 Yongyan Chen
Yongyan Chen Wenwen Cui†
Wenwen Cui† Xiao Li
Xiao Li Huan Yang
Huan Yang- Xuzhou Key Laboratory of Laboratory Diagnostics, School of Medical Technology, Xuzhou Medical University, Xuzhou, China
In inflammatory bowel disease (IBD), intestinal mucosa cell and intestinal epithelial cell are severely damaged, and then their susceptibility to bacteria increases, so many commensal bacteria become pathogenic. The pathogenic commensal bacteria can stimulate a series of compensatory immune responses in the intestine. However, the immune response prevents the intestinal tract from restoring homeostasis, which in turn produces an indispensable inflammatory response. On the contrary, in IBD, the fierce inflammatory response contributes to the development of IBD. However, the effect of commensal bacteria on inflammation in IBD has not been clearly studied. Therefore, we further summarize the changes brought about by the changes of commensal bacteria to the inflammation of the intestines and their mutual influence. This article reviews the protective mechanism of commensal bacteria in healthy people and the mechanism of commensal bacteria and immune response to the destruction of the intestinal barrier when IBD occurs. The treatment and prevention of IBD are also briefly summarized.
Introduction
Inflammatory bowel disease (IBD) is chronic and recurrent and it mainly affects the ileum, rectum, and colon. It includes ulcerative colitis (UC) and Crohn’s disease (CD). The global burden of IBD remains a persistent health problem. The prevalence of IBD in Europe, North America, and other Western countries exceeds 0.3% and is increasing in many newly industrialized countries (1, 2).
The affected bowel in UC and CD are quite different. UC is limited to the colon, it mainly invades the lamina propria and crypts (3, 4). CD can cause transmural enteritis, it can affect any part of the stomach and intestine, especially the terminal ileum and colon (5). The damage caused by IBD can be attributed to the destruction of the intestinal barrier (3, 6). The etiology of IBD is not yet fully understood, and it has been proven to be related to complex factors such as genetics, environment, intestinal microbes, and immunity. Among them, intestinal commensal bacteria play a pivotal role, including bacteria and fungi (3, 7).
Intestinal commensal bacteria do not play an independent role in host health. Under normal circumstances, commensal bacteria and the intestinal immunity of the host are in a balanced state, and they resist the invasion of pathogenic microorganisms and maintain homeostasis together (8). The human intestinal barrier is the first line of defense for the invasion of pathogenic microorganisms (9). It is associated with many diseases, such as IBD, acute pancreatitis, colon cancer (10–12). The direct communication between the intestinal symbiotic flora and intestinal immunity controls the development of the disease. Once the balance is broken, enteritis will be induced in the form of positive feedback (13).
The commensal bacteria in IBD always have non-negligible changes, including their quantity or products, and the coordination of the immune response with the intestinal tract exposes the intestinal barrier to danger (14). It causes the activation of immune cells and an overload of cytokines, and the activation of a series of receptors and proteins ultimately promotes the occurrence and development of IBD (15). In addition, oral commensal bacteria also contribute to the development of IBD. They will be highly colonized in the unhealthy and fragile intestines after swallowing. This has been verified. The pathogenic bacteria in these conditions will further threaten the intestinal tract (16). Many risk factors for IBD, such as diet, smoking, and antibiotics (3), can change the state of normal intestinal commensal bacteria and immunity and increase the risk of IBD.
In this review, we focus on discussing the lesions of the colonic mucosa of IBD, which are both present in CD and UC (17). We describe the barrier protection of commensal bacteria and intestinal immunity in a healthy state and focus on the changes in commensal bacteria in IBD and the damage of commensal bacteria-dependent immune responses to the intestinal barrier. Each of these mechanisms is closely linked, with feedback mechanisms to promote the final outcome, IBD. We then discuss the treatments and prevention that rely on this series of mechanisms.
Commensal Bacteria Maintain the Homeostasis of the Intestinal Mucosal Barrier
The intestinal barrier is mainly composed of mucus and epithelial cells. Commensal bacteria and the immune system in the intestine work together to regulate the homeostasis of the intestinal barrier. The gastrointestinal tract provides 150-200 m2 of surface area for microorganisms (18), and about 1013 bacteria are colonized, which is comparable to the number of human cells (19). Because of pH and other factors, a small number of microorganisms are colonized above the upper end of the small intestine. The most abundant commensal bacteria are found in the colon, accounting for about 70% of all bacteria in the human body. Most are Bacteroidetes, Firmicutes, Actinobacteria, Proteobacteria, and Verrucomicrobia (20, 21). There are large differences in individual flora, which are closely related to dietary and living habits (22).
Mucosal Barrier Isolates Commensal Bacteria and Epithelial Tissue
The intestinal barrier is in constant dynamic renewal (Figure 1). Stem cells continue to divide and proliferate, replenishing the epithelial cells shedding into the intestinal lumen and becoming epithelial cells (23). The secretory activity of goblet cells (secretion of mucin, especially MUC2) gives intestinal epithelial cells an extra protective gel mucus layer (24). Commensal bacteria can pass through the mucus in the small intestine (25). When a small amount of commensal bacteria pass through the mucus, they are presented to T cells and B cells by dendritic cells in the intestine and induce B cells to produce IgA, which targets intestinal bacteria. At this time, the macrophages in the lamina propria are activated, and the macrophages exert their phagocytosis and secretion functions. With the production of antibacterial substances such as defensins, the commensal bacteria that are about to touch the epithelial cells can be completely eliminated (26, 27). The defensins secreted by the Paneth cells in the small intestine enhance this effect (28). The mucus in the colon can directly prevent the contact of commensal bacteria, and the outer mucus layer provides a living environment for the commensal bacteria (24, 25), and IgA also exists in a large amount in the outer mucus layer. The IgA of glycosylation, however, no longer resists commensal bacteria. The IgA-MAFF (Mucus-Associated Functional Factor) system can promote the growth of Bacteroidetes, induce the proliferation of Clostridium, promote the renewal of epithelial cells, and deal with the damage in time, stabilizing the intestinal barrier (29).
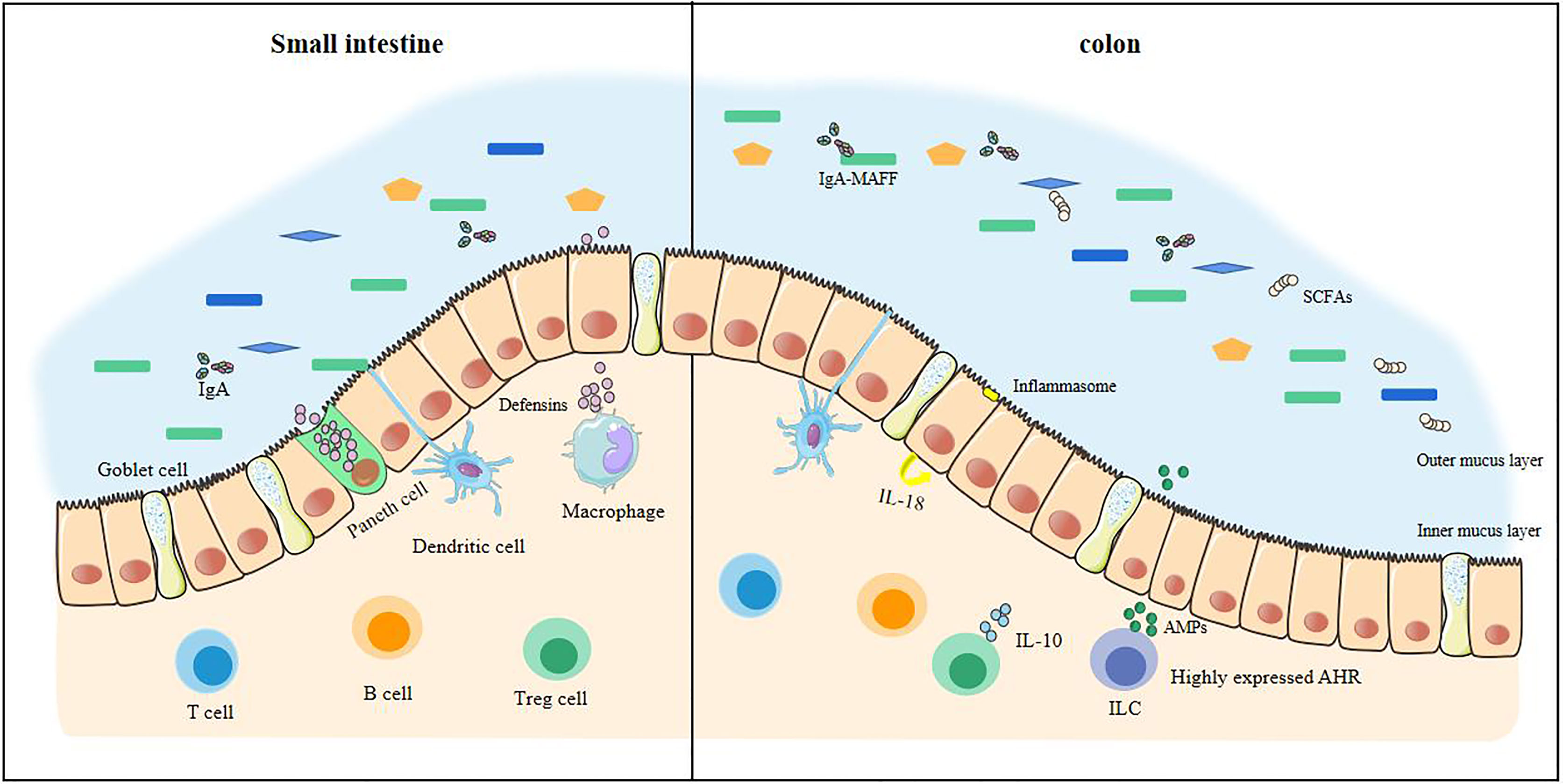
Figure 1 The stable intestinal barrier of a healthy small intestine and colon. The homeostasis of the intestinal tract of a healthy body depends on the regulation of many factors, including commensal bacteria, short-chain fatty acids, and the immune system. In health, commensal bacteria are diverse. The antibacterial substances produced in the small intestine are sufficient to resist pathogenic bacteria, making it difficult to cross the intestinal epithelial cells. There is also an internal mucus layer in the colon that cannot be crossed by these bacteria. The production of short-chain fatty acids keeps the colon in a hypoxic state and maintains tight epithelial junctions, increasing anti-inflammatory effects. At this time, the immune system has a large anti-inflammatory effect, which is enough to prevent the inflammatory load caused by the invasion of pathogenic bacteria.
Short-Chain Fatty Acids Are Positive for Stabilizing the Intestines
Short-chain fatty acids (SCFAs) play a pivotal role in maintaining intestinal homeostasis, especially butyrate, the main source of energy for colon cells (30). SCFAs are produced when the commensal bacteria metabolize dietary fiber. Bacteroidetes and Firmicutes are the most abundant bacteria in the intestine. The Bacteroidetes mainly produce acetic acid and propionic acid, while Firmicutes mainly produce butyrate in the human intestine (31, 32). Butyrate can maintain the proliferation of small intestinal epithelial cells (33). As butyrate is produced by anaerobic bacteria, butyrate stabilizes hypoxia inducible factor (HIF, which is able to coordinate barrier protection) during the process of being absorbed and metabolized in an hypoxic environment (34, 35) and can induce innate lymphocyte (ILCs) to produce antimicrobial peptides (AMPs) to regulate commensal bacteria (36). The β-oxidation of butyrate can maintain the low-oxygen environment in the intestine so that butyrate in a healthy intestine can be continuously produced (37). SCFAs activate and release IL-18 through inflammasomes to maintain the integrity of the intestinal epithelium (38). In addition, SCFAs also have anti-inflammatory effects (39). They can increase the IgA in the intestine, maintain the development of B cells, and play an important role in promoting the differentiation and expansion of Treg cells (38, 40).
Therefore, commensal bacteria help the intestines resist pathogens at any time, and its function depends on a good ratio of commensal bacteria.
The Main Immune Factors in a Healthy Intestine That Inhibit Inflammation
In a healthy gut, immunity is the main factor that maintains homeostasis. The proliferation and differentiation of T cells are inhibited, but Treg cells are active. IL-10 may be produced by most immune cells, especially Treg cells. IL-10 directly inhibit the production of IL-12 and IL-23, which is equivalent to limiting the differentiation of Th1 cells and the response of pathogenic Th17 cells (41). IL-10 deficiency mainly induce Th17 cells, and then Th17 could promote colitis (42). The activation of the aryl hydrocarbon receptor (AHR) is another mechanism of intestinal homeostasis. Once activated, AHR releases IL-22, induces IL-10R expression, and strengthens the tight junctions of the intestinal epithelium to maintain the integrity of the intestine (43). AHR is highly expressed in ILC2 and can inhibit the expression of IL-33 receptor ST2 [this receptor is highly expressed in IBD (44)] and the production of some pro-inflammatory factors, such as IL-13 (45). Overall, the homeostasis of the intestine needs to be maintained by inhibiting inflammatory factors to prevent excessive inflammation. When the intestinal commensal bacteria change, the “balance” of immunity will be broken and tilted toward proinflammatory effects, and the body will be in a state of inflammatory stress.
Interaction Between Commensal Bacteria and the Intestinal Barrier in IBD
Commensal Bacteria Degrade More Mucus
During IBD, the microbial genes detected in feces were reduced by about 25%, and the abundance and diversity of commensal bacteria decreased overall (46, 47). Patients with IBD have more harmful bacteria and fewer beneficial bacteria. In fact, under the protection of the mucus barrier, the intestinal tract can maintain a steady state even though the bacteria in the intestinal tract undergo minor changes (48). However, the total abundance of mucus-degrading bacteria in IBD patients is increased significantly (49). The mucus-degrading bacteria in healthy humans degrade the skeletal structure of mucus-mucin (MUC2) to release the product for use by other bacteria (50), but the sharp increase in mucus-degrading bacteria, such as Ruminococcus gnavus (R. gnavus) and Ruminococcus torques (R. torque), will dissolve more mucus and increase the exposure of epithelial cells (49). In addition, IBD is usually accompanied by truncated O-glycans in intestinal epithelial cells, which may be related to glycosidase produced by microorganisms and oxidative stress caused by inflammation (51). The truncated intestinal sugar chain makes the mucosa thinner, and the SCFAs are reduced in IBD and cannot regulate mucin glycosylation normally (52). This makes mucin lose its stability. The decreased Bacteroides and Faecalibacterium prausnitzii also failed to promote the differentiation of goblet cells, and they could not regulate the glycosylation of the mucosa (Figure 2) (53–55).
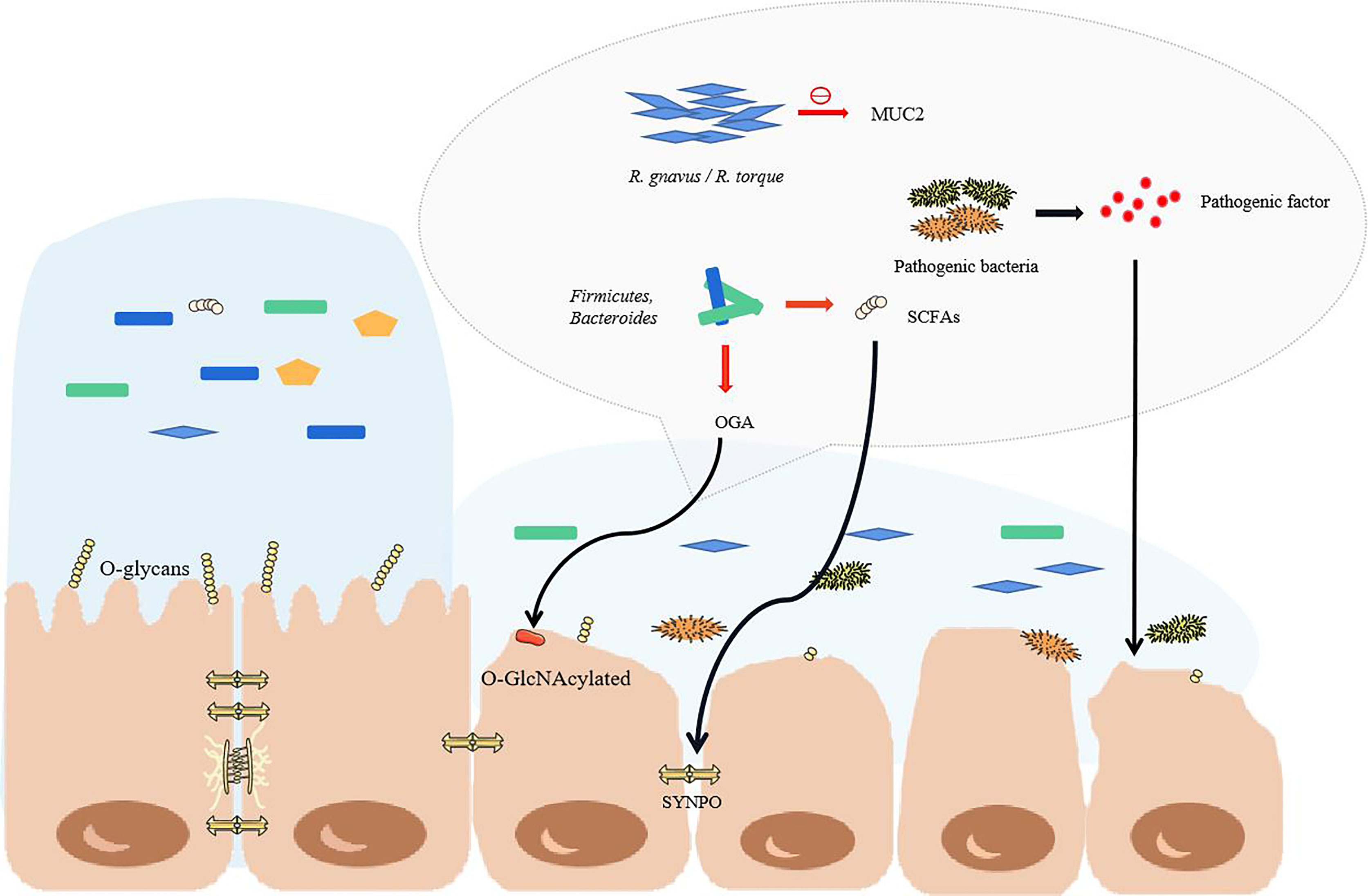
Figure 2 The structural changes of commensal bacteria during IBD lead to the destruction of the intestinal barrier. When the structure of the commensal bacteria changes, the mucus in the colon becomes thinner, mainly due to the increase in the proportion of some major mucus-degrading bacteria. The O-glycans on the intestinal epithelial cells would normally stabilize the mucus, but they are reduced in IBD. At this time, goblet cells cannot differentiate normally, so the mucus becomes thinner, and pathogenic bacteria enter the inner mucus layer and contact the epithelium. As the proportion of pathogenic bacteria greatly increases, the pathogenic factors produced directly attack the intestinal cells. With the reduction of short-chain fatty acid-producing bacteria, the tight junctions of the intestine cannot be maintained. The red arrow highlights the reduction.
The Structure of Commensal Bacteria Is Changed in IBD
With the increase of R. gnavus and R. torque, more mucosa is broken down, and the inflammation of IBD makes the intestinal mucosa become thinner. The change in oxygen gradient is conducive to the survival of intestinal facultative aerobes and reduces the proportion of anaerobic bacteria. This should be the main reason for the changes in the structure of intestinal commensal bacteria (Figure 2) (56, 57). Therefore, the thin intestinal mucus cannot stop the invasion of the increased pathogenic bacteria. Proteobacteria, Escherichia coli, Fusobacteria, Klebsiella pneumoniae, Clostridium difficile and other pathogenic commensal bacteria in the intestine of patients with IBD are increased significantly (58–61). This is an extremely serious phenomenon for intestinal homeostasis. Among them, Klebsiella pneumoniae is most likely swallowed through the mouth to the intestines and colonized, which may be before or after IBD. In fact, oral commensal bacteria take advantage of IBD to take advantage of it. At this time, the colonization resistance of the intestinal tract of a healthy body no longer exists. Ectopic colonization of conditional pathogens, including Porphyromonas gingivalis, Streptococcus mutans, Fusobacterium nucleatum, Campylobacter concisus, and Klebsiella pneumoniae, will aggravate the development of IBD (15, 62).
Proteus has a low abundance in healthy intestines and are the first reported pathogenic bacteria in gastrointestinal diseases. Because of its adhesion and ability to produce urease, hemolysin, and virulence factors, its pathogenicity is significantly manifested in IBD with the increase in number (63, 64). Adherent invasive Escherichia coli (AIEC) is a well-known pathogen. AIEC penetrates the mucus layer and resists antibacterial proteins, adheres to intestinal epithelial cells to release enterotoxins, and can block the autophagy process of lysosomes (autophagy plays an important role in maintaining the body’s immune homeostasis) (65, 66). For patients with IBD who lack PTPN2 (an autoimmune susceptibility gene) or mutations in PTPN2, this is more like a help. Macrophages won’t against AIEC (67). At the same time as the increase in pathogenic bacteria, the proportion of beneficial commensal bacteria such as Firmicutes, Clostridiales, Bacteroides, Ruminococcaceae, Lactobacillus, Bifidobacteria, Faecalibacterium prausnitzii and other butyrate- producing bacteria in the intestine is significantly reduced, which is also an important factor in the development of IBD (53, 54, 68). O-GlcNAcase (OGA) that is enriched in Bacteroides and Firmicutes can hydrolyze O-GlcNAcylated protein in epithelial cells and immune cells, which can inhibit the development of colitis. However, because of the reduction of bacteria, the level of OGA in IBD decreases, which is also an important mechanism for the continuous development of IBD (69). Indeed, the diversity of intestinal commensal bacteria decreases, especially anaerobic bacteria. In this case, there is no surprise that SCFAs, well-known nutritional and anti-inflammatory substances in the intestine, are drastically reduced, especially butyrate (37, 40). The actin-binding protein synaptopodin (SYNPO) is an important protein that maintains the tight junctions of the intestinal epithelium and is interdependent with butyrate. Therefore, SYNPO in IBD is naturally lost with the decrease of butyrate, which directly affects the connections intestinal epithelium (70).
In recent years, scientists have discovered that fungi also coordinates commensal bacteria, and there is an intricate relationship between them. Although the bacterial changes of UC and CD are similar, fungi are very different. This is mainly related to the ileum due to the unique function of the ileum, it can produce antimicrobial peptides and absorb bile acids. The ileum CD is conducive to the growth of fungi, while the diversity of UC and CD fungi that do not involve the ileum is reduced. This is also inseparable from some commensal bacteria in the intestine. The abundance of bifidobacteria and brucella in IBD is positively correlated with yeast, but these bacteria are reduced in IBD. In fact, bacteria and fungi are two mechanisms in CD, but they are closely related in UC (71, 72). The common mechanism in IBD is that the ratio of Basidiomycetes/Ascomycetes increases, the ratio of Candida albicans increases, and the ratio of Saccharomyces cerevisiae decreases, which reduces AIEC-induced colitis (72, 73).
Interaction Between Commensal Bacteria and Immune Response in IBD
Innate Immune Response
Dendritic cells form an extensive network under the intestinal epithelium. After a large number of Proteobacteria pass through the mucosal barrier, pathogen-associated molecular patterns (PAMP), lipopolysaccharide and flagellin on the surface of the proteobacteria, are recognized by the toll-like receptors on the surface of dendritic cells (74, 75). The immature dendritic cells produce IL-23, which causes local intestinal inflammation (76). At the same time, macrophages have a phagocytic effect. In addition, the production of cytokines such as IL-1β, IL-6, IL-18, and TNF by dendritic cells and macrophages further aggravates inflammation (77). In addition, IL-10, which inhibits inflammation, is produced by intestinal dendritic cells. Bifidobacteria can increase its release, but this is inhibited in IBD by the decrease of these bacteria. Its related Saccharomyces cerevisiae also promotes this effect (72, 78). The inflammatory overload caused by macrophages plays a pivotal role in the immune response in IBD, and treatment for it may be able to effectively alleviate the inflammation of IBD (79).
Natural killer (NK) cells are activated by cytokines, bind to infected epithelial cells, release toxic particles, and induce apoptosis. IL-1β further promotes the production of IFN-γ, IL-17, and other cytokines by ILCs, and promotes the development of inflammation (80). γδ T cells are another way to produce IL-17 (81), but it can be inhibited by propionate, a metabolite of symbiotic bacteria. However, because of the lack of SCFA-producing bacteria, this effect is also inhibited, which is also one of the reasons for the rampant inflammatory factors. IL-13 can be produced by ILC2 dependent on commensal bacteria (82). A large number of cytokines produced by antigen-presenting cells (APC) not only bring about an overload of inflammation but also provide conditions for specific immune responses (Figure 3).
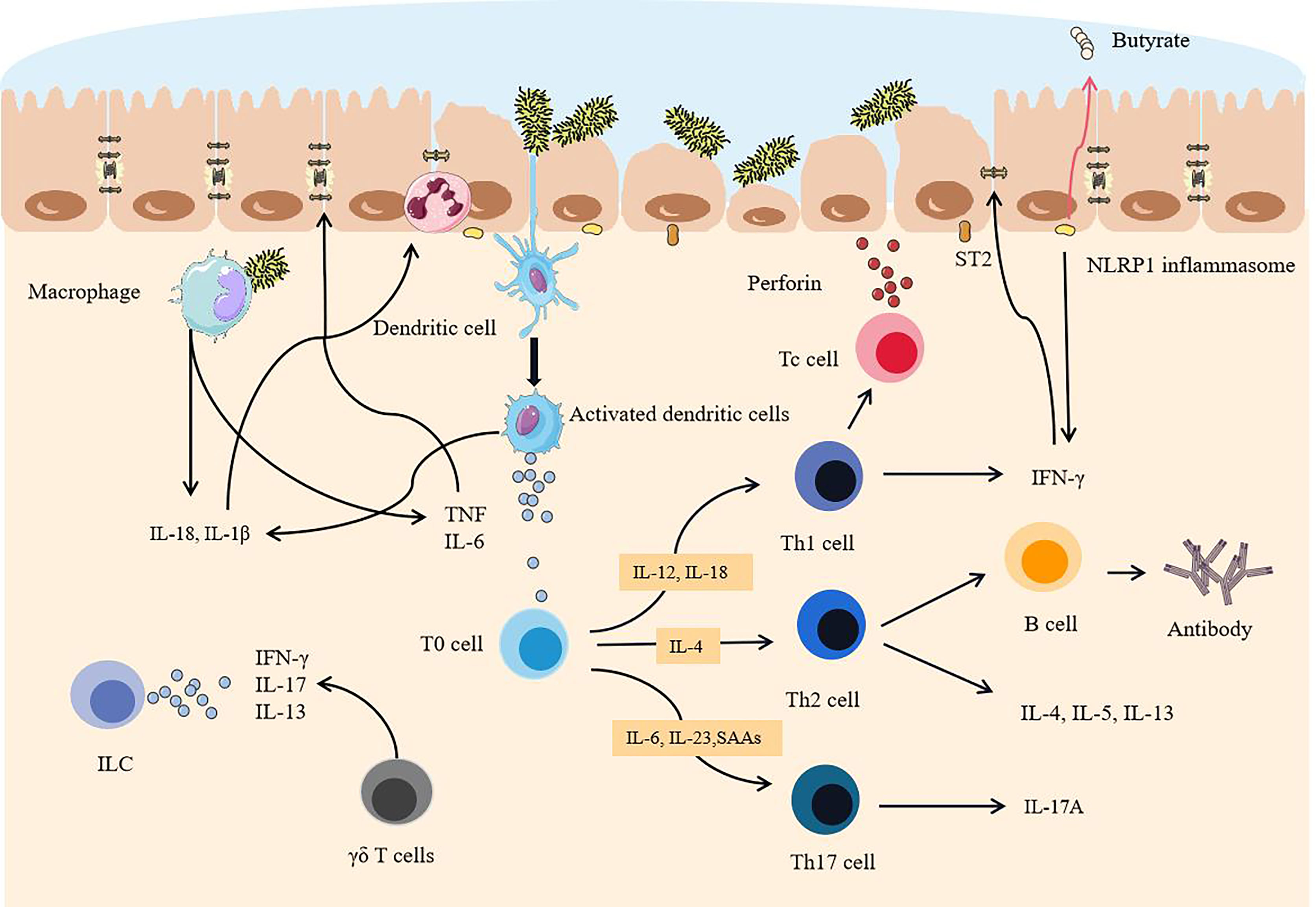
Figure 3 After the intestinal mucosa are degraded, pathogenic commensal bacteria trigger intestinal overload immunity. When the mucosa become thinner, a sharp increase in pathogenic commensal bacteria activates dendritic cells. Because of the destruction of the tight junctions of the intestine, pathogenic commensal bacteria cross the epithelial layer and reach the lamina propria, activating macrophages and NK cells and releasing a large number of cytokines. APC binds antigens to T cells and B cells and differentiates and matures in an environment rich in cytokines. Eventually, a large number of activated immune cells and a large number of proinflammatory factors are dominant, exceeding the body’s ability to inhibit inflammatory effects, epithelial cells are severely attacked, and tight junctions are further destroyed.
Adaptive Immune Response
Dendritic cells activate T cells after ingesting antigens, and Th0 cells differentiate into Th1 cells, Th2 cells, and Th17 cells. Th1 cells induce cytotoxic T cells to activate, proliferate, and attack the infected intestinal epithelial cells. Th2 cells activate B cells, proliferate and differentiate into plasma cells, and secrete antibodies to neutralize pathogens. Th17 cells mainly secrete IL-17A to mediate inflammation (83). Moreover, Th1 cells are induced to produce a large amount of IFN-γ by IL-12 and IL-18, which mainly induces CD (84). Th2 cells release IL-4, IL-5, and IL-13 (85), which mainly play a role in UC. Th2 cells can also activate B cells as well and secrete antibodies (mainly IgG) to attack infected epithelial cells, but this response seems to be secondary (84, 86). SCFAs can induce the production of anti-inflammatory Treg cells and the c-MAF transcription factor produced by Treg cells that can maintain the function of Tregs and inhibit the activation of Th17 cells (87). However, because of the reduction of anaerobic bacteria and the lack of anaerobic environment, SCFAs are greatly reduced. Not only that, the colonization of segmented filamentous bacteria in IBD can trigger the secretion of serum amyloid A proteins (SAA1 and SAA2). The increase of these two has been confirmed in IBD, which also promotes the differentiation of pathogenic Th17 cells (88, 89). Conversely, RORγt+Treg in IBD was reduced, which is all due to changes in commensal bacteria (90). For example, Bacteroides fragilis is reduced in IBD. It normally acts on immune cells through outer membrane vesicles and relies on the IBD-related gene ATG16L1. Because of the lack of this gene and these bacteria, the Treg cell response is defective, which also leads to the inhibition of IL-10 release (91–93). Therefore, once inflammation develops, the axis of Th17 and Treg is destined to tilt, biasing the role of Th17 (94). When oral inflammation such as periodontitis occurs, the pathogenic commensal bacteria expand and at the same time produce Th17 cells and migrate to the intestine (95), which will only aggravate the development of IBD and cause the inflammation of the intestines to persist. Recently, studies have found that tissue-resident memory T cells (TRM cells) play an important role in promoting the development of inflammation in IBD (96). Although the mechanism is not yet clear, it is still very likely to become an effective therapeutic target in the future. In general, as the immune system of the lamina propria is successively activated, the body can no longer regulate inflammatory effect brought by a large number of inflammatory cells and inflammatory factors, which is accompanied by irreversible destruction of the intestinal barrier (97).
Cytokines Destroy Tight Junctions
A large number of proinflammatory factors produced at this time not only increase the burden of inflammation but also seriously affect the tight junctions of cells. IL-1β can recruit granulocytes to infiltrate the infection foci and directly destroy the connection and tightness of intestinal epithelial cells (98). IL-13 can activate STAT6 in epithelial cells and affect the tight junctions of the intestinal epithelium (82). In addition, TNF-α and IL-1β induce endoplasmic reticulum stress, affect Caco-2 cells (intestinal epithelial cells), and significantly change key proteins from the apical and basolateral membranes, such as E-cadherin. This further destroys the tight junctions of the intestinal epithelium (99). TNF will promote the expression of myosin light chain kinase (MLCK) in the intestinal epithelium, which is also one of the mechanisms by which the permeability of intestinal epithelial tight junctions increases (100). The NLRP1 inflammasome in the intestinal inflammation area of IBD patients increases sharply, which is a known negative factor for butyrate-producing commensal bacteria, and the promoted IFN-γ will promote IBD (101). IFN-γ and IL-13 can induce apoptosis of intestinal epithelial cells and further increase intestinal permeability (102, 103). In addition, after AIEC colonization, AIEC can also induce the expression of IL-33 receptor ST2 in intestinal epithelial cells, thereby enhancing the IL-33 signaling pathway, up-regulating TGF-β, promoting the expression of collagen in fibroblasts, and promoting the development of intestinal fibrosis (44). Once the immune system is activated, the cytokines produced will further attack the tight junctions of intestinal epithelial cells, severely exceeding the self-healing ability of the intestinal barrier.
Treatment and Prevention
In order to improve the balance of commensal bacteria, fecal microbiota transplantation has great potential (104). It seems that the direct replacement of the microbial system in the patient’s body can directly treat IBD, using the healthy microbiota to reestablish a more perfect intestinal barrier. However, because the underlying mechanism of IBD-related core microbiota and pathogenesis is not fully clear, this method still needs further research. Most importantly, this method is not completely safe. Two IBD patients had persistent diarrhea after Fecal microbiota transplantation (FMT) treatment, and enterotoxigenic C. perfringens type A was detected in the stool (105). At the end of 2018, a patient was infected with extended-spectrum beta-lactamase-producing Escherichia coli after FMT treatment and eventually died (106). In addition, probiotics can also be used as therapeutic agents to treat IBD, but it seems that its efficacy on CD still needs to be explored (107). Scientists are trying to use the metabolites of commensal bacteria as therapeutic targets, such as the use of commensal bacteria that specifically produce butyrate, which has very great therapeutic potential (108). Researchers are also trying to block the overloaded immune response in IBD. Many new drugs reduce the inflammatory response by preventing immune cell migration and communication, such as anti-integrins, anti-MAdCAM-1, and anticytokines. Antibody blocking of the IL-6 signal shows considerable therapeutic effect in the treatment of CD patients (109, 110). ILCs that release a variety of proinflammatory cytokines may also be a new therapeutic target (111). Recently, studies have found that the calcium channel TRPV1 is significantly upregulated in the inflammatory colon of IBD, which may also be a therapeutic target for IBD (112). In addition, proteins that affect intestinal epithelial connections could become therapeutic targets, including NLRP3 inflammasome, STAT6, and MLCK (82, 100, 113). Methods that directly target the mucosa are also being explored, such as stem cell transplantation (114). A high-fiber diet is extremely beneficial for glycosylation of the intestinal mucosa. Strengthening the intestinal barrier seems to be more promising than simply inhibiting the development of inflammation (55). More scientists are also paying attention to dietary therapy and improving the composition of commensal bacteria in the intestine through food intake, which could become an effective treatment plan in the future (Figure 4) (115).
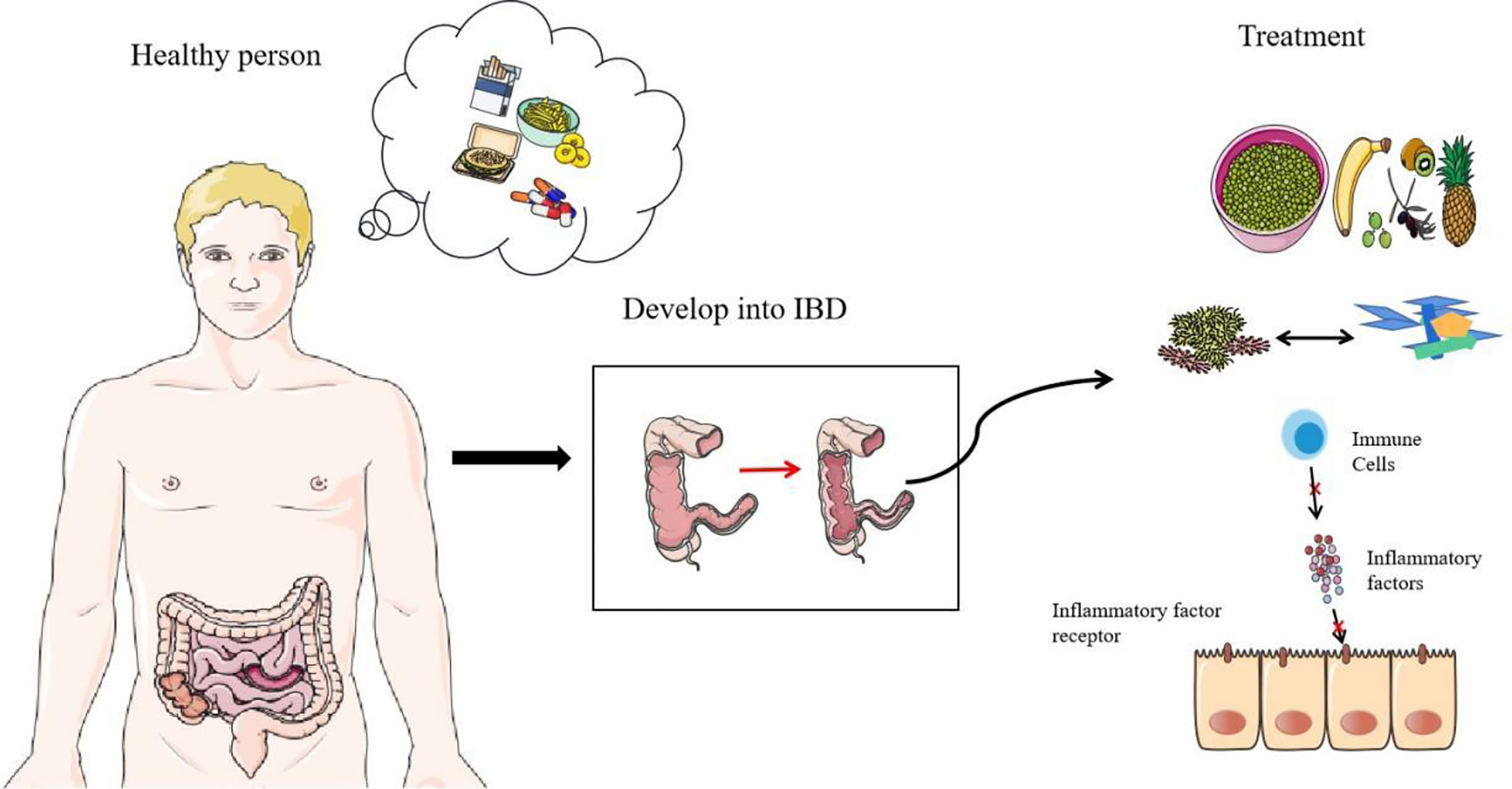
Figure 4 Prevention and treatment of IBD. Diet, smoking, and antibiotics are all predisposing factors for IBD. The first thing they destroy is the normal structure of commensal bacteria. With the research on the mechanism of IBD, there are some therapeutic targets of IBD, including the regulation of diet, the replacement of flora (including FMT and probiotics), and the suppression of immunity (including antagonistic cytokines, antagonistic cytokine receptors and other measures).
The main risk factors for IBD are diet, smoking, and antibiotics (Figure 4). Diet is a key factor in the development of IBD. High-calorie food rich in saturated fats, carbohydrates, and animal protein has a negative impact on the composition of intestinal microorganisms (116). A decrease in fiber intake leads to a decrease in the number of commensal bacteria that ferment dietary fiber, and SCFAs are derived from the decomposition of dietary fiber and resistant starch. Diet’s positive effect on anti-inflammatories is well known (117). Regional differences in diet highlight its effect in IBD. The incidence of IBD in many western countries with low-fiber diets is significantly higher than incidence in the eastern countries with high-fiber diets (118, 119). In addition, a high-salt diet will reduce the proportion of Enterobacteriaceae and affect the intestinal ecological balance (120). Smoking and antibiotics are both inducers and promoters of IBD. Studies have shown that they all start by affecting commensal bacteria and disrupting the balance of the intestinal barrier (121–123). Smoking has made a great contribution to increasing Clostridium, reducing Firmicutes, segmented filamentous bacteria. Smoking has a significant effect on mucin and inflammation genes (124). Besides, antibiotics are susceptible to AIEC in host (125). Overall, the best way to prevent IBD is to form good habits. Quitting smoking and adopting high-fiber diets need to be promoted. A healthy diet may be the best prevention. The correct use of antibiotics also requires our focus. In addition, oral health also requires our attention. Avoid causing or advancing the development of IBD due to tooth decay or periodontitis (62).
Summary and Outlook
Intestinal mucus separates commensal bacteria from intestinal epithelial cells, which keeps intestinal epithelial cells tightly aligned. The commensal bacteria degrade food in the intestine to supply energy to cells, and the degradation products SCFAs play a major role in immune regulation and reducing inflammation in the intestine. However, when some risk factors destroy the balance of the intestinal tract, the polymorphism and abundance of commensal bacteria reduce, the mucus cannot maintain the normal thickness, and the permeability increases, which allows invasive bacteria to contact intestinal epithelial cells, and induce a series of immune reactions. A large number of immune cells are activated, and the production of immune factors exceeds the limit that the body can maintain. The most prominent effect of IBD is the decrease of beneficial bacteria and the increase of pathogenic bacteria. Understanding the effect of commensal bacteria in the healthy gut and IBD and the dysregulated immune response can lead to developing new therapeutic strategies. In addition, we should pay attention to the risk factors of IBD, promote healthy diet and lifestyle, and establish healthy and strong intestinal barriers and commensal bacteria systems. In the future, scientists should pay more attention to the mutual regulation between commensal bacteria and immune system and explore more effective treatment options. Additional mechanisms of commensal bacteria and the immune system need to be studied.
Author Contributions
These authors contributed equally to this work. YC wrote the article, WC revised the article, HY provided the theme and made revisions, and XL provided revision opinions. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the National Natural Science Foundation of China (82102408), the Xuzhou Science and Technology planning Project (KC20116), Natural Science Research Project of Higher Education Institutions in Jiangsu Province (20KJB310013) and Xuzhou Medical University Excellent Talent Introduction Project (D2019030, RC20552061).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kaplan GG. The Global Burden of IBD: From 2015 to 2025. Nat Rev Gastroenterol Hepatol (2015) 12:720–7. doi: 10.1038/nrgastro.2015.150
2. Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide Incidence and Prevalence of Inflammatory Bowel Disease in the 21st Century: A Systematic Review of Population-Based Studies. Lancet (2018) 390:2769–78. doi: 10.1016/S0140-6736(17)32448-0
3. Zhang YZ, Li YY. Inflammatory Bowel Disease: Pathogenesis. World J Gastroenterol (2014) 20:91–9. doi: 10.3748/wjg.v20.i1.91
4. Ungar B, Kopylov U. Advances in the Development of New Biologics in Inflammatory Bowel Disease. Ann Gastroenterol (2016) 29:243–8. doi: 10.20524/aog.2016.0027
5. Schaubeck M, Clavel T, Calasan J, Lagkouvardos I, Haange SB, Jehmlich N, et al. Dysbiotic Gut Microbiota Causes Transmissible Crohn’s Disease-Like Ileitis Independent of Failure in Antimicrobial Defence. Gut (2016) 65:225–37. doi: 10.1136/gutjnl-2015-309333
6. Dulai PS, Levesque BG, Feagan BG, D’Haens G, Sandborn WJ. Assessment of Mucosal Healing in Inflammatory Bowel Disease: Review. Gastrointest Endosc (2015) 82:246–55. doi: 10.1016/j.gie.2015.03.1974
7. Guan Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J Immunol Res (2019) 2019:7247238. doi: 10.1155/2019/7247238
8. Leung CY, Weitz JS. Not by (Good) Microbes Alone: Towards Immunocommensal Therapies. Trends Microbiol (2019) 27:294–302. doi: 10.1016/j.tim.2018.12.006
9. Turner JR. Intestinal Mucosal Barrier Function in Health and Disease. Nat Rev Immunol (2009) 9:799–809. doi: 10.1038/nri2653
10. Cai R, Cheng C, Chen J, Xu X, Ding C, Gu B. Interactions of Commensal and Pathogenic Microorganisms With the Mucus Layer in the Colon. Gut Microbes (2020) 11:680–90. doi: 10.1080/19490976.2020.1735606
11. Wu LM, Sankaran SJ, Plank LD, Windsor JA, Petrov MS. Meta-Analysis of Gut Barrier Dysfunction in Patients With Acute Pancreatitis. Br J Surg (2014) 101:1644–56. doi: 10.1002/bjs.9665
12. Puppa MJ, White JP, Sato S, Cairns M, Baynes JW, Carson JA. Gut Barrier Dysfunction in the Apc(Min/+) Mouse Model of Colon Cancer Cachexia. Biochim Biophys Acta (2011) 1812:1601–6. doi: 10.1016/j.bbadis.2011.08.010
13. Kelly D, Conway S, Aminov R. Commensal Gut Bacteria: Mechanisms of Immune Modulation. Trends Immunol (2005) 26:326–33. doi: 10.1016/j.it.2005.04.008
14. Ahmed I, Roy BC, Khan SA, Septer S, Umar S. Microbiome, Metabolome and Inflammatory Bowel Disease. Microorganisms (2016) 4:20. doi: 10.3390/microorganisms4020020
15. Mehandru S, Colombel JF. The Intestinal Barrier, an Arbitrator Turned Provocateur in IBD. Nat Rev Gastroenterol Hepatol (2021) 18:83–4. doi: 10.1038/s41575-020-00399-w
16. Read E, Curtis MA, Neves JF. The Role of Oral Bacteria in Inflammatory Bowel Disease. Nat Rev Gastroenterol Hepatol (2021) 18:731–42. doi: 10.1038/s41575-021-00488-4
17. Stange EF, Schroeder BO. Microbiota and Mucosal Defense in IBD: An Update. Expert Rev Gastroenterol Hepatol (2019) 13:963–76. doi: 10.1080/17474124.2019.1671822
18. Berg RD. The Indigenous Gastrointestinal Microflora. Trends Microbiol (1996) 4:430–5. doi: 10.1016/0966-842x(96)10057-3
19. Sender R, Fuchs S, Milo R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PloS Biol (2016) 14:e1002533. doi: 10.1371/journal.pbio.1002533
20. Huse SM, Dethlefsen L, Huber JA, Mark Welch D, Relman DA, Sogin ML. Exploring Microbial Diversity and Taxonomy Using SSU Rrna Hypervariable Tag Sequencing. PloS Genet (2008) 4:e1000255. doi: 10.1371/journal.pgen.1000255
21. Hillman ET, Lu H, Yao T, Nakatsu CH. Microbial Ecology Along the Gastrointestinal Tract. Microbes Environ (2017) 32:300–13. doi: 10.1264/jsme2.ME17017
22. Flint HJ, Duncan SH, Scott KP, Louis P. Interactions and Competition Within the Microbial Community of the Human Colon: Links Between Diet and Health. Environ Microbiol (2007) 9:1101–11. doi: 10.1111/j.1462-2920.2007.01281.x
23. Darwich AS, Aslam U, Ashcroft DM, Rostami-Hodjegan A. Meta-Analysis of the Turnover of Intestinal Epithelia in Preclinical Animal Species and Humans. Drug Metab Dispos (2014) 42:2016–22. doi: 10.1124/dmd.114.058404
24. Johansson ME, Larsson JM, Hansson GC. The Two Mucus Layers of Colon are Organized by the MUC2 Mucin, Whereas the Outer Layer Is a Legislator of Host-Microbial Interactions. Proc Natl Acad Sci U S A (2011) 108 Suppl 1:4659–65. doi: 10.1073/pnas.1006451107
25. Ermund A, Schütte A, Johansson ME, Gustafsson JK, Hansson GC. Studies of Mucus in Mouse Stomach, Small Intestine, and Colon. I. Gastrointestinal Mucus Layers Have Different Properties Depending on Location as Well as Over the Peyer’s Patches. Am J Physiol Gastrointest Liver Physiol (2013) 305:G341–7. doi: 10.1152/ajpgi.00046.2013
26. Macpherson AJ, Uhr T. Induction of Protective Iga by Intestinal Dendritic Cells Carrying Commensal Bacteria. Science (2004) 303:1662–5. doi: 10.1126/science.1091334
27. Hooper LV, Littman DR, Macpherson AJ. Interactions Between the Microbiota and the Immune System. Science (2012) 336:1268–73. doi: 10.1126/science.1223490
28. Lueschow SR, McElroy SJ. The Paneth Cell: The Curator and Defender of the Immature Small Intestine. Front Immunol (2020) 11:587. doi: 10.3389/fimmu.2020.00587
29. Nakajima A, Vogelzang A, Maruya M, Miyajima M, Murata M, Son A, et al. Iga Regulates the Composition and Metabolic Function of Gut Microbiota by Promoting Symbiosis Between Bacteria. J Exp Med (2018) 215:2019–34. doi: 10.1084/jem.20180427
30. Rivière A, Selak M, Lantin D, Leroy F, De Vuyst L. Bifidobacteria and Butyrate-Producing Colon Bacteria: Importance and Strategies for Their Stimulation in the Human Gut. Front Microbiol (2016) 7:979. doi: 10.3389/fmicb.2016.00979
31. Louis P, Flint HJ. Formation of Propionate and Butyrate by the Human Colonic Microbiota. Environ Microbiol (2017) 19:29–41. doi: 10.1111/1462-2920.13589
32. Louis P, Flint HJ. Diversity, Metabolism and Microbial Ecology of Butyrate-Producing Bacteria From the Human Large Intestine. FEMS Microbiol Lett (2009) 294:1–8. doi: 10.1111/j.1574-6968.2009.01514.x
33. Park JH, Kotani T, Konno T, Setiawan J, Kitamura Y, Imada S, et al. Promotion of Intestinal Epithelial Cell Turnover by Commensal Bacteria: Role of Short-Chain Fatty Acids. PloS One (2016) 11:e0156334. doi: 10.1371/journal.pone.0156334
34. Kelly CJ, Colgan SP. Breathless in the Gut: Implications of Luminal O2 for Microbial Pathogenicity. Cell Host Microbe (2016) 19:427–8. doi: 10.1016/j.chom.2016.03.014
35. Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, et al. Crosstalk Between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe (2015) 17:662–71. doi: 10.1016/j.chom.2015.03.005
36. Zhao Y, Chen F, Wu W, Sun M, Bilotta AJ, Yao S, et al. GPR43 Mediates Microbiota Metabolite SCFA Regulation of Antimicrobial Peptide Expression in Intestinal Epithelial Cells via Activation of Mtor and STAT3. Mucosal Immunol (2018) 11:752–62. doi: 10.1038/mi.2017.118
37. Gasaly N, Hermoso MA, Gotteland M. Butyrate and the Fine-Tuning of Colonic Homeostasis: Implication for Inflammatory Bowel Diseases. Int J Mol Sci (2021) 22:3061. doi: 10.3390/ijms22063061
38. Kim M, Qie Y, Park J, Kim CH. Gut Microbial Metabolites Fuel Host Antibody Responses. Cell Host Microbe (2016) 20:202–14. doi: 10.1016/j.chom.2016.07.001
39. Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, et al. Metabolite-Sensing Receptors GPR43 and GPR109A Facilitate Dietary Fibre-Induced Gut Homeostasis Through Regulation of the Inflammasome. Nat Commun (2015) 6:6734. doi: 10.1038/ncomms7734
40. Lavelle A, Sokol H. Gut Microbiota-Derived Metabolites as Key Actors in Inflammatory Bowel Disease. Nat Rev Gastroenterol Hepatol (2020) 17:223–37. doi: 10.1038/s41575-019-0258-z
41. Zhou L, Sonnenberg GF. Essential Immunologic Orchestrators of Intestinal Homeostasis. Sci Immunol (2018) 3:eaao1605. doi: 10.1126/sciimmunol.aao1605
42. Xu M, Pokrovskii M, Ding Y, Yi R, Au C, Harrison OJ, et al. C-MAF-Dependent Regulatory T Cells Mediate Immunological Tolerance to a Gut Pathobiont. Nature (2018) 554:373–7. doi: 10.1038/nature25500
43. Stockinger B, Shah K, Wincent E. AHR in the Intestinal Microenvironment: Safeguarding Barrier Function. Nat Rev Gastroenterol Hepatol (2021) 18:559–70. doi: 10.1038/s41575-021-00430-8
44. Imai J, Kitamoto S, Sugihara K, Nagao-Kitamoto H, Hayashi A, Morhardt TL, et al. Flagellin-Mediated Activation of IL-33-ST2 Signaling by a Pathobiont Promotes Intestinal Fibrosis. Mucosal Immunol (2019) 12:632–43. doi: 10.1038/s41385-019-0138-4
45. Li S, Bostick JW, Ye J, Qiu J, Zhang B, Urban JF Jr, et al. Aryl Hydrocarbon Receptor Signaling Cell Intrinsically Inhibits Intestinal Group 2 Innate Lymphoid Cell Function. Immunity (2018) 49:915–28.e5. doi: 10.1016/j.immuni.2018.09.015
46. Hansen R, Thomson JM, El-Omar EM, Hold GL. The Role of Infection in the Aetiology of Inflammatory Bowel Disease. J Gastroenterol (2010) 45:266–76. doi: 10.1007/s00535-009-0191-y
47. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A Human Gut Microbial Gene Catalogue Established by Metagenomic Sequencing. Nature (2010) 464:59–65. doi: 10.1038/nature08821
48. Caenepeel C, Sadat Seyed Tabib N, Vieira-Silva S, Vermeire S. Review Article: How the Intestinal Microbiota may Reflect Disease Activity and Influence Therapeutic Outcome in Inflammatory Bowel Disease. Aliment Pharmacol Ther (2020) 52:1453–68. doi: 10.1111/apt.16096
49. Png CW, Lindén SK, Gilshenan KS, Zoetendal EG, McSweeney CS, Sly LI, et al. Mucolytic Bacteria With Increased Prevalence in IBD Mucosa Augment In Vitro Utilization of Mucin by Other Bacteria. Am J Gastroenterol (2010) 105:2420–8. doi: 10.1038/ajg.2010.281
50. Sicard JF, Le Bihan G, Vogeleer P, Jacques M, Harel J. Interactions of Intestinal Bacteria With Components of the Intestinal Mucus. Front Cell Infect Microbio (2017) 7:387. doi: 10.3389/fcimb.2017.00387
51. Kudelka MR, Stowell SR, Cummings RD, Neish AS. Intestinal Epithelial Glycosylation in Homeostasis and Gut Microbiota Interactions in IBD. Nat Rev Gastroenterol Hepatol (2020) 17:597–617. doi: 10.1038/s41575-020-0331-7
52. Qu D, Wang G, Yu L, Tian F, Chen W, Zhai Q. The Effects of Diet and Gut Microbiota on the Regulation of Intestinal Mucin Glycosylation. Carbohydr Polym (2021) 258:117651. doi: 10.1016/j.carbpol.2021.117651
53. Matsuoka K, Kanai T. The Gut Microbiota and Inflammatory Bowel Disease. Semin Immunopathol (2015) 37:47–55. doi: 10.1007/s00281-014-0454-4
54. Shiomi Y, Nishiumi S, Ooi M, Hatano N, Shinohara M, Yoshie T, et al. GCMS-Based Metabolomic Study in Mice With Colitis Induced by Dextran Sulfate Sodium. Inflamm Bowel Dis (2011) 17:2261–74. doi: 10.1002/ibd.21616
55. Gamage HKAH, Chong RWW, Bucio-Noble D, Kautto L, Hardikar AA, Ball MS, et al. Changes in Dietary Fiber Intake in Mice Reveal Associations Between Colonic Mucin O-Glycosylation and Specific Gut Bacteria. Gut Microbes (2020) 12:1802209. doi: 10.1080/19490976.2020.1802209
56. Rivera-Chávez F, Lopez CA, Bäumler AJ. Oxygen as a Driver of Gut Dysbiosis. Free Radic Biol Med (2017) 105:93–101. doi: 10.1016/j.freeradbiomed.2016.09.022
57. Fornelos N, Franzosa EA, Bishai J, Annand JW, Oka A, Lloyd-Price J, et al. Growth Effects of N-Acylethanolamines on Gut Bacteria Reflect Altered Bacterial Abundances in Inflammatory Bowel Disease. Nat Microbiol (2020) 5:486–97. doi: 10.1038/s41564-019-0655-7
58. Mukhopadhya I, Hansen R, El-Omar EM, Hold GL. IBD-What Role do Proteobacteria Play? Nat Rev Gastroenterol Hepatol (2012) 9:219–30. doi: 10.1038/nrgastro.2012.14
59. Nazareth N, Magro F, Machado E, Ribeiro TG, Martinho A, Rodrigues P, et al. Prevalence of Mycobacterium Avium Subsp. Paratuberculosis and Escherichia Coli in Blood Samples From Patients With Inflammatory Bowel Disease. Med Microbiol Immunol (2015) 204:681–92. doi: 10.1007/s00430-015-0420-3
60. Mondot S, Kang S, Furet JP, Aguirre de Carcer D, McSweeney C, Morrison M, et al. Highlighting New Phylogenetic Specificities of Crohn’s Disease Microbiota. Inflamm Bowel Dis (2011) 17:185–92. doi: 10.1002/ibd.21436
61. Sartor RB. Therapeutic Correction of Bacterial Dysbiosis Discovered by Molecular Techniques. Proc Natl Acad Sci U S A (2008) 105:16413–4. doi: 10.1073/pnas.0809363105
62. Qi Y, Zang SQ, Wei J, Yu HC, Yang Z, Wu HM, et al. High-Throughput Sequencing Provides Insights Into Oral Microbiota Dysbiosis in Association With Inflammatory Bowel Disease. Genomics (2021) 113:664–76. doi: 10.1016/j.ygeno.2020.09.063
63. Dobrzanska DA, Lamaudière MTF, Rollason J, Acton L, Duncan M, Compton S, et al. Proteobacteria: Microbial Signature of Dysbiosis in Gut Microbiota. Microb Biotechnol (2020) 13:669–82. doi: 10.1111/1751-7915.13496
64. Hamilton AL, Kamm MA, Ng SC, Morrison M. Proteus Spp. As Putative Gastrointestinal. Clin Microbiol Rev (2018) 31:e00085–17. doi: 10.1128/CMR.00085-17
65. Palmela C, Chevarin C, Xu Z, Torres J, Sevrin G, Hirten R, et al. Adherent-Invasive Escherichia Coli in Inflammatory Bowel Disease. Gut (2018) 67:574–87. doi: 10.1136/gutjnl-2017-314903
66. Larabi A, Barnich N, Nguyen HTT. New Insights Into the Interplay Between Autophagy, Gut Microbiota and Inflammatory Responses in IBD. Autophagy (2020) 16:38–51. doi: 10.1080/15548627.2019.1635384
67. Spalinger MR, Shawki A, Chatterjee P, Canale V, Santos A, Sayoc-Becerra A, et al. Autoimmune Susceptibility Gene PTPN2 is Required for Clearance of Adherent-Invasive Escherichia Coli by Integrating Bacterial Uptake and Lysosomal Defence. Gut (2021) gutjnl-2020. doi: 10.1136/gutjnl-2020-323636. gutjnl-2020-323636.
68. Kostic AD, Xavier RJ, Gevers D. The Microbiome in Inflammatory Bowel Disease: Current Status and the Future Ahead. Gastroenterology (2014) 146:1489–99. doi: 10.1053/j.gastro.2014.02.009
69. He X, Gao J, Peng L, Hu T, Wan Y, Zhou M, et al. Bacterial O-Glcnacase Genes Abundance Decreases in Ulcerative Colitis Patients and Its Administration Ameliorates Colitis in Mice. Gut (2020) 70:1872–83. doi: 10.1136/gutjnl-2020-322468. gutjnl-2020-322468.
70. Wang RX, Lee JS, Campbell EL, Colgan SP. Microbiota-Derived Butyrate Dynamically Regulates Intestinal Homeostasis Through Regulation of Actin-Associated Protein Synaptopodin. Proc Natl Acad Sci U S A (2020) 117:11648–57. doi: 10.1073/pnas.1917597117
71. Glassner KL, Abraham BP, Quigley EMM. The Microbiome and Inflammatory Bowel Disease. J Allergy Clin Immunol (2020) 145:16–27. doi: 10.1016/j.jaci.2019.11.003
72. Sokol H, Leducq V, Aschard H, Pham HP, Jegou S, Landman C, et al. Fungal Microbiota Dysbiosis in IBD. Gut (2017) 66:1039–48. doi: 10.1136/gutjnl-2015-310746
73. Sivignon A, de Vallée A, Barnich N, Denizot J, Darcha C, Pignède G, et al. Saccharomyces Cerevisiae CNCM I-3856prevents Colitis Induced by AIEC Bacteria in the Transgenic Mouse Model Mimicking Crohn’s Disease. Inflammation Bowel Dis (2015) 21:276–86. doi: 10.1097/MIB.0000000000000280
74. Kawai T, Akira S. Toll-Like Receptors and Their Crosstalk With Other Innate Receptors in Infection and Immunity. Immunity (2011) 34:637–50. doi: 10.1016/j.immuni.2011.05.006
75. Wells JM, Rossi O, Meijerink M, van Baarlen P. Epithelial Crosstalk at the Microbiota-Mucosal Interface. Proc Natl Acad Sci U S A (2011) 108 Suppl 1:4607–14. doi: 10.1073/pnas.1000092107
76. Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, et al. Interleukin-23 Drives Innate and T Cell-Mediated Intestinal Inflammation. J Exp Med (2006) 203:2473–83. doi: 10.1084/jem.20061099
77. Ng SC, Benjamin JL, McCarthy NE, Hedin CR, Koutsoumpas A, Plamondon S, et al. Relationship Between Human Intestinal Dendritic Cells, Gut Microbiota, and Disease Activity in Crohn’s Disease. Inflamm Bowel Dis (2011) 17:2027–37. doi: 10.1002/ibd.21590
78. Benjamin JL, Hedin CR, Koutsoumpas A, Ng SC, McCarthy NE, Hart AL, et al. Randomised, Double-Blind, Placebo-Controlled Trial of Fructo-Oligosaccharides in Active Crohn’s Disease. Gut (2011) 60(7):923–9. doi: 10.1136/gut.2010.232025
79. Na YR, Stakenborg M, Seok SH, Matteoli G. Macrophages in Intestinal Inflammation and Resolution: A Potential Therapeutic Target in IBD. Nat Rev Gastroenterol Hepatol (2019) 16:531–43. doi: 10.1038/s41575-019-0172-4
80. Bishop JL, Roberts ME, Beer JL, Huang M, Chehal MK, Fan X, et al. Lyn Activity Protects Mice From DSS Colitis and Regulates the Production of IL-22 From Innate Lymphoid Cells. Mucosal Immunol (2014) 7:405–16. doi: 10.1038/mi.2013.60
81. Dupraz L, Magniez A, Rolhion N, Richard ML, Da Costa G, Touch S, et al. Gut Microbiota-Derived Short-Chain Fatty Acids Regulate IL-17 Production by Mouse and Human Intestinal γδ T Cells. Cell Rep (2021) 36:109332. doi: 10.1016/j.celrep.2021.109332
82. Lin Y, Li B, Yang X, Liu T, Shi T, Deng B, et al. Non-Hematopoietic STAT6 Induces Epithelial Tight Junction Dysfunction and Promotes Intestinal Inflammation and Tumorigenesis. Mucosal Immunol (2019) 12:1304–15. doi: 10.1038/s41385-019-0204-y
83. Lee SH, Kwon JE, Cho ML. Immunological Pathogenesis of Inflammatory Bowel Disease. Intest Res (2018) 16:26–42. doi: 10.5217/ir.2018.16.1.26
84. Chang JT. Pathophysiology of Inflammatory Bowel Diseases. N Engl J Med (2020) 383:2652–64. doi: 10.1056/NEJMra2002697
85. Romagnani S. Lymphokine Production by Human T Cells in Disease States. Annu Rev Immunol (1994) 12:227–57. doi: 10.1146/annurev.iy.12.040194.001303
86. Armstrong H, Alipour M, Valcheva R, Bording-Jorgensen M, Jovel J, Zaidi D, et al. Host Immunoglobulin G Selectively Identifies Pathobionts in Pediatric Inflammatory Bowel Diseases. Microbiome (2019) 7:1. doi: 10.1186/s40168-018-0604-3
87. Caruso R, Lo BC, Núñez G. Host-Microbiota Interactions in Inflammatory Bowel Disease. Nat Rev Immunol (2020) 20:411–26. doi: 10.1038/s41577-019-0268-7
88. Ye RD, Sun L. Emerging Functions of Serum Amyloid a in Inflammation. J Leukoc Biol (2015) 98:923–9. doi: 10.1189/jlb.3VMR0315-080R
89. Lee JY, Hall JA, Kroehling L, Wu L, Najar T, Nguyen HH, et al. Serum Amyloid a Proteins Induce Pathogenic TH17 Cells and Promote Inflammatory Disease. Cell (2020) 180:79–91.e16. doi: 10.1016/j.cell.2019.11.026
90. Britton GJ, Contijoch EJ, Mogno I, Vennaro OH, Llewellyn SR, Ng R, et al. Microbiotas From Humans With Inflammatory Bowel Disease Alter the Balance of Gut Th17 and Rorγt+ Regulatory T Cells and Exacerbate Colitis in Mice. Immunity (2019) 50:212–24.e4. doi: 10.1016/j.immuni.2018.12.015
91. Round JL, Mazmanian SK. Inducible Foxp3+ Regulatory T-Cell Development by a Commensal Bacterium of the Intestinal Microbiota. Proc Natl Acad Sci U S A (2010) 107:12204–9. doi: 10.1073/pnas.0909122107
92. Lee YK, Mehrabian P, Boyajian S, Wu WL, Selicha J, Vonderfecht S, et al. The Protective Role of Bacteroides Fragilis in a Murine Model of Colitis-Associated Colorectal Cancer. mSphere (2018) 3:e00587–18. doi: 10.1128/mSphere.00587-18
93. Chu H, Khosravi A, Kusumawardhani IP, Kwon AH, Vasconcelos AC, Cunha LD, et al. Gene-Microbiota Interactions Contribute to the Pathogenesis of Inflammatory Bowel Disease. Science (2016) 352:1116–20. doi: 10.1126/science.aad9948
94. Ueno A, Jeffery L, Kobayashi T, Hibi T, Ghosh S, Jijon H. Th17 Plasticity and its Relevance to Inflammatory Bowel Disease. J Autoimmun (2018) 87:38–49. doi: 10.1016/j.jaut.2017.12.004
95. Kitamoto S, Nagao-Kitamoto H, Jiao Y, Gillilland MG 3rd, Hayashi A, Imai J, et al. The Intermucosal Connection Between the Mouth and Gut in Commensal Pathobiont-Driven Colitis. Cell (2020) 182:447–62.e14. doi: 10.1016/j.cell.2020.05.048
96. Zundler S, Becker E, Spocinska M, Slawik M, Parga-Vidal L, Stark R, et al. Hobit- and Blimp-1-Driven CD4 Tissue-Resident Memory T Cells Control Chronic Intestinal Inflammation. Nat Immunol (2019) 20:288–300. doi: 10.1038/s41590-018-0298-5
97. Plichta DR, Graham DB, Subramanian S, Xavier RJ. Therapeutic Opportunities in Inflammatory Bowel Disease: Mechanistic Dissection of Host-Microbiome Relationships. Cell (2019) 178:1041–56. doi: 10.1016/j.cell.2019.07.045
98. Coccia M, Harrison OJ, Schiering C, Asquith MJ, Becher B, Powrie F, et al. Il-1β Mediates Chronic Intestinal Inflammation by Promoting the Accumulation of IL-17A Secreting Innate Lymphoid Cells and CD4+ Th17 Cells. J Exp Med (2012) 209:1595–609. doi: 10.1084/jem.20111453
99. Chotikatum S, Naim HY, El-Najjar N. Inflammation Induced ER Stress Affects Absorptive Intestinal Epithelial Cells Function and Integrity. Int Immunopharmacol (2018) 55:336–44. doi: 10.1016/j.intimp.2017.12.016
100. He WQ, Wang J, Sheng JY, Zha JM, Graham WV, Turner JR. Contributions of Myosin Light Chain Kinase to Regulation of Epithelial Paracellular Permeability and Mucosal Homeostasis. Int J Mol Sci (2020) 21:993. doi: 10.3390/ijms21030993
101. Tye H, Yu CH, Simms LA, de Zoete MR, Kim ML, Zakrzewski M, et al. NLRP1 Restricts Butyrate Producing Commensals to Exacerbate Inflammatory Bowel Disease. Nat Commun (2018) 9:3728. doi: 10.1038/s41467-018-06125-0
102. Ceponis PJ, Botelho F, Richards CD, McKay DM. Interleukins 4 and 13 Increase Intestinal Epithelial Permeability by a Phosphatidylinositol 3-Kinase Pathway. J Biol Chem (2000) 275:29132–7. doi: 10.1074/jbc.M003516200
103. He L, Liu T, Shi Y, Tian F, Hu H, Deb DK, et al. Gut Epithelial Vitamin D Receptor Regulates Microbiota-Dependent Mucosal Inflammation by Suppressing Intestinal Epithelial Cell Apoptosis. Endocrinology (2018) 159:967–79. doi: 10.1210/en.2017-00748
104. Khan I, Ullah N, Zha L, Bai Y, Khan A, Zhao T, et al. Alteration of Gut Microbiota in Inflammatory Bowel Disease (IBD): Cause or Consequence? IBD Treatment Targeting the Gut Microbiome. Pathogens (2019) 8:126. doi: 10.3390/pathogens8030126
105. Azimirad M, Yadegar A, Asadzadeh Aghdaei H, Kelly CR. Enterotoxigenic Clostridium Perfringens Infection as an Adverse Event After Faecal Microbiota Transplantation in Two Patients With Ulcerative Colitis and Recurrent Clostridium Difficile Infection: A Neglected Agent in Donor Screening. J Crohns Colitis (2019) 13:960–1. doi: 10.1093/ecco-jcc/jjz006
106. U.S.Food and Drug Administration. Important Safety Alert Regarding Use Offecal Microbiota for Transplantation and Risk of Serious Adverse Reactions Dueto Transmission of Multi-Drug Resistant Organisms (2019). Available at: https://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/important-safety-alert-regarding-use-fecal-microbiota-transplantation-and-risk-serious-adverse (Accessed August 2, 2021).
107. Abraham BP, Quigley EMM. Probiotics in Inflammatory Bowel Disease. Gastroenterol Clin North Am (2017) 46:769–82. doi: 10.1016/j.gtc.2017.08.003
108. Geirnaert A, Calatayud M, Grootaert C, Laukens D, Devriese S, Smagghe G, et al. Butyrate-Producing Bacteria Supplemented In Vitro to Crohn’s Disease Patient Microbiota Increased Butyrate Production and Enhanced Intestinal Epithelial Barrier Integrity. Sci Rep (2017) 7:11450. doi: 10.1038/s41598-017-11734-8
109. Na SY, Moon W. Perspectives on Current and Novel Treatments for Inflammatory Bowel Disease. Gut Liver (2019) 13:604–16. doi: 10.5009/gnl19019
110. Ito H, Takazoe M, Fukuda Y, Hibi T, Kusugami K, Andoh A, et al. A Pilot Randomized Trial of a Human Anti-Interleukin-6 Receptor Monoclonal Antibody in Active Crohn’s Disease. Gastroenterology (2004) 126:989–96; discussion 947. doi: 10.1053/j.gastro.2004.01.012
111. Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial Type 1 Innate Lymphoid Cells Are a Unique Subset of IL-12- and IL-15-Responsive IFN-γ-Producing Cells. Immunity (2013) 38:769–81. doi: 10.1016/j.immuni.2013.02.010
112. Duo L, Wu T, Ke Z, Hu L, Wang C, Teng G, et al. Gain-of-Function of Ion Channel TRPV1 Exacerbates Experimental Colitis by Promoting Dendritic Cell Activation. Mol Ther Nucleic Acids (2020) 22:924–36. doi: 10.1016/j.omtn.2020.10.006
113. Zhao Y, Guo Q, Zhu Q, Tan R, Bai D, Bu X, et al. Flavonoid VI-16 Protects Against DSS-Induced Colitis by Inhibiting Txnip-Dependent NLRP3 Inflammasome Activation in Macrophages via Reducing Oxidative Stress. Mucosal Immunol (2019) 12:1150–63. doi: 10.1038/s41385-019-0177-x
114. Na SY, Moon W. Evolving Therapeutic Strategies in the Inflammatory Bowel Disease. Korean J Gastroenterol (2018) 71:61–8. doi: 10.4166/kjg.2018.71.2.61
115. Wędrychowicz A, Zając A, Tomasik P. Advances in Nutritional Therapy in Inflammatory Bowel Diseases: Review. World J Gastroenterol (2016) 22:1045–66. doi: 10.3748/wjg.v22.i3.1045
116. Chassaing B, Koren O, Goodrich JK, Poole AC, Srinivasan S, Ley RE, et al. Dietary Emulsifiers Impact the Mouse Gut Microbiota Promoting Colitis and Metabolic Syndrome. Nature (2015) 519:92–6. doi: 10.1038/nature14232
117. Yoo JY, Groer M, Dutra SVO, Sarkar A, McSkimming DI. Gut Microbiota and Immune System Interactions. Microorganisms (2020) 8:1587. doi: 10.3390/microorganisms8101587
118. Chiba M, Nakane K, Komatsu M. Westernized Diet is the Most Ubiquitous Environmental Factor in Inflammatory Bowel Disease. Perm J (2019) 23:18–107. doi: 10.7812/TPP/18-107
119. Rizzello F, Spisni E, Giovanardi E, Imbesi V, Salice M, Alvisi P, et al. Implications of the Westernized Diet in the Onset and Progression of IBD. Nutrients (2019) 11:1033. doi: 10.3390/nu11051033
120. Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, et al. Salt-Responsive Gut Commensal Modulates TH17 Axis and Disease. Nature (2017) 551:585–9. doi: 10.1038/nature24628
121. Vedamurthy A, Ananthakrishnan AN. Influence of Environmental Factors in the Development and Outcomes of Inflammatory Bowel Disease. Gastroenterol Hepatol (N Y) (2019) 15:72–82.
122. Lee SH, Yun Y, Kim SJ, Lee EJ, Chang Y, Ryu S, et al. Association Between Cigarette Smoking Status and Composition of Gut Microbiota: Population-Based Cross-Sectional Study. J Clin Med (2018) 7:282. doi: 10.3390/jcm7090282
123. Ianiro G, Tilg H, Gasbarrini A. Antibiotics as Deep Modulators of Gut Microbiota: Between Good and Evil. Gut (2016) 65:1906–15. doi: 10.1136/gutjnl-2016-312297
124. Allais L, Kerckhof FM, Verschuere S, Bracke KR, De Smet R, Laukens D, et al. Chronic Cigarette Smoke Exposure Induces Microbial and Inflammatory Shifts and Mucin Changes in the Murine Gut. Environ Microbiol (2016) 18(5):1352–63. doi: 10.1111/1462-2920.12934
Keywords: commensal bacteria, immune response, intestinal barrier, IBD, SCFAs
Citation: Chen Y, Cui W, Li X and Yang H (2021) Interaction Between Commensal Bacteria, Immune Response and the Intestinal Barrier in Inflammatory Bowel Disease. Front. Immunol. 12:761981. doi: 10.3389/fimmu.2021.761981
Received: 20 August 2021; Accepted: 26 October 2021;
Published: 11 November 2021.
Edited by:
James Cheng-Chung Wei, Chung Shan Medical University Hospital, TaiwanReviewed by:
Meng-Che Wu, Taichung Veterans General Hospital, TaiwanMing-Shiou Jan, Chung Shan Medical University, Taiwan
Copyright © 2021 Chen, Cui, Li and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huan Yang, eWFuZ2h1YW4yMDE1QHRtdS5lZHUuY24=; Xiao Li, YWhseHhpYW9AeWVhaC5uZXQ=
†These authors share first authorship