 Junhua Xie1,2
Junhua Xie1,2 Lien Van Hoecke
Lien Van Hoecke Roosmarijn E. Vandenbroucke
Roosmarijn E. Vandenbroucke- 1VIB Center for Inflammation Research, VIB, Ghent, Belgium
- 2Department of Biomedical Molecular Biology, Ghent University, Ghent, Belgium
Alzheimer’s disease (AD) is a devastating age-related neurodegenerative disorder with an alarming increasing prevalence. Except for the recently FDA-approved Aducanumab of which the therapeutic effect is not yet conclusively proven, only symptomatic medication that is effective for some AD patients is available. In order to be able to design more rational and effective treatments, our understanding of the mechanisms behind the pathogenesis and progression of AD urgently needs to be improved. Over the last years, it became increasingly clear that peripheral inflammation is one of the detrimental factors that can contribute to the disease. Here, we discuss the current understanding of how systemic and intestinal (referred to as the gut-brain axis) inflammatory processes may affect brain pathology, with a specific focus on AD. Moreover, we give a comprehensive overview of the different preclinical as well as clinical studies that link peripheral Inflammation to AD initiation and progression. Altogether, this review broadens our understanding of the mechanisms behind AD pathology and may help in the rational design of further research aiming to identify novel therapeutic targets.
Introduction
Dementia is a devastating age-related neurodegenerative disorder that affects over 40 million people worldwide. The speed of disease progression is subjective to individual variability but patients are estimated to live from a few up to 20 years after their diagnosis. Dementia is a very burdensome disorder for patients, their family, caretakers and the health care system as a whole (1). The most prevalent cause of dementia is Alzheimer disease (AD), which is a fatal neurodegenerative disorder that is characterized by progressive cognitive and functional impairment and memory loss. Most AD patients are late-onset and sporadic cases with no proven Mendelian pattern of inheritance. The prevalence of the disease increases with life expectancy and affects 10-30% of people aged over 65 years (2). Recently, the FDA approved Aducanumab which is the first medication that aims at treating AD via targeting (one of) the cause(s) of the disease, namely amyloid β (Aβ). However, its therapeutic effect is not yet conclusively proven (3) and next to that only symptomatic medication that is effective for some AD patients is available. Therefore, there is still an urgent need to develop new effective therapies that slow or prevent the progression of AD.
During the past years, the Aβ cascade hypothesis has been the most influential model explaining the pathogenesis of AD. This hypothesis proposes that the extracellular deposition of Aβ in the form of neuritic plaques is the initial pathological event in AD that also leads to the intracellular accumulation of abnormal tau proteins in neurofibrillary tangles (NFTs). These pathological changes, directly or indirectly, induce synaptic and neuronal dysfunction, and ultimately, clinical dementia (4). The steady progress in the understanding of the etiopathogenesis of AD has led to the evaluation of therapies aiming to reduce pathological aggregates of either Aβ or phosphorylated tau (pTau). Unfortunately, none of these strategies has led to clinical success (5–10). As the number of people affected with AD is rising every year, we urgently need to improve our understanding of non-amyloid components and their role in AD pathogenesis. Such new insight may help to identify novel pathways that can be targeted in novel AD therapies.
Over the last years, it became increasingly clear that innate immune activation plays a crucial role in the pathogenesis and progression of AD (11–13). For example, genome-wide association studies (GWASs) show that genes encoding immune receptors such as CR1, CLU, CD33, and TREM2 are linked with AD development (14). Moreover, a new study also identified SYK, GRN, SLC2A5, PYDC1, HEXB, and BLNK (15), genes involved in the regulation of the immune function within and outside the central nervous system (CNS), as risk genes. Taken together, these studies suggest the role of both the central as well as the peripheral immune system in the development and/or progression of AD.
In this review, we provide a short summary of the different pathways by which the periphery communicates with the brain. Next, we give a comprehensive overview of the different preclinical as well as clinical studies that link peripheral inflammation to initiation and progression of AD. This is followed by an in-depth analysis of the mechanisms of AD pathology that are affected by peripheral inflammation. Lastly, we elaborate on the growing body of evidence indicating that AD may have an underlying intestinal inflammatory process, to which alterations in gut microbiota plays an important role. These insights are essential to broaden our understanding of the mechanisms behind AD pathology and to rationally design new strategies to treat or even prevent the disease.
How Can the Periphery Communicate With the Brain?
The brain is protected from invading substances by tight barriers, including the blood-brain barrier (BBB), the blood- cerebrospinal fluid (CSF) barriers and the arachnoid barrier. These barriers assure a balanced and well-controlled micro-environment in the CNS and they provide protection against external insults such as toxins, infectious agents and peripheral pro-inflammatory cytokines. For this reason, the CNS has long been considered as an ‘immune privileged’ site. However, over the years this idea is been challenged as it is now clear that acute systemic bacterial or viral infections do affect brain functioning (16). Preclinical as well as clinical studies provide evidence that systemic generated inflammatory mediators signal to the brain via alternative pathways, namely via neural and humoral pathways (Figure 1) (17).
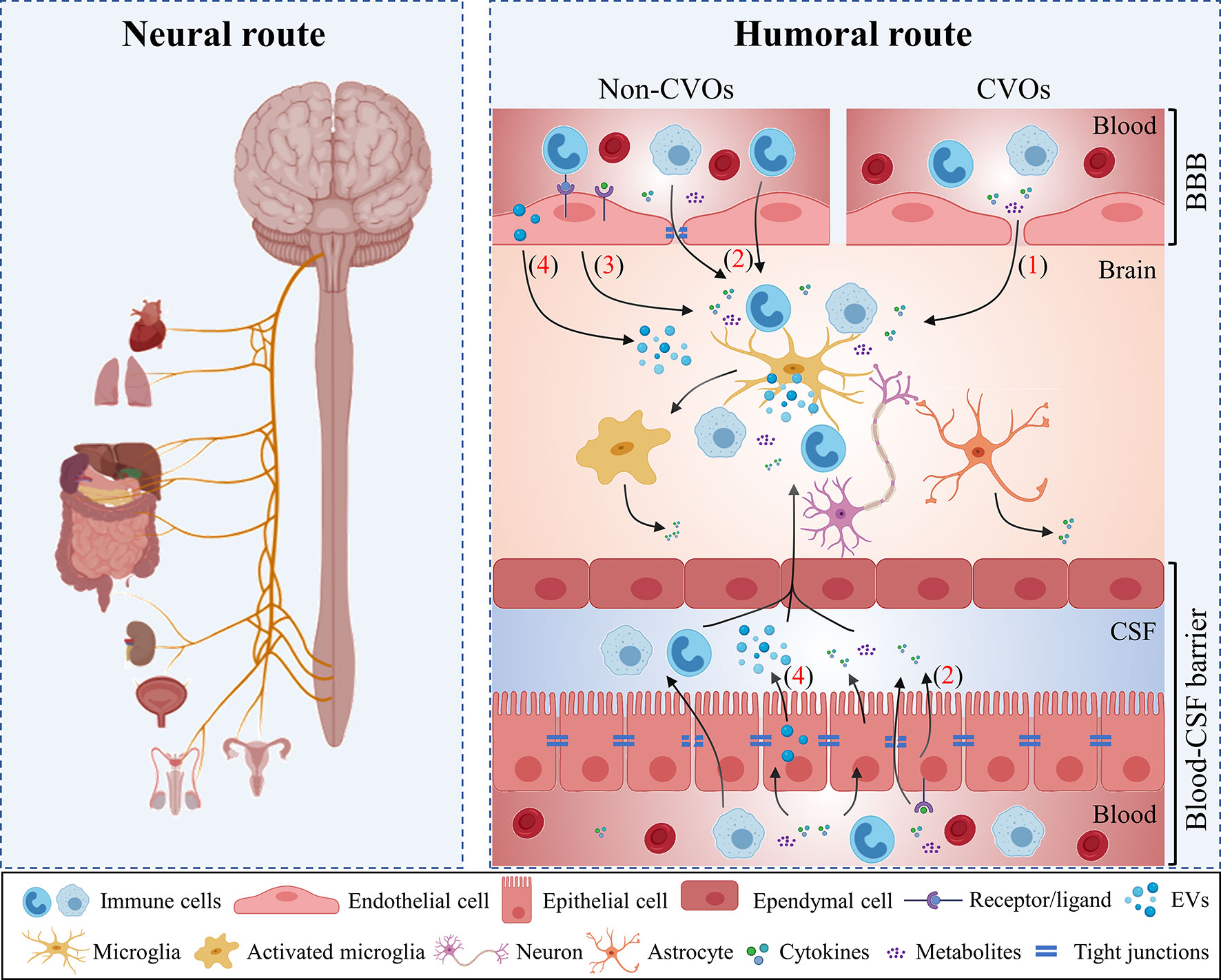
Figure 1 Periphery-to-brain communication pathways. The periphery can communicate to the brain via neural and humoral routes. Peripheral organs project signals to varied cerebral regions via the vagus nerve (the neural route). Additionally, different humoral routes are used by the periphery to communicate to the brain: (1) Circulating immune mediators access the brain via the circumventricular organs (CVOs); (2) Peripheral immune cells cytokines and metabolites interact with their transporters on cerebral endothelial cells and choroid plexus epithelium (CPE) cells and subsequently enter to the brain; (3) Periphery-to-brain communication can occur via cell-mediated interactions between peripheral signals and brain cells which in turn lead to microglial activation and neuroinflammation. (4) Peripheral immune mediators activate choroid plexus epithelial cells and induces the release of extracellular vesicles (EVs). EVs enter the brain and can be engulfed by microglia to induce pro-inflammatory response. BBB, blood-brain barrier; CPE, choroid plexus epithelium; CVOs, circumventricular organs; CSF, cerebrospinal fluid; EVs, extracellular vesicles.
Neural Pathways
Peripheral signals such as cytokines or prostaglandins from the thoracic-abdominal cavity (e.g. Kupffer cells in the liver) directly activate the afferent vagal nerve by binding with the corresponding receptors located at the vagus nerve fiber terminals (16, 18). Subsequently, the vagal nerve signals to the medulla oblongata that on its turn signals to the hypothalamus. This cascade of signaling ultimately leads to changes in neural activity inducing physiological and behavioral responses such as fever and somnolence (16, 18). Important to note is that the efferent vagal nerves can also secrete acetylcholine which on its turn acts on α7 nicotinic receptors expressed on macrophages leading to a downregulation of inflammatory cytokines such as TNF (19). Preclinical evidence shows that vagotomy attenuates the expression of pro-inflammatory cytokines in the brain induced by peripherally administered of LPS or TNF (20). Taken together, these findings underlie the importance of vagal afferents and efferents in the communication between the periphery and the brain.
Humoral Pathways
Humoral signals, including microbial metabolites, cytokines and immune cells, circulate in the blood and signal to the CNS mainly through the circumventricular organs (CVOs), across the brain barriers or via activating vascular cells at the brain barriers (21). Via these (in)direct pathways, humoral signals can potentially influence the inflammatory reactions within the brain by modulating the microglial activation, affecting myelination and neurogenesis (22).
CVOs are specialized CNS regions that lack a typical barrier structure as no tight junctions (TJs) between the capillary endothelial cells are present. Consequently, circulating substances from the blood can enter the brain via these CVOs (23).
Next to the CVOs, inflammatory molecules and immune cells can also enter the CNS by crossing one of the brain barriers via an active transport system (24) or via a disrupt barrier (25). Under physiological conditions, only soluble lipid molecules with a low molecular weight (under 0.4-0.6 kDa) and with a positive charge can cross (26). However, during systemic inflammation brain barrier dysfunction may occur and may allow the infiltration of peripheral molecules and immune cells into the CNS. This can then ultimately lead to neuroinflammation (27). Next to the well-known BBB, also the blood-CSF barrier forms an important brain barrier in health and disease. This latter barrier is formed by TJs located between the apical parts of the choroid plexus epithelium (CPE) cells and prevents the accumulation of noxious compounds into the CSF and the brain (28). Recently, it is shown that CPE cells play an important role in the transmission of peripheral signals to the brain (29). In accordance with the BBB, systemic inflammation can disrupt the blood-CSF barrier integrity (29) and leads to the invasion of peripheral immune cells (30). In this regard, Marques et al. showed that the choroid plexus displays an altered expression of genes related to cell entry pathways and innate immune responses upon repeated inflammatory stimuli (31). Interestingly, systemic inflammation also induces the release of pro-inflammatory miRNA-containing extracellular vesicles (EVs) into the CSF. Upon entry into the brain parenchyma, those EVs can be engulfed by e.g. microglia and astrocytes and subsequently regulate gene expression thereby transmitting signals from the periphery to the brain (32).
In a third possible humoral route, systemic perivascular macrophages (PVMs) or inflammatory mediators directly activate signaling pathways in vascular cells. This e.g. leads to the release of prostaglandins that are implicated in the development of sickness symptoms such as fever (33).
Of course, the above described pathways are not mutually exclusive, so the net effect of systemic inflammation on the brain can be the result of a combination of the different routes.
An Increasing Body of Evidence Links Systemic Inflammation to AD Development
Over the last years, it became increasingly clear that systemic inflammation affects the brain in multiple ways and that it is key in the development of neurodegenerative diseases. Below, we look into preclinical and clinical studies that hint to this latter association.
Evidence From Preclinical Studies
To be able to investigate the effect of systemic inflammatory processes on AD pathology, different mouse models using different insults have been designed. Below we look deeper into these different models and their effect on AD pathogenesis.
The majority of animal models use peripheral injection of immune-stimulating molecules such as lipopolysaccharide (LPS), polyinosinic-polycytidylic acid (poly I:C) or pro-inflammatory cytokines to evoke systemic inflammation. For example, systemic LPS challenge induces a number of sickness symptoms in rodents including fever, loss of appetite and decreased activity (16). Remarkable is that peripheral systemic LPS or increased levels of peripheral pro-inflammatory cytokines do not lead to a prolonged induction of neuronal death (34, 35). It however does lead to temporal induction of neuronal death. This temporary effect can be explained by a whole repertoire of regulatory mechanisms that limit the effects of pro-inflammatory responses in the brain. E.g. increased production of a large variety of proteins from microglial cells including antimicrobial peptides, anti-inflammatory cytokines (IL-10), transforming growth factor (TGF)-β, prostaglandin E2 (PGE2), anti-nuclear factor-kappa B (NF-κB) proteins, mitogen-activated protein and suppressor of cytokine signaling proteins (36). Next to this, the CNS regulates the peripheral inflammatory response through feedback loops in hypothalamic-pituitary-adrenal (HPA) axis and vagal reflex, and neurochemical changes (16). Despite the limited and temporal induction of neuronal damage, a single systemic LPS challenge does lead to increased deposition of Aβ1-42 and phospho-tau (p-tau) levels in the brain of wild type rodents (37). Moreover, repeated systemic LPS injections even lead to a prolonged elevation of Aβ levels and cognitive deficits (38–40). Strikingly, pregnant mice exposed to repeated systemic LPS causes AD‐related features including behavioral and neuropathological changes in their offspring (41).
To better understand the impact of systemic inflammation on AD pathology, researcher looked into the effect of systemic inflammation on the removal or the deposition of Aβ plaques in the brain (42). Studies in Tg2576 (43), PDAPP (44), APPSwe (45), APP/PS1 (40, 46) and AppNL-G-F (47) mice show an increase in Aβ deposition upon LPS-induced systemic inflammation. Similarly, poly I:C induced systemic inflammation in 4 months old 3xTg-AD mice increases Aβ deposition in the brain (48). Also, TNF-induced systemic inflammation in 15 months old APP/PS1 mice increases Aβ plaques formation (49). Unfortunately, there are several contradictory results regarding the impact of systemic inflammation on Aβ burden in the mouse brain (Table 1). For example, experiments similar to those mentioned above in 3xTg-AD and 5XFAD mice show elevated C-terminal APP fragments (β-CTF) and tau hyperphosphorylation while Aβ deposition is unchanged (59, 60). Even studies in Tg2576 (64, 65, 74), APP/PS1 (50–53) and tgSwe (66) mice show reduced Aβ burden upon systemic LPS challenge. Important to keep in mind is that none of these latter studies analyzed the impact on cognitive impairment. This is important as LPS-induced systemic inflammation affects more than only the Aβ pathology. For example, a study in 3xTg-AD mice demonstrates that LPS challenge affects key neuropathological features of the AD-like phenotype including behavior, microgliosis and astrocytosis (62). In addition, we recently found that low-grade systemic inflammation induced by LPS also causes the microgliosis and neuronal dysfunction in a second-generation mouse model (AppNL-G-F) of AD (47).
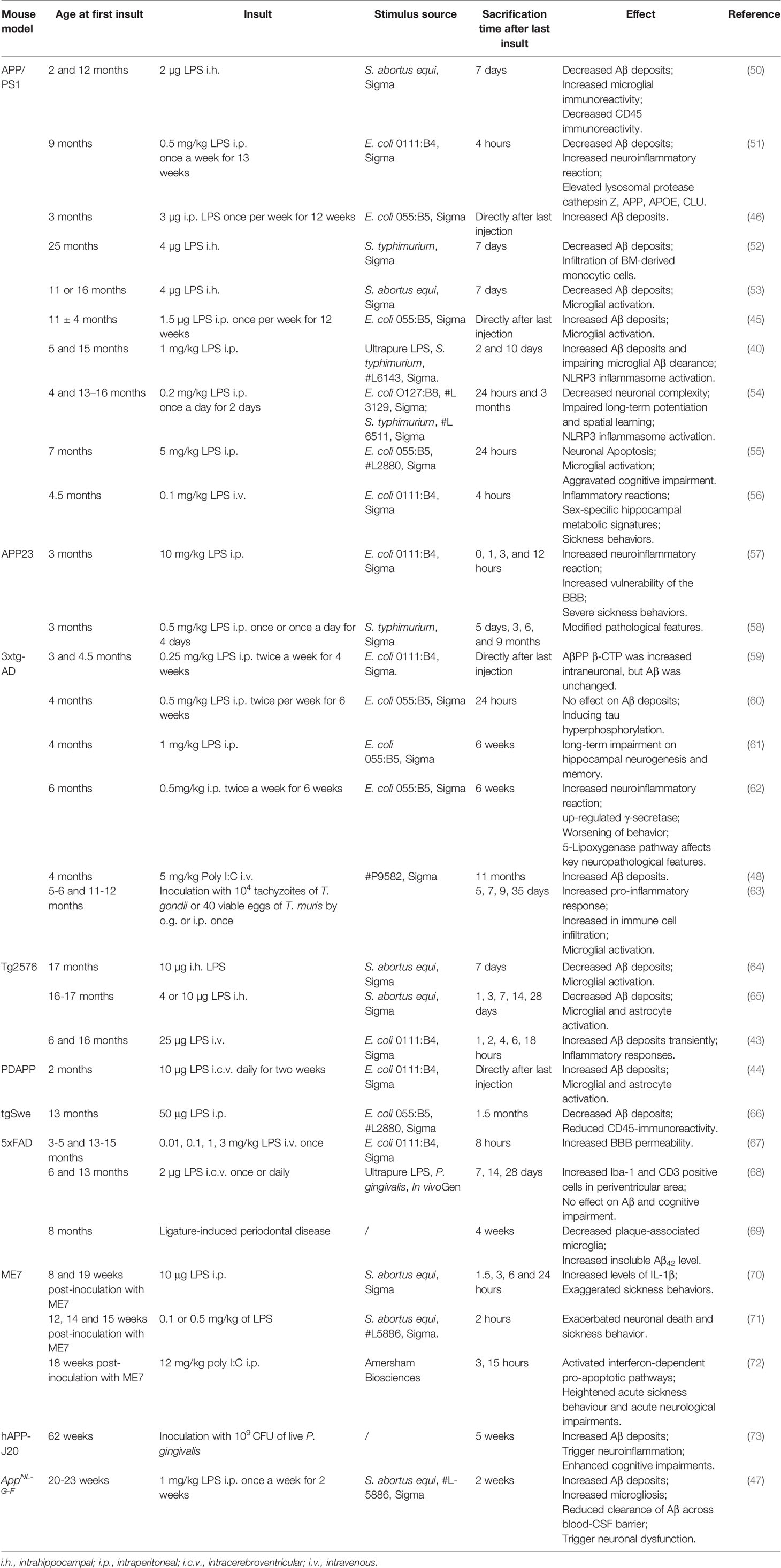
Table 1 Preclinical studies on the effect of systemic inflammation in mouse models for AD.
The contradictory results in literature regarding the effect of systemic inflammation on Aβ pathology can be due to the experimental setup, such as the genetic background of the used mouse model, age, LPS injection procedure such as number of injections, LPS dosage, source and route of administration, and the time between injection and sacrification. Table 1 gives an overview of preclinical mouse studies in which the effect of systemic inflammation on AD was studied.
Next to the above described genetic mouse models for AD, also ME7 prion mice are used as a model for AD. These mice are characterized by synaptic loss, microglial and astrocyte activation, neuronal death and an age-independent induction of AD pathology (75). A systemic LPS challenge of these ME7 prion mice evokes exaggerated AD related sickness (70). Moreover, a single systemic LPS challenge is enough to cause abnormal CNS functioning including increased pro-inflammatory responses, neuronal apoptosis, cognitive decline, motor symptoms and an earlier onset of AD in the ME7 compared to control mice (35, 71).
Increasing evidence supports the association of systemic inflammation induced by bacterial infections and AD progression in humans. For example, Helicobacter pylori (H. pylori) and periodontal bacteria like Porphyromonas gingivalis (P. gingivalis) are potential risk factors for the development of AD (16). As the above described immune-stimulating molecules are only mimicking parts of a ‘real’ infection, several studies also use peripheral bacterial infections to induce systemic inflammation.
An experimental study demonstrates that P. gingivalis infected mice evoke increased expression of IL-1β, AβPP770, cathepsin B (CatB) and Aβ in the liver (76). Additionally, P. gingivalis infected-hAPP-J20 AD mice, show exacerbated cognitive impairment with increased Aβ deposits and neuroinflammation (73). A variety of in vivo studies show that also H. pylori infection plays a role in AD development. For example, the intraperitoneal injection of H. pylori TN2GF4 leads to elevated Aβ levels and induces spatial learning and memory impairment in wild type rats (77). The same research group showed that H. pylori TN2GF4 infection induces significant tau hyperphosphorylation with activation of glycogen synthase kinase-3β (GSK-3β) and that treatment with GSK-3 inhibitors significantly attenuated H. pylori-induced AD pathology (78). Important to underscore is that these results are obtained upon intraperitoneal injection of H. pylori while normally these germs live in the digestive tract. To explore the long-term effect of H. pylori infection on brain function, H. pylori TN2GF4 was given this time by oral gavage to rats once a week during a period of four weeks. Despite the reported short-term effects upon intraperitoneal injection, 4 weeks H. pylori TN2GF4-infected rats via oral gavage showed no tauopathy or cognitive impairment. Compared to other H. pylori strains such as B47 and SS1, the colonization capacity of TN2GF4 is significant lower (79). Consequently, TN2GF4 may not be able to induce a strong inflammatory response resulting in the recovery of the observed short-term pathology (40). However, the route of infection may also play a crucial role on the outcome of the experiments.
Additionally, also the used strain has an impact on the outcome of the infection. For example, infection with H. pylori B47 or H. felis CS1 increases neuroinflammation but has no effect on brain Aβ deposition (80) while infection with H. pylori SS1 or H. felis has no effect on neuroinflammation nor on Aβ deposition (81). This contradiction can be explained by the fact that different H. pylori strains and H. pylori SS1 lacks some important virulence factors such as functional cag pathogenicity island (cagPAI) and vacuolating cytotoxin A (VacA) (82).
Next to bacterial infections, also viruses and parasites are known to induce systemic inflammation in human and several of them such as herpes viruses and Toxoplasma have been proposed as triggers of AD (83, 84). Similarly, also a few mouse studies using viruses or parasites to induce systemic inflammation have been reported in the context of AD. For example, local gut inflammation induced by infection with the parasites Toxoplasma gondii and Trichuris muris significantly enhances neuroinflammation in 3xTg-AD mice. Unfortunately, no cognitive tests were performed in this study (63). Also Chlamydophila pneumonia (C. pneumoniae) (85), fungi (86), herpes simplex viruses (HSV) (87) and cytomegalovirus (CMV) (88) are reported to promote the development of AD. Most recently, coronavirus disease 2019 (COVID-19) has been reported to cause various neurologic symptoms including cognitive impairment that may ultimately result in AD, either directly through the invasion of the coronavirus into the CNS or indirectly via virus-induced inflammation (89). In rhesus monkey, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was shown to invade primarily via the olfactory bulb followed by spreading into CNS thereby inducing neuroinflammation possibly by targeting neurons, microglia, and astrocytes (90).
Surgical interventions are sometimes necessary in human patients for a variety of reasons. In some cases, the immune system may not effectively distinguish between stimuli elicited by surgery and those elicited by trauma or pathogenic infection (91). Surgical procedures thus represent a potential trigger for systemic inflammation and are consequently also used in animal research. A study that uses cecal ligation and puncture (CLP) to induce polymicrobial sepsis in rats shows escalated levels of Aβ, p-tau protein and receptor for advanced glycation end products (RAGE) markers with simultaneous cognitive impairment (92). Similarly, laparotomy-induced systemic inflammation in wild-type mice induced abnormal tau phosphorylation and memory impairment (93). Furthermore, orthopedic surgery causes hippocampal-dependent memory impairment, which is associated with increased levels of IL-1β both in plasma and hippocampus (94).
Also for several non-infectious diseases such as atherosclerosis, obesity, diabetes and depressive illness, there is clear evidence for being risk factors in the development of AD and all these diseases are associated with a chronic pro-inflammatory phenotype (16). For example, in B6Tg2576 mice, a transgenic mouse model of AD was produced by back-crossing Tg2576 mice, early atherosclerosis lesions were detected and were positively correlated with brain Aβ accumulation when mice were fed a normal diet or atherogenic diets (95, 96). Diabetic mouse models include streptozotocin-induced Type 1 diabetes and high-fat and/or sugar diet-induced Type 2 diabetes have been showed to aggravate Aβ pathology in APP transgenic mice and even in non-transgenic rodents (97). Similarly, high fat diet-induced obesity in mice promotes systemic inflammation and impairs cognitive functioning through increased Aβ accumulation and BBB disruption (98, 99). For the studies on depression in mouse models of AD, anti-depressive treatment can decrease Aβ burden and cognitive impairment (100).
Altogether, the above described preclinical studies form a large body of evidence showing an association between peripheral inflammation and AD pathology. Rodent studies suggest that Aβ and neuroinflammation are the result of a direct response to systemic infections and are part of the brains innate immune response to inflammatory insults. Of note, every mouse model has limitations in replicating the full AD pathology, and further side-by-side comparisons between different mouse models and observations from human AD patients are required to move towards the development of effective treatments.
Evidence From Clinical Studies
Next to the preclinical studies, there are also a large number of clinical studies reporting that systemic inflammation is associated with the increase in cognitive decline in AD (101–103). This systemic inflammation can be caused by specific environmental factors, bacterial and viral infections and the presence of a chronic disease, such as diabetes.
Environmental Risk Factors
It is well known that several of the environmental risk factors for the development of AD have systemic pro-inflammation as a common characteristic. For example, ageing is a major risk factor and is accompanied by a low-grade systemic inflammation and a relative decline in adaptive immunity and T helper 2 (Th2) cell response. This concept is better known as inflammaging (16) and is caused by an imbalance between pro- and anti-inflammatory mediators. An example of inflammaging is the age-related endocrine dyscrasia with loss of sex steroids and elevation of gonadotrophins that is associated with an increased pro-inflammatory state and is remarkably also associated with an increased development of AD (104, 105).
There is also a build-up of amyloid plaques throughout life. However, it is not clear if this build-up is primarily caused by the occurrence of systemic inflammatory events or if it is due to other ageing mechanisms or even a combination of both. Cross-sectional studies show that patients with cognitive impairment and evidence of Aβ exhibit an increased systemic inflammatory response and increased microglial activation compared to healthy subjects (106). Nevertheless, it is also known that not all these patients have activated microglia (107) and that one-third of the healthy population older than 80 years have Aβ loads comparable to the load found in AD patients (108). Next to this, persons with high Aβ loads but without dementia have lower level of pro-inflammatory cytokines than AD patients (16). Additionally, analysis of AD and mild cognitive impairment (MCI) patients showed a significant correlation between cognition and microglial activation but not with Aβ loads (109, 110). These findings support the idea that microglial activation rather than Aβ alone may be the key change leading to initiation and progression of AD.
Bacterial and Viral Infections
Along with preclinical studies, also epidemiologic studies demonstrate an association between periodontal bacteria or H. pylori infection and MCI and AD (111–113). GWAS show that P. gingivalis infection significantly enrich the expression of genes related to cognitive decline (114). Moreover, AD patients show elevated levels of antibodies against periodontal bacteria (115, 116), increased levels of periodontopathic virulence factors (117) and P. gingivalis in postmortem brain tissue (118). Also, gingipains, i.e. proteases secreted by P. gingivalis, are found in the brain of 90% of AD patients and this is correlated with the present tau and Aβ levels (118). Taken together, all these data suggest that periodontitis leads to cognitive decline that may be mediated by systemic inflammation (119). In contrast, a recent cross-sectional study reports that only 11 of the 29 inflammatory biomarkers are associated with cognitive impairment in patients with severe periodontitis. However, the inflammatory response to severe periodontitis was more reduced (lower biomarker concentrations) in patients with cognitive impairment or dementia than in cognitively healthy controls (120). These contradictory or inconsistent results may be caused by differences in study design, diagnostic criteria, length of follow-up, controls, and appropriate analytical approach (121).
Next to periodontal bacteria, also H. pylori is a possible risk factor for the development of AD, although not all studies are consistent about this (122). Clinical studies confirm that AD and MCI patients have higher anti-H. pylori IgG titers in their blood and brain (111, 123) and indicate that AD patients have more gastric inflammation (112). Moreover, the presence of H. pylori IgG antibodies is associated with a 1.46 times increased risk for the development of dementia compared to non-infected controls (124). Additionally, two independent surveys show that H. pylori eradication may improve AD manifestation at 2- and 5-year clinical endpoints (125, 126). In contrast to the above studies is the study of Roubaud-Baudron et al. that shows that there is no correlation between H. pylori infection and the occurrence of AD (127). Important to keep in mind is that the latter study has a small patient cohort and lack non-AD dementia patients or control patients. Moreover, serum antibody detection was used to diagnose H. pylori infection but this method has a high rate of false negative result (128).
Next to the effect of bacterial infections, increasing clinical evidence indicates that also viral and fungal infections may be causative factors for the development of AD. For example, neurotropic pathogens such as HSV-1 have been repeatedly isolated from the brains of AD patients and HSV infection is associated with a higher risk for dementia in some studies (129–132). In contrast to these studies, a genetic analysis study using Mendelian randomization found no association between gene-predicted risk for herpes infection and subsequent cognitive decline or AD (133). The same holds true for the association between an infection with cytomegalovirus (CMV) and the development of dementia (134).
Chronic Diseases
The chronic diseases including obesity, diabetes, Atherosclerosis and depressive illness have also an epidemiological basis for being proposed as risk factors in the development of AD and are all associated with a chronic pro-inflammatory state. The abnormal metabolism inherent to a chronic disease induces a general pro-inflammatory response in peripheral organs. For example, dysfunction of lipid metabolism in obese patients leads to inappropriate overflow of circulating free fatty acids (FFAs), which can activate pro-inflammatory pathways through cell surface pattern recognition receptors (PRRs) (135). Consequently, this sustained chronic inflammatory situation can amongst others disrupt blood-brain interfaces, allowing peripheral mediators to enter the brain. This on its term might induce persistent chronic brain inflammation resulting in problems with brain function, including cognitive impairment in AD (22). In obese and diabetic patients, they show a greater cognitive decline over last decades compared to healthy subjects (136–139). The further investigation indicates the higher blood glucose levels in the preceding 5 years correlate with an increased risk of dementia among participants with and without diabetes (140). Epidemiological studies also report that the incidence cardiovascular disease affects Aβ metabolism and leads to accumulation of Aβ in peripheral and brain (141). Depression is common in AD patients, but the individual with depression is also associated with higher chance in dementia risk (100). While their individual attributable risk is likely to be small, their combined cumulative effects over time might be considerable (16).
Systemic Inflammation Affects Different Characteristics of AD Pathology
As discussed above there is a huge amount of (pre)clinical data supporting the hypothesis that systemic inflammation is correlated with AD pathology. Below, we look deeper into the mechanisms of AD pathology that are affected by systemic inflammation. More precisely, we elaborate on how systemic inflammation induces neuroinflammation, promotes Tau hyperphosphorylation, impairs Aβ clearance and brain barrier integrity (Figure 2). The mechanism studies included below are mainly from preclinical experiments, but were applicable also clinical studies are mentioned.
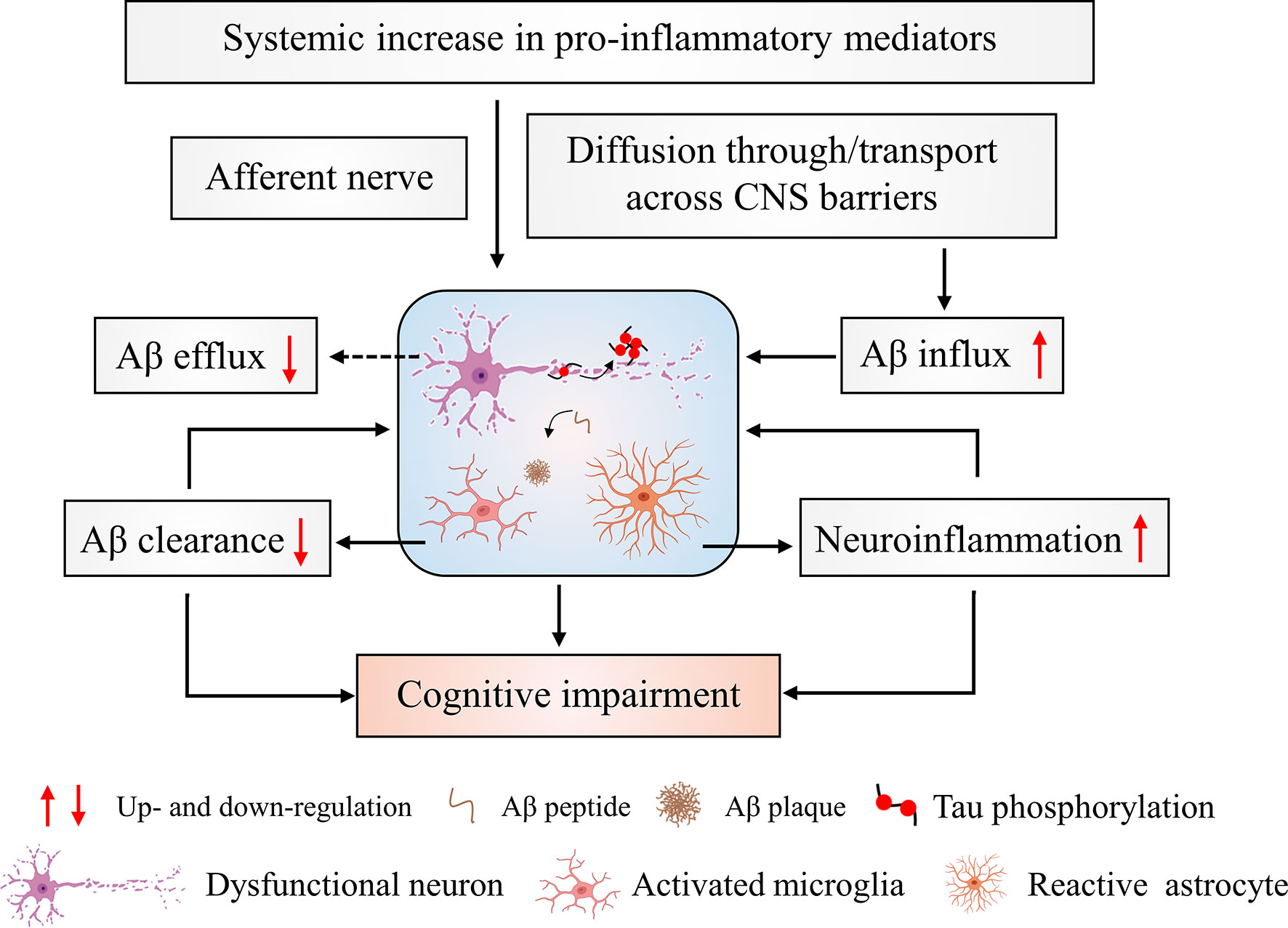
Figure 2 Cerebral changes in response to systemic inflammation. Systemic inflammation leads to increased levels of pro-inflammatory mediators. These signals can project to the brain via nerve afferents and the brain barriers. This can directly and/or indirectly induce neuronal cytotoxicity and affect Aβ transport resulting in increased Aβ aggregation. The aggregated Aβ induces an initial activation of microglial cells that leads to activated microglial cells with an impaired Aβ clearing ability. Additionally, the activated microglial cells produce a large amount of pro-inflammatory cytokines that further exacerbate neuroinflammation. This worsening of neuroinflammation promotes the development of brain pathology and ultimately leads to cognitive impairments.
Systemic Inflammation Triggers Neuroinflammation
Peripheral inflammation results in the activation of the innate immune system and the production of pro-inflammatory cytokines. These immune factors circulate in the blood and ultimately affect neurons and glial cells in the brain via the neural and humoral pathways mentioned above. In the context of chronic inflammation present in AD patients, evidence suggests that peripheral immune cells infiltrate in the CNS and accumulate near areas of pathology (142). On their term, these cells induce increased microglial activation and Aβ deposition (143). Microglia, the primary immune effector cells of the CNS, are key cellular mediators of the neuroinflammatory responses in AD (144). In absence of inflammatory stimuli, microglial cells are in a “resting” or inactive state in which they continually survey their immediate environment without interfering with neurons and neuronal activities. However, when activated, the morphological and biological functions of microglial cells are altered and they migrate to the site of injury to initiate an innate immune response (145, 146). Unfortunately, when microglia remain for a long time in such an activated state, they release cytokines and neurotoxic agents such as IL-1β, TNF, IL-6, nitric oxide (NO) and reactive oxygen species (ROS) that can directly or indirectly cause neuronal cell death (144).
Microglia express various cell surface receptors, such as PRRs that recognize misfolded and aggregated proteins such as Aβ and subsequently trigger an innate immune response (147). Activated microglia that are persistent in the release of pro-inflammatory mediators are involved in the suppression of axonal transport and adult neurogenesis (148). Moreover, activated microglia are characterized by the retraction of their processes, which is a phenotypic change that correlates with an impaired ability to remodel synapses (47, 149). This effect contributes to impaired synaptic plasticity seen in AD. Additionally, prolonged microglia impairment caused by pro-inflammatory cytokines can also damage neurons by reducing trophic factors such as brain-derived neurotrophic factor (BDNF) and insulin-like growth factor (IGF) (149–151). Studies on signaling mechanisms indicate that microglial myeloid differentiation primary response 88 (MyD88) (152) and p38 mitogen activated protein kinase (MAPK) (153, 154) signaling are involved in the release of neurotoxins leading to neuronal damage. These inflammatory effects are specifically initiated by the microglia-derived pro-inflammatory cytokine TNF (153, 155). In addition, neuronal mitochondria play an important role in the regulation of microglial activation and the neuronal protein Mitofusin-2 is likely the mechanistic linker between neuronal mitochondria dysfunction and neuroinflammation (156).
Next to microglia also inflammasomes, i.e. multiprotein complexes that control the production of pro-inflammatory cytokines, play an important role in neuroinflammation (157). The NOD-, LRR- and pyrin domain-containing 3 (NLRP3) inflammasome is one of the best defined and most widely implicated regulators of IL-1β and IL-18 production (157). Inflammasomes function as intracellular sensors for both foreign and host-derived danger signals. For example, soluble Aβ oligomers and protofibrils can induce NLRP3 inflammasome activation in microglia (158). Ablation of NLRP3 inflammasomes protects mice from age-related increases of innate immune activation in the periphery and the CNS and attenuates functional decline (159).
In addition, also other molecular and cellular players are involved in the institution of neuroinflammation upon peripheral inflammation. For example, a study demonstrates that IL-32 and IL-32β affect neuroinflammation and Aβ formation by activating signal transducers and activators of transcription 3 (STAT3) and NF-κB (160).
Systemic Inflammation Impairs Aβ Clearance
Aβ load is the net result of Aβ production and Aβ clearance and small changes in this equilibrium result in abnormal accumulation of the protein. Aβ clearance depends on several potential pathways (1): phagocytosis (2), endocytosis and macropinocytosis by professional phagocytes and microglia, as well as by astrocytes, oligodendrocytes and neurons (3), Aβ degradation by Aβ-degrading enzymes like neprilysin, insulin-degrading enzyme (IDE) and matrix metalloproteinases (MMPs) and (4) Aβ efflux from the brain to the blood and influx from the blood to the brain via transport across the BBB and blood-CSF barrier, interstitial fluid bulk flow and CSF egress pathways, including arachnoid villi and glymphatic-lymphatic pathways (47, 161).
In aged AD mice, microglial cells have lower expression of Aβ-phagocytic receptors and Aβ-degrading enzymes, but their ability to produce pro-inflammatory cytokines is maintained (162). Furthermore, AD mice crossed with Tg197 mice (163), mice carrying a modified human TNF-globin transgene, show deregulated patterns of human TNF gene expression that develop chronic inflammatory polyarthritis and amyloid deposition. The increased amyloid deposition can be explained by the fact that systemic TNF indirectly modulates Aβ pathology by regulating peripheral immune cell trafficking and glial responses in the brain (164). Our recent study has also indicated that sustained exposure to LPS leads to impaired microglial phagocytosis of Aβ and increased Aβ deposition in AppNL-G-F mice (47).
Recently, it has been shown that inflammasome signaling is involved in microglial Aβ clearance. Indeed, NLRP3 deficient APP/PS1 mice are partially protected from Aβ pathology and neuronal dysfunction under LPS-induced systemic inflammation (40, 54). Importantly, these mice show almost normal cognitive function and they are protected from Aβ-induced suppression of synaptic plasticity (165). ASC (apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain) is a key adaptor molecule required for the inflammatory processes and represents an essential step in the activation of NLRP3 inflammasomes (166). Aβ can bind to ASC and consequently amplifies NLRP3 inflammasome activity which on his turn impairs Aβ clearance by microglia. The resulting accumulated Aβ binds to more ASC and in this way a vicious circle is established (167).
Next, low Aβ levels within the healthy brain are also maintained through transport across the BBB (161). Unfortunately, LPS-induced systemic inflammation causes lipoprotein receptor-related protein-1 (LRP-1)-dependent decreased Aβ entry into the blood (168, 169) and increased Aβ influx into the brain (170). Moreover, polymicrobial infection-induced RAGE accumulation facilitates the transport of Aβ across the BBB and increases the central Aβ load (92). In addition, the blood-CSF barrier is also involved in processes that clear substances from the CSF and the blood. Moreover, the main transporters of Aβ are also found in choroid plexus epithelium, including LRP1, LRP2, P-glycoprotein (P-gp) and RAGE (47, 171). Interestingly, using primary cells to mimic the blood-CSF barrier, we recently showed that the transporter LRP2 is involved in Aβ efflux from the CSF to blood side and that this is impaired in response to inflammation (47).
Systemic Inflammation Induces Changes at the Brain Barriers
As already mentioned above, the brain parenchyma is enclosed by different structures including multilayered meninges, the BBB, the blood-CSF barrier and the glia limitans. The BBB and blood-CSF barrier are the two main brain barriers to impede free diffusion between brain and blood and to provide transport processes for essential nutrients, ions and metabolic waste products (172). Although research is mainly focused on the BBB, more and more research is now emphasizing the important role of the blood-CSF barrier in CNS homeostasis and neurological disorders. The BBB is formed by endothelial cells that are tightly linked by tight junctions (TJs). On the basement side of the membrane, pericytes and astrocytes perform supporting and regulatory functions (173). The blood-CSF barrier is formed by TJs between neighboring choroid plexus epithelial cells (174).
As described above, a number of in vitro and in vivo studies show that systemic inflammation has disruptive effects on BBB integrity leading to the diffusion of peripheral inflammatory factors into the brain (25). These factors then further induce changes in the brain and are associated with an increased cognitive decline in AD patients (16). Next to the diffusion of peripheral factors, also Tau transmission in the brain via the disrupted BBB is observed upon peripheral inflammation (175).
A number of mechanisms are described to explain the BBB disruptive effect of systemic inflammation. Central to these mechanisms are prostanoids and NO, which are both synthesized by LPS-stimulated cerebrovascular endothelium and surrounding cells (25). The mediators, including MMPs (176, 177) and ROS (178), are also involved in the destruction of BBB integrity by activating intracellular pathways such as MAPK signaling (176) and inducing mitochondrial dysfunction (179) while blood-CSF barrier integrity loss was linked to MMP-dependent collagen cleavage (173). In addition, some miRNAs play a role in BBB structure and function (180). E.g. the expression of miR-155 in microvessels is strongly and rapidly upregulated by inflammatory cytokines and alters BBB function by affecting expression of TJs and adhesion components (181).
Next to these molecules, also different cell types influence BBB integrity. Abbott and colleagues showed that astrocytes and pericytes secrete a number of molecules that enhance and maintain BBB integrity. Moreover, the end-feet of these cells form a lacework of fine lamellae closely linked to the outer surface of the BBB endothelium and basement membrane (182). Previous studies have shown that systemic inflammation induces astrocyte proliferation and activation followed by astrocyte loss (183, 184) and changes in astrocytic end-feet structures (185). During sustained systemic inflammation, astrocytes produce a range of substances including pro-inflammatory cytokines and prostaglandins that are associated with disruption of the BBB (25). Next, also microglia are important players of the neurovascular unit although their ablation in mature mice does not directly leads to an increase in BBB permeability (150). However, a recent study shows that vessel-associated microglia initially maintain BBB integrity via expression of claudin-5 and make physical contact with endothelial cells. Yet, under LPS-induced systemic inflammation, brain resident microglia migrate in a CCR5-dependent manner to the cerebral vasculature and phagocytose astrocytic end-feet. In this way microglia then anyway impair BBB integrity (186).
The crossing of leukocytes through the BBB upon systemic inflammation primarily occurs at postcapillary venules and may occur in a paracellular or transcellular way (25). TNF has been shown to promote expression of brain microvessel P- and E-selectin, which are both required for cellular recruitment (187). In addition, systemic inflammation stimulates endothelial production of chemokines such as CCL2, which lead to conformational changes in leukocyte integrins and enhance their binding to endothelial ligands (188). Similarly, the expression of endothelial cell adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1) are promoted upon inflammation. Important is that leukocytes need to cross the glia limitans to enter the brain parenchyma after passing though the endothelium and this crossing depends on the degradation of basement membrane components by MMP-2 and -9 (189). In vitro studies show that LPS stimulation increases expression of MMP-2 in the endothelium (176) and pericytes (190).
Next to direct morphological changes in the BBB, systemic inflammation can also cause non-disruptive changes that affect BBB functionality. For example, there are different studies indicating that BBB transport pathways are affected by systemic inflammation. Including the downregulation of the multi-functional efflux transporter P-gp (191) and the upregulation of influx carriers responsible for TNF (192). Correspondingly, systemic inflammation also results in a reduced bulk flow of CSF and interstitial fluid (ISF) across the BBB, which can further impair Aβ clearance (168). Additionally, non-disruptive BBB changes during systemic inflammation may also promote neuroinvasion of pathogens. For example, systemic LPS enhances the transcellular transport of the human immunodeficiency virus (HIV) without disrupting the BBB though luminal stimulation by IL-6 and granulocyte-macrophage colony-stimulating factor (GM-CSF) and MAPK signaling (193).
As mentioned above, systemic inflammation not only has an impact on the integrity of the BBB, but LPS-induced systemic inflammation in mice also disrupts the blood-CSF barrier (47, 177) and induces the release of extracellular vesicles (EVs) (32); both resulting in increased neuroinflammation. Interestingly, also Aβ on its own has a direct impact on the blood-CSF barrier: induction of morphological changes of the CPE cells, decreased expression of TJ components, loss of barrier integrity, and EV release into the CSF (47, 194, 195). Analysis of human gene expression data comparing control and AD patient choroid plexus tissue revealed that TNF/TNFR1 signaling was upregulated in AD suggesting an involvement of this pathway in the blood-CSF barrier associated changes in AD (196). In agreement with this, both TNFR1 deficiency and treatment with a TNFR1 inhibitor prevented the Aβ-induced cognitive decline (196). Additionally, also treatment with an inhibitor of EV production had the same effect (195). Based on these results, it is tempting to speculate that the combined effect of systemic inflammation and the presence of Aβ might further worsen AD disease progression among others via specific mechanisms at the blood-CSF barriers, such as loss of barrier integrity and EV release, which on their turn further aggravate neuroinflammation. However, in our study in which APPNL-G-F mice were challenged twice with a low dose of LPS no additive effect on blood-CSF barrier disruption was observed (47), which might indicate that high LPS levels are needed to disrupt this barrier.
Gut Inflammation Affects AD Pathology
Despite the anatomical separation between the CNS and the gastrointestinal system, a bidirectional network between the two, known as the ‘gut-brain axis’, exists. A growing body of evidence indicates that AD may have an underlying intestinal inflammatory process, to which altered gut microbiota plays an important role. The gut-brain interaction can occur via two routes: via vagal transmission and via systemic circulation (197).
Thousands of sensor and motor fibers from the vagus nerve connect the gut with the brainstem and serve as a conduit for neural signals. These signals are governed by changes in enteric neuron activity and the behavior of gut microbes. Such gut bacteria activate the vagus nerve either directly or indirectly through their metabolites and neuroactive substances. Moreover, the vagus nerve interacts extensively with different components of the systemic immune system and in this way continuously monitors the inflammatory state of the gut. Upon sensing inflammatory signals by binding to specific receptors in the vagus nerve fiber such as the proinflammatory tachykinin/neurokinin receptors (198), the vagal afferents transmit signals to the dorsal vagal complex (DVC) where most sensory information is relayed to nucleus tractus solitarii (NTS) (199). Neurons in the NTS that receive the vagal sensory inputs can modulate microglial state and activity by specific ligand-receptor pairs (e.g. CX3CL1-CX3CR1 and CD200-CD200R), neurotransmitters (e.g. glutamate and GABA), and purinergic signaling (200). Activated microglia on their turn produce pro-inflammatory (e.g. nitric oxide and PGE2) and cytokines (e.g. TNF, IL-1β and IL-6) and ultimately influence the neuroinflammatory state (201). Concurrently, vagal efferent nerves hand over information about the immune status of the brain back to the gut, with increased CNS inflammation feeding back to inhibit further release of peripheral pro-inflammatory cytokines through acetylcholine-mediated signaling (202). Effective vagal nerve signaling is critical for sending appropriate signals to microglia in order to modulate levels of neuroinflammation (201). Moreover, vagus nerve stimulation combined with LPS challenge leads to a decrease in microglial production of pro-inflammatory cytokines in the brain, an effect no longer observed after vagotomy (203). Taken together, the vagus nerve is a physical conduit between gut microbial activity and neuroinflammation.
Interestingly, intestinal inflammation induced by gut microbiota perturbation is directly associated with intestinal barrier dysfunction. The enhancement of intestinal permeability allows the entrance of pathogenic, immune-stimulating and neuroactive substances into the systemic circulation. Increased systemic pro-inflammatory cytokines and neurotoxic compounds may contribute to an increased microglial activation and production of pro-inflammatory cytokines in the brain as described above (204, 205). Calprotectin as a marker of intestinal inflammation and has intrinsically amyloidogenic amino acid sequences that can form amyloid oligomers and fibrils (206). Interestingly, calprotectin levels are significantly increased in the CSF and the brain of AD patients, which promotes its amyloid aggregation and co-aggregation with Aβ (207). It is possible that this intestinal source of calprotectin may contribute to amyloid fibril formation in the gut or directly in the brain.
Along with affecting the level of intestinal permeability, gut microbiota can indirectly influence the state of systemic inflammation through interactions with nearby immune cells. When pathogen-associated molecular patterns (PAMPs) produced by pathogenic invaders bind to PRRs, such as TLRs, inflammatory cytokine production is altered (208). The circulation and subsequent potential entry of these cytokines into the brain act locally on CNS cells, including microglia, thereby influencing the state of inflammation in the brain (209). Indeed, increased intestinal inflammation driven by either LPS or bacterial infection correlates with elevated levels of microglial activation and release of pro-inflammatory cytokines (124, 210).
Gut microbiota-derived metabolites are additional contributors to the gut-brain crosstalk. Circulation of microbial metabolites, including neurotransmitters, short-chain fatty acids (SCFAs), and trimethylamines can potentially influence microglial activation through direct and indirect means (197, 211). For example, binding of serotonin to 5-hydroxytryptamine receptors expressed on microglia induces the release of cytokine-carrying exosomes and induces neuroinflammation (212). However, the exact SCFA signaling pathways that modulate microglial activation are not yet fully understood. Mice lacking the free fatty acid receptor 2 (FFAR2), a GPCR required for SCFA signaling in the gut, exhibit a microglial phenotype similar to that observed in germ-free mice (209). The absence of FFAR2 expression on microglia suggests that SCFAs may influence microglial activation through signaling that originate in the gut and SCFAs may have potential direct influences on microglia. Indeed, treatment of microglial cells in vitro with various SCFAs, including valproic acid and butyric acid, elevates the levels of acetylation of histone H3 (213). This suggests that SCFAs influence microglial behavior in vivo through a combination of GPCR signaling and histone deacetylase inhibition.
Bacterial extracellular vesicles (bEVs), including Gram-negative bacteria derived outer membrane vesicles (OMVs) and Gram-positive bacteria secreted membrane vesicles (MVs), carry molecular cargo from paternal bacteria to target cells. bEVs contain numerous PAMPs, including DNA, RNA, lipoproteins, LPS and peptidoglycan. The PAMP content of bEVs enables them to engage with host PRRs and consequently initiate pro-inflammatory signaling cascades that lead to the production of cytokines and chemokines. A growing body of evidence suggests that bEVs play a key role in the communication and regulation of the host and even manipulate the host immune response. Interestingly, this may ultimately affect AD progression (214, 215). For example, OMVs from H. pylori were shown to modulate pro-inflammatory responses in gut epithelial cells with a dose-dependent production of the pro-inflammatory cytokine IL-8 (216). Also bEVs from other bacteria have immunostimulatory abilities in mice and human (214). Taken together, bEVs have the abilities to modulate systemic inflammation and disrupt epithelial barrier integrity and ultimately increase development of AD though the pathways mentioned above. For example, extracellular RNAs (exRNAs) in periodontal bacteria Aggregatibacter actinomycetemcomitans OMVs were shown to cross the BBB in mice and promote TNF production in human macrophages via activating TLR-8 and NF-κB signaling pathways (217). In addition, intravenous administration of bEVs isolated from faeces of AD patients increases the BBB permeability and promotse glial cell activation in wild-type mice, thereby inducing an inflammatory response and tau hyperphosphorylation by activating the GSK‐3β pathway and finally leading to cognitive impairment (218).
All the above mentioned findings further underscore the associated between systemic inflammation, influenced in part by the gut microbiota, and BBB, microglial activation and neuroinflammation, and ultimately AD.
Conclusion
Increasing evidence indicates that systemic inflammation might drive the initiation and progression of AD. It is becoming increasingly clear that the brain cannot longer be viewed as an immune-privileged region and that CNS inflammation and systemic inflammation are connected to each other. Systemic inflammation rather than Aβ or tau alone may be a key player in AD pathology and its role may precede Aβ deposition. In addition, whether the build-up of Aβ plaques and tau hyperphosphorylation as we age is primarily due to the occurrence of systemic inflammatory events or to other ageing mechanisms is still unclear but it is likely that the combination of different factors, such as inflammaging and microglial priming are crucial. Clearly, continued research in this area is needed to further unravel the effects of systemic inflammation on AD and its mechanisms. Furthermore, this review strengthens the believe that peripheral inflammation worsens AD progression and this opens up a wide range of possible therapeutic strategies for AD via the modulation of peripheral inflammation.
Author Contributions
JX wrote the manuscript. LVH and RV critically revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
JX is supported by the Chinese Scholarship Council (CSC) grant number: 201808360194. Research in the authors’ lab is funded by the Foundation for Alzheimer’s Research Belgium (SAO-FRA) grant number: 20200032, the Baillet Latour Fund and the Research Foundation Flanders (FWO Vlaanderen) G055121N.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wong W. Economic Burden of Alzheimer Disease and Managed Care Considerations. Am J Manag Care (2020) 26:S177–83. doi: 10.37765/ajmc.2020.88482
2. Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer’s Disease. Nat Rev Dis Primers (2015) 1:15056. doi: 10.1038/nrdp.2015.56
3. Rabinovici GD. Controversy and Progress in Alzheimer’s Disease — FDA Approval of Aducanumab. N Engl J Med (2021) 385:771–4. doi: 10.1056/NEJMp2111320
4. Hardy J, Selkoe DJ. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science (2002) 297:353–6. doi: 10.1126/science.1072994
5. Arvanitakis Z, Grodstein F, Bienias JL, Schneider JA, Wilson RS, Kelly JF, et al. Relation of NSAIDs to Incident AD, Change in Cognitive Function, and AD Pathology. Neurology (2008) 70:2219–25. doi: 10.1212/01.wnl.0000313813.48505.86
6. Breitner JC, Haneuse SJ, Walker R, Dublin S, Crane PK, Gray SL, et al. Risk of Dementia and AD With Prior Exposure to NSAIDs in an Elderly Community-Based Cohort. Neurology (2009) 72:1899–905. doi: 10.1212/WNL.0b013e3181a18691
7. Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. A Phase 3 Trial of Semagacestat for Treatment of Alzheimer’s Disease. N Engl J Med (2013) 369:341–50. doi: 10.1056/NEJMoa1210951
8. Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, et al. Clinical Effects of Abeta Immunization (AN1792) in Patients With AD in an Interrupted Trial. Neurology (2005) 64:1553–62. doi: 10.1212/01.WNL.0000159740.16984.3C
9. Group AR, Martin BK, Szekely C, Brandt J, Piantadosi S, Breitner JC, et al. Cognitive Function Over Time in the Alzheimer’s Disease Anti-Inflammatory Prevention Trial (ADAPT): Results of a Randomized, Controlled Trial of Naproxen and Celecoxib. Arch Neurol (2008) 65:896–905. doi: 10.1001/archneur.2008.65.7.nct70006
10. Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, et al. Subacute Meningoencephalitis in a Subset of Patients With AD After Abeta42 Immunization. Neurology (2003) 61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8
11. Heneka MT, Golenbock DT, Latz E. Innate Immunity in Alzheimer’s Disease. Nat Immunol (2015) 16:229–36. doi: 10.1038/ni.3102
12. Frost GR, Jonas LA, Li YM. Friend, Foe or Both? Immune Activity in Alzheimer’s Disease. Front Aging Neurosci (2019) 11:337. doi: 10.3389/fnagi.2019.00337
13. Labzin LI, Heneka MT, Latz E. Innate Immunity and Neurodegeneration. Annu Rev Med (2018) 69:437–49. doi: 10.1146/annurev-med-050715-104343
14. Kamboh MI, Demirci FY, Wang X, Minster RL, Carrasquillo MM, Pankratz VS, et al. Genome-Wide Association Study of Alzheimer’s Disease. Transl Psychiatry (2012) 2:e117. doi: 10.1038/tp.2012.45
15. Sierksma A, Lu A, Mancuso R, Fattorelli N, Thrupp N, Salta E, et al. Novel Alzheimer Risk Genes Determine the Microglia Response to Amyloid-Beta But Not to TAU Pathology. EMBO Mol Med (2020) 12:e10606. doi: 10.15252/emmm.201910606
16. Holmes C. Review: Systemic Inflammation and Alzheimer’s Disease. Neuropathol Appl Neurobiol (2013) 39:51–68. doi: 10.1111/j.1365-2990.2012.01307.x
17. Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Dolan RJ, et al. Neural Origins of Human Sickness in Interoceptive Responses to Inflammation. Biol Psychiatry (2009) 66:415–22. doi: 10.1016/j.biopsych.2009.03.007
18. Perry VH. Contribution of Systemic Inflammation to Chronic Neurodegeneration. Acta Neuropathol (2010) 120:277–86. doi: 10.1007/s00401-010-0722-x
19. Pavlov VA, Tracey KJ. The Cholinergic Anti-Inflammatory Pathway. Brain Behav Immun (2005) 19:493–9. doi: 10.1016/j.bbi.2005.03.015
20. Zielinski MR, Dunbrasky DL, Taishi P, Souza G, Krueger JM. Vagotomy Attenuates Brain Cytokines and Sleep Induced by Peripherally Administered Tumor Necrosis Factor-Alpha and Lipopolysaccharide in Mice. Sleep (2013) 36:1227–38. doi: 10.5665/sleep.2892
21. Paouri E, Gcorgopoulos S. Systemic and CNS Inflammation Crosstalk: Implications for Alzheimer’s Disease. Curr Alzheimer Res (2019) 16:559–74. doi: 10.2174/1567205016666190321154618
22. Kempuraj D, Thangavel R, Selvakumar GP, Zaheer S, Ahmed ME, Raikwar SP, et al. Brain and Peripheral Atypical Inflammatory Mediators Potentiate Neuroinflammation and Neurodegeneration. Front Cell Neurosci (2017) 11:216. doi: 10.3389/fncel.2017.00216
23. Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From Inflammation to Sickness and Depression: When the Immune System Subjugates the Brain. Nat Rev Neurosci (2008) 9:46–57. doi: 10.1038/nrn2297
24. Banks WA. Blood-Brain Barrier Transport of Cytokines: A Mechanism for Neuropathology. Curr Pharm Des (2005) 11:973–84. doi: 10.2174/1381612053381684
25. Varatharaj A, Galea I. The Blood-Brain Barrier in Systemic Inflammation. Brain Behav Immun (2017) 60:1–12. doi: 10.1016/j.bbi.2016.03.010
26. Bellettato CM, Scarpa M. Possible Strategies to Cross the Blood-Brain Barrier. Ital J Pediatr (2018) 44:131. doi: 10.1186/s13052-018-0563-0
27. Jiao Z, Zhang W, Chen C, Zhu X, Chen X, Zhou M, et al. Gene Dysfunction Mediates Immune Response to Dopaminergic Degeneration in Parkinson’s Disease. ACS Chem Neurosci (2019) 10:803–11. doi: 10.1021/acschemneuro.8b00373
28. Ghersi-Egea JF, Strazielle N, Catala M, Silva-Vargas V, Doetsch F, Engelhardt B. Molecular Anatomy and Functions of the Choroidal Blood-Cerebrospinal Fluid Barrier in Health and Disease. Acta Neuropathol (2018) 135:337–61. doi: 10.1007/s00401-018-1807-1
29. Gorle N, Blaecher C, Bauwens E, Vandendriessche C, Balusu S, Vandewalle J, et al. The Choroid Plexus Epithelium as a Novel Player in the Stomach-Brain Axis During Helicobacter Infection. Brain Behav Immun (2018) 69:35–47. doi: 10.1016/j.bbi.2017.12.010
30. Kooij G, Kopplin K, Blasig R, Stuiver M, Koning N, Goverse G, et al. Disturbed Function of the Blood-Cerebrospinal Fluid Barrier Aggravates Neuro-Inflammation. Acta Neuropathol (2014) 128:267–77. doi: 10.1007/s00401-013-1227-1
31. Marques F, Sousa JC, Coppola G, Geschwind DH, Sousa N, Palha JA, et al. The Choroid Plexus Response to a Repeated Peripheral Inflammatory Stimulus. BMC Neurosci (2009) 10:135. doi: 10.1186/1471-2202-10-135
32. Balusu S, Van Wonterghem E, De Rycke R, Raemdonck K, Stremersch S, Gevaert K, et al. Identification of a Novel Mechanism of Blood-Brain Communication During Peripheral Inflammation via Choroid Plexus-Derived Extracellular Vesicles. EMBO Mol Med (2016) 8:1162–83. doi: 10.15252/emmm.201606271
33. Matsumura K, Kobayashi S. Signaling the Brain in Inflammation: The Role of Endothelial Cells. Front Biosci (2004) 9:2819–26. doi: 10.2741/1439
34. Nadeau S, Rivest S. Endotoxemia Prevents the Cerebral Inflammatory Wave Induced by Intraparenchymal Lipopolysaccharide Injection: Role of Glucocorticoids and CD14. J Immunol (2002) 169:3370–81. doi: 10.4049/jimmunol.169.6.3370
35. Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and Systemic Endotoxin Challenges Exacerbate the Local Inflammatory Response and Increase Neuronal Death During Chronic Neurodegeneration. J Neurosci (2005) 25:9275–84. doi: 10.1523/Jneurosci.2614-05.2005
36. Rivest S. Regulation of Innate Immune Responses in the Brain. Nat Rev Immunol (2009) 9:429–39. doi: 10.1038/nri2565
37. Wang LM, Wu Q, Kirk RA, Horn KP, Ebada Salem AH, Hoffman JM, et al. Lipopolysaccharide Endotoxemia Induces Amyloid-Beta and P-Tau Formation in the Rat Brain. Am J Nucl Med Mol Imaging (2018) 8:86–99.
38. Kahn MS, Kranjac D, Alonzo CA, Haase JH, Cedillos RO, McLinden KA, et al. Prolonged Elevation in Hippocampal Abeta and Cognitive Deficits Following Repeated Endotoxin Exposure in the Mouse. Behav Brain Res (2012) 229:176–84. doi: 10.1016/j.bbr.2012.01.010
39. Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW, et al. Neuro-Inflammation Induced by Lipopolysaccharide Causes Cognitive Impairment Through Enhancement of Beta-Amyloid Generation. J Neuroinflamm (2008) 5:37. doi: 10.1186/1742-2094-5-37
40. Tejera D, Mercan D, Sanchez-Caro JM, Hanan M, Greenberg D, Soreq H, et al. Systemic Inflammation Impairs Microglial A Beta Clearance Through NLRP3 Inflammasome. EMBO J (2019) 38:e101064. doi: 10.15252/embj.2018101064
41. Wang F, Zhang ZZ, Cao L, Yang QG, Lu QF, Chen GH. Lipopolysaccharide Exposure During Late Embryogenesis Triggers and Drives Alzheimer-Like Behavioral and Neuropathological Changes in CD-1 Mice. Brain Behav (2020) 10:e01546. doi: 10.1002/brb3.1546
42. Dawson TM, Golde TE, Lagier-Tourenne C. Animal Models of Neurodegenerative Diseases. Nat Neurosci (2018) 21:1370–9. doi: 10.1038/s41593-018-0236-8
43. Sly LM, Krzesicki RF, Brashler JR, Buhl AE, McKinley DD, Carter DB, et al. Endogenous Brain Cytokine mRNA and Inflammatory Responses to Lipopolysaccharide Are Elevated in the Tg2576 Transgenic Mouse Model of Alzheimer’s Disease. Brain Res Bull (2001) 56:581–8. doi: 10.1016/S0361-9230(01)00730-4
44. Qiao X, Cummins DJ, Paul SM. Neuroinflammation-Induced Acceleration of Amyloid Deposition in the APPV717F Transgenic Mouse. Eur J Neurosci (2001) 14:474–82. doi: 10.1046/j.0953-816x.2001.01666.x
45. Sheng JG, Bora SH, Xu G, Borchelt DR, Price DL, Koliatsos VE. Lipopolysaccharide-Induced-Neuroinflammation Increases Intracellular Accumulation of Amyloid Precursor Protein and Amyloid Beta Peptide in APPswe Transgenic Mice. Neurobiol Dis (2003) 14:133–45. doi: 10.1016/s0969-9961(03)00069-x
46. Michaud JP, Halle M, Lampron A, Theriault P, Prefontaine P, Filali M, et al. Toll-Like Receptor 4 Stimulation With the Detoxified Ligand Monophosphoryl Lipid A Improves Alzheimer’s Disease-Related Pathology. Proc Natl Acad Sci USA (2013) 110:1941–6. doi: 10.1073/pnas.1215165110
47. Xie J, Gorlé N, Vandendriessche C, Van Imschoot G, Van Wonterghem E, Van Cauwenberghe C, et al. Low-Grade Peripheral Inflammation Affects Brain Pathology in the AppNL-G-FMouse Model of Alzheimer’s Disease. Acta Neuropathol Commun (2021) 9:163. doi: 10.1186/s40478-021-01253-z
48. Krstic D, Madhusudan A, Doehner J, Vogel P, Notter T, Imhof C, et al. Systemic Immune Challenges Trigger and Drive Alzheimer-Like Neuropathology in Mice. J Neuroinflamm (2012) 9:151. doi: 10.1186/1742-2094-9-151
49. Hennessy E, Gormley S, Lopez-Rodriguez AB, Murray C, Murray C, Cunningham C. Systemic TNF-alpha Produces Acute Cognitive Dysfunction and Exaggerated Sickness Behavior When Superimposed Upon Progressive Neurodegeneration. Brain Behav Immun (2017) 59:233–44. doi: 10.1016/j.bbi.2016.09.011
50. Go M, Kou JH, Lim JE, Yang JL, Fukuchi KI. Microglial Response to LPS Increases in Wild-Type Mice During Aging But Diminishes in an Alzheimer’s Mouse Model: Implication of TLR4 Signaling in Disease Progression. Biochem Biophys Res Commun (2016) 479:331–7. doi: 10.1016/j.bbrc.2016.09.073
51. Thygesen C, Ilkjaer L, Kempf SJ, Hemdrup AL, Von Linstowl CU, Babcock AA, et al. Diverse Protein Profiles in CNS Myeloid Cells and CNS Tissue From Lipopolysaccharide- and Vehicle-Injected APP(SWE)/PS1(Delta E9) Transgenic Mice Implicate Cathepsin Z in Alzheimer’s Disease. Front Cell Neurosci (2018) 12:397. doi: 10.3389/fncel.2018.00397
52. Malm TM, Koistinaho M, Parepalo M, Vatanen T, Ooka A, Karlsson S, et al. Bone-Marrow-Derived Cells Contribute to the Recruitment of Microglial Cells in Response to Beta-Amyloid Deposition in APP/PS1 Double Transgenic Alzheimer Mice. Neurobiol Dis (2005) 18:134–42. doi: 10.1016/j.nbd.2004.09.009
53. DiCarlo G, Wilcock D, Henderson D, Gordon M, Morgan D. Intrahippocampal LPS Injections Reduce Abeta Load in APP+PS1 Transgenic Mice. Neurobiol Aging (2001) 22:1007–12. doi: 10.1016/s0197-4580(01)00292-5
54. Beyer MMS, Lonnemann N, Remus A, Latz E, Heneka MT, Korte M. Enduring Changes in Neuronal Function Upon Systemic Inflammation Are NLRP3 Inflammasome Dependent. J Neurosci (2020) 40:5480–94. doi: 10.1523/JNEUROSCI.0200-20.2020
55. Zhou J, Yu WH, Zhang M, Tian X, Li Y, Lu Y. Imbalance of Microglial TLR4/TREM2 in LPS-Treated APP/PS1 Transgenic Mice: A Potential Link Between Alzheimer’s Disease and Systemic Inflammation. Neurochem Res (2019) 44:1138–51. doi: 10.1007/s11064-019-02748-x
56. Agostini A, Ding YC, Bai L, Kendall DA, Pardon MC. Sex-Specific Hippocampal Metabolic Signatures at the Onset of Systemic Inflammation With Lipopolysaccharide in the APPswe/PS1dE9 Mouse Model of Alzheimer’s Disease. Brain Behav Immun (2020) 83:87–111. doi: 10.1016/j.bbi.2019.09.019
57. Takeda S, Sato N, Ikimura K, Nishino H, Rakugi H, Morishita R. Increased Blood-Brain Barrier Vulnerability to Systemic Inflammation in an Alzheimer Disease Mouse Model. Neurobiol Aging (2013) 34:2064–70. doi: 10.1016/j.neurobiolaging.2013.02.010
58. Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate Immune Memory in the Brain Shapes Neurological Disease Hallmarks. Nature (2018) 556:332–8. doi: 10.1038/s41586-018-0023-4
59. McAlpine FE, Lee JK, Harms AS, Ruhn KA, Blurton-Jones M, Hong J, et al. Inhibition of Soluble TNF Signaling in a Mouse Model of Alzheimer’s Disease Prevents Pre-Plaque Amyloid-Associated Neuropathology. Neurobiol Dis (2009) 34:163–77. doi: 10.1016/j.nbd.2009.01.006
60. Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-Induced Inflammation Exacerbates Tau Pathology by a Cyclin-Dependent Kinase 5-Mediated Pathway in a Transgenic Model of Alzheimer’s Disease. J Neurosci (2005) 25:8843–53. doi: 10.1523/JNEUROSCI.2868-05.2005
61. Valero J, Mastrella G, Neiva I, Sanchez S, Malva JO. Long-Term Effects of an Acute and Systemic Administration of LPS on Adult Neurogenesis and Spatial Memory. Front Neurosci (2014) 8:83. doi: 10.3389/fnins.2014.00083
62. Joshi YB, Giannopoulos PF, Chu J, Pratico D. Modulation of Lipopolysaccharide-Induced Memory Insult, Gamma-Secretase, and Neuroinflammation in Triple Transgenic Mice by 5-Lipoxygenase. Neurobiol Aging (2014) 35:1024–31. doi: 10.1016/j.neurobiolaging.2013.11.016
63. Montacute R, Foley K, Forman R, Else KJ, Cruickshank SM, Allan SM. Enhanced Susceptibility of Triple Transgenic Alzheimer’s Disease (3xtg-AD) Mice to Acute Infection. J Neuroinflamm (2017) 14:50. doi: 10.1186/s12974-017-0826-5
64. Herber DL, Mercer M, Roth LM, Symmonds K, Maloney J, Wilson N, et al. Microglial Activation Is Required for A Beta Clearance After Intracranial Injection of Lipopolysaccharide in APP Transgenic Mice. J Neuroimmune Pharmacol (2007) 2:222–31. doi: 10.1007/s11481-007-9069-z
65. Herber DL, Roth LM, Wilson D, Wilson N, Mason JE, Morgan D, et al. Time-Dependent Reduction in A Beta Levels After Intracranial LPS Administration in APP Transgenic Mice. Exp Neurol (2004) 190:245–53. doi: 10.1016/j.expneurol.2004.07.007
66. Jendresen C, Digre A, Cui H, Zhang X, Vlodavsky I, Li JP, et al. Systemic LPS-Induced A Beta-Solubilization and Clearance in A Beta PP-Transgenic Mice Is Diminished by Heparanase Overexpression. Sci Rep (2019) 9:4600. doi: 10.1038/s41598-019-40999-4
67. Barton SM, Janve VA, McClure R, Anderson A, Matsubara JA, Gore JC, et al. Lipopolysaccharide Induced Opening of the Blood Brain Barrier on Aging 5XFAD Mouse Model. J Alzheimers Dis (2019) 67:503–13. doi: 10.3233/Jad-180755
68. Hayashi K, Hasegawa Y, Takemoto Y, Gao C, Takeya H, Komohara Y, et al. Continuous Intracerebroventricular Injection of Porphyromonas Gingivalis Lipopolysaccharide Induces Systemic Organ Dysfunction in a Mouse Model of Alzheimer’s Disease. Exp Gerontol (2019) 120:1–5. doi: 10.1016/j.exger.2019.02.007
69. Kantarci A, Tognoni CM, Yaghmoor W, Marghalani A, Stephens D, Ahn JY, et al. Microglial Response to Experimental Periodontitis in a Murine Model of Alzheimer’s Disease. Sci Rep (2020) 10:18561. doi: 10.1038/s41598-020-75517-4
70. Combrinck MI, Perry VH, Cunningham C. Peripheral Infection Evokes Exaggerated Sickness Behaviour in Pre-Clinical Murine Prion Disease. Neuroscience (2002) 112:7–11. doi: 10.1016/s0306-4522(02)00030-1
71. Cunningham C, Campion S, Lunnon K, Murray CL, Woods JF, Deacon RM, et al. Systemic Inflammation Induces Acute Behavioral and Cognitive Changes and Accelerates Neurodegenerative Disease. Biol Psychiatry (2009) 65:304–12. doi: 10.1016/j.biopsych.2008.07.024
72. Field R, Campion S, Warren C, Murray C, Cunningham C. Systemic Challenge With the TLR3 Agonist Poly I:C Induces Amplified IFN Alpha/Beta and IL-1 Beta Responses in the Diseased Brain and Exacerbates Chronic Neurodegeneration. Brain Behav Immun (2010) 24:996–1007. doi: 10.1016/j.bbi.2010.04.004
73. Ishida N, Ishihara Y, Ishida K, Tada H, Funaki-Kato Y, Hagiwara M, et al. Periodontitis Induced by Bacterial Infection Exacerbates Features of Alzheimer’s Disease in Transgenic Mice. NPJ Aging Mech Dis (2017) 3:15. doi: 10.1038/s41514-017-0015-x
74. Ray S, Britschgi M, Herbert C, Takeda-Uchimura Y, Boxer A, Blennow K, et al. Classification and Prediction of Clinical Alzheimer’s Diagnosis Based on Plasma Signaling Proteins. Nat Med (2007) 13:1359–62. doi: 10.1038/nm1653
75. Chouhan JK, Fowler SB, Webster CI, Teeling JL. The ME7 Prion Model of Neurodegeneration as a Tool to Understand and Target Neuroinflammation in Alzheimer’s Disease. Drug Discov Today Dis Models (2017) 25-26:45–52. doi: 10.1016/j.ddmod.2018.10.004
76. Nie R, Wu Z, Ni J, Zeng F, Yu W, Zhang Y, et al. Porphyromonas Gingivalis Infection Induces Amyloid-Beta Accumulation in Monocytes/Macrophages. J Alzheimers Dis (2019) 72:479–94. doi: 10.3233/JAD-190298
77. Wang XL, Zeng J, Feng J, Tian YT, Liu YJ, Qiu M, et al. Helicobacter Pylori Filtrate Impairs Spatial Learning and Memory in Rats and Increases Beta-Amyloid by Enhancing Expression of Presenilin-2. Front Aging Neurosci (2014) 6:66. doi: 10.3389/fnagi.2014.00066
78. Wang XL, Zeng J, Yang Y, Xiong Y, Zhang ZH, Qiua M, et al. Helicobacter Pylori Filtrate Induces Alzheimer-Like Tau Hyperphosphorylation by Activating Glycogen Synthase Kinase-3 Beta. J Alzheimers Dis (2015) 43:153–65. doi: 10.3233/Jad-140198
79. Laur AM, Floch P, Chambonnier L, Benejat L, Korolik V, Giese A, et al. Regulatory T Cells May Participate in Helicobacter Pylori Persistence in Gastric MALT Lymphoma: Lessons From an Animal Model. Oncotarget (2016) 7:3384–92. doi: 10.18632/oncotarget.6492
80. Albaret G, Sifre E, Floch P, Laye S, Aubert A, Dubus P, et al. Alzheimer’s Disease and Helicobacter Pylori Infection: Inflammation From Stomach to Brain? J Alzheimers Dis (2020) 73:801–9. doi: 10.3233/Jad-190496
81. Roubaud Baudron C, Chambonnier L, Buissionnière A, Giese A, Macrez N, Cho Y, et al. An Eighteen-Month Helicobacter Infection Does Not Induce Amyloid Plaques or Neuroinflammation in Brains of Wild Type C57BL/6J Mice. J Alzheimers Dis (2015) 45:1045–50. doi: 10.3233/JAD-143129
82. Allison CC, Ferrero RL. Role of Virulence Factors and Host Cell Signaling in the Recognition of Helicobacter Pylori and the Generation of Immune Responses. Future Microbiol (2010) 5:1233–55. doi: 10.2217/fmb.10.84
83. Cairns DM, Rouleau N, Parker RN, Walsh KG, Gehrke L, Kaplan DL. A 3D Human Brain-Like Tissue Model of Herpes-Induced Alzheimer’s Disease. Sci Adv (2020) 6:eaay8828. doi: 10.1126/sciadv.aay8828
84. Ngo HM, Zhou Y, Lorenzi H, Wang K, Kim TK, Zhou Y, et al. Toxoplasma Modulates Signature Pathways of Human Epilepsy, Neurodegeneration & Cancer. Sci Rep (2017) 7:11496. doi: 10.1038/s41598-017-10675-6
85. Hammond CJ, Hallock LR, Howanski RJ, Appelt DM, Little CS, Balin BJ. Immunohistological Detection of Chlamydia Pneumoniae in the Alzheimer’s Disease Brain. BMC Neurosci (2010) 11:121. doi: 10.1186/1471-2202-11-121
86. Pisa D, Alonso R, Juarranz A, Rabano A, Carrasco L. Direct Visualization of Fungal Infection in Brains From Patients With Alzheimer’s Disease. J Alzheimers Dis (2015) 43:613–24. doi: 10.3233/Jad-141386
87. Civitelli L, Marcocci ME, Celestino I, Piacentini R, Garaci E, Grassi C, et al. Herpes Simplex Virus Type 1 Infection in Neurons Leads to Production and Nuclear Localization of APP Intracellular Domain (AICD): Implications for Alzheimer’s Disease Pathogenesis. J Neurovirol (2015) 21:480–90. doi: 10.1007/s13365-015-0344-0
88. Lovheima H, Olsson J, Weidung B, Johansson A, Erikssona S, Hallmans G, et al. Interaction Between Cytomegalovirus and Herpes Simplex Virus Type 1 Associated With the Risk of Alzheimer’s Disease Development. J Alzheimers Dis (2018) 61:939–45. doi: 10.3233/Jad-161305
89. Xia X, Wang Y, Zheng J. COVID-19 and Alzheimer’s Disease: How One Crisis Worsens the Other. Transl Neurodegener (2021) 10:15. doi: 10.1186/s40035-021-00237-2
90. Jiao L, Yang Y, Yu W, Zhao Y, Long H, Gao J, et al. The Olfactory Route Is a Potential Way for SARS-CoV-2 to Invade the Central Nervous System of Rhesus Monkeys. Signal Transduct Target Ther (2021) 6:169. doi: 10.1038/s41392-021-00591-7
91. Margraf A, Ludwig N, Zarbock A, Rossaint J. Systemic Inflammatory Response Syndrome After Surgery: Mechanisms and Protection. Anesth Analg (2020) 131:1693–707. doi: 10.1213/ANE.0000000000005175
92. Gasparotto J, Girardi CS, Somensi N, Ribeiro CT, Moreira JCF, Michels M, et al. Receptor for Advanced Glycation End Products Mediates Sepsis-Triggered Amyloid-Beta Accumulation, Tau Phosphorylation, and Cognitive Impairment. J Biol Chem (2018) 293:226–44. doi: 10.1074/jbc.M117.786756
93. Huang C, Irwin MG, Wong GTC, Chang RCC. Evidence of the Impact of Systemic Inflammation on Neuroinflammation From a Non-Bacterial Endotoxin Animal Model. J Neuroinflamm (2018) 15:147. doi: 10.1186/s12974-018-1163-z
94. Fidalgo AR, Cibelli M, White JPM, Nagy I, Maze M, Ma DQ. Systemic Inflammation Enhances Surgery-Induced Cognitive Dysfunction in Mice. Neurosci Lett (2011) 498:63–6. doi: 10.1016/j.neulet.2011.04.063
95. Li L, Cao D, Garber DW, Kim H, Fukuchi K. Association of Aortic Atherosclerosis With Cerebral Beta-Amyloidosis and Learning Deficits in a Mouse Model of Alzheimer’s Disease. Am J Pathol (2003) 163:2155–64. doi: 10.1016/s0002-9440(10)63572-9
96. Franciosi S, Gama Sosa MA, English DF, Oler E, Oung T, Janssen WG, et al. Novel Cerebrovascular Pathology in Mice Fed a High Cholesterol Diet. Mol Neurodegener (2009) 4:42. doi: 10.1186/1750-1326-4-42
97. Kimura N. Diabetes Mellitus Induces Alzheimer’s Disease Pathology: Histopathological Evidence From Animal Models. Int J Mol Sci (2016) 17:503. doi: 10.3390/ijms17040503
98. Tucsek Z, Toth P, Sosnowska D, Gautam T, Mitschelen M, Koller A, et al. Obesity in Aging Exacerbates Blood-Brain Barrier Disruption, Neuroinflammation, and Oxidative Stress in the Mouse Hippocampus: Effects on Expression of Genes Involved in Beta-Amyloid Generation and Alzheimer’s Disease. J Gerontol A Biol Sci Med Sci (2014) 69:1212–26. doi: 10.1093/gerona/glt177
99. Theriault P, ElAli A, Rivest S. High Fat Diet Exacerbates Alzheimer’s Disease-Related Pathology in APPswe/PS1 Mice. Oncotarget (2016) 7:67808–27. doi: 10.18632/oncotarget.12179
100. Dafsari FS, Jessen F. Depression-An Underrecognized Target for Prevention of Dementia in Alzheimer’s Disease. Transl Psychiatry (2020) 10:160. doi: 10.1038/s41398-020-0839-1
101. Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, et al. Systemic Inflammation and Disease Progression in Alzheimer Disease. Neurology (2009) 73:768–74. doi: 10.1212/WNL.0b013e3181b6bb95
102. Leung R, Proitsi P, Simmons A, Lunnon K, Guntert A, Kronenberg D, et al. Inflammatory Proteins in Plasma Are Associated With Severity of Alzheimer’s Disease. PloS One (2013) 8:e64971. doi: 10.1371/journal.pone.0064971
103. Motta M, Imbesi R, Di Rosa M, Stivala F, Malaguarnera L. Altered Plasma Cytokine Levels in Alzheimer’s Disease: Correlation With the Disease Progression. Immunol Lett (2007) 114:46–51. doi: 10.1016/j.imlet.2007.09.002
104. Clark IA, Atwood CS. Is TNF a Link Between Aging-Related Reproductive Endocrine Dyscrasia and Alzheimer’s Disease? J Alzheimers Dis (2011) 27:691–9. doi: 10.3233/Jad-2011-110887
105. Butchart J, Birch B, Bassily R, Wolfe L, Holmes C. Male Sex Hormones and Systemic Inflammation in Alzheimer Disease. Alzheimer Dis Assoc Disord (2013) 27:153–6. doi: 10.1097/WAD.0b013e318258cd63
106. Surendranathan A, Su L, Mak E, Passamonti L, Hong YT, Arnold R, et al. Early Microglial Activation and Peripheral Inflammation in Dementia With Lewy Bodies. Brain (2018) 141:3415–27. doi: 10.1093/brain/awy265
107. Okello A, Edison P, Archer HA, Turkheimer FE, Kennedy J, Bullock R, et al. Microglial Activation and Amyloid Deposition in Mild Cognitive Impairment A PET Study. Neurology (2009) 72:56–62. doi: 10.1212/01.wnl.0000338622.27876.0d
108. Savva GM, Wharton SB, Ince PG, Forster G, Matthews FE, Brayne C, et al. Age, Neuropathology, and Dementia. N Engl J Med (2009) 360:2302–9. doi: 10.1056/NEJMoa0806142
109. Edison P, Archer HA, Gerhard A, Hinz R, Pavese N, Turkheimer FE, et al. Microglia, Amyloid, and Cognition in Alzheimer’s Disease: An [11c](R)PK11195-PET and [11C]PIB-PET Study. Neurobiol Dis (2008) 32:412–9. doi: 10.1016/j.nbd.2008.08.001
110. Yokokura M, Mori N, Yagi S, Yoshikawa E, Kikuchi M, Yoshihara Y, et al. In Vivo Changes in Microglial Activation and Amyloid Deposits in Brain Regions With Hypometabolism in Alzheimer’s Disease. Eur J Nucl Med Mol Imaging (2011) 38:343–51. doi: 10.1007/s00259-010-1612-0
111. Kountouras J, Tsolaki M, Boziki M, Gavalas E, Zavos C, Stergiopoulos C, et al. Association Between Helicobacter Pylori Infection and Mild Cognitive Impairment. Eur J Neurol (2007) 14:976–82. doi: 10.1111/j.1468-1331.2007.01827.x
112. Kountouras J, Tsolaki M, Gavalas E, Boziki M, Zavos C, Karatzoglou P, et al. Relationship Between Helicobacter Pylori Infection and Alzheimer Disease. Neurology (2006) 66:938–40. doi: 10.1212/01.wnl.0000203644.68059.5f
113. Dioguardi M, Crincoli V, Laino L, Alovisi M, Sovereto D, Mastrangelo F, et al. The Role of Periodontitis and Periodontal Bacteria in the Onset and Progression of Alzheimer’s Disease: A Systematic Review. J Clin Med (2020) 9:495. doi: 10.3390/jcm9020495
114. Carter CJ, France J, Crean S, Singhrao SK. The Porphyromonas Gingivalis/Host Interactome Shows Enrichment in GWASdb Genes Related to Alzheimer’s Disease, Diabetes and Cardiovascular Diseases. Front Aging Neurosci (2017) 9:408. doi: 10.3389/fnagi.2017.00408
115. Kamer AR, Craig RG, Pirraglia E, Dasanayake AP, Norman RG, Boylan RJ, et al. TNF-Alpha and Antibodies to Periodontal Bacteria Discriminate Between Alzheimer’s Disease Patients and Normal Subjects. J Neuroimmunol (2009) 216:92–7. doi: 10.1016/j.jneuroim.2009.08.013
116. Farhad SZ, Amini S, Khalilian A, Barekatain M, Mafi M, Barekatain M, et al. The Effect of Chronic Periodontitis on Serum Levels of Tumor Necrosis Factor-Alpha in Alzheimer Disease. Dent Res J (Isfahan) (2014) 11:549–52.
117. Poole S, Singhrao SK, Kesavalu L, Curtis MA, Crean S. Determining the Presence of Periodontopathic Virulence Factors in Short-Term Postmortem Alzheimer’s Disease Brain Tissue. J Alzheimers Dis (2013) 36:665–77. doi: 10.3233/Jad-121918
118. Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas Gingivalis in Alzheimer’s Disease Brains: Evidence for Disease Causation and Treatment With Small-Molecule Inhibitors. Sci Adv (2019) 5:eaau3333. doi: 10.1126/sciadv.aau3333
119. Ide M, Harris M, Stevens A, Sussams R, Hopkins V, Culliford D, et al. Periodontitis and Cognitive Decline in Alzheimer’s Disease. PloS One (2016) 11:e0151081. doi: 10.1371/journal.pone.0151081
120. Montoya JAG, Barrios R, Sanchez-Lara I, Ramos P, Carnero C, Fornieles F, et al. Systemic Inflammatory Impact of Periodontitis on Cognitive Impairment. Gerodontology (2019) 37:11–8. doi: 10.1111/ger.12431
121. Wu B, Fillenbaum GG, Plassman BL, Guo L. Association Between Oral Health and Cognitive Status: A Systematic Review. J Am Geriatr Soc (2016) 64:739–51. doi: 10.1111/jgs.14036
122. Doulberis M, Kotronis G, Thomann R, Polyzos SA, Boziki M, Gialamprinou D, et al. Review: Impact of Helicobacter Pylori on Alzheimer’s Disease: What Do We Know So Far? Helicobacter (2018) 23:e12454. doi: 10.1111/hel.12454
123. Kountouras J, Boziki M, Gavalas E, Zavos C, Deretzi G, Grigoriadis N, et al. Increased Cerebrospinal Fluid Helicobacter Pylori Antibody in Alzheimer’s Disease. Int J Neurosci (2009) 119:765–77. doi: 10.1080/00207450902782083
124. Roubaud-Baudron C, Krolak-Salmon P, Quadrio I, Megraud F, Salles N. Impact of Chronic Helicobacter Pylori Infection on Alzheimer’s Disease: Preliminary Results. Neurobiol Aging (2012) 33:1009.e11–9. doi: 10.1016/j.neurobiolaging.2011.10.021
125. Kountouras J, Boziki M, Gavalas E, Zavos C, Deretzi G, Chatzigeorgiou S, et al. Five-Year Survival After Helicobacter Pylori Eradication in Alzheimer Disease Patients. Cognit Behav Neurol (2010) 23:199–204. doi: 10.1097/WNN.0b013e3181df3034
126. Kountouras J, Boziki M, Gavalas E, Zavos C, Grigoriadis N, Deretzi G, et al. Eradication of Helicobacter Pylori May Be Beneficial in the Management of Alzheimer’s Disease. J Neurol (2009) 256:758–67. doi: 10.1007/s00415-009-5011-z
127. Shiota S, Murakami K, Yoshiiwa A, Yamamoto K, Ohno S, Kuroda A, et al. The Relationship Between Helicobacter Pylori Infection and Alzheimer’s Disease in Japan. J Neurol (2011) 258:1460–3. doi: 10.1007/s00415-011-5957-5
128. Graham DY, Miftahussurur M. Helicobacter Pylori Urease for Diagnosis of Helicobacter Pylori Infection: A Mini Review. J Adv Res (2018) 13:51–7. doi: 10.1016/j.jare.2018.01.006
129. Wozniak MA, Mee AP, Itzhaki RF. Herpes Simplex Virus Type 1 DNA Is Located Within Alzheimer’s Disease Amyloid Plaques. J Pathol (2009) 217:131–8. doi: 10.1002/path.2449
130. Tsai MC, Cheng WL, Sheu JJ, Huang CC, Shia BC, Kao LT, et al. Increased Risk of Dementia Following Herpes Zoster Ophthalmicus. PloS One (2017) 12:e0188490. doi: 10.1371/journal.pone.0188490
131. Chen VC, Wu SI, Huang KY, Yang YH, Kuo TY, Liang HY, et al. Herpes Zoster and Dementia: A Nationwide Population-Based Cohort Study. J Clin Psychiatry (2018) 79:16m11312. doi: 10.4088/JCP.16m11312
132. Tzeng NS, Chung CH, Lin FH, Chiang CP, Yeh CB, Huang SY, et al. Anti-Herpetic Medications and Reduced Risk of Dementia in Patients With Herpes Simplex Virus Infections-A Nationwide, Population-Based Cohort Study in Taiwan. Neurotherapeutics (2018) 15:417–29. doi: 10.1007/s13311-018-0611-x
133. Kwok MK, Schooling CM. Herpes Simplex Virus and Alzheimer’s Disease: A Mendelian Randomization Study. Neurobiol Aging (2021) 99:101.e11–3. doi: 10.1016/j.neurobiolaging.2020.09.025
134. Warren-Gash C, Forbes HJ, Williamson E, Breuer J, Hayward AC, Mavrodaris A, et al. Human Herpesvirus Infections and Dementia or Mild Cognitive Impairment: A Systematic Review and Meta-Analysis. Sci Rep (2019) 9:4743. doi: 10.1038/s41598-019-41218-w
135. McArdle MA, Finucane OM, Connaughton RM, McMorrow AM, Roche HM. Mechanisms of Obesity-Induced Inflammation and Insulin Resistance: Insights Into the Emerging Role of Nutritional Strategies. Front Endocrinol (Lausanne) (2013) 4:52. doi: 10.3389/fendo.2013.00052
136. Spauwen PJ, Kohler S, Verhey FR, Stehouwer CD, van Boxtel MP. Effects of Type 2 Diabetes on 12-Year Cognitive Change: Results From the Maastricht Aging Study. Diabetes Care (2013) 36:1554–61. doi: 10.2337/dc12-0746
137. Ding J, Strachan MW, Reynolds RM, Frier BM, Deary IJ, Fowkes FG, et al. Diabetic Retinopathy and Cognitive Decline in Older People With Type 2 Diabetes: The Edinburgh Type 2 Diabetes Study. Diabetes (2010) 59:2883–9. doi: 10.2337/db10-0752
138. Hassing LB, Dahl AK, Pedersen NL, Johansson B. Overweight in Midlife Is Related to Lower Cognitive Function 30 Years Later: A Prospective Study With Longitudinal Assessments. Dement Geriatr Cognit Disord (2010) 29:543–52. doi: 10.1159/000314874
139. Qizilbash N, Gregson J, Johnson ME, Pearce N, Douglas I, Wing K, et al. BMI and Risk of Dementia in Two Million People Over Two Decades: A Retrospective Cohort Study. Lancet Diabetes Endocrinol (2015) 3:431–6. doi: 10.1016/S2213-8587(15)00033-9
140. Crane PK, Walker R, Hubbard RA, Li G, Nathan DM, Zheng H, et al. Glucose Levels and Risk of Dementia. N Engl J Med (2013) 369:540–8. doi: 10.1056/NEJMoa1215740
141. Stakos DA, Stamatelopoulos K, Bampatsias D, Sachse M, Zormpas E, Vlachogiannis NI, et al. The Alzheimer’s Disease Amyloid-Beta Hypothesis in Cardiovascular Aging and Disease: JACC Focus Seminar. J Am Coll Cardiol (2020) 75:952–67. doi: 10.1016/j.jacc.2019.12.033
142. Dionisio-Santos DA, Olschowka JA, O’Banion MK. Exploiting Microglial and Peripheral Immune Cell Crosstalk to Treat Alzheimer’s Disease. J Neuroinflamm (2019) 16:74. doi: 10.1186/s12974-019-1453-0
143. McManus RM, Higgins SC, Mills KHG, Lynch MA. Respiratory Infection Promotes T Cell Infiltration and Amyloid-Beta Deposition in APP/PS1 Mice. Neurobiol Aging (2014) 35:109–21. doi: 10.1016/j.neurobiolaging.2013.07.025
144. Solito E, Sastre M. Microglia Function in Alzheimer’s Disease. Front Pharmacol (2012) 3:14. doi: 10.3389/fphar.2012.00014
145. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol (2015) 14:388–405. doi: 10.1016/S1474-4422(15)70016-5
146. Schwabenland M, Bruck W, Priller J, Stadelmann C, Lassmann H, Prinz M. Analyzing Microglial Phenotypes Across Neuropathologies: A Practical Guide. Acta Neuropathol (2021) 142:923–36. doi: 10.1007/s00401-021-02370-8
147. Heppner FL, Ransohoff RM, Becher B. Immune Attack: The Role of Inflammation in Alzheimer Disease. Nat Rev Neurosci (2015) 16:358–72. doi: 10.1038/nrn3880
148. Monje ML, Toda H, Palmer TD. Inflammatory Blockade Restores Adult Hippocampal Neurogenesis. Science (2003) 302:1760–5. doi: 10.1126/science.1088417
149. Siskova Z, Tremblay ME. Microglia and Synapse: Interactions in Health and Neurodegeneration. Neural Plast (2013) 2013:425845. doi: 10.1155/2013/425845
150. Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, Lafaille JJ, et al. Microglia Promote Learning-Dependent Synapse Formation Through Brain-Derived Neurotrophic Factor. Cell (2013) 155:1596–609. doi: 10.1016/j.cell.2013.11.030
151. Brown GC, Vilalta A. How Microglia Kill Neurons. Brain Res (2015) 1628:288–97. doi: 10.1016/j.brainres.2015.08.031
152. Dean JM, Wang X, Kaindl AM, Gressens P, Fleiss B, Hagberg H, et al. Microglial MyD88 Signaling Regulates Acute Neuronal Toxicity of LPS-Stimulated Microglia In Vitro. Brain Behav Immun (2010) 24:776–83. doi: 10.1016/j.bbi.2009.10.018
153. Xing B, Bachstetter AD, Van Eldik LJ. Microglial P38 Alpha MAPK Is Critical for LPS-Induced Neuron Degeneration, Through a Mechanism Involving TNF Alpha. Mol Neurodegener (2011) 6:84. doi: 10.1186/1750-1326-6-84
154. Bachstetter AD, Xing B, de Almeida L, Dimayuga ER, Watterson DM, Van Eldik LJ. Microglial P38 Alpha MAPK Is a Key Regulator of Proinflammatory Cytokine Up-Regulation Induced by Toll-Like Receptor (TLR) Ligands or Beta-Amyloid (A Beta). J Neuroinflamm (2011) 8:79. doi: 10.1186/1742-2094-8-79
155. Nimmervoll B, White R, Yang JW, An SM, Henn C, Sun JJ, et al. LPS-Induced Microglial Secretion of TNF Alpha Increases Activity-Dependent Neuronal Apoptosis in the Neonatal Cerebral Cortex. Cereb Cortex (2013) 23:1742–55. doi: 10.1093/cercor/bhs156
156. Harland M, Torres S, Liu J, Wang X. Neuronal Mitochondria Modulation of LPS-Induced Neuroinflammation. J Neurosci (2020) 40:1756–65. doi: 10.1523/JNEUROSCI.2324-19.2020
157. Heneka MT, McManus RM, Latz E. Inflammasome Signalling in Brain Function and Neurodegenerative Disease. Nat Rev Neurosci (2018) 19:610–21. doi: 10.1038/s41583-018-0055-7
158. Luciunaite A, McManus RM, Jankunec M, Racz I, Dansokho C, Dalgediene I, et al. Soluble Abeta Oligomers and Protofibrils Induce NLRP3 Inflammasome Activation in Microglia. J Neurochem (2020) 155:650–61. doi: 10.1111/jnc.14945
159. Youm YH, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A, et al. Canonical Nlrp3 Inflammasome Links Systemic Low-Grade Inflammation to Functional Decline in Aging. Cell Metab (2013) 18:519–32. doi: 10.1016/j.cmet.2013.09.010
160. Yun HM, Kim JA, Hwang CJ, Jin P, Baek MK, Lee JM, et al. Neuroinflammatory and Amyloidogenic Activities of IL-32beta in Alzheimer’s Disease. Mol Neurobiol (2015) 52:341–52. doi: 10.1007/s12035-014-8860-0
161. Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, et al. Clearance Systems in the Brain-Implications for Alzheimer Disease. Nat Rev Neurol (2015) 11:457–70. doi: 10.1038/nrneurol.2015.119
162. Hickman SE, Allison EK, El Khoury J. Microglial Dysfunction and Defective Beta-Amyloid Clearance Pathways in Aging Alzheimer’s Disease Mice. J Neurosci (2008) 28:8354–60. doi: 10.1523/JNEUROSCI.0616-08.2008
163. Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, et al. Transgenic Mice Expressing Human Tumour Necrosis Factor: A Predictive Genetic Model of Arthritis. EMBO J (1991) 10:4025–31. doi: 10.1002/j.1460-2075.1991.tb04978.x
164. Paouri E, Tzara O, Kartalou GI, Zenelak S, Georgopoulos S. Peripheral Tumor Necrosis Factor-Alpha (TNF-Alpha) Modulates Amyloid Pathology by Regulating Blood-Derived Immune Cells and Glial Response in the Brain of AD/TNF Transgenic Mice. J Neurosci (2017) 37:5155–71. doi: 10.1523/Jneurosci.2484-16.2017
165. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 Is Activated in Alzheimer’s Disease and Contributes to Pathology in APP/PS1 Mice. Nature (2013) 493:674–8. doi: 10.1038/nature11729
166. Compan V, Martin-Sanchez F, Baroja-Mazo A, Lopez-Castejon G, Gomez AI, Verkhratsky A, et al. Apoptosis-Associated Speck-Like Protein Containing a CARD Forms Specks But Does Not Activate Caspase-1 in the Absence of NLRP3 During Macrophage Swelling. J Immunol (2015) 194:1261–73. doi: 10.4049/jimmunol.1301676
167. Friker LL, Scheiblich H, Hochheiser IV, Brinkschulte R, Riedel D, Latz E, et al. Beta-Amyloid Clustering Around ASC Fibrils Boosts Its Toxicity in Microglia. Cell Rep (2020) 30:3743–54.e6. doi: 10.1016/j.celrep.2020.02.025
168. Erickson MA, Hartvigson PE, Morofuji Y, Owen JB, Butterfield DA, Banks WA. Lipopolysaccharide Impairs Amyloid Beta Efflux From Brain: Altered Vascular Sequestration, Cerebrospinal Fluid Reabsorption, Peripheral Clearance and Transporter Function at the Blood-Brain Barrier. J Neuroinflamm (2012) 9:150. doi: 10.1186/1742-2094-9-150
169. Erickson MA, Hansen K, Banks WA. Inflammation-Induced Dysfunction of the Low-Density Lipoprotein Receptor-Related Protein-1 at the Blood-Brain Barrier: Protection by the Antioxidant N-Acetylcysteine. Brain Behav Immun (2012) 26:1085–94. doi: 10.1016/j.bbi.2012.07.003
170. Jaeger LB, Dohgu S, Sultana R, Lynch JL, Owen JB, Erickson MA, et al. Lipopolysaccharide Alters the Blood-Brain Barrier Transport of Amyloid Beta Protein: A Mechanism for Inflammation in the Progression of Alzheimer’s Disease. Brain Behav Immun (2009) 23:507–17. doi: 10.1016/j.bbi.2009.01.017
171. Gonzalez-Marrero I, Gimenez-Llort L, Johanson CE, Carmona-Calero EM, Castaneyra-Ruiz L, Brito-Armas JM, et al. Choroid Plexus Dysfunction Impairs Beta-Amyloid Clearance in a Triple Transgenic Mouse Model of Alzheimer’s Disease. Front Cell Neurosci (2015) 9:17. doi: 10.3389/fncel.2015.00017
172. Redzic Z. Molecular Biology of the Blood-Brain and the Blood-Cerebrospinal Fluid Barriers: Similarities and Differences. Fluids Barriers CNS (2011) 8:3. doi: 10.1186/2045-8118-8-3
173. Van Dyken P, Lacoste B. Impact of Metabolic Syndrome on Neuroinflammation and the Blood-Brain Barrier. Front Neurosci (2018) 12:930. doi: 10.3389/fnins.2018.00930
174. Liddelow SA. Development of the Choroid Plexus and Blood-CSF Barrier. Front Neurosci (2015) 9:32. doi: 10.3389/fnins.2015.00032
175. Liu Y, Zhang S, Li X, Liu E, Wang X, Zhou Q, et al. Peripheral Inflammation Promotes Brain Tau Transmission via Disrupting Blood-Brain Barrier. Biosci Rep (2020) 40:BSR20193629. doi: 10.1042/BSR20193629
176. Qin LH, Huang W, Mo XA, Chen YL, Wu XH. LPS Induces Occludin Dysregulation in Cerebral Microvascular Endothelial Cells via MAPK Signaling and Augmenting MMP-2 Levels. Oxid Med Cell Longev (2015) 2015:120641. doi: 10.1155/2015/120641
177. Vandenbroucke RE, Dejonckheere E, Van Lint P, Demeestere D, Van Wonterghem E, Vanlaere I, et al. Matrix Metalloprotease 8-Dependent Extracellular Matrix Cleavage at the Blood-CSF Barrier Contributes to Lethality During Systemic Inflammatory Diseases. J Neurosci (2012) 32:9805–16. doi: 10.1523/JNEUROSCI.0967-12.2012
178. Yu HY, Cai YB, Lu Z. Activation of AMPK Improves Lipopolysaccharide-Induced Dysfunction of the Blood-Brain Barrier in Mice. Brain Inj (2015) 29:777–84. doi: 10.3109/02699052.2015.1004746
179. Doll DN, Hu H, Sun JH, Lewis SE, Simpkins JW, Ren XF. Mitochondrial Crisis in Cerebrovascular Endothelial Cells Opens the Blood-Brain Barrier. Stroke (2015) 46:1681–9. doi: 10.1161/Strokeaha.115.009099
180. Ma FF, Zhang XJ, Yin KJ. MicroRNAs in Central Nervous System Diseases: A Prospective Role in Regulating Blood-Brain Barrier Integrity. Exp Neurol (2020) 323:113094. doi: 10.1016/j.expneurol.2019.113094
181. Lopez-Ramirez MA, Wu D, Pryce G, Simpson JE, Reijerkerk A, King-Robson J, et al. MicroRNA-155 Negatively Affects Blood-Brain Barrier Function During Neuroinflammation. FASEB J (2014) 28:2551–65. doi: 10.1096/fj.13-248880
182. Abbott NJ. Astrocyte-Endothelial Interactions and Blood-Brain Barrier Permeability. J Anat (2002) 200:629–38. doi: 10.1046/j.1469-7580.2002.00064.x
183. Cardoso FL, Herz J, Fernandes A, Rocha J, Sepodes B, Brito MA, et al. Systemic Inflammation in Early Neonatal Mice Induces Transient and Lasting Neurodegenerative Effects. J Neuroinflamm (2015) 12:82. doi: 10.1186/s12974-015-0299-3
184. Biesmans S, Meert TF, Bouwknecht JA, Acton PD, Davoodi N, De Haes P, et al. Systemic Immune Activation Leads to Neuroinflammation and Sickness Behavior in Mice. Mediators Inflamm (2013) 2013:271359. doi: 10.1155/2013/271359
185. Fan L, Wang TL, Chang LR, Song YZ, Wu Y, Ma DQ. Systemic Inflammation Induces a Profound Long Term Brain Cell Injury in Rats. Acta Neurobiol Exp (Wars) (2014) 74:298–306.
186. Haruwaka K, Ikegami A, Tachibana Y, Ohno N, Konishi H, Hashimoto A, et al. Dual Microglia Effects on Blood Brain Barrier Permeability Induced by Systemic Inflammation. Nat Commun (2019) 10:5816. doi: 10.1038/s41467-019-13812-z
187. Carvalho-Tavares J, Hickey MJ, Hutchison J, Michaud J, Sutcliffe IT, Kubes P. A Role for Platelets and Endothelial Selectins in Tumor Necrosis Factor-Alpha-Induced Leukocyte Recruitment in the Brain Microvasculature. Circ Res (2000) 87:1141–8. doi: 10.1161/01.Res.87.12.1141
188. Chui R, Dorovini-Zis K. Regulation of CCL2 and CCL3 Expression in Human Brain Endothelial Cells by Cytokines and Lipopolysaccharide. J Neuroinflamm (2010) 7:1. doi: 10.1186/1742-2094-7-1
189. Agrawal S, Anderson P, Durbeej M, van Rooijen N, Ivars F, Opdenakker G, et al. Dystroglycan Is Selectively Cleaved at the Parenchymal Basement Membrane at Sites of Leukocyte Extravasation in Experimental Autoimmune Encephalomyelitis. J Exp Med (2006) 203:1007–19. doi: 10.1084/jem.20051342
190. Pieper C, Pieloch P, Galla HJ. Pericytes Support Neutrophil Transmigration via Interleukin-8 Across a Porcine Co-Culture Model of the Blood-Brain Barrier. Brain Res (2013) 1524:1–11. doi: 10.1016/j.brainres.2013.05.047
191. Hartz AMS, Bauer B, Fricker G, Miller DS. Rapid Modulation of P-Glycoprotein-Mediated Transport at the Blood-Brain Barrier by Tumor Necrosis Factor-Alpha and Lipopolysaccharide. Mol Pharmacol (2006) 69:462–70. doi: 10.1124/mol.105.017954
192. Osburg B, Peiser C, Domling D, Schomburg L, Ko YT, Voigt K, et al. Effect of Endotoxin on Expression of TNF Receptors and Transport of TNF-Alpha at the Blood-Brain Barrier of the Rat. Am J Physiol Endocrinol Metab (2002) 283:E899–908. doi: 10.1152/ajpendo.00436.2001
193. Dohgu S, Fleegal-DeMotta MA, Banks WA. Lipopolysaccharide-Enhanced Transcellular Transport of HIV-1 Across the Blood-Brain Barrier Is Mediated by Luminal Microvessel IL-6 and GM-CSF. J Neuroinflamm (2011) 8:167. doi: 10.1186/1742-2094-8-167
194. Brkic M, Balusu S, Van Wonterghem E, Gorle N, Benilova I, Kremer A, et al. Amyloid Beta Oligomers Disrupt Blood-CSF Barrier Integrity by Activating Matrix Metalloproteinases. J Neurosci (2015) 35:12766–78. doi: 10.1523/JNEUROSCI.0006-15.2015
195. Vandendriessche C, Balusu S, Van Cauwenberghe C, Brkic M, Pauwels M, Plehiers N, et al. Importance of Extracellular Vesicle Secretion at the Blood-Cerebrospinal Fluid Interface in the Pathogenesis of Alzheimer’s Disease. Acta Neuropathol Commun (2021) 9:143. doi: 10.1186/s40478-021-01245-z
196. Steeland S, Gorle N, Vandendriessche C, Balusu S, Brkic M, Van Cauwenberghe C, et al. Counteracting the Effects of TNF Receptor-1 Has Therapeutic Potential in Alzheimer’s Disease. EMBO Mol Med (2018) 10:e8300. doi: 10.15252/emmm.201708300
197. Abdel-Haq R, Schlachetzki JCM, Glass CK, Mazmanian SK. Microbiome-Microglia Connections via the Gut-Brain Axis. J Exp Med (2019) 216:41–59. doi: 10.1084/jem.20180794
198. Goehler LE, Gaykema RP, Hansen MK, Anderson K, Maier SF, Watkins LR. Vagal Immune-to-Brain Communication: A Visceral Chemosensory Pathway. Auton Neurosci (2000) 85:49–59. doi: 10.1016/S1566-0702(00)00219-8
199. Fernandez R, Nardocci G, Navarro C, Reyes EP, Acuna-Castillo C, Cortes PP. Neural Reflex Regulation of Systemic Inflammation: Potential New Targets for Sepsis Therapy. Front Physiol (2014) 5:489. doi: 10.3389/fphys.2014.00489
200. Eyo UB, Wu LJ. Bidirectional Microglia-Neuron Communication in the Healthy Brain. Neural Plast (2013) 2013:456857. doi: 10.1155/2013/456857
201. Kaczmarczyk R, Tejera D, Simon BJ, Heneka MT. Microglia Modulation Through External Vagus Nerve Stimulation in a Murine Model of Alzheimer’s Disease. J Neurochem (2018) 146:76–85. doi: 10.1111/jnc.14284
202. Breit S, Kupferberg A, Rogler G, Hasler G. Vagus Nerve as Modulator of the Brain-Gut Axis in Psychiatric and Inflammatory Disorders. Front Psychiatry (2018) 9:44. doi: 10.3389/fpsyt.2018.00044
203. Meneses G, Bautista M, Florentino A, Diaz G, Acero G, Besedovsky H, et al. Electric Stimulation of the Vagus Nerve Reduced Mouse Neuroinflammation Induced by Lipopolysaccharide. J Inflamm Lond (2016) 13:33. doi: 10.1186/s12950-016-0140-5
204. Riazi K, Galic MA, Kuzmiski JB, Ho W, Sharkey KA, Pittman QJ. Microglial Activation and TNFalpha Production Mediate Altered CNS Excitability Following Peripheral Inflammation. Proc Natl Acad Sci USA (2008) 105:17151–6. doi: 10.1073/pnas.0806682105
205. Qin L, He J, Hanes RN, Pluzarev O, Hong JS, Crews FT. Increased Systemic and Brain Cytokine Production and Neuroinflammation by Endotoxin Following Ethanol Treatment. J Neuroinflamm (2008) 5:10. doi: 10.1186/1742-2094-5-10
206. Kowalski K, Mulak A. Brain-Gut-Microbiota Axis in Alzheimer’s Disease. J Neurogastroenterol Motil (2019) 25:48–60. doi: 10.5056/jnm18087
207. Wang C, Klechikov AG, Gharibyan AL, Warmlander SKTS, Jarvet J, Zhao LN, et al. The Role of Pro-Inflammatory S100A9 in Alzheimer’s Disease Amyloid-Neuroinflammatory Cascade. Acta Neuropathol (2014) 127:507–22. doi: 10.1007/s00401-013-1208-4
208. Mogensen TH. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin Microbiol Rev (2009) 22:240–73. doi: 10.1128/CMR.00046-08
209. Erny D, de Angelis ALH, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host Microbiota Constantly Control Maturation and Function of Microglia in the CNS. Nat Neurosci (2015) 18:965–77. doi: 10.1038/nn.4030
210. Hoogland ICM, Westhoff D, Engelen-Lee JY, Melief J, Seron MV, Houben-Weerts JHMP, et al. Microglial Activation After Systemic Stimulation With Lipopolysaccharide and Escherichia Coil. Front Cell Neurosci (2018) 12:110. doi: 10.3389/fncel.2018.00110
211. Colombo AV, Sadler RK, Llovera G, Singh V, Roth S, Heindl S, et al. Microbiota-Derived Short Chain Fatty Acids Modulate Microglia and Promote Abeta Plaque Deposition. Elife (2021) 10:e59826. doi: 10.7554/eLife.59826
212. Glebov K, Lochner M, Jabs R, Lau T, Merkel O, Schloss P, et al. Serotonin Stimulates Secretion of Exosomes From Microglia Cells. Glia (2015) 63:626–34. doi: 10.1002/glia.22772
213. Chen PS, Wang CC, Bortner CD, Peng GS, Wu X, Pang H, et al. Valproic Acid and Other Histone Deacetylase Inhibitors Induce Microglial Apoptosis and Attenuate Lipopolysaccharide-Induced Dopaminergic Neurotoxicity. Neuroscience (2007) 149:203–12. doi: 10.1016/j.neuroscience.2007.06.053
214. Kaparakis-Liaskos M, Ferrero RL. Immune Modulation by Bacterial Outer Membrane Vesicles. Nat Rev Immunol (2015) 15:375–87. doi: 10.1038/nri3837
215. Aguayo S, Schuh CMAP, Vicente B, Aguayo LG. Association Between Alzheimer’s Disease and Oral and Gut Microbiota: Are Pore Forming Proteins the Missing Link? J Alzheimers Dis (2018) 65:29–46. doi: 10.3233/Jad-180319
216. Ismail S, Hampton MB, Keenan JI. Helicobacter Pylori Outer Membrane Vesicles Modulate Proliferation and Interleukin-8 Production by Gastric Epithelial Cells. Infect Immun (2003) 71:5670–5. doi: 10.1128/iai.71.10.5670-5675.2003
217. Han EC, Choi SY, Lee Y, Park JW, Hong SH, Lee HJ. Extracellular RNAs in Periodontopathogenic Outer Membrane Vesicles Promote TNF-Alpha Production in Human Macrophages and Cross the Blood-Brain Barrier in Mice. FASEB J (2019) 33:13412–22. doi: 10.1096/fj.201901575R
Keywords: Alzheimer’s disease, neuroinflammation, peripheral inflammation, systemic inflammation, gut-brain axis
Citation: Xie J, Van Hoecke L and Vandenbroucke RE (2022) The Impact of Systemic Inflammation on Alzheimer’s Disease Pathology. Front. Immunol. 12:796867. doi: 10.3389/fimmu.2021.796867
Received: 17 October 2021; Accepted: 15 December 2021;
Published: 06 January 2022.
Edited by:
Liwei Xie, Guangdong Academy of Science, ChinaReviewed by:
Marcella Reale, University of Studies G. d’Annunzio Chieti and Pescara, ItalyMarta Olah, Columbia University Irving Medical Center, United States
Copyright © 2022 Xie, Van Hoecke and Vandenbroucke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roosmarijn E. Vandenbroucke, Um9vc21hcmlqbi5WYW5kZW5icm91Y2tlQGlyYy5WSUItVUdlbnQuYmU=
†These authors have contributed equally to this work and share senior authorship