 Jasmin Scheurer1
Jasmin Scheurer1 Kerstin Kitt2Heinrich J. Huber3,4
Kerstin Kitt2Heinrich J. Huber3,4 Katrin Fundel-Clemens3Stefan Pflanz2
Katrin Fundel-Clemens3Stefan Pflanz2 Klaus-Michael Debatin1
Klaus-Michael Debatin1 Gudrun Strauss1*
Gudrun Strauss1*- 1Department of Pediatrics and Adolescent Medicine, University Medical Center Ulm, Ulm, Germany
- 2Department of Cancer Immunology and Immune Modulation, Boehringer Ingelheim Pharma Co KG, Biberach an der Riss, Germany
- 3Global Computational Biology and Digital Sciences, Boehringer Ingelheim Pharma Co KG, Biberach an der Riss, Germany
- 4Drug Discovery Services, Boehringer Ingelheim Regional Center Vienna (RCV) GmbH & Co KG, Vienna, Austria
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of myeloid progenitor cells that dampen overwhelming adaptive immune responses through multiple mechanisms and are recognized as an attractive novel immune intervention therapy for counteracting the destructive effects of graft-versus-host disease (GVHD) developing after allogeneic bone marrow transplantation (BMT). MDSCs can be produced in great numbers for cellular therapy, but they present a mixture of subsets whose functions in GVHD prevention are undefined. Here, we generated MDSCs in vitro from murine BM cells in the presence of GM-CSF and defined the integrin CD11c as a marker to subdivide MDSCs into two functional subgroups: CD11b+CD11c+ and CD11b+CD11c− MDSCs. Isolated CD11b+CD11c+ and CD11b+CD11c− MDSCs both inhibited alloantigen-stimulated T-cell proliferation in vitro, although CD11b+CD11c+ MDSCs were more efficient and expressed higher levels of different immunosuppressive molecules. Likewise, expression of surface markers such as MHC class II, CD80, CD86, or PD-L1 further delineated both subsets. Most importantly, only the adoptive transfer of CD11b+CD11c+ MDSCs into a single MHC class I-disparate allogeneic BMT model prevented GVHD development and strongly decreased disease-induced mortality, while CD11b+CD11c− MDSCs were totally ineffective. Surprisingly, allogeneic T-cell homing and expansion in lymphatic and GVHD target organs were not affected by cotransplanted CD11b+CD11c+ MDSCs indicating a clear contradiction between in vitro and in vivo functions of MDSCs. However, CD11b+CD11c+ MDSCs shifted immune responses towards type 2 immunity reflected by increased Th2-specific cytokine expression of allogeneic T cells. Induction of type 2 immunity was mandatory for GVHD prevention, since CD11b+CD11c+ MDSCs were ineffective if recipients were reconstituted with STAT6-deficient T cells unable to differentiate into Th2 cells. Most importantly, the beneficial graft-versus-tumor (GVT) effect was maintained in the presence of CD11b+CD11c+ MDSCs since syngeneic tumor cells were efficiently eradicated. Strong differences in the transcriptomic landscape of both subpopulations underlined their functional differences. Defining CD11b+CD11c+ MDSCs as the subset of in vitro-generated MDSCs able to inhibit GVHD development might help to increase efficiency of MDSC therapy and to further delineate relevant target molecules and signaling pathways responsible for GVHD prevention.
Introduction
In the year 2007, myeloid-derived suppressor cells (MDSCs) were introduced as a heterogeneous population of myeloid progenitors with potent immunosuppressive functions (1, 2) that expand under various inflammatory pathological conditions such as chronic inflammation, autoimmune diseases, infections, and cancer. Under inflammatory conditions, MDSCs fail to complete their regular differentiation into mature macrophages, granulocytes, or dendritic cells and are phenotypically and functionally distinct from normal myeloid cells. T cells are the preferred and major targets of MDSCs. MDSCs inhibit T-cell responses by versatile mechanisms including nutrient depletion, nitrosylation, apoptosis, or blockade of lymphocyte homing or induction of Tregs (3, 4). By studying the role of MDSCs in different disease entities, it turned out that they strongly contribute to the decision whether immune responses develop towards type 1 or type 2 immunity. MDSCs shift the balance towards Th2 immunity in pathologies such as sepsis, viral infections, or certain types of cancers (5–8), while they support Th1 immunity in Th2-driven asthma-related airway inflammation (9, 10).
Considering their immunomodulatory functions, MDSCs represent attractive candidates to counterbalance overwhelming immune responses associated with T-cell-mediated diseases. Graft-versus-host disease (GVHD) represents a disease which develops after allogeneic bone marrow (BMT) transplantation and is induced by activation and expansion of alloantigen-activated mature transplant-derived T cells. These donor T cells attack and destroy recipient tissue finally leading to life-threatening posttransplantation complications, which dramatically limit the success of allogeneic stem cell transplantation for treatment of hematological malignancies and genetic disorders (11). However, transplanted allogeneic T cells also mediate the graft-versus-tumor effect (GVT), which ensures eradication of alloantigen expressing residual tumor cells. Therefore, GVHD treatment strategies aiming to interfere with allogenic T-cell activation, proliferation, and function should be balanced in terms to prevent allogeneic T-cell-mediated tissue destruction while simultaneously guaranteeing efficient T-cell immunity to cope with infections and destroy residual tumor cells (12).
MDSCs for adoptive cell therapy of GVHD can be successfully generated in mice. MDSCs can be directly isolated from tumor-bearing mice (13) or after in vivo administration of 3′5′-cytidylylguanosine (CpG), granulocyte-colony stimulating factor (G-CSF), or a synthetic G-CSF/Flt-3 ligand. Subsequent adoptive transfer of the isolated MDSCs in allogeneic BM recipients efficiently prevents GVHD development (13–16). Different precursor cells and cytokine combinations are suitable for in vitro induction and expansion of MDSCs. While Zhou et al. used mouse embryonic stem cells activated with a mixture of cytokines in a three-step differentiation strategy (17), MDSCs currently are mostly induced by culturing unseparated BM cells in GM-CSF alone or in combinations with cytokines such as G-CSF or IL-13 (18–20). Independent of the cytokine combination used, adoptive transfer of in vitro-generated MDSCs efficiently prevents GVHD induction, while tumor reactivity in MDSC-treated mice is maintained. Although randomized trials proving therapeutic potential of MDSCs in humans are lacking, promising results are obtained from humanized mouse models receiving in vitro-expanded human MDSCs for prevention of xenogeneic GVHD (21, 22).
MDSCs either isolated ex vivo from tissues or generated in vitro from hematopoietic precursor cells always present a mixture of cells. Classically, unseparated murine MDSCs coexpress CD11b and Gr-1 and expression of Ly-6C and Ly-6G further subdivides MDSCs into the two major subpopulations: monocytic (M) MDSCs (CD11b+Ly-6G-Ly-6Chigh) and polymorphonuclear (PMN) MDSCs (CD11b+Ly-6G-Ly-6Chigh) (23, 24). Since Ly-6G and Ly-6C are also expressed on differentiated monocytes and mature neutrophils, further marker panels have been designed including transcription factors, cytokines, and effector molecules to distinguish MDSC subsets from fully matured myeloid cells (25), but the final identification as MDSCs is always designated by their ability to mediate T-cell suppressive functions (26, 27).
Currently, it is not defined which subset of MDSCs contribute to immunosuppression and GVHD prevention in the context of allogeneic BMT. In recent work, we could show that adoptive transfer of CD11b+Gr-1+ MDSCs induced from BM cells in the presence of GM-CSF efficiently prevented GVHD development in two different allogeneic BMT models. GVHD inhibition was mostly attributed to the ability of MDSCs to shift the immune response in the transplanted recipients towards type 2 immunity (18). Interestingly, phenotypic characterization of the in vitro-generated MDSCs indicated that these cells were not a unique population but could be distinguished by the expression of the integrin CD11c. CD11b+CD11c+ MDSC subpopulations exhibited increased expression of CD301b, which expression is linked to the Th2-inducing abilities of DCs (28). Additionally, transcription factors IRF4 and Klf4 also associated with Th2 induction (29, 30) were upregulated compared with CD11b+CD11c− MDSCs indicating that both subpopulations might exhibit different properties in GVHD prevention. In the current study, we therefore aimed to further characterize CD11b+CD11c+ and CD11b+CD11c− MDSCs for their T-cell suppressive capacities and their function in GVHD prevention. Although both subsets suppressed T-cell proliferation in vitro, only CD11b+CD11c+ MDSCs prevented GVHD development after allogeneic BMT while maintaining tumor cytotoxicity. GVHD prevention was totally dependent on the ability of CD11b+CD11c+ MDSCs to shift the immune response towards type 2 immunity. Strong differences in the transcriptomic landscape of both MDSC subsets further underlined their functional differences and might be used in further studies to delineate molecules and pathways responsible for MDSC-mediated GVHD inhibition.
Material and Methods
Tissue Preparation
Bone Marrow
Bone marrow (BM) cells were isolated with 26-gauge needle from femurs and tibias. Single-cell suspensions were prepared using a syringe with 20-gauge needle, and erythrocytes were depleted.
Spleen
Splenic single-cell suspensions were prepared by pouring the spleen through a 70-µm cell strainer followed by erythrocyte depletion.
Liver
Liver was perfused by the injection of 5 ml liver perfusion medium (Gibco, Carlsbad, CA, USA), followed by 5 ml liver digest medium (Gibco) into the vena cava inferior. Without the gall bladder, liver was digested for 30 min at 37°C in 10 ml liver digest medium. Single-cell suspensions were prepared by pouring the liver through a 70-µm cell strainer. Liver cells were suspended in 35% Percoll (Sigma-Aldrich, St. Louis, MO, USA), followed by overlaying cells onto 70% Percoll. The gradient was centrifuged at 2,000 rpm for 20 min. Interfaces containing liver leukocytes were collected, and residual erythrocytes were depleted.
Serum
Serum was collected from submandibular blood. Serum was stored at −80°C in cytokine stabilization buffer (U-CyTech Biosciences, Utrecht, Netherlands) (1:20 of collected serum volume) until ProcartaPlex Multiplex immunoassays (ThermoFisher Scientific, Waltham, MA, USA) were performed.
MDSC In Vitro Generation
MDSCs were generated in vitro by incubating freshly isolated BM cells with 250 U/ml murine GM-CSF for 4 days at 37°C in an atmosphere with 7.5% CO2. BM cells at 9 × 106–1 × 107 were cultured in α-minimum essential medium (Lonza, Basel, Switzerland), 10% fetal calf serum (Sigma Aldrich), 2 mM l-glutamine (Gibco), 1 mM sodium-pyruvate (Gibco), 100 U/ml penicillin-streptomycin (Gibco), and 0.05 mM 2-mercaptoethanol (Gibco) in Ø 15 cm culture dishes “Cell+” (Sarstedt, Germany).
Isolation of CD11b+CD11c+ and CD11b+CD11c− MDSCs
CD11b+CD11c+ MDSCs were positively isolated by magnetic-activated cell sorting using anti-CD11c MicroBeads (Miltenyi, Bergisch Gladbach, Germany) according to manufacturer’s protocol. CD11b+CD11c- MDSCs were isolated from the flow-through of CD11c isolation by loading the flow-through on a depleting LD column (Miltenyi). Purity of both MDSC subpopulations ranged between 85% and 99%.
Isolation of CD3+ T Cells
CD3+ T cells were positively isolated from splenic single-cell suspensions by magnetic-activated cell sorting using the CD3ϵ MicroBead Kit (Miltenyi) according to manufacturer’s protocol. Purity of isolated T cells was over 70%.
Mice and Bone Marrow Transplantation
Mice
Mouse strains used are listed in Supplementary Table S1.
BMT
One day before BMT, B6.bm1 recipient mice received total body irradiation with 12 Gy split in two doses 3 h apart from a 137Cs source. BM cells were depleted from T cells as described previously (18, 19). Mice were intravenously reconstituted with 5 × 106 T-cell-depleted BM (TCD-BM) in the presence or absence of 2 × 107.spleen cells (SC). In vitro-generated CD11b+CD11c+ or CD11b+CD11c− MDSCs at 1 × 106 were coinjected with the transplant. In studies analyzing the GVT effect 5 × 104 JM6 thymoma (18) were coinjected with the transplant. Clinical GVHD was evaluated according to Cooke et al. (31) by evaluating the parameters weight loss, activity, posture, fur texture, and skin integrity. Animals euthanized during the experiment due to their moribund state remained included in the calculation until the end of experiment with their final GVHD scores. All animal experiments were performed according to the international regulations for the care and use of laboratory animals and were approved by the local ethical committee Regierungspräsidium Tübingen, Germany.
Carboxyfluorescein Diacetate Succinimidyl Ester Labeling
Cells at 2 × 107 in 10 ml PBS containing 5% FCS were labeled with 50 µM carboxyfluorescein diacetate succinimidyl ester (CFSE) (ThermoFisher Scientific, MA, USA) for 10 min at 37°C in the dark.
Mixed Lymphocyte Reaction
CFSE-labeled B6.SJL-derived SCs at 2.5 × 105 were stimulated with 2.5 × 105 irradiated (33 Gy) DBA/2-derived SCs in the absence or presence of B6-derived CD11b+CD11c+ or CD11b+CD11c− MDSCs. iNOS was inhibited using 500 µM L-NG-monomethyl-arginine-citrate (L-NMMA) (Merck, Darmstadt, Germany) and PD-L2 was blocked using 10 μg/ml antimouse PD-L2 antibodies (Biocell, St. Irvine, CA, USA). Mixed lymphocyte reactions (MLRs) were cultured in α-MEM medium (Lonza) supplemented with 10% FCS (Sigma Aldrich), 2 mM l-glutamine (Gibco), 1 mM sodium-pyruvate (Gibco), 100 U/ml penicillin-streptomycin (Gibco), and 0.05 mM 2-mercaptoethanol (Gibco) for 4 days at 37°C in an atmosphere with 7.5% CO2. After 4 days, T-cell proliferation was determined using flow cytometry and percentage of T-cell suppression was calculated.
Flow Cytometry
Cells at 5 × 105–1 × 106 were stained with respective fluorochrome-conjugated antibodies. Antibodies used are listed in Supplementary Table S2. Flow cytometric analyses were performed on a LSR II flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Quantitative Reverse-Transcription Polymerase Chain reaction
Quantitative reverse-transcription polymerase chain reaction (qRT-PCR) was performed using the SsoAdvanced™ Universal SYBR® Green Supermix (BIO-RAD, Irvine, CA, USA) and analysis was performed on a CFX Connect Optics Module (BIO-RAD). Relative expression was determined using the comparative CT method. Mouse aryl hydrocarbon receptor-interacting protein (AIP) was used as a housekeeping gene. Primer sets used are listed in Supplementary Table S3.
Cytokine Analysis Using ProcartaPlex™ Multiplex Immunoassay
Cytokine concentrations of 12.5 µl blood serum or 25 µl cell culture supernatant were analyzed by ProcartaPlex™ multiplex immunoassays (ThermoFisher Scientific) according to manufacturer’s protocol. Analyses were performed on a BIO-RAD Bioplex 200 system (BIO-RAD).
RNA Isolation and Quality Control
FACS-sorted CD11b+CD11c+ and CD11b+CD11c− MDSCs were collected in RLT buffer (QIAGEN, Hilden, Germany) supplemented with 2-mercaptoethanol followed by RNA extraction using the RNeasy Mini Kit (Qiagen) and adding an on-column DNA digestion step according to manufacturer’s instructions. Total RNA was quantitatively and qualitatively assessed using the absorbance-based Take3 microvolume plate system on a Cytation 5 instrument (BioTek, Bad Friedrichshall, Germany) and the Standard Sensitivity RNA Analysis DNF-471 Kit on a 12-channel Fragment Analyzer (Agilent Technologies, Santa Clara, CA, USA), respectively. Concentrations averaged at 310 ng/µl while RIN values ranged from 8.6 to 10, with an average of 9.8.
Whole Transcriptome Profiling With PolyA Enrichment (mRNA-Seq)
MDSC-derived RNA samples were normalized, and a RNA input of 100 ng was used for library construction with the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina #E7760, together with the NEBNext Poly(A) mRNA Magnetic Isolation Module #E7490 upstream and the NEXNext Multiplex Oligos for Illumina #E7600 downstream (New England Biolabs, Frankfurt am Main, Germany). Ampure XP beads (Beckman Coulter, Brea, CA, USA) were used for double-stranded cDNA purification. mRNA sequencing libraries were quantified by the High Sensitivity dsDNA Quanti-iT Assay Kit (ThermoFisher Scientific) on a Synergy HTX (BioTek). Library molarity averaged at 134 nM. Final library size distribution was assessed (smear analysis of 364 bp average and adapter dimer presence <0.5%) by the High Sensitivity Small Fragment DNF-477 Kit on a 12-channel Fragment Analyzer (Agilent Technologies). All sequencing libraries passed quality check, were normalized, pooled, and spiked in with PhiX Control v3 (Illumina, San Diego, CA, USA). The library pool was subsequently clustered with the HiSeq 3000/4000 SR Cluster Kit on a cBot and sequenced on a HiSeq 3000 Sequencing System (Illumina) with single index, single read at 85 bp length (Read parameters: Rd1: 85, Rd2: 8), reaching an average depth of 29 million Pass-Filter reads per sample (11% CV).
mRNA-Seq Computational Analysis
Illumina reads were converted to FASTQ files and aligned to the mouse reference genomes from Ensembl 70 (http://www.ensembl.org) using the STAR v2.5.2 program on default settings (32). SAM files were converted by samtools v0.1.18 (33) to BAM files. Sequenced read quality was checked with FastQC v0.11.2 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and alignment quality metrics were calculated using the RNASeQC v1.1.8 (34). Duplication rates were assessed with bamUtil v1.0.11 (35) and dupRadar v1.4 (36). Gene expression levels were quantified by Cufflinks v2.2.1 (37) to get reads per kilobase per million mapped reads (RPKM) as well as FeatureCounts (38) to get read counts. Differential expression analysis was performed based on voom-normalized (39) read counts as input for the Bioconductor R package LIMMA (40). The batch number was used as a factor in the LIMMA linear regression model. p-values were corrected for multiple testing by Benjamini–Hochberg. Complete mRNA-sequencing (mRNA-Seq) data are available at Gene Expression Omnibus (GEO accession number: GSE182262).
Statistics
Data were analyzed using Mann-Whitney U test or unpaired Student’s t-test. For multiple comparisons ANOVA Tukey multiple comparison test or Kruskal-Wallis test were used. Survival studies were analyzed using Log-Rank (Mantel-Cox) test. Results were considered significant if p < 0.05. Statistical tests were performed with GraphPad Prism 8.
Results
Expression of CD11b and CD11c Distinguishes Two Subpopulations of In Vitro-Generated MDSCs
MDSCs were generated from BM cells in the presence of GM-CSF. After 4 days, more than 90% of cells expressed CD11b and Gr-1 indicative for successful MDSC generation in vitro (Figure 1A). By staining CD11b and CD11c in vitro-generated MDSCs could be separated into two major subpopulations. Eighty percent of MDSCs exhibited solely CD11b positivity, while 20% coexpressed CD11b and CD11c (Figure 1B). CD11b+CD11c+ MDSCs could be distinguished from CD11b+CD11c− MDSCs by decreased Gr-1 expression. To assign both MDSC subsets to M-MDSCs (Ly-6ChighLy6G−) or PMN-MDSCs (Ly-6ClowLy6G+), we costained for Ly-6C and Ly-6G. CD11b+CD11c+ MDSCs consist of about 60% M-MDSCs and low percentage of PMN-MDSCs, while 20% of the cells neither expresses Ly-6G and Ly-6C. CD11b+CD11c− MDSCs represent a mixture of M-MDSCs (about 50%) and PMN-MDSCs (40%) (Figure 1C). To further define differences between both MDSC subpopulations, we analyzed expression of surface markers often coexpressed on CD11c-positive cells. CD11b+CD11c+ MDSCs exhibited increased expression of antigen-presenting cell (APC)-associated markers MHC class II (I-Ab), F4/80, CD40, the activating costimulatory molecules CD80 and CD86, as well as the inhibitory molecules PD-L1 (CD274) and PD-L2 (CD273) (Figure 1D). These results clearly show that by using GM-CSF for MDSC generation in vitro, most of the cells exhibit the classical CD11b+CD11c− phenotype, while about 20% of cells showed coexpression of CD11c and APC-associated markers.
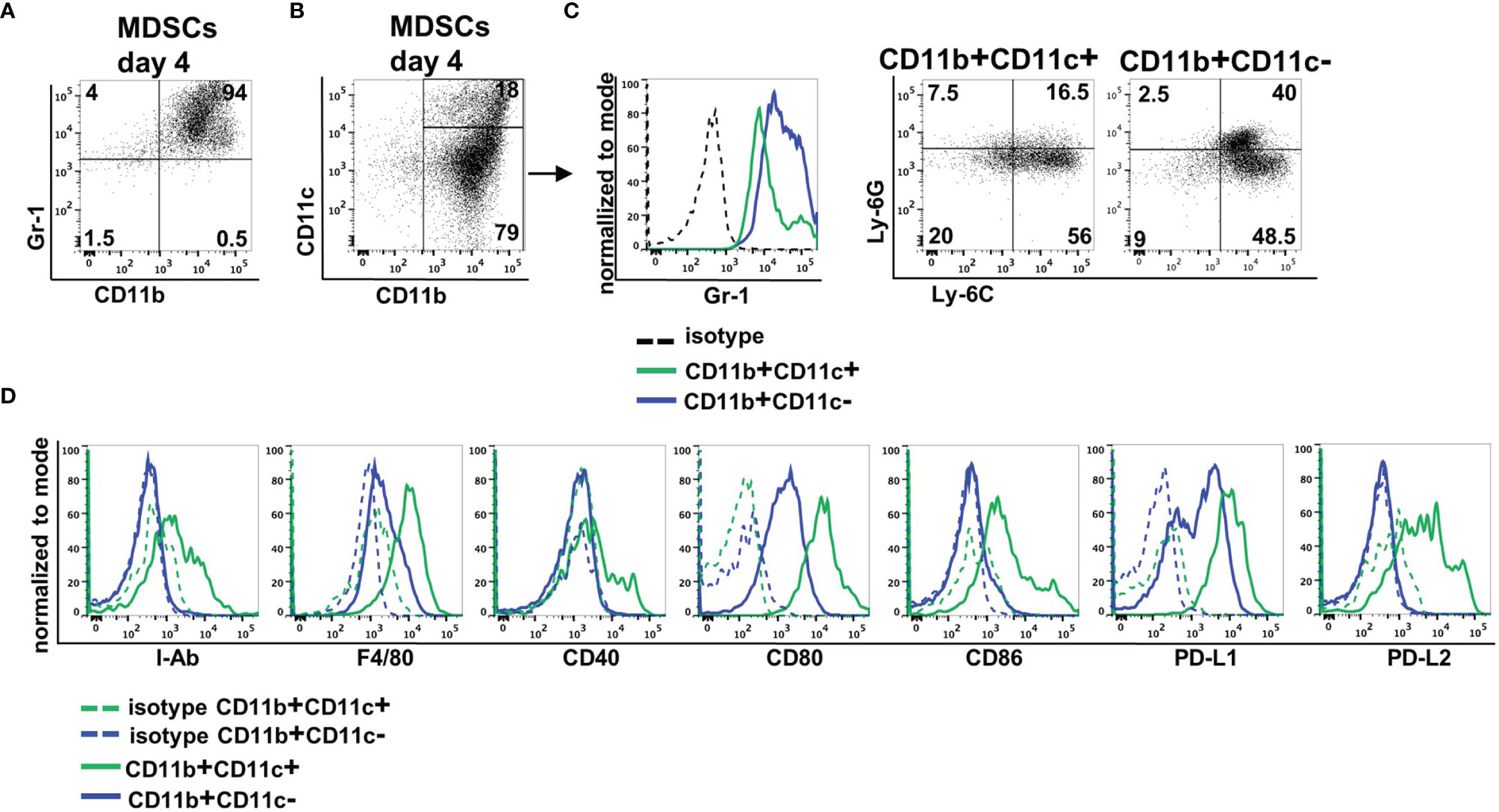
Figure 1 In vitro-generated MDSCs consist of CD11b+CD11c+ and CD11b+CD11c− MDSCs. MDSCs were generated from BM cells in the presence of GM-CSF. (A) After 4 days, MDSCs were stained for CD11b and Gr-1 expression or (B) for CD11b and CD11c expression. (C) Expression of Gr-1 was determined on CD11b+CD11c+ and CD11b+CD11c− MDSCs, and CD11b+CD11c+ and CD11b+ and CD11c+ MDSCs were designated to M-MDSCs or PMN-MDSCs by staining Ly-6C and Ly-6G. (D) Staining of various surface markers to define differences in the expression on CD11b+CD11c+ and CD11b+CD11c− MDSCs. FACS diagrams show one representative experiment out of at least three experiments performed.
CD11b+CD11c+ MDSCs Exhibit Increased Immunosuppressive Capacity Compared With CD11b+CD11c− MDSCs In Vitro and Shift the T-Cell Response Towards Type 2 Immunity
Since CD11c is expressed on APCs such as dendritic cells, macrophages, and a small subset of B cells, we next defined whether CD11b+CD11c+ MDSCs exhibit immune-activating or suppressing functions. Therefore, B6-derived in vitro-generated MDSCs were separated by CD11c Micro Beads into CD11b+CD11c+ and CD11b+CD11c− MDSCs (Supplementary Figure S1A) with a purity of about 95% for both populations (Supplementary Figure 1B). Purified MDSCs were added at different numbers to CFSE-labeled B6-SJL (H-2b, CD45.1+) spleen cells, which were activated by DBA/2-derived (H-2d, CD45.2+) irradiated spleen cells. Using the congenic marker CD45.1 expressed solely on CFSE-labeled effector cells, proliferation of CD45.1+CD4+ and CD45.1+CD8+ T cells was determined. Both MDSC subpopulations efficiently suppressed T-cell proliferation, but CD11b+CD11c+ MDSCs exhibited strongly increased inhibitory capacity especially towards CD8+ T-cell proliferation (Figure 2A). Due to the differences in the immunosuppressive capacity, expression of molecules attributed to mediate suppression was defined in the isolated MDSC subsets by qRT-PCR. iNOS, IDO, and HO-1 expression were increased in CD11b+CD11c+ MDSCs while expression of arginase-1 and the anti-inflammatory modulators TGF-β and IL-10 were similar in both MDSC subsets (Figure 2B). By using the iNOS inhibitor L-NMMA, the immunosuppressive capacity of CD11b+CD11c− MDSCs towards CD4+ and CD8+ T cells was abolished to nearly 100%. L-NMMA-treated CD11b+CD11c+ MDSCs, however, maintained about 40% of their inhibitory function towards both T-cell subsets (Figure 2C). IDO or HO-1 inhibitors, however, did not affect the inhibitory capacity of CD11b+CD11c+ MDSCs (data not shown), indicating that immunosuppression is mediated by iNOS activity and a not yet identified mechanism. Since PD-L1 and PD-L2 were strongly upregulated on CD11b+CD11c+ MDSCs (Figure 1A) and are known to inhibit T-cell activation by binding to PD-1, impact of PD-L1 and PD-L2 on the suppressive function of CD11b+CD11c+ MDSCs was defined. PD-L1 function was abrogated by using isolated CD11b+CD11c+ MDSCs generated from BM cells of PD-L1−/− mice and PD-L2 blocking was achieved by antagonistic antibodies. Purified PD-L1−/− CD11b+CD11c+ MDSC added to allogeneic-activated spleen cells suppressed T-cell proliferation of CD4+ and CD8+ T cells comparable with PD-L1 expressing MDSCs derived from B6 wildtype (WT) mice (Figure 2D). Likewise, adding PD-L2 antagonistic antibodies to allogeneic-activated T cells in the presence of B6-derived CD11b+CD11c+ MDSCs did not impair T-cell suppression (Figure 2E) clearly showing that neither PD-L1 or PD-L2 contribute to CD11b+CD11c+ MDSC-mediated immunosuppression in vitro.
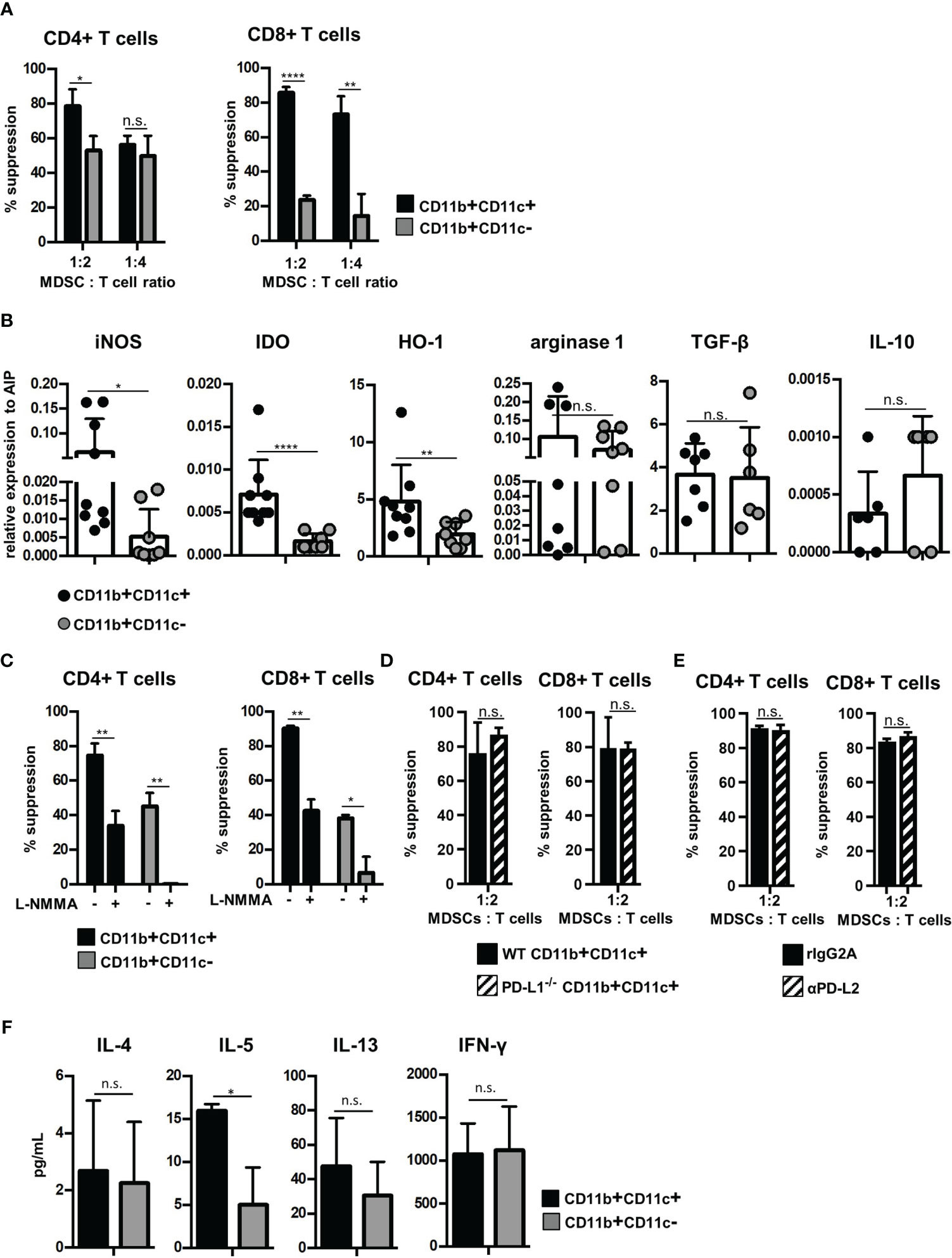
Figure 2 CD11b+CD11c+ MDSCs exhibit increased immunosuppressive capacity than CD11b+CD11c− MDSCs and induce type 2 immunity in vitro. CD11b+CD11c+ and CD11b+CD11c− MDSCs were isolated from B6-derived (H-2b, CD45.2+) in vitro-generated MDSCs. (A) CD11b+CD11c+ or CD11b+CD11c− MDSCs were cocultivated with B6.SJL-derived (H-2b, CD45.1+) CFSE-labeled spleen cells stimulated by irradiated allogeneic DBA/2-derived (H-2d, CD45.2+) spleen cells. After 4 days, CD45.1+ T cells were stained for CD3, CD4, and CD8 and suppression of CD4+ and CD8+ T-cell proliferation was calculated. (B) CD11b+CD11c+ and CD11b+CD11c− MDSC subpopulations were analyzed for relative expression of immunosuppressive molecules by qRT-PCRs. (C) CD11b+CD11c+ or CD11b+CD11c− MDSCs were cocultivated with B6.SJL-derived CFSE-labeled spleen cells stimulated by irradiated allogeneic DBA/2-derived spleen cells in the absence or presence of iNOS inhibitor L-NMMA (500 µM). After 4 days, suppression of CD4+ and CD8+ T-cell proliferation was determined. (D) B6-derived wildtype (WT) and PD-L1−/− CD11b+CD11c+ or CD11b+CD11c− MDSCs were cocultured with CFSE-labeled B6.SJL-derived spleen cells stimulated with irradiated allogeneic DBA/2 spleen cells. (E) To block PD-L2, antagonistic PD-L2 antibodies or recombinant isotype control were added to MLRs, in which CFSE-labeled B6.SJL-derived spleen cells were stimulated with irradiated allogeneic DBA/2 spleen cells in the presence of CD11b+CD11c+ MDSCs. (F) CD11b+CD11c+ or CD11b+CD11c− MDSCs were cocultivated with B6.SJL-derived CFSE-labeled spleen cells stimulated by irradiated allogeneic DBA/2-derived spleen cells. After 4 days, secretion of cytokines associated with type 2 T-cell immunity (IL-4, IL-5, and IL-13) or type 1 T-cell immunity (IFN-γ) were determined in the supernatants. (A) Data represent the mean value ± SD of triplicates of one representative experiment out of four experiments performed. (B) Data represent the mean value ± SD of six to nine samples. (C) Data represent the mean value ± SD of triplicates of one representative experiment out of three experiments performed. (D) Data represent the mean value ± SD of n = 3 PD-L1−/− and WT mice. (E) One experiment out of two experiments performed. Values present the mean value ± SD of triplicates. (F) Data represent the mean value ± SD of n = 3 experiments. (A, C–F) Student’s t-test. (B) Mann-Whitney U test. *p ≤ 0.05; **p ≤ 0.01; ****p ≤ 0.0001. n.s., not significant.
Although inhibition of T-cell expansion designates the main feature of MDSCs, MDSCs functions are also attributed to modulate the Th1/Th2 induction especially in vivo. To define the T-cell polarizing capacity of MDSC subsets in vitro, supernatants of allogeneic MLRs performed in the presence of CD11b+CD11c+ or CD11b+CD11c− were analyzed for composition of type 1- and type 2-asssociated cytokines. Of the type 2-specific cytokines analyzed, IL-5 secretion was strongly upregulated by CD11b+CD11c+ MDSCs, while IL-4 and IL-13 was unaffected. Th1-specific IFN-γ production was similar in CD11b+CD11c+- and CD11b+CD11c−-treated cultures (Figure 2F). In summary, these results show that CD11b+CD11c+ and CD11b+CD11c− MDSCs can be distinguished phenotypically and functionally.
Exclusively the CD11b+CD11c+ MDSC Subset Prevents GVHD While Maintain the GVT Effect
Due to functional differences between both MDSC subsets in vitro, we tested their potential to block GVHD development after allogeneic BMT. We used the single MHC class I-disparate allogeneic BMT model, B6 (H-2Kb)→B6.bm1 (H-Kbm1), in which lethally irradiated B6.bm1 mice were reconstituted with TCD-BM and SCs from B6 mice. At the day of BMT, isolated CD11b+CD11c+ or CD11b+CD11c− MDSCs were cotransplanted together with TCD-BM and SCs. While 52% of mice transplanted with TCD-BM and SCs succumbed to the disease associated with high GVHD scores and weight loss of about 20%, CD11b+CD11c+ co-transplantation rescued 78% of the mice from disease-induced mortality reflected by a reduced GVHD score and less weight loss. In contrast, cotransplantation of CD11b+CD11c− MDSCs totally failed to prevent GVHD development. Surviving rates and GVHD scores were undistinguishable in mice receiving TCD-BM and SC and mice cotreated with CD11b+CD11c− MDSCs. Control mice receiving TCD-BM survived and did not develop GVHD (Figures 3A–C).
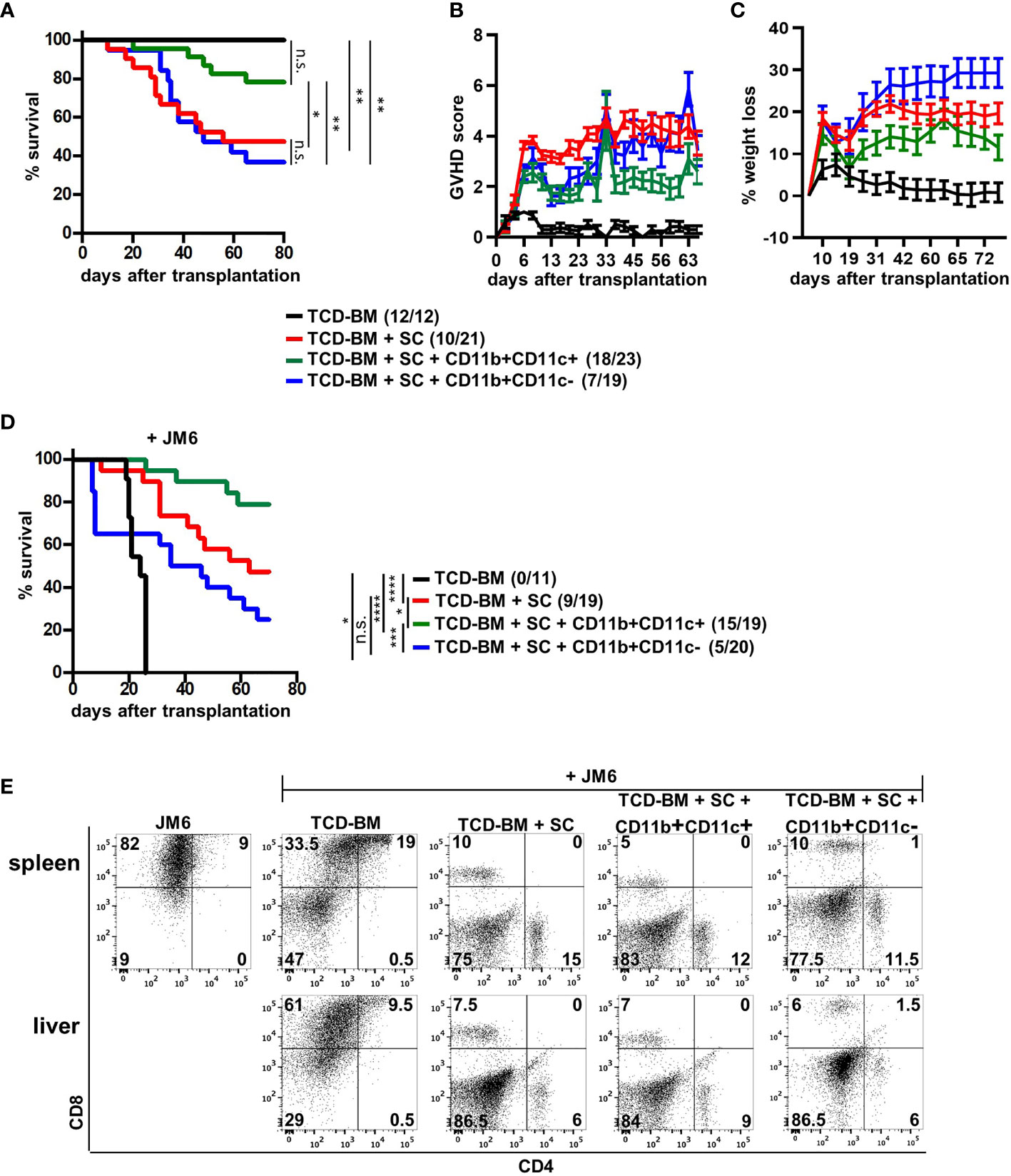
Figure 3 CD11b+CD11c+ expression defines the subpopulation of MDSCs able to prevent GVHD without disabling the GVT effect. (A–E) Lethally irradiated B6.bm1 mice (H-2Kbm1) were reconstituted with B6-derived (H-2Kb) TCD-BM and SCs with or without B6-derived CD11b+CD11c+ or CD11b+CD11c− MDSCs. (D, E) Mice were additionally coinjected with the CD8+CD4− syngeneic thymoma tumor cell line JM6 at day of transplantation. (A, D) Survival was determined. Surviving animals/total animals treated are indicated in brackets. (B) Clinical GVHD scores (C) and percentage of weight loss were determined. (E) Presence of tumor cells was analyzed in spleens and livers by staining for CD4 and CD8 at day mice were sacrificed due to their moribund state or at the end of the experiment. (A, C) Kaplan-Meier method and Log-rank test. (B, C) Data represent the mean value ± SEM. (D) Representative FACS diagrams of one mouse/group are displayed. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001; n.s., not significant.
Maintenance of the GVT effect is a basic requirement for the application of allogeneic stem cell transplantation in the treatment of hematological malignancies. Therefore, the impact of MDSC subpopulations on the GVT effect was determined by coinjecting the CD8+CD4− syngeneic thymoma cell line JM6 in BM-reconstituted mice. All mice receiving only BM cells and JM6 died between 20 and 24 days after BMT from tumor development (Figure 3D) reflected by high numbers of tumor cells in spleen and liver (Figure 3E). Although transplantation of TCD-BM and SC totally prevented tumor growth in all mice due to the presence of tumor-reactive splenic mature T cells, 50% of the mice died by GVHD development. Most importantly, about 80% of the mice cotreated with CD11b+CD11c+ MDSCs survived reflected by the absence of tumor cells in spleen and liver. Five mice from this group died during the experiment. They were all tumor free but succumbed GVHD-induced death. Although all mice transplanted with CD11b+CD11c− MDSCs did not develop spleen or liver tumors, only 50% of the mice survived due to GVHD development, as shown in Figure 3A. Thus, our experiments define CD11b+CD11c+ MDSCs as the subpopulation of in vitro-generated MDSCs able to protect BMT mice from GVHD development without impairing antitumor cytotoxicity.
CD11b+CD11c+-Mediated GVHD Inhibition Does Not Prevent Expansion and Homing of Allogeneic T Cells In Vivo But Requires Induction of Type 2 Immunity
Next, we questioned whether GVHD prevention by CD11b+CD11c+ MDSCs was due to impaired expansion of allogenic GVHD-inducing T cells, since CD11b+CD11c+ MDSCs most efficiently blocked allogeneic T-cell proliferation in vitro. By transplanting SCs from B6.SJL (CD45.1+) mice together with B6-derived TCD-BM (CD45.2+) into irradiated B6.bm1 (CD45.2+) mice, homing and expansion of allogeneic GVHD-inducing T cells were followed by staining the congenic marker CD45.1 in spleen and liver of transplanted mice. CD45.1+ T cells were detectable in spleen and the GVHD target organ liver already at day 3 after BMT in mice transplanted with TCD-BM and SCs. An increase of about 200-fold was achieved 10 days after BMT in both organs. However, cotransplantation of MDSCs did not prevent invasion and expansion of allogeneic T cells independent whether isolated CD11b+CD11c+ or CD11b+CD11c− MDSCs were transferred (Figure 4A). Ten days after BMT, allogeneic T-cell numbers continuously decreased and mice became lymphopenic at the time when clinical signs of GVHD were manifested (data not shown).
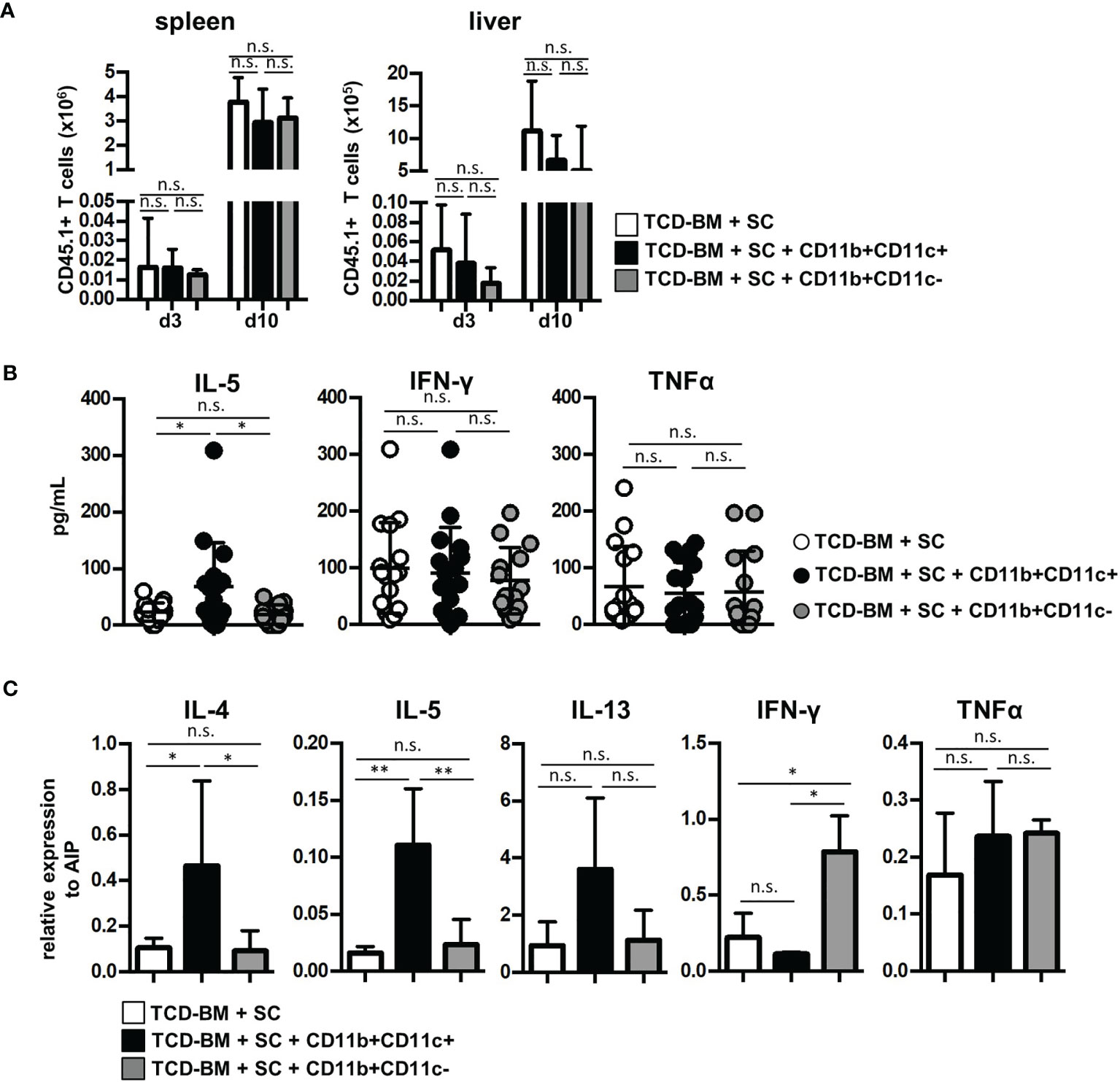
Figure 4 Cotransplantation of CD11b+CD11c+ MDSCs does not prevent allogeneic T-cell expansion and homing but induces type 2 immunity. (A–C) Lethally irradiated B6.bm1 (H-2Kbm1, CD45.2) mice were reconstituted with B6-derived (H-2Kb, CD45.2) TCD-BM and B6.SJL-derived (H-2Kb, CD45.1) spleen cells in the presence or absence of B6-derived (H-2Kb; CD45.2) CD11b+CD11c+ or CD11b+CD11c− MDSCs. (A) Spleen and liver were analyzed for infiltrated allogeneic CD45.1+ T cells 3 and 10 days after transplantation. (B) Ten days after transplantation, serum cytokine concentrations of Th2- (IL-5) and Th1-associated (IFN-γ and TNF-α) cytokines were determined. (C) Ten days after transplantation, splenic T cells were isolated and relative mRNA expression of Th2- (IL-4, IL-5, IL-13) and Th1-associated (TNF-α, IFN-γ) cytokines was analyzed by qRT-PCRs. (A) Data represent the mean value ± SD of three mice/group. Kruskal-Wallis test. (B) Data represent the mean value ± SD of 14–16 mice/group. (C) Data represent the mean value ± SD of n = 3–5 samples with cells from three to five pooled mice/sample. (B, C) ANOVA Tukey multiple comparison test. *p ≤ 0.05; **p ≤ 0.01. n.s., not significant.
Since CD11b+CD11c+-mediated inhibition of GVHD did not impair allogeneic T-cell expansion, we determined whether CD11b+CD11c+ MDSCs support T-cell polarization towards Th2 immunity known to be advantageous for GVHD inhibition. Serum level of Th2-specific cytokine IL-5 was only elevated in mice treated with CD11b+CD11c+ MDSC. Type 2-specific cytokines such as IL-4 and IL-13 were not detectable, probably due to concentrations below the detection level of the kit used or degradation after freezing and thawing (41). Serum levels of Th1-associated cytokines IFN-γ or TNF-α were not altered by MDSC treatment (Figure 4B). To further prove CD11b+CD11c+-mediated type 2 polarization, mRNA expression of allogeneic T cells isolated from mice reconstituted with TCD-BM and SCs or cotransplanted with either CD11b+CD11c+ or CD11b+CD11c− MDSCs was determined. T cells isolated from CD11b+CD11c+ MDSC-treated mice expressed significantly increased levels of IL-4 and IL-5, while IL-13 was only slightly upregulated (Figure 4C). While TNF-α expression was unaffected by MDSC treatment, IFN-γ levels increased in T cells from CD11b+CD11c-treated mice further indicating that CD11b+CD11c− MDSCs support Th1 immunity and GVHD induction.
To prove the indispensability of type 2 polarization for GVHD prevention in mice treated with CD11b+CD11c+ MDSCs, we reconstituted B6.bm1 mice with TCD-BM and allogeneic SCs either derived from STAT6-deficient (STAT6−/−) or B6 WT mice and CD11b+CD11c+ MDSCs. STAT6−/− splenic T cells are unable to differentiate into type 2 T cells but retain their ability to turn into type 1 T cells and to induce GVHD indistinguishable from STAT6-expressing WT T cells as shown previously (18). Cotransplantation of CD11b+CD11c+ MDSCs in mice reconstituted with STAT6−/− SCs failed to prevent GVHD and 64% of the mice succumbed to the disease, while only 18% of the mice receiving B6-derived WT SCs and CD11b+CD11c+ MDSCs developed lethal GVHD associated with increased GVHD scores (Figures 5A, B). In accordance to the survival data, IL-5 levels were only increased in mice reconstituted with B6-derived WT SCs, while IFN-γ levels were elevated in serum from mice reconstituted with STAT6−/− SCs. Differences in the concentration of TNF-α levels were not detected (Figure 5C). In summary, these results clearly show that CD11b+CD11c+ MDSCs do not impair the expansion and homing of allogeneic T cells in lymphatic and GVHD target organs but prevent GVHD induction by shifting the T-cell response towards type 2 immunity.
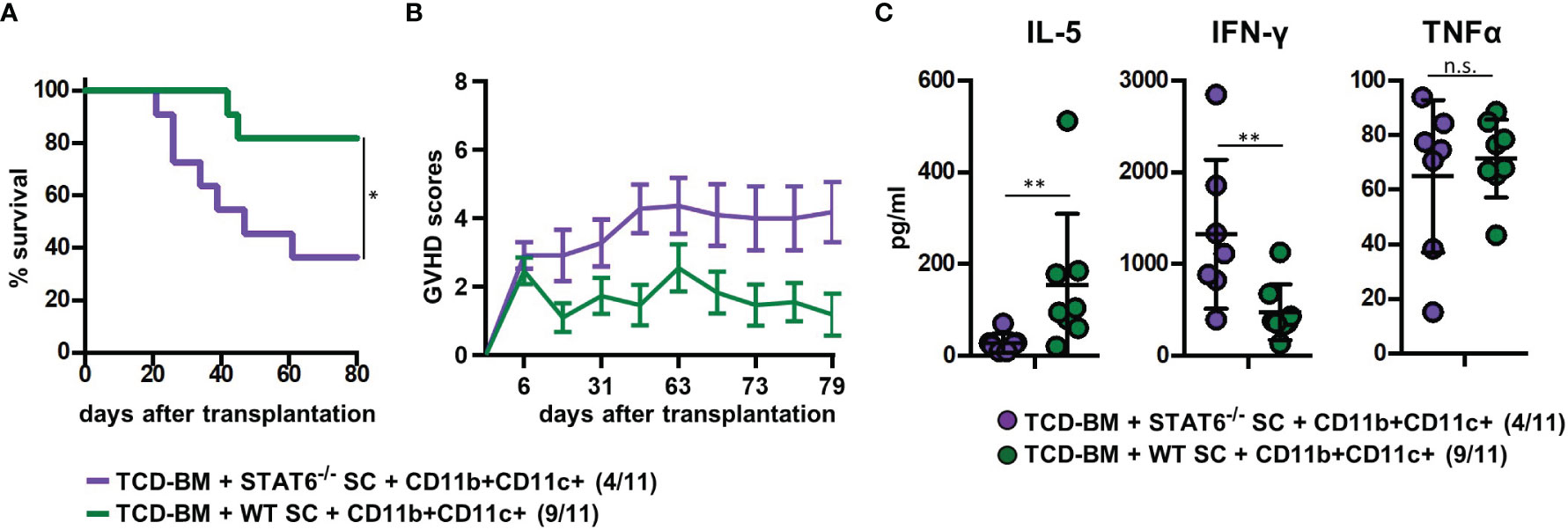
Figure 5 Type 2 immune induction by cotransplanted CD11b+CD11c+ MDSCs is required for GVHD prevention. Lethally irradiated B6.bm1 (H-2Kbm1) recipient mice were reconstituted with B6-dervied (H-2Kb) T-cell-depleted bone marrow (TCD-BM) and SCs either derived from B6 wild-type (WT) mice (H-2Kb) or STAT6−/− mice (H-2Kb). B6-derived (H-2Kb) CD11b+CD11c+ MDSCs were cotransplanted at day of transplantation. (A) Survival and (B) GVHD scores were analyzed. Surviving animals/total animals treated are indicated in brackets. (C) Ten days after transplantation, serum cytokine concentrations of Th2- (IL-5) and Th1-associated (IFN-γ and TNF-α) cytokines were determined. (A) Data represent the mean value ± SEM. (B) Kaplan-Meier method and Log-rank test. (C) Mann-Whitney U test. *p ≤ 0.05; **p ≤ 0.01; n.s., not significant.
Comparative Transcriptome Analysis Between CD11b+CD11c+ and CD11b+CD11c− MDSCs
Since CD11b+CD11c+ and CD11b+CD11c− MDSCs can be clearly distinguished by their ability to interfere with GVHD development, we aimed to define genes and signaling pathways mediating immunosuppressive functions of CD11b+CD11c+ MDSCs in the context of BMT. Comparison of the transcriptome between CD11b+CD11c+ and CD11b+CD11c− MDSCs was done by mRNA-Seq. Principal component analysis (PCA) of two experiments displays the degree to which the transcriptome of CD11b+CD11c+ MDSCs differs from CD11b+CD11c− MDSCs. In both experiments, PCA analysis clearly separated two clusters corresponding to the CD11b+CD11c+ and CD11b+CD11c− MDSCs (Supplementary Figure S2), revealing a totally different transcriptome of both MDSC subpopulations which further underlies the functional differences between both subpopulations. In total, 2,783 differentially expressed genes (p-value <0.01, RPKM >5) were identified, from which 1,443 genes were upregulated and 1,340 genes were downregulated in CD11b+CD11c+ MDSCs compared with CD11b+CD11c− MDSCs. Focusing on the transcripts that were highly upregulated or downregulated in the CD11b+CD11c+ MDSC subpopulation, we performed enrichment analysis using GO database. Target genes upregulated in CD11b+CD11c+ MDSCs can be largely grouped into the biological and functional categories (Table 1): 1. cell movement and migration, 2. cell adhesion, 3. leukocyte activation and immune response, 4. ERK1 and ERK2 cascade, 5. response to cytokine, and 6. Stress response. Identified GO terms and linked target genes are listed in Table 1 and Supplementary Table S4. Target genes downregulated in CD11b+CD11c+ MDSCs are mostly related to immune and defense response against other organisms such as bacteria or fungi, which might be related to their immunosuppressive phenotype. Identified GO terms and linked target genes are listed in Table 2 and Supplementary Table S5. Focusing on the transcripts that were highly upregulated in CD11b+CD11c+ MDSCs, we ascertained the target genes that were upregulated more than 15-fold in CD11b+CD11c+ compared with CD11b+CD11c− MDSCs (Table 3). With a fold change of 57.96, CCL17 was the highest expressed target gene in CD11b+CD11c+ compared with CD11b+CD11c− MDSCs. Together with CCL22, which showed a 24-fold overexpression in CD11b+CD11c+ MDSCs, both chemokines are known to attract CCR4-bearing Th2 cells and serve as markers for the severity of Th2-mediated atopic dermatitis (42, 43). Furthermore, the fatty acid translocase CD36 is 20-fold stronger expressed in CD11b+CD11c+ MDSCs than in CD11b+CD11c− MDSCs and serves in association with the platelet-activating factor receptor as an important mediator of Th2-mediated house dust mite allergy development (44). Increased expression of CCL17, CCl22, and CD36 by CD11b+CD11c+ MDSC in comparison with CD11b+CD11c− MDSCs was confirmed by qRT-PCR. Additionally, CD36 was found to be strongly expressed on the surface of CD11b+CD11c+ MDSCs (Supplementary Figure S3). In summary, transcriptome analysis further underlines the functional differences between CD11b+CD11c+ and CD11b+CD11c− MDSCs and indicates candidate genes and pathways, which might contribute to the therapeutic potential of CD11b+CD11c+ MDSCs.
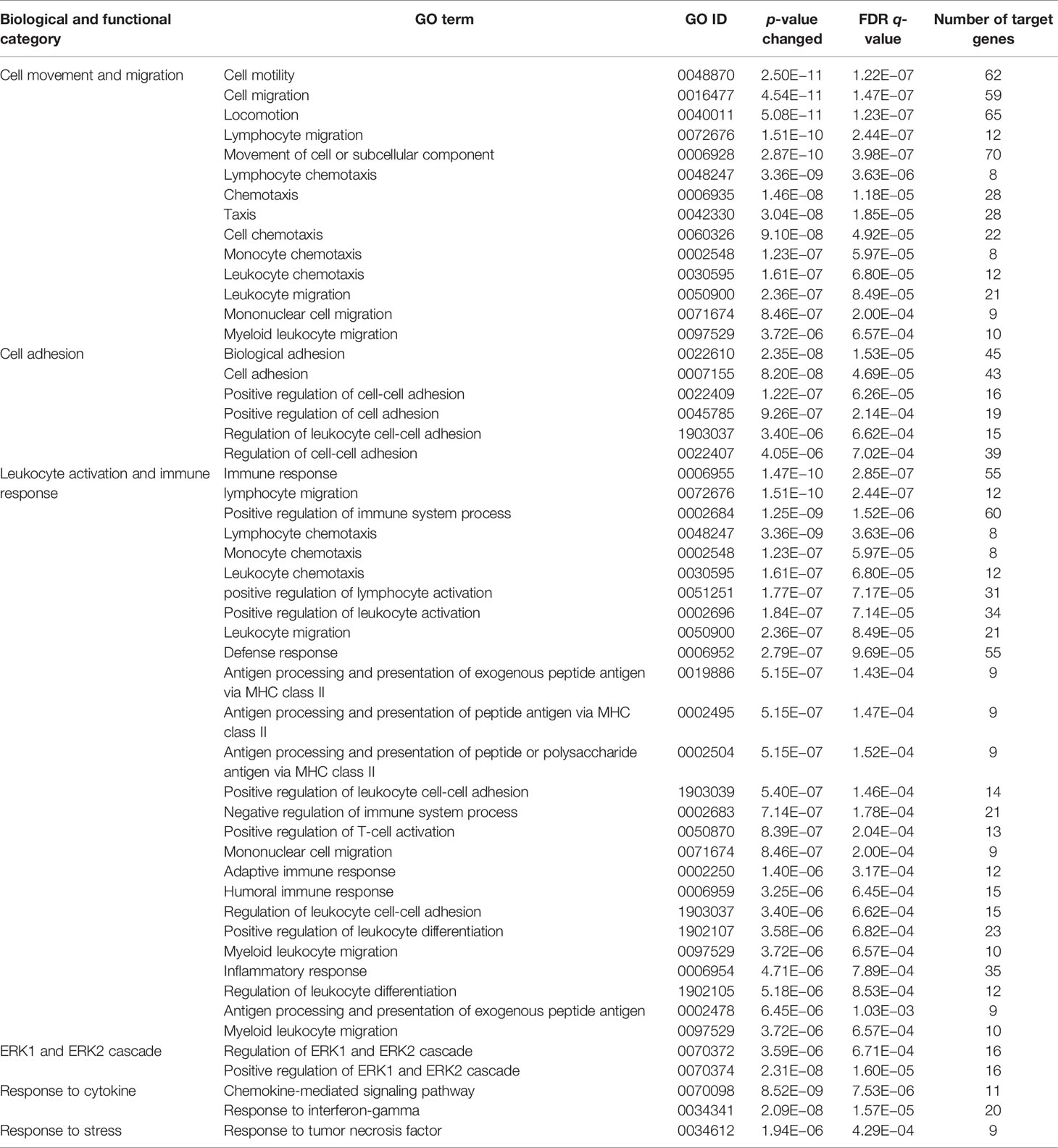
Table 1 GO term analysis and identification of biological and functional processes activated in CD11b+CD11c+ MDSCs compared with CD11b+CD11c− MDSCs.
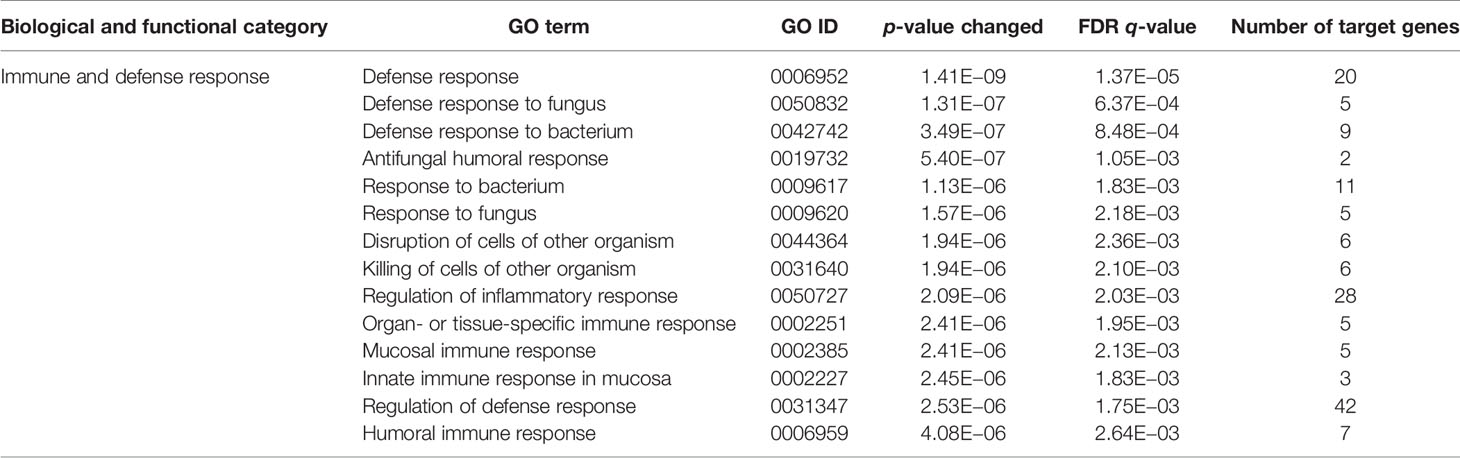
Table 2 GO term analysis and identification of biological and functional processes downregulated in CD11b+CD11c+ MDSCs compared with CD11b+CD11c− MDSCs.
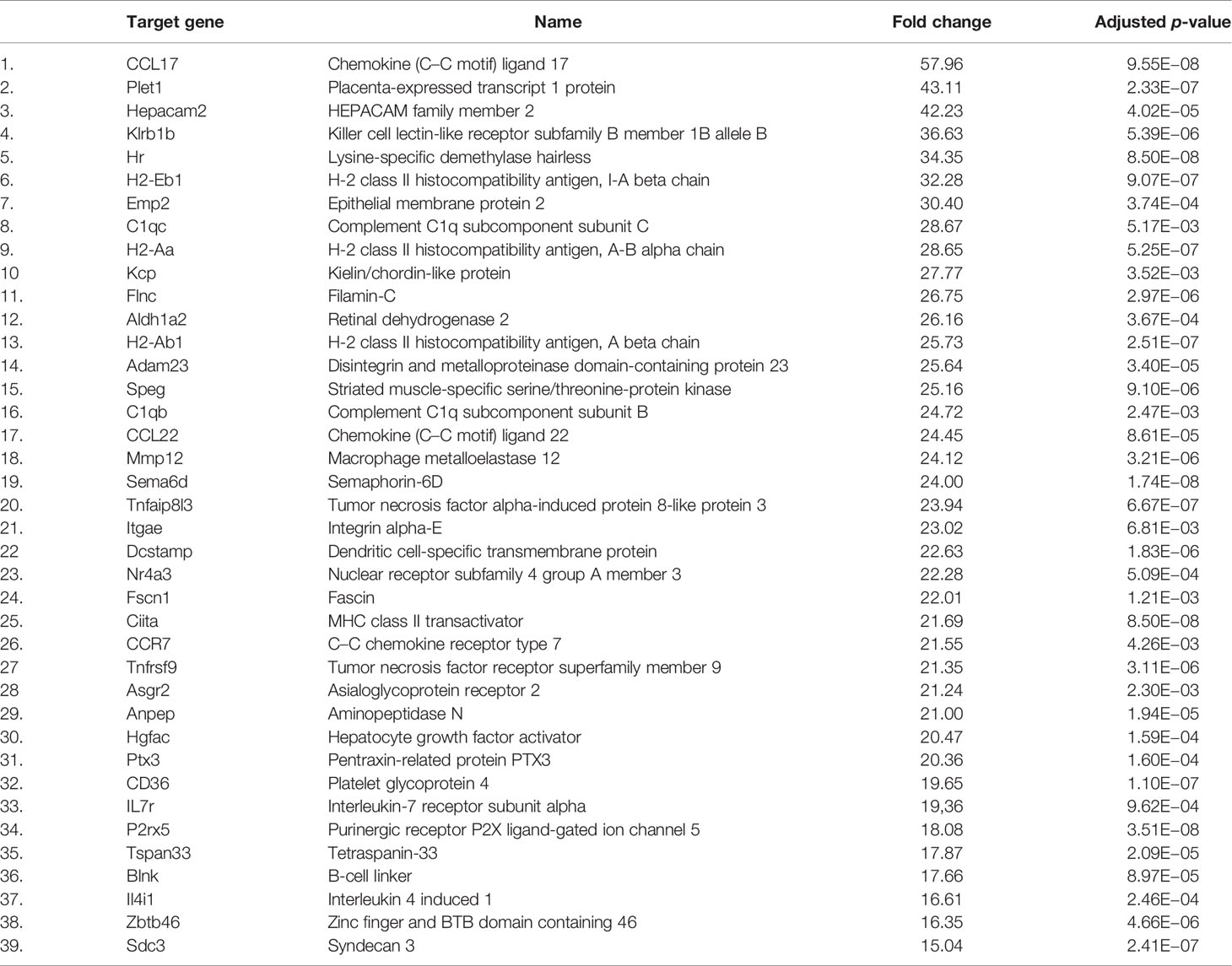
Table 3 Most strongly upregulated genes (>15-fold increase) in CD11b+CD11c+ MDSCs compared with CD11b+CD11c− MDSCs.
Discussion
Allogeneic hematopoietic cell transplantation is considered an important treatment strategy to cure life-threatening malignant hematological diseases, however, with the limitation of GVHD development. Initial treatment comprises steroid therapy, while second-line treatment often includes immunomodulatory therapies to dampen the destructive capacity of allogeneic T cells. MDSCs are recognized as strong modulators of T-cell functions and were already applied in preclinical models as cellular therapy for GVHD prevention. Considering the heterogenicity of in vitro-generated MDSCs, we aimed to define the MDSC subset responsible for GVHD prevention. To our knowledge, we show here for the first time that only a small proportion of MDSCs, which have been generated in vitro form BM cells, fulfills GVHD-inhibiting functions. This subset is characterized by the coexpression of Gr-1, CD11b, and CD11c. Gr-1+CD11b+CD11c+ MDSCs effectively prevent GVHD development and maintain antitumor cytotoxicity of allogeneic T cells, while the majority of the in vitro-generated MDSCs expressing Gr-1+CD11b+CD11c− are totally inefficient to dampen GVHD, although they block T-cell expansion in vitro. Extensive differences in the transcriptomic landscape of both populations underlined their various in vivo functions, indicating that the success of cellular therapies using MDSCs requires a thoughtful characterization of MDSC subset functions in vitro and in vivo.
In a clinically relevant BMT model with disparity in only one MHC molecule (B6 into B6.bm1), we defined which subset of in vitro-generated MDSCs prevents GVHD. MDSC were generated from BM cells in the presence of GM-CSF. More than 90% of the cells exhibited Gr-1 and CD11b expression, but only a small proportion of about 10%–25% cells showed coexpression of the integrin alphaX CD11c, which is also found at high levels not only on the surface of dendritic cell, but also on monocytes, macrophages, neutrophils, and subsets of NK, B, and T cells. By separating MDSCs into CD11b+CD11c+ and CD11b+CD11c− subsets, a clear correlation with the classically defined M-MDSCs and PMN-MDSCs was not observed. While CD11b+CD11c− MDSCs represented a mixture of M-MDSCs and PMN-MDSCs, CD11b+CD11c+ MDSCs consisted mainly of M-MDSCs, a small proportion of PMN-MDSCs and cells, which neither expressed Ly-6C nor Ly-6G. CD11b+CD11c+ MDSCs further expressed higher levels of APC-associated markers such as CD80, CD86, MHC class II, and F4/80 compared with their CD11c-negative counterparts. Importantly, a single injection of CD11b+CD11c+ MDSCs inhibited GVHD development in about 80% of the BM-transplanted mice, while adoptive transfer of CD11b+CD11c− MDSCs had no impact on disease development. CD11b+CD11c+-treated mice, however, remain immunosufficient since syngeneic tumor cells were efficiently eradicated in 100% of the mice. Although BMTs are routinely applied to abrogate residual B-cell lymphoma cells, we used the CD8+CD4− JM6 thymoma cell line. To our knowledge, JM6 is currently the only available syngeneic tumor cell line for B6.bm1 mice. By using JM6 cells, we cannot totally exclude that MDSCs interact with JM6 tumor growth in transplanted mice. However, in a previous work, unseparated MDSCs, which represent a mixture of CD11b+CD11c+ and CD11b+CD11c− MDSCs, did not abrogate the GVT effect in a parent into F1 BMT model (18), indicating that none of the MDSC subpopulations reduce the capacity of allogeneic T cells to attack residual tumor cells. Furthermore, the GVHD-inhibiting capacity of CD11b+CD11c+ MDSC requires confirmation in other BMT models with disparities also in only MHC class II genes or disparities in MHC class I and II genes to exclude that the observed effects are model dependent.
In vitro or in vivo induction of MDSCs for cellular therapy of GVHD have been performed by using various approaches with different effectiveness (45); however, the ability of different MDSC subpopulations have not been analyzed for their GVHD-inhibiting potential. Treatment of donor mice with CpG and incomplete Freund’s adjuvant (IFA), G-CSF, or recombinant G-CSF/Flt-3 ligand + G-CSF results in increase of splenic CD11b+Gr1+ cells preventing GVHD after cotransplantation with allogeneic T cells (14–16). While a proportion of CpG+IFA-induced Gr-1 cells coexpress CD80, CD86, and CD11c (15), CD11c expression is absent on in vivo-generated G-CSF-or G-CSF/Flt-3 + G-CSF-induced MDSCs (14, 16). MDSCs induced in vitro from BM cells by GM-CSF, G-CSF, and IL-13 exhibited expression of CD11c on about 15% of the cells, but adoptive transfer into BMT mice was performed solely with unseparated MDSCs (20). However, GM-CSF+G-CSF+IL-13-induced MDSCs upregulated CD11c, MHC class II, and F4/80 in the inflammatory GVHD environment. Re-isolation of CD11c+, MHC class IIhigh, and F4/80high cells from GVHD mice showed a loss in immunosuppressivity in vitro (46). Despite the expression of surface makers similar to in vitro-generated CD11b+CD11c+ MDSCs, functional properties are different. While ex vivo-isolated CD11c+ MDSCs mediate T-cell suppression by arginase-1, suppressive ability by in vitro-generated CD11b+CD11c+ MDSCs was mainly attributed to iNOS activity and a not yet defined mechanism, which does not involve IDO, arginase-1, or HO-I activity. Even the role of PMN-MDSCs and M-MDSCs for GVHD development is not defined since we are not aware of BMT experiments using isolated PMN- or M-MDSCs as suppressor cells. G-CSF treatment of donor mice induced low-density splenic granulocytes, which inhibit experimental GVHD (47) and the administration of GVHD-suppressing drug rapamycin, results in expansion of PMN-MDSCs (48) indicating that PMN-MDSCs are the major suppressor population. On the other hand, the presence of G-CSF-induced M-MDSCs correlates with a lower GVHD incidence in humans and humanized GVHD models (21, 49, 50). These findings might reflect species-specific differences in the dependence on MDSC subsets for GVHD inhibition, but extracorporeal photopheresis promotes protective PMN-MDSC expansion in GVHD patients (51).
Striking differences were observed in the in vitro and in vivo activity of MDSC subsets. While CD11b+CD11c− MDSCs suppressed allogeneic T-cell expansion in vitro although to a lesser extent than CD11b+CD11c+ MDSCs, they totally failed to prevent GVHD induction. Immunosuppressive mechanisms differ in both subpopulations since CD11b+CD11c− MDSCs inhibit T-cell proliferation in vitro exclusively by iNOS activity, while function of CD11b+CD11c+ MDSCs depends only half on iNOS. Despite upregulation of PD-L1 and PD-L2, both molecules are neglectable for immunosuppression in vitro by CD11b+CD11c+ MDSCs. The discrepancy of in vitro and in vivo action of MDSCs is supported by our work. Unseparated in vitro-generated MDSCs induced from BM cells by GM-CSF strongly suppressed T-cell proliferation in vitro, but act immunostimulatory in mice receiving blunt chest trauma (TxT). MDSC treatment of TxT mice strongly increased splenic T-cell numbers and proliferative capacity without impairing antigen reactivity (52). Studies by Schmidt et al. also show that tumor-induced MDSCs prevent cytotoxic T lymphocyte (CTL) functions in vitro but not in vivo following adoptive transfer (53) strongly indicating an important effect on MDSC functions by the interacting microenvironment. Likewise, MDSCs isolated from septic mice at different time points after sepsis induction and transferred into septic mice either deteriorate or ameliorate disease development (54).
Microenvironmental influence on MDSC function is further underlined by the finding that CD11b+CD11c+ MDSCs prevent GVHD development by inducing Th2 immunity without altering allogeneic T-cell expansion and homing, although T-cell expansion was severely blocked by this subpopulation in vitro. MDSC-mediated type 2 immunity induction is reported also in the context of cancer, sepsis, pregnancy, and virus infection (5, 6, 8, 55). On the other hand, the transfer of MDSCs in models of Th2-mediated diseases such as asthma-related airway inflammation dampens disease development by shifting immune responses towards Th1 immunity (9, 10). Interestingly, Th1 immunity induction by MDSCs in asthma-related models is found independent whether MDSCs were derived from LPS-treated or tumor-bearing mice, although MDSCs in the context of cancers are known to promote Th2 immunity. Inhibiting T-cell proliferation in vitro is indispensable for their assignment as MDSCs (26) but is not necessarily indicative for their in vivo functions.
Defining MDSC subsets either able to prevent GVHD or being totally inefficient in blocking GVHD development opens up the possibility to define molecules and molecular pathways contributing to MDSC-mediated GVHD inhibition. mRNA-Seq analysis showed that CD11b+CD11c+ and CD11b+CD11c− MDSCs had a totally different transcriptomic landscape differing in more than 2,500 genes. Upon the most strongly upregulated genes (>15-fold increase), the fatty acid translocase CD36 or the chemokines CCL17 and CCL22 were identified. Although CD36 expression is not directly linked to elevated immunosuppressivity, increased lipid contents are reported to augment the immunosuppressive functions of MDSCs (56–58), and Baumann et al. recently reported that human MDSCs derived from isolated CD14+ blood monocytes downregulate glycolysis-related enzymes (59). CCL17 and CCL22 are key chemokines inducing Th2 chemotaxis and are strongly elevated in the serum of patients with Th2-driven atopic dermatitis (42, 43). Possibly, Th2 cells are attracted into lymphatic areas invaded by CCL17/CCL22 expressing CD11b+CD11c+ MDSCs and stimulated for increased expansion. However, only adoptive transfer experiments with MDSCs derived from CD36 or CCL17/CCL22-deficient mice will clarify their substantial role in GVHD prevention. Due to the high numbers of differentially expressed genes, it might be worthwhile to re-isolate adoptively transferred CD11b+CD11c+ and CD11b+CD11c− MDSCs from BM-transplanted mice for transcriptome analysis. Defining the intersection of genes differentially expressed by in vitro and ex vivo isolated CD11b+CD11c+ MDSCs might narrow down the number of possible candidates responsible for GVHD prevention.
Taken together, we could define a small subset of GM-CSF-induced MDSCs characterized by the coexpression of Gr-1+CD11b+CD11c+ as the MDSC subpopulation able to prevent GVHD while maintaining T-cell reactivity and cytotoxicity. This might offer the possibility to identify key molecules and signaling pathways involved in disease prevention with the future perspective to substitute cellular MDSC therapy by pharmacological approaches. Furthermore, the clear discrepancy between in vitro and in vivo functions of MDSCs requires thoughtful testing of MDSC functions in the relevant disease context.
Data Availability Statement
The original contributions presented in the study are publicly available and are based on JS's dissertation: (60). mRNA Seq data can be found here: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE182262.
Ethics Statement
The animal study was reviewed and approved by the ethical committee Regierungspräsidium Tübingen, Germany.
Author Contributions
JS performed experiments, analyzed data, and generated figures. KK, HH, SP, and KF-C performed mRNA-Seq analysis and analyzed transcriptomic data. K-MD contributed to the study design and interpreted data. GS created the study design, performed data interpretation, and wrote the manuscript together with JS. All authors approved the final version of the manuscript, revised the manuscript, and are accountable for all respects of the work.
Funding
This study was supported by Boehringer Ingelheim Ulm University BioCenter (TPI2) and the International Graduate School in Molecular Medicine, Ulm, Germany.
Conflict of Interest
KK, HH, KF-C, and SP are employed by Boehringer Ingelheim Pharma Co. KG and Boehringer Ingelheim RCV GmbH & Co. KG.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Linda Wolf and Ingrid Knape (Department of Pediatrics and Adolescent Medicine, University Medical Center Ulm, Ulm, Germany) for excellent technical assistance, Prof. Dr. Reinhold Schirmbeck (Internal Medicine I, University Medical Center Ulm, Ulm, Germany) for providing PD-L1−/− mice, and Dagmar Knebel and Werner Rust from Boehringer Ingelheim for assisting in RNA preparation and mRNA Seq.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.754316/full#supplementary-material
References
1. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The Terminology Issue for Myeloid-Derived Suppressor Cells. Cancer Res (2007) 67:425; author reply 26. doi: 10.1158/0008-5472.CAN-06-3037
2. Talmadge JE, Gabrilovich DI. History of Myeloid-Derived Suppressor Cells. Nat Rev Cancer (2013) 13:739–52. doi: 10.1038/nrc3581
3. Gabrilovich DI, Nagaraj S. Myeloid-Derived Suppressor Cells as Regulators of the Immune System. Nat Rev Immunol (2009) 9:162–74. doi: 10.1038/nri2506
4. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated Regulation of Myeloid Cells by Tumours. Nat Rev Immunol (2012) 12:253–68. doi: 10.1038/nri3175
5. Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, et al. MyD88-Dependent Expansion of an Immature GR-1(+)CD11b(+) Population Induces T Cell Suppression and Th2 Polarization in Sepsis. J Exp Med (2007) 204:1463–74. doi: 10.1084/jem.20062602
6. Jeisy-Scott V, Davis WG, Patel JR, Bowzard JB, Shieh WJ, Zaki SR, et al. Increased MDSC Accumulation and Th2 Biased Response to Influenza A Virus Infection in the Absence of TLR7 in Mice. PloS One (2011) 6:e25242. doi: 10.1371/journal.pone.0025242
7. Gao J, Wu Y, Su Z, Amoah Barnie P, Jiao Z, Bie Q, et al. Infiltration of Alternatively Activated Macrophages in Cancer Tissue Is Associated With MDSC and Th2 Polarization in Patients With Esophageal Cancer. PloS One (2014) 9:e104453. doi: 10.1371/journal.pone.0104453
8. Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated Myeloid-Derived Suppressor Cells in Pancreatic, Esophageal and Gastric Cancer Are an Independent Prognostic Factor and Are Associated With Significant Elevation of the Th2 Cytokine Interleukin-13. Cancer Immunol Immunother (2011) 60:1419–30. doi: 10.1007/s00262-011-1028-0
9. Arora M, Poe SL, Oriss TB, Krishnamoorthy N, Yarlagadda M, Wenzel SE, et al. TLR4/MyD88-Induced CD11b+Gr-1 Int F4/80+ Non-Migratory Myeloid Cells Suppress Th2 Effector Function in the Lung. Mucosal Immunol (2010) 3:578–93. doi: 10.1038/mi.2010.41
10. Song C, Yuan Y, Wang XM, Li D, Zhang GM, Huang B, et al. Passive Transfer of Tumour-Derived MDSCs Inhibits Asthma-Related Airway Inflammation. Scand J Immunol (2014) 79:98–104. doi: 10.1111/sji.12140
11. Ghimire S, Weber D, Mavin E, Wang XN, Dickinson AM, Holler E. Pathophysiology of GvHD and Other HSCT-Related Major Complications. Front Immunol (2017) 8:79. doi: 10.3389/fimmu.2017.00079
12. Blazar BR, Hill GR, Murphy WJ. Dissecting the Biology of Allogeneic HSCT to Enhance the GvT Effect Whilst Minimizing GvHD. Nat Rev Clin Oncol (2020) 17:475–92. doi: 10.1038/s41571-020-0356-4
13. Zhang J, Chen HM, Ma G, Zhou Z, Raulet D, Rivera AL, et al. The Mechanistic Study Behind Suppression of GVHD While Retaining GVL Activities by Myeloid-Derived Suppressor Cells. Leukemia (2019) 33:2078–89. doi: 10.1038/s41375-019-0394-z
14. Joo YD, Lee SM, Lee SW, Lee WS, Lee SM, Park JK, et al. Granulocyte Colony-Stimulating Factor-Induced Immature Myeloid Cells Inhibit Acute Graft-Versus-Host Disease Lethality Through an Indoleamine Dioxygenase-Independent Mechanism. Immunology (2009) 128:e632–40. doi: 10.1111/j.1365-2567.2009.03048.x
15. Morecki S, Gelfand Y, Yacovlev E, Eizik O, Shabat Y, Slavin S. CpG-Induced Myeloid CD11b+Gr-1+ Cells Efficiently Suppress T Cell-Mediated Immunoreactivity and Graft-Versus-Host Disease in a Murine Model of Allogeneic Cell Therapy. Biol Blood Marrow Transplant (2008) 14:973–84. doi: 10.1016/j.bbmt.2008.06.018
16. MacDonald KP, Rowe V, Clouston AD, Welply JK, Kuns RD, Ferrara JL, et al. Cytokine Expanded Myeloid Precursors Function as Regulatory Antigen-Presenting Cells and Promote Tolerance Through IL-10-Producing Regulatory T Cells. J Immunol (2005) 174:1841–50. doi: 10.4049/jimmunol.174.4.1841
17. Zhou Z, French DL, Ma G, Eisenstein S, Chen Y, Divino CM, et al. Development and Function of Myeloid-Derived Suppressor Cells Generated From Mouse Embryonic and Hematopoietic Stem Cells. Stem Cells (2010) 28:620–32. doi: 10.1002/stem.301
18. Messmann JJ, Reisser T, Leithauser F, Lutz MB, Debatin KM, Strauss G. In Vitro-Generated MDSCs Prevent Murine GVHD by Inducing Type 2 T Cells Without Disabling Antitumor Cytotoxicity. Blood (2015) 126:1138–48. doi: 10.1182/blood-2015-01-624163
19. Scheurer J, Reisser T, Leithauser F, Messmann JJ, Holzmann K, Debatin KM, et al. Rapamycin-Based Graft-Versus-Host Disease Prophylaxis Increases the Immunosuppressivity of Myeloid-Derived Suppressor Cells Without Affecting T Cells and Anti-Tumor Cytotoxicity. Clin Exp Immunol (2020) 202:407–22. doi: 10.1111/cei.13496
20. Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, et al. Bone Marrow Myeloid-Derived Suppressor Cells (MDSCs) Inhibit Graft-Versus-Host Disease (GVHD) via an Arginase-1-Dependent Mechanism That Is Up-Regulated by Interleukin-13. Blood (2010) 116:5738–47. doi: 10.1182/blood-2010-06-287839
21. Wang K, Lv M, Chang YJ, Zhao XY, Zhao XS, Zhang YY, et al. Early Myeloid-Derived Suppressor Cells (HLA-DR(-)/(Low)CD33(+)CD16(-)) Expanded by Granulocyte Colony-Stimulating Factor Prevent Acute Graft-Versus-Host Disease (GVHD) in Humanized Mouse and Might Contribute to Lower GVHD in Patients Post Allo-HSCT. J Hematol Oncol (2019) 12:31. doi: 10.1186/s13045-019-0710-0
22. Park MY, Lim BG, Kim SY, Sohn HJ, Kim S, Kim TG. GM-CSF Promotes the Expansion and Differentiation of Cord Blood Myeloid-Derived Suppressor Cells, Which Attenuate Xenogeneic Graft-Vs.-Host Disease. Front Immunol (2019) 10:183. doi: 10.3389/fimmu.2019.00183
23. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. J Immunol (2008) 181:5791–802. doi: 10.4049/jimmunol.181.8.5791
24. Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of Discrete Tumor-Induced Myeloid-Derived Suppressor Cell Subpopulations With Distinct T Cell-Suppressive Activity. Blood (2008) 111:4233–44. doi: 10.1182/blood-2007-07-099226
25. Eckert I, Ribechini E, Lutz MB. In Vitro Generation of Murine Myeloid-Derived Suppressor Cells, Analysis of Markers, Developmental Commitment, and Function. Methods Mol Biol (2021) 2236:99–114. doi: 10.1007/978-1-0716-1060-2_10
26. Cassetta L, Baekkevold ES, Brandau S, Bujko A, Cassatella MA, Dorhoi A, et al. Deciphering Myeloid-Derived Suppressor Cells: Isolation and Markers in Humans, Mice and non-Human Primates. Cancer Immunol Immunother (2019) 68:687–97. doi: 10.1007/s00262-019-02302-2
27. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for Myeloid-Derived Suppressor Cell Nomenclature and Characterization Standards. Nat Commun (2016) 7:12150. doi: 10.1038/ncomms12150
28. Kumamoto Y, Linehan M, Weinstein JS, Laidlaw BJ, Craft JE, Iwasaki A. CD301b(+) Dermal Dendritic Cells Drive T Helper 2 Cell-Mediated Immunity. Immunity (2013) 39:733–43. doi: 10.1016/j.immuni.2013.08.029
29. Gao Y, Nish SA, Jiang R, Hou L, Licona-Limon P, Weinstein JS, et al. Control of T Helper 2 Responses by Transcription Factor IRF4-Dependent Dendritic Cells. Immunity (2013) 39:722–32. doi: 10.1016/j.immuni.2013.08.028
30. Tussiwand R, Everts B, Grajales-Reyes GE, Kretzer NM, Iwata A, Bagaitkar J, et al. Klf4 Expression in Conventional Dendritic Cells Is Required for T Helper 2 Cell Responses. Immunity (2015) 42:916–28. doi: 10.1016/j.immuni.2015.04.017
31. Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte J Jr, Crawford JM, et al. An Experimental Model of Idiopathic Pneumonia Syndrome After Bone Marrow Transplantation: I. The Roles of Minor H Antigens and Endotoxin. Blood (1996) 88:3230–9. doi: 10.1182/blood.V88.8.3230.bloodjournal8883230
32. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics (2013) 29:15–21. doi: 10.1093/bioinformatics/bts635
33. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map Format and SAMtools. Bioinformatics (2009) 25:2078–9. doi: 10.1093/bioinformatics/btp352
34. DeLuca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire MD, Williams C, et al. RNA-SeQC: RNA-Seq Metrics for Quality Control and Process Optimization. Bioinformatics (2012) 28:1530–2. doi: 10.1093/bioinformatics/bts196
35. Jun G, Wing MK, Abecasis GR, Kang HM. An Efficient and Scalable Analysis Framework for Variant Extraction and Refinement From Population-Scale DNA Sequence Data. Genome Res (2015) 25:918–25. doi: 10.1101/gr.176552.114
36. Sayols S, Scherzinger D, Klein H. Dupradar: A Bioconductor Package for the Assessment of PCR Artifacts in RNA-Seq Data. BMC Bioinf (2016) 17:428. doi: 10.1186/s12859-016-1276-2
37. Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential Analysis of Gene Regulation at Transcript Resolution With RNA-Seq. Nat Biotechnol (2013) 31:46–53. doi: 10.1038/nbt.2450
38. Liao Y, Smyth GK, Shi W. Featurecounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics (2014) 30:923–30. doi: 10.1093/bioinformatics/btt656
39. Law CW, Chen Y, Shi W, Smyth GK. Voom: Precision Weights Unlock Linear Model Analysis Tools for RNA-Seq Read Counts. Genome Biol (2014) 15:R29. doi: 10.1186/gb-2014-15-2-r29
40. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res (2015) 43:e47. doi: 10.1093/nar/gkv007
41. Simpson S, Kaislasuo J, Guller S, Pal L. Thermal Stability of Cytokines: A Review. Cytokine (2020) 125:154829. doi: 10.1016/j.cyto.2019.154829
42. Imai T, Nagira M, Takagi S, Kakizaki M, Nishimura M, Wang J, et al. Selective Recruitment of CCR4-Bearing Th2 Cells Toward Antigen-Presenting Cells by the CC Chemokines Thymus and Activation-Regulated Chemokine and Macrophage-Derived Chemokine. Int Immunol (1999) 11:81–8. doi: 10.1093/intimm/11.1.81
43. Nakazato J, Kishida M, Kuroiwa R, Fujiwara J, Shimoda M, Shinomiya N. Serum Levels of Th2 Chemokines, CCL17, CCL22, and CCL27, Were the Important Markers of Severity in Infantile Atopic Dermatitis. Pediatr Allergy Immunol (2008) 19:605–13. doi: 10.1111/j.1399-3038.2007.00692.x
44. Patel PS, Kearney JF. CD36 and Platelet-Activating Factor Receptor Promote House Dust Mite Allergy Development. J Immunol (2017) 199:1184–95. doi: 10.4049/jimmunol.1700034
45. Demosthenous C, Sakellari I, Douka V, Papayanni PG, Anagnostopoulos A, Gavriilaki E. The Role of Myeloid-Derived Suppressor Cells (MDSCs) in Graft-Versus-Host Disease (GVHD). J Clin Med (2021) 10:2050. doi: 10.3390/jcm10102050
46. Koehn BH, Apostolova P, Haverkamp JM, Miller JS, McCullar V, Tolar J, et al. GVHD-Associated, Inflammasome-Mediated Loss of Function in Adoptively Transferred Myeloid-Derived Suppressor Cells. Blood (2015) 126:1621–8. doi: 10.1182/blood-2015-03-634691
47. Vasconcelos ZF, Dos Santos BM, Farache J, Palmeira TS, Areal RB, Cunha JM, et al. G-CSF-Treated Granulocytes Inhibit Acute Graft-Versus-Host Disease. Blood (2006) 107:2192–9. doi: 10.1182/blood-2005-08-3239
48. Lin Y, Wang B, Shan W, Tan Y, Feng J, Xu L, et al. mTOR Inhibitor Rapamycin Induce Polymorphonuclear Myeloid-Derived Suppressor Cells Mobilization and Function in Protecting Against Acute Graft-Versus-Host Disease After Bone Marrow Transplantation. Clin Immunol (2018) 187:122–31. doi: 10.1016/j.clim.2017.11.005
49. D'Aveni M, Rossignol J, Coman T, Sivakumaran S, Henderson S, Manzo T, et al. G-CSF Mobilizes CD34+ Regulatory Monocytes That Inhibit Graft-Versus-Host Disease. Sci Transl Med (2015) 7:281ra42. doi: 10.1126/scitranslmed.3010435
50. Lv M, Zhao XS, Hu Y, Chang YJ, Zhao XY, Kong Y, et al. Monocytic and Promyelocytic Myeloid-Derived Suppressor Cells may Contribute to G-CSF-Induced Immune Tolerance in Haplo-Identical Allogeneic Hematopoietic Stem Cell Transplantation. Am J Hematol (2015) 90:E9–E16. doi: 10.1002/ajh.23865
51. Rieber N, Wecker I, Neri D, Fuchs K, Schafer I, Brand A, et al. Extracorporeal Photopheresis Increases Neutrophilic Myeloid-Derived Suppressor Cells in Patients With GvHD. Bone Marrow Transplant (2014) 49:545–52. doi: 10.1038/bmt.2013.236
52. Kustermann M, Klingspor M, Huber-Lang M, Debatin KM, Strauss G. Immunostimulatory Functions of Adoptively Transferred MDSCs in Experimental Blunt Chest Trauma. Sci Rep (2019) 9:7992. doi: 10.1038/s41598-019-44419-5
53. Schmidt K, Zilio S, Schmollinger JC, Bronte V, Blankenstein T, Willimsky G. Differently Immunogenic Cancers in Mice Induce Immature Myeloid Cells That Suppress CTL In Vitro But Not In Vivo Following Transfer. Blood (2013) 121:1740–8. doi: 10.1182/blood-2012-06-436568
54. Brudecki L, Ferguson DA, McCall CE, El Gazzar M. Myeloid-Derived Suppressor Cells Evolve During Sepsis and can Enhance or Attenuate the Systemic Inflammatory Response. Infect Immun (2012) 80:2026–34. doi: 10.1128/IAI.00239-12
55. Kostlin N, Hofstadter K, Ostermeir AL, Spring B, Leiber A, Haen S, et al. Granulocytic Myeloid-Derived Suppressor Cells Accumulate in Human Placenta and Polarize Toward a Th2 Phenotype. J Immunol (2016) 196:1132–45. doi: 10.4049/jimmunol.1500340
56. Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, et al. Fatty Acid Transport Protein 2 Reprograms Neutrophils in Cancer. Nature (2019) 569:73–8. doi: 10.1038/s41586-019-1118-2
57. Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, et al. Exogenous Lipid Uptake Induces Metabolic and Functional Reprogramming of Tumor-Associated Myeloid-Derived Suppressor Cells. Oncoimmunology (2017) 6:e1344804. doi: 10.1080/2162402X.2017.1344804
58. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol Res (2015) 3:1236–47. doi: 10.1158/2326-6066.CIR-15-0036
59. Baumann T, Dunkel A, Schmid C, Schmitt S, Hiltensperger M, Lohr K, et al. Regulatory Myeloid Cells Paralyze T Cells Through Cell-Cell Transfer of the Metabolite Methylglyoxal. Nat Immunol (2020) 21:555–66. doi: 10.1038/s41590-020-0666-9
Keywords: graft-versus-host disease, prophylaxis, myeloid-derived suppressor cells, allogeneic bone marrow transplantation, mouse model, type 2 immune response, GVT effect
Citation: Scheurer J, Kitt K, Huber HJ, Fundel-Clemens K, Pflanz S, Debatin K-M and Strauss G (2021) Graft-Versus-Host Disease Prevention by In Vitro-Generated Myeloid-Derived Suppressor Cells Is Exclusively Mediated by the CD11b+CD11c+ MDSC Subpopulation. Front. Immunol. 12:754316. doi: 10.3389/fimmu.2021.754316
Received: 06 August 2021; Accepted: 21 September 2021;
Published: 14 October 2021.
Edited by:
Andreas Beilhack, Julius Maximilian University of Würzburg, GermanyReviewed by:
Shigeo Fuji, Osaka International Cancer Institute, JapanDaniel Peltier, University of Michigan, United States
Copyright © 2021 Scheurer, Kitt, Huber, Fundel-Clemens, Pflanz, Debatin and Strauss. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gudrun Strauss, Z3VkcnVuLnN0cmF1c3NAdW5pa2xpbmlrLXVsbS5kZQ==