 Nina Worel1†
Nina Worel1† Katharina Grabmeier-Pfistershammer2†
Katharina Grabmeier-Pfistershammer2† Bernhard Kratzer2†
Bernhard Kratzer2† Martina Schlager3
Martina Schlager3 Andreas Tanzmann1Arno Rottal2Ulrike Körmöczi2Edit Porpaczy3Philipp B. Staber3Cathrin Skrabs3
Andreas Tanzmann1Arno Rottal2Ulrike Körmöczi2Edit Porpaczy3Philipp B. Staber3Cathrin Skrabs3 Harald Herkner4
Harald Herkner4 Venugopal Gudipati5
Venugopal Gudipati5 Johannes B. Huppa5Benjamin Salzer6
Johannes B. Huppa5Benjamin Salzer6 Manfred Lehner6
Manfred Lehner6 Nora Saxenhuber3Eleonora Friedberg3Philipp Wohlfarth7Georg Hopfinger7Werner Rabitsch7Ingrid Simonitsch-Klupp8
Nora Saxenhuber3Eleonora Friedberg3Philipp Wohlfarth7Georg Hopfinger7Werner Rabitsch7Ingrid Simonitsch-Klupp8 Ulrich Jäger3
Ulrich Jäger3 Winfried F. Pickl2*
Winfried F. Pickl2*- 1Department of Blood Group Serology and Transfusion Medicine, Medical University of Vienna, Vienna, Austria
- 2Institute of Immunology, Center for Pathophysiology, Infectiology and Immunology, Medical University of Vienna, Vienna, Austria
- 3Department of Medicine I, Division of Hematology and Hemostaseology, Medical University of Vienna, Vienna, Austria
- 4Department of Emergency Medicine, Medical University of Vienna, Vienna, Austria
- 5Institute for Hygiene and Applied Immunology, Center for Pathophysiology, Infectiology and Immunology, Medical University of Vienna, Vienna, Austria
- 6Christian Doppler Laboratory for Next Generation CAR T Cells, St. Anna Children´s Cancer Research Institute, Vienna, Austria
- 7Department of Medicine I, Division of Blood and Bone Marrow Transplantation, Medical University of Vienna, Vienna, Austria
- 8Department of Pathology, Medical University of Vienna, Vienna, Austria
Background: Chimeric antigen receptor T (CART) cell therapy targeting the B cell specific differentiation antigen CD19 has shown clinical efficacy in a subset of relapsed/refractory (r/r) diffuse large B cell lymphoma (DLBCL) patients. Despite this heterogeneous response, blood pre-infusion biomarkers predicting responsiveness to CART cell therapy are currently understudied.
Methods: Blood cell and serum markers, along with clinical data of DLBCL patients who were scheduled for CART cell therapy were evaluated to search for biomarkers predicting CART cell responsiveness.
Findings: Compared to healthy controls (n=24), DLBCL patients (n=33) showed significant lymphopenia, due to low CD3+CD4+ T helper and CD3-CD56+ NK cell counts, while cytotoxic CD3+CD8+ T cell counts were similar. Although lymphopenic, DLBCL patients had significantly more activated HLA-DR+ (P=0.005) blood T cells and a higher frequency of differentiated CD3+CD27-CD28- (28.7 ± 19.0% versus 6.6 ± 5.8%; P<0.001) T cells. Twenty-six patients were infused with CART cells (median 81 days after leukapheresis) and were analyzed for the overall response (OR) 3 months later. Univariate and multivariate regression analyses showed that low levels of differentiated CD3+CD27-CD28- T cells (23.3 ± 19.3% versus 35.1 ± 18.0%) were independently associated with OR. This association was even more pronounced when patients were stratified for complete remission (CR versus non-CR: 13.7 ± 11.7% versus 37.7 ± 17.4%, P=0.001). A cut-off value of ≤ 18% of CD3+CD27-CD28- T cells predicted CR at 12 months with high accuracy (P<0.001). In vitro, CD3+CD8+CD27-CD28- compared to CD3+CD8+CD27+CD28+ CART cells displayed similar CD19+ target cell-specific cytotoxicity, but were hypoproliferative and produced less cytotoxic cytokines (IFN-γ and TNF-α). CD3+CD8+ T cells outperformed CD3+CD4+ T cells 3- to 6-fold in terms of their ability to kill CD19+ target cells.
Interpretation: Low frequency of differentiated CD3+CD27-CD28- T cells at leukapheresis represents a novel pre-infusion blood biomarker predicting a favorable response to CART cell treatment in r/r DLBCL patients.
Introduction
Diffuse large B cell lymphoma (DLBCL) represents the most frequent form of non-Hodgkin’s lymphoma (NHL). Five-year survival rates range from 55% to 64% (1, 2); however, patients who experience early relapse, or who are refractory to initial immunochemotherapy have a poor prognosis (3). In fact, salvage therapy for patients with refractory NHL has been associated with frequent therapy failures (>70%) and poor long-term outcome with an overall survival of only 6 months (3). Even consolidation therapy with subsequent autologous stem cell transplantation leads to only 50% long-term survival (4, 5).
Chimeric antigen receptor T (CART) cells represent a novel treatment option for patients with refractory/relapsing (r/r) DLBCL (6, 7). CART cell therapy takes advantage of autologous peripheral blood (PB) T cells, which are genetically modified ex vivo to express a chimeric antigen receptor (CAR) designed to target the CD19 antigen on the surface of the malignant B cell clone (8). Despite initial promising results with tisagenlecleucel (formerly CTL019) (7, 9), axicabtagene ciloleucel (formerly KTE-C19) (10) and lisocabtagene maraleucel (formerly JCAR017) (11), leading to overall response (OR) and complete remission (CR) rates of 83% to 52% and 58% to 40% (7, 11, 12), respectively, clearly not all patients benefit from CART cell therapy in the long-term (7). In fact, response rates decline to approximately 32% after one year (7). However, the identification of patients most likely to benefit from CART cell therapy is difficult to achieve by solely using clinical and basic laboratory criteria. Therefore, a reliable predictor of response to CART cell therapy at the time of enrollment, e.g., by a simple blood test, is an unmet need for optimal patient selection (13).
Currently, the best predictors of responsiveness to CART cell therapy are low lactate dehydrogenase (LDH) levels after lymphodepletion before CART cell infusion, a low tumor volume and a low Eastern Cooperative Oncology Group (ECOG) performance status (7, 14–16). Owing to the mode of action of CART cells, the immune system most likely plays a major role in its effectiveness. However, all three markers (LDH, tumor volume, ECOG) are not directly related to the immune system, and thus can, at best, represent surrogate markers for future tumor-immune surveillance by the gene modified autologous CART cells. More recently, other factors strongly linked to the immune system and possibly impacting on the response to CART cell therapy have been suggested. These factors include, but are not restricted to: i) defective T cell function (poor initial “pre-CAR” T cell quality or decreasing “post-CAR” T cell function) (17); ii) microenvironmental suppression (check point inhibition and suppressive cytokines) (18); and iii) antigen escape (target antigen modulation (19) or myeloid lineage switch) (20). In addition, increased frequencies of CD27+CD45RO-CD8+ T cells at the time of leukapheresis have been implicated to correlate with sustained remission in patients with chronic lymphocytic leukemia treated with CD19 CART cells (21), in multiple myeloma patients treated with B cell maturation antigen (BCMA)-specific CART cells (22), and recently in patients with DLBCL (23). It has been suggested that CD27+CD45RO-CD8+ T cells belong to the group of antigen-experienced CD3+CD8+ T lymphocytes that have long-lasting memory capabilities and improved ability to expand in vitro and in vivo (21, 22, 24). While of interest, the respective marker combination does not define a single cellular phenotype since CD45RO negativity may identify both naïve CD8+ T cells as well as antigen-experienced “stem cell memory” cells (23). Moreover, focusing the analyses exclusively on CD8+ T cells has the problem of potentially underestimating the cytotoxic potency of CD4+ T cells turned into CART cells during the manufacturing process. However, it has been clearly shown in adoptive T cell transfer studies in preclinical melanoma models that more differentiated CD8+ effector T cells are less effective for in vivo tumor treatment and that the renewal capacity of CD8+ T cells as determined by their telomer length plays an important role in that respect (25). In line with these studies, adoptive T cell transfer studies with autologous CD8+CD27+ T cells led to durable responses in heavily pretreated patients with metastatic melanoma (26). Apart from phenotypic data, a recent study suggested that germline mutations in UNC13D and compound heterozygous forms of CXCR1 may represent additional resistance factors to CART therapy (17). Whether and how they correlate with the cell surface phenotype of CD3+ T cells remains to be shown in the future. Furthermore, it should also be noted that for many patients, it is not possible to generate a suitable CART cell product due to prolonged lymphopenia and the associated inability to isolate a sufficient number of functional T cells (27).
Lymphopenia, as well as poor T cell quality and function, may reflect the intensity of previous immuno-chemotherapies, but may also result from hyperactivation of T cells, a process well-known to lead to their subsequent hypoproliferation and reduced life expectancy. Hyperactivated HLA-DR+ T cells have been shown to down-modulate cell surface expression of the co-stimulatory molecules CD27 and CD28 (28–31), which are otherwise decisively involved in the regulation of T cell activation (32, 33), the formation and maintenance of antigen-experienced T cells (34) and tumor immune surveillance (35). However, increased frequencies of HLA-DR+ T cells may also be the result of homeostatic proliferation (36). The expression levels of CD27 and CD28 as well as those of the high molecular weight form of CD45, i.e., CD45RA, and the chemokine receptor CCR7 allow, in principle, the determination of the position of a given T cell within the linear T cell differentiation model proposed by Romero et al. (37). In that model, CD3+CD27-CD28- T cells are mainly composed of T effector memory cells re-expressing CD45RA (TEMRA) cells and to a lower degree also contain effector memory type 3 (EM3) cells. While CCR7 is a robust marker for distinguishing between central and effector memory T cells, CD45RA is somewhat problematic because it is expressed on both naïve and terminally differentiated TEMRA cells and is overexpressed in 1 of 20 Caucasian individuals due to the C77G mutation (38), making it much more difficult to distinguish between bona fide CD45RA+ and CD45RA- cell subsets. Therefore, we here analyzed leukocyte subset distribution, T cell activation, and focused on CD27 and CD28 expression of bulk CD3+ T cells in the blood and corresponding leukapheresis products of adult r/r DLBCL patients and correlated the results with 3 months OR to CART cell therapy.
Patients and methods
Patients and clinical trial conduct
Between January 2016 and January 2022, 33 patients diagnosed with r/r DLBCL and scheduled for treatment with CART cells at our institution were enrolled into this study to investigate the composition of leukocyte subpopulations, their activation and differentiation status, together with serum markers in peripheral blood (PB) and leukapheresis samples. Patients gave their written informed consent in accordance with the Declaration of Helsinki. Patients received CART cells in clinical trials with tisagenlecleucel (n=15; Ethics Committee (EC) No.: 1422/2015, 1607/2018), YTB323 [n=2; EC No.: 2055/2019 (39)], or in routine applications of tisagenlecleucel (n=6) or axicabtagene ciloleucel (n=3). Analysis of data was approved by the EC of the Medical University of Vienna (EC No.: 1290/2020). Patient characteristics are presented in Table 1 and S1. Of the 33 enrolled patients, 26 already received CART cells, more than 3 months previously, at the time of data cut-off of this study. Seven patients were excluded from the study because they died before CART cell infusion (n=5), or received another treatment (n=2). The patients included into this study were heavily pretreated, showing failure to respond to two or more treatment lines, thus representing the subpopulation of patients with relapsed DLBCL eligible for CART therapy. The healthy control subjects (n=24) were age- (median 60 years; range 33-77 years) and sex- (10 women; 41.7%) matched and similar to the patients of Caucasian ethnicity.
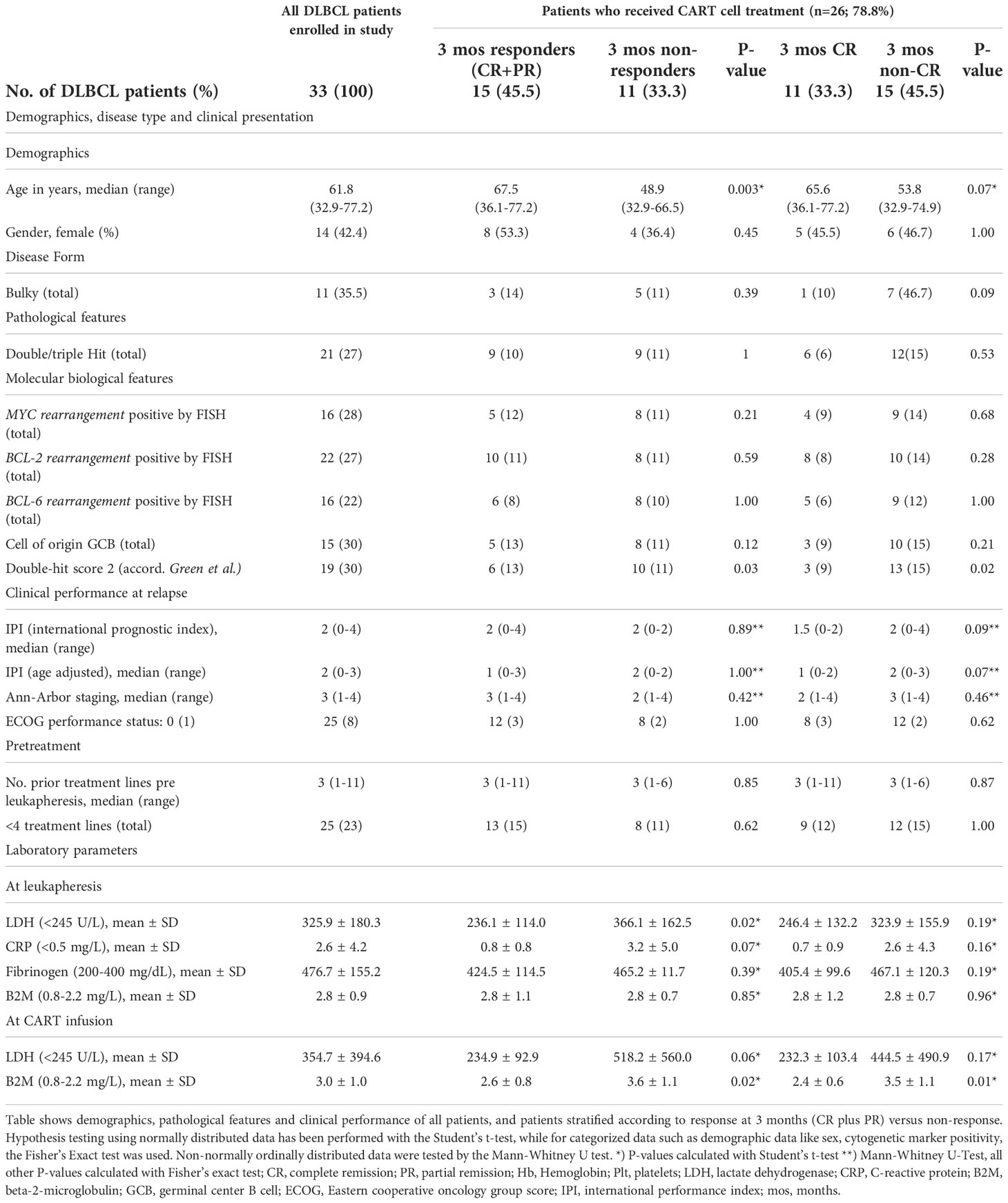
Table 1 Demographics, pathological features and clinical performance of patients.
Flow cytometric analyzes
Immunophenotyping of PB and the leukapheresis products was performed with fresh samples according to standard procedures (40) using directly conjugated monoclonal antibodies (Supplemental Table S2). To keep numbers of flow cytometric parameters low, the CD27 and CD28 expression status was analyzed on bulk CD45+CD3+ T cells. Acquisition was performed on flow cytometers (FACS Calibur and LSR Fortessa, Becton Dickinson, San Jose, CA; Navios or Cytoflex, Beckman Coulter, Krefeld, Germany) supported by the Cellquest, Diva and Kaluza software, respectively. Acquired data were analyzed with Flow Jo software (BD).
Generation of CART cells for in vitro studies
Buffy coats from anonymous healthy donor’s blood were purchased from the Austrian Red Cross, Vienna. CD3+ primary human T cells were isolated using the RosetteSep Human T cell Enrichment Cocktail (STEMCELL Technologies, Vancouver Canada) and immediately cryopreserved in RPMI-1640 GlutaMAX medium (Thermo Fisher Scientific, Waltham, MA) supplemented with 20% FCS and 10% DMSO (both from Merck, Darmstadt, Germany). Primary human T cells were thawed in RPMI-1640 GlutaMAX medium, supplemented with 10% FCS, 1% penicillin-streptomycin (Thermo Fisher Scientific) and 200 IU mL-1 recombinant human IL-2 (Peprotech, Waltham, MA) and activated with Dynabeads Human T-Activator CD3/CD28 beads (Thermo Fisher Scientific) at a 1:1 ratio according to the manufacturer’s instructions. Twenty-four hours after stimulation, T cells were transduced in cell culture plates, which were coated with RetroNectin (Takara, Shiga, Japan), according to the manufacturer’s instructions. Thawed lentiviral supernatant was added to the T cells at a final dilution of 1:2, yielding a cell concentration of 0.5 x 106 cells mL-1. Forty-eight hours after transduction, selection of CART cells was initiated by treatment with 1 µg mL-1 puromycin (Merck, Germany) for two days. Transduced T cells were cultivated in AIM V medium (Thermo Fisher Scientific) supplemented with 2% Octaplas (Octapharma, Vienna, Austria), 1% L-glutamine, 2.5% HEPES (both from Thermo Fisher Scientific) and 200 IU mL-1 recombinant human IL-2 for 14 days and then frozen in liquid nitrogen in IMDM medium containing 20% FB and 10% DMSO until further use.
Construction of lentiviral vector
VSV-G pseudotyped lentivirus was generated by co-transfection of Lenti-X 293T cells (Takara) with a puromycin-selectable pCDH expression vector (System Biosciences, USA) encoding the second-generation anti-CD19-CAR (FMC63.4-1BB.ζ) and viral packaging plasmids pMD2.G and psPAX2 (Addgene plasmids #12259 and #12260, respectively; kind gifts from Didier Trono) using the PureFection Transfection Reagent (System Biosciences, Palo Alto, CA) according to the manufacturer’s instructions. Viral supernatants were collected on day 2 and 3 after transfection and were concentrated 100-fold using the Lenti-X Concentrator (Takara) according to the manufacturer’s instructions. Viral suspensions were frozen at -80°C until further use.
Functional in vitro assays with CART cells
For in vitro experiments, CART cells were gently thawed and cultured in AIMV medium (Thermo Fisher Scientific, USA) supplemented with 2% Octaplas (Octapharma), 1% L-glutamine, 2.5% HEPES (both from Thermo Fisher Scientific, USA) and 50 IU mL-1 recombinant human IL-2 (Peprotech). One day after thawing, CART cells were expanded by adding five times the number of irradiated (120 Gray) TM-LCL cells, a human B lymphocyte cell line immortalized by Epstein-Barr virus infection (41), which have been optimized as feeder cells for CD19 CART cell expansion (42). Expansion of CD19 CART cells after removal of CD3CD28-beads with CD19+LCL cells has been used in the past and represents an accepted procedure for CART cell expansion and propagation (43). After three days, cells were further expanded every two to three days by adding fresh medium in a 1:2 ratio. Ten days after expansion, CART cells were FACS sorted with the antibodies listed in Table S2 to obtain CD3+CD8+CD27+CD28+, CD3+CD8+CD27-CD28- T, CD3+CD4+CD27+CD28+ and CD3+CD4+CD27-CD28- cell populations on a Sony SH800 Sorter (Sony Biotechnology, San Jose, CA) and cultured in the presence of IL-2 in medium as described above. Five to seven days later, cells were used for in vitro assays. For proliferation assays, 1 x 105 CART cells were incubated with the indicated amounts of irradiated (120 Gray) CD19+ TM-LCL cells (ranging from 2 x 105 to 1 x 104 cells) in triplicates in 96-well round-bottom tissue culture plates (Sarstedt, Nümbrecht, Germany) in a total volume of 200 µl for 48 h. Cells were pulsed with [methyl-3H]-thymidine (1 µCi per well) for 18 hours and thymidine up-take was analyzed as previously described (44). For analysis of T cell activation and cytokine production, 1 x 105 CART cells were incubated with the indicated amounts of CD19+ TM-LCL cells (ranging from 2 x 105 to 1 x 104 cells) in triplicates in 96-well round-bottom plates in a total volume of 200 µl for 72 hours. Subsequently, cell suspensions were transferred to 1.5 ml microcentrifuge tubes, centrifuged at 600 g for 5 minutes, supernatants were collected and subjected to cytokine analyses with a cytometric bead array (Luminex, Austin, TX) as described previously (45). Cells were stained as described (44), acquired on a Cytoflex flow cytometer (Beckmann Coulter) and data analyzed with the Flow Jo software package (Becton Dickinson).
For cytotoxicity assays, 1 x 106 CD19+ TM-LCL or CD19- K562 cells were resuspended in 50 µl of culture medium each and labelled with 50 µl of Na51CrO4 (Perkin Elmer, Boston, MA) at 37°C for 1 hour. After four subsequent washes, 5 x 103 TM-LCL and K562 cells were seeded into individual wells of 96-well round-bottom tissue-culture plates and incubated with the indicated amounts of sorted CART cells in duplicates/triplicates. Medium or 2% triton-X100 was added to target cells to determine spontaneous and maximum release, respectively. Subsequently, plates were centrifuged at 100 g for 5 minutes and incubated at 37°C for 5 hours. Supernatants were then collected with the Skatron system (Molecular Devices, Biberach an der Riss, Germany) and radioactivity was determined on a Cobra II gamma-counter (Packard, Meriden, CT). The percentage of specific release was determined as follows [CART cell induced release (cpm) – spontaneous release (cpm)]/[maximum release (cpm) - spontaneous release (cpm)]x100.
Statistics
The study was designed as a cohort study. Response to CART cell treatment was defined as complete response (CR) or partial remission (PR) at three months after CART cell infusion. No response was defined as stable disease (SD) or progressive disease (PD) after receiving CART cells. We present categorized data as absolute counts and relative frequencies, continuous data as mean and standard deviation, or median and range. Where applicable, we log-transformed variables to yield approximate normal distributions. To test the H0 of no association of T cell subsets with the outcome to CART cell therapy, the Fisher’s exact test and the independent sample t-test was used. To quantify the association between the outcome overall response at 3 months and the percentage of CD3+CD27-CD28- T cells, we used exact logistic regression, owing to the limited sample size. We also assessed other predefined variables and added these variables as co-variables into the main model separately. Generally, a two-sided P-value <0.05 was considered statistically significant.
Data Sharing
Please contact Dr. Nina Worel for sharing of data at bmluYS53b3JlbEBtZWR1bml3aWVuLmFjLmF0.
Results
Enrollment and clinical characterization of r/r DLBCL patients
Our study aimed to identify robust pre-infusion biomarkers in the blood and leukapheresis samples of r/r DLBCL patients, as possible predictors to the subsequent response to CART cell therapy. Accordingly, between January 2016 and January 2022, 33 patients with r/r DLBCL were enrolled into this cohort study (Figure S1). Patients consisted of 19 males and 14 females, with a median age of 61.8 years (range, 32.9 to 77.2 years, Table 1) and a median disease duration at leukapheresis of 18.0 months (range, 3.7-266.4 months) (Table S1). Median time from PB assessment at the time of leukapheresis to CART cell infusion was 3.3 months (range, 1.2 to 14.1 months). These patients had a median follow-up time of 15.5 months (range, 6.1 to 57.1 months). OR at 3 months was observed in 15 patients (57.7%), with 11 patients achieving a CR (42.3%).
Cellular parameters of r/r DLBCL patients at leukapheresis
First, we assessed PB leukocyte subpopulations at the time of leukapheresis (Table 2 and Figure 1). Remarkably, r/r DLBCL patients had significant lymphopenia compared to healthy controls (HC) (1009 ± 927 x106/L versus 1785 ± 478 x106/L; P<0.001), due to reduced CD3+CD4+ T helper (297 ± 236 x106/L versus 735 ± 229 x106/L; P<0.001) and CD3-CD56+ NK cell numbers (164 ± 218 x106/L versus 313 ± 176 x106/L; P=0.009). CD3+CD8+ T cell, NKT cell, neutrophil and overall leukocyte numbers were similar to HC (Table 2 and Figure S2). The CD3+CD4+ lymphopenia led to a significantly lower CD4/CD8-ratio (0.9 ± 0.6 versus 2.1 ± 1.1, P<0.001) in DLBCL patients. Moreover, patients’ T cells had clear signs of activation, as determined by HLA-DR co-expression (315 ± 322 x106/L versus 113 ± 116 x106/L; P=0.005). Notably, chronic activation of T cells may lead to cell differentiation and replicative senescence, which is frequently accompanied by downregulation of the co-stimulatory molecules CD27 and CD28 (30, 31), the acquisition of memory (CD45RO/RA) and the loss of lymphnode homing (CCR7) markers (37). Indeed, when we examined the overall study population of r/r DLBCL patients in that regard, we found significantly higher percentages of differentiated CD3+CD27-CD28- (28.7 ± 19.0% versus 6.6 ± 5.8%; P<0.001), CD3+CD27- (38.6 ± 19.2% versus 19.6 ± 11.9%; P<0.001) and CD3+CD28- (41.7 ± 19.6% versus 15.5 ± 8.5%; P<0.001) PB T cells when compared to age-matched HC (Table 3; Figures 2A and S3). CD3+CD27-CD28- consisted exclusively of highly differentiated CCR7- CD45RA-/+ T effector memory (EM)/T effector memory RA cells (TEMRA) (Figure S4). Not unexpectedly, almost complete B cell aplasia was seen in most DLBCL patients (P<0.001).
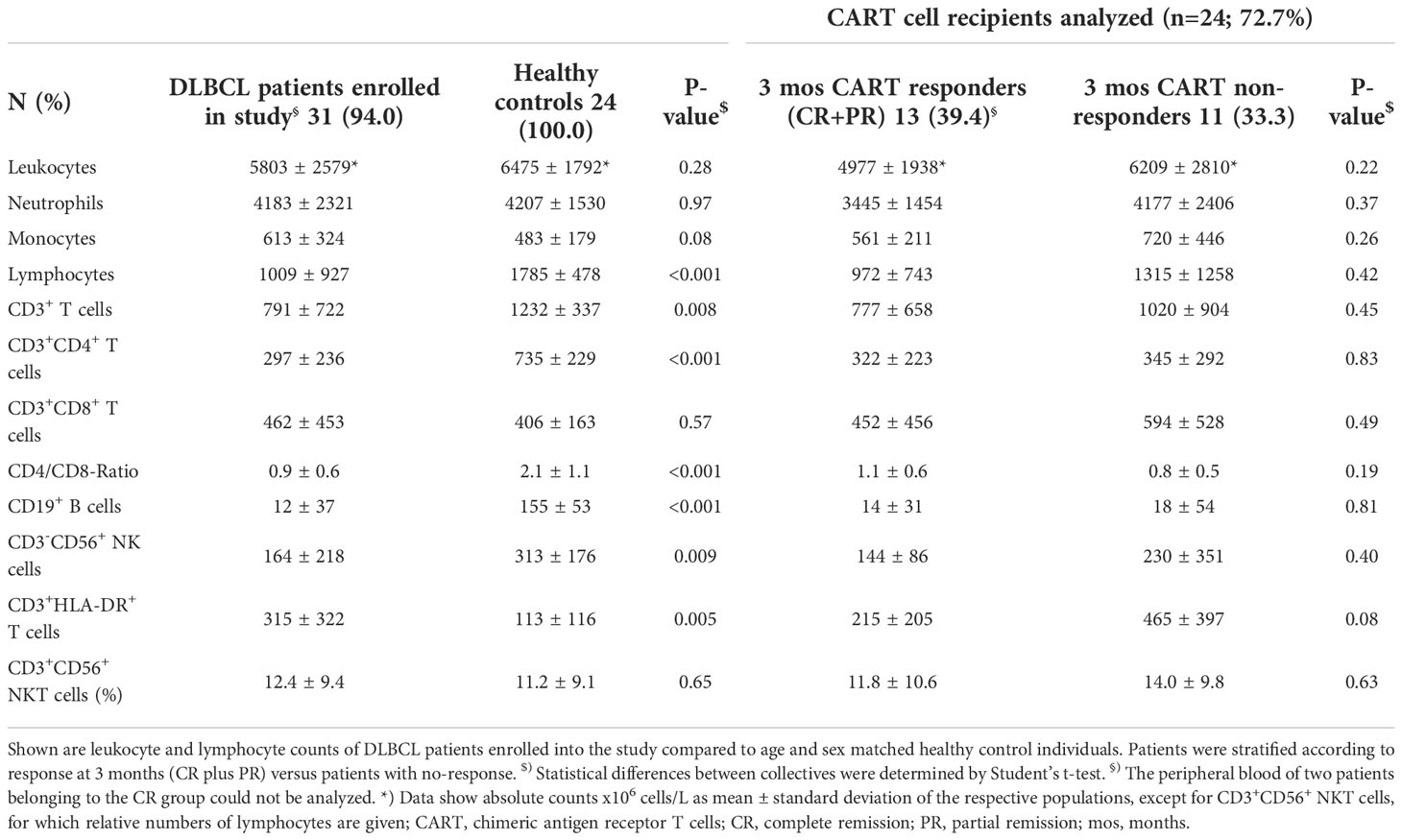
Table 2 Differences in lymphocyte populations between r/r DLBCL patients and healthy controls.
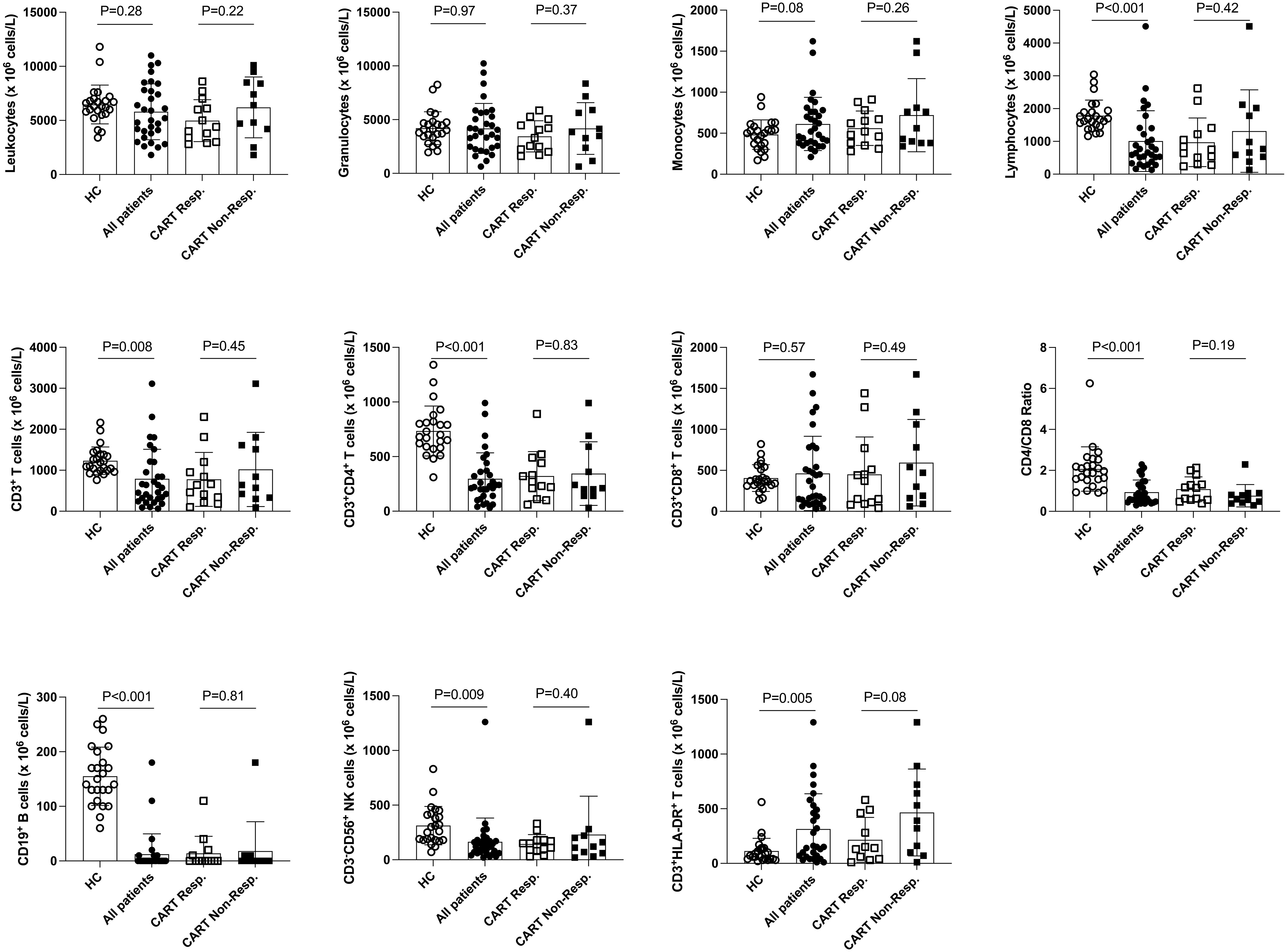
Figure 1 Distribution of leukocyte populations in the PB of healthy control subjects, total r/r DLBCL patients and CART cell recipients. Here, the distribution of PB cell populations of 24 healthy control subjects (HC) and 31 of 33* r/r DLBCL patients is shown. Data of 13 of 15* CART cell responders (except 11 for CD3+HLA-DR+ T cells) and 11 CART non-responders 3 months after CART therapy are shown separately. P-values (unpaired t-test) are indicated. *) PB of two patients belonging to the CART responders was not available for analyses at this stage.
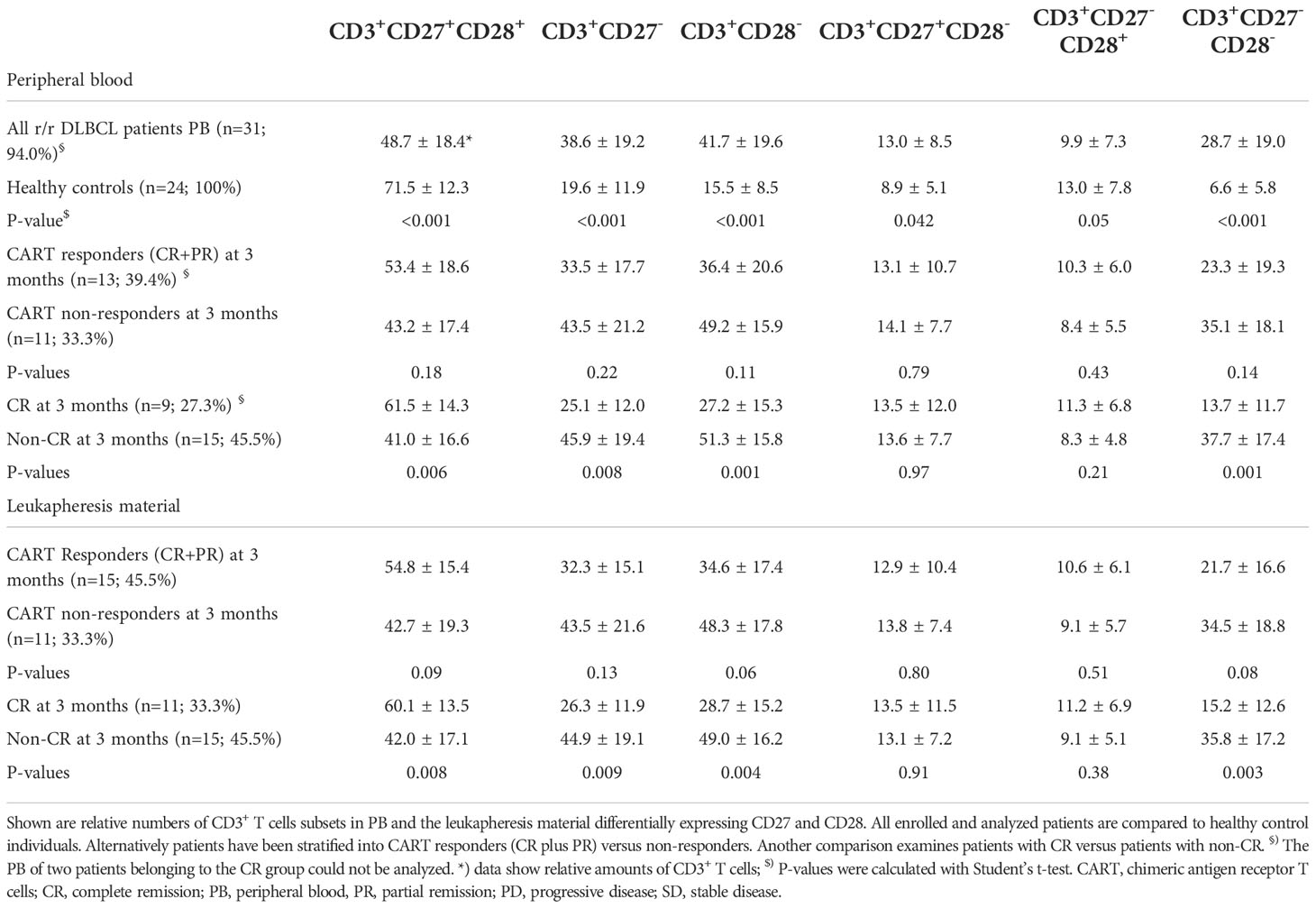
Table 3 PB and leukapheresis material of r/r DLBCL patients scheduled for CART cell therapy contain significantly more CD3+ T cells with a differentiated CD27-CD28- phenotype when compared to healthy control individuals.
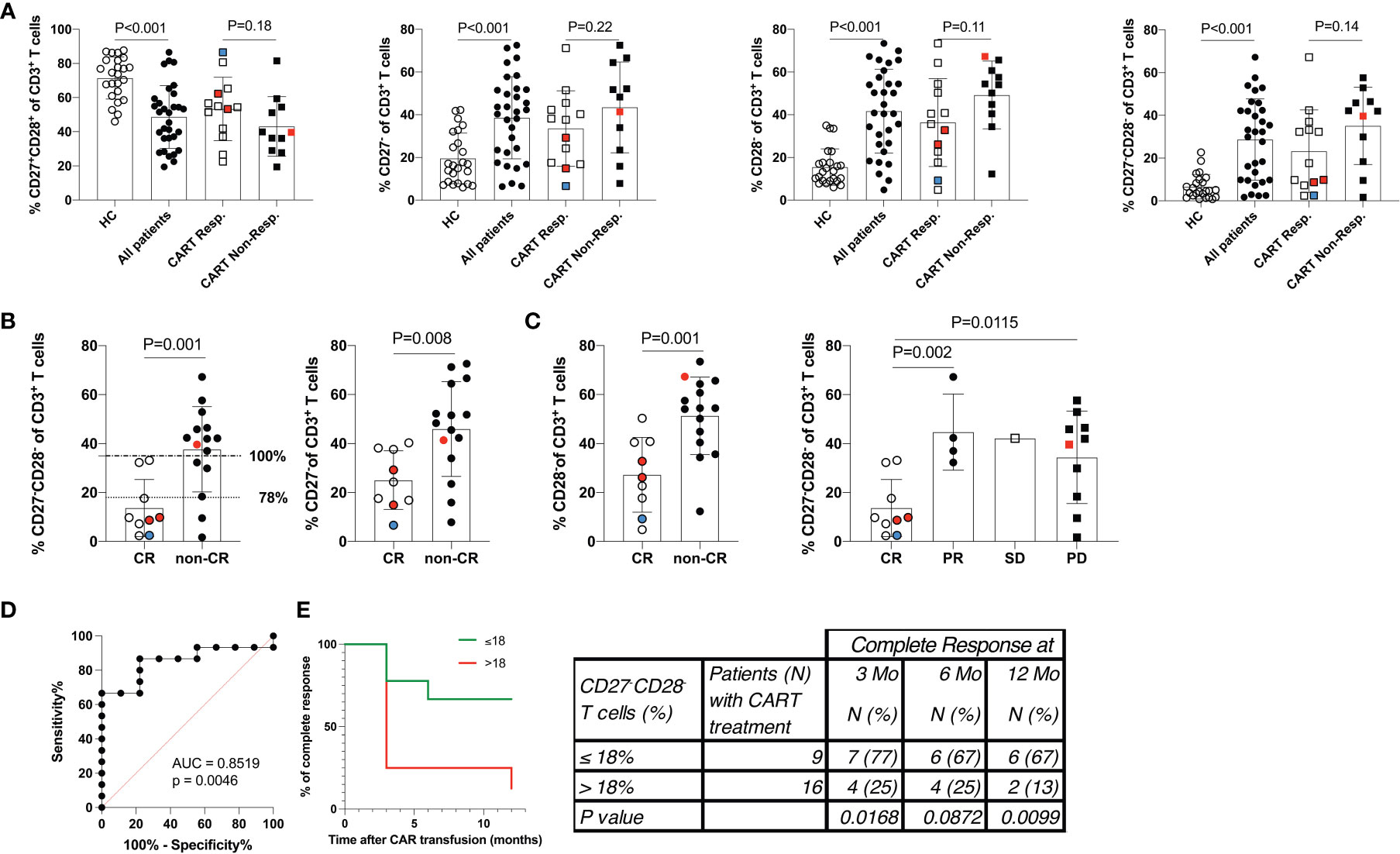
Figure 2 Low frequency of differentiated CD3+CD27-CD28- T cells predicts a favorable response to CART cell therapy. (A) The distribution of PB CD3+ T cell populations stratified by the CD27 and CD28 expression status is given. Data show 24 healthy control subjects (HC), 31 of 33* r/r DLBCL patients scheduled for CART cell therapy, and more detailed data for 13 of 15* CART cell responders and all non-responders (n=11). (B) Shows the distribution of PB CD3+ T cell populations stratified by the CD27 and CD28 expression status in patients who were further separated into 9 of 11* complete responders and compared to 15 non-complete responders. Horizontal lines at 18% of CD3+CD27-CD28- T cells indicate the 78% (dotted line) and at 35% of CD3+CD27-CD28- T cells indicate 100% (dashed-and-dotted line) specificity levels (at sensitivity levels of 87% and 67%, respectively) of numbers of CD3+CD27-CD28- T cell numbers to predict CR (C) Shown are the numbers of CD3+CD27-CD28- T cells of patients who achieved complete remission (CR), partial remission (PR), stable disease (SD) or progressive disease (PD). P-values (unpaired Student’s t-test) are indicated. Cell frequencies were determined in 31 of 33* patients. Patients were treated with tisagenlecleucel (white/black symbols), axicabtagene ciloleucel (red symbols) or YTB323 (blue symbols). *) PB of two patients belonging to the CART responders was not available for analyses at this stage. (D) ROC (receiver operator characteristics) curve indicating the performance of numbers CD3+CD27-CD28- T cells for classifying CR. (E) Duration of complete remission (CR) after CART cell therapy. Shown are the percent of patients presenting with CR over the observational period of 12 months (Mo) with staging at 0, 3, 6 and 12 months. Patients were stratified according to those with >18% or ≤18% of CD3+CD27-CD28- T cells at the time of leukapheresis. Table shows the number (N) of patients within each group at each time point (percent of group in parenthesis). P values were calculated with Fisher’s exact test.
Low frequency of differentiated CD3+CD27-CD28- PB T cells in r/r DLBCL patients at leukapheresis correlates with OR
Stratification of patients into CART cell responders at 3 months after CART infusion (CR and PR) versus non-responders (SD and PD) revealed that the T cells of the latter group were in particular more activated, as indicated by HLA-DR co-expression (215 ± 205 x106/L versus 465 ± 397 x106/L; P<0.08) (Table 2). Accordingly, a higher frequency of differentiated CD3+CD27-CD28- PB T cells was also associated with non-responsiveness, while a lower frequency of differentiated CD3+CD27-CD28- PB T cells was a salient feature of patients with OR (35.1 ± 18.1% versus 23.3 ± 19.3%; P=0.14) (Table 3 and Figure 2). This was due to a trend towards lower frequencies of CD3+CD27- PB T cells (33.5 ± 17.7% versus 43.5 ± 21.2%; P=0.22) and CD3+CD28- PB T cells (36.4 ± 20.6 versus 49.2 ± 15.9; P=0.11) (Table 3 and Figure 2A). We found a tendency of low numbers of CD3+CD27-CD28- T cells being associated with month 3 OR (odds-ratio 0.97; 95% confidence interval 0.92-1.01; P=0.14; Figure S5A). This association remained virtually unchanged after pairwise adjustment for clinical (international prognostic index, double/triple hit mutation, cell of origin, gender, age at leukapheresis, NOS mutations) and PB parameters (LDH levels at CART cell infusion, frequency of CD3+CD27-CD28- T cells).
Low frequency of differentiated CD3+CD27-CD28- PB T cells at leukapheresis identifies patients with a high likelihood for CR
Next, we compared the CD27 and CD28 expression status on PB T cells of 9 of 11 CR patients to 15 patients presenting with non-CR (PR, SD and PD). From two CR patients no PB was available. Notably, a low frequency of CD3+CD27-CD28- T cells at the time of leukapheresis (13.7 ± 11.7% versus 37.7 ± 17.4%) was significantly associated with CR at month 3 (P=0.001) (Figure 2B and Table 3). Inclusion of CD3+CD27-CD28- values of the two patients with missing PB data but available values of the leukapheresis products (i.e., 16.5% and 37.4% of CD3+CD27-CD28-, respectively) changed the strength of the statistical comparison between CR and non-CR only very slightly (p-values 0.002 versus 0.001, respectively). For ease of comparison, the type of CAR used is given in Figure 2. Patients with low or high numbers of CD3+CD27-CD28- T cells were equally distributed in the subgroups treated with different CAR products suggesting that the type of CAR used did not appear to affect CR rates.
Similar to the above analyses obtained with CART cell responders versus non-responders, pairwise adjustment for clinical and PB parameters did not significantly change this association (Figure S5B). Both CD3+CD27- (25.1 ± 12.0% versus 45.9 ± 19.4%; P=0.008) and CD3+CD28- T cells (27.2 ± 15.3 versus 51.3 ± 15.8; P=0.001) contributed to this association (Table 3; Figures 2C and S3). Of note, the residual CD27 expression on the CD27+ T cells within the CD3+CD28- subset was found to be reduced compared to the one within the CD3+CD27+CD28+ subset. This indicated that the CD3+CD28- subgroup had already begun to downregulate also CD27 expression (data not shown). Therefore, determining the double-negative CD27-CD28- status of CD3+ T cells appeared to be the most robust strategy for enumerating differentiated T cells and also resulted in a moderately better statistical discrimination between CR and non-CR groups (P=0.001 versus P=0.006) when compared to the CD3+CD27+CD28+ subset. To exclude a sampling bias due to the lack of PB samples from the two CR patients, in addition we compared the leukapheresis products of the CR patients with those of the non-CR patients, for whom the full dataset of 11 and 15 patients was available, in terms of their CD3+CD27-CD28- T cell counts (Table 3). Very similar to PB, we found that low frequencies of CD3+CD27-CD28- T cells (15.2 ± 12.6% versus 35.8 ± 17.2%) were significantly associated with CR at month 3 (P=0.003) also in the leukapheresis product. Receiver operator characteristics (ROC) curve was used to determine the cut-off above which non-CR could be expected. Numbers of CD3+CD27-CD28- T cells greater 18% or 35% predicted non-CR with 78% or 100% specificity, (Figure 2D). Moreover, the cut-off value of ≤ 18% CD3+CD27-CD28- T cells predicted the duration of response over the subsequent 12-month follow-up period with high accuracy (p<0.001) (Figure 2E).
CD3+CD8+CD27-CD28- are inferior to CD3+CD8+CD27+CD28+ CART cells in terms of proliferation and cytotoxic cytokine production, but not regarding target-cell cytotoxicity
CD19 CART cells kill malignant and normal CD19+ B cells without MHC restriction. CD3+CD8+ CD19 CART cells have been reported to be able to perform serial killings with higher efficiency and speed than CD3+CD4+ CD19 CART cells (46). Our above finding that patients with lower numbers of CD3+CD27-CD28- T cells at leukapheresis have a much better chance of achieving CR, when undergoing CD19-directed CART cell therapy, prompted us to test whether CD3+CD8+CD27+CD28+ are, in fact, functionally superior to CD3+CD8+CD27-CD28- CD19 CART cells. Accordingly, we analyzed their cytotoxic, proliferative and cytokine-producing capabilities. Remarkably, CD3+CD8+CD27+CD28+ and CD3+CD8+CD27-CD28- CD19 CART cells (expressing the CD19-specific CART cell receptor on 91.6 ± 0.1% % and 91.4 ± 0.1% of CD8+ T cells, respectively, Figure S7) killed CD19+ B cells (TM-LCL) with nearly identical efficacies over the entire range of effector to target (E:T) ratios tested, while no such killing of CD19- K562 cells was observed with either of the two CD3+CD8+ CD19 CART cell subsets (Figure 3A). Notably, also CD3+CD4+ CART cells (expressing the CD19-specific CART cell receptor on 95.3 ± 0.8% of CD27-CD28- and 94.9 ± 2.2% of CD27+CD28+ CD4+ T cells, respectively, Figure S8) killed the CD19+ B cells (TM-LCL), however, with at least 3- to 6-fold lower efficacy when compared to their CD3+CD8+ counterparts (Figure 3A). Notably, CD4+CD27-CD28- outperformed CD4+CD27+CD28+ T cells in the killing of CD19+ target cells by a factor of 2. However, both CD3+CD8+CD27+CD28+ and CD3+CD4+CD27+CD28+ CART cells proliferated significantly more efficiently than CD3+CD8+CD27-CD28- and CD3+CD4+CD27-CD28- CART cells, respectively, when co-incubated with CD19+ TM-LCL cells at all E:T-ratios tested, with differences ranging between 1.5 ± 0.4 and 2.7 ± 1.7-fold for CD3+CD8+ and 1.0 ± 0.1 and 1.9 ± 1.1-fold for CD3+CD4+ T cells (Figure 3B). Moreover, CD3+CD8+CD27+CD28+ CART cells secreted higher levels of the Th1 cytokines IL-2, IFN-γ and TNF-α, while CD3+CD8+CD27-CD28- CART cells seemed to overproduce the Th2 cytokine IL-13 (Figure 3C). The situation was similar for CD3+CD4+ CART cells, with the sole exception that CD3+CD4+CD27-CD28- as compared to CD3+CD4+CD27+CD28+ CART cells produced higher levels of IFN-γ. Notably, the elevated IL-2 secretion levels of CD3+CD8+CD27+CD28+ CART cells were paralleled by their increased high-affinity IL-2R (CD25) expression when compared to CD3+CD8+CD27-CD28- CART cells (Figure 3D). The limited functional capabilities (i.e., proliferation, IL-2 and TNF-α production both subsets; IFN-γ production for CD8+ T cells) of CD27-CD28- T cells can be explained by their belonging to the TEMRA and EM3 subsets of memory cells (CCR7-CD45RA+/-), which are known to have limited renewal capacity (Figure S4) (37).
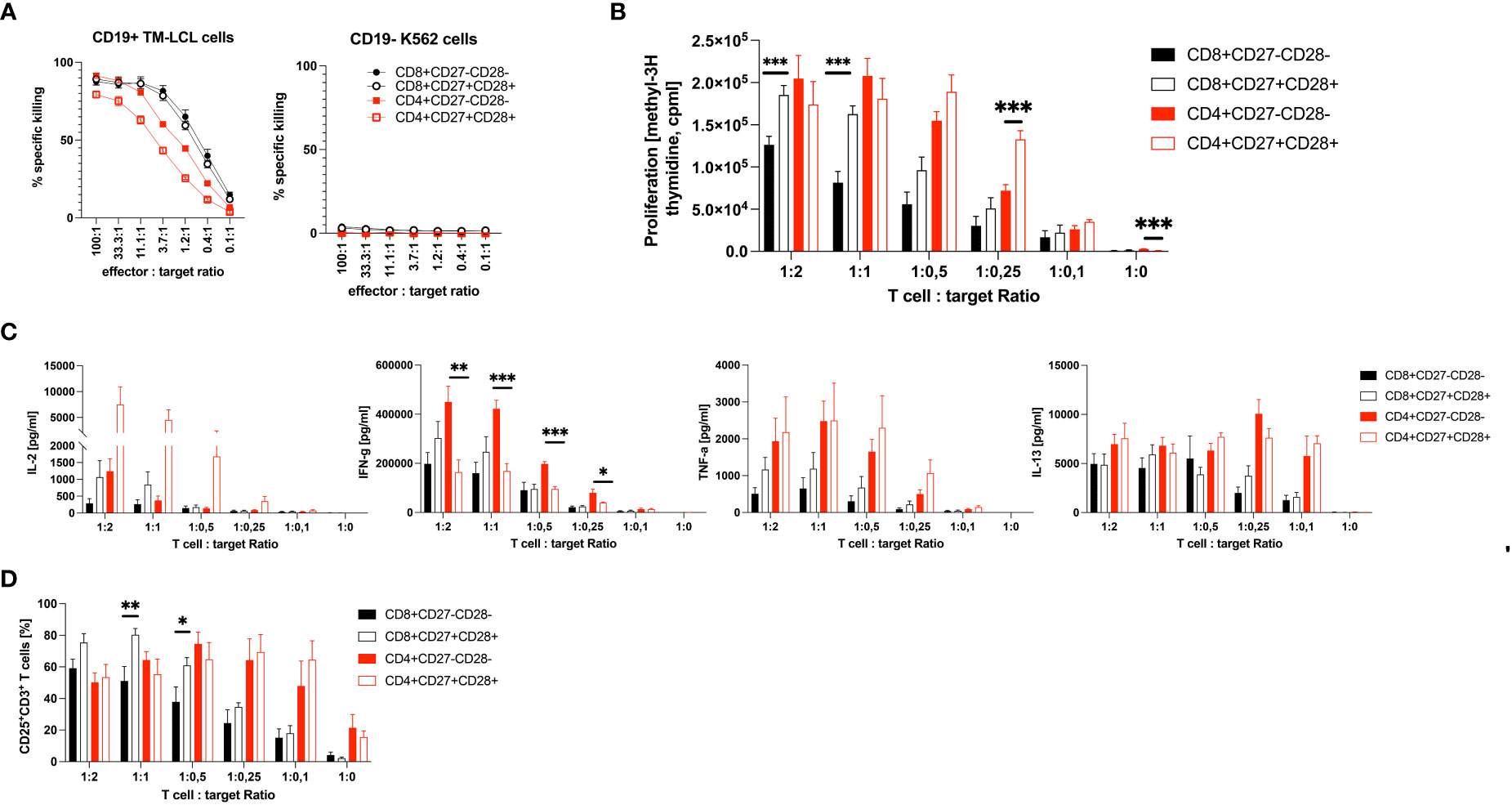
Figure 3 Functional comparison of CD3+CD8+ and CD3+CD4+ CD27+CD28+ to CD3+CD8+ and CD3+CD4+ CD27-CD28- CD19 CART cells in vitro. Shown is (A) the cytotoxic potential as percent specific killing in 5-hour 51Cr-release assays, (B) the proliferation as count per minutes (cpm) (C) the cytokine production in pg/ml and (D) the percent expression of the high-affinity IL-2R (CD25) on CD27-CD28- CD3+CD8+ and CD3+CD4+ T cells in comparison to CD27+CD28+ CD3+CD8+ and CD3+CD4+ T cells upon co-culture with CD19+ TM-LCL cells. The cytotoxic potential of CD27-CD28- and CD27+CD28+ T cells in (A) is also shown against CD19- K562 cells. X-axes show the effector to target ratios with either a constant amount of 5 x 103 target cells (A), or a constant amount of effector cells of 1 x 105 (B–D). Data are shown as means plus SEM (whiskers). Numbers of CD19 CAR positive T cells were 91.5 ± 0.1% and 96.0 ± 0.3%, respectively. Data show the summary of three (A, D) or four (B, C) independent experiments with three different donors in biological triplicates for CD8+ T cells, except one donor in duplicates for Cytotoxicity tests, and two independent experiments with two different donors in biological triplicates for CD4+ T cells. *P < 0.05, **P < 0.01, ***P < 0.001 as determined by unpaired Student’s t-test.
Discussion
In this cohort study, we aimed to identify a simple and robust pre-infusion blood biomarker to predict the future response to CART cell treatment in r/r DLBCL patients. Compared to HC, r/r DLBCL patients presented with significantly more activated HLA-DR-expressing PB T cells, indicating cellular activation and/or homeostatic proliferation (36), as well as pathologically increased, frequencies of CD3+CD27-CD28- T cells. According to the linear T cell differentiation model proposed by Romero et al. and substantiated by our own analyses (Figure S4), T cells with this phenotype belong to the CCR7-CD45RA+/- terminally differentiated T effector memory RA (TEMRA) and effector memory type 3 (EM3) cells, respectively (37). We stratified patients according to OR (CR and PR) versus non-response (SD and PD), or CR versus non-CR (PR, SD and PD) 3 months after CART cell treatment, respectively. This revealed that the pathologically high levels of CD3+CD27-CD28- T cells were associated with non-CR (37.7 ± 17.4%), while patients with CR presented with low, almost physiological, levels of CD3+CD27-CD28- T cells compared to HC (13.7 ± 11.7% versus 6.6 ± 5.8%). A numeric predictor of CR was determined by plotting a ROC curve, which showed that a cut-off value of ≤18% CD3+CD27-CD28- T cells (Figure 2B) predicted CR with high accuracy even 12 months after CART cell transfusion (Figure 2E). This is the first study identifying low numbers of CD3+CD27-CD28- T cells as a valuable pre-infusion blood biomarker for long-term response to CART cell treatment in r/r DLBCL. Our clinical data corroborate previous in vitro findings indicating that both CD27 and CD28 are functionally important co-stimulatory molecules on T cells, which are critically involved in cellular activation programs (32, 47). Moreover, we have demonstrated herein that CD3+CD8+CD27-CD28- CART cells have comparable CD19+ target cell killing activity when compared to CD3+CD8+CD27+CD28+ CART cells, however, they are clearly inferior regarding CD19+ target cell-dependent proliferation and cytotoxic cytokine production, such as IFN-γ and TNF-α (48, 49). Interferon-γ is well-known to contribute to the CART cells’ cytotoxicity by targeting and destroying the tumor stroma (48), while TNF-α has been shown to sensitize tumor cells themselves for getting killed by CD8+ T cells (49). In addition, the elevated CD25 (high-affinity IL-2R) expression levels, along with their increased IL-2 secretion, speaks for a better overall fitness of CD3+CD8+CD27+CD28+ compared to CD3+CD8+CD27-CD28- CART cells.
We show here that CD3+CD4+ T cells can also be turned into CART killer cells, however, they have a 3- to 6-fold lower killing efficacy when compared to CD3+CD8+ T cells. Similar to CD8+CD27+CD28+ T cells, CD4+CD27+CD28+ T cells proliferated better and produced more IL-2 and TNF-α when compared to CD4+CD27-CD28- T cells. However, it is noteworthy that CD4+CD27-CD28- T cells produced significantly more IFN-γ than CD4+CD27+CD28+ T cells, which may explain their moderately superior killing activity compared with CD4+CD27+CD28+ T cells.
Accordingly, our findings also provide an explanation as to why the lack of CD27 and/or CD28 on T cells has been described to be associated with impaired immuno-surveillance capabilities of non-CART cells, previously (35). While the engagement of CD28 with an agonistic CD28 monoclonal antibody was, in fact “too potent in vivo” and induced a highly problematic cytokine storm in six participants of a fist-in-human phase I clinical trial in a previous study (50), engagement of CD27 by varlilumab (CDX-1127), a novel, agonistic, fully human CD27 monoclonal antibody, revealed durable antigen-specific antitumor efficacy (51), by increasing effector T cell numbers with an activated phenotype which was at the expense of naïve and Treg cell numbers in pre-clinical and human phase I and II immunotherapy trials (52). Moreover, conditioning treatment with CD27 mAb in a preclinical model enhanced the expansion and anti-tumor activity of adoptively transferred T cells (53) and by activating T cells recruits and stimulates myeloid cells for enhanced killing of CD27 mAb-opsonized tumors (54). In some CD27 mAb-treated melanoma patients, increased numbers of T cells that recognize melanoma-related antigens were revealed (52). Thus, the expression and active engagement on T cells of CD27 by mAbs has the potential to positively affect adaptive immunotherapy against cancer (55), suggesting conversely that the pathological increase of T cells which lack CD27 expression could be a gradually increasing disadvantage.
Until recently, the best predictors for response to CART cell treatment in r/r DLBCL have been factors not related to immune system function, such as low tumor volume, number of extranodal sites, low serum LDH levels immediately prior to CART cell infusion and a low ECOG performance status (7, 14–16). However, tumor volume/burden lacks specificity because it is a predictor of therapeutic success for the treatment of a large collection of different disease entities and therapies (56). The same holds true for serum LDH levels, which are an established marker of tumor burden, metabolic activity and thus aggressiveness of NHL. Very similar to tumor volume, the serum LDH level has been established as a prognostic factor for the disease course and treatment success of NHL since the 1970s and therefore is also included in the IPI score. Accordingly, while we found elevated serum LDH levels in the overall r/r DLBCL study group (325.9 ± 180.3 U/L), they were lower in OR (236.1 ± 114.0 versus 366.1 ± 162.5; P=0.08) and CR (246.4 ± 132.2 versus 323.9 ± 155.9; P=0.19) patients as compared to non-OR and non-CR patients, respectively, especially when determined at leukapheresis (Table 1), although, without reaching statistical significance.
More recently, the search for new biomarkers has turned to studying the nature of the tumor microenvironment, with the intention to identify the mechanistic basis of putative inhibitory factors, followed by the development of strategies for their inhibition/neutralization with, e.g., checkpoint inhibitors (57). These experimental approaches will help us to understand how to pave the way for the facilitated tumor invasion by the infused CART cells and to ultimately steer and support the activation and cytotoxicity of the latter. However, access to the site of tumor cell accumulation in DLBCL for diagnostic purposes, i.e., the bone marrow and/or lymph nodes, demands utterly invasive and thus burdensome procedures (e.g., bone marrow and/or lymph node biopsies). In contrast, the herein described assessment of the levels of peripheral blood CD3+CD27-CD28- T cells is easy to perform and standardize, also in sequential series of biological samples and thus suitable for daily clinical laboratory routine. In addition, numbers of CD3+CD27-CD28- T cells can reliably be determined in the leukapheresis product, as well with similar accuracy to peripheral blood (Figure S6 and Table S3).
Which mechanism(s) could be responsible for the down-regulation of CD27 and CD28 on the surface of CD3+ T cells in the PB of CART cell non-responders?
The fact that almost 90% of patients expressed elevated levels of HLA-DR+ T cells in their circulation (Table 2), indicates a possible hyperactivation of the immune system (58), which may be a reflection of the lengthy disease course (35.9 ± 53.8 months) and the associated microbial pressure on the lymphodepleted patients and/or the number of prior therapy lines given (median 3, range 1-11) to our patients. In this study, we found no correlation between the total number of different treatment lines and the number of CD3+CD27-CD28- T cells in the PB at the time of leukapheresis. However, we found a weak correlation between the number of R-CHOP cycles administered and the number of CD3+CD27-CD28- T cells (r=0.3931, P=0.0287).
Alternatively, the increased number of HLA-DR+ T cells could also be a sign of homeostatic proliferation due to treatment-induced lymphopenia. In this regard, it is important to note that homeostatically proliferating CD8+ T cells have been shown to neo-express HLA-DR, but always in conjunction with telomerase activity (36).
One mechanism explaining the loss of CD27 on activated T cells is that these cells tend to upregulate CD70, which is the ligand for CD27 (59). In turn, CD70 up-regulation and interaction with its ligand CD27, either on the same or on adjacent cells, may then lead to reactive downregulation of the latter (60). Similarly, CD28 modulation is known to be the result of cellular activation and replicative senescence (31, 61). Notably, the molecular mechanism(s) leading to CD70 upregulation on T cells during chronic systemic inflammation, such as in lupus erythematosus, are governed by epigenetic changes in T cells, such as histone modifications at the TNFSF7 (CD70) promoter (62) with subsequent downregulation of CD27 on terminally differentiated T effector memory RA cells (TEMRA) (63). CD28null cells were also found to exhibit significant changes in their whole-genome methylation pattern (64) and to receive less signaling through the ERK and JNK pathways, reducing the expression of the DNA methyltransferases Dnmt1 and Dnmt3a, which in turn contributes to the epigenetic downregulation of CD28 expression (65). Taken together, both CD27 and CD28 modulation seem to be governed by several factors, including ligand- and epigenetic/promoter-driven downregulation, all supported by chronic hyperactivation of the immune system.
The fact that low lymphocyte counts are frequently detected in DLBCL patients at initial presentation (66) and that lymphocytopenia after first-line therapy is a predictor of relapse (67) is well known. Therefore, it was not entirely surprising that our patient population suffered from significant lymphopenia. Several reasons can be suggested for the intrinsic activation-induced lymphocyte depletion, such as i) canonical tumor antigen-specific activation by lymphoma cells, ii) cytokine-dependent bystander activation caused by DLBCL-secreted and T cell tropic cytokines like IL-2 and IL-6 (68), or iii) reactivation of latent viruses such as CMV or EBV, which have been shown to be associated with the increased appearance of CD3+CD27-CD28- PB T cells previously (28). While the first two explanations are the matter of intense research, the latter can be excluded since no CMV and EBV reactivation was observed in our patients.
Previous studies suggested that an increased frequency of CD27+CD45RO-CD8+ T cells at the time of leukapheresis may correlate with sustained remission in patients with chronic lymphocytic leukemia treated with CD19 CART cells (21), in multiple myeloma patients treated with B cell maturation antigen (BCMA)-specific CART cells (22), and very recently in patients with DLBCL (23). The authors suggested that CD27+CD45RO-CD8+ T cells belong to the group of antigen-experienced CD3+CD8+ T lymphocytes that have long-lasting memory capabilities and improved ability to expand in vitro and in vivo (21, 22, 24). However, this T cell subset, which according to our algorithm belongs to T cells with a CD3+CD27+CD28- phenotype (29), was not found to be associated with OR and/or CR in our study (Table 3). We considered it important to focus on a combination of T lymphocyte surface markers with proven importance during the T cell activation process, i.e., well-established co-stimulatory molecules, such as CD27 and CD28, rather than the combination of one such marker (CD27) with a purely phenotypic marker, such as CD45RO negativity, which may, in fact, identify more than one T cell phenotype, e.g., naïve T cells and antigen-experienced “stem cell memory” cells (23). Romero et al., showed in healthy individuals that the majority of CD3+CD8+CD27-CD28- T cells is composed of CCR7-CD45RA+ terminally differentiated T effector memory RA cells (TEMRA), while they clearly also contain a smaller 10-20% fraction of CD27-CD28- T cells which belongs to the effector memory (EM) subset. The latter subset is commonly referred to as EM3 cells (37). Indeed, in CART patients at leukapheresis and healthy controls, it turned out that CD3+CD8+CD27-CD28- T cells are also highly enriched for CD45RA+CCR7- TEMRA cells (72.3±18.8% in healthy donors vs. 59.2±19.2% in lymphoma patients), the rest of the cells presented with a CD45RA-CCR7- EM phenotype, which is compatible with their relationship to EM3 cells (Figure S4). Within the CD3+CD4+ T cell subset, the picture was different. Herein, CD27-CD28- T cells are mainly composed of CCR7-CD45RA- EM cells belonging to the EM3 phenotype, while the number of TEMRA is usually low to non- existent among CD3+CD4+ T cells in healthy individuals (Figure S4). Thus, when gating on CD3+CD27-CD28- T cells one reads out the “sum of TEMRA and EM3 T cells” of both CD8+ and CD4+ T cells, with CD8+ T cells mainly contributing to the CD27-CD28- phenotype (65.4 ± 23.3% for healthy controls and 59.1 ± 23.8% for patients). A similar picture is seen in typical DLBCL patients (Figure S4).
Furthermore, analyzes of activation marker expression on T cells used for in vitro experiments confirmed that HLA-DR was clearly expressed on all cell types with a tendency for up-regulation on CD27-CD28- T cells as compared to CD27+CD28+ T cells on CD4 and CD8 T subsets. Moreover,CD69 was upregulated on both CD4+ and CD8+ CD27-CD28- T cells as compared to their CD27+CD28+ counterparts (Table S4). The picture was different for CD25 expression, which was downregulated on CD4+CD27-CD28- T cells as compared to CD4+CD27+CD28+ T cells. However, no clear sign for the upregulation of exhaustion markers (LAG-3, TIM-3 and PD-1) was evident on in vitro tested CART cells (Table S4), except TIM-3 on CD8+ T cells.
Comparable albeit slightly different changes were found on cells of patients undergoing leukapheresis. Here, HLA-DR was generally more up-regulated on CD27-CD28- T cells when compared to CD27+CD28+ T cells in patients. CD69 was found to be upregulated more on CD8+CD27-CD28- T cells as compared to CD8+CD27+CD28+ T cells, while no significant expression of CD69 was found on CD4+ T cells (Figure S9). CD25 expression was lower in all patients on the CD27-CD28- when compared to the CD27+CD28+ subset. Notably, PD-1 was clearly upregulated on CD4+CD27-CD28- as compared to CD4+CD27+CD28+ T cells which was in clear contrast to the CD8+ subset, in which PD-1 expression was higher on the CD27+CD28+ T cells when compared to CD27-CD28- T cells (Figure S9). The latter findings points to a remarkable and potentially functionally relevant dissociation of the expression of co-stimulatory and exhaustion marker molecules in DLBCL patients.
The significant association of low numbers of CD3+CD27-CD28- T cells in PB at the time of leukapheresis with CR at 3 months with the cut-off of ≤ 18% CD3+CD27-CD28- T cells to predict CR at 12 months after CART cell treatment seems to be a promising new predictive biomarker. Although our study shows that patients with high numbers of CD3+CD27-CD28- T cells may not respond as well to CART cell therapy as patients with low numbers of CD3+CD27-CD28- T cells, we are far from claiming that this circumstance is irreversible. For example, it may well turn out that administration of checkpoint inhibitors at the time of CART cell administration, e.g., against PD-1, could improve the inferior outcome of this group of patients. Of note in that respect, two of our patients with high numbers of differentiated T cells responded to CART cells when pretreated with pembrolizumab (69). Moreover, recent studies have shown that the use of the Bruton’s tyrosine kinase inhibitor ibrutinib (70), or the phosphoinositide-3 kinase inhibitor idelalisib (71, 72) can improve CART cell production in patients with chronic lymphocytic leukemia. Similar effects may be realized in r/r DLBCL in the future.
The better in vivo performance of CART cell products containing a low baseline amount of CD3+CD27-CD28- T cells may also have adverse effects. Patients receiving such T cells may suffer from more treatment-related toxicities after CART cell transfusion because the CART cells may exhibit greater CD19 target cell-dependent proliferation and cytotoxic factor (IFN-γ, TNF-α) production in vivo and thus a likely higher killing rate. However, no significant associations were found between the number of CD3+CD27-CD28- T cells in the leukapheresis product and the occurrence of i) cytokine release syndrome (CRS, r=0.1 and P=0.072), ii) clinical requirements for tocilizumab therapy (r=0.14 and P=0.51), or iii) long-term cytopenias (r=0.16 and P=0.57) (Spearman’s r-tests) in the present study.
Several important limitations of this trial should be considered. During the planning and recruitment phase of this trial no validated flow cytometric assay was available to monitor CART cell expansion in vivo and respective binding reagents for reliable monitoring had only become available very recently (73). Therefore, the relationship between the CD3+CD27-CD28- T cell status determined at leukapheresis and the kinetics of CART cell expansion in vivo could not be monitored.
In addition, our study is limited by a small sample size of only 33 patients with 26 patients who received CART cells at least three-month before response assessment. Therefore, larger multi-center studies are certainly needed to confirm our findings in the future. Due to the limited sample size, we were not able to test our biomarker in an independent validation cohort.
It has to be noted that the ethical permission did not include to test CART cells from patients in in vitro studies.
Therefore, in the CD19 CART cell in vitro studies shown here, T cells of healthy donors were transduced with a CD19-CAR. For that purpose, PBMC from healthy donors were processed for CART cell production using a protocol comparable to that used for the processing of the leukapheresis products from patients, without prior sorting into CD4+ and CD8+ T cells subsets before transduction and expansion. Accordingly, CD3CD28-bead stimulated PBMC were transduced with the CD19 CAR and further expanded for 14 days. Upon cryopreservation and recultivation, CART cells were further expanded by incubation with irradiated (120 Gy) CD19+ TM-LCL cells for 10 days followed by FACS-sorting for CD27 and CD28 expression. TM-LCL cells, while being non-proliferative, are still able to provide the CD19 antigen necessary for antigen-dependent proliferation of CD19 CART cells. They have been successfully used in the past for CD19 CART cell expansion (42). In fact, upon co-culturing with irradiated TM-LCL cells, the authors of this report routinely observed a 18-20-fold expansion of CD19 CART cells within 10 days. Expanded and sorted CART cells were than rested for 7 days followed by determination of their CD27 and CD28 expression status, their antigen-dependent cytotoxicity, proliferative capacity and factor production capabilities. While we did not observe a significant difference in the killing capacity between CD27+CD28+ and CD27-CD28- CART cells, we consider the differences in the proliferative capacity of CD3+CD27+CD28+ CD19 CART cells versus CD3+CD27-CD28- CD19 CART cells worth reporting, especially since previous studies had already shown that the ability to proliferate and expand well is associated with the expression of T cell clusters harboring upregulated proliferation-associated genes (74). Our study now shows that a similar stratification of T cells can be achieved by virtue of separating T cells according to their surface-expression status of the co-stimulatory molecules CD27 and CD28. It is in line with the linear differentiation model of T cells which has shown previously that CD3+CD27-CD28- T cells consist of TEMRA and EM3 cells, both belong to the terminally differentiated T effector memory cells with undetectable TREC numbers and short telomers (37). Elevated numbers of this phenotype are not found in healthy individuals (Figure 2A), but are a salient feature of individuals with considerable immunological dysregulation (chronic inflammation), such as the one found in r/r DLBCL patients.
In summary, our study has identified that a low number of CD3+CD27-CD28- T cells is a new biomarker associated with better treatment response to CART cell therapy. This novel insight has the potential to contribute to an improved selection of patients with a high chance of CR after CART cell treatment and/or to form the rational basis for co-medications, such as ibrutinib, at the time of leukapheresis or administration of checkpoint-inhibitors at the time of CART transfusion. Such findings may thus provide the basis for further increasing the success rates of this innovative and potentially curative therapy.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors. Requests to access the datasets should be directed to Dr. Nina Worel at bmluYS53b3JlbEBtZWR1bml3aWVuLmFjLmF0.
Ethics statement
The studies involving human participants were reviewed and approved by Ethics Committee of the Medical University of Vienna, EC No.:1422/2015, 1607/2018, 2055/2019, 1290/2020. The patients/participants provided their written informed consent to participate in this study.
Author contributions
UJ, NW, and WP designed research; NW, KG-P, BK, MS, AT, AR, UK, EP, PS, CS, HH, VG, JH, BS, ML, NS, EF, PW, WR, and IS-K performed research and analyzed data; UJ, GH, PW, NW, and WP supervised experiments and clinical study; WP, NW, KG-P, and UJ wrote the paper. All authors contributed to the article and approved the submitted version.
Funding
UJ, VG and JH were supported by the Innovative Medicines Initiative (IMI) T2EVOLVE. This project has received funding from the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 945393. This Joint Undertaking receives support from the European Union ı́s Horizon 2020 research and innovation program and EFPIA. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We want to thank Beate Pribitzer, Karin Feldmann, Martina Muck, Martina Preslmayr, Verena Raggl and Marie-Christin Röxeisen for their laboratory support, the team of nurses for taking excellent care of patients and patient’s samples, and the physicians who referred the patents for further treatment with CART cells. The authors are indebted to Dr. Mairi McGrath for expert proofreading.
Conflict of interest
With regards to the authors’ disclosure of potential conflicts of interest we would like to indicate that WP receives honoraria from Novartis, Astra Zeneca and Roche. UJ reports honoraria and advisory roles for Novartis, BMS/Celgene, Gilead, Miltenyi. NW, PW, and GH report honoraria from Novartis, BMS/Celgene and Gilead. WR reports honoraria from BMS/Celgene. JH reports honoraria from Novartis.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1004703/full#supplementary-material
References
1. Howlader N, Noone AM, Krapcho M, Miller D, Brest A, Yu M, et al. SEER cancer statistics review. Bethesda, MD: National Cancer Institute (2020).
2. Sant M, Minicozzi P, Mounier M, Anderson LA, Brenner H, Holleczek B, et al. Survival for haematological malignancies in Europe between 1997 and 2008 by region and age: Results of EUROCARE-5, a population-based study. Lancet Oncol (2014) 15:931–42. doi: 10.1016/S1470-2045(14)70282-7
3. Crump M, Neelapu SS, Farooq U, Van Den Neste E, Kuruvilla J, Westin J, et al. Outcomes in refractory diffuse large b-cell lymphoma: Results from the international SCHOLAR-1 study. Blood (2017) 130:1800–8. doi: 10.1182/blood-2017-03-769620
4. Hamadani M, Hari PN, Zhang Y, Carreras J, Akpek G, Aljurf MD, et al. Early failure of frontline rituximab-containing chemo-immunotherapy in diffuse large b cell lymphoma does not predict futility of autologous hematopoietic cell transplantation. Biol Blood Marrow Transplant (2014) 20:1729–36. doi: 10.1016/j.bbmt.2014.06.036
5. Gisselbrecht C, Schmitz N, Mounier N, Singh Gill D, Linch DC, Trneny M, et al. Rituximab maintenance therapy after autologous stem-cell transplantation in patients with relapsed CD20(+) diffuse large b-cell lymphoma: Final analysis of the collaborative trial in relapsed aggressive lymphoma. J Clin Oncol (2012) 30:4462–9. doi: 10.1200/JCO.2012.41.9416
6. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med (2018) 379:64–73. doi: 10.1056/NEJMra1706169
7. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse Large b-cell lymphoma. N Engl J Med (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
8. Hopfinger G, Jager U, Worel N. CAR-T cell therapy in diffuse Large b cell lymphoma: Hype and hope. Hemasphere (2019) 3:e185. doi: 10.1097/HS9.0000000000000185
9. Awasthi R, Pacaud L, Waldron E, Tam CS, Jager U, Borchmann P, et al. Tisagenlecleucel cellular kinetics, dose, and immunogenicity in relation to clinical factors in relapsed/refractory DLBCL. Blood Adv (2020) 4:560–72. doi: 10.1182/bloodadvances.2019000525
10. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory Large b-cell lymphoma. N Engl J Med (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
11. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large b-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet (2020) 396:839–52. doi: 10.1016/S0140-6736(20)31366-0
12. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large b-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1-2 trial. Lancet Oncol (2019) 20:31–42. doi: 10.1016/S1470-2045(18)30864-7
13. Mirzaei HR, Mirzaei H, Namdar A, Rahmati M, Till BG, Hadjati J. Predictive and therapeutic biomarkers in chimeric antigen receptor T-cell therapy: A clinical perspective. J Cell Physiol (2019) 234:5827–41. doi: 10.1002/jcp.27519
14. Nastoupil LJ, Jain MD, Feng L, Spiegel JY, Ghobadi A, Lin Y, et al. Standard-of-Care axicabtagene ciloleucel for relapsed or refractory Large b-cell lymphoma: Results from the US lymphoma CAR T consortium. J Clin Oncol (2020) 38:3119–28. doi: 10.1200/JCO.19.02104
15. Vercellino L, Di Blasi R, Kanoun S, Tessoulin B, Rossi C, D'Aveni-Piney M, et al. Predictive factors of early progression after CAR T-cell therapy in relapsed/refractory diffuse large b-cell lymphoma. Blood Adv (2020) 4:5607–15. doi: 10.1182/bloodadvances.2020003001
16. Westin JR, Tam CS, Borchmann P, Jaeger U, McGuirk JP. Correlative analyses of patient and clinical characteristics associated with efficacy in tesagenlecleucel-treated relapsed/refractory diffuse large b-cell lymphoma patients in the Juliet trial. Blood (2019) 134:4103. doi: 10.1182/blood-2019-129107
17. Wang X, Riviere I. Clinical manufacturing of CAR T cells: Foundation of a promising therapy. Mol Ther Oncolytics (2016) 3:16015. doi: 10.1038/mto.2016.15
18. Rodriguez-Garcia A, Palazon A, Noguera-Ortega E, Powell DJ Jr., Guedan S. CAR-T cells hit the tumor microenvironment: Strategies to overcome tumor escape. Front Immunol (2020) 11:1109. doi: 10.3389/fimmu.2020.01109
19. Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M, et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med (2018) 24:1504–6. doi: 10.1038/s41591-018-0146-z
20. Perna F, Sadelain M. Myeloid leukemia switch as immune escape from CD19 chimeric antigen receptor (CAR) therapy Transl Cancer Res (2016) 5:S221-S225. doi: 10.21037/tcr.2016.08.15
21. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med (2018) 24:563–71. doi: 10.1038/s41591-018-0010-1
22. Cohen AD, Garfall AL, Stadtmauer EA, Melenhorst JJ, Lacey SF, Lancaster E, et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J Clin Invest (2019) 129:2210–21. doi: 10.1172/JCI126397
23. Cuffel A, Allain V, Faivre L, Di Blasi R, Morin F, Vercellino L, et al. Real-world characteristics of T-cell apheresis and clinical response to tisagenlecleucel in b-cell lymphoma. Blood Adv (2022) 6:4657–60. doi: 10.1182/bloodadvances.2022007057
24. Garfall AL, Dancy EK, Cohen AD, Hwang WT, Fraietta JA, Davis MM, et al. T-Cell phenotypes associated with effective CAR T-cell therapy in postinduction vs relapsed multiple myeloma. Blood Adv (2019) 3:2812–5. doi: 10.1182/bloodadvances.2019000600
25. Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest (2005) 115:1616–26. doi: 10.1172/JCI24480
26. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res (2011) 17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116
27. Allen ES, Stroncek DF, Ren J, Eder AF, West KA, Fry TJ, et al. Autologous lymphapheresis for the production of chimeric antigen receptor T cells. Transfusion (2017) 57:1133–41. doi: 10.1111/trf.14003
28. Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med (2002) 8:379–85. doi: 10.1038/nm0402-379
29. Appay V, van Lier RA, Sallusto F, Roederer M. Phenotype and function of human T lymphocyte subsets: Consensus and issues. Cytometry A (2008) 73:975–83. doi: 10.1002/cyto.a.20643
30. van Baarle D, Tsegaye A, Miedema F, Akbar A. Significance of senescence for virus-specific memory T cell responses: Rapid ageing during chronic stimulation of the immune system. Immunol Lett (2005) 97:19–29. doi: 10.1016/j.imlet.2004.10.003
31. Effros RB. Loss of CD28 expression on T lymphocytes: A marker of replicative senescence. Dev Comp Immunol (1997) 21:471–8. doi: 10.1016/S0145-305X(97)00027-X
32. Hintzen RQ, Lens SM, Lammers K, Kuiper H, Beckmann MP, van Lier RA. Engagement of CD27 with its ligand CD70 provides a second signal for T cell activation. J Immunol (1995) 154:2612–23.
33. Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol (1996) 14:233–58. doi: 10.1146/annurev.immunol.14.1.233
34. Hendriks J, Gravestein LA, Tesselaar K, van Lier RA, Schumacher TN, Borst J. CD27 is required for generation and long-term maintenance of T cell immunity. Nat Immunol (2000) 1:433–40. doi: 10.1038/80877
35. Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science (2017) 355:1423–7. doi: 10.1126/science.aaf0683
36. Speiser DE, Migliaccio M, Pittet MJ, Valmori D, Lienard D, Lejeune F, et al. Human CD8(+) T cells expressing HLA-DR and CD28 show telomerase activity and are distinct from cytolytic effector T cells. Eur J Immunol (2001) 31:459–66. doi: 10.1002/1521-4141(200102)31:2<459::AID-IMMU459>3.0.CO;2-Y
37. Romero P, Zippelius A, Kurth I, Pittet MJ, Touvrey C, Iancu EM, et al. Four functionally distinct populations of human effector-memory CD8+ T lymphocytes. J Immunol (2007) 178:4112–9. doi: 10.4049/jimmunol.178.7.4112
38. Tchilian EZ, Wallace DL, Imami N, Liao HX, Burton C, Gotch F, et al. The exon a (C77G) mutation is a common cause of abnormal CD45 splicing in humans. J Immunol (2001) 166:6144–8. doi: 10.4049/jimmunol.166.10.6144
39. Flinn IW, Jaeger U, Shah NN, Blaise D, Briones J, Shune L, et al. A first-in-Human study of YTB323, a novel, autologous CD19-directed CAR-T cell therapy manufactured using the novel T-charge TM platform, for the treatment of patients (Pts) with Relapsed/Refractory (r/r) diffuse Large b-cell lymphoma (DLBCL). Blood (2021) 138:740. doi: 10.1182/blood-2021-146268
40. Cossarizza A, Chang HD, Radbruch A, Acs A, Adam D, Adam-Klages S, et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). Eur J Immunol (2019) 49:1457–973. doi: 10.1002/eji.201970107
41. Pelloquin F, Lamelin JP, Lenoir GM. Human b lymphocytes immortalization by Epstein-Barr virus in the presence of cyclosporin a. In Vitro Cell Dev Biol (1986) 22:689–94. doi: 10.1007/BF02621085
42. Terakura S, Yamamoto TN, Gardner RA, Turtle CJ, Jensen MC, Riddell SR. Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood (2012) 119:72–82. doi: 10.1182/blood-2011-07-366419
43. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult b cell ALL patients. J Clin Invest (2016) 126:2123–38. doi: 10.1172/JCI85309
44. Kratzer B, Trapin D, Gattinger P, Oberhofer T, Sehgal ANA, Waidhofer-Sollner P, et al. Lack of induction of RBD-specific neutralizing antibodies despite repeated heterologous SARS-CoV-2 vaccination leading to seroconversion and establishment of T cell-specific memory in a patient in remission of multiple myeloma. Vaccines (Basel) (2022) 10:374. doi: 10.3390/vaccines10030374
45. Harrison N, Grabmeier-Pfistershammer K, Graf A, Trapin D, Tauber P, Aberle JH, et al. Tick-borne encephalitis specific lymphocyte response after allogeneic hematopoietic stem cell transplantation predicts humoral immunity after vaccination. Vaccines (Basel) (2021) 9:908. doi: 10.3390/vaccines9080908
46. Liadi I, Singh H, Romain G, Rey-Villamizar N, Merouane A, Adolacion JR, et al. Individual motile CD4(+) T cells can participate in efficient multikilling through conjugation to multiple tumor cells. Cancer Immunol Res (2015) 3:473–82. doi: 10.1158/2326-6066.CIR-14-0195
47. June CH, Ledbetter JA, Linsley PS, Thompson CB. Role of the CD28 receptor in T-cell activation. Immunol Today (1990) 11:211–6. doi: 10.1016/0167-5699(90)90085-N
48. Textor A, Listopad JJ, Wuhrmann LL, Perez C, Kruschinski A, Chmielewski M, et al. Efficacy of CAR T-cell therapy in large tumors relies upon stromal targeting by IFNgamma. Cancer Res (2014) 74:6796–805. doi: 10.1158/0008-5472.CAN-14-0079
49. Kearney CJ, Vervoort SJ, Hogg SJ, Ramsbottom KM, Freeman AJ, Lalaoui N, et al. Tumor immune evasion arises through loss of TNF sensitivity. Sci Immunol (2018) 3:eaar3451. doi: 10.1126/sciimmunol.aar3451
50. Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med (2006) 355:1018–28. doi: 10.1056/NEJMoa063842
51. He LZ, Prostak N, Thomas LJ, Vitale L, Weidlick J, Crocker A, et al. Agonist anti-human CD27 monoclonal antibody induces T cell activation and tumor immunity in human CD27-transgenic mice. J Immunol (2013) 191:4174–83. doi: 10.4049/jimmunol.1300409
52. Burris HA, Infante JR, Ansell SM, Nemunaitis JJ, Weiss GR, Villalobos VM, et al. Safety and activity of varlilumab, a novel and first-in-Class agonist anti-CD27 antibody, in patients with advanced solid tumors. J Clin Oncol (2017) 35:2028–36. doi: 10.1200/JCO.2016.70.1508
53. Wasiuk A, Weidlick J, Sisson C, Widger J, Crocker A, Vitale L, et al. Conditioning treatment with CD27 ab enhances expansion and antitumor activity of adoptively transferred T cells in mice. Cancer Immunol Immunother (2022) 71:97–109. doi: 10.1007/s00262-021-02958-9
54. Turaj AH, Hussain K, Cox KL, Rose-Zerilli MJJ, Testa J, Dahal LN, et al. Antibody tumor targeting is enhanced by CD27 agonists through myeloid recruitment. Cancer Cell (2017) 32:777–91.e776. doi: 10.1016/j.ccell.2017.11.001
55. Bullock TN. Stimulating CD27 to quantitatively and qualitatively shape adaptive immunity to cancer. Curr Opin Immunol (2017) 45:82–8. doi: 10.1016/j.coi.2017.02.001
56. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer (2009) 45:228–47. doi: 10.1016/j.ejca.2008.10.026
57. Chen DS, Mellman I. Oncology meets immunology: The cancer-immunity cycle. Immunity (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
58. Ko HS, Fu SM, Winchester RJ, Yu DT, Kunkel HG. Ia determinants on stimulated human T lymphocytes. occurrence on mitogen- and antigen-activated T cells. J Exp Med (1979) 150:246–55. doi: 10.1084/jem.150.2.246
59. Hintzen RQ, Lens SM, Koopman G, Pals ST, Spits H, van Lier RA. CD70 represents the human ligand for CD27. Int Immunol (1994) 6:477–80. doi: 10.1093/intimm/6.3.477
60. Arens R, Tesselaar K, Baars PA, van Schijndel GM, Hendriks J, Pals ST, et al. Constitutive CD27/CD70 interaction induces expansion of effector-type T cells and results in IFNgamma-mediated b cell depletion. Immunity (2001) 15:801–12. doi: 10.1016/S1074-7613(01)00236-9
61. Vallejo AN, Brandes JC, Weyand CM, Goronzy JJ. Modulation of CD28 expression: Distinct regulatory pathways during activation and replicative senescence. J Immunol (1999) 162:6572–9.
62. Zhou Y, Qiu X, Luo Y, Yuan J, Li Y, Zhong Q, et al. Histone modifications and methyl-CpG-binding domain protein levels at the TNFSF7 (CD70) promoter in SLE CD4+ T cells. Lupus (2011) 20:1365–71. doi: 10.1177/0961203311413412
63. Fritsch RD, Shen X, Sims GP, Hathcock KS, Hodes RJ, Lipsky PE. Stepwise differentiation of CD4 memory T cells defined by expression of CCR7 and CD27. J Immunol (2005) 175:6489–97. doi: 10.4049/jimmunol.175.10.6489
64. Suarez-Alvarez B, Rodriguez RM, Schlangen K, Raneros AB, Marquez-Kisinousky L, Fernandez AF, et al. Phenotypic characteristics of aged CD4(+) CD28(null) T lymphocytes are determined by changes in the whole-genome DNA methylation pattern. Aging Cell (2017) 16:293–303. doi: 10.1111/acel.12552
65. Chen Y, Gorelik GJ, Strickland FM, Richardson BC. Decreased ERK and JNK signaling contribute to gene overexpression in "senescent" CD4+CD28- T cells through epigenetic mechanisms. J Leukoc Biol (2010) 87:137–45. doi: 10.1189/jlb.0809562
66. Talaulikar D, Choudhury A, Shadbolt B, Brown M. Lymphocytopenia as a prognostic marker for diffuse large b cell lymphomas. Leuk Lymphoma (2008) 49:959–64. doi: 10.1080/10428190801959026
67. Aoki T, Nishiyama T, Imahashi N, Kitamura K. Lymphopenia following the completion of first-line therapy predicts early relapse in patients with diffuse large b cell lymphoma. Ann Hematol (2012) 91:375–82. doi: 10.1007/s00277-011-1305-1
68. Gupta M, Han JJ, Stenson M, Maurer M, Wellik L, Hu G, et al. Elevated serum IL-10 levels in diffuse large b-cell lymphoma: A mechanism of aberrant JAK2 activation. Blood (2012) 119:2844–53. doi: 10.1182/blood-2011-10-388538
69. Jaeger U, Worel N, McGuirk JP, Riedell PA, Fleury I, Du Y, et al. Safety and efficacy of tisagenlecleucel plus pembrolizumab in patients with r/r DLBCL: Results from the phase ib PORTIA study. Blood Adv (2022). doi: 10.1182/bloodadvances.2022007779
70. Fan F, Yoo HJ, Stock S, Wang L, Liu Y, Schubert ML, et al. Ibrutinib for improved chimeric antigen receptor T-cell production for chronic lymphocytic leukemia patients. Int J Cancer (2021) 148:419–28. doi: 10.1002/ijc.33212
71. Stock S, Ubelhart R, Schubert ML, Fan F, He B, Hoffmann JM, et al. Idelalisib for optimized CD19-specific chimeric antigen receptor T cells in chronic lymphocytic leukemia patients. Int J Cancer (2019) 145:1312–24. doi: 10.1002/ijc.32201
72. Funk CR, Wang S, Chen KZ, Waller A, Sharma A, Edgar CL, et al. PI3Kdelta/gamma inhibition promotes human CART cell epigenetic and metabolic reprogramming to enhance antitumor cytotoxicity. Blood (2022) 139:523–37. doi: 10.1182/blood.2021011597
73. Laurent E, Sieber A, Salzer B, Wachernig A, Seigner J, Lehner M, et al. Directed evolution of stabilized monomeric CD19 for monovalent CAR interaction studies and monitoring of CAR-T cell patients. ACS Synth Biol (2021) 10:1184–98. doi: 10.1021/acssynbio.1c00010
Keywords: diffuse large B cell lymphoma, chimeric antigen receptor T cells therapy, CD27, CD28, biomarker
Citation: Worel N, Grabmeier-Pfistershammer K, Kratzer B, Schlager M, Tanzmann A, Rottal A, Körmöczi U, Porpaczy E, Staber PB, Skrabs C, Herkner H, Gudipati V, Huppa JB, Salzer B, Lehner M, Saxenhuber N, Friedberg E, Wohlfarth P, Hopfinger G, Rabitsch W, Simonitsch-Klupp I, Jäger U and Pickl WF (2023) The frequency of differentiated CD3+CD27-CD28- T cells predicts response to CART cell therapy in diffuse large B-cell lymphoma. Front. Immunol. 13:1004703. doi: 10.3389/fimmu.2022.1004703
Received: 27 July 2022; Accepted: 28 November 2022;
Published: 09 January 2023.
Edited by:
Catherine Sautes-Fridman, U1138 Centre de Recherche des Cordeliers (CRC) (INSERM), FranceReviewed by:
Vijay Bhoj, University of Pennsylvania, United StatesSarwish Rafiq, School of Medicine, Emory University, United States
Stephen Gottschalk, St. Jude Children’s Research Hospital, United States
Copyright © 2023 Worel, Grabmeier-Pfistershammer, Kratzer, Schlager, Tanzmann, Rottal, Körmöczi, Porpaczy, Staber, Skrabs, Herkner, Gudipati, Huppa, Salzer, Lehner, Saxenhuber, Friedberg, Wohlfarth, Hopfinger, Rabitsch, Simonitsch-Klupp, Jäger and Pickl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Winfried F. Pickl, d2luZnJpZWQucGlja2xAbWVkdW5pd2llbi5hYy5hdA==
†These authors have contributed equally to this work