 Sandrine Luce1,2
Sandrine Luce1,2 Sophie Guinoiseau1,2
Sophie Guinoiseau1,2 Alexis Gadault1,2
Alexis Gadault1,2 Franck Letourneur1
Franck Letourneur1 Patrick Nitschke2Marc Bras2Michel Vidaud3Pierre Charneau4
Patrick Nitschke2Marc Bras2Michel Vidaud3Pierre Charneau4 Etienne Larger1,5
Etienne Larger1,5 Maikel L. Colli6
Maikel L. Colli6 Decio L. Eizirik6,7François Lemonnier1,2Christian Boitard1,2,5*
Decio L. Eizirik6,7François Lemonnier1,2Christian Boitard1,2,5*- 1Laboratory Immunology of Diabetes, INSERMU1016, Department EMD, Cochin Institute, Paris, France
- 2Medical Faculty, Paris University, Paris, France
- 3Biochemistry and Molecular Genetics Department, Cochin Hospital, Paris, France
- 4Molecular Virology and Vaccinology, Pasteur Institute, Paris, France
- 5Diabetology Department, Cochin Hospital, Paris, France
- 6Université Libre de Bruxelles (ULB) Center for Diabetes Research, Medical Faculty, Université Libre de Bruxelles, Brussels, Belgium
- 7Diabetes Center, Indiana Biosciences Research Institute (IBRI), Indianapolis, IN, United States
To circumvent the limitations of available preclinical models for the study of type 1 diabetes (T1D), we developed a new humanized model, the YES-RIP-hB7.1 mouse. This mouse is deficient of murine major histocompatibility complex class I and class II, the murine insulin genes, and expresses as transgenes the HLA-A*02:01 allele, the diabetes high-susceptibility HLA-DQ8A and B alleles, the human insulin gene, and the human co-stimulatory molecule B7.1 in insulin-secreting cells. It develops spontaneous T1D along with CD4+ and CD8+ T-cell responses to human preproinsulin epitopes. Most of the responses identified in these mice were validated in T1D patients. This model is amenable to characterization of hPPI-specific epitopes involved in T1D and to the identification of factors that may trigger autoimmune response to insulin-secreting cells in human T1D. It will allow evaluating peptide-based immunotherapy that may directly apply to T1D in human and complete preclinical model availability to address the issue of clinical heterogeneity of human disease.
Introduction
Type 1 diabetes (T1D) is a multifactorial autoimmune disease that remains a major health challenge (1). Its incidence increases by 3% to 4% yearly. There is presently no therapy to definitively revert or stop the autoimmune process responsible for the destruction of β cells (2). Upstream of therapies, immunological markers for the autoimmune response to β cells have shown limitations in predicting the development of type 1 diabetes in subjects with prediabetes, especially when a single autoantibody is detected (3). Following their use in preclinical models of T1D, antigen and peptide-specific immunotherapies have been proposed as strategies with a low risk/benefit ratio in human. Early attempts have shown minimal efficacy in human, as in using glutamic acid decarboxylase-65 (GAD65) (4). However, the use of an immunodominant proinsulin peptide has proven to be well-tolerated and to delay C-peptide decline in human (5). From both a diagnostic and a therapeutic standpoint, preclinical models of T1D have fallen short of translating into human. Current models do not allow testing peptides derived from human autoantigens that may directly apply to the human situation in vaccination strategies. A new preclinical model to study T1D in a humanized mouse model would be amenable to evaluate the relevance of T-cell assays or peptide immunotherapy that would directly apply to human diabetes.
Our aim in this study was to create a preclinical model that would develop spontaneous T1D and allow characterizing HLA-A class I and class II MHC-restricted peptides that directly apply to human. We chose human preproinsulin (hPPI) as a major T1D autoantigen (6–8), the most common class I MHC HLA-A*02:01 allele in the three major ethnic groups (50% in Caucasian and Asian and 30% in African) (9) and the high T1D susceptibility class II DQ8 A and B alleles (10). We previously generated the YES mouse that expresses the HLA-A*02:01, the HLA-DQ8, and the human insulin (hINS) genes and fails to develop spontaneous T1D, but develops T1D when challenged with polyinosonic-polycytidylic acid (pI:C) (11). As the expression of B7.1 in pancreatic β cells has been shown to trigger the development of T1D in conventional mice (12) and accelerate diabetes in the NOD mouse (13), we introduced the human costimulatory molecule B7.1 (hB7.1) under the control of the rat insulin promoter in pancreatic β cells onto the YES background to enforce the development of spontaneous T1D. The first objective was to identify autoreactive epitopes from hPPI involved in the T1D autoimmune response and evaluate their relevance in human. The second objective was to identify external factors that may accelerate the development of T1D within the frame of human MHC presenting molecules (14).
This new humanized mouse expressing HLA-A*02:01, HLA-DQ8, hINS, and the RIP-hB7.1 transgenes, thereafter called YES-RIP-hB7.1 mice, develops spontaneous T1D and shares immunological features with human T1D. This new model of spontaneous diabetes completes the previously reported YES model. According to the clinical complexity of human T1D that is likely to be a heterogeneous set of diseases, these new models altogether provide a larger set of preclinical tools to study human T1D.
Materials and Methods
Patients
Patients (Table 1) were recruited under a human protocol of clinical trial for the access of T1D and healthy donor peripheral blood mononuclear cells (PBMCs) in agreement with the Research Ministry Authorization (MESR number DC-2015-2536/IDRCB number 2015-A01875-44). Recent-onset T1D patients (18 males, 12 females, aged 35.3 ± 17.5 years) were studied within 3 months from diagnosis (9.4 ± 14.5 days, range 1–75 days). HLA genotyping was performed by AmbiSolv genotyping (Dynal/Invitrogen) on DNA with Gentra Puregene Blood kit A (Qiagen). Eleven recent-onset T1D patients expressed HLA-DQ8 (DQA*03:01, DQB*03:02) and 19 co-expressed HLA-DQ8 and HLA-DQ2 (DQA*05:01, DQB*02:01). Long-standing HLA-DQ8-expressing T1D patients (3 males, 3 females, ages 55.25 ± 21.93 years) had a median of diabetes duration of 5.34 ± 2.6 years (range 0.7 to 7 years). Control subjects were healthy donor (n = 9, 3 males, 6 females, aged 34 ± 18.6 years) and T2D controls (n = 4, 1 male, 3 females, aged 67.7 ± 4.57 years) and only express HLA-DQ8 molecule.
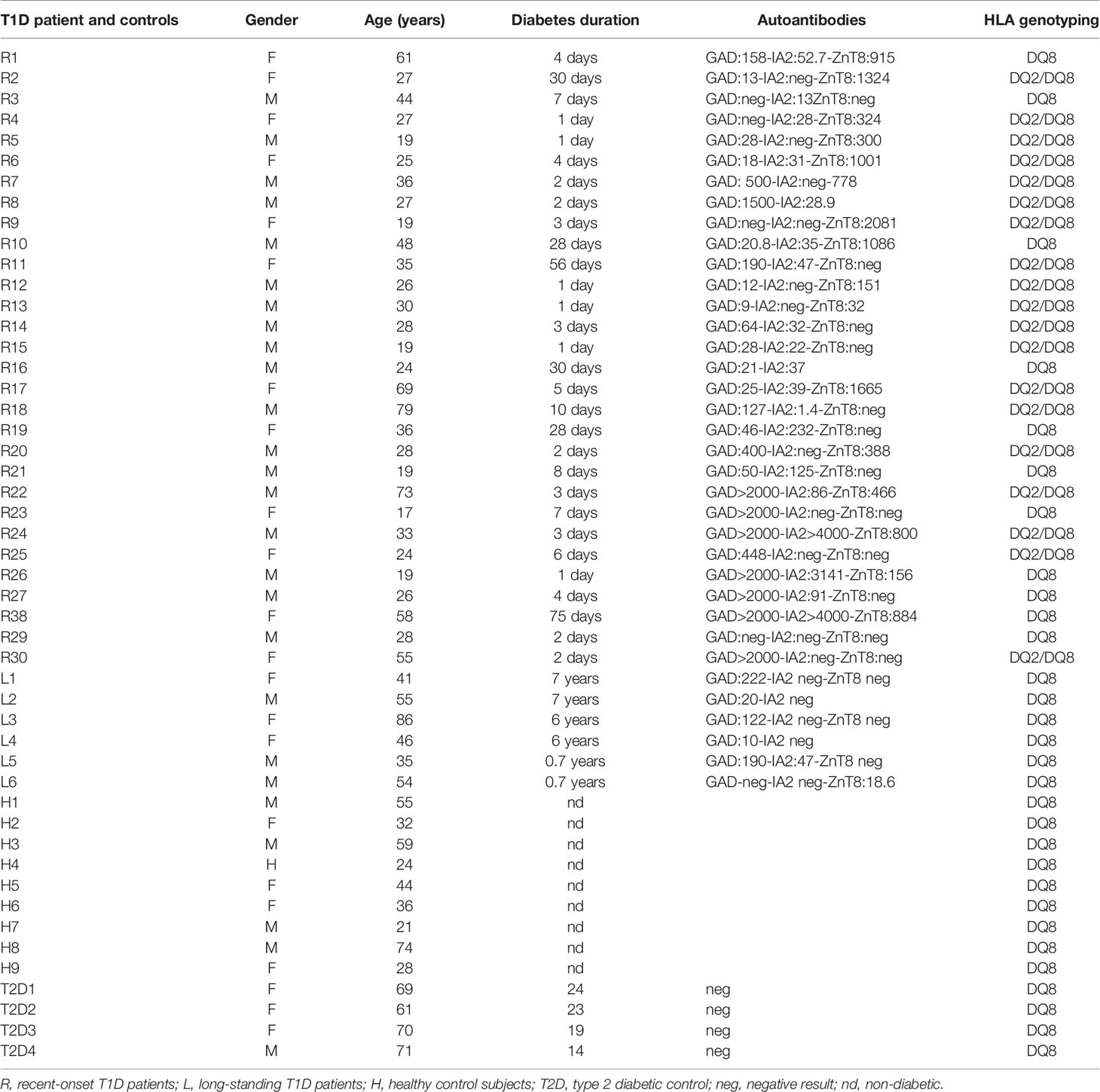
Table 1 Patients and healthy donors.
Mice
YES-RIP-hB7.1 mice were obtained by lentiviral transgenesis of YES mice (H-2 Db, mouse β2 microglobulin, IAαb, βb, IEβb quintuple KO mice expressing a chimeric-α3 H-2 Db domain, human β2 microglobulin-HLA-A*02:01 monochain molecule named HHD- and the HLA-DQB1*03.02 and HLA-DQA1*03.01 genes) (15, 16). The RIP-hB7.1 transgene was inserted in a HIV-derived recombinant lentiviral vector (LV-RIP-hB7.1; insert of 4,640 bp) as previously reported into YES mice (17, 18). Retroviral pseudotypes were injected into fertilized eggs obtained from super-ovulated female YES mice mated with male YES mice (16). Fertile and transduced eggs were reimplanted into pseudo-pregnant C57BL/6xCBA F1 mice. All mice were maintained under specific pathogen-free conditions, and experimental studies were performed in accordance with the Institutional Animal Care and Use Guidelines and accredited by the Ethics Committee n°34 of Paris Descartes under number CEEA34.CB.024.11. Mice were monitored three times a week for glycosuria. When glycosuria was detected, diabetes was diagnosed when two successive glycemic values >250 mg/dl were detected at 24 h interval. Diabetes incidences curves correspond to the percentage of mice diagnosed as diabetic referring to the aforementioned criteria.
Molecular Characterization of LV-RIP-hB7.1 Transgene
YES mice submitted to lentiviral transgenesis were genotyped using the following primers: 5′end (AGGGAACATCACCATCCAAG) and 3′end (TGCCAGTAGATGCGAGTTTG), annealing temperature: 62°C, amplicon: 181 bp. A positive RIP-hB7.1 mouse was selected to perform the molecular characterization of the LV-RIP-hB7.1 transgene. To characterize insertion sites in genomic DNA of the founder, we realized a sequence capture design using the SeqCap EZ system (Roche NimbleGen) targeting 97.6% of the transgene. After nebulization, fragmented genomic DNA was end-repaired and ligated with adapters. A double capture was realized to obtain a GS Junior library ready for emulsion PCR (emPCR). Fragments were then annealed to capture beads and clonally amplified by emPCR (emPCR Amplification Method Manual LibL; GS Junior Titanium Series, Roche). Beads with the cloned amplicons were then enriched, loaded on a 454 picotiter plate, and sequenced on the Roche GS Junior Sequencer according to the protocol of the manufacturer (Sequencing Method Manual, GS Junior Titanium Series, Roche). Image analysis and base calling of the raw sequencing data were performed using the default “shotgun” Roche GS Junior data analysis pipeline. To obtain the position of transgene insertions in genomic DNA, sequence reads were aligned to the 50 bp of each end of the transgene primary sequence using BWASW, an algorithm designed for long reads with more errors. Aligned sequences were then aligned against C57BL/6NJ genomic sequences to obtain scaffold at both ends of the transgene sequence. A 10 kb for each region was extracted and amplified by long range PCR (LR-PCR). A nested PCR targeting the hB7.1 transgene was performed on the long range PCR product to highlight the presence of the transgene and identify the sequence of the insertion site of the LV-RIP-hB7.1 transgene on genomic DNA. A genotyping PCR was designed from the region overlapping genomic DNA and LV-RIP-hB7.1 transgene using the following primers: 5′end (TAAATGCAGGGCTCCAGACT) and 3′end (TAGTGTGTGCCCGTCTGTTG), annealing temperature: 62°C, amplicon: 645 bp.
To compare the genetic background of YES-RIP-hB7.1 and YES mice, we performed a GenScan SNP Affymetrix. Briefly, high-quality genomic DNAs (250 ng) were digested with NspI and StyI enzymes. NspI and StyI adaptors were then ligated to restricted fragments followed by PCR using the universal primer PCR002. Each amplicon was purified, pooled, and used for fragmentation and end-labeling with biotin using terminal transferase. Labeled targets were then hybridized overnight to Genechip® Mouse Diversity Genotyping Array (Affymetrix, ref: 901615) at 49°C. Chips were washed on the fluidic station FS450 following specific Affymetrix protocols, scanned with the GCS3000 7G, and analyzed using GCOS software to obtain raw data (.cel files). Genotypes were identified by the Affymetrix Genotyping Console tool using Dynamic Model (DM) and Bayesian Robust Linear Model with Mahalanobis (BRLMM) mapping algorithms. Genotypes were then extracted for each SNP of the chip. Big data were exploited in R console software. We submitted the data generated by the Affymetrix SNP Array detection to a public repository (http://www.ncbi.nlm.nih.gov/geo) and the GEO Series accession numbers were GSE101551 and GSE151644.
In Vivo T-Cell Depletion Treatment and pI:C Stimulation
For in vivo T-cell depletion, YES-RIP-hB7.1 mice were injected with four weekly injections (i.p.) of 1 mg of anti-CD4 (GK1.5 clone) (19) and 1 mg of anti-CD8 (YTS 169.4 clone) (20) starting at 2 or 8 weeks of age and monitored for diabetes development. For in vivo poly(I:C) stimulation, YES and YES-RIP-hB7.1 mice (8 weeks old of age) were submitted to seven daily injections (i.p.) of 100 µg of poly(I:C) VacciGrade (vac-pIC, Invivogen) and monitored for diabetes development (16).
Cytotoxic Assay
T-cell cytotoxicity assay was performed on HHD-transfected P815 cells prepulsed with 10 μg peptide for 2 h at 37°C using the LDH Cytotoxicity Detection KitPLUS (Roche) or hPPI/HHD-doubled transfected P815 cells. High control of lysis corresponded to cell killing with a lysis solution (Tween-20), which provide the maximum LDH release. Low control of lysis, which provide the spontaneous cytotoxicity, was evaluated on cells that were not submitted to additional treatment. Specific lysis was calculated as optical density of (targeted condition − spontaneous lysis)/(maximum LDH release − spontaneous lysis) × 100.
Enzyme-Linked Immunospot Assay
IFNγ-ELISpot assays were achieved as previously reported (21). Spots were counted using Bioreader 5000 Pro Sf (BioSys GmbH). Splenocytes from mice were stimulated overnight with peptides and Il-2 (5 U/ml final). The enzyme-linked immunospot (ELISpot) assay was performed using mouse γ-ELISpot antibody pair, from U-CyTech biosciences. Data are the mean of triplicate wells. The background of IFNγ responses was evaluated in the absence of peptide. Specific IFNγ responses were expressed as spot-forming cells (SFC) per 106 cells after normalization of the background. Positive controls were triplicates of cells stimulated by 1 μg/ml ConA (Sigma-Aldrich) and negative controls by pyruvate dehydrogenase (PDHase208-216) irrelevant peptide.
Human Recombinant-PPI Protein Production
The cDNA sequence of preproinsulin was mutated to convert the Ala codon in position 3 of the leader DNA sequence to Asp by site-directed mutagenesis (NEB) to abolish the signal-sequence site cleavage of PPI (22). Mutated PPI was cloned in pFastBac vector to generate the PPI-recombinant bacmid, then PPI-recombinant Baculovirus to produce human PPI protein into Bac-to-Bac Baculovirus Expression System (Invitrogen) according to the instruction of the manufacturer. Purification was performed on pelleted infected insect cells, following preparation and extraction procedures for insoluble proteins (inclusion body) from Escherichia coli (23). Purity was confirmed on SDS/PAGE gel and quantification was performed using the BCA Protein assay kit (Pierce).
Cell Proliferation Assay
Spleen cells (105/well) were incubated with 0.5 μg antigen/well or 0.1 µg peptide/well for 72 h at 37°C in triplicate. Proliferation was evaluated with BrdU Cell Proliferation Assay Kit (Cell Signaling) and expressed as proliferation index (PI). Background and positive controls were evaluated in triplicate wells containing 105 cells/well incubated without antigen or in the presence of 10 μg/ml final concentration of anti-mouse CD3ϵ antibody, respectively.
Immunohistochemistry and Fluorescence
Immunofluorescence stainings were performed on formalin-fixed paraffin pancreas sections that were deparaffinized in xylene and dehydrated by ethanol. After washing, antigen retrieval was realized by hot incubation in citrate buffer, followed by permeabilization (20 min in PBS 1×/0.4% Triton X-100) and saturation (30 min PBS 1×/1% horse serum) before immunostaining with biotinylated rat anti-human CD3ε(AbD Serotec) and polyclonal rabbit anti-glucagon (DAKO) antibodies overnight. Slides were washed with PBS 1×/1% BSA/0.1% Triton X-100 and stained with an anti-rabbit Ig-FITC antibody (Abcam) and SAV-Cy3 (Abcam) at RT. Sections were mounted in Vectashield mounting medium for fluorescence with DAPI (Vector Laboratories). Observations were made with a spinning disk confocal apparatus at the Cochin Institute Imaging Platform and pictures analyzed with the ImageJ software.
Stable immunohistochemical stainings were performed on paraffin-embedded pancreas sections and stained with polyclonal guinea-pig anti-human insulin (DAKO) or rat anti-mouse CD4 biotin (eBioscience), followed by incubation with peroxidase-labeled antibodies. All reactions were revealed with diaminobenzidine (DAB, Genemed). Sections were counterstained with hematoxylin and mounted. Observations were made with Zeiss AxioObserver Z1 microscope coupled with MRm Axiocam Zeiss and pictures analyzed with the ImageJ software.
Assessment of total β-cell mass was performed on scan-stained microscope slides with Inform software using a guinea pig anti-human insulin antibody (DAKO) as the ratio between β-cell surface (µm2)/pancreas surface (µm2) multiplied by pancreas weight (mg) (24). Assessment of total α-cell mass was performed on scan-stained microscope slides with Inform software using a rabbit anti-glucagon antibody (DAKO) as the ratio between α-cell surface (µm2)/pancreas surface (µm2) multiplied by pancreas weight (mg). Quantification of insulin-positive or glucagon-positive areas was performed from the entire pancreas on serially cut 8-µm-thick sections. Five to 10 pancreatic sections were processed for immunostaining. Cellular mass obtained for each group was compared using the non-parametric Mann–Whitney statistical test.
Tetramer Assays and Single-Cell RT-PCR
Tetramers (TMrs) associating the chimeric HLA-A*02:01-H-2Kb and the human β2-microglobulin to hPPI peptides were obtained from the NIH Tetramer Core Facility. Stainings were performed as previously reported (11). Incubation of cells with TMrs was performed for 30 min at RT. The following antibodies were used: anti-mouse CD3ε-AlexaFluo700, anti-mouse CD8α-APC, anti-mouse CD19-PerCP-Cy5, and anti-mouse CD14-PercP-Cy5 antibodies (BD/Pharmingen and eBioscience). Cells were analyzed with a BD LSR-Fortessa flow cytometer and the FlowJo 9.2 software (Tree Star Inc.). For single-cell RT-PCR assays, CD8+/TMr+ T cells were sorted with a FACSAria II at 1 cell/well into 96-well PCR plates containing 5 μl PBS 1× treated with diethyl pyrocarbonate (DEPC) (Sigma) and immediately frozen at −80°C. Single-cell RT-PCR was performed as previously described (11). Perforin (prf1), granzyme A (gzma), granzyme B (gzmb), Fas ligand (tnfsf6), IFNγ (ifng), TGFβ (tgfb), TGFβ R1 (tgfbr1), TGFβ R2 (tgfbr2), TGFβ R3 (tgfbr3), TNFα (lta), IL2 (il2), IL7R (il7r), IL10Rα (il10ra), IL15 (il15), IL15R (il15r), IL21 (il21), CCR7 (ccr7), and CD3ϵ (cd3e) mRNAs were analyzed (Table S15). Naive and memory CD8+/TMr+ T cells were differentiated by the Foxo1 expression using 5′ (GGA CAG CCG CGC AAG ACC AG) and 3′ (ACT GTT GTT GTC CAT GGA CGC) first PCR primers and 5′ (ATC ACC AAG GCC ATC GAG AGC) and 3′ (TTG AAT TCT TCC AGC CCG CCG A) second PCR primers.
Islet Isolation Procedure
Islets of Langerhans were isolated as described previously (25). Islets were dissociated with Cell Dissociation Solution Non-Enzymatic (Sigma) for 30 min at RT. Cells were filtered and stained with anti-HLA-DQ-FITC (BD Biosciences), anti-human β2m-PE (BD Biosciences), guinea pig anti-human insulin (DAKO), anti-guinea pig Ig-Biot and SAV-PECy7, anti-mouse CD4-AlexaFluo700, anti-mouse CD8α-APC, anti-mouse CD19-Percp-Cy5.5, and/or anti-mouse TCRβ-efluo450 antibodies (eBioscience). Acquisition was done with a BD FACSAria flow cytometer and analyzed using FlowJo 10.7.1 software.
Islet Infiltrate Staining
Handpicked islets were pooled in 24-well plates for 24 h in RPMI supplemented with 10% FCS, penicillin–streptomycin, and 5.10−5 M β-mercaptoethanol allowing extrusion of infiltrating lymphocytes (25). Infiltrating lymphocytes were recovered. Cells recovered were used for the analysis of T cells and antigen-presenting cells. T-cell analysis was performed using the Foxp3 intra-staining buffer set according to the recommendation of the manufacturer (eBioscience) with the following combination: anti-mouse CD45-efluo450, anti-mouse CD3ϵ-AlexaFluo700, anti-mouse B220-HorizonViolet500, anti-mouse CD8α-Percp-Cy5.5, anti-mouse CD4-PE, anti-mouse Foxp3-APC, and anti-mouse CD25-BrillantViolet711 mAbs (eBioscience). β Cells and dendritic cells were analyzed with anti-mouse CD45-AlexaFluo700, anti-mouse TCRβ-APC-Cy7, anti-mouse B220-HorizonViolet500, anti-mouse CD11b-efluor450, anti-mouse CD11c-APC, anti-mouse CD8α-BrillantViolet605, and anti-mouse CD4-PE mAbs (eBioscience). Acquisitions were performed with a BD LSR-Fortessa flow cytometer and analyzed using FlowJo 10.7.1 software.
Statistics
The biostatistic method used to compare diabetes incidences between different groups of mice was the log-rank Mantel–Cox test. Comparison of distribution scores between the different mouse strains used the non-parametric Mann–Whitney test. T-cell reactivity was compared using non-parametric Mann–Whitney test and non-parametric Kruskal–Wallis test. ns, non-significant, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. For T-cell proliferation assays, we used the Bland and Altman test to determine the threshold value for a global reliability statistic for significant proliferation response.
Study Approval
Experimental studies were performed in accordance with the Institutional Animal Care and Use Guidelines and accredited by the ethics committee under number CEEA34.CB.024.11. Clinical trial: MESR number DC-2015-2536/IDRCB number 2015-A01875-44.
Accession Numbers
We submitted the data generated by the Affymetrix SNP Array detection to a public repository (http://www.ncbi.nlm.nih.gov/geo) and the GEO Series accession numbers were GSE101551 and GSE151644.
Results
Characterization of the YES-RIP-hB7.1 Mice
In order to enforce the development of T1D in YES mice, we introduced the hB7.1 gene under the control of the rat insulin gene promoter (RIP) using the LV-RIP-hB7.1 vector (17). We selected three mice that were positive for hB7.1 for backcrossing onto the YES background. Throughout the crosses, one founder progeny was selected. To further stabilize its lineage, we realized a NimbleGen Sequence Capture of the RIP-hB7.1 transgene of the founder and one of its offspring. Inserted hB7.1 sequences were identified as indicated in Table S2, matching with regions located on chr. 11, 14, 16, and 19 in the founder mouse. Nevertheless, the only region located in chr.19 remained detectable in the offspring, a chromosome with no Idd known to associate with TID. We did not evidence hot spots of enhancer transcripts close to the LV-RIP-hB7.1 insertions using the mouse ENCODE database from UCSC Genome Bioinformatics (GSE101551 and GSE151644 GEO data accession numbers). We screened 128 mice from the progeny for the selected insertion sites in order to stabilize the lineage at a homozygote status using a classical progeny test.
YES-RIP-hB7.1 Mice Develop Spontaneous Type 1 Diabetes
Insulitis and spontaneous diabetes developed in founder YES-RIP-hB7.1 offspring that were submitted to 20 brother–sister mattings. In the founder progeny, 46 out of 128 YES-RIP-hB7.1 mice developed diabetes, while diabetes was not observed in non-transgenic YES mice. Diabetes incidence of stabilized YES-RIP-hB7.1 mice is shown in Figure 1A. Age at onset varied from one mouse to another, spanning from 9 to 51 weeks of age. The overall prevalence of diabetes was similar in female (53.8%) and male (50.0%) YES-RIP-hB7.1 mice. The difference in diabetic incidence was not statistically different between males and females. The average glycemia in YES-RIP-hB7.1 mice when diagnosed as diabetic was 520 ± 115 and 532 ± 99 mg/dl in female and male, respectively. The average glycemia in non-diabetic YES-RIP-hB7.1 mice at the end of the experiment (40 weeks) was 143 ± 35 and 125 ± 18 mg/dl in female and male, respectively. The average glycemia in YES mice was 107 ± 13 and 102 ± 12 mg/dl in female and male, respectively. In order to address whether diabetes was immune-related, we analyzed hematoxylin–eosin-stained pancreas paraffin sections from diabetic and non-diabetic YES-RIP-hB7.1 mice. As shown in Figure 1B, insulitis was detected by immunofluorescence staining using an anti-CD3ϵ antibody and a rabbit anti-glucagon antibody to locate remnant islets, showing the infiltration of islets by CD3+ T lymphocytes (Figure 1C). Glucagon-positive cells were dispersed in remnant islets that showed a dislocated architecture in YES-RIP-hB7.1 diabetic mice (Figure 1D) compared with the YES mice control (Figure 1E). Stable immunohistochemistry staining with an anti-CD4 antibody confirmed the detection of CD4+ T cells within the infiltrate (Figure 1F). As shown in Figure 2, the number of islets expressing insulin was decreased by 70.9% in diabetic YES-RIP-hB7.1 mice as compared with YES mice or non-diabetic YES-RIP-hB7.1 mice. Islet size (Figure 2B) was decreased by over 60% in diabetic YES-RIP-hB7.1 mice as compared with YES controls. A dramatic decrease of the β-cell mass was observed in diabetics YES-RIP-hB7.1 mice as compared with age-matched YES mice (0.60 ± 0.49 versus 3.93 ± 0.65, respectively, p ≤ 0.02) and non-diabetic YES-RIP-hB7.1 mice (7.01 ± 2.06) (Figure 2C). The α-cell mass (Figure 2D) also showed a significant decrease in diabetic YES-RIP-hB7.1 mice as compared with non-diabetic YES-RIP-hB7.1 mice and YES controls (0.60 ± 0.49 versus 3.93 ± 0.65, respectively, p ≤ 0.02). In non-diabetic YES-RIP-hB7.1 mice, the α-cell mass was heterogeneous (7.01 ± 2.06). Infiltrates from three diabetic mice were recovered and pooled to be analyzed (Figure S1). They were composed of 52% T cells, among which 81% were CD8+ T cells, 8% were CD4+ T cells, 21% were β cells, 0.8% were dendritic cells, and 0.6% were macrophages. Among CD4+ T cells, 7% were CD4+CD25+FoxP3+ T cells. Infiltrates recovered from three non-diabetic YES-RIP-hB7.1 mice (Figure S1) were composed of 56% T cells, among which 56% were CD8+ T cells, 34% were CD4+ T cells, 34% were β cells, and 2.3% were dendritic cells or macrophages. Among CD4+ T cells, 1% were CD25+FoxP3+. These data demonstrate the presence of an immune response along the development of diabetes in YES-RIP-hB7.1 mice, a dramatic decrease in β cells and a reduced α-cell mass. As the extent of infiltration was milder than the infiltrate seen in the NOD mouse model, we addressed whether autoimmune development could be delayed by a transient treatment with anti-CD4 and anti-CD8 monoclonal antibodies. As shown in Figure 3A, in vivo depletion of T cells by anti-CD4 and anti-CD8 antibodies from either 2 to 5 or 8 to 11 weeks of age significantly delayed T1D development in YES-RIP-hB7.1 mice. There was no difference in the protection observed in early-treated and late-treated mice. As previously described in the YES mouse model (16), we addressed whether diabetes development could be triggered in YES-RIP-hB7.1 mice by poly(I:C). As shown in Figure 3B, diabetes was induced within 6 to 17 days following the first poly(I:C) injection.
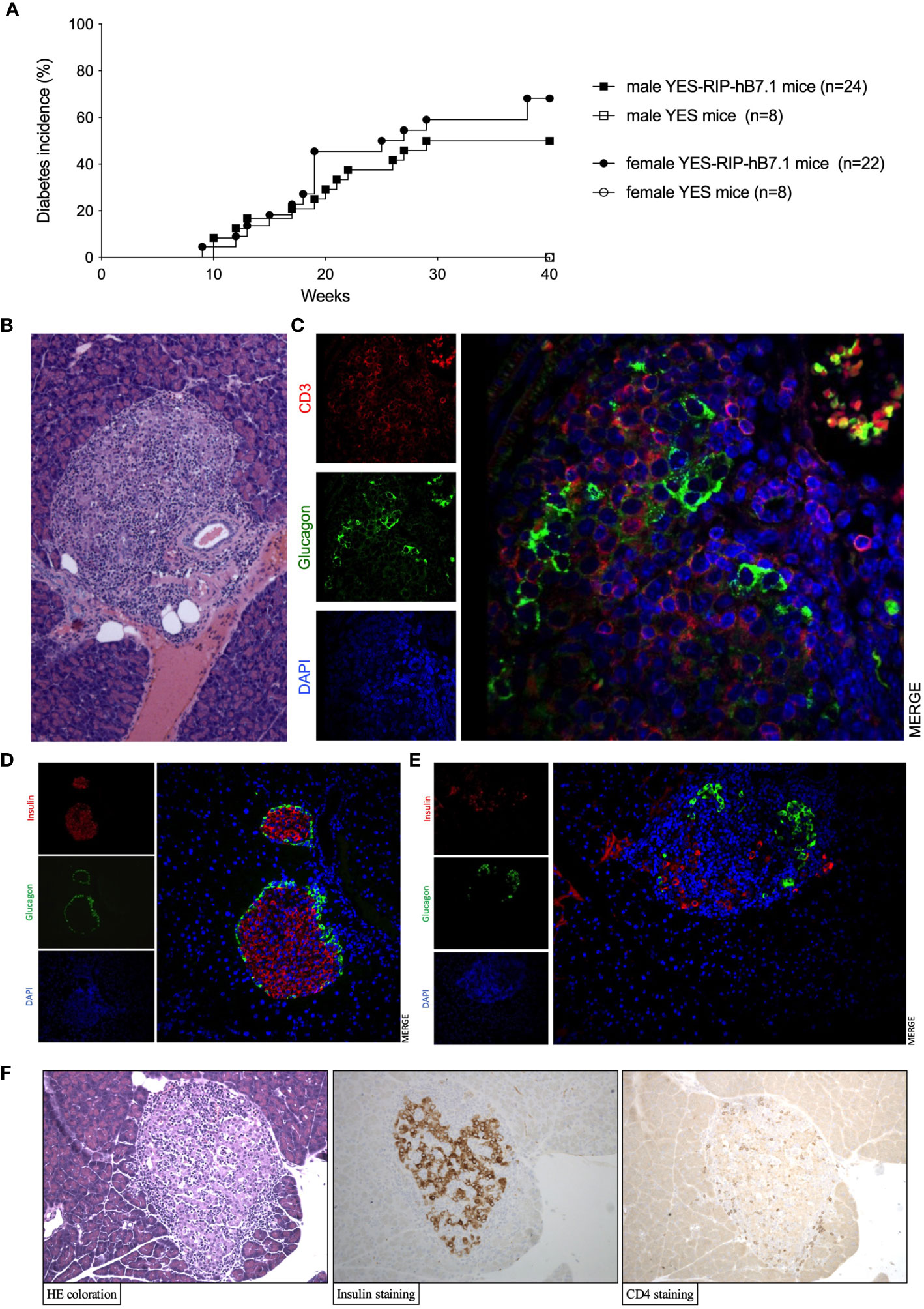
Figure 1 Diabetes incidence, islet infiltration in YES-RIP-hB7.1 mice. (A) Diabetes incidence in female ( , n = 22) and male (■, n = 24) YES-RIP-hB7.1 mice as compared with female (
, n = 22) and male (■, n = 24) YES-RIP-hB7.1 mice as compared with female ( , n = 8) and male (□, n = 6) YES mice as controls. (B) Pancreas paraffin sections stained by hematoxylin–eosin in diabetic YES-RIP-hB7.1 mice, ×20. (C) Immunofluorescent staining of an islet from a diabetic YES-RIP-hB7.1 mouse with anti-glucagon (green) and anti-CD3ε(red) antibodies, ×40. Immunofluorescent staining of an islet from a diabetic YES mice control (D) or YES-RIP-hB7.1 diabetic mouse (E) with anti-glucagon (green) and anti-insulin (red) antibodies, ×20. (F) Immunohistochemical staining of an islet from a diabetic YES-RIP-hB7.1 mouse with anti-insulin and anti-CD4 antibodies, ×20.
, n = 8) and male (□, n = 6) YES mice as controls. (B) Pancreas paraffin sections stained by hematoxylin–eosin in diabetic YES-RIP-hB7.1 mice, ×20. (C) Immunofluorescent staining of an islet from a diabetic YES-RIP-hB7.1 mouse with anti-glucagon (green) and anti-CD3ε(red) antibodies, ×40. Immunofluorescent staining of an islet from a diabetic YES mice control (D) or YES-RIP-hB7.1 diabetic mouse (E) with anti-glucagon (green) and anti-insulin (red) antibodies, ×20. (F) Immunohistochemical staining of an islet from a diabetic YES-RIP-hB7.1 mouse with anti-insulin and anti-CD4 antibodies, ×20.
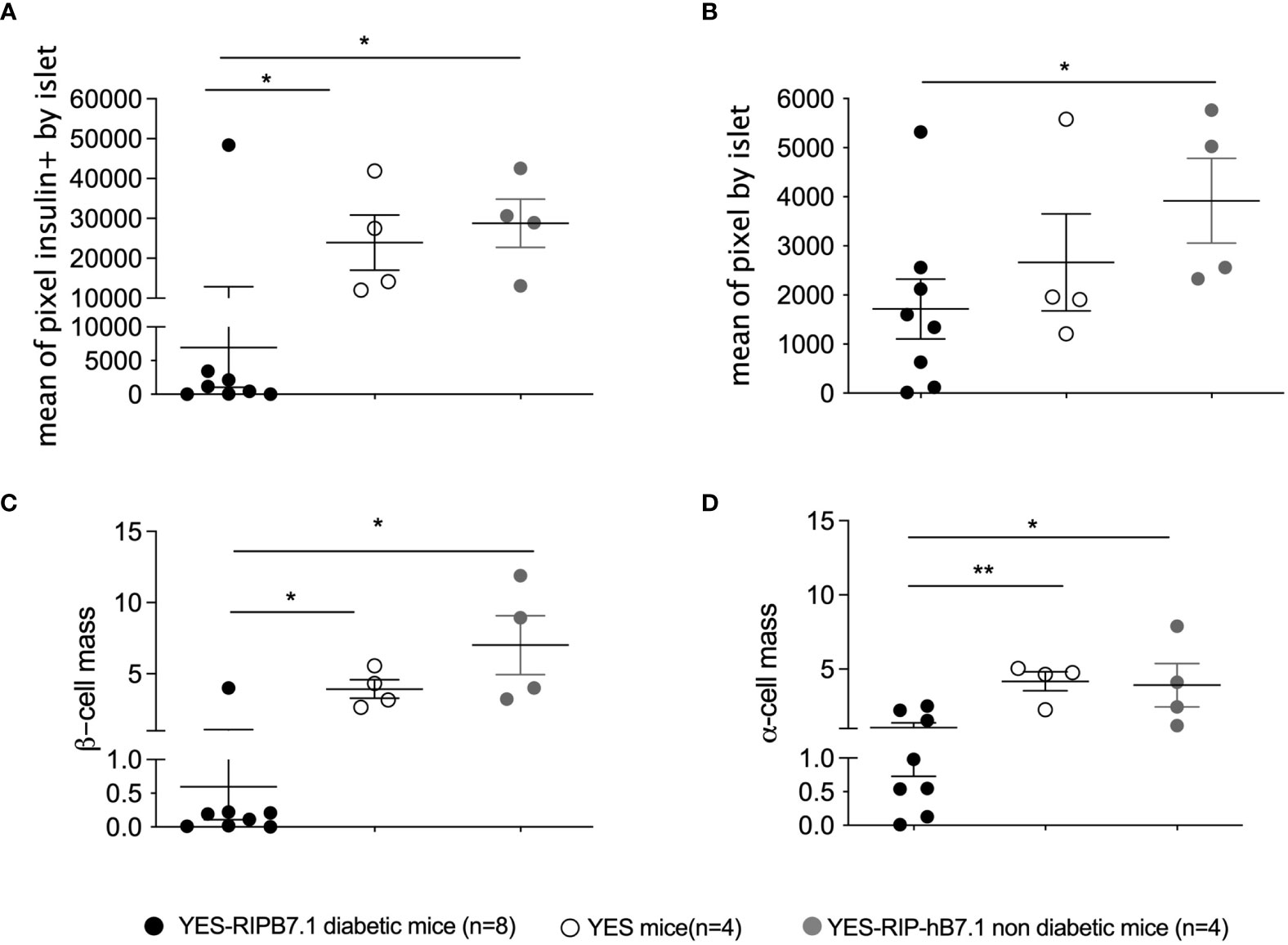
Figure 2 β-Cell and α-cell number and mass in YES-RIP-hB7.1 mice. (A) Mean of pixel insulin positive by level of pancreatic section of diabetic YES-RIP-hB7.1 (, n = 8), non-diabetic YES-RIP-hB7.1 ( , n = 4), and control YES (, n = 4) mice. (B) Mean of pixels per islet, representing the estimation of the islet size in pancreas of diabetic YES-RIP-hB7.1 (, n = 8), non-diabetic YES-RIP-hB7.1 (, n = 4), and control YES (, n = 4) mice. (C) β-Cell mass of diabetic YES-RIP-hB7.1 (, n = 8), non-diabetic YES-RIP-hB7.1 (, n = 4), and control YES (, n = 4) mice. (D) α-Cell mass of diabetic YES-RIP-hB7.1 (), non-diabetic YES-RIP-hB7.1 (), and control YES () mice. Each dot corresponds to an individual mouse. ns, non-significant, *p ≤ 0.05, **p ≤ 0.01; Mann–Whitney test.
, n = 4), and control YES (, n = 4) mice. (B) Mean of pixels per islet, representing the estimation of the islet size in pancreas of diabetic YES-RIP-hB7.1 (, n = 8), non-diabetic YES-RIP-hB7.1 (, n = 4), and control YES (, n = 4) mice. (C) β-Cell mass of diabetic YES-RIP-hB7.1 (, n = 8), non-diabetic YES-RIP-hB7.1 (, n = 4), and control YES (, n = 4) mice. (D) α-Cell mass of diabetic YES-RIP-hB7.1 (), non-diabetic YES-RIP-hB7.1 (), and control YES () mice. Each dot corresponds to an individual mouse. ns, non-significant, *p ≤ 0.05, **p ≤ 0.01; Mann–Whitney test.
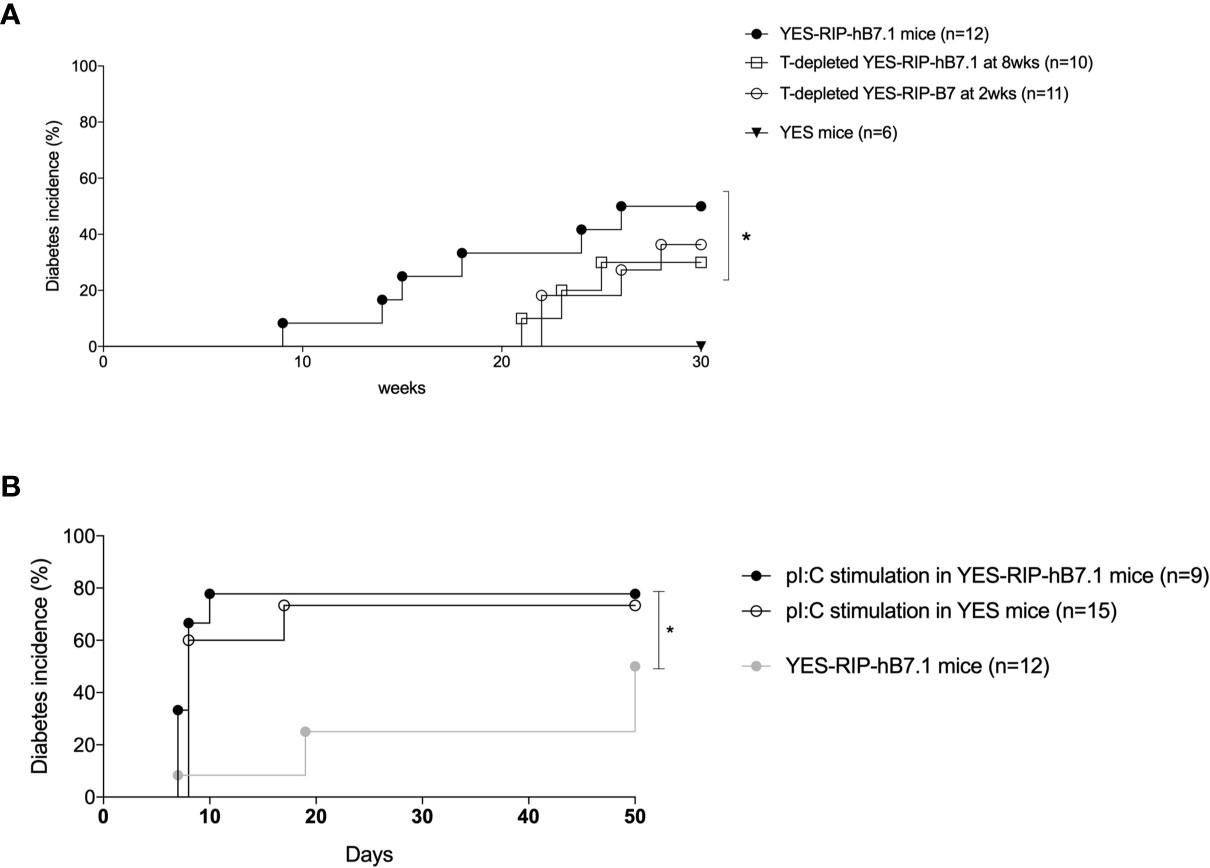
Figure 3 Diabetes incidence after in vivo T-cell depletion and poly(I:C) stimulation in YES-RIP-hB7.1 mice. (A) Diabetes incidence in untreated YES-RIP-hB7.1 mice (, n = 12), after in vivo T-cell depletion (four i.p. weekly injections of anti-CD4 and anti-CD8 antibodies) in YES-RIP-hB7.1 mice treated from 8 to 11 weeks of age (□, n = 10) or from to 2 weeks of age (, n = 11) and in YES mice (▼, n = 6) as controls. (B) Diabetes incidence in YES-RIP-hB7.1 mice (, n = 12), poly(I:C)-stimulated YES-RIP-hB7.1 mice (, n = 15), and poly(I:C)-stimulated YES mice (, n = 15) as controls. PolyI:C stimulation were performed by seven i.p. daily injections of 100 µg pI:C/mice. *p ≤ 0.05; Mann-Withney test.
hPPI-Specific CD8+ T lymphocytes Are Detected in YES-RIP-hB7.1 Mice
We previously characterized preproinsulin-specific CD8+ T cells in human T1D diabetes (11). In order to validate the YES-RIP-hB7.1 mice as a model to study T cells in T1D, we characterized CD8+ T-cell responses in 12- to 20-week-old diabetic mice and control mice that remained diabetes-free up to 30 weeks of age as previously reported (11). Using an IFNγ-ELISpot assay, we evaluated CD8+ T-cell responses to a library of hPPI peptides that were selected for presentation by HLA-A*02:01. Most hPPI peptides that were previously defined as recognized by human CD8+ T cells (11, 26) were recognized by T cells obtained from diabetic YES-RIP-hB7.1 mice (Figure 4A). Responses were detected against hPPI2-11 (p ≤ 0.03), hPPI6-14 (p ≤ 0.001), hPPI15-24 (p ≤ 0.02), and hPPI33-42 (p ≤ 0.013). No responses were observed against any of these hPPI peptides in control YES mice and in non-diabetic YES-RIP-hB7.1 mice. When considering responses that were over the mean ± 3SD of responses seen in control YES mice, significant responses were observed against hPPI6-14 (9/26, p ≤ 0.016), hPPI15-24 (11/26, p ≤ 0.004), and hPPI33-42 (9/26, p ≤ 0.016). Considering individual mice, responses were observed against hPPI2-11 (19.2%), hPPI30-39 (23.1%), hPPI34-42 (19.2%), hPPI42-51 (15.4%), and hPPI101-109 (15.4%), although they did not reach statistical significance in the whole population of diabetic mice analyzed (Table S3). As a whole, 93.75% YES-RIP-hB7.1 diabetic mice showed a response to at least one of the HLA-A*02:01-restricted hPPI peptides studied (defined as > mean ± 3SD of the response of YES mice against each given peptide, Table S3). An IFNγ-ELISpot response of pancreatic infiltrating cells was also documented against pooled hPPI peptides (Figure 4A, in red symbol) as the limited number of infiltrating cells prevented testing individual peptides. Using chimeric HLA-A*02:01 tetramers, we detected significant expansions of CD8+ T cells against hPPI6-14 (47.1%), hPPI15-24 (29.4%), and hPPI33-42 (47.1%) in YES-RIP-hB7.1 mice as compared with YES mice (Figure 4B). Considering individual mice, an expansion of CD8+ T cells was seen in 70.6% YES-RIP-hB7.1 mice against either hPPI6-14, hPPI15-24, or hPPI33-42. Single hPPI-specific CD8+ T cells showed gene expression profiles that were characteristic of memory T cells (Figure 4C). hPPI-specific CD8+ T cells from diabetic YES-RIP-hB7.1 mice expressed gzmA (59.38%), among which 100% co-expressed ccr7, while only 4.33% hPPI-specific CD8+ T cells from both YES-RIP-hB7.1 non-diabetic mice and YES control mice expressed gzmA, of which 76.7% co-expressed ccr7. To confirm a naive T-cell profile of cells from non-diabetic mice, we performed gene expression analysis for foxo1, which is expressed in basal naive T cells (27). An increased percentage of CD8+ T cells expressed foxo1 in non-diabetic as compared with diabetic mice (Figure S2A). A significant level of foxo1 expression was detected in 10-cell and single-cell batches (Figure S2B) in non-diabetics YES-RIP-hB7.1 mice when normalized to cd3ϵ expression. Finally, we determined ex vivo cytotoxic effects of hPPI-specific CD8+ T cells (Figure S3) in five diabetic YES-RIP-hB7.1 mice as compared with three YES controls in a LDH-release assay using peptide-pulsed HLA-A*02.01-transfected P815 target cells. Cytotoxic responses were detected against all hPPI peptides in diabetic YES-RIP-hB7.1 mice. Mice considered individually responded to different hPPI peptides. All mice that responded against at least one hPPI peptide also responded to full-length hPPI. Three mice showed cytotoxic responses against several hPPI peptides and two mice showed no cytotoxicity against any of the eight hPPI peptides. A heterogeneity of the cytotoxic responses in diabetic YES-RIP-hB7.1 was observed against hPPI.
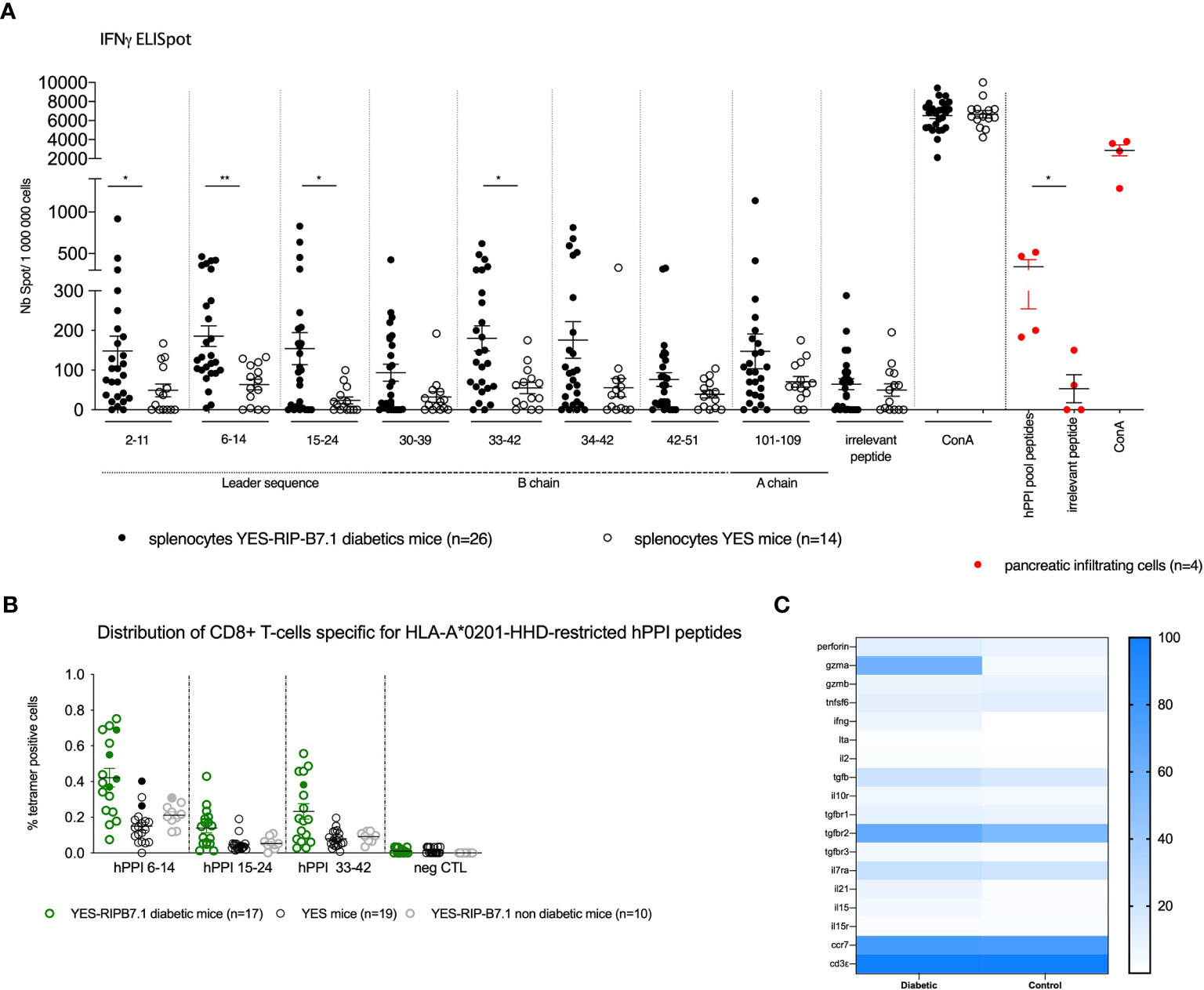
Figure 4 hPPI responses of YES-RIP-hB7.1 CD8+ T cells. (A) IFNγ responses to hPPI peptides. Splenocytes from individual diabetic (), non-diabetic () YES-RIP-hB7.1 mice, and control YES mice () were restimulated overnight with HLA-A*02:01-restricted hPPI peptides (20 µg/ml). Red cells correspond to cells from pancreatic infiltration. Each dot corresponds to an individual mouse for each peptide studied. (B) hPPI-specific CD8+ T-cell expansions in YES-RIP-hB7.1 mice. Splenic CD8+ T cells were stained with hPPI6-14, hPPI15-24, and hPPI33-42 or control tetramers comparing diabetic ( ), non-diabetic YES-RIP-hB7.1 mice (), and control YES mice (). Each dot corresponds to an individual mouse. Filled symbols correspond to the sorting cells for multiparametric RT-PCR. (C) Heat map of single-cell multiparametric RT-PCR in hPPI6-14, hPPI15-24, or hPPI33-42-specific CD8+ T cells from diabetic YES-RIP-hB7.1 or non-diabetic YES-RIP-hB7.1 mice and control YES mice. ns, non-significant, *p ≤ 0.05, **p ≤ 0.01; Mann–Whitney test.
), non-diabetic YES-RIP-hB7.1 mice (), and control YES mice (). Each dot corresponds to an individual mouse. Filled symbols correspond to the sorting cells for multiparametric RT-PCR. (C) Heat map of single-cell multiparametric RT-PCR in hPPI6-14, hPPI15-24, or hPPI33-42-specific CD8+ T cells from diabetic YES-RIP-hB7.1 or non-diabetic YES-RIP-hB7.1 mice and control YES mice. ns, non-significant, *p ≤ 0.05, **p ≤ 0.01; Mann–Whitney test.
hPPI-Specific CD4+ T-Cell Responses in YES-RIP-hB7.1 Mice
We studied HLA-DQ8-restricted T-cell responses to an overlapping hPPI peptide library. Significant T-cell proliferative responses were detected against hPPI1-15 (p ≤ 0.003), hPPI8-23 (p ≤ 0.008), hPPI16-30 (p ≤ 0.015), hPPI25-40 (p ≤ 0.014), hPPI33-47 (p ≤ 0.05), hPPI55-70 (p ≤ 0.029), hPPI61-76 (p ≤ 0.032), hPPI80-97 (p ≤ 0.022), and hPPI92-110 (p ≤ 0.028) peptides in YES-RIP-hB7.1 diabetic mice as compared with YES controls (Figure 5A). A non-significant trend for proliferative responses was detected against the same peptides in a few non-diabetic YES-RIP-hB7.1 mice. As in the case of CD8+ T cells, individual responses were observed against peptides covering the whole hPPI sequence. After determination of the threshold value for each peptide in controls using pairs of measurements and the Bland and Altman test (Figure S4, Table S4), proliferative responses were observed in a significant number of mice against all peptides except for hPPI16-30, hPPI40-55, and hPPI46-61. A significant response to full-length hPPI was observed in 51.5% of mice (p ≤ 0.009). Significant responses were observed in more than 30% diabetic YES-RIP-hB7.1 mice against hPPI1-15 (30.3% mice, p ≤ 0.008), hPPI8-23 (36.4% mice, p ≤ 0.002), hPPI18-30 (30.3% mice, p ≤ 0.04), hPPI25-40 (36.4% mice, p ≤ 0.002), hPPI33-47 (30.3% mice, p ≤ 0.009), hPPI55-70 (39.4% mice, p ≤ 0.009), hPPI61-76 (30.3% mice, p ≤ 0.04), hPPI70-86 (30.3% mice, p ≤ 0.04), and hPPI92-110 (45.4% mice, p ≤ 0.002) (Table S5). We observed a significant correlation between proliferative responses against the hPPI protein and several hPPI peptides in diabetic YES-RIP-hB7.1 mice, including hPPI16-30, hPPI25-40, hPPI33-47, hPPI55-70, hPPI61-76, and hPPI92-110 (Figure S5). Proliferative responses were seen against at least one hPPI peptide in 87.9% diabetic mice.
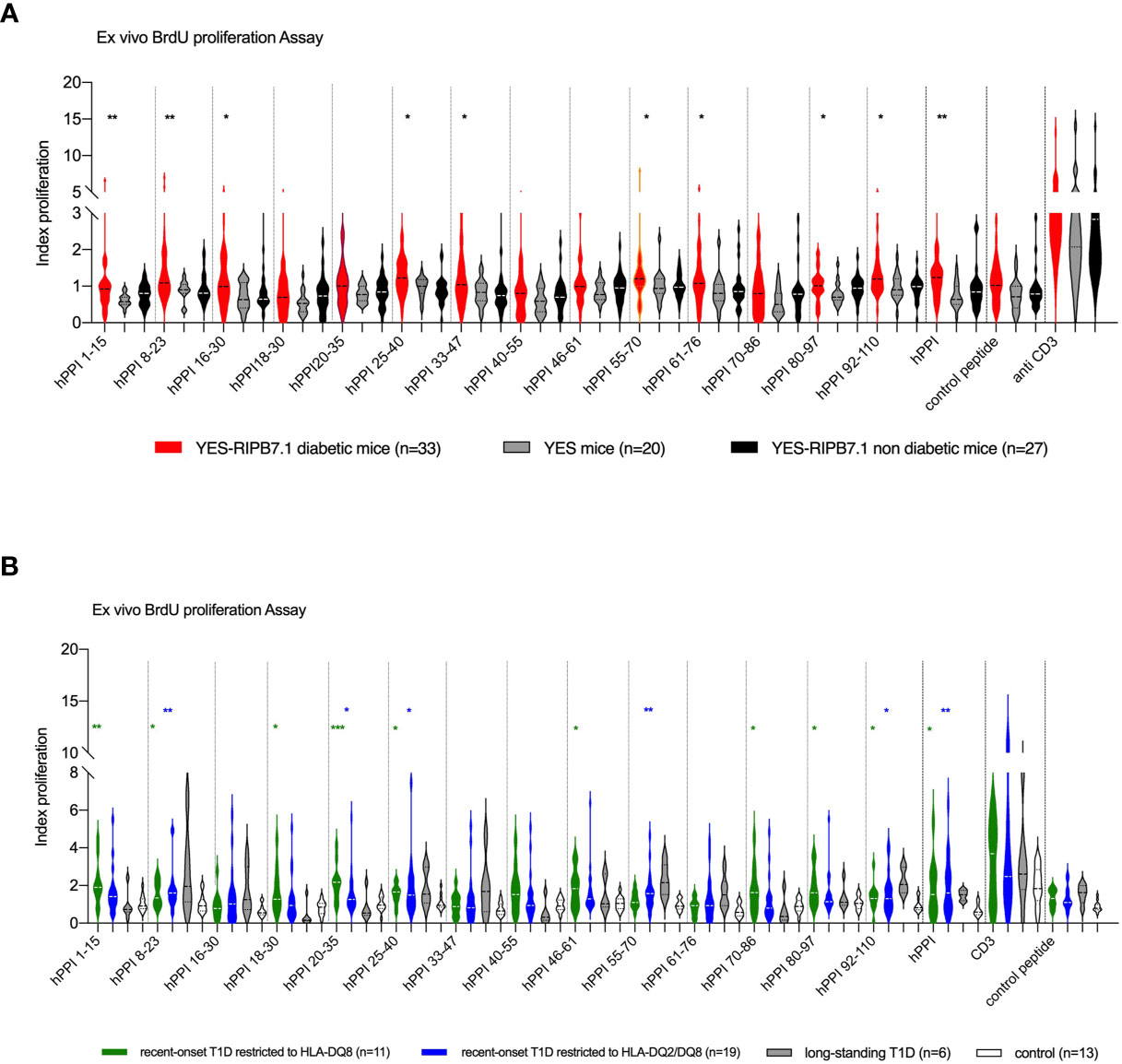
Figure 5 CD4+ T-cell responses against hPPI in YES-RIP-hB7 mice and T1D patients. (A) Proliferative responses to full-length hPPI and hPPI peptides hPPI1-15, hPPI8-23, hPPI16-30, hPPI18-30, hPPI20-35, hPPI25-40, hPPI33-47, hPPI40-55, hPPI46-61, hPPI55-70, hPPI61-76, hPPI70-86, hPPI80-97, and hPPI92-110. Splenocytes from diabetic YES-RIP-hB7.1 (), non-diabetic YES-RIP-hB7.1 (), and YES mice () were stimulated in vitro for 3 days, and proliferation was evaluated by measuring BrdU incorporation. (B) Proliferative responses to full-length hPPI and hPPI peptides hPPI1-15, hPPI8-23, hPPI16-30, hPPI18-30, hPPI20-35, hPPI25-40, hPPI33-47, hPPI40-55, hPPI46-61, hPPI55-70, hPPI61-76, hPPI70-86, hPPI80-97, and hPPI92-110. PBMCs from new-onset T1D patients restricted to HLA-DQ8 () or restricted to HLA-DQ2/DQ8 (■), long-standing T1D patients (), and controls () were stimulated in vitro for 3 days, and proliferation was evaluated by measuring BrdU incorporation. Statistic significances were obtained with data from control compared with HLA-DQ8 restriction patients (black bar) or with HLA-DQ2/DQ8 restriction patients (gray bar). Each dot represents individual mouse. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001; Mann–Whitney test.
hPPI-Specific CD4+ T Lymphocytes Are Detected in the Peripheral Blood of T1D Patients
The presence of hPPI-specific CD4+ T cells in the peripheral blood of T1D diabetic patients was analyzed by ex vivo proliferative assay and compared with HLA-DQ8 healthy donor and type 2 diabetes (T2D) patients as controls in order to address whether hPPI regions recognized in human matched with peptides recognized in YES-RIP-hB7.1 mice. Clinical features of T1D patients and control subjects (who were negative for detection of anti-GAD, anti-IA2, and anti-ZnT8 autoantibodies) are summarized in Table 1. Considering the possible trans-complementation between HLA-DQ8 and HLA-DQ2 class II MHC molecules, T1D patients were included according to their HLA-DQ8 or HLA-DQ2/DQ8 genotype. A proliferative response was observed against full-length recombinant hPPI in HLA-DQ8 T1D patients (p ≤ 0.04) and in HLA-DQ2/DQ8 T1D patients (p ≤ 0.003) as compared with controls (Figure 5B). When dissecting epitopes recognized, significant proliferative responses were observed against leader sequence hPPI1-15 (p ≤ 0.007), hPPI8-23 (p ≤ 0.003) and hPPI18-30 (p ≤ 0.02) peptides, overlapping leader sequence and B-chain hPPI20-35 peptide (p ≤ 0.007), B-chain hPPI25-40 (p ≤ 0.03) and hPPI46-61 (p ≤ 0.03) peptides, C-peptide hPPI70-86 (p ≤ 0.05) peptide, overlapping C-peptide and A-chain hPPI80-97 peptide (p ≤ 0.02), and the A-chain hPPI92-110 peptide (p ≤ 0.03) in HLA-DQ8 patients (Figure 4B). Significant proliferative responses were observed against leader sequence hPPI8-23 peptide (p ≤ 0.009), B-chain hPPI20-35 (p ≤ 0.05) and hPPI25-40 (p ≤ 0.035) peptides, C-peptide hPPI55-70 (p ≤ 0.003) peptide, and A-chain hPPI92-110 (p ≤ 0.04) peptide in HLA-DQ2/DQ8 T1D patients. After determination of the threshold value for each peptide in controls using pairs of measurements and the Bland and Altman test (Figure S6, Table S6), proliferative responses against hPPI1-15 (72.75%, p ≤ 0.01), hPPI18-30 (45.4%, p ≤ 0.041), hPPI20-35 (72.7% of T1D, p ≤ 0.04), hPPI40-55 (54.4%, p ≤ 0.003), hPPI46-61 (54.4%, p ≤ 0.02), and hPPI80-97 (63.6%, p ≤ 0.03) were seen in a significant number of HLA-DQ8 recent-onset T1D patients (Table S7). Significant proliferative responses to hPPI33-47 (42.1%, p ≤ 0.05), hPPI40-55 (36.8%, p ≤ 0.03), hPPI55-70 (57.9%, p ≤ 0.0006), and hPPI92-110 (47.4%, p ≤ 0.03) were observed in HLA-DQ2/DQ8 recent-onset T1D patients (Table S8). Distribution of proliferative responses to hPPI peptides was different in HLA-DQ8 and HLA-DQ2/DQ8 patients. As a total, proliferative responses were seen against at least one hPPI peptide in 94.44% T1D patients. Overall, the proliferative responses in HLA-DQ8 T1D patients largely matched the responses in YES-RIP-hB7.1 mice.
Reactivity Against Spliced hPPI in YES-RIP-hB7.1 Mice and HLA-DQ8 and DQ2/DQ8 Patients
Based on the observation of insulin mRNA alternative splicing events when reanalyzing databases generated in the context of human islet inflammation (28, 29), we designed an HLA-A*02:01-restricted spliced hPPI peptide, sp-hPPI60-68, and three HLA-DQ8-restricted spliced peptides, named sp-hPPI55-74, sp-hPPI52-74, and sp-hPPI51-77 using HLA-restrictor and NetMHCII 2.3 server, respectively (Figure 6A). Using an IFNγ-ELISpot assay, we detected HLA-A*02:01-restricted IFNγ responses against sp-hPPI60-68 peptide (Figure 6B) and HLA-DQ8-restricted responses to the sp-hPPI51-77 peptide (p ≤ 0.05) in splenocytes from diabetic YES-RIP-hB7.1 mice as compared with non-diabetic YES-RIP-hB7.1 or YES mouse controls (Figure 6C). Proliferative responses were further observed against the sp-hPPI52-74 and sp-hPPI51-77 peptides (p ≤ 0.009 and p ≤ 0.049, respectively, Figure 6C) in diabetic YES-RIP-hB7.1 mice as compared with non-diabetic YES-RIP-hB7.1 or YES controls. Seemingly, we addressed whether proliferative responses were detected in T1D patient PBMCs. Significant proliferative responses were seen against the sp-hPPI55-74 and sp-hPPI52-74 peptides in recent-onset HLA-DQ8 patients (p ≤ 0.0001 and p ≤ 0.001, respectively, black circle, Figure 6D) and in recent-onset HLA-DQ2/DQ8 diabetic patients (p ≤ 0.008 and p ≤ 0.03, respectively, black square, Figure 6D) as compared with control donors. Threshold values were determined in YES mice (Table S9) and in human controls (Table S10) using pairs of measurements and the Bland and Altman test (Figure S7). The frequency of recognition of sp-hPPI55-74, sp-hPPI52-74, and sp-hPPI51-77 peptides was not significantly different in diabetic YES-RIP-hB7.1 mice (Table S11). By contrast, the frequency of recognition of sp-hPPI55-74 and sp-hPPI52-74 peptides was significant in recent-onset HLA-DQ8 (57.9%, p ≤ 0.009 and 52.6%, p ≤ 0.02, respectively, Table 2) and HLA-DQ2/DQ8 (63.6%, p ≤ 0.009 and 72.5%, p ≤ 0.003, respectively, Table 2) T1D patients.
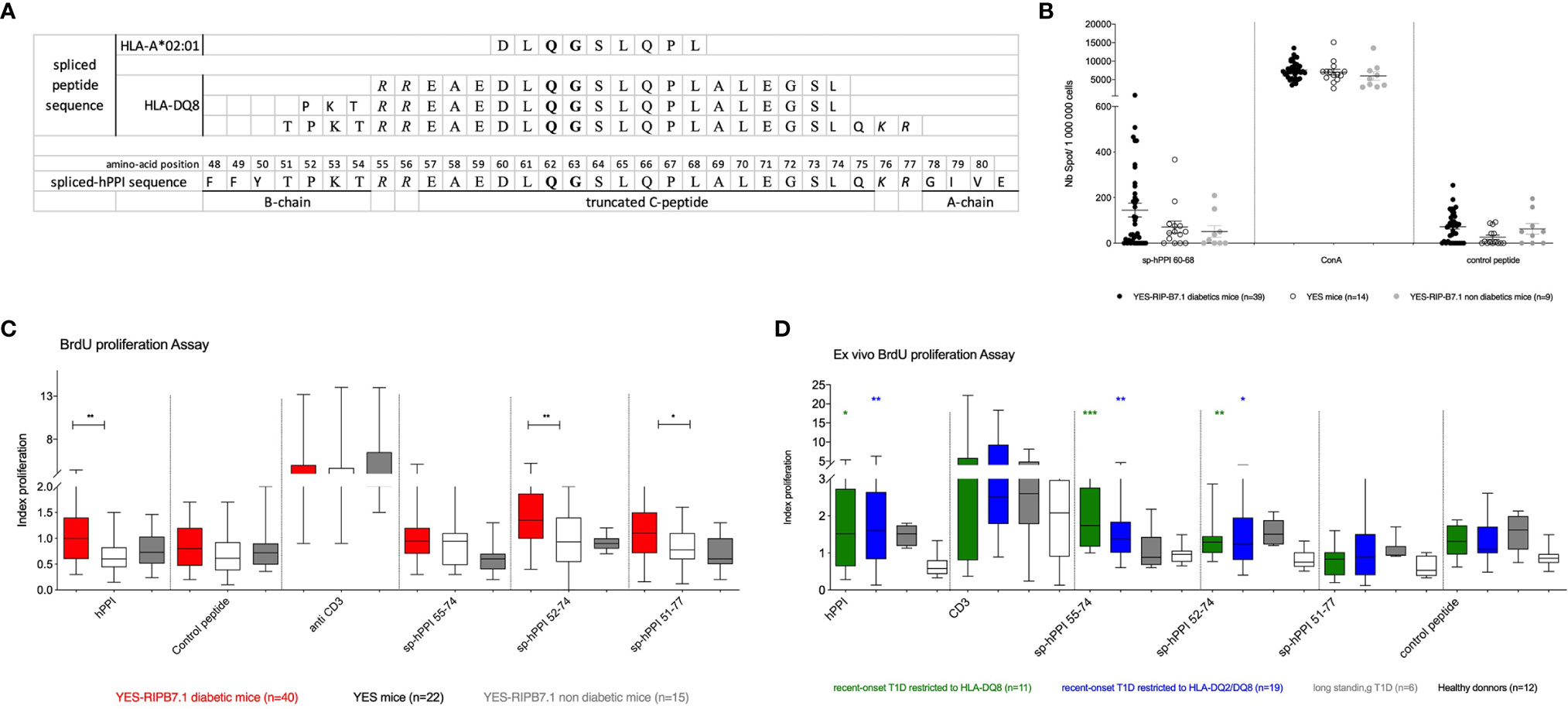
Figure 6 Responses to spliced hPPI. (A) Sequence of the spliced hPPI protein and spliced peptides restricted to HLA-A*02:01 and HLA-DQ8. Bold delimitates the truncated region of the alternatively spliced insulin protein sequence and italic defines bond-disulfur sites. (B) IFNγ responses to spliced hPPI peptides restricted to HLA-A*02:01. Splenocytes from individual diabetic (), non-diabetic () YES-RIP-hB7.1 mice, and control YES mice () were restimulated overnight with sp-hPPI60-68 peptide (20 µg/ml). Each dot corresponds to an individual mouse for each condition studied. (C) Proliferative responses to sp-hPPI55-74, sp-hPPI52-74, and sp-hPPI51-77 peptides. Splenocytes from diabetic YES-RIP-hB7.1 (), non-diabetic YES-RIP-hB7.1 (), and YES mice () were stimulated in vitro for 3 days, and proliferation was evaluated by measuring BrdU incorporation. (D) Proliferative responses to sp-hPPI55-74, sp-hPPI52-74, and sp-hPPI51-77 peptides. PBMC from new-onset T1D patients restricted to HLA-DQ8 () or restricted to HLA-DQ2/DQ8 (■), long-standing T1D patients (), and controls () were stimulated in vitro for 3 days, and proliferation was evaluated by measuring BrdU incorporation. Statistic significances were obtained with data from control compared with HLA-DQ8 restriction patients (black bar) or with HLA-DQ2/DQ8 restriction patients (gray bar). Each dot represents individual mouse. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001; Mann–Whitney test.
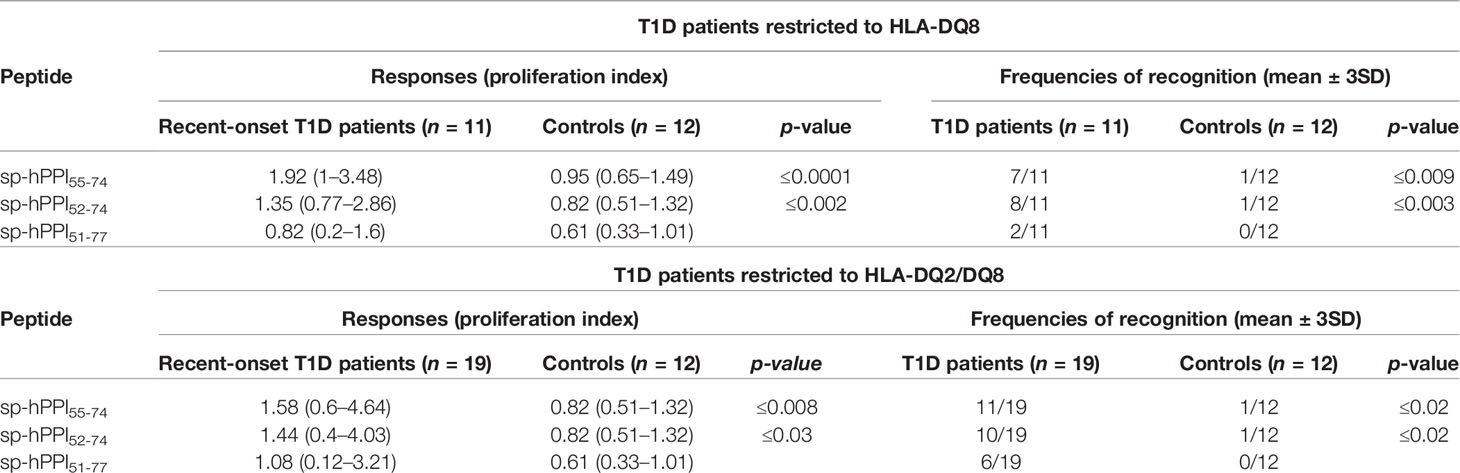
Table 2 Anti-spliced hPPI proliferative responses in T1D HLA-DQ8 patients.
Discussion
We developed a new preclinical model of spontaneous T1D in YES mice engineered to express the human co-activation hB7.1 gene in β cells by injecting a HIV-derived recombinant lentiviral vector in which a RIP-hB7.1 transgene has been inserted as previously reported (17), in addition to expression of human MHC and insulin genes instead of the corresponding mouse genes (16). We obtained a founder in which four insertions were detected and one stabilized as homozygous in the progeny. This stable insertion was located at a distance from any known Idd loci. The YES genetic background on which the C57BL/6 background is dominant was previously described (16). This likely indicates that the main genetic constraint that favors diabetes development, beyond expression of hB7.1, is the expression of the human class II HLA-DQ8 and, to a lower extent, class I HLA-A*02:01 alleles. While less than 2% conventional RIP-hB7.1 transgenic mice developed diabetes by 8 months of age (30), spontaneous diabetes was commonly observed in transgenic mice that co-expressed RIP-hB7.1 and the human insulin gene β cells (12, 31). Co-expression of RIP-hB7.1 in addition to HLA-DQ8 has been shown to allow the development of diabetes on a C57BL/6 genetic background (32–35). Along with the development of diabetes and islet infiltration by CD4+ and CD8+ T cells, a dramatic decrease of β-cell mass and a decrease in α-cell mass were observed in diabetic YES-RIP-hB7.1 mice, as previously been reported in NOD mice (36). In addition, RNA sequencing of human islet cells obtained from T1D patients showed a decrease in the expression of glucagon and other α-cell genes (37, 38), which is confirmed by a recent study on T1D patients based on the network for Pancreatic Organ Donors repository (39). In diabetic YES-RIP-hB7.1 mice in which the islet infiltrate was recovered from the pancreas, the islet infiltrate was predominantly composed of lymphocytes as observed in human T1D insulitis (40). CD8+ T cells were largely predominant, suggesting that they were a driving force in the diabetes process. We previously reported an increased percentage of single positive CD8+ T cells in the YES mouse, likely related to a lower efficiency of class II HLA-DQ8 to select CD4+ T cells than of class I HLA-A*02:01 to select CD8+ T cells (10). However, CD4+ T cells were recovered from non-diabetic YES-RIP-hB7.1 infiltrates in addition to CD8+ T cells. Spontaneous diabetes was significantly delayed in YES-RIP-hB7.1 mice following transient treatment with depleting anti-CD4 and anti-CD8 monoclonal antibodies, leaving open the issue of the predominant role of either CD4+ or CD8+ T cells in our model. In most T1D preclinical models, CD4+ T cells have been reported as dominant although a major role of CD8+ T cells has been reported in some models (41). In addition, an acute form of diabetes was induced in 8-week-old, prediabetic, YES-RIP-hB7.1 mice by seven daily poly(I:C) injections, as reported in the YES funders (16), indicating that different triggering events may concur to autoimmune T1D, as is probably the case in human disease. These data are reminiscent of data involving T1D induction by Coxsackie B4 virus (42–44), pointing to islet–environment interactions through signals carried by pattern recognition receptors (PPRs) (45, 46) in induction of T1D (47, 48).
In YES-RIP-hB7.1 mice, CD8+ T cells were detected against a wide array of epitopes covering the hPPI sequence. Responses were observed against two leader sequence epitopes, i.e., hPPI6-14 and hPPI15-24, and the B-chain peptide hPPI33-42 using an IFNγ-ELISpot assay, as was previously reported in human (11, 49). We and others have previously provided evidence that they are naturally processed as hPPI6-14-, hPPI15-24-, and hPPI33-42-specific CD8+ T cells are detected (11, 50), which are cytotoxic either to hPPI-HLA-A*02:01 doubled transfected-P815 target cells (26) or to human HLA-A*02:01 islets (49). As in human, however, the repertoire of hPPI epitopes recognized by CD8+ T cells was highly diversified from one mouse to another, with CD8+ T-cell responses observed against a wide array of hPPI epitopes spanning the whole hPPI sequence, beyond the three aforementioned epitopes. No correlation was observed between the affinity of peptides for soluble HLA-A*02:01 (11, 21, 26) and the prevalence of CD8+ T-cell responses in diabetic mice. Using HLA-A*02:01-restricted tetramers, we further detected expansions of CD8+ T cells against the three dominant HLA-A*02:01-restricted hPPI peptides. An expansion of hPPI-specific hPPI6-14-, hPPI15-24-, and hPPI34-42-CD8+ T cells was seen in diabetic mice and in some non-diabetic mice, as we also reported in human T1D. The study of gene expression in hPPI6-14-, hPPI15-24-, and hPPI33-42-specific CD8+ T cells allowed discriminating diabetic mice, in which 40% to 80% hPPI-specific single CD8+ T cells expressed gzma, from non-diabetic YES-RIP-hB7.1 mice, in which gzma expression was absent. Fifty-five percent to 95% CD8+ T cells were shown to express ccr7, suggesting that CD8+ T cells were mostly central memory cells in diabetic YES-RIP-hB7.1 mice, while CD8+ T cells mostly showed a naive phenotype in non-diabetic YES-RIP-hB7.1 mice, as confirmed by increased expression of foxo1, a gene that has been implicated in the regulation of T-cell homeostasis (51).
The study of class I HLA-A*02:01-restricted CD8+ T-cell responses in YES-RIP-hB7.1 diabetic mice has shown to broadly match epitope recognition that we previously reported in HLA-A*02:01 T1D patients. The YES-RIP-hB7.1 model is thus likely relevant to study recognition of a major human β-cell autoantigen, namely preproinsulin, presented by human susceptibility MHC molecules. In contrast with class-I-restricted epitopes, HLA-class II-restricted CD4+ T-cell responses remain ill-defined (52). CD4+ T-cell responses were observed in diabetic YES-RIP-hB7.1 mice against full-length hPPI and a wide array of hPPI peptides spanning the whole hPPI sequence, both in mouse and human, without any evidence for recognition of a dominant region. This is in contrast with previous reports of selective IAg7-restricted and HLA-DQ8-restricted T-cell responses observed in NOD mouse (7, 53, 54) and in human (7) against the B-chain peptide B9-23 (hPPI33-47). In NOD mouse, although an insulin B-chain epitope has been proposed as dominant along the autoimmune response to β cells, we showed that hybridomas directly generated from the islet-infiltrating CD4+ T cells recognized a large array of insulin epitopes (25). Significant frequencies of proliferative responses were observed against hPPI1-15, hPPI33-47, hPPI70-86, and hPPI92-110 peptides in both T1D induced by polyI:C in YES mice and spontaneous T1D observed in YES-RIP-hB7.1 mice. YES-RIP-hB7.1 mice showed significant proliferative responses to additional hPPI peptides, possibly reflecting an immune heterogeneity that is likely to apply to human disease (55, 56). Proliferative responses were observed against hPPI1-15, hPPI8-23, hPPI25-40, hPPI80-97, and hPPI92-110 both in HLA-DQ8 T1D patients and in diabetic YES-RIP-hB7.1 mice (16). In previous studies, epitopes located in the C-peptide region have been characterized as recognized by T-cell clones obtained from patients with T1D (57, 58). The hPPI55-69 epitope located in the junction of B-chain and C-peptide region was identified as a deaminated proinsulin peptide with cross-reactivity with native proinsulin peptide upon restimulation (59). Epitopes located in the peptide signal region and in the B-chain–C-peptide overlapping region have been identified as natural epitopes presented by high-risk HLA-DQ2/DQ8 heterozygous molecules (60, 61). HLA-DQ8-restricted CD4+ T-cell clones obtained from a T1D pancreatic infiltrate have been shown to recognize an hPPI epitope located in the C-peptide (57) that covers hPPI61-76 and hPPI80-97 for which we found responses in YES-RIP-hB7.1 mice and HLA-DQ8 T1D patients. We further found responses to hPPI61-76 in diabetic YES mice upon polyI:C injections (16). CD4+ T cells specific of the InsB30-C13 peptide (hPPI55-69) have been reported in 68% of HLA-DQ2/DQ8-restricted T1D patients versus 37% in healthy donors (59). This epitope is similar to our hPPI55-70 epitope for which we found significant proliferative responses in spontaneous-diabetic YES-RIP-hB7.1 mice (39.4%) and in T1D HLA-DQ2/DQ8 patients (57.9% in T1D patients versus 0% in control). In our study, HLA-DQ8-restricted patients showed responses against signal peptide hPPI epitopes which were not observed in HLA-DQ2/DQ8 patients. Reactivity against the B-chain or A-chain was observed in both HLA-DQ8 and HLA-DQ2/DQ8 patients. C-peptide recognition was mostly observed in HLA-DQ2/DQ8 patients. The frequency of recognition of hPPI peptides observed in diabetic YES-RIP-hB7.1 mice showed similarly significant responses against the hPPI signal peptide.
According to the description of spliced epitopes generated along inflammation development in the islets of Langerhans (62), we addressed whether T cells were responsive to these neoepitopes. Following evidence for CD4+ T-cell recognition of hPPI-spliced peptides in diabetic YES-RIP-hB7.1 mice, we evaluated corresponding responses in T1D patients. As in the mouse, we observed responses in both HLA-DQ8 and HLA-DQ2/DQ8 patients against an hPPI-spliced sequence that joins spliced hPPI55-74 and spliced hPPI52-74 peptides. This points to the interest of the YES-RIP-hB7.1 model in the study of modified hPPI peptides, which will be interesting to explore for other modifications such as citrullinated peptides (63) or epitopes issued from defective ribosomal insulin gene products (64).
Mouse models have been developed that express T1D susceptibility HLA class I (65, 66) or class II genes (31, 67). They allow defining epitopes on murine autoantigens that possibly correspond to epitopes recognized on human autoantigen along human T1D (37, 54). Sequence differences between murine and human MHC presenting molecules cannot exclude, however, that sets of epitopes defined on murine autoantigen differ from those recognized in human T1D. Models have further been reported that express T1D susceptibility HLA class I A*02:01 and/or the high-susceptibility DQ8 class II gene along with either the human preproinsulin or GAD genes (68, 69). These models are likely to allow characterizing autoantigen epitopes that may directly apply to human T1D (11, 70). Among these models, the YES-RIP-hB7.1 mouse is expected to allow the characterization of hPPI-specific CD8+ and CD4+ hPPI epitopes on a major autoantigen targeted in T1D, including new epitopes, such as spliced or modified epitopes, in this proinflammatory context (70, 71). Beyond allowing the identification of HLA-A02*01 and DQ8-restricted epitopes, it allows exemplifying different mechanisms of induction of T1D in the context of human disease that is likely heterogeneous (55, 56). Such models may prove valuable in developing T-cell assays in T1D and evaluating strategies to induce immune tolerance in T1D patients using peptides targeted by the autoimmune response to β cells.
Data Availability Statement
The original contributions presented in the study are publicly available. These data can be found here: http://www.ncbi.nlm.nih.gov/geo, GSE101551 and GSE151644.
Ethics Statement
The studies involving human participants were reviewed and approved by the Research Ministry Authorization MESR under number DC-2015-2536/IDRCB number 2015-A01875-44. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by the Ethics Committee n°34 of Paris Descartes under number CEEA34.CB.024.11.
Author Contributions
SL performed the experiments, was involved in the discussion, and contributed to the writing of the manuscript. SG was in charge of the transgenic animals. AG performed DAB staining. FLet and MV were involved in Affymetrix genotyping array discussion and RIP-hB7.1 capture strategy. PN and MB performed big data analysis. PC participated in the production of lentiviral particles for the lentiviral transgenesis. EL was responsible for patient recruitment and follow-up. MC and DE were implicated and collaborated in the alternative spliced hPPI section. FLem was involved in the discussion and manuscript editing. CB designed the experiments, chaired discussions, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was performed within the Département Hospitalo-Universitaire (DHU) AUToimmune and HORmonal diseaseS and supported by ANR grant R11189KK-RPV11189KKA, ANR2010-Biot-00801, EFSD grant 1-2008-106, and INNODIA grant E15179KK. DE was supported by grants from the Fonds National de la Recherche 420 Scientifiques (FNRS) - Welbio CR-2015A-06, Belgium, and the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No. 115797 (INNODIA). This joint undertaking receives support from the Union’s Horizon 2020 research and innovation program and EFPIA, JDRF, and The Leona M. and Harry B. Helmsley Charitable Trust.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We acknowledge the NIH tetramer core facility for provision of MHC class I tetramers; Francina Langa Vives and the CIGM platform at Institut Pasteur, Paris; Béatrice Durel and the imaging platform at Institut Cochin, Paris; Yannick Marie and the genotyping and sequencing platform at ICM, Paris; Sébastien Jacques, Florent Dumont, and the genotyping platform at Institut Cochin; and Camille Huet for the staining to evaluate the alpha and beta masses.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.748679/full#supplementary-material
References
1. Pyatak EA, Carandang K, Vigen C, Blanchard J, Sequeira PA, Wood JR, et al. Resilient, Empowered, Active Living With Diabetes (REAL Diabetes) Study: Methodology and Baseline Characteristics of a Randomized Controlled Trial Evaluating an Occupation-Based Diabetes Management Intervention for Young Adults. Contemp Clin Trials (2017) 54:8–17. doi: 10.1016/j.cct.2016.12.025
2. Beik P, Ciesielska M, Kucza M, Kurczewska A, Kuźmińska J, Maćkowiak B, et al. Prevention of Type 1 Diabetes: Past Experiences and Future Opportunities. J Clin Med (2020) 9:2805. doi: 10.3390/jcm9092805
3. Ziegler AG, Rewers M, Simell O, Simell T, Lempainen J, ASteck A, et al. Seroconversion to Multiple Islet Autoantibodies and Risk of Progression to Diabetes in Children. JAMA (2013) 309:2473–9. doi: 10.1001/jama.2013.6285
4. Ludvigsson J, Krisky D, Casas R, Battelino T, Castaño L, Greening J, et al. GAD65 Antigen Therapy in Recently Diagnosed Type 1 Diabetes Mellitus. Clin Trial (2012) 366:433–42. doi: 10.1056/NEJMoa1107096
5. Thrower SL, James L, Hall W, Green KM, Arif S, Allen JS, et al. Proinsulin Peptide Immunotherapy in Type 1 Diabetes: Report of a First-In-Man Phase I Safety Study. Clin Trial (2009) 155:156–65. doi: 10.1111/j.1365-2249.2008.03814.x
6. Thebault-Baumont K, Dubois-Laforgue D, Krief P, Briand JP, Halbout P, Vallon-Geoffroy K, et al. Acceleration of Type 1 Diabetes Mellitus in Proinsulin 2-Deficient NOD Mice. J Clin Invest (2003) 111:851–7. doi: 10.1172/JCI16584
7. Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, et al. Prime Role for an Insulin Epitope in the Development of Type 1 Diabetes in NOD Mice. Nature (2005) 435:220–3. doi: 10.1038/nature03523
8. Harrison LC. The Dark Side of Insulin: A Primary Autoantigen and Instrument of Self-Destruction in Type 1 Diabetes. Mol Metab (2021) 52:101288–94. doi: 10.1016/j.molmet.2021.101288
9. Noble JA, Valdes AM, Varney MD, Carlson JA, Moonsamy P, Fear AL, et al. And Genetic Susceptibility to Type 1 Diabetes: Results From the Type 1 Diabetes Genetics Consortium. Diabetes (2010) 59:2972–9. doi: 10.2337/db10-0699
10. Jones EY, Fugger L, Strominger JL, Siebold C. MHC Class II Proteins and Disease: A Structural Perspective. Nat Rev Immunol (2006) 6:271–82. doi: 10.1038/nri1805
11. Luce S, lemonnier F, Briand JP, Coste J, Lahlou N, Muller S, et al. Single Insulin-Specific CD8+ T Cells Show Characteristic Gene Expression Profiles in Human Type 1 Diabetes. Diabetes (2011) 60:3289–99. doi: 10.2337/db11-0270
12. von Herrath MG, Guerder S, Lewicki H, Flavell RA, Oldstone MB. Coexpression of B7-1 and Viral (“Self”) Transgenes in Pancreatic Beta Cells can Break Peripheral Ignorance and Lead to Spontaneous Autoimmune Diabetes. Immunity (1995) 3:727–38. doi: 10.1016/1074-7613(95)90062-4
13. Wong S, Guerder S, Visintin I, Reich E-P, Swenson KE, Flavell RA, et al. Expression of the Co-Stimulator Molecule B7-1 in Pancreatic (5-Cells Accelerates Diabetes in the NOD Mouse. Diabetes (1995) 44:326–9. doi: 10.2337/diab.44.3.326
14. Green J, Casabonne D, Newton R. Coxsackie B Virus Serology and Type 1 Diabetes Mellitus: A Systematic Review of Published Case-Control Studies. Diabetes Med (2004) 21:507–14. doi: 10.1111/j.1464-5491.2004.01182.x
15. Nabozny GH, Baisch J, Cheng S, Cosgrove D, Griffiths MM, Luthra HS, et al. HLA-DQ8 Transgenic Mice Are Highly Susceptible to Collagen-Induced Arthritis: A Novel Model for Human Polyarthritis. J Exp Med (1996) 183:27–37. doi: 10.1084/jem.183.1.27
16. Luce S, Guinoiseau S, Gadault A, Letourneur F, Blondeau B, Nitschke P, et al. Humanized Mouse Model to Study Type 1 Diabetes. Diabetes (2018) 67:1816–29. doi: 10.2337/db18-0202
17. Zennou V, Petit C, Guetard D, Nerhbass U, Montagnier L, Charneau P. HIV-1 Genome Nuclear Import is Mediated by a Central DNA Flap. Cell (2000) 101:173–85. doi: 10.1016/S0092-8674(00)80828-4
18. Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline Transmission and Tissue-Specific Expression of Transgenes Delivered by Lentiviral Vectors. Science (2002) 295:868–72. doi: 10.1126/science.1067081
19. Kurasawa K, Sakamoto A, Maeda T, Sumida T, Ito I, Tomioka H, et al. Short-Term Administration of Anti-L3T4 MoAb Prevents Diabetes in NOD Mice. Clin Exp Immunol (1992) 91:376–80. doi: 10.1111/j.1365-2249.1993.tb05912.x
20. Grcević D, Lee SK, Marusić A, Lorenzo JA. Depletion of CD4 and CD8 T Lymphocytes in Mice In Vivo Enhances 1,25-Dihydroxyvitamin D3-Stimulated Osteoclast-Like Cell Formation In Vitro by a Mechanism That is Dependent on Prostaglandin Synthesis. J Immunol (2000) 165:4231–8. doi: 10.4049/jimmunol.165.8.4231
21. Toma A, Haddouk S, Briand JP, Camoin L, Gahery H, Connan F, et al. Recognition of a Subregion of Human Proinsulin by Class I-Restricted T Cells in Type 1 Diabetic Patients. Proc Natl Acad Sci USA (2005) 102:10581–6. doi: 10.1073/pnas.0504230102
22. Congia M, Patel S, Cope AP, De Virgiliis S, Sønderstrup G. T Cell Epitopes of Insulin Defined in HLA-DR4 Transgenic Mice are Derived From Preproinsulin and Proinsulin. Proc Natl Acad Sci USA (1998) 95:3833–8. doi: 10.1073/pnas.95.7.3833
23. Palmer I, Wingfield PT. Preparation and Extraction of Insoluble (Inclusion-Body) Proteins From Escherichia Coli. Curr Protoc Protein Sci (2012) 6(6.3). doi: 10.1002/0471140864.ps0603s70
24. Rachdi L, Aiello V, Duvillié B, Scharfmann R. L-Leucine Alters Pancreatic β-Cell Differentiation and Function via the Mtor Signaling Pathway. Diabetes (2012) 61:409–17. doi: 10.2337/db11-0765
25. Halbout P, Briand JP, Bécourt C, Muller S, Boitard C. T Cell Response to Preproinsulin I and II in the Nonobese Diabetic Mouse. J Immunol (2002) 169:2436–43. doi: 10.4049/jimmunol.169.5.2436
26. Toma A, Laika T, Haddouk S, Luce S, Briand JP, Camoin L, et al. Recognition of Human Proinsulin Leader Sequence by Class I-Restricted T-Cells in HLA-A*0201 Transgenic Mice and in Human Type 1 Diabetes. Diabetes (2009) 58:394–402. doi: 10.2337/db08-0599
27. Ouyang W, Beckett O, Flavell RA, Li MO. An Essential Role of the Forkhead-Box Transcription Factor Foxo1 in Control of T Cell Homeostasis and Tolerance. Immunity (2009) 30:358–71. doi: 10.1016/j.immuni.2009.02.003
28. Villate O, Turatsinze JV, Mascali LG, Grieco FA, Nogueira TC, Cunha DA, et al. Nova1 is a Master Regulator of Alternative Splicing in Pancreatic Beta Cells. Nucleic Acids Res (2014) 42:11818–30. doi: 10.1093/nar/gku861
29. Alvelos MI, Juan-Mateu J, Colli ML, Turatsinze J-V, Eizirik DL. When One Becomes Many - Alternative Splicing in Beta Cell Function and Failure. Diabetes Obes Metab (2018) 20:77–87. doi: 10.1111/dom.13388
30. Guerder S, Picarella DE, Linsley PS, Flavell RA. Costimulator B7-1 Confers Antigen-Presenting-Cell Function to Parenchymal Tissue and in Conjunction With Tumor Necrosis Factor Alpha Leads to Autoimmunity in Transgenic Mice. Proc Natl Acad Sci USA (1994) 91:5138–42. doi: 10.1073/pnas.91.11.5138
31. Verhagen J, Yusuf N, Smith EL, Whettlock EM, Naran K, Arif S, et al. Proinsulin Peptide Promotes Autoimmune Diabetes in a Novel HLA-DR3-DQ2-Transgenic Murine Model of Spontaneous Disease. Diabetologia (2019) 62:2252–61. doi: 10.1007/s00125-019-04994-8
32. Wen L, Wong FS, Tang J, Chen NY, Altieri M, David C, et al. In Vivo Evidence for the Contribution of Human Histocompatibility Leukocyte Antigen (HLA)-DQ Molecules to the Development of Diabetes. J Exp Med (2000) 191:97–104. doi: 10.1084/jem.191.1.97
33. Wen L, Chen NY, Tang J, Sherwin R, Wong FS. The Regulatory Role of DR4 in a Spontaneous Diabetes DQ8 Transgenic Model. J Clin Invest (2001) 107:871–80. doi: 10.1172/JCI11708
34. Rajagopalan G, Kudva YC, Chen L, Wen L, David CS. Autoimmune Diabetes in HLA-DR3/DQ8 Transgenic Mice Expressing the Co-Stimulatory Molecule B7-1 in the Beta Cells of Islets of Langerhans. Int Immunol (2003) 15:1035–44. doi: 10.1093/intimm/dxg103
35. Gioia L, Holt M, Costanzo A, Sharma S, Abe B, Kain L, et al. Position β57 of I-Ag7 Controls Early Anti-Insulin Responses in NOD Mice, Linking an MHC Susceptibility Allele to Type 1 Diabetes Onset. Sci Immunol (2019) 4:eaaw6329. doi: 10.1126/sciimmunol.aaw6329
36. Pechhold K, Zhu X, Harrison VS, Lee J, Chakrabarty S, Koczwara K, et al. Dynamic Changes in Pancreatic Endocrine Cell Abundance, Distribution, and Function in Antigen-Induced and Spontaneous Autoimmune Diabetes. Diabetes (2009) 58:1175–84. doi: 10.2337/db08-0616
37. Brissova M, Haliyur R, Saunders D, Shrestha S, Dai C, Blodgett DM, et al. α Cell Function and Gene Expression Are Compromised in Type 1 Diabetes. Cell Rep (2018) 22:2667–76. doi: 10.1016/j.celrep.2018.02.032
38. Mastracci TL, Turatsinze J-V, Book BK, Restrepo IA, Pugia MJ, Wiebke EA, et al. Distinct Gene Expression Pathways in Islets From Individuals With Short- And Long-Duration Type 1 Diabetes. Diabetes Obes Metab (2018) 20:1859–67. doi: 10.1111/dom.13298
39. Bonnet-Serrano F, Diediesheim M, Mallone R, Larger E. Decreased α-Cell Mass and Early Structural Alterations of the Exocrine Pancreas in Patients With Type 1 Diabetes: An Analysis Based on the nPOD Repository. PloS One (2018) 13:1–16. doi: 10.1371/journal.pone.0191528
40. Campbell-Thompson M, Fu A, Kaddis JS, Wasserfall C, Schatz DA, Pugliese A, et al. Insulitis and β-Cell Mass in the Natural History of Type 1 Diabetes. Diabetes (2016) 65:719–31. doi: 10.2337/db15-0779
41. von Herrath MG, Evans CF, Horwitz MS, Oldstone MB. Using Transgenic Mouse Models to Dissect the Pathogenesis of Virus-Induced Autoimmune Disorders of the Islets of Langerhans and the Central Nervous System. Immunol Rev (1996) 152:111–43. doi: 10.1111/j.1600-065X.1996.tb00913.x
42. Ghazarian L, Diana J, Simoni Y, Beaudoin L, Lehuen A. Prevention or Acceleration of Type 1 Diabetes by Viruses. Cell Mol Life Sci (2013) 70:257. doi: 10.1007/s00018-012-1191-2
43. Ghazarian L, Diana J, Beaudoin L, Larsson PG, Puri RK, van Rooijen N, et al. Protection Against Type 1 Diabetes Upon Coxsackievirus B4 Infection and iNKT-Cell Stimulation: Role of Suppressive Macrophages. Diabetes (2013) 62:3785–96. doi: 10.2337/db12-0958
44. McCall KD, Thuma JR, Courreges MC, Benencia F, James CB, Malgor R, et al. Toll-Like Receptor 3 Is Critical for Coxsackievirus B4-Induced Type 1 Diabetes in Female NOD Mice. Endocrinology (2015) 156:453–61. doi: 10.1210/en.2013-2006
45. Hutton MJ, Soukhatcheva G, Johnson JD, Verchere CB. Role of the TLR Signaling Molecule TRIF in β-Cell Function and Glucose Homeostasis. Islets (2010) 2:104–11. doi: 10.4161/isl.2.2.11209
46. Giarratana N, Penna G, Amuchastegui S, Mariani R, Daniel KC, Adorini L. A Vitamin D Analog Down-Regulates Proinflammatory Chemokine Production by Pancreatic Islets Inhibiting T Cell Recruitment and Type 1 Diabetes Development. J Immunol (2004) 173:2280–7. doi: 10.4049/jimmunol.173.4.2280
47. Swiecki M, McCartney SA, Wang Y, Colonna M. TLR7/9 Versus TLR3/MDA5 Signaling During Virus Infections and Diabetes. J Leukoc Biol (2011) 90:691–701. doi: 10.1189/jlb.0311166
48. Alkanani AK, Hara N, Lien E, Ir D, Kotter CV, Robertson CE, et al. Induction of Diabetes in the RIP-B7.1 Mouse Model is Critically Dependent on TLR3 and MyD88 Pathways and Is Associated With Alterations in the Intestinal Microbiome. Diabetes (2014) 63:619–31. doi: 10.2337/db13-1007
49. Skowera A, Ellis RJ, Varela-Calviño R, Arif S, Huang GC, Van-Krinks C, et al. CTLs are Targeted to Kill Beta Cells in Patients With Type 1 Diabetes Through Recognition of a Glucose-Regulated Preproinsulin Epitope. J Clin Invest (2008) 118:3390–402. doi: 10.1172/JCI35449
50. Rodriguez-Calvo T, Krogvold L, Amirian N, Dahl-Jorgensen K, von Herrath M. One in Ten CD81 Cells in the Pancreas of Living Individuals With Recent-Onset Type 1 Diabetes Recognizes the Preproinsulin Epitope PPI15-24. Diabetes (2021) 70:752–8. doi: 10.2337/db20-0908
51. Tejera MM, Kim EH, Sullivan JA, Plisch EH, Suresh M. FoxO1 Controls Effector-To-Memory Transition and Maintenance of Functional CD8 T Cell Memory. J Immunol (2013) 191:187–99. doi: 10.4049/jimmunol.1300331
52. Landry LG, Anderson AN, Russ HA, Yu L, Kent SC, Atkinson MA, et al. Proinsulin-Reactive CD4 T Cells in the Islets of Type 1 Diabetes Organ Donors. Front Endocrinol (2021). doi: 10.3389/fendo.2021.622647
53. Wegmann DR, Gill RG, Norbury-Glaser M, Schloot N, Daniel D. Analysis of the Spontaneous T Cell Response to Insulin in NOD Mice. J Autoimmun (1994) 7:833–43. doi: 10.1006/jaut.1994.1066
54. Tan S, Yang l, Xia J, Jin C-H, Hu Z, Duinkerken G, et al. Type 1 Diabetes Induction in Humanized Mice. Proc Nati Acad Sci USA (2017) 114:10954–9. doi: 10.1073/pnas.1710415114
55. Battaglia M, Ahmed S, Anderson MS, Atkinson MA, Becker D, Bingley PJ, et al. Introducing the Endotype Concept to Address the Challenge of Disease Heterogeneity in Type 1 Diabetes. Diabetes Care (2020) 43:5–12. doi: 10.2337/dc19-0880
56. Claessens LA, Wesselius J, van Lummel M, Laban S, Mulder F, Mul D, et al. Clinical and Genetic Correlates of Islet-Autoimmune Signatures in Juvenile-Onset Type 1 Diabetes. Diabetologia (2020) 63:351–61. doi: 10.1007/s00125-019-05032-3
57. Pathiraja V, Kuehlich JP, Campbell PD, Krishnamurthy B, Loudovaris T, Coates PT, et al. Proinsulin Specific, HLA-DQ8 and HLA-DQ8 Transdimer Restricted, CD4+ T Cells Infiltrate the Islets in Type 1 Diabetes. Diabetes (2014) 64:172–82. doi: 10.2337/db14-0858
58. van Lummel M, Buis DTP, Ringeling C, de Ru AH, Pool J, Papadopoulos GK, et al. Epitope Stealing as Mechanism of Dominant Protection by HLA-DQ6 in Type 1 Diabetes. Diabetes (2019) 68:787–95. doi: 10.2337/db18-0501
59. van Lummel M, Duinkerken G, van Veelen PA, de Ru A, Cordfunke R, Zaldumbide A, et al. Posttranslational Modification of HLA-DQ Binding Islet Autoantigens in Type 1 Diabetes. Diabetes (2014) 63:237–47. doi: 10.2337/db12-1214
60. van Lummel M, van Veelen PA, de Ru AH, Pool J, Nikolic T, Laban S, et al. Discovery of a Selective Islet Peptidome Presented by the Highest-Risk HLA-DQ8trans Molecule. Diabetes (2016) 65:732–41. doi: 10.2337/db15-1031
61. Chow IT, Gates TJ, Papadopoulos GK, Moustakas AK, Kolawole EM, Notturno RJ, et al. Discriminative T Cell Recognition of Cross-Reactive Islet-Antigens is Associated With HLA-DQ8 Transdimer-Mediated Autoimmune Diabetes. Sci Adv (2019) 5:eaw9336. doi: 10.1126/sciadv.aaw9336
62. Gonzalez-Duque S, Azoury ME, Colli ML, Afonso G, Turatsinze J-V, Nigi L, et al. Conventional and Neo-Antigenic Peptides Presented by β Cells Are Targeted by Circulating Naïve CD8+ T Cells in Type 1 Diabetic and Healthy Donors. Cell Metab (2018) 28:946–60. doi: 10.1016/j.cmet.2018.07.007
63. Yang M-L, Sodré FMC, Mamula MJ, Overbergh L. Citrullination and PAD Enzyme Biology in Type 1 Diabetes - Regulators of Inflammation, Autoimmunity, and Pathology. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.678953
64. Kracht MJ, van Lummel M, Nikolic T, Joosten AM, Laban S, van der Slik AR, et al. Autoimmunity Against a Defective Ribosomal Insulin Gene Product in Type 1 Diabetes. Nat Med (2017) 23:501–7. doi: 10.1038/nm.4289
65. Schloss J, Ali R, Racine JJ, Chapman HD, Serreze DV, DiLorenzo TP. HLA-B*39:06 Efficiently Mediates Type 1 Diabetes in a Mouse Model Incorporating Reduced Thymic Insulin Expression. J Immunol (2018) 200:3353–63. doi: 10.4049/jimmunol.1701652
66. Racine JJ, Stewart I, Ratiu J, Christianson G, Lowell E, Helm K, et al. Improved Murine MHC-Deficient HLA Transgenic NOD Mouse Models for Type 1 Diabetes Therapy Development. Diabetes (2018) 67:923–35. doi: 10.2337/db17-1467
67. Verhagen J, Smith EL, Whettlock EM, Macintyre B, Peakman M. Proinsulin-Mediated Induction of Type 1 Diabetes in HLA-DR4-Transgenic Mice. Sci Rep (2018) 8:14106. doi: 10.1038/s41598-018-32546-4
68. Elagin RB, Balijepalli S, Diacovo MJ, Baekkeskov S, Jaume JC. Homing of GAD65 Specific Autoimmunity and Development of Insulitis Requires Expression of Both DQ8 and Human GAD65 in Transgenic Mice. J Autoimmun (2009) 33:50–7. doi: 10.1016/j.jaut.2009.02.004
69. Elagin RB, Jaume JC. Glucose Intolerance and Diabetes Following Antigen-Specific Insulitis in Diabetes-Susceptible “Humanized” Transgenic Mice. Biochem Biophys Res Commun (2010) 395:99–103. doi: 10.1016/j.bbrc.2010.03.146
70. Juan-Mateu J, Villate O, Eizirik DL. Mechanisms in Endocrinology: Alternative Splicing: The New Frontier in Diabetes Research. Eur J Endocrinol (2016) 174:R225–38. doi: 10.1530/EJE-15-0916
Keywords: autoimmunity, type 1 diabetes (T1D), humanized mouse, HLA-DQ8, epitopes, preproinsulin
Citation: Luce S, Guinoiseau S, Gadault A, Letourneur F, Nitschke P, Bras M, Vidaud M, Charneau P, Larger E, Colli ML, Eizirik DL, Lemonnier F and Boitard C (2021) A Humanized Mouse Strain That Develops Spontaneously Immune-Mediated Diabetes. Front. Immunol. 12:748679. doi: 10.3389/fimmu.2021.748679
Received: 28 July 2021; Accepted: 22 September 2021;
Published: 14 October 2021.
Edited by:
Urs Christen, Goethe University Frankfurt, GermanyReviewed by:
Shun-Fen Tzeng, National Cheng Kung University, TaiwanMyung-Shik Lee, Yonsei University Health System, South Korea
Copyright © 2021 Luce, Guinoiseau, Gadault, Letourneur, Nitschke, Bras, Vidaud, Charneau, Larger, Colli, Eizirik, Lemonnier and Boitard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Boitard, Y2hyaXN0aWFuLmJvaXRhcmRAaW5zZXJtLmZy