the production of the potent anti-inflammatory cytokine IL-10 (18, 19). IL-10 producing B cell subsets (Breg) that are CD1d high were first identified by Mizuguchi et al. in 2002 in the study of chronic intestinal inflammation (20). Breg was shown to regulate autoimmunity in murine models of systemic lupus erythematosus (SLE), type 1 diabetes, arthritis and chronic intestinal inflammation (21–25). Previous studies have also reported that Breg suppress anti-tumor T cell responses through induction of Treg (10).
The significance of Bregs in anti-tumor immunity became clear when improved therapeutic efficacy was observed in lung tumor-bearing B cell-deficient mice following treatment with a combination of chemotherapy and IL-15 (26). A significant increase in tumor burden was also reported following the adoptive transfer of Breg in a murine model of lymphoma with anti-CD20 antibody treatment (27, 28). Moreover, Breg contribute to immunosuppression in pulmonary metastasis during breast cancer progression (29). However, the underlying mechanisms of Breg development during the progression of lung tumors still remain mostly unknown.
Lineage-specific transcription factors have so far not been identified for Breg, suggesting that Breg are reactive rather than lineage specific. It is well known that immature B cells, mature B cells and plasmablasts may differentiate into IL-10 producing Breg through activation of Toll-like receptor (TLR) and CD40 (30, 31). Immunosuppressive properties of B cells, especially the production of IL-10 in experimental autoimmune encephalomyelitis (EAE) mice model is dependent on toll-like receptor-2 (TLR-2), toll-like receptor-4 (TLR-4)and myeloid differentiation primary response 88 (MyD88) signaling (32). Moreover, the development of Breg in thymoma patients with myasthenia gravis was regulated by TLR9/MyD88/NF- kappa B signaling pathway (33). These studies collectively support the hypothesis that Breg differentiation occurs in response to positive stimuli rather than by regulation via a specific lineage factor. Thus, investigating novel mechanisms of regulation of Breg will lead to the identification of promising targets for cancer therapeutics.
Recently we showed that while MDSCs inhibit B cell responses during lung tumor progression, with increased infiltration of Breg in the lung (34). MDSCs are heterogeneous and immature myeloid cells that are established drivers of immune suppression during tumor progression (35–38). While MDSCs are known to induce Treg expansion and activation (39, 40), recent studies suggest that MDSCs may promote Treg through Breg (15). In our studies, we also identified that MDSCs are major contributors of Indoleamine 2, 3-dioxygenase (IDO) activity in the lung TME (41). IDO is a rate-limiting tryptophan (Trp) metabolizing enzyme that is expressed in MDSCs and tumor cells. IDO has emerged as a potent immunosuppressive pathway for MDSCs for suppression of anti-tumor CD8+ T cells and the expansion of Treg (42–45). Trp metabolite L-Kynurenine (L-Kyn), a well-known endogenous ligand of aryl hydrocarbon receptor (AhR), activates AhR to induce immunosuppression through Treg expansion (46). Furthermore, L-Kyn promotes tumor growth by inhibiting anti-tumor immune responses via AhR in an autocrine/paracrine fashion (47). A potential role for Trp metabolism as a regulator of Breg differentiation and function has not been investigated yet.
In this study, we provide direct evidence for Trp-metabolite L-Kyn in modulating differentiation of Breg in vitro. Our studies with a syngeneic model of lung cancer show impaired infiltration of Breg in TME and spleens of IDO deficient (IDO-/-) mice suggesting an important role for IDO pathway in Breg infiltration and differentiation. Further, using mice with conditional deletion of IDO in myeloid lineage cells, we provide evidence that MDSC-associated IDO contribute significantly to Breg infiltration during lung cancer progression. Additionally, in-vivo and ex-vivo studies with AhR deficient mice (AhR-/-) indicate a potential role for AhR in Breg differentiation and corroborate our hypothesis that L-Kyn induced differentiation of Breg precursors is AhR independent whereas IL-10 producing Breg require AhR signaling. In addition, our ex-vivo data generated from TLR-4, MyD88, TLR-2, and TLR-2, 4 deficient B cells suggest a crucial role of TLR-4-MyD88 signaling in L-Kyn induced Breg differentiation. Collectively, our present study reveals a novel role of IDO and the Trp metabolite L-Kyn in Breg differentiation during the progression of lung cancer.
Material and Methods
Cell Culture and Mice
Lewis lung carcinoma cells (LLCs) (American Type Culture Collection (ATCC) (Manassas, VA)) were propagated in Dulbecco’s Modified Eagle Medium (DMEM) with supplementation of 10% fetal bovine serum (FBS), 2 mM L-glutamine, 1 mM sodium pyruvate, 10 μg/ml penicillin-streptomycin and 0.1 mM non-essential amino acids (Life Technologies, Waltham, MA). LLCs (1 x106 LLC cells per mouse in 100 μl PBS) at 70-80% confluency were injected via intracardiac (i.c.) or tail vein (i.v.) route into. C57BL/6 and B6129-Ido1/J (IDO-/-) mice (Jackson Laboratories (Bar Harbor, ME). Tumor weights at early time points in IDO deficient mice are not accurately measurable and hence these measurements were made 9 days post tumor implantation as we have described before (41). AhR-/- mice were obtained from a UAB colony maintained by John F. Kearney. TLR-2-/-, TLR-4-/- TLR-2, 4-/-, and MyD88-/- mice were obtained from UAB colony maintained by Dr. Suzanne M. Michalek.
IDO f MCre Mice
MCre and IDO f MCre mice were obtained from Robert H. McCusker, University of Illinois at Urbana-Champaign. Ido1 floxed mice (Ido1fl) were generated by inserting a 75bp loxP cassette 5’ of the second exon of the reference gene, NM_008324.2, by Ingenous targeting laboratory (Ronkonkuma, NY). A second loxP site was inserted 3’ of the fourth exon. Targeting was performed with C57BL/6 embryonic stem cells and microinjected into Balb/c blastocysts. Chimeras with a high percentage black coat color were mated to C57BL/6 FLP mice to remove the Neo cassette. Cre recombinase excises a 2.37 kb region of the Ido1 gene. Ido1M-Cre mice were generated by crossbreeding Ido1fl mice to M-Cre mice (B6.129P2-Lyz2tm1(cre)Ifo/J; JAX 004781) to target Ido1 inactivation in myeloid-derived cells. Mice were bred to homozygosity for ease of colony propagation. Both male and female mice of 6-8 weeks old were used for in-vitro and in-vivo experiments. All mice used in this study were bred and maintained in pathogen-free animal facility at Research Support Building, UAB at Birmingham, AL, and handled in accordance with the Guidelines for Animal Experiments at the University of Alabama at Birmingham, AL, USA.
Antibodies and Reagents
We used the following antibodies and reagents: anti-CD19 (clone: eBio1D3)-PE-Cy7, anti-CD5 (clone: 53-7.3)-APC, anti-CD1d (clone: 1B1)-eFluor 450, anti-IL-10 (clone: JES5-16E3)-PE, anti-Gr-1 (clone: RB6-8C5)-PE-Cy5, anti-CD11b (clone: M1/70)-APC-Cy7)-PE, anti-TCRαβ (clone: 145-2C11)-PE-Cy5,anti-IgM (clone: II/41)-FITC, anti-IgD (clone: 11-26C)-PerCPeFluor710, anti-B220 (clone: RA3-6B2)-APC-eFluor780, anti-CD21 (clone: eBio4E3)-FITC, anti-CD43 (clone: eBioR2/60)-PE, anti-CD23 (clone: B3B4)-PE, anti-CD93 (clone: AA4.1)-APC, anti-CD24 (clone: M1/69)-APC, and anti-IgM (clone II/41)-eFluor 450. All the antibodies were purchased from ebiosciences, USA. Anti-CD3 (clone: 145-2C11)-APC-Cy7 and the purified rat anti-mouse CD16/CD32 (Mouse BD Fc Block™) (clone#2.4G2) was from BD Pharmingen, USA. Polyclonal AhR antibody (cat# BML-SA210-0025) was obtained from Enzo Life Sciences, USA. Histone H4 Antibody (cat# SC25260) and GAPDH Antibody (cat # SC365062) from Santa Cruz Biotechnology, USA. Anti-Rabbit IgG HRP conjugate (cat # W401B) and Anti-mouse IgG HRP conjugate (cat # W402B) from Promega, USA. L-Kyn (cat# K8625), Lipopolysaccharide (LPS) (cat# L4391), AhR antagonist CH-223191 (cat# C8124) and 1-Methyl-D-tryptophan (D-1MT) (cat# 452483) from Sigma-Aldrich. Recombinant murine- rIFN-ϒ (cat# 315-05) from PeproTech and CellTrace CFSE (cat# C34554) from invitrogen USA. Easy-Sep™ Mouse B Cell Isolation Kit (cat# 19854) from Stem Cell Technology, USA, Cell fractionation kit-Standard (cat# ab109719) from abcam, USA, Pierce BCA Protein Assay Kit (cat# 23225) from Thermo Scientific, USA, Pure-Link™ RNA Mini Kit (cat# 12183018A) from Invitrogen, USA, Prime script 1st strand cDNA synthesis kit (cat# 6110A) from TaKaRa Biotechnology, USA, VeriQuest SYBR Green qPCR master mix from Affymetrix, USA and Mouse IL-10 ELISA kit (cat# 88-7105-22) from ebioscience, USA.
Flow Cytometry Analyses
Spleens were isolated from mice and macerated in RPMI 1640 (Gibco, Invitrogen, UK) supplemented with 10% FBS to prepare a single-cell suspension. Isolated tumor tissues were first macerated in serum-free Iscove’s Dulbecco’s Modified Eagle Medium (IDMEM) (Corning) and then digested in serum-free IDMEM contained with 2 mg/ml of collagenase B (Roche) and 0.02 mg/ml of deoxyribonuclease from bovine pancreas (Sigma) at 37°C for 30 min. After digestion, suspensions were neutralized with sterile IDMEM supplemented with 10% FBS filtered through 0.45 mm strainers. Red blood cells (RBCs) were lysed with ACK lysis buffer (Quality Biological, Inc.) incubated at room temperature for 1 minute and washed with 10% RPMI 1640. The cells were counted, and 0.5-1×106 cells were used for surface staining post Fc blocking (1μg/ml) in 3% BSA or FBS for 30 minutes over ice. For intracellular IL-10 staining 0.5-1×106 cells were cultured per well in 96-well plates (Nunc, USA) then incubated for 5hrs in stimulating RPMI media containing Phorbol 12-myristate 13-acetate, or PMA, (0.1 mg/ml, Sigma), ionomycin (1 mg/ml, Sigma), LPS (10 µg/ml, Sigma) and a 1:1000 dilution of Golgistop/Golgiplug (BD Biosciences). Following stimulation, cells were first surface stained for 30 minutes at ice. After washing with PBS cells were fixed and permeabilized using the BD cytofix/cytoperm kit (BD Biosciences) for 30 minutes at 4°C then washed again with BD perm wash kit (BD Biosciences) and stained with intracellular fluorescently labeled anti-IL-10 antibodies. Fluorescence intensity of fluorochrome-labeled cells was measured by flow cytometry (BD LSR-II) while required sorting was done by BD FACS Aria III cytometer (UAB Flow Cytometry Core Facility). FACS Diva was used for acquiring the cells, and final data analysis was performed by Flow Jo (Tree star, USA). We included the whole nuclear cell population, including the tumor cells (CD45neg) for the analyses of % total Breg.
B Cells Purification and In-Vitro Breg Differentiation
Spleens or Bone marrow (BM) were isolated from mice and macerated in RPMI 1640 (Gibco, Invitrogen, UK) supplemented with 10% FBS to prepare a single-cell suspension. Red blood cells (RBCs) were lysed with ACK lysis buffer (Quality Biological, Inc.), then washed with 10% RPMI 1640. Cells were rewashed again in 1x PBS, and B cells were purified following manufacturer’s protocol. Briefly, 1x108/ml splenocytes were resuspended in RoboSep buffer (Stem cell technology, USA) followed by the addition of rat serum (50µl/ml) (Stem cell technology, USA) to the sample. Samples were transferred then to 5ml polystyrene round-bottom tube and B cell isolation Antibodies cocktail (50µl/ml) was added and mixed by pipetting, then incubated for 10 minutes at room temperature (RT). Rapid sphere (75µl/ml) (Stem cell technology, USA) was then added to the sample after vortexing it for 30 sec then mixed and incubated for 2.5 minutes at RT. RoboSep buffer was topped up to sample to make total volume 2.5ml and pipetted gently. Sample tubes were then placed in magnet (Easy Sep Magnet,cat# 18000, stem cell technology, USA) and incubated for 2.5 minutes at RT. Magnet was picked up and inverted the magnet tube pouring off the enriched cell suspension into a new tube. Purified B cells were washed twice with 10% RPMI 1640 and counted. B cells were disaggregated and stained with CFSE if needed and then 5 x105 cells were seeded in 200 µL per well in a 96 round bottom well plate with LPS (10μg/ml), increasing concentrations of L-Kyn (50µM and 100μM) and LPS + L-Kyn + CH 223191(10μM) an aryl hydrocarbon receptor antagonist (AhRA) for 72 hrs. Bregs were identified as CD19+CD5+CD1dhiIL-10+ by flow cytometry.
In-Vitro Co-Culture
For the in-vitro co-culture of B cells and MDSCs, CD19+ B cells were purified from splenocytes of naïve mice and co-cultured in a 5:1 ratio with MDSCs (CD11b+Gr-1+) sorted from the spleens and tumors of tumor-bearing mice and incubated for 72 hrs. For the in-vitro co-culture of B and T cells, T cells (CD3+TCRαβ+) were sorted from splenocytes of naïve mice and co-cultured in 1:1 ratio with purified B cells and incubated for 72 hrs. Bregs were identified in the co-culture as CD19+CD5+CD1dhiIL-10+ by flow cytometry.
Cell Fractionation and Immunoblotting
Cytoplasmic and nucleoplasmic fractions were extracted from cell pellets collected from ex-vivo experiments following stimulation with LPS and LPS+L-Kyn using protocols provided with the Cell fractionation kit-Standard (cat# ab109719, Abcam, USA). Protein concentrations were measured using Pierce BCA Protein Assay Kit (cat# 23225, Thermo Scientific, USA). 20-40µg of the cytoplasmic and nucleoplasmic fractions were used for western blot analysis to detect AhR expression on 10% SDS-PAGE gels. Gels were transferred overnight on to 0.45 μm Immobilon-P PVDF membranes (EMD Millipore). Blots were blocked in 5% non-fat dry milk (Lab Scientific) for 1 hour and incubated with primary antibody (manufacturer’s recommended concentrations) overnight at 4°C and then incubated in secondary anti-rabbit or anti-mouse HRP-conjugated antibodies (Promega, City, State), and protein bands detected using chemiluminescence (ECL, Santa Cruz Biotechnology).
RNA Isolation and qRT-PCR
Total RNA was isolated from cell pellets collected from ex-vivo experiments following stimulation with LPS, LPS+L-Kyn, LPS+L-Kyn+AhRA, and LPS+AhRA using Pure-Link™ RNA Mini Kit (cat# 12183018A, Invitrogen, USA) and by following manufacturer’s recommendations. cDNA was made using Prime script 1st strand cDNA synthesis kit (cat# 6110A, TaKaRa Biotechnology, USA). Reverse transcription was conducted with Veri Quest SYBR Green qPCR master mix (Affymetrix, USA). The primer sequences used were as follows: AhR (forward-AGGCCAGGACCAGTGTAGAG, reverse-CTTGGATAGTGGAGGAAGCA), Cyp1A1 (forward-TGGATGCCTTCAAGGACTTG, reverse- CAGCTTCCTGTCCTGACAAT) and RPS-18 was selected as reference or housekeeping genes (forward-GAACTCACGGAGGATGAGGT, reverse-TCTAGACCGTTGGCCAGAAC).
In-Vitro B Cell Proliferation Assays
Splenocytes or sorted CD19+B220+ B cells from tumor-bearing WT or IDO deficient mice were labeled with CFSE (Molecular Probes, Eugen, OR) and culture for 4 days with 20 ug/ml LPS (Sigma-Aldrich, St. Louis, MO) and 10 ng/ml IL-4 (PeproTech, Rocky Hills, NJ). The percentages of CD19+CFSElow cells (proliferating cells) were determined by FACS analysis.
Quantitation of IL-10 and IgG by ELISA
The mouse sera were collected from naive and tumor-bearing mice. The levels of IL-10 were determined by ELISA kits from ebioecience in sera following the manufacturer protocol. The supernatants from splenocytes or purified B cells from tumor-bearing WT or IDO deficient mice were collected on day 4 of culture. The levels of IgG were determined by ELISA kit from Life Technologies (Grand Island, NY) in the supernatants following the instruction of the manufacturer.
Statistical Analyses
Statistical analyses were performed using Graph pad prism 5 software (La Jolla, CA, USA) and values were presented as mean ± SD unless indicated otherwise in the figure legend. Significant differences between the group means were determined by One-way ANOVA with Tukey’s multiple comparisons test for multiple groups. Comparisons between two groups were evaluated by an unpaired parametric student’s t-test. A value of p<0.05 was considered statistically significant.
Results
Tumors and MDSC-Mediated IDO Promotes Breg Differentiation in Lung Cancer
We first investigated if Breg play an important role in the progression of lung cancer. Using a syngeneic murine model of lung cancer with intravenous implant LLCs, we first studied the kinetics of infiltration of CD19+CD1dhiCD5+IL-10+ Bregs which significantly increased in a time-dependent manner (Figure 1A). We and others have previously reported that immunosuppressive MDSCs are important drivers of tumor growth in this tumor model (41, 45). Additionally, we have delineated using tumor implanted IDO deficient mice, a critical role for MDSC-associated IDO in dampening anti-tumor T cell responses (42). Recent evidence also suggests that MDSC-mediated regulatory T cell induction is through Bregs (48, 49). Therefore, we first evaluated a potential role for IDO in Breg infiltration during tumor progression using our syngeneic lung cancer model (intracardiac, i.c) in B6129-Ido1/J (IDO-/-) and wild type C57BL/6 (WT) control mice. Significantly reduced Breg frequency was observed in spleens and tumors of IDO-/- compared to WT control mice at 9 days following in-vivo establishment of lung tumors (Figures 1B–E and Supplementary Figure 1A).
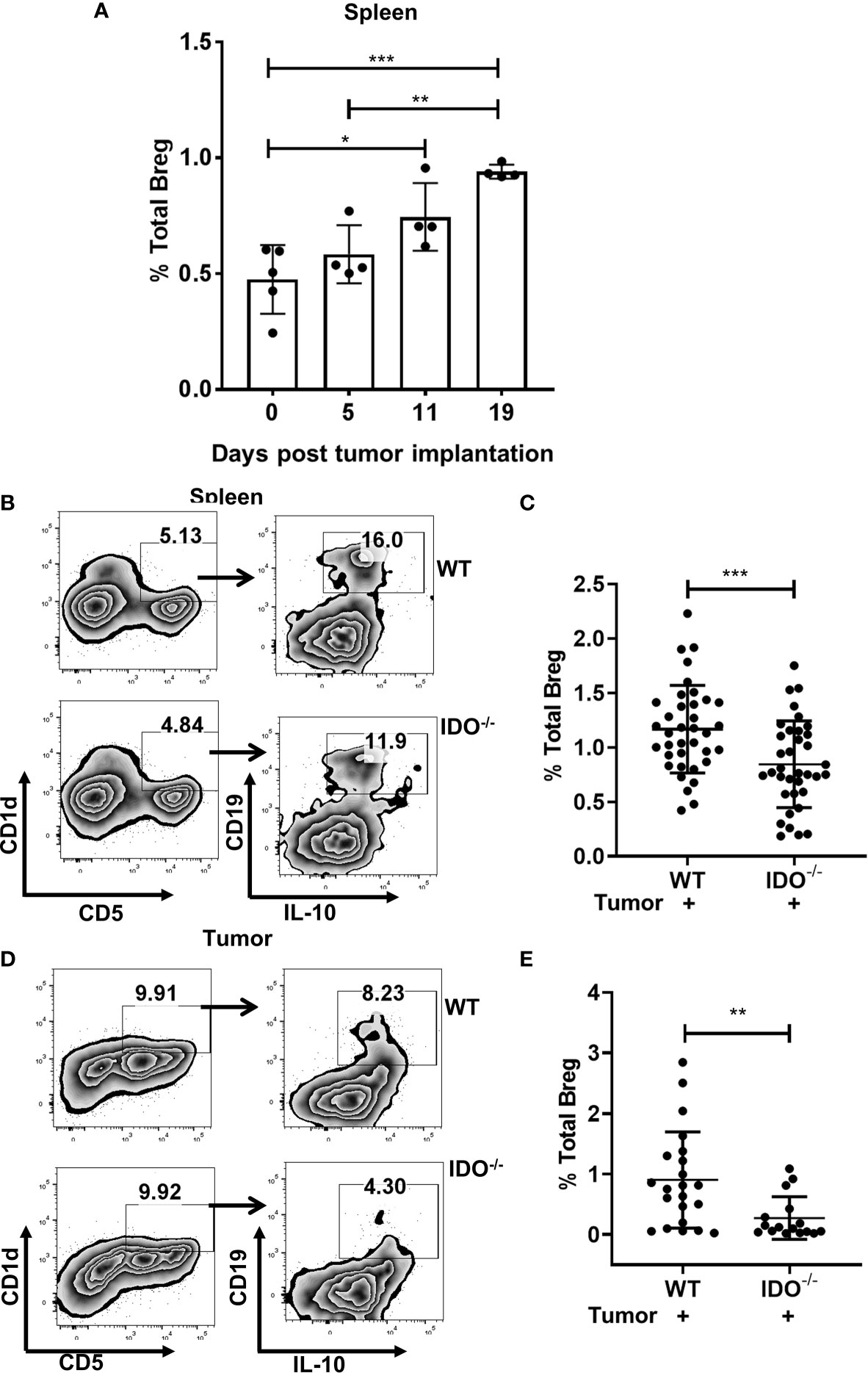
Figure 1 IDO deficiency reduces Breg frequencies in lung cancer. (A) Time kinetics of infiltration of Bregs (CD19+CD5+CD1dhiIL-10+) during lung cancer progression in tumor-bearing wild type (WT) on Day 5,11 and 19 post i.v. injection of LLC compared to naïve WT mice (n = 4-5 mice per group). (B) Representative figure of FACS analyses of splenic tissue confirmed the reduction of Breg (CD19+CD5+CD1dhiIL-10+) in tumor-bearing IDO-/- on day 9 post tumor implant. (C) Total percentage of Bregs (CD19+CD5+CD1dhiIL-10+) in spleens of tumor-bearing WT and IDO-/- mice on Day 9 post tumor implant (n = 36-37 mice per group). (D) Representative figure of FACS analyses of lung tumor tissue confirmed reduction of Breg (CD19+CD5+CD1dhiIL-10+) in tumor-bearing IDO-/- on day 9 post tumor implant. (E) Total percentage of Breg (CD19+CD5+CD1dhiIL-10+) in lung tumors of WT and IDO-/- mice on Day 9 post tumor implant (n = 16-22 mice per group). *p < 0.05, **p < 0.001, ***p < 0.0001.
The immunosuppressive properties of Bregs have been attributed to IL-10 (18, 50, 51). We quantitated serum levels of IL-10, which was elevated in tumor-bearing WT compared to IDO deficient mice (Supplementary Figure 1B). We then examined a potential direct role for MDSC-associated IDO as a driver of Breg differentiation in co-cultures of immune sorted MDSCs (CD11b+Gr-1+) from splenocytes and tumors of tumor implanted WT and IDO-/- mice with CD19+ purified B cells in 5:1 ratio (5 B cells: MDSC) (Figures 2A, B). Impairment of Breg differentiation was observed in B cells co-cultured with MDSCs from IDO-/- compared to WT mice (Figures 2A, B). Breg differentiation was significantly reduced when MDSCs (CD11b+Gr-1+) sorted from splenocytes of tumor-bearing IDO-/- mice were co-cultured with total splenocytes (5:1 ratio) compared to WT (Supplementary Figures 1C, D). Further, we validated the contribution of MDSC-associated IDO in Breg differentiation using IDO f MCre mice with conditional deletion of IDO in myeloid-lineage cells (Supplementary Figure 1E) and their MCre control mice. At day-9 post tumor implant, IDO f MCre mice exhibited significantly lowered tumor burden compared to controls (Figure 2C). Importantly, we observed a significant reduction in Breg in tumor-bearing IDO f MCre compared to their MCre control (Supplementary Figure 2A and Figures 2D–G). Collectively, these data suggest that Trp catabolizing enzyme IDO plays a major role in Breg differentiation in the lung TME with a significant contribution from MDSC-associated IDO.
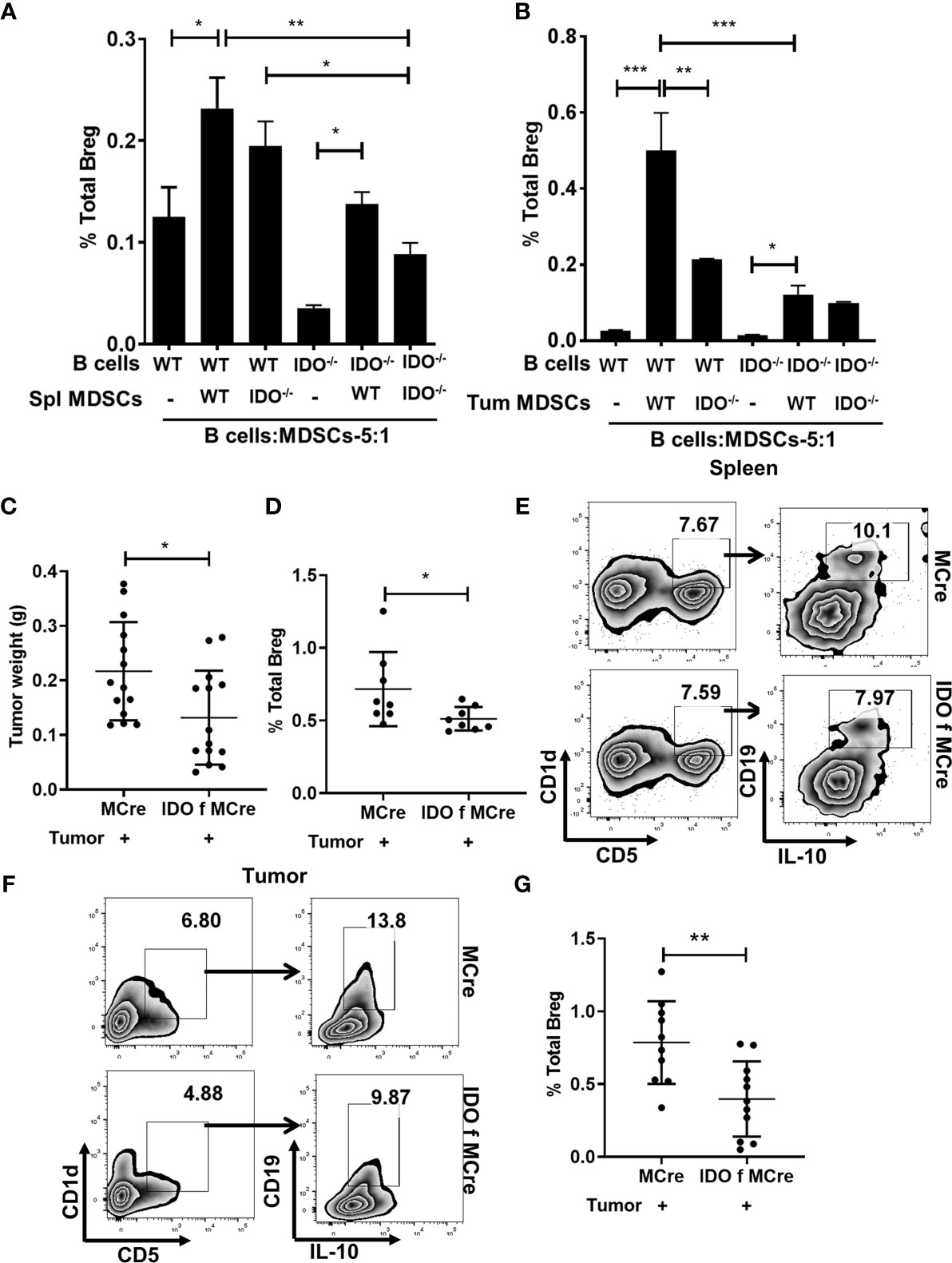
Figure 2 MDSCs expressing IDO promotes Breg differentiation. (A) CD19+ B cells were purified from splenocytes of WT and IDO-/- naïve mice and co-cultured in 5:1 ratio with MDSCs sorted from the spleen of tumor-bearing WT and IDO-/- and incubated for 72 hrs. Bregs were identified in the co-culture as CD19+CD5+CD1dhiIL-10+ by flow cytometry. Data shown here are representative of three independent experiments with three replicates for each group and represent the mean ± STDEV values. (B) CD19+ B cells were purified from splenocytes of WT and IDO-/- naïve mice and co-cultured in 5:1 ratio with MDSCs sorted from tumor of WT and IDO-/- and incubated for 72 hrs. Bregs were identified in the co-culture as CD19+CD5+CD1dhiIL-10+ by flow cytometry. Data shown here are representative of three independent experiments with three replicates for each group and represent the mean ± STDEV values. (C) Conditional deletion of IDO in myeloid cells (IDO f MCre) reduced tumor burden (g) compared to control MCre mice on Days 9 of tumor implant (n = 14 mice per group). (D) Representative figure of FACS analyses of splenic tissue confirmed reduction of Breg (CD19+CD5+CD1dhiIL-10+) in tumor-bearing IDO f Mcre on day 9 post tumor implant. (E) Diminished Breg frequency in IDO f Mcre mice with lung cancer compared to MCre control on Days 9 post tumor implant (n = 8 mice per group). Total percentage of Bregs (CD19+CD5+CD1dhiIL-10+) were determined by flow cytometry analysis. (F) Representative FACS plot of tumor tissue confirming reduction of Breg in IDO f MCre on day 9 of tumor implant. (G) Reduced Breg frequency in lung tumor of IDO f MCre mice compared to MCre control on day 9 of tumor implant (n = 10-11 mice per group). *p < 0.05, **p < 0.001, ***p < 0.0001.
Trp-Metabolite L-Kyn Promotes Breg Differentiation Through AhR Pathway
IDO pathway metabolite L-Kyn has been reported to be immunosuppressive by promoting Treg through AhR activation (45, 52). Since IDO deficiency resulted in impaired Bregs frequency in lung TME, we evaluated whether L-Kyn promotes Breg differentiation through AhR. In ex-vivo cultures of CD19+ B cells purified from splenocytes of naïve WT mice stimulated with no LPS controls and LPS+Kyn and LPS+Kyn+AhRA (AhR antagonist, CH-223191, AhRA) treatments, we observed induction of CD19+CD5+CD1dhiIL-10+ Bregs frequency in response to stimulation with LPS+L-Kyn while Breg differentiation was inhibited with the addition of AhRA (Supplementary Figures 3A, B and Figure 3A). In addition, we detected elevated levels of IL-10 in the culture supernatant from LPS+Kyn treated samples (Figure 3B). These data suggest that L-Kyn promotes Breg through AhR pathway. Furthermore, CD19+CD5+CD1dhi Breg precursors were elevated following treatment with LPS+L-Kyn in a dose-dependent manner. But the expansion of these Breg precursors was present even in the presence of AhRA compared to LPS only stimulation ex-vivo (Supplementary Figure 4A), demonstrating that L-Kyn induced Breg precursor differentiation was AhR independent. We then evaluated the role of L-Kyn alone without LPS, and found that Breg precursors (CD19+CD5+CD1dhi) were enhanced, but L-Kyn alone was unable to transform them into IL-10 producing Breg, suggesting that LPS stimulation was essential to induce Breg precursors to become IL-10 producing Breg ex-vivo (Supplementary Figures 4B, C). Our qRT-PCR data validated this finding (Supplementary Figure 3C). Furthermore, we observed enhanced Breg frequency also in ex-vivo cultures of whole splenocytes stimulated with LPS+L-Kyn compared to only LPS controls (Supplementary Figure 4D). We confirmed that the viability of CD19+ purified B cells was not affected following stimulation with either L-Kyn over a range of 50 to 500µM concentration with or without LPS and AhRA (Supplementary Figure 5). Thus taken together, inhibition in Breg frequency was due to the inactivation of AhR rather than the toxicity of L-Kyn or AhRA. Further, we validated the contribution of AhR in Breg differentiation using AhR-/- mice at day-9 post tumor implant, AhR-/- mice exhibited significantly lowered Breg frequency compared to WT control mice (Supplementary Figure 2B and Figures 3C, D).
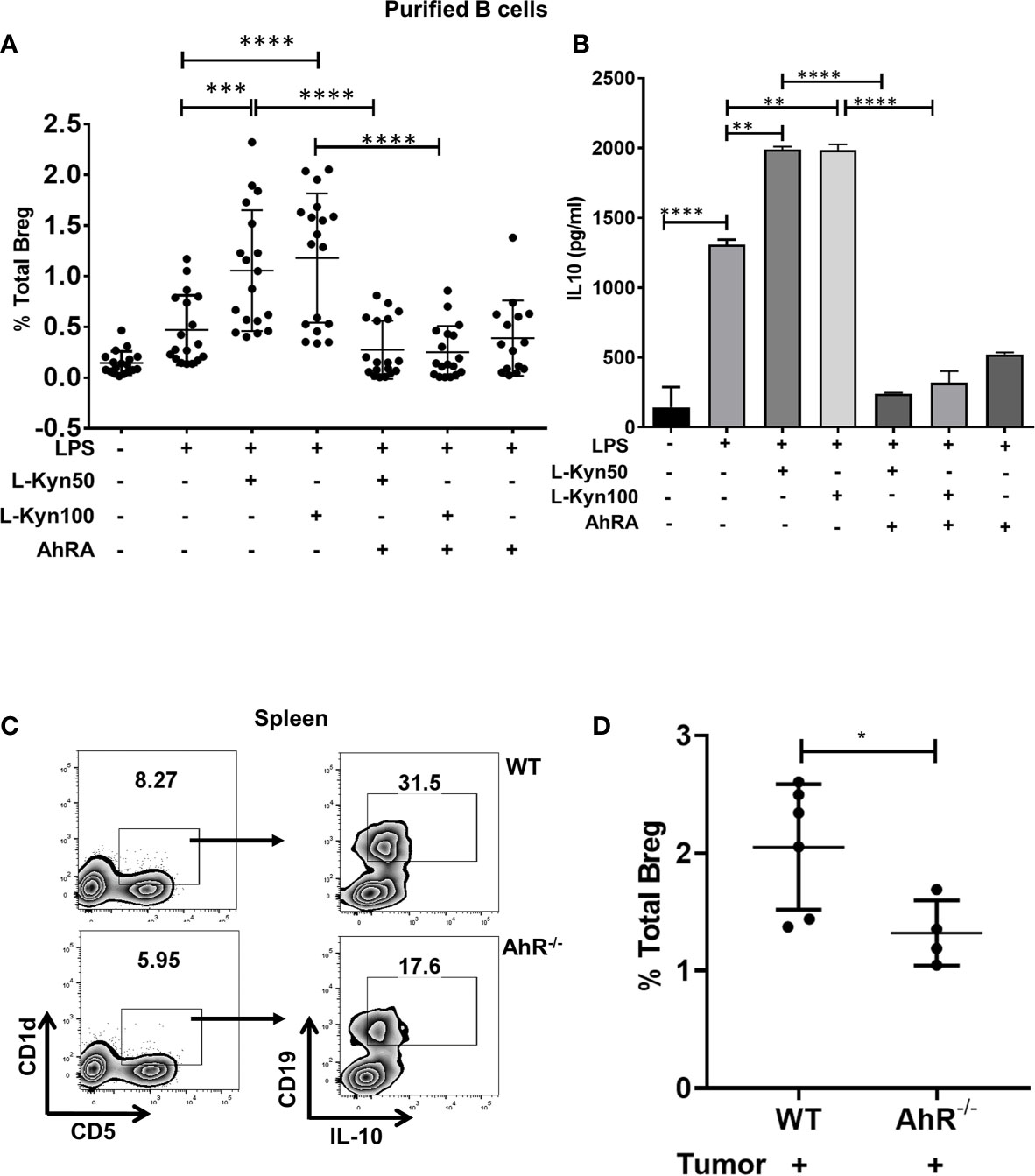
Figure 3 Trp metabolite L-Kyn promoted Breg differentiation through AhR pathway. CD19+ B cells were negatively selected from whole splenocytes of WT mice, disaggregated and seeded with LPS (10μg/ml), increasing concentrations of L-Kyn (50µM and 100μM) and LPS + L-Kyn + CH 223191(10μM) an aryl hydrocarbon receptor antagonist (AhRA) for 72 hrs. Bregs were identified as CD19+CD5+CD1dhiIL-10+ by flow cytometry. (A) Total percentage of Bregs (CD19+CD5+CD1dhiIL-10+) in ex-vivo experiments performed with negatively selected purified B cells following stimulation with no LPS, LPS, LPS+L-Kyn, LPS+AhRA+L-Kyn, and LPS+AhRA. Data shown here are pooled from six independent experiments with 3 technical replicates in each condition. (B) IL-10 levels in the media collected from above described experiments. (C) Representative figure of FACS analyses of splenic tissue confirmed the reduction of Breg (CD19+CD5+CD1dhiIL-10+) in tumor-bearing AhR-/- on day 9 post tumor implant. (D) Total percentage of Bregs (CD19+CD5+CD1dhiIL-10+) in spleens of tumor-bearing WT and AhR-/- mice on Day 9 post tumor implant (n = 4-6 mice per group). *p < 0.05, **p < 0.001, ***p < 0.0001, ****p < 0.00001.
To further ascertain the role of AhR, we determined the expression of AhR and its target gene Cyp1A1 by qRT-PCR in purified CD19+ B cells stimulated with either LPS, LPS+L-Kyn or LPS+L-Kyn+AhRA. Both AhR and Cyp1A1 gene expression was upregulated in B cells stimulated with LPS+L-Kyn (Figures 4A, B) compared to controls. Additionally, we confirmed the role of AhR in Breg differentiation in lung cancer, by validating the upregulation of AhR expression in immune sorted CD19+ B cells from splenocytes of tumor-bearing WT compared to tumor-bearing IDO-/- and WT naïve (Figure 4C). L-Kyn is an endogenous ligand of cytoplasmic transcription factor AhR, which after binding with AhR makes AhR complex, translocates into the nucleus to bind further with AhR nuclear translocator (Arnt). TheAhR-Arnt heterodimers then finally bind with dioxin-responsive elements (DREs) to regulate gene expression in a variety of cells (47). To obtain mechanistic understanding behind L-Kyn induced Breg differentiation, we assessed if stimulation of B cells with L-Kyn resulted in translocation of AhR to the nucleus. Immunoblot analyses performed with cytoplasmic and nucleoplasmic fractions obtained from LPS and LPS+ L-Kyn stimulated B cells showed induction of AhR expression (Figure 4D), suggesting a critical role of AhR in L-Kyn induced Breg differentiation. We then investigated if AhR deficiency in B cells impairs L-Kyn induced Breg differentiation. Purified CD19+ B cells from splenocytes of naïve AhR-/- were cultured to perform ex-vivo experiments followed by LPS+L-Kyn stimulation (53).
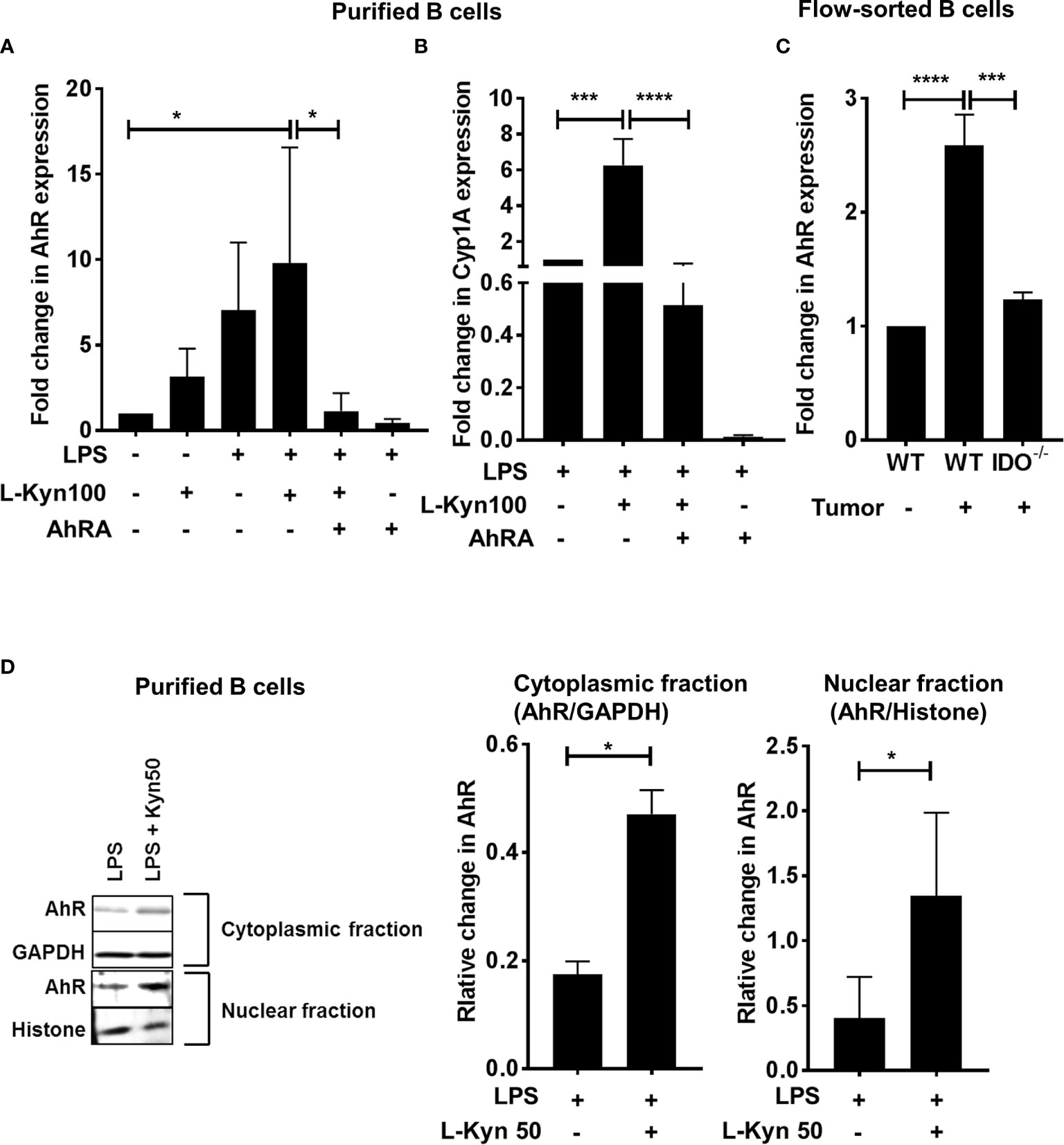
Figure 4 Trp metabolite L-Kyn activated AhR pathway. CD19+ B cells were negatively selected from whole splenocytes of WT mice, disaggregated and seeded with LPS (10μg/ml), L-Kyn (50µM and 100μM) and LPS + L-Kyn + CH 223191(10μM) an aryl hydrocarbon receptor antagonist (AhRA) for 72 hrs (A) AhR expression was assessed by qRT-PCR in the cells collected from ex-vivo experiments performed with purified B cells in experimental conditions described above (B) AhR target gene Cyp1A1 expression was assessed by qRT-PCR in purified B cells ex-vivo experiments following stimulation of LPS, LPS+L-Kyn, LPS+L-Kyn+AhRA and LPS+AhRA (C) AhR expression was assessed by qRT-PCR in B cells (CD19+) sorted from naïve WT, tumor-bearing WT and tumor-bearing IDO-/- mice on Day 9 post tumor implant (D) Cytoplasmic and nucleoplasmic fraction were collected from purified B cells from ex-vivo experiments following stimulation with LPS and LPS+L-Kyn Western blot analysis was performed to detect AhR expression in cytoplasmic and nucleoplasmic fractions and the relative expression was normalized with GAPDH and histone respectively. *p < 0.05, ***p < 0.0001, ****p < 0.00001.
Interestingly LPS+ L-Kyn was unable to induce Breg differentiation in AhR-/- B cells, and in addition, a significant reduction in Bregs frequency was also observed in AhR-/- compared to WT control mice (Figure 5A). Furthermore, we found diminished ex-vivo proliferation in CFSE stained Bregs of AhR-/- compared to their WT controls followed by LPS and LPS+L-Kyn stimulation, as described above (Figure 5B). Additionally, consistent with our previous finding (Supplementary Figure 4A), we observed induction in frequency of Breg precursors (CD19+CD5+CD1dhi) in AhR deficient CD19+ B cells upon stimulation with LPS+L-Kyn compared to LPS only stimulation (Figure 5C), strongly supporting our hypothesis that L-Kyn induced Breg precursor differentiation is AhR independent.
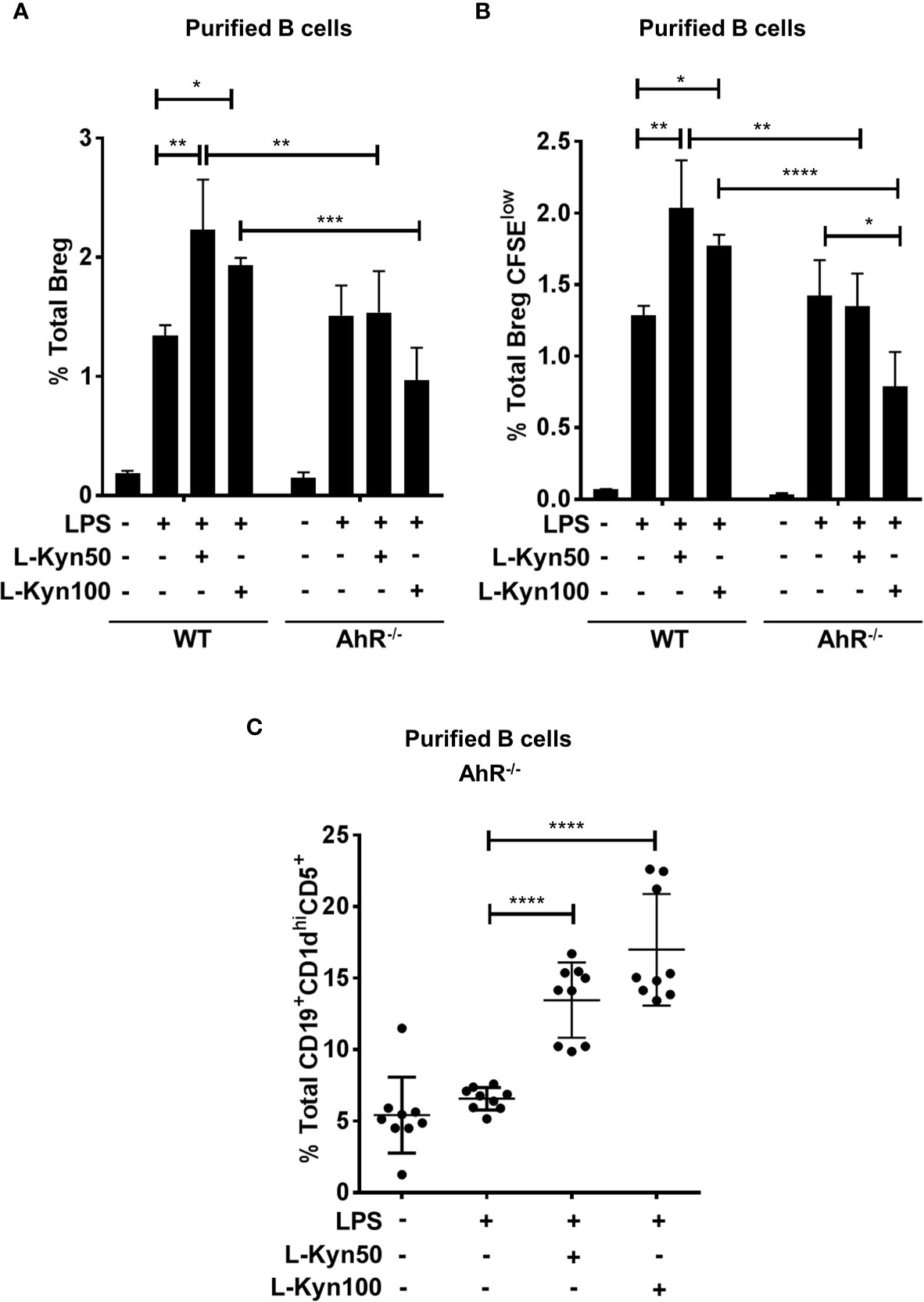
Figure 5 AhR deficiency in B cells impaired Breg differentiation ex-vivo. CD19+ B cells were negatively selected from whole splenocytes of wild-type (WT), and AhR-/-, mice were disaggregated and seeded with LPS (10μg/ml), and increasing concentrations of L-Kyn (50µM and 100μM) for 72 hrs. Bregs were identified as CD19+CD5+CD1dhiIL-10+ by flow cytometry. (A) AhR deficiency inhibited L-Kyn induced Breg differentiation following stimulation of B cells with LPS+L-Kyn. Data shown here are representative of three independent experiments with three replicates for each group and represent the mean ± STDEV values. (B) AhR deficiency altered the proliferation of Breg. CD19+ cells were negatively selected from whole splenocytes of WT, and AhR-/- mice and labeled with CFSE and then stimulated with LPS and increasing concentrations of L-Kyn for 72 hrs. Total percentages of Breg+CFSElow (CD19+CD5+CD1dhiIL-10+CFSElow) cells were determined by flow cytometry analysis. Data shown here are representative of two independent experiments with 3 technical replicates in each condition. (C) Breg precursors (CD19+CD1dhiCD5+) induced by LPS+L-Kyn were not impaired in AhR-/-. Total percentages of CD19+CD1dhighCD5 in AhR-/- were determined by flow cytometry analysis. Data shown here are pooled of three independent experiments with 3 technical replicates in each condition. *p < 0.05, **p < 0.001, ***p < 0.0001, ****p < 0.00001.
CD40,TLRs, or cytokine-mediated activation turns immature B cells directly into Breg or immature B cells may first differentiate into mature B cells and then into IL-10 producing Breg (54). Therefore we investigated if L-Kyn modulated specific B cell subsets to differentiate into Breg. Interestingly, we observed that only mature B cells (CD19+IgM+IgD+) upon stimulation with LPS+L-Kyn induced frequency of CD19+CD5+CD1dhi Breg precursors (Supplementary Figure 6). Collectively these findings suggest that L-Kyn, an IDO-derived Trp metabolite activates AhR and translocate AhR complex into the nucleus of B cells to drive their differentiation into IL-10, producing Breg. At the same time, L-Kyn alone enhances the Breg precursor pool ex-vivo, which upon stimulation with LPS transform them into IL10 producing Bregs.
L-Kyn Promotes Breg Differentiation in TLR-4-MyD88 Dependent but TLR-2 Independent Manner While Breg Precursor Differentiation Is Independent of TLR-2 and TLR-4
It has been already established that LPS dependent activation in immune cells is mainly mediated by TLR-4 through CD14 via transcription factor NF- κB to regulate several genes associated with the expression of antigen-presenting molecules, cytokines, transcription factors and enzymes including IDO (55, 56). We next elucidated the potential role of TLRs in L-Kyn induced Breg differentiation. To functionally characterize these using naïve TLR-4 deficient mice, we analyzed Breg as well as Breg precursors in ex-vivo co-cultures of CD19+ B cells purified from splenocytes of TLR-4-/- mice. We observed no significant difference in Breg but a significant increase in Breg precursors in cultures with TLR-4 deficient cells (Figures 6A, B), suggesting that TLR-4 signaling may not be necessary for differentiation of B reg precursors. But TLR-4 signaling was important for transforming Breg precursors into IL-10 producing Breg. Additionally, LPS-TLR-4 interaction resulted in MyD88 dependent activation of L-Kyn induced Breg differentiation (Supplementary Figures 7A, B), collectively demonstrating that LPS+L-Kyn induced Breg differentiation is TLR-4-MyD88 dependent, but Breg precursor differentiation is TLR-4 independent.
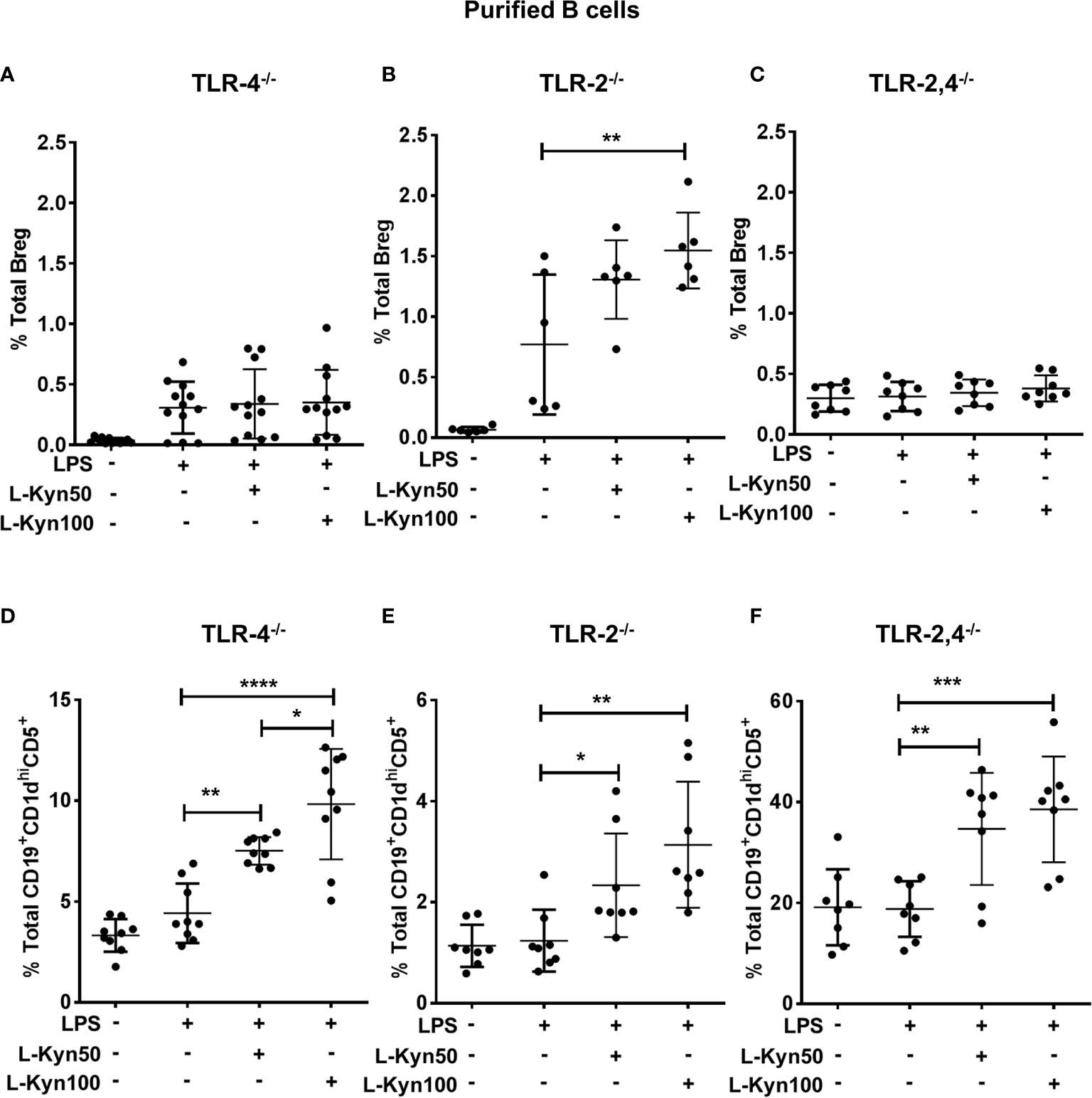
Figure 6 L-Kyn induced Breg differentiation was TLR-4 dependent but TLR-2 independent, the differentiation of Breg precursor was TLR-2 and TLR-4 independent. CD19+ B cells were negatively selected from whole splenocytes of TLR-4-/-, TLR-2-/- and TLR-2,4-/- mice were disaggregated and seeded with LPS (10μg/ml), and increasing concentrations of L-Kyn (50µM and 100μM) for 72 hrs. Bregs were identified as CD19+CD5+CD1dhiIL-10+ by flow cytometry. (A, C, E) Upper panel- L-Kyn induced Breg differentiation was impaired in B cells of TLR-4-/- and TLR-2, 4-/- but induction in TLR-2-/- following stimulation with LPS+L-Kyn. Data shown here are pooled from four independent experiments with 3 technical replicates in each condition. (B, D, F) Lower panel- L-Kyn promoted Breg precursors (CD19+CD1dhiCD5+) in B cells of TLR-4-/- TLR-2-/- and TLR-2, 4-/- following stimulation with LPS+L-Kyn. Data shown here are pooled from three independent experiments with 3 technical replicates in each condition *p < 0.05, **p < 0.001, ***p < 0.0001, ****p < 0.00001.
Previously, LPS induced TLR-2 activation and TLR-2 dependent Breg induction have been reported (55, 57). To determine whether TLR-2 influences L-Kyn induced Breg differentiation ex-vivo, we stimulated CD19+ purified B cells from TLR-2-/- mice with LPS+L-Kyn. Both Breg as well as Breg precursors increased in response to LPS+L-Kyn compared to only LPS stimulation (Figures 6C, D). These data suggested that L-Kyn induced Breg precursors as well as Breg differentiation, may be TLR-2 independent. Importantly this was confirmed with CD19+ purified B cells from TLR-2,4 double knock out (TLR-2,4-/-) mice (Figures 6E, F). It is unclear if in the absence of TLR-2, compensatory signaling may enhance B reg precursor and B reg differentiation. The total percentage of Breg (CD19+CD5+CD1dhiIL-10+) was analyzed in ex-vivo experiments performed with negatively selected purified B cells from WT, MYD88-/- and TLR2-/- following stimulation with high concentration of L-Kyn (100μM) alone compared to LPS alone. No significant change in Breg was noted among WT, MYD88-/- and TLR2-/- in absence of LPS (Supplementary Figure 7B). While the Breg precursors at baseline were significantly higher in the TLR-2,4-/- deficient mice compared to the individual KO, this did not correlate with baseline B reg numbers (Figure 6F).
IDO Negatively Regulates B Cell Proliferation and Function
We then investigated if the reduction in B reg differentiation is due to alteration of overall B cell differentiation resulting from IDO deficiency. As we had reported earlier that B cell differentiation is impaired during tumor growth in IDO sufficient mice (34), we evaluated this in IDO deficient mice. We determined if IDO deficiency affect B cell development in tumor-bearing mice. Both percentages and absolute numbers of total B220+ cells, B220+IgD-IgM-CD24intCD43+ pro-B cells, B220+IgD-IgM- CD24hiCD43- pre-B and B220+IgD+IgM+ mature B cells were decreased in BM of tumor-bearing IDO deficient mice compared with naïve IDO deficient mice (Supplementary Figures 8A, B, 9A, B). Furthermore, both the percentages and numbers of B220+CD93+ immature B cells were increased, whereas B220+CD93-CD21intCD23+ follicular B cells were decreased in tumor-bearing IDO deficient mice compared with naïve IDO deficient mice (Supplementary Figures 10A, B). However, we observed no difference in total B cells, pro-B cells, pre-B cells, immature B cells and mature B cells in BM (Supplementary Figures 9A, B), as well as total B cells, immature B cells, marginal zone B cells and follicular B cells in spleens (Supplementary Figures 10A, B) between WT and IDO naïve mice or tumor-bearing mice. These results indicate that IDO deficiency does not affect B cells development in BM and spleen both in naïve tumor-free mice and tumor-bearing mice.
It has been reported that B cell-intrinsic IDO regulates humoral immunity to T cell-independent antigens (58). To investigate whether IDO regulates B cell proliferation in vitro, splenocytes or sorted CD19+B220+ B cells from WT or IDO-/- tumor-bearing mice were labelled with CFSE and cultured in the presence of LPS and IL-4. We observed that the percentages of CD19+CFSElow cells were increased in splenocytes (Supplementary Figure 11 and Figure 7A) or purified B cells (Figure 7B) from IDO mice compared with that from WT mice. IgG production in culture supernatants was elevated in activated splenocytes (Figure 7C) and/or purified B cells (Figure 7D) from IDO-/- mice compared with WT controls. These results suggest that IDO negatively regulates B cell proliferation and IgG production in tumor-bearing mice.
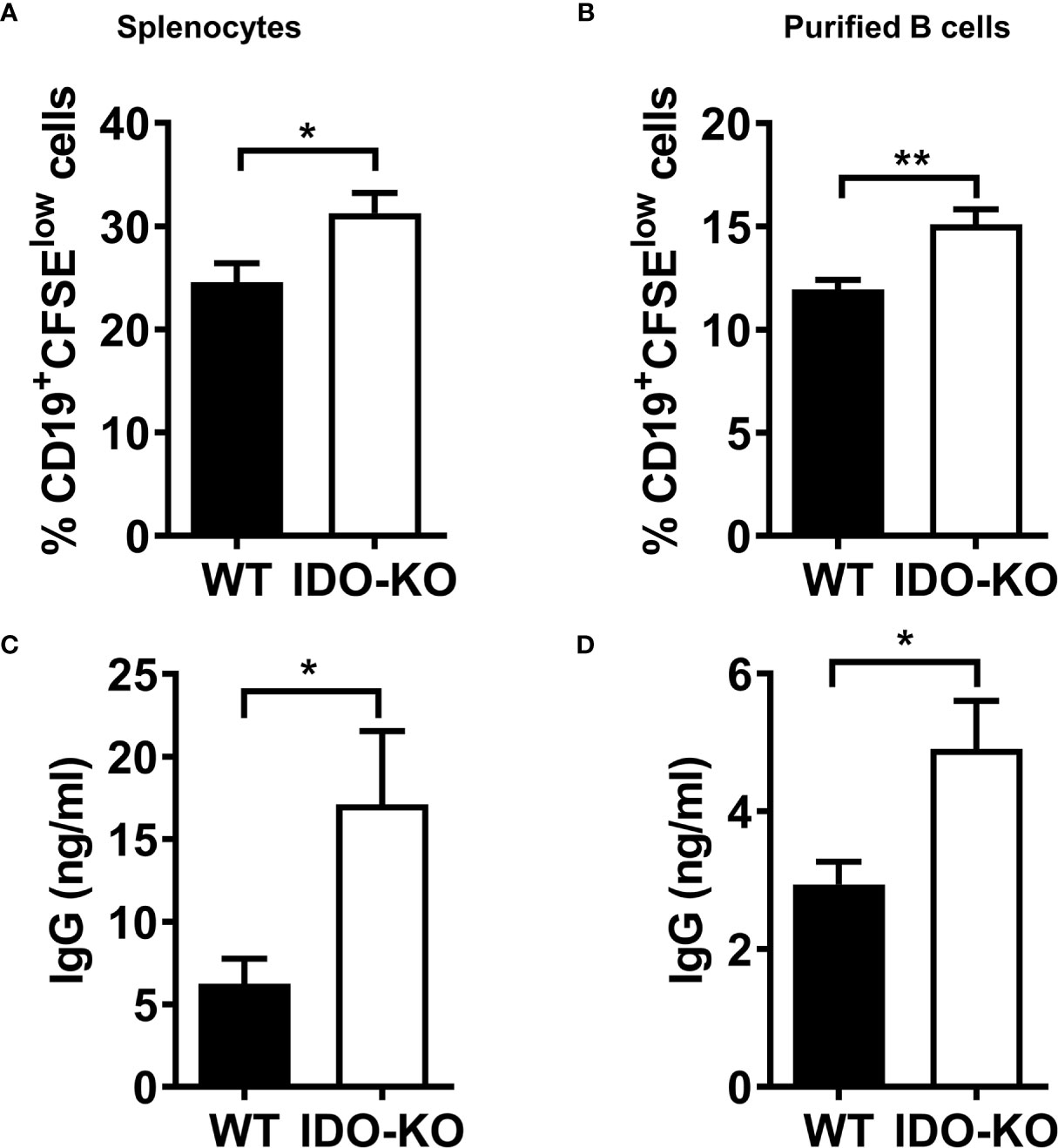
Figure 7 IDO negatively regulate B cell proliferation an IgG production. (A, B) Splenocytes (A) or CD19+B220+ B cells sorted (B) from tumor-bearing mice on day 9 post-LLC i.c. challenge were labeled with CFSE and cultured with LPS (20 µg/ml) and IL-4 (10 ng/ml). The percentages of CD19+CFSElow cells were determined by FACS analysis on day 4 of culture. (C, D) culture (C) or purified B cells (D) on day 4 of culture. The levels of IgG in the culture supernatants were determined by ELISA. *P < 0.05, **P < 0.01.
Discussion
In this study, we demonstrate that splenic Breg frequency increases with the progression of lung tumor growth in a syngeneic model of lung cancer. Additionally, we report for the first time that IDO associated with tumors and MDSCs promote Breg differentiation during lung cancer progression. Our investigations suggest that IDO-induced Trp metabolite L-Kyn mediates the development of Breg precursors that differentiate into IL-10 producing Breg upon stimulation with LPS ex-vivo. Further, we report an uncharacterized role for AhR in regulating L-Kyn induced Breg differentiation. Our ex-vivo data generated from TLR-2, TLR-4, TLR-2, 4, and MyD88 deficient B cells indicate that L-Kyn induces Breg differentiation through TLR-4-MyD88 pathway ex-vivo while L-Kyn promotes Breg precursors independent of TLR-2 and TLR-4 signaling. Furthermore, our ex-vivo B and T cells co-culture studies support our conclusion that L-Kyn induces Breg differentiation independent of T cells. Importantly, we provide evidence from ex-vivo experiments that tumor antigens modulated by IFN-γ induces IDO, which then promotes antigen-specific Breg differentiation through AhR pathway. Collectively, these data conclude that IDO, a Trp metabolizing enzyme, and L-Kyn, a major metabolite of Trp promote Breg differentiation through AhR and TLR-4-MyD88 pathways.
Immune cell expression of IDO is dominated by immunosuppressive cell types including Tregs, MDSCs, and pDCs and very recently reported also by macrophages (59–62). IDO+ dendritic cells can impair antigen presentation from antigen-presenting cells (APCs) in the tumor-draining lymph nodes to recruited effector T cells. Furthermore, tumor cells that express IDO not only promote Treg differentiation but also disable effector T cell function (45). The concept that Breg orchestrate immune modulation was first suggested 4 decades ago, when B cell depleted splenocytes were associated with the increased hypersensitivity, suggesting a positive correlative loop between Breg and Treg (63, 64).
The mechanisms underlying the augmentation of Breg infiltration with lung cancer progression are largely unknown. Previously, researchers have reported corroborative evidence regarding correlation of Breg infiltration with cancer progression (65, 66). Breg have been implicated in the clinical progression of various types of cancers and have been proposed to promote tumor growth through IL-10 production (67–69).To date, relatively little is known about Breg differentiation in the lung TME. Previous studies have shown that chemoattractant may attract naïve B cells into TME and promote their differentiation into Breg through a series of soluble mediators (IL-1β, IL-6, IL-21 and IL-35) and cell interaction dependent mechanism (CD40-CD40-L) (54, 70, 71). The induction of Breg was not completely suppressed upon blockade of CD40 (71), thereby indicating that some other factors may likely be involved in Breg differentiation.
We and others have reported previously that in addition to tumor cells, immunosuppressive MDSCs are the major contributors of IDO, a rate-limiting enzyme of Trp catabolism in the TME (41, 42, 72, 73). The impact of MDSC- associated IDO on T cells is well known, but its effect on several subtypes of B cells remains to be determined. It has been hypothesized that MDSCs and tumor cell-associated IDO impairs effector T cell function by promoting differentiation of Treg that support tumor progression (43, 44). Moreover, recent evidence shows that Breg promotes tumor growth by recruiting Treg that inhibit the infiltration of CD8+ T cells and CD49+ NK cells within the TME (15). There is increasing evidence that MDSCs regulate B cells indirectly by expansion of Treg through Breg (21, 48, 49, 74–76). We recently reported that MDSCs impair B cells proliferation and function during lung tumor progression while differentiation of immunosuppressive Breg was increased (34). It is not known whether the MDSC supported milieu suppresses normal B cell differentiation, instead drives Breg differentiation. Park et al. assessed the impact of MDSCs in Breg differentiation in the murine model of systemic lupus erythematosus (SLE). They proposed that MDSCs induce Breg differentiation via iNOS and ameliorate autoimmunity (77). Interestingly, we observed that MDSCs suppress B cell responses via iNOS, and it is possible that the iNOS induction while suppressing B cell proliferation promotes Breg (34). To the best of our knowledge, MDSCs-mediated regulation of the function of Breg has not been studied in the context of cancer. In addition, none of these prior studies have identified that IDO produced from MDSCs and tumor cells is involved in Breg expansion. Our co-culture data obtained from IDO deficient MDSCs and B cells demonstrate for the first time that IDO associated MDSCs induces Breg ex-vivo. In addition, reduced Breg frequency in a syngeneic tumor model with conditional deletion of IDO in myeloid cells (IDO f MCre) further support the positive regulation by MDSC- associated IDO in Breg differentiation in-vivo. Therefore our current studies and previous evidence are in resonance with our observations that IDO from MDSCs and tumor cells promote Breg during lung cancer progression.
Induction of IDO activity in the TME results in the depletion of Trp, an essential amino acid needed for T cell proliferation, thus regulating T cell responses (45, 78). IDO-dependent Trp metabolite L-Kyn is a well-known endogenous ligand of cytoplasmic transcription factor AhR (47). Ligand mediated activation of AhR performs a crucial role in regulating gene expression in a variety of cells (47). AhR has long been established as an environmental receptor for dioxins. Additionally, it has been previously reported that L-Kyn activates AhR to induce selective expansion of Treg by activation of forkhead box p3 (Foxp3) in naïve T-cells and also biasing macrophages and dendritic cells (DCs) towards an immunosuppressive phenotype (46, 52, 79, 80). Importantly, AhR activation positively regulates IDO induction in DCs and consequently promotes L-Kyn production (81, 82). L-Kyn mediated AhR activation promotes tumor growth with elevated levels of inflammatory cytokines (IL-6, IL-8, and IL-1β) and a decreased frequency of infiltrating cytotoxic CD8+ T cells in TME (47). AhR is also highly induced in subsets of B cells and leads to suppression of humoral immune response upon activation with environmental pollutants (83, 84). There is emerging evidence that AhR plays a critical role in activation-induced cell fates resulting in class switching of B cells (85). Recently, Matteo et al. have shown that mature B cells express AhR upon ligand-specific activation by AhR translocation to the nucleus resulting in induction of downstream target gene Cyp1A1 (86). Some of tumor-derived metabolites such as 5-lipoxygenase in a breast cancer model and placental growth factor (PIGF) in gliomas have been proposed to promote Breg (87, 88). To date, prior studies have not identified a role for L-Kyn mediated AhR activation in Breg differentiation. Consistent with these prior studies, we put forth new evidence that L-Kyn activates AhR in mature B cell subsets by translocating AhR into their nucleus and helping them differentiate into Breg precursors, and furthermore, TLR-4 signaling transforms them into IL-10 producing Breg.
Immune cell activation by LPS is mainly mediated by TLR-4 (55). TLR ligands, such as LPS and CpG are potent AhR agonists in various cell types (82). Previously TLRs signaling was thought to be directly involved in modulating the regulatory function of B cells (32). Evidences suggest that B cells produce IL-10 upon stimulation with LPS (51, 89). It has been previously reported that TLR-4, TLR-2, and adaptor MyD88 are directly involved in modulating the regulatory function of B cells in a murine model of experimental autoimmune encephalomyelitis (EAE) (32). Our ex-vivo data obtained from TLR-4, TLR-2 and MyD88 deficient B cells are consistent with these prior studies and strongly support our current hypothesis that L-Kyn induces Breg precursors (CD19+CD1dhiCD5+) independent of TLR-2 and TLR-4 signaling but depends on TLR-4-MyD88 pathway to transform them into IL-10 producing Breg cells. Furthermore, our hypothesis is also strongly proven by another ex-vivo data showing LPS (TLR-4 ligand) is required to transform Breg precursors into Breg cells.
We reported earlier that Breg cells were increased in the spleens and lungs of wild type tumor-bearing mice (34). The reduction in percentages of Breg in in the spleen and tumor tissue of IDO-/- mice, that we report here, is not due to overall reduction B cell development or differentiation. Although we and others (58) show that IDO deficiency does not affect B cell development in BM and spleen, IDO negatively regulates B cell proliferation and IgG production in tumor-bearing mice. The functional enhancement of B cells with IDO deficiency is also consistent with the reduction in tumor burden in IDO-/- mice (41).
Thus our current study provides valuable insights into mechanisms underlying Breg differentiation, specifically, regulation by IDO (from MDSCs and tumor cells) and by Trp metabolite L-Kyn via AhR pathway and highlight a potential link between IDO and Breg development in TME. Enhanced tumor protection was achieved in B cells deficient mice, and the anti-tumor immune response was reduced significantly when B cells were adoptively transferred in those murine models of tumor (15, 90). Considering the immunosuppressive effect of B cells, enhanced efficacy of melanoma vaccine has been observed in the absence of B cells (91). Consistent with these prior studies, our recent most data on Breg raises the new possibility of immunotherapeutic intervention for lung cancer.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee the University of Alabama at Birmingham, AL, USA.
Author Contributions
ST, YW, JJ, KH, and JD designed the experiments. ST, YW, JJ, KH, and JS and performed the experiments. ST, YW, and JD prepared figures and supplementary material sections. ST, YW, and JD wrote the manuscript. ST, YW JJ, KH, JS, MA, VT, RM, SP, and JD reviewed and revised the manuscript. All the authors contributed to final data analysis, discussions, and manuscript preparation.
Funding
This study was supported by institutional support through ACS– IRG-60-001-53-IRG.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors wish to acknowledge Dr. John F. Kearney, University of Alabama at Birmingham for giving us AhR-/- mice and Dr. Suzanne M. Michalek, University of Alabama at Birmingham for providing us TLR-2-/-, TLR-4-/- TLR-2, 4-/- and MyD88-/- mice. We thank Dr. Robert H. McCusker, University of Illinois at Urbana-Champaign for giving us MCre and IDO f MCre mice. We acknowledge Joo Hyoung Lee, University of Alabama at Birmingham for assistance in immunoblot analyses. We acknowledge Santosh Shah, University of Alabama at Birmingham for assistance in immuno staining. We thank Marion Spell, Research Core Facility Mgr, Center for AIDS Research, Flow cytometry core, University of Alabama at Birmingham for his assistance and support in immunosorting.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.747780/full#supplementary-material
References
1. Siegel R, Naishadham D, Jemal A. Cancer Statistics, 2013. CA Cancer J Clin (2013) 63(1):11–30. doi: 10.3322/caac.21166
2. Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, et al. Adult-Onset Pulmonary Fibrosis Caused by Mutations in Telomerase. Proc Natl Acad Sci USA (2007) 104(18):7552–7. doi: 10.1073/pnas.0701009104
3. Armanios M. Syndromes of Telomere Shortening. Annu Rev Genomics Hum Genet (2009) 10:45–61. doi: 10.1146/annurev-genom-082908-150046
4. Busch SE, Hanke ML, Kargl J, Metz HE, MacPherson D, Houghton AM. Lung Cancer Subtypes Generate Unique Immune Responses. J Immunol (2016) 197(11):4493–503. doi: 10.4049/jimmunol.1600576
5. Wood SL, Pernemalm M, Crosbie PA, Whetton AD. The Role of the Tumor-Microenvironment in Lung Cancer-Metastasis and Its Relationship to Potential Therapeutic Targets. Cancer Treat Rev (2014) 40(4):558–66. doi: 10.1016/j.ctrv.2013.10.001
6. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific Recruitment of Regulatory T Cells in Ovarian Carcinoma Fosters Immune Privilege and Predicts Reduced Survival. Nat Med (2004) 10(9):942–9. doi: 10.1038/nm1093
7. DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, et al. CD4(+) T Cells Regulate Pulmonary Metastasis of Mammary Carcinomas by Enhancing Protumor Properties of Macrophages. Cancer Cell (2009) 16(2):91–102. doi: 10.1016/j.ccr.2009.06.018
8. de Visser KE, Eichten A, Coussens LM. Paradoxical Roles of the Immune System During Cancer Development. Nat Rev Cancer (2006) 6(1):24–37. doi: 10.1038/nrc1782
9. Schreiber RD, Old LJ, Smyth MJ. Cancer Immunoediting: Integrating Immunity's Roles in Cancer Suppression and Promotion. Science (2011) 331(6024):1565–70. doi: 10.1126/science.1203486
10. Olkhanud PB, Damdinsuren B, Bodogai M, Gress RE, Sen R, Wejksza K, et al. Tumor-Evoked Regulatory B Cells Promote Breast Cancer Metastasis by Converting Resting CD4(+) T Cells to T-Regulatory Cells. Cancer Res (2011) 71(10):3505–15. doi: 10.1158/0008-5472.CAN-10-4316
11. Inoue S, Leitner WW, Golding B, Scott D. Inhibitory Effects of B Cells on Antitumor Immunity. Cancer Res (2006) 66(15):7741–7. doi: 10.1158/0008-5472.CAN-05-3766
12. Fremd C, Schuetz F, Sohn C, Beckhove P, Domschke C. B Cell-Regulated Immune Responses in Tumor Models and Cancer Patients. Oncoimmunology (2013) 2(7):e25443. doi: 10.4161/onci.25443
13. de Visser KE, Korets LV, Coussens LM. De Novo Carcinogenesis Promoted by Chronic Inflammation Is B Lymphocyte Dependent. Cancer Cell (2005) 7(5):411–23. doi: 10.1016/j.ccr.2005.04.014
14. Schioppa T, Moore R, Thompson RG, Rosser EC, Kulbe H, Nedospasov S, et al. B Regulatory Cells and the Tumor-Promoting Actions of TNF-Alpha During Squamous Carcinogenesis. Proc Natl Acad Sci USA (2011) 108(26):10662–7. doi: 10.1073/pnas.1100994108
15. Zhang Y, Eliav Y, Shin SU, Schreiber TH, Podack ER, Tadmor T, et al. B Lymphocyte Inhibition of Anti-Tumor Response Depends on Expansion of Treg But Is Independent of B-Cell IL-10 Secretion. Cancer Immunol Immunother (2013) 62(1):87–99. doi: 10.1007/s00262-012-1313-6
16. Mauri C, Bosma A. Immune Regulatory Function of B Cells. Annu Rev Immunol (2012) 30:221–41. doi: 10.1146/annurev-immunol-020711-074934
17. Zhou J, Min Z, Zhang D, Wang W, Marincola F, Wang X. Enhanced Frequency and Potential Mechanism of B Regulatory Cells in Patients With Lung Cancer. J Transl Med (2014) 12:304. doi: 10.1186/s12967-014-0304-0
18. O'Garra A, Stapleton G, Dhar V, Pearce M, Schumacher J, Rugo H, et al. Production of Cytokines by Mouse B Cells: B Lymphomas and Normal B Cells Produce Interleukin 10. Int Immunol (1990) 2(9):821–32. doi: 10.1093/intimm/2.9.821
19. Couper KN, Blount DG, Riley EM. IL-10: The Master Regulator of Immunity to Infection. J Immunol (2008) 180(9):5771–7. doi: 10.4049/jimmunol.180.9.5771
20. Mizoguchi A, Mizoguchi E, Takedatsu H, Blumberg RS, Bhan AK. Chronic Intestinal Inflammatory Condition Generates IL-10-Producing Regulatory B Cell Subset Characterized by CD1d Upregulation. Immunity (2002) 16(2):219–30. doi: 10.1016/S1074-7613(02)00274-1
21. Mauri C, Gray D, Mushtaq N, Londei M. Prevention of Arthritis by Interleukin 10–Producing B Cells. J Exp Med (2003) 197(4):489–501. doi: 10.1084/jem.20021293
22. Hussain S, Delovitch TL. Intravenous Transfusion of BCR-Activated B Cells Protects NOD Mice From Type 1 Diabetes in an IL-10-Dependent Manner. J Immunol (2007) 179(11):7225–32. doi: 10.4049/jimmunol.179.11.7225
23. Haas KM, Watanabe R, Matsushita T, Nakashima H, Ishiura N, Okochi H, et al. Protective and Pathogenic Roles for B Cells During Systemic Autoimmunity in NZB/W F1 Mice. J Immunol (2010) 184(9):4789–800. doi: 10.4049/jimmunol.0902391
24. Watanabe R, Ishiura N, Nakashima H, Kuwano Y, Okochi H, Tamaki K, et al. Regulatory B Cells (B10 Cells) Have a Suppressive Role in Murine Lupus: CD19 and B10 Cell Deficiency Exacerbates Systemic Autoimmunity. J Immunol (2010) 184(9):4801–9. doi: 10.4049/jimmunol.0902385
25. Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B Cells Regulate Autoimmunity by Provision of IL-10. Nat Immunol (2002) 3(10):944–50. doi: 10.1038/ni833
26. Chapoval AI, Fuller JA, Kremlev SG, Kamdar SJ, Evans R. Combination Chemotherapy and IL-15 Administration Induce Permanent Tumor Regression in a Mouse Lung Tumor Model: NK and T Cell-Mediated Effects Antagonized by B Cells. J Immunol (1998) 161(12):6977–84.
27. Horikawa M, Minard-Colin V, Matsushita T, Tedder TF. Regulatory B Cell Production of IL-10 Inhibits Lymphoma Depletion During CD20 Immunotherapy in Mice. J Clin Invest (2011) 121(11):4268–80. doi: 10.1172/JCI59266
28. Minard-Colin V, Xiu Y, Poe JC, Horikawa M, Magro CM, Hamaguchi Y, et al. Lymphoma Depletion During CD20 Immunotherapy in Mice Is Mediated by Macrophage FcgammaRI, FcgammaRIII, and FcgammaRIV. Blood (2008) 112(4):1205–13. doi: 10.1182/blood-2008-01-135160
29. Vitale G, Mion F, Pucillo C. Regulatory B Cells: Evidence, Developmental Origin and Population Diversity. Mol Immunol (2010) 48(1-3):1–8. doi: 10.1016/j.molimm.2010.09.010
30. Lundy SK. Killer B Lymphocytes: The Evidence and the Potential. Inflammation Res (2009) 58(7):345–57. doi: 10.1007/s00011-009-0014-x
31. Rosser EC, Oleinika K, Tonon S, Doyle R, Bosma A, Carter NA, et al. Regulatory B Cells are Induced by Gut Microbiota-Driven Interleukin-1beta and Interleukin-6 Production. Nat Med (2014) 20(11):1334–9. doi: 10.1038/nm.3680
32. Lampropoulou V, Hoehlig K, Roch T, Neves P, Calderon Gomez E, Sweenie CH, et al. TLR-Activated B Cells Suppress T Cell-Mediated Autoimmunity. J Immunol (2008) 180(7):4763–73. doi: 10.4049/jimmunol.180.7.4763
33. Zhang YLaP. Regulation of TLR9/MyD88/NF-Kappa B Signaling Pathway on Regulatory B Cells (Breg) in Patients With Thymoma and Myasthenia Gravis. Arch Cancer Res (2018). doi: 10.21767/2254-6081-C1-006
34. Wang Y, Schafer CC, Hough KP, Tousif S, Duncan SR, Kearney JF, et al. Myeloid-Derived Suppressor Cells Impair B Cell Responses in Lung Cancer Through IL-7 and STAT5. J Immunol (2018) 201(1):278–95. doi: 10.4049/jimmunol.1701069
35. Gabrilovich DI, Nagaraj S. Myeloid-Derived Suppressor Cells as Regulators of the Immune System. Nat Rev Immunol (2009) 9(3):162–74. doi: 10.1038/nri2506
36. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. J Immunol (2008) 181(8):5791–802. doi: 10.4049/jimmunol.181.8.5791
37. Ostrand-Rosenberg S, Sinha P. Myeloid-Derived Suppressor Cells: Linking Inflammation and Cancer. J Immunol (2009) 182(8):4499–506. doi: 10.4049/jimmunol.0802740
38. Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, et al. Recommendations for Myeloid-Derived Suppressor Cell Nomenclature and Characterization Standards. Nat Commun (2016) 7:12150. doi: 10.1038/ncomms12150
39. Yang R, Cai Z, Zhang Y, t. Yutzy WH, Roby KF, Roden RB. CD80 in Immune Suppression by Mouse Ovarian Carcinoma-Associated Gr-1+CD11b+ Myeloid Cells. Cancer Res (2006) 66(13):6807–15. doi: 10.1158/0008-5472.CAN-05-3755
40. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ Immature Myeloid Suppressor Cells Mediate the Development of Tumor-Induced T Regulatory Cells and T-Cell Anergy in Tumor-Bearing Host. Cancer Res (2006) 66(2):1123–31. doi: 10.1158/0008-5472.CAN-05-1299
41. Schafer CC, Wang Y, Hough KP, Sawant A, Grant SC, Thannickal VJ, et al. Indoleamine 2,3-Dioxygenase Regulates Anti-Tumor Immunity in Lung Cancer by Metabolic Reprogramming of Immune Cells in the Tumor Microenvironment. Oncotarget (2016) 7(46):75407–24. doi: 10.18632/oncotarget.12249
42. Sawant A, Schafer CC, Jin TH, Zmijewski J, Tse HM, Roth J, et al. Enhancement of Antitumor Immunity in Lung Cancer by Targeting Myeloid-Derived Suppressor Cell Pathways. Cancer Res (2013) 73(22):6609–20. doi: 10.1158/0008-5472.CAN-13-0987
43. Zoso A, Mazza EM, Bicciato S, Mandruzzato S, Bronte V, Serafini P, et al. Human Fibrocytic Myeloid-Derived Suppressor Cells Express IDO and Promote Tolerance via Treg-Cell Expansion. Eur J Immunol (2014) 44(11):3307–19. doi: 10.1002/eji.201444522
44. Zhang H, Maric I, DiPrima MJ, Khan J, Orentas RJ, Kaplan RN, et al. Fibrocytes Represent a Novel MDSC Subset Circulating in Patients With Metastatic Cancer. Blood (2013) 122(7):1105–13. doi: 10.1182/blood-2012-08-449413
45. Munn DH. Indoleamine 2,3-Dioxygenase and Tumor-Induced Tolerance. J Clin Invest (2007) 117: (5):1147–54. doi: 10.1172/jci31178
46. Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah AM, Burns EJ, et al. An Endogenous Aryl Hydrocarbon Receptor Ligand Acts on Dendritic Cells and T Cells to Suppress Experimental Autoimmune Encephalomyelitis. Proc Natl Acad Sci USA (2010) 107(48):20768–73. doi: 10.1073/pnas.1009201107
47. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An Endogenous Tumour-Promoting Ligand of the Human Aryl Hydrocarbon Receptor. Nature (2011) 478(7368):197–203. doi: 10.1038/nature10491
48. Özkan B, Lim H, Park SG. Immunomodulatory Function of Myeloid-Derived Suppressor Cells During B Cell-Mediated Immune Responses. Int J Mol Sci (2019) 19(5)1468. doi: 10.3390/ijms19051468
49. Iikuni N, Lourenco EV, Hahn BH, La Cava A. Cutting Edge: Regulatory T Cells Directly Suppress B Cells in Systemic Lupus Erythematosus. J Immunol (2009) 183(3):1518–22. doi: 10.4049/jimmunol.0901163
50. Mauri C, Ehrenstein MR. The 'Short' History of Regulatory B Cells. Trends Immunol (2008) 29(1):34–40. doi: 10.1016/j.it.2007.10.004
51. O'Garra A, Chang R, Go N, Hastings R, Haughton G, Howard M. Ly-1 B (B-1) Cells are the Main Source of B Cell-Derived Interleukin 10. Eur J Immunol (1992) 22(3):711–7. doi: 10.1002/eji.1830220314
52. Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. The Combined Effects of Tryptophan Starvation and Tryptophan Catabolites Down-Regulate T Cell Receptor Zeta-Chain and Induce a Regulatory Phenotype in Naive T Cells. J Immunol (2006) 176(11):6752–61. doi: 10.4049/jimmunol.176.11.6752
53. Ziv-Gal A, Gao L, Karman BN, Flaws JA. In Vitro Re-Expression of the Aryl Hydrocarbon Receptor (Ahr) in Cultured Ahr-Deficient Mouse Antral Follicles Partially Restores the Phenotype to That of Cultured Wild-Type Mouse Follicles. Toxicol In Vitro (2015) 29(2):329–36. doi: 10.1016/j.tiv.2014.11.011
54. Rosser EC, Mauri C. Regulatory B Cells: Origin, Phenotype, and Function. Immunity (2015) 42(4):607–12. doi: 10.1016/j.immuni.2015.04.005
55. Darveau RP, Pham TT, Lemley K, Reife RA, Bainbridge BW, Coats SR, et al. Porphyromonas Gingivalis Lipopolysaccharide Contains Multiple Lipid A Species That Functionally Interact With Both Toll-Like Receptors 2 and 4. Infect Immun (2004) 72(9):5041–51. doi: 10.1128/IAI.72.9.5041-5051.2004
56. Pahl HL. Activators and Target Genes of Rel/NF-kappaB Transcription Factors. Oncogene (1999) 18(49):6853–66. doi: 10.1038/sj.onc.1203239
57. Siewe B, Stapleton JT, Martinson J, Keshavarzian A, Kazmi N, Demarais PM, et al. Regulatory B Cell Frequency Correlates With Markers of HIV Disease Progression and Attenuates Anti-HIV CD8(+) T Cell Function In Vitro. J Leukoc Biol (2013) 93(5):811–8. doi: 10.1189/jlb.0912436
58. Shinde R, Shimoda M, Chaudhary K, Liu H, Mohamed E, Bradley J, et al. B Cell-Intrinsic IDO1 Regulates Humoral Immunity to T Cell-Independent Antigens. J Immunol (2015) 195(5):2374–82. doi: 10.4049/jimmunol.1402854
59. Sucher R, Kurz K, Weiss G, Margreiter R, Fuchs D, Brandacher G. IDO-Mediated Tryptophan Degradation in the Pathogenesis of Malignant Tumor Disease. Int J Tryptophan Res (2010) 3:113–20. doi: 10.4137/ijtr.s4157
60. Yang B, Wang X, Ren X. Amino Acid Metabolism Related to Immune Tolerance by MDSCs. Int Rev Immunol (2012) 31(3):177–83. doi: 10.3109/08830185.2012.679989
61. Zamanakou M, Germenis AE, Karanikas V. Tumor Immune Escape Mediated by Indoleamine 2,3-Dioxygenase. Immunol Lett (2007) 111(2):69–75. doi: 10.1016/j.imlet.2007.06.001
62. Choe JY, Yun JY, Jeon YK, Kim SH, Park G, Huh JR, et al. Indoleamine 2,3-Dioxygenase (IDO) Is Frequently Expressed in Stromal Cells of Hodgkin Lymphoma and Is Associated With Adverse Clinical Features: A Retrospective Cohort Study. BMC Cancer (2014) 14:335. doi: 10.1186/1471-2407-14-335
63. Katz SI, Parker D, Turk JL. B-Cell Suppression of Delayed Hypersensitivity Reactions. Nature (1974) 251(5475):550–1. doi: 10.1038/251550a0
64. Neta R, Salvin SB. Specific Suppression of Delayed Hypersensitivity: The Possible Presence of a Suppressor B Cell in the Regulation of Delayed Hypersensitivity. J Immunol (1974) 113(6):1716–25.
65. Liu J, Wang H, Yu Q, Zheng S, Jiang Y, Liu Y, et al. Aberrant Frequency of IL-10-Producing B Cells and Its Association With Treg and MDSC Cells in Non Small Cell Lung Carcinoma Patients. Hum Immunol (2016) 77(1):84–9. doi: 10.1016/j.humimm.2015.10.015
66. Shao Y, Lo CM, Ling CC, Liu XB, Ng KT, Chu AC, et al. Regulatory B Cells Accelerate Hepatocellular Carcinoma Progression via CD40/CD154 Signaling Pathway. Cancer Lett (2014) 355(2):264–72. doi: 10.1016/j.canlet.2014.09.026
67. Lindner S, Dahlke K, Sontheimer K, Hagn M, Kaltenmeier C, Barth TF, et al. Interleukin 21-Induced Granzyme B-Expressing B Cells Infiltrate Tumors and Regulate T Cells. Cancer Res (2013) 73(8):2468–79. doi: 10.1158/0008-5472.CAN-12-3450
68. Qian L, Bian GR, Zhou Y, Wang Y, Hu J, Liu X, et al. Clinical Significance of Regulatory B Cells in the Peripheral Blood of Patients With Oesophageal Cancer. Cent Eur J Immunol (2015) 40(2):263–5. doi: 10.5114/ceji.2015.52840
69. Zhang C, Xin H, Zhang W, Yazaki PJ, Zhang Z, Le K, et al. CD5 Binds to Interleukin-6 and Induces a Feed-Forward Loop With the Transcription Factor STAT3 in B Cells to Promote Cancer. Immunity (2016) 44(4):913–23. doi: 10.1016/j.immuni.2016.04.003
70. Maseda D, Candando KM, Smith SH, Kalampokis I, Weaver CT, Plevy SE, et al. Peritoneal Cavity Regulatory B Cells (B10 Cells) Modulate IFN-Gamma+CD4+ T Cell Numbers During Colitis Development in Mice. J Immunol (2013) 191(5):2780–95. doi: 10.4049/jimmunol.1300649
71. Zhou X, Su YX, Lao XM, Liang YJ, Liao GQ. CD19(+)IL-10(+) Regulatory B Cells Affect Survival of Tongue Squamous Cell Carcinoma Patients and Induce Resting CD4(+) T Cells to CD4(+)Foxp3(+) Regulatory T Cells. Oral Oncol (2016) 53:27–35. doi: 10.1016/j.oraloncology.2015.11.003
72. Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a Tumoral Immune Resistance Mechanism Based on Tryptophan Degradation by Indoleamine 2,3-Dioxygenase. Nat Med (2003) 9(10):1269–74. doi: 10.1038/nm934
73. Lob S, Konigsrainer A, Zieker D, Brucher BL, Rammensee HG, Opelz G, et al. IDO1 and IDO2 are Expressed in Human Tumors: Levo- But Not Dextro-1-Methyl Tryptophan Inhibits Tryptophan Catabolism. Cancer Immunol Immunother (2009) 58(1):153–7. doi: 10.1007/s00262-008-0513-6
74. Rosser EC, Blair PA, Mauri C. Cellular Targets of Regulatory B Cell-Mediated Suppression. Mol Immunol (2014) 62(2):296–304. doi: 10.1016/j.molimm.2014.01.014
75. Gray M, Miles K, Salter D, Gray D, Savill J. Apoptotic Cells Protect Mice From Autoimmune Inflammation by the Induction of Regulatory B Cells. Proc Natl Acad Sci USA (2007) 104(35):14080–5. doi: 10.1073/pnas.0700326104
76. Lim HW, Hillsamer P, Banham AH, Kim CH. Cutting Edge: Direct Suppression of B Cells by CD4+ CD25+ Regulatory T Cells. J Immunol (2005) 175(7):4180–3. doi: 10.4049/jimmunol.175.7.4180
77. Park MJ, Lee SH, Kim EK, Lee EJ, Park SH, Kwok SK, et al. Myeloid-Derived Suppressor Cells Induce the Expansion of Regulatory B Cells and Ameliorate Autoimmunity in the Sanroque Mouse Model of Systemic Lupus Erythematosus. Arthritis Rheumatol (2016) 68(11):2717–27. doi: 10.1002/art.39767
78. Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ. Kynurenines In The Mammalian Brain: When Physiology Meets Pathology. Nat Rev Neurosci (2012) 13(7):465–77. doi: 10.1038/nrn3257
79. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An Interaction Between Kynurenine and the Aryl Hydrocarbon Receptor can Generate Regulatory T Cells. J Immunol (2010) 185(6):3190–8. doi: 10.4049/jimmunol.0903670
80. Ravishankar B, Liu H, Shinde R, Chaudhary K, Xiao W, Bradley J, et al. The Amino Acid Sensor GCN2 Inhibits Inflammatory Responses to Apoptotic Cells Promoting Tolerance and Suppressing Systemic Autoimmunity. Proc Natl Acad Sci USA (2015) 112(34):10774–9. doi: 10.1073/pnas.1504276112
81. Jux B, Kadow S, Esser C. Langerhans Cell Maturation and Contact Hypersensitivity are Impaired in Aryl Hydrocarbon Receptor-Null Mice. J Immunol (2009) 182(11):6709–17. doi: 10.4049/jimmunol.0713344
82. Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl Hydrocarbon Receptor Negatively Regulates Dendritic Cell Immunogenicity via a Kynurenine-Dependent Mechanism. Proc Natl Acad Sci USA (2010) 107(46):19961–6. doi: 10.1073/pnas.1014465107
83. Marcus RS, Holsapple MP, Kaminski NE. Lipopolysaccharide Activation of Murine Splenocytes and Splenic B Cells Increased the Expression of Aryl Hydrocarbon Receptor and Aryl Hydrocarbon Receptor Nuclear Translocator. J Pharmacol Exp Ther (1998) 287(3):1113–8.
84. Kerkvliet NI, Steppan LB, Brauner JA, Deyo JA, Henderson MC, Tomar RS, et al. Influence of the Ah Locus on the Humoral Immunotoxicity of 2,3,7,8-Tetrachlorodibenzo-P-Dioxin: Evidence for Ah-Receptor-Dependent and Ah-Receptor-Independent Mechanisms of Immunosuppression. Toxicol Appl Pharmacol (1990) 105(1):26–36. doi: 10.1016/0041-008X(90)90356-Y
85. Vaidyanathan B, Chaudhry A, Yewdell WT, Angeletti D, Yen W-F, Wheatley AK, et al. The Aryl Hydrocarbon Receptor Controls Cell-Fate Decisions in B Cells. J Exp Med (2016) 85:197–208. doi: 10.1084/jem.20160789
86. Villa M, Gialitakis M, Tolaini M, Ahlfors H, Henderson CJ, Wolf CR, et al. Aryl Hydrocarbon Receptor Is Required for Optimal B-Cell Proliferation. EMBO J (2017) 36(1):116–28. doi: 10.15252/embj.201695027
87. Wejksza K, Lee-Chang C, Bodogai M, Bonzo J, Gonzalez FJ, Lehrmann E, et al. Cancer-Produced Metabolites of 5-Lipoxygenase Induce Tumor-Evoked Regulatory B Cells via Peroxisome Proliferator-Activated Receptor Alpha. J Immunol (2013) 190(6):2575–84. doi: 10.4049/jimmunol.1201920
88. Han S, Feng S, Ren M, Ma E, Wang X, Xu L, et al. Glioma Cell-Derived Placental Growth Factor Induces Regulatory B Cells. Int J Biochem Cell Biol (2014) 57:63–8. doi: 10.1016/j.biocel.2014.10.005
89. Burger C, Vitetta ES. The Response of B Cells in Spleen, Peyer's Patches, and Lymph Nodes to LPS and IL-4. Cell Immunol (1991) 138(1):35–43. doi: 10.1016/0008-8749(91)90130-4
90. Shah S, Divekar AA, Hilchey SP, Cho HM, Newman CL, Shin SU, et al. Increased Rejection of Primary Tumors in Mice Lacking B Cells: Inhibition of Anti-Tumor CTL and TH1 Cytokine Responses by B Cells. Int J Cancer (2005) 117(4):574–86. doi: 10.1002/ijc.21177
Keywords: IDO, L-Kynurenine, Breg cells, MDSC (myeloid-derived suppressor cells), TME (tumor microenvironment), immunosuppression, lung cancer, aryl hydrocarbon receptor (AhR)
Citation: Tousif S, Wang Y, Jackson J, Hough KP, Strenkowski JG, Athar M, Thannickal VJ, McCusker RH, Ponnazhagan S and Deshane JS (2021) Indoleamine 2, 3-Dioxygenase Promotes Aryl Hydrocarbon Receptor-Dependent Differentiation Of Regulatory B Cells in Lung Cancer. Front. Immunol. 12:747780. doi: 10.3389/fimmu.2021.747780
Received: 26 July 2021; Accepted: 02 November 2021;
Published: 19 November 2021.
Edited by:
Elisa Wirthgen, Rostock University Medical Center, GermanyReviewed by:
Masood A. Shammas, Dana–Farber Cancer Institute, United StatesSatoshi Ueha, Tokyo University of Science, Japan
Ollie Haworth, University of Westminster, United Kingdom
Copyright © 2021 Tousif, Wang, Jackson, Hough, Strenkowski, Athar, Thannickal, McCusker, Ponnazhagan and Deshane. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jessy S. Deshane, amVzc3lkZXNoYW5lQHVhYm1jLmVkdQ==