 Blake L. Torrance
Blake L. Torrance Laura Haynes
Laura Haynes- UConn Center on Aging and Department of Immunology, School of Medicine, University of Connecticut, Farmington, CT, United States
Aging results in systemic changes that leave older adults at much higher risk for adverse outcomes following respiratory infections. Much work has been done over the years to characterize and describe the varied changes that occur with aging from the molecular/cellular up to the organismal level. In recent years, the systemic accumulation of senescent cells has emerged as a key mediator of many age-related declines and diseases of aging. Many of these age-related changes can impair the normal function of the respiratory system and its capability to respond appropriately to potential pathogens that are encountered daily. In this review, we aim to establish the effects of cellular senescence on the disruption of normal lung function with aging and describe how these effects compound to leave an aged respiratory system at great risk when exposed to a pathogen. We will also discuss the role cellular senescence may play in the inability of most vaccines to confer protection against respiratory infections when administered to older adults. We posit that cellular senescence may be the point of convergence of many age-related immunological declines. Enhanced investigation into this area could provide much needed insight to understand the aging immune system and how to effectively ameliorate responses to pathogens that continue to disproportionately harm this vulnerable population.
1 Introduction
With age, there are many changes that occur throughout the body and affect nearly every organ system, including the respiratory and immune systems. The primary function of the lungs is to facilitate effective gas exchange, but another critical function is serving as a mucosal barrier site that is adequately able to resist infection despite being exposed to a legion of microbes and potential pathogens on a daily basis. The complex organization of various tissues and cell types in the lungs, non-immune and immune alike, are responsible for coordinating these important functions. Aging will affect all facets of this system and will go on to cause age-related dysfunction both in the normal physiological function of the lungs as well as leaving the host at great risk for infections and subsequent morbidity and mortality. While there are many processes of aging that can cause dysfunction in these areas, our focus in this review is the contribution of cellular senescence and the accumulation of senescent cells in causing and exacerbating age-related declines in the lungs.
Cellular senescence is characterized by irreversible growth arrest that occurs when cells experience a stressor (1). Senescence has two main roles in an adult: one is to suppress cells that have incurred DNA or other damage to prevent them from transitioning into cancer (2), the other is to aid in wound healing (3). The stimuli that lead to senescence are well reviewed elsewhere (4), but include DNA damage, replicative exhaustion, oxidative or metabolic stress, oncogene expression, phototoxic stress, shear stress, as well as inflammation. The key mediators of the cell cycle arrest include p21Cip1 (p21) and p16INK4A (p16), which are cyclin dependent kinase inhibitors (5). Often, expression of these molecules is used as a marker for senescence but they should be combined with other assays for DNA damage, telomere shortening, or senescence associated beta-galactosidase activity to definitively conclude that a cell is senescent. Importantly, after senescent cells perform their physiological functions, they are typically cleared by the innate immune system, including macrophages and natural killer (NK) cells (6). Effective clearance is necessarily reliant on a well-functioning immune system and therefore, with age, senescent cells are more likely to evade clearance due to age-related changes in the immune system.
When senescent cells fail to be detected and cleared, they begin to cause dysfunction. Senescent cells engage various pathways to enforce resistance to apoptosis via a variety of pathways as reviewed by others (7), collectively termed senescent cell anti-apoptotic pathways (SCAPs). Engagement of SCAPs furthers the accumulation of these cells, which can have deleterious effects on the microenvironments where senescent cells are found and beyond. Aside from the cell-intrinsic dysfunction of senescent cells, a key hallmark of senescence is the secretion of a generally-proinflammatory heterogenous cocktail of soluble factors including cytokines and chemokines, termed the senescence associated secretory phenotype (SASP). Not only does this alter the cytokine composition of the microenvironment which will disrupt physiological cell signaling networks, but it can be a source of inflammation resulting in the induction of senescence in nearby cells in a paracrine manner (8). This cycle of senescence induction can wreak havoc on dynamic and cell-rich tissues, such as those that comprise the respiratory system. In this review, we aim to highlight work that links cellular senescence and chronic diseases of aging in the lung as well as the ability of the immune system to adequately respond to respiratory infections.
1.1 Effects of aging and senescence on development of chronic lung pathologies
In order for the lungs to function properly, many different tissue structures, cell types, signaling mechanisms, and other processes need to be tightly regulated. Air is conducted through the trachea and branches into bronchi that infiltrate into the lungs and continue to branch into smaller bronchioles that terminate in alveoli wherein gas exchange occurs with blood vessels in close association. A complex community of cells facilitates these processes at each distinct tissue region. In the bronchi, goblet cells secrete mucus that trap debris and potential pathogens while ciliated cells work to move the mucus and its contents up and away from the lower respiratory tract. Found in the alveolus, type I and II pneumocytes are specialized epithelial cells that line the majority of the alveolar space to perform gas exchange or secrete necessary surfactants, respectively. Nearby, endothelial cell tight junctions in the blood vessel achieve limited permeability to protect the airway from blood infiltrate. Fibroblasts are also present in the alveolus and provide important structural and barrier integrity functions. Various resident immune cells are also found in the airway during homeostasis (in the absence of infection), notably alveolar macrophages that surveil the airway and sample incoming antigens taken in from the airway. Two major chronic diseases of aging, chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF), have been linked to cellular senescence. Here, we briefly discuss recent efforts linking COPD and senescence. IPF has been more definitively shown to be primarily driven by senescence (9) via similar mechanisms and are comprehensively reviewed elsewhere (10). Further, targeting senescence in patients with IPF has already been shown effective in an early stage clinical trial (11). Both pathologies serve as a case in point for our discussion of mechanisms by which senescence can cause baseline dysfunctions in the respiratory system that can be a detrimental risk for developing severe respiratory infections.
1.1.1 Senescence and COPD development
Chronic obstructive pulmonary disease (COPD) is a chronic illness that can occur as a result of chronic bronchitis and/or emphysema. Induction is typically caused by chronic exposure to cigarette smoke or other toxins. Bronchitis is characterized by increased inflammation of the bronchi while emphysema is characterized by severely damaged alveoli (12). Common symptoms include shortness of breath, persistent cough, overproduction of mucus, and wheezing. While the majority of COPD patients can be linked to smoking, aging processes in the lung, particularly cellular senescence, certainly accelerate the development of the disease. For years, COPD has been described as a disease of accelerated aging in the lungs (13). The exact contribution of age versus environmental exposure to the development of COPD is not entirely clear. There are a substantial number of early-onset COPD patients who develop symptoms under the age of 53 and often are not heavy smokers (14). The etiology of these age-independent cases of COPD has remained elusive. However, recent work has pointed towards cellular senescence a driver of COPD onset and severity.
A number of hallmarks of senescence can be identified in COPD development. Increased inflammatory cytokine and chemokine expression is commonly observed (15). IL-6 and other molecules related to IL-6 signaling are among the most consistent (16, 17). Interestingly, IL-6 is also among the most consistent factors present in the SASP of senescent cells (18). Mouse models of cigarette smoke induced COPD have indicated smoke-induced senescence as a mediator of pathogenesis (19). More recently, the first concrete link between senescence and COPD was described by demonstrating a greater frequency of senescent lung fibroblasts from COPD patients (including early onset) compared to non-COPD controls (20). This study utilized expression of both p21 and p16, γ-H2A.X (DNA damage marker), and SA-β-Gal. Fibroblasts in the lung are principally associated with mediating extracellular matrix (ECM) remodeling, as reviewed elsewhere (21). Increased secretion of TGF-β by fibroblasts been associated with COPD development as well (22). When senescence occurs in these cells, it can cause aberrant repair mechanisms similar to a wound healing or tissue remodeling response. These responses have been long since linked to senescence and the SASP (23), including enhanced TGF-β production. Clearly, this indicates that senescent cell participation can lead to COPD, but these same mechanisms can also severely disrupt the ability of the immune system to respond effectively to an incoming pathogen.
Disruptions of basal cytokine signaling will certainly alter the response of immune cells. Increased production of SASP factors can also alter the composition of the immune cell compartment in the lungs. It has been reported that increased chronic inflammation with age, likely exacerbated by SASP, results in a change in homeostatic CD4+ T-helper cell (Th) subset balances. Notably, there is a shift towards circulating IL-17 producing Th17 cells over regulatory T cells (Tregs) with age (24). This same Th17/Treg ratio dysfunction has also been linked to COPD (25). Dysfunctional cytokine signaling will also alter the responsiveness of innate immune cells like alveolar macrophages, dendritic cells, and neutrophils, all of which are critical first line responders to respiratory pathogens. Therefore, mechanisms of senescence highlighted by chronic disease development can alter the function of both non-immune and immune cell compartments in the absence of an infection, which will have severe consequences when a pathogen enters and requires a highly coordinated response to effectively control.
2 Aging and senescence in regulating immune responses to infections
Chronological aging is associated with a wide-ranging decline in overall efficacy of the immune response. Unfortunately, the COVID-19 pandemic has made this fact abundantly clear. At the time of preparing this submission, over 700,000 people aged 65 or older have succumbed due to COVID-19 infection in the United States alone (26). Our group and others have extensively studied the effects of age on responses to influenza (flu) infection, which similarly is a disproportionate concern for individuals over 65. This population, on average, accounts for 70-85% of flu-related deaths each season (27). These vulnerabilities are mostly caused by deficits in the immune system and the ability to mount an appropriate response to an infection. Much work has been done characterizing the changes in the immune system associated with chronological age, but we wish to highlight and discuss a few key points.
2.1 The innate immune compartment
Chief among the age-associated changes in the immune system is the increase in chronic, low-grade, sterile inflammation. This phenomenon, termed inflammaging, was originally described a number of years ago (28–30) and has been broadly defined as increased systemic and local concentrations of pro-inflammatory cytokines including IL-6, TNF-α, IL-1β, and others. Aside from influencing the tightly regulated immune responses to pathogens, acute inflammation and inflammaging has been associated with the progression and development of many diseases of aging including Alzheimer’s disease (31), type 2 diabetes (32), and osteoarthritis (33). Interestingly, some studies focusing on healthy older adults have found no increased systemic IL-6 or TNF-α in their cohorts (34, 35). Utilizing a sophisticated computational deep-learning model, CXCL9 was identified as a more informative biomarker for inflammaging (35). The exclusion criteria of these studies were relatively strict compared to other studies and perhaps this can explain some of the discrepancy. It is plausible that senescent cell burden may influence the degree of inflammaging across individuals due to SASP production. Further, this may highlight the pathogenic role of these cytokines in developing chronic diseases of aging. The source of this systemic accumulation of cytokines has been an area of active research and has yielded a wide spectrum of cell types and signaling mechanisms that could be the source this inflammation. Considering the pro-inflammatory phenotype exhibited by senescent cells, they are a major contributor to inflammaging, however, age-related changes in innate immune signaling are also responsible for a large portion of this phenomenon.
Toll-like receptor (TLR) stimulation undergoes changes with age. In general with aging, TLRs are subject to more chronic stimulation, perhaps through increased leaking of microbial products from the microbiome, which has been demonstrated in older adults (36) and aged mice (37). Chronic stimulation of TLRs in both immune and non-immune cells will engage canonical cytokine signaling mechanisms through NF-κB, which is also known to be hyperactive with aging (38). Hyperresponsiveness to pathogen-associated molecular patterns (PAMPs) via commensal microbial products will impair the ability of both infected cells and cells of the innate immune system to appropriately respond to invading pathogens. Importantly, for reasons we will discuss in depth later, pathogen clearance is impaired with aging. This will result in lingering stimulation of TLRs which then leads to persistent cytokine secretion and enhanced recruitment of phagocytic cells. This initiates a vicious cycle of abundant cytokine production, which impairs the ability to resolve the infection and the return to homeostasis. While these mechanisms can impact the function of all innate immune cells, the effects of aging on antigen presenting cells (APCs) can exacerbate dysfunctions in the adaptive arm of the immune system as well.
When mounting a response to a pathogen in the respiratory tract, dendritic cells (DCs) are the key nexus of innate-adaptive immune crosstalk. Aside from participating in and being affected by increased tonic cytokine signaling, DCs perform an integral function in coordinating the activity of first line innate defenses and the robust response of T and B lymphocytes. While a number of mechanisms have been identified by which age affects DCs, it is still unclear how exactly the overall state of DC function changes with age.
Similar to other innate cell types, DCs from older adults have a propensity to secrete proinflammatory cytokines tonically in the airway (39). This chronic inflammation will leave an individual at higher risk for developing chronic conditions, as discussed earlier, but it will also disrupt the threshold of inflammation necessary to induce a robust immune response. Because the baseline levels of inflammation in the airway are increased, it will require higher levels of newly produced proinflammatory cytokines to create the acute spike of inflammation required to kickstart the infiltration of phagocytes and T cells to initiate pathogen control. One of the most important roles of the DC during an infection is their ability to upregulate MHC class II and costimulatory molecules in order to provide a strong signal 1 and 2, priming and costimulation, respectively, to CD4 T cells. This costimulation is typically delivered via enhanced surface expression of CD40 and CD80 which will bind to their ligands on the T cell within the context of the immune synapse. In mice, aging has been demonstrated to decrease expression of MHC II as well as costimulatory molecules upon activation (40, 41). However, there exists some cross-species discrepancy when DCs isolated from older adults are analyzed along the same lines: human DCs, even in advanced age, have not been observed to have these specific declines (42–44). It is likely that these older studies were unable to differentiate between the many DC subsets that have been identified and this particular question regarding the effects of age on DC presentation to and costimulation of T cells merits further examination.
The effect of cellular senescence on the function of other cells of the innate immune system with age remains unclear. It has been shown that macrophages can upregulate senescence markers (45) and recent work has linked senescent macrophages to the development of pulmonary fibrosis (46), the disease most intimately linked to cellular senescence. Type II pneumocytes have also been shown to take on a senescent phenotype and can be effectively cleared by ABT-263, a senolytic drug targeting SCAP pathway mediator Bcl-xl (47). Some of these studies utilized a radiation model of inducing senescence, which can be clinically relevant for studying the effects of certain cancer treatments on respiratory function. However, they may not fully capture the landscape of naturally accumulation of senescent cells with chronological age or exposure to common environmental stimuli. Airway fibroblasts, epithelial, and endothelial cells are the most common senescent cell types identified in the lungs (48–51). Because many of these cells are integral for the migration of dendritic cells between the airway and the lymphatics, increased senescent cell burden could be the explanation for the well documented inability of DCs to migrate appropriately with age (43). Increased expression of prostaglandins with age from lung endothelial cells (52) has been demonstrated to impair the lymph node migration of DCs in pulmonary infections (53) and it is possible that increased senescence in the endothelium may be responsible for this phenomenon. Typically senescence causes a change in cell morphology (1), which can disrupt the tight barriers found between cells in the airway among fibroblasts, epithelial, and endothelial cells. Although not yet directly studied, it is likely that senescence could increase permeability and leaking of signaling molecules across disparate tissues, causing dysfunction in cell-to-cell communication. There remains much to uncover in the relationship between cellular senescence and age-associated declines in the innate immune system, it is clear that increased senescent cell burden can influence the function and migration of these cells and alter their ability to respond effectively to a given pathogen.
2.2 The adaptive immune compartment
The effects of age on the adaptive arm of the immune system, namely T and B cells, have been extensively studied. These effects range from cell intrinsic changes as well as cell extrinsic environmental influences. The direct interactions between senescence and T and B cell function are rapidly evolving, drawing upon work by our group and many others currently focusing on this problem. Here, we aim to highlight some of the most important age-related changes to T and B cells during homeostasis and how they leave the host unprepared for a pathogen encounter.
2.2.1 B cells
B cells are affected by age across their developmental lifespan. Starting with their generation in the bone marrow, it has been appreciated for nearly 30 years that hematopoiesis of B cells declines with aging (54). Interestingly, this does not result in an overall decrease in frequency of B cells or any subset in the circulation of older adults (note that these types of human studies are limited to the periphery) (55). While the frequency of various peripheral B cell subsets may not be altered, B cells in older individuals remain ill equipped to respond robustly to an infection.
In order for B cell activation to occur, the B cell receptor (BCR) repertoire of naïve B cells must be diverse enough to capture a wide variety of epitopes to initiate a broad and effective antibody response. In recent years, the technology available to comprehensively study the diversity of these receptors, as well as those of T cells, has advanced considerably. Analyses of BCR repertoires taken from lymphoid tissue biopsies of older adults has revealed that blood and lymph node BCR repertoire diversity is markedly lower than that found in young adults (56).
Aside from repertoire diversity, B cells employ affinity maturation via somatic hypermutation (SHM) of immunoglobulin variable region genes to fine tune antibody avidity and thus enhance the ability of an antibody to bind to its specific antigen. This process is critical for generating antibodies that have potent neutralizing activity, especially when it comes to stopping the spread of viruses and other pathogens. This process is tightly regulated: upon recognition of cognate antigen by the BCR in a secondary lymphoid organ, along with help given by T follicular helper cells (Tfh), B cells can form germinal centers which are highly organized structures in which BCRs undergo affinity maturation and selection, resulting in antibodies with enhanced antigen binding capacity. SHM is the process that introduces mutations in immunoglobulin variable region genes to achieve this higher binding affinity. In order to perform SHM, B cells must upregulate activation-induced cytidine deaminase (AID) which actually executes the DNA modification (57). It has been observed that, with age, murine B cells are unable to robustly express this key enzyme, causing deficits in SHM and affinity maturation (58). This was also observed in B cells isolated from older adults (59). These enzymes are also involved with class-switch recombination (CSR), which is the means by which B cells which express one immunoglobulin isotype (such as IgM) can switch to express another isotype (such as IgG or IgE). As with SHM, this process is chiefly mediated by activity of AID and is similarly affected by age when AID expression is decreased upon activation. Thus, three key features of B cell responses are impacted by aging: repertoire diversity, affinity maturation, and isotype class switching. Taken together, this results in age-related reductions in humoral responses.
We have mentioned a number of instances where aging, perhaps exacerbated by accumulation of senescent cells, results in tissue disorganization and changes in morphology of critical structural cells. The same applies to the germinal center. The germinal center is composed to two regions: the dark zone, in which SMH and rapid proliferation occurs, and the light zone where B cells interact with antigen-presenting follicular DCs and follicular helper T cells and the highest affinity BCRs are selected for. B cells will cycle from the dark to light zones for a number of rounds until a high affinity BCR is generated. Because these mechanisms are disparately compartmentalized, any disruption in the boundaries can be disastrous. Surrounding the germinal centers in the spleen, the marginal zone is the protective barrier between the rapidly proliferating B cell follicle and the rest of the organ. Expression of cell adhesion molecules along the sinus of this compartment is decreased with age, causing disruptions in trafficking and sequestration of B cells and cells involved with the germinal center reaction (60). Work in our lab demonstrated that the delineation between the disparate B and T cell zones in the spleen is also compromised (61). Recently, a similar study found that stromal cells expressing cell adhesion molecules in the lymph nodes are dysfunctional with age and result in poor germinal center formation and function (62). It is possible these cells may be undergoing cellular senescence which may explain their altered expression levels of cell adhesion molecules. Senescence can alter the morphology of these stromal cells, which may compromise the architecture of these tightly regulated structures. Even if the stromal cells themselves do not become senescent, the altered functionality and basal inflammaging certainly would contribute to their dysfunction. Some reports have even concluded that B cells can take on a senescent phenotype themselves, including SASP factor production and an altered metabolic state (63, 64). Importantly, there remains much to be discovered in order to understand how B cells age, whether senescence can alter their functionality intrinsically, and how this relates to their decreased function with age.
One particular subset of B cells entirely unique to aging has been identified in both mice (65) and humans (66). These age associated B cells (ABCs) are generally proinflammatory and are a subject of great interest in recent years. Since their discovery, they have been most greatly associated with increased incidence of autoimmunity (66, 67) and chronic viral infections like HIV (68). ABCs are more innate-like and typically are activated via TLR signaling (typically TLR7 and 9) independent of their BCR and express the transcription factor Tbet, which is typically expressed by antiviral Th1 CD4 T cells (69). ABCs are often functional in the context of antiviral responses and are able to strongly activate T cells and generate highly specific antibodies in response to infections (70). These cells represent an altered functionality of immune aging as opposed to declines. Much work is needed to understand how these cells emerge and the various ways they contribute to immune responses.
2.2.2 T Cells
T cells are also affected by aging in a wide variety of cell intrinsic and extrinsic ways. These cells are primarily responsible for pathogen clearance, especially in dynamic mucosal sites like the respiratory system. Prior to an infection or vaccination, naïve CD4 and CD8 T cells circulate through the lymphatics awaiting activation from an APC, typically a DC. This activation requires DCs to have been adequately polarized to deliver appropriate costimulation to the T cell, a key factor we have already discussed as being less effective with age. Increased inflammaging and circulating SASP from nearby senescent cells will also disrupt “signal 3” of T cell activation, the cytokine microenvironment. After activation, most T cells will respond to the infection and then die while a small percentage will become long lived memory cells that can respond better and faster upon a subsequent encounter with the same antigen. From the start of activation, aged T cells are at a disadvantage for responding appropriately. However, there are notable intrinsic defects that are also important to note.
Similar to the B cell compartment, naïve T cell output sharply declines throughout life, reaching its apex around puberty and decreasing thereafter, occurring concomitantly with thymic involution (71). By age 75, nearly all naïve T cell output is halted entirely (72). The diversity of the T cell receptor (TCR) also undergoes rapid changes with age, with some studies characterizing a general shrinking of the overall diversity, as reviewed elsewhere (72). Many of the early studies relied on flow cytometric analysis of various T cell receptor isoforms. While this approach was capable of detecting broad differences with age, it was not able to comprehensively describe the antigen specificities affected by these differences. Other studies have found that the overall diversity of human TCRs are altered but not in terms of diversity, concluding that older adults can retain highly diverse repertoires (73). These authors suggest that the common finding of reduced diversity may be influenced by perturbation in relative abundance of certain clones, which can be delineated using deeper sequencing methods. In recent years, sophisticated sequencing technologies have begun to tease apart more detailed and nuanced differences revealing that CD4 T cells tend to increase the frequency of shared specificities with age (74). This indicates that aged CD4 T cells are predisposed to respond to a more limited pool of antigens. Interestingly, this phenomenon was not observed in the CD8 compartment. These studies reveal that there may be more nuances to how aging impacts certain aspects of TCR diversity, which have yet to be fully connected to effects on the functionality of responses in older populations.
While age limits the capability of T cells to recognize antigen, it also affects the functionality of these cells upon their activation. We first turn to the CD4 compartment which is indispensable in offering “help” in the form of cytokines to CD8 T cells and B cells. CD4 T cells recognize antigen in the context of MHC class II expressed on DCs (and other APCs) and then they differentiate into T helper subsets depending on the cytokine environment. The goal of this differentiation is to generate T helper subsets that are best suited to combating certain classes of pathogens (for example: Th1 for viral infections and Th2 for parasitic infections). In the absence of infection, age causes dramatic shifts in CD4 subset activity during homeostasis. As mentioned previously, circulating CD4 T cells in older adults take on a pro-inflammatory IL-17-producing Th17 phenotype (24, 75). Th17 cells can be pathogenic in many autoimmune diseases and studies have linked this Th17 induction with osteoporosis in mice and humans (76, 77) and mouse models of colitis (78). Interestingly, recent evidence has emerged pointing towards a role for senescent cells in driving Th17 activity. In a model of osteoarthritis, a study demonstrated that increased senescent cell burden caused naïve CD4 T cells to skew towards a Th17 phenotype which then increased senescent cell burden in a feedforward loop (79). This work raises some important questions, including whether this mechanism can be applicable in other situations. If so, it may provide interesting therapeutic strategies to diminish the effects of senescent cells and rescue CD4 T cell subset balance in the absence of infection. This also suggests that treatment with IL-17 inhibitors may mimic the effects of senolytics, which are drugs that clear senescent cells from the body. Future studies elucidating these points may prove fruitful as the field searches for interventions that can rejuvenate T cell function with age.
Regulatory T cells (Treg), expressing the transcription factor FoxP3, also undergo complex changes with age, perhaps exacerbated by SASP. Canonically, Tregs are important for homeostasis by enforcing tolerance to self-antigens and preventing improperly robust responses to innocuous antigen (80). A subset of Tregs develop naturally from the thymus (nTregs) and are thus affected by thymic involution and declines in T cell production like their conventional T cell counterparts. In mouse models, it has been demonstrated that Treg production actually declines faster and more severely than conventional T cells, perhaps via suppressive activity of peripheral Tregs migrating back to the thymus (81). Aside from age-related changes in production, Treg activity is also altered with age. Functional changes in Tregs are very heterogenous and still require much work to fully understand. The controversies are well reviewed elsewhere (82), but the consensus is that aged Tregs are more suppressive and preferentially secrete IL-10 over TGF-β, which is preferred by young Tregs. This creates a rather complicated landscape balancing between being intensely regulatory while remaining systemically pro-inflammatory. Future studies should focus on the function of Tregs in various tissue niches with age, which may reveal more heterogeneity and may explain why the systemic view remains pro-inflammatory generally.
CD8 T cells likewise are deleteriously affected by aging. Decreased frequency of naïve cells (83) and narrowing of the TCR repertoire (84) have been known for many years now. During homeostasis, CD8 T cells undergo functional changes similar to CD4s. Especially in humans, peripheral CD8 T cells take on a suppressive phenotype resembling that of Foxp3+ CD4 Tregs (85). Because many of the age-related changes to CD8s are in the context of how they respond to pathogens, we will further discuss the effects of age on the function of CD8s in the next section.
A key feature of immune aging, especially in humans, is the inflation of the memory CD8 T cell compartment. Over time, virus specific CD8 memory cells predominate among total T cells in the periphery at homeostasis in older adults (86). In the past decade, much work has been done to categorize memory T cell subsets on the basis of surface marker expression and trafficking pattern. They can be broadly characterized as T central memory (Tcm), T effector memory (Tem), tissue resident memory T cells (Trm), and peripheral memory T cells (Tpm) (87). The expansion of memory T cells usually is found in the Tem compartment and has been mostly attributed to antigen stimulation by latent viral infections like cytomegalovirus (CMV) (88). In humans, these cells are typically described as Tem cells expressing CD45RA (TEMRA) (89). However these effects are not unique to CMV and they have also been linked to other common herpesviruses (90). These cells should not be conflated with exhausted T cells that can accumulate with chronic infections that result from consistently high levels of antigen presentation. Aged memory-inflated cells do not seem to follow an exhausted phenotype as defined by inhibitory receptor expression. TEMRA cells do not highly express PD-1 and they retain their effector function upon reactivation (91). Distinct from TEMRA cells, a recent report has described a novel subset of memory CD8 T cells that are associated with age (92). Using single cell RNA sequencing approaches, this novel subset, termed age associated T cells (Taa), uniquely express granzyme K which was associated with higher SASP factor secretion. They also found that the GzmK+ Taa cells expressed common exhaustion markers such as PD-1 and the transcription factor Tox. In contrast with many previous studies, this study did not observe increases in the TEMRA population. This reveals an emerging link between senescent cells and the exacerbation of immune aging which, with further work, may provide a means to rejuvenate some T cell responses with age.
2.2.3 Immune-structural cell interactions
Structural cells like fibroblasts, endothelial and epithelial cells are likely the senescent cells present in the lung. Evidence for immune cell senescence remains controversial and dependent on the definitions of senescence. Some groups have identified subsets of senescent T cells in humans (93) while there is stronger evidence for senescent B cells, reviewed elsewhere (94). Interestingly, fewer studies have identified senescent T and B cells in mice, perhaps due to lack of consistent pathogen encounter. Senescent structural cells create two problems: 1) SASP production altering the tissue microenvironment and 2) morphological and functional changes in the structural cells. We’ve discussed at length the possible downstream effects of SASP production on the function of immune cells during homeostasis and will continue to touch upon how it affects responses to pathogens. Generally, proinflammatory SASP production can cause dysregulated responses in nearby immune cells simply by tonic cytokine signaling in the absence of an infection. When cells engage the senescence pathway, their morphology changes (95). When considering cells that maintain tight barriers in tissues like epithelial and endothelial cells, senescent morphological changes could result in loss of barrier integrity. While this has not yet been studied, it is likely that permeability in these barriers, especially endothelial barriers will result in improper trafficking of immune cells into the tissue and exacerbation of immunopathology. Future work should aim to address crosstalk between immune and structural cells and how senescence, via the two effects mentioned, may cause dysfunction in these communication networks. Certainly, in the context on an infection, all of these age and senescence-associated changes can prove to be deleterious during an infection.
2.3 The aged response to respiratory pathogens
In prior sections, we have discussed some of the myriad of ways age affects the homeostatic functions that are relevant for initiating and fostering immune responses to respiratory infections. Now we consider how these changes, including cellular senescence, integrate to leave older adults vulnerable to adverse outcomes following an infection. Influenza (flu) infection is of particular interest to our group and many others because it is among the greatest threats to older adults, who make up between 70 and 85% of flu related deaths (96). This includes individuals who succumb to secondary bacterial pneumonias, which we will also discuss. Many of these concepts also apply to SARS-CoV-2 infection and COVID-19 outcomes as well, which is well reviewed by others (97).
2.3.1 The aged response to influenza infection
Work done by our group and many others has revealed a striking effect of age on outcomes of flu infection. This is particularly well characterized in mouse models, which have demonstrated that aged mice lose more body mass and are substantially delayed in the clearance of a sublethal flu infection when compared to young (98). There are numerous factors, many of which have been mentioned earlier, contributing to this deficit. The flu virion infects lung epithelial cells with high specificity (99). While not confirmed in the context of natural aging in vivo, epithelial cells are known to undergo senescence in response to radiation (100). It also has been suggested that viral infection itself can induce senescence: a recent study characterized a SASP phenotype that was pathogenic in the progression of COVID-19 and increased immunopathology (101). It is therefore possible that senescence can also be induced by flu infection.
The heightened inflammation with aging and SASP results in the aberrant function of many cell types already discussed. Resident alveolar macrophages are among the first responders to any respiratory pathogen, including flu. These cells are developed very early on in gestation and populate the lungs by responding to cytokines secreted by fetal lung epithelial cells (102, 103). With age, these cells begin to be replenished by bone marrow-derived monocytes rather than by self-renewal (104). A recent study in mice (105) examined the consequences of this in the context of flu infection and found that while fetal derived alveolar macrophages are able to self-renew effectively over time, they are outcompeted by monocyte-derived counterparts that are more glycolytic and proliferative. The monocyte-derived alveolar macrophages are in a more proinflammatory state during flu infection and cause immunopathology and prolong the course of infection, delaying adequate healing. While beyond the scope of this study, the consequences are likely not limited to a primary infection. If the wound healing phase of the antiviral response is limited, the generation of T and B cell memory is likely affected as well. Memory T cell differentiation is heavily reliant on TGF-β, a key molecule for effective wound healing (106). Therefore, this heightened inflammatory state can have downstream consequences outside of the activity of alveolar macrophages. As with alveolar macrophages, DCs are an integral part of mounting a robust flu response and are also affected by alterations in inflammatory states.
In order to initiate a robust antiviral response, type I interferons (IFN-α and -β) are secreted by infected cells as well as dendritic cells and macrophages/monocytes in response to exposure to viral components (107). Type I IFN signaling activates APCs and allows for effective antigen presentation to T cells. Importantly, it has been shown that in older adults, monocytes express less of the type I IFN signaling machinery (108) and that DCs do not effectively express costimulatory molecules (40, 41). Both of these factors could then contribute to reduced T cell priming and effective viral clearance (109).
When responding to an infection, CD4 T cells are critically important for providing help to CD8 T cells and B cells as well as mediating cytotoxic activity directly (110). A hallmark of CD4 T cells is their ability to differentiate into specialized T helper subsets that are more effective at directing responses towards specific classes of pathogens. We have discussed the changes during homeostasis in basal CD4 subset balance and now we turn to the various deficits in CD4 subsets during an aged response to flu infection. This topic has been a focus of our group for many years now and evidence is emerging that the senescent environment is a driving force in causing these deficits.
Work done by our group demonstrated that, in mice, some of the age-related deficits in CD4 T cell function during infection are cell extrinsic and thus regulated by the aged microenvironment (111). Transfer of young CD4 T cells into aged hosts results in reduced trafficking into secondary lymphoid organs, reduced efficacy of priming by APCs, and reduced differentiation in to Bcl-6-expressing T follicular helper (Tfh) subsets. Less robust Tfh differentiation will deleteriously affect the formation and function of germinal centers and coordination of antibody-mediated responses. While these experiments were performed a few years before cellular senescence was being implicated in chronic pathologies of aging like IPF, osteoporosis, Alzheimer’s, and cardiovascular disease (9, 112–114), A theory is emerging in which senescent cells may be at the root of the aged microenvironment that was driving deficits in CD4 function during infection (Figure 1).
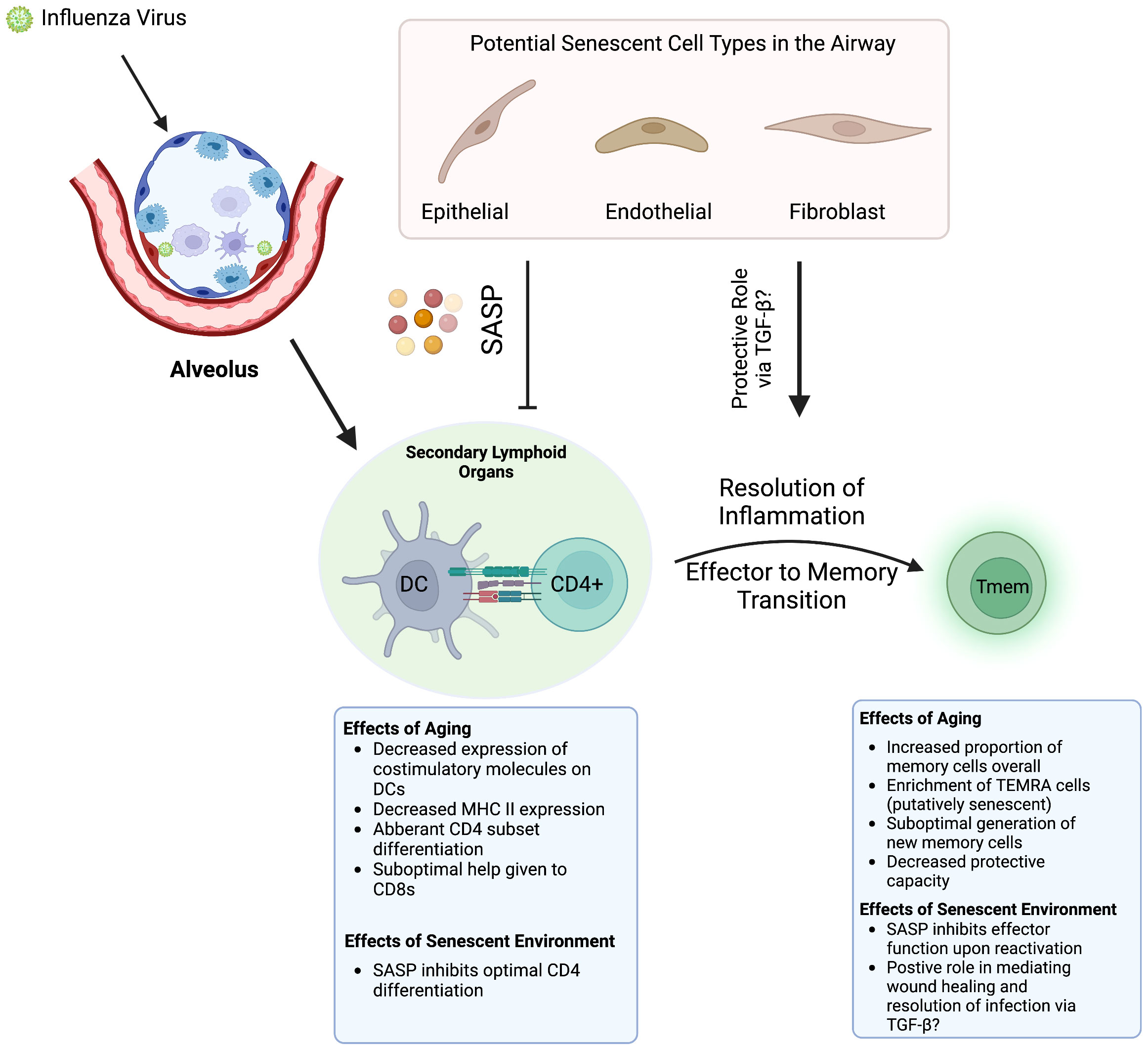
Figure 1 How Aging and Senescence Regulate Antiviral T Cell Responses. When a flu infection occurs, APCs such as DCs present antigen to naïve CD4 T cells, which requires costiumulation and a conducive cytokine milieu. Aging results in various defects in this process and senescent cells alter the milieu by secreting SASP factors. These perturbations result in suboptimal responses in both the CD4 and CD8 T cell compartments. Whenever the pathogen is cleared, the return to homeostasis and the effector to memory transition is also affected. Especially in the CD8 compartment, memory cells generated following infection are also deleteriously affected by aging. It is possible that SASP may play a role in regulating memory differentiation via its wound healing activities and secretion of TGF-β. Created with BioRender.com.
CD8 T cells are also extremely important for mediating an effective antiviral response to flu however the ways in which CD8s are affected by age are more nuanced. In general, deficits in aged CD8 function during flu infection have been mostly attributed to decreased TCR repertoire diversity (84), the function of APCs (115), and increased proportion of Tregs in the CD4 compartment (116), all from mechanistic mouse studies. This would explain why transferring young CD8 T cells into aged hosts still results in deficits during an infection but when isolated, aged CD8s can function as well as their young counterparts in vitro (115). However, in light of reports pointing towards the accumulation of various dysfunctional age-associated CD8 subsets in humans like GzmK-expressing Taa and virtual memory CD8s, which do show some signs of cellular senescence (117), there remains much more to be done to understand how the CD8 compartment is affected directly by age across species and what can be done to potentiate their responses independent of other cells.
There is also an emerging role for nonconventional immune cell types and how they can affect immune responses with age. Innate lymphoid cells (ILCs) derive developmentally from the same common lymphoid progenitor cell as B and T cells but lack adaptive antigen receptors and function similar to CD4 subsets in secreting type I, II, or III cytokines and expressing Tbet, GATA3, or RORγT, respectively (118). Type II ILCs (ILC2s), have been linked most strongly to regulating responses in the lungs, especially viral infection as reviewed elsewhere (119). Studies have shown that following flu infection, ILC2 secretion of IL-33 causes an asthma-like phenotype in young mice (120). Other studies have similarly found that ILC2s can drive a fibrotic phenotype following pathogen challenge in mice and observed increased ILC2 frequency in lungs of human idiopathic fibrosis patients (121). In this sense, ILC2s mirror senescent cells in exacerbating pro-fibrotic phenotypes. A more recent study in aged mice revealed a broad heterogeneity in the ILC2 compartment in the lungs. Aging was associated with decreased overall frequency of ILC2s in the lungs and were more transcriptionally diverse than young ILC2s. Interestingly, transfer of young ILC2s into aged mice prior to influenza infection significantly enhanced protection against morbidity and mortality (122). The implication of this study has yet to be fully recapitulated in humans, but it is likely, as with most cell types discussed, that aging results in nuanced changes in ILC2 function that results in an overall decline in functionality following an infection.
A dysfunctional B cell response is another hallmark of an aged response to flu infection. Most of these changes are not flu-specific and are a result of the various homeostatic alterations to antibody production that we discussed previously. Importantly, most of these deficits have consequences following a subsequent encounter either following a primary infection or a vaccine response, which is the focus of the next section.
Taken together, these factors contribute to delayed viral clearance and could have serious effects on tissue repair, exacerbation of immunopathology and failure to return to homeostasis (98). For example, this is clearly illustrated by the various inflammation-driven adverse outcomes following COVID-19 infection in older adults (123). This failure is also one of the factors involved in the enhanced susceptibility of older adults to develop secondary bacterial pneumonia following an influenza infection. The mechanisms of susceptibility to bacterial infection are well reviewed elsewhere (124), but the main contributors are damage to the lung epithelium following flu infection as well as dysfunctional cytokine signaling by innate cells. Although we have mentioned various ways innate cells in the lung are affected by age, there have been few studies connecting flu infection, bacterial pneumonia, and the effects of age together. One study, utilizing a Pseudomonas aeruginosa pneumonia model demonstrated that aged mice, similar to what is overserved for older adults, were more severely affected by the bacterial infection after flu challenge (125). Clearly, more work is needed in this clinically important area.
3 Aged vaccine/memory responses
One goal of the generation of immunological memory is to reduce the pathology associated with a subsequent respiratory infection and limit lung damage. Importantly, apart from the primary response to infection, aging also diminishes the capacity of T and B cells to form durable and protective memory following either an infection or a vaccination. This is made evident by the fact that in spite of widespread and successful vaccination campaigns among older adults, they remain the most vulnerable group in terms of morbidity and mortality following infection. When considering how age is affecting the ability of older adults to effectively respond to an infection, immune memory is a key factor to consider, especially in the context of flu or COVID-19.
A key driver of dysfunctional memory responses in T cells, especially in older adults, is the reality of chronic herpesvirus infections such as cytomegalovirus, Epstein-Barr virus, and varicella (126). Estimates have ranged as high as 90% of individuals over 80 years age in developed countries are CMV positive (127). Although we have discussed how CMV status can affect memory cell populations over time, the consequences of this in terms of vaccination and infection is controversial. CMV-induced accumulation of CD4 memory T cells expressing a TEMRA phenotype was correlated with poor antibody production following flu vaccination in older adults (128). This highlights the central role of T cells in coordinating effective B cell responses to vaccination, which are also well reviewed elsewhere (129). A similar study found that CMV positive older adults who failed to respond to vaccination had higher expression of TNF-α and IL-6 (130). Conversely, a similarly structured study has found that CMV positivity was associated with heightened immune responses to vaccination in young individuals while CMV status did not correspond to any differences in the response among older participants (131). Mouse studies have found that while CMV diminishes the frequency of CD8 T cells responding to influenza, overall mortality is not affected (132, 133). There remains much to be understood regarding the role of CMV status in altering responses to infections or vaccinations, especially in humans. This may reveal some of the great heterogeneity seen with older populations (134) and some discrepancy could be attributed to differences in the cohorts utilized. and more studies should be performed to build consensus on this controversial but certainly relevant aspect of immune aging.
While CMV status may affect flu vaccine responses with age, there are many other factors that also contribute. CD4 T cell subsets, as with response to infection, are also deleteriously affected following vaccination. Work in our lab utilizing a crossprotective vaccine model against the internal flu nucleoprotein (NP) demonstrated that while vaccination improves excessive lung infiltration of innate inflammatory mediators, it fails to improve differentiation of CD4 T cells towards an antiviral Th1 polarized state in aged mice (135). This study also implicated a role for aged CD4 T cells in the susceptibility to secondary bacterial pneumonia since aged vaccinated mice remained severely vulnerable to mortality when coinfected with Streptococcus pneumoniae.
The effects of age on CD8 responses to flu vaccinations, especially in older adults, have been comprehensively reviewed elsewhere (136). Notably, the most reliable correlate of protection following flu vaccination in older adults can be found in the T cell compartment (137) and especially in the ability to generate IFN-γ preferentially over IL-10 upon stimulation (138). This observation is important for two reasons: (1) it demonstrates an easy and reliable correlate of protection that can be used to assess any proposed intervention, and (2) it demonstrates that interventions that target T cell responses may be the most beneficial for older adults.
Aging results in changes in the generation of memory T cells and we have shown that the ability of T cells from aged mice to generate fully functional memory T cells is significantly impacted (139). Furthermore, a recent study in mice demonstrated that increased establishment of flu-specific tissue resident T cells (Trms) following flu infection resulted in nonresolving immunopathology in the lungs of aged mice (140). Interestingly, this was linked to increased TGF-β (a known SASP factor) levels in the lung (8). While too much TGF-β may exacerbate this pathology, it is certainly required to form protective Trm cells. Thus, further work should elucidate how the dynamics of TGF-β signaling differ between young versus aged and how those differences result in either protective or pathogenic memory cell differentiation. In addition, future studies should also focus on how senescence impacts the generation of protective immune memory, including changes to the microenvironment that support the appropriate differentiation of memory T cells.
4 Role of senolytics in potentiating aged immune responses
Throughout this review, we have mentioned the various ways that senescent cells may influence the function of the immune system in response to infection and vaccination. As senescence emerges as a key driver of diseases of aging, we now have the tools to assess the impact of senescence on these age-related changes. One of the most effective tools is treatment with senolytics, which are drugs that can selectively target and induce apoptosis in senescent cells (141). The first senolytic treatment described was a combination treatment with dasatinib and quercetin (141) and is the most widely characterized. Others have also been identified including fisetin (142) and navitoclax (143). Generally, these drugs inhibit the anti-apoptotic pathways engaged by senescent cells thus inducing apoptosis. While these drugs have been shown in mouse studies to increase measures of healthspan and extend longevity (142, 144), very few studies have utilized this approach to ameliorate immune responses with age.
Recently, studies from our lab and others have begun to apply senolytic strategies to overcome age-related deficits during an infection. Our work with flu infection demonstrated that treatment of aged mice with D+Q decreased the proportion of Tregs in the lungs during infection in favor of a more healing GATA-3 expressing Th2 T cell population by mitigating the senescent environment (145). Strikingly, D+Q treatment completely abrogated TGF-β production in the lung following infection. Similar studies utilizing a model of nonspecific polymicrobial exposure concluded that treatment with fisetin in aged mice significantly protected them from mortality following microbial exposure via pet-store mice or their dirty bedding (146). While this study clearly implicates senescent cells play some role in age-related immune deficits to infection, more specialized experiments are needed to reveal exactly which arms of the immune system are sensitive to senolytic treatment.
Although the nuances of senolytics and immune responses have yet to be fully characterized in mechanistic pre-clinical animal studies, currently there are a number of clinical trials administering senolytics to older adults. D+Q has been demonstrated to be effective in clearing human senescent cells in vivo and improved symptoms of IPF in older adults (11, 147). Fisetin is being investigated in an ongoing trial to improve COVID-19 disease severity among nursing home residents (NCT04537299). The evidence that senescent cells are solely deleterious in modulating immune responses is far from clear. Some components of SASP, like TGF- β, can have consequences for the formation of memory T cells as previously discussed (106). The important role of senescent cells in mediating wound healing and the return to homeostasis cannot be ignored either (3). As the reports begin to emerge from these clinical trials, it is likely that the role of senescent cells in immune responses may be more complicated than previously appreciated.
5 Discussion
It is clear that aging has a significant impact on immune function, which can be detrimental when attempting to fight a respiratory infection. The root causes of these age-related changes are still not clear, but the accumulation of senescent cells likely plays a role. In the past, it has been quite difficult to identify truly senescent cells in vivo since this requires a preponderance of evidence to definitively determine that a cell is truly senescent. As mentioned earlier, this typically involves expression of senescence enforcement molecules p16 and/or p21, SA-β-Gal, SASP factors, and more. Nevertheless, new tools including transgenic mouse models (3, 148, 149) and the use of senolytic drugs (144) have recently enabled researchers to manipulate senescent cells in vivo. These tools will enable researchers to further investigate the role that senescent cells play in the lungs both at homeostasis and during an immune response. These studies will have the potential to move the field forward substantially and could result in novel approaches to improving immunity in older adults.
Author contributions
BLT and LH both wrote, edited, and approved the final version of this manuscript.
Funding
LH is supported by NIH grants P30AG067988 and R21AG071292. BLT is supported by NIH grant R21AG071292.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Campisi J, d'Adda di Fagagna F. Cellular senescence: When bad things happen to good cells. Nat Rev Mol Cell Biol (2007) 8(9):729–40. doi: 10.1038/nrm2233
2. Wyld L, Bellantuono I, Tchkonia T, Morgan J, Turner O, Foss F, et al. Senescence and cancer: A review of clinical implications of senescence and senotherapies. Cancers (Basel) (2020) 12(8):2134. doi: 10.3390/cancers12082134
3. Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. An essential role for senescent cells in optimal wound healing through secretion of pdgf-aa. Dev Cell (2014) 31(6):722–33. doi: 10.1016/j.devcel.2014.11.012
4. Khosla S, Farr JN, Tchkonia T, Kirkland JL. The role of cellular senescence in ageing and endocrine disease. Nat Rev Endocrinol (2020) 16(5):263–75. doi: 10.1038/s41574-020-0335-y
5. Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. Involvement of the cyclin-dependent kinase inhibitor P16 (Ink4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci USA (1996) 93(24):13742–7. doi: 10.1073/pnas.93.24.13742
6. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by P53 restoration in murine liver carcinomas. Nature (2007) 445(7128):656–60. doi: 10.1038/nature05529
7. Hu L, Li H, Zi M, Li W, Liu J, Yang Y, et al. Why senescent cells are resistant to apoptosis: An insight for senolytic development. Front Cell Dev Biol (2022) 10:822816. doi: 10.3389/fcell.2022.822816
8. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol (2013) 15(8):978–90. doi: 10.1038/ncb2784
9. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun (2017) 8:14532. doi: 10.1038/ncomms14532
10. Kellogg DL, Musi N, Nambiar AM. Cellular senescence in idiopathic pulmonary fibrosis. Curr Mol Biol Rep (2021) 7(3):31–40. doi: 10.1007/s40610-021-00145-4
11. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-Human, open-label, pilot study. EBioMedicine (2019) 40:554–63. doi: 10.1016/j.ebiom.2018.12.052
12. Brandsma CA, de Vries M, Costa R, Woldhuis RR, Königshoff M, Timens W. Lung ageing and copd: Is there a role for ageing in abnormal tissue repair? Eur Respir Rev (2017) 26(146):170073. doi: 10.1183/16000617.0073-2017
13. Ito K, Barnes PJ. Copd as a disease of accelerated lung aging. Chest (2009) 135(1):173–80. doi: 10.1378/chest.08-1419
14. Silverman EK, Chapman HA, Drazen JM, Weiss ST, Rosner B, Campbell EJ, et al. Genetic epidemiology of severe, early-onset chronic obstructive pulmonary disease. risk to relatives for airflow obstruction and chronic bronchitis. Am J Respir Crit Care Med (1998) 157(6 Pt 1):1770–8. doi: 10.1164/ajrccm.157.6.9706014
15. Chung KF. Cytokines in chronic obstructive pulmonary disease. Eur Respir J Suppl (2001) 34:50s–9s. doi: 10.1183/09031936.01.00229701
16. Donaldson GC, Seemungal TA, Patel IS, Bhowmik A, Wilkinson TM, Hurst JR, et al. Airway and systemic inflammation and decline in lung function in patients with copd. Chest (2005) 128(4):1995–2004. doi: 10.1378/chest.128.4.1995
17. Kiszałkiewicz JM, Majewski S, Piotrowski WJ, Górski P, Pastuszak-Lewandoska D, Migdalska-Sęk M, et al. Evaluation of selected Il6/Stat3 pathway molecules and mirna expression in chronic obstructive pulmonary disease. Sci Rep (2021) 11(1):22756. doi: 10.1038/s41598-021-01950-8
18. Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic ras and the P53 tumor suppressor. PloS Biol (2008) 6(12):2853–68. doi: 10.1371/journal.pbio.0060301
19. Rashid K, Sundar IK, Gerloff J, Li D, Rahman I. Lung cellular senescence is independent of aging in a mouse model of Copd/Emphysema. Sci Rep (2018) 8(1):9023. doi: 10.1038/s41598-018-27209-3
20. Woldhuis RR, de Vries M, Timens W, van den Berge M, Demaria M, Oliver BGG, et al. Link between increased cellular senescence and extracellular matrix changes in copd. Am J Physiol Lung Cell Mol Physiol (2020) 319(1):L48–60. doi: 10.1152/ajplung.00028.2020
21. Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol (2009) 4:435–59. doi: 10.1146/annurev.pathol.4.110807.092145
22. Togo S, Holz O, Liu X, Sugiura H, Kamio K, Wang X, et al. Lung fibroblast repair functions in patients with chronic obstructive pulmonary disease are altered by multiple mechanisms. Am J Respir Crit Care Med (2008) 178(3):248–60. doi: 10.1164/rccm.200706-929OC
23. Wilkinson HN, Hardman MJ. Senescence in wound repair: Emerging strategies to target chronic healing wounds. Front Cell Dev Biol (2020) 8:773. doi: 10.3389/fcell.2020.00773
24. Schmitt V, Rink L, Uciechowski P. The Th17/Treg balance is disturbed during aging. Exp Gerontol (2013) 48(12):1379–86. doi: 10.1016/j.exger.2013.09.003
25. Silva LEF, Lourenço JD, Silva KR, Santana FPR, Kohler JB, Moreira AR, et al. Th17/Treg imbalance in copd development: Suppressors of cytokine signaling and signal transducers and activators of transcription proteins. Sci Rep (2020) 10(1):15287. doi: 10.1038/s41598-020-72305-y
26. Provisional covid-19 death counts by age in years 2020-2022. In: National center for health statistics. Available at: https://data.cdc.gov/d/3apk-4u4f.
27. Estimated influenza disease burden, by season — united states, 2010-11 through 2019-20 influenza seasons. In: Centers for disease control and prevention , national center for immunization and respiratory diseases (NCIRD). Available at: https://www.cdc.gov/flu/about/burden/past-seasons.html.
28. Salminen A, Kaarniranta K, Kauppinen A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging (2012) 4(3):166–75. doi: 10.18632/aging.100444
29. Frasca D, Blomberg BB. Inflammaging decreases adaptive and innate immune responses in mice and humans. Biogerontology (2016) 17(1):7–19. doi: 10.1007/s10522-015-9578-8
30. Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci (2000) 908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x
31. Chang R, Yee KL, Sumbria RK. Tumor necrosis factor α inhibition for alzheimer's disease. J Cent Nerv Syst Dis (2017) 9:1179573517709278. doi: 10.1177/1179573517709278
32. Prattichizzo F, De Nigris V, Spiga R, Mancuso E, La Sala L, Antonicelli R, et al. Inflammageing and metaflammation: The yin and yang of type 2 diabetes. Ageing Res Rev (2018) 41:1–17. doi: 10.1016/j.arr.2017.10.003
33. Rezuș E, Cardoneanu A, Burlui A, Luca A, Codreanu C, Tamba BI, et al. The link between inflammaging and degenerative joint diseases. Int J Mol Sci (2019) 20(3):614. doi: 10.3390/ijms20030614
34. Whiting CC, Siebert J, Newman AM, Du H-W, Alizadeh AA, Goronzy J, et al. Large-Scale and comprehensive immune profiling and functional analysis of normal human aging. PloS One (2015) 10(7):e0133627. doi: 10.1371/journal.pone.0133627
35. Sayed N, Huang Y, Nguyen K, Krejciova-Rajaniemi Z, Grawe AP, Gao T, et al. An inflammatory aging clock (Iage) based on deep learning tracks multimorbidity, immunosenescence, frailty and cardiovascular aging. Nat Aging (2021) 1(7):598–615. doi: 10.1038/s43587-021-00082-y
36. Claesson MJ, Jeffery IB, Conde S, Power SE, O'Connor EM, Cusack S, et al. Gut microbiota composition correlates with diet and health in the elderly. Nature (2012) 488(7410):178–84. doi: 10.1038/nature11319
37. Conley MN, Wong CP, Duyck KM, Hord N, Ho E, Sharpton TJ. Aging and serum mcp-1 are associated with gut microbiome composition in a murine model. PeerJ (2016) 4:e1854. doi: 10.7717/peerj.1854
38. Tilstra JS, Clauson CL, Niedernhofer LJ, Robbins PD. Nf-κb in aging and disease. Aging Dis (2011) 2(6):449–65.
39. Prakash S, Agrawal S, Vahed H, Ngyuen M, BenMohamed L, BenMohamad L, et al. Dendritic cells from aged subjects contribute to chronic airway inflammation by activating bronchial epithelial cells under steady state. Mucosal Immunol (2014) 7(6):1386–94. doi: 10.1038/mi.2014.28
40. Li G, Smithey MJ, Rudd BD, Nikolich-Žugich J. Age-associated alterations in Cd8α+ dendritic cells impair Cd8 T-cell expansion in response to an intracellular bacterium. Aging Cell (2012) 11(6):968–77. doi: 10.1111/j.1474-9726.2012.00867.x
41. Zacca ER, Crespo MI, Acland RP, Roselli E, Núñez NG, Maccioni M, et al. Aging impairs the ability of conventional dendritic cells to cross-prime Cd8+ T cells upon stimulation with a Tlr7 ligand. PloS One (2015) 10(10):e0140672. doi: 10.1371/journal.pone.0140672
42. Steger MM, Maczek C, Grubeck-Loebenstein B. Morphologically and functionally intact dendritic cells can be derived from the peripheral blood of aged individuals. Clin Exp Immunol (1996) 105(3):544–50. doi: 10.1046/j.1365-2249.1996.d01-790.x
43. Agrawal A, Agrawal S, Cao JN, Su H, Osann K, Gupta S. Altered innate immune functioning of dendritic cells in elderly humans: A role of phosphoinositide 3-Kinase-Signaling pathway. J Immunol (2007) 178(11):6912–22. doi: 10.4049/jimmunol.178.11.6912
44. Lung TL, Saurwein-Teissl M, Parson W, Schönitzer D, Grubeck-Loebenstein B. Unimpaired dendritic cells can be derived from monocytes in old age and can mobilize residual function in senescent T cells. Vaccine (2000) 18(16):1606–12. doi: 10.1016/s0264-410x(99)00494-6
45. Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, et al. Aging of mice is associated with P16(Ink4a)- and β-Galactosidase-Positive macrophage accumulation that can be induced in young mice by senescent cells. Aging (Albany NY) (2016) 8(7):1294–315. doi: 10.18632/aging.100991
46. Su L, Dong Y, Wang Y, Guan B, Lu Y, Wu J, et al. Potential role of senescent macrophages in radiation-induced pulmonary fibrosis. Cell Death Dis (2021) 12(6):527. doi: 10.1038/s41419-021-03811-8
47. Pan J, Li D, Xu Y, Zhang J, Wang Y, Chen M, et al. Inhibition of bcl-2/Xl with abt-263 selectively kills senescent type ii pneumocytes and reverses persistent pulmonary fibrosis induced by ionizing radiation in mice. Int J Radiat Oncol Biol Phys (2017) 99(2):353–61. doi: 10.1016/j.ijrobp.2017.02.216
48. Lin Y, Xu Z. Fibroblast senescence in idiopathic pulmonary fibrosis. Front Cell Dev Biol (2020) 8:593283. doi: 10.3389/fcell.2020.593283
49. Álvarez D, Cárdenes N, Sellarés J, Bueno M, Corey C, Hanumanthu VS, et al. Ipf lung fibroblasts have a senescent phenotype. Am J Physiol Lung Cell Mol Physiol (2017) 313:L1164 – L1173. doi: 10.1152/ajplung.00220.2017
50. Papadopoulou A, Kletsas D. Human lung fibroblasts prematurely senescent after exposure to ionizing radiation enhance the growth of malignant lung epithelial cells in vitro and in vivo. Int J Oncol (2011) 39(4):989–99. doi: 10.3892/ijo.2011.1132
51. von Kobbe C. Targeting senescent cells: Approaches, opportunities, challenges. Aging (Albany NY) (2019) 11(24):12844–61. doi: 10.18632/aging.102557
52. Qian H, Luo N, Chi Y. Aging-shifted prostaglandin profile in endothelium as a factor in cardiovascular disorders. J Aging Res (2012) 2012:121390. doi: 10.1155/2012/121390
53. Zhao J, Legge K, Perlman S. Age-related increases in Pgd(2) expression impair respiratory dc migration, resulting in diminished T cell responses upon respiratory virus infection in mice. J Clin Invest (2011) 121(12):4921–30. doi: 10.1172/JCI59777
54. Zharhary D. Age-related changes in the capability of the bone marrow to generate b cells. J Immunol (1988) 141(6):1863–9.
55. Blanco E, Pérez-Andrés M, Arriba-Méndez S, Contreras-Sanfeliciano T, Criado I, Pelak O, et al. Age-associated distribution of normal b-cell and plasma cell subsets in peripheral blood. J Allergy Clin Immunol (2018) 141(6):2208–19.e16. doi: 10.1016/j.jaci.2018.02.017
56. Tabibian-Keissar H, Hazanov L, Schiby G, Rosenthal N, Rakovsky A, Michaeli M, et al. Aging affects b-cell antigen receptor repertoire diversity in primary and secondary lymphoid tissues. Eur J Immunol (2016) 46(2):480–92. doi: 10.1002/eji.201545586
57. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (Aid), a potential rna editing enzyme. Cell (2000) 102(5):553–63. doi: 10.1016/s0092-8674(00)00078-7
58. Frasca D, van der Put E, Riley RL, Blomberg BB. Reduced ig class switch in aged mice correlates with decreased E47 and activation-induced cytidine deaminase. J Immunol (2004) 172(4):2155–62. doi: 10.4049/jimmunol.172.4.2155
59. Frasca D, Landin AM, Lechner SC, Ryan JG, Schwartz R, Riley RL, et al. Aging down-regulates the transcription factor E2a, activation-induced cytidine deaminase, and ig class switch in human b cells. J Immunol (2008) 180(8):5283–90. doi: 10.4049/jimmunol.180.8.5283
60. Birjandi SZ, Ippolito JA, Ramadorai AK, Witte PL. Alterations in marginal zone macrophages and marginal zone b cells in old mice. J Immunol (2011) 186(6):3441–51. doi: 10.4049/jimmunol.1001271
61. Masters AR, Jellison ER, Puddington L, Khanna KM, Haynes L. Attrition of T cell zone fibroblastic reticular cell number and function in aged spleens. Immunohorizons (2018) 2(5):155–63. doi: 10.4049/immunohorizons.1700062
62. Denton AE, Dooley J, Cinti I, Silva-Cayetano A, Fra-Bido S, Innocentin S, et al. Targeting Tlr4 during vaccination boosts madcam-1. Sci Immunol (2022) 7(71):eabk0018. doi: 10.1126/sciimmunol.abk0018
63. Rinaldi S, Pallikkuth S, George VK, de Armas LR, Pahwa R, Sanchez CM, et al. Paradoxical aging in hiv: Immune senescence of b cells is most prominent in young age. Aging (Albany NY) (2017) 9(4):1307–25. doi: 10.18632/aging.101229
64. Frasca D, Diaz A, Romero M, Blomberg BB. Human peripheral Late/Exhausted memory b cells express a senescent-associated secretory phenotype and preferentially utilize metabolic signaling pathways. Exp Gerontol (2017) 87(Pt A):113–20. doi: 10.1016/j.exger.2016.12.001
65. Hao Y, O'Neill P, Naradikian MS, Scholz JL, Cancro MP. A b-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood (2011) 118(5):1294–304. doi: 10.1182/blood-2011-01-330530
66. Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, et al. Toll-like receptor 7 (Tlr7)-driven accumulation of a novel Cd11c+ b-cell population is important for the development of autoimmunity. Blood (2011) 118(5):1305–15. doi: 10.1182/blood-2011-01-331462
67. Saadoun D, Terrier B, Bannock J, Vazquez T, Massad C, Kang I, et al. Expansion of autoreactive unresponsive Cd21-/Low b cells in sjögren's syndrome-associated lymphoproliferation. Arthritis Rheum (2013) 65(4):1085–96. doi: 10.1002/art.37828
68. Moir S, Ho J, Malaspina A, Wang W, DiPoto AC, O'Shea MA, et al. Evidence for hiv-associated b cell exhaustion in a dysfunctional memory b cell compartment in hiv-infected viremic individuals. J Exp Med (2008) 205(8):1797–805. doi: 10.1084/jem.20072683
69. Johnson JL, Rosenthal RL, Knox JJ, Myles A, Naradikian MS, Madej J, et al. The transcription factor T-bet resolves memory b cell subsets with distinct tissue distributions and antibody specificities in mice and humans. Immunity (2020) 52(5):842–55.e6. doi: 10.1016/j.immuni.2020.03.020
70. Mouat IC, Horwitz MS. Age-associated b cells in viral infection. PloS Pathog (2022) 18(3):e1010297. doi: 10.1371/journal.ppat.1010297
71. Palmer DB. The effect of age on thymic function. Front Immunol (2013) 4:316. doi: 10.3389/fimmu.2013.00316
72. Goronzy JJ, Lee WW, Weyand CM. Aging and T-cell diversity. Exp Gerontol (2007) 42(5):400–6. doi: 10.1016/j.exger.2006.11.016
73. Qi Q, Liu Y, Cheng Y, Glanville J, Zhang D, Lee J-Y, et al. Diversity and clonal selection in the human T-cell repertoire. Proc Natl Acad Sci (2014) 111(36):13139–44. doi: 10.1073/pnas.1409155111
74. Egorov ES, Kasatskaya SA, Zubov VN, Izraelson M, Nakonechnaya TO, Staroverov DB, et al. The changing landscape of naive T cell receptor repertoire with human aging. Front Immunol (2018) 9:1618. doi: 10.3389/fimmu.2018.01618
75. De Martinis M, Modesti M, Ginaldi L. Phenotypic and functional changes of circulating monocytes and polymorphonuclear leucocytes from elderly persons. Immunol Cell Biol (2004) 82(4):415–20. doi: 10.1111/j.0818-9641.2004.01242.x
76. Bhadricha H, Patel V, Singh AK, Savardekar L, Patil A, Surve S, et al. Increased frequency of Th17 cells and il-17 levels are associated with low bone mineral density in postmenopausal women. Sci Rep (2021) 11(1):16155. doi: 10.1038/s41598-021-95640-0
77. Zhu L, Hua F, Ding W, Ding K, Zhang Y, Xu C. The correlation between the Th17/Treg cell balance and bone health. Immun Ageing (2020) 17:30. doi: 10.1186/s12979-020-00202-z
78. Ouyang X, Yang Z, Zhang R, Arnaboldi P, Lu G, Li Q, et al. Potentiation of Th17 cytokines in aging process contributes to the development of colitis. Cell Immunol (2011) 266(2):208–17. doi: 10.1016/j.cellimm.2010.10.007
79. Faust HJ, Zhang H, Han J, Wolf MT, Jeon OH, Sadtler K, et al. Il-17 and immunologically induced senescence regulate response to injury in osteoarthritis. J Clin Invest (2020) 130(10):5493–507. doi: 10.1172/JCI134091
80. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133(5):775–87. doi: 10.1016/j.cell.2008.05.009
81. Thiault N, Darrigues J, Adoue V, Gros M, Binet B, Perals C, et al. Peripheral regulatory T lymphocytes recirculating to the thymus suppress the development of their precursors. Nat Immunol (2015) 16(6):628–34. doi: 10.1038/ni.3150
82. Rocamora-Reverte L, Melzer FL, Würzner R, Weinberger B. The complex role of regulatory T cells in immunity and aging. Front Immunol (2020) 11:616949. doi: 10.3389/fimmu.2020.616949
83. Cicin-Sain L, Messaoudi I, Park B, Currier N, Planer S, Fischer M, et al. Dramatic increase in naive T cell turnover is linked to loss of naive T cells from old primates. Proc Natl Acad Sci USA (2007) 104(50):19960–5. doi: 10.1073/pnas.0705905104
84. Ahmed M, Lanzer KG, Yager EJ, Adams PS, Johnson LL, Blackman MA. Clonal expansions and loss of receptor diversity in the naive Cd8 T cell repertoire of aged mice. J Immunol (2009) 182(2):784–92. doi: 10.4049/jimmunol.182.2.784
85. Simone R, Zicca A, Saverino D. The frequency of regulatory Cd3+Cd8+Cd28- Cd25+ T lymphocytes in human peripheral blood increases with age. J Leukoc Biol (2008) 84(6):1454–61. doi: 10.1189/jlb.0907627
86. O'Hara GA, Welten SP, Klenerman P, Arens R. Memory T cell inflation: Understanding cause and effect. Trends Immunol (2012) 33(2):84–90. doi: 10.1016/j.it.2011.11.005
87. Martin MD, Badovinac VP. Defining memory Cd8 T cell. Front Immunol (2018) 9:2692. doi: 10.3389/fimmu.2018.02692
88. Holtappels R, Pahl-Seibert MF, Thomas D, Reddehase MJ. Enrichment of immediate-early 1 (M123/Pp89) peptide-specific Cd8 T cells in a pulmonary Cd62l(Lo) memory-effector cell pool during latent murine cytomegalovirus infection of the lungs. J Virol (2000) 74(24):11495–503. doi: 10.1128/jvi.74.24.11495-11503.2000
89. Wertheimer AM, Bennett MS, Park B, Uhrlaub JL, Martinez C, Pulko V, et al. Aging and cytomegalovirus infection differentially and jointly affect distinct circulating T cell subsets in humans. J Immunol (2014) 192(5):2143–55. doi: 10.4049/jimmunol.1301721
90. Lang A, Brien JD, Nikolich-Zugich J. Inflation and long-term maintenance of Cd8 T cells responding to a latent herpesvirus depend upon establishment of latency and presence of viral antigens. J Immunol (2009) 183(12):8077–87. doi: 10.4049/jimmunol.0801117
91. Akbar AN, Henson SM. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat Rev Immunol (2011) 11(4):289–95. doi: 10.1038/nri2959
92. Mogilenko DA, Shpynov O, Andhey PS, Arthur L, Swain A, Esaulova E, et al. Comprehensive profiling of an aging immune system reveals clonal gzmk. Immunity (2021) 54(1):99–115.e12. doi: 10.1016/j.immuni.2020.11.005
93. Martínez-Zamudio RI, Dewald HK, Vasilopoulos T, Gittens-Williams L, Fitzgerald-Bocarsly P, Herbig U. Senescence-associated β-galactosidase reveals the abundance of senescent Cd8+ T cells in aging humans. Aging Cell (2021) 20(5):e13344. doi: 10.1111/acel.13344
94. Frasca D. Senescent b cells in aging and age-related diseases: Their role in the regulation of antibody responses. Exp Gerontol (2018) 107:55–8. doi: 10.1016/j.exger.2017.07.002
95. Cho KA, Ryu SJ, Oh YS, Park JH, Lee JW, Kim H-P, et al. Morphological adjustment of senescent cells by modulating caveolin-1 status*. J Biol Chem (2004) 279(40):42270–8. doi: 10.1074/jbc.M402352200
96. [Dataset] estimated influenza disease burden, by season — united states, 2010-11 through 2019-20 influenza seasons . Available at: https://www.cdc.gov/flu/about/burden/past-seasons.html.
97. Lynch SM, Guo G, Gibson DS, Bjourson AJ, Rai TS. Role of senescence and aging in sars-Cov-2 infection and covid-19 disease. Cells (2021) 10(12):3367. doi: 10.3390/cells10123367
98. Bartley JM, Pan SJ, Keilich SR, Hopkins JW, Al-Naggar IM, Kuchel GA, et al. Aging augments the impact of influenza respiratory tract infection on mobility impairments, muscle-localized inflammation, and muscle atrophy. Aging (Albany NY) (2016) 8(4):620–35. doi: 10.18632/aging.100882
99. Kuiken T, Taubenberger JK. Pathology of human influenza revisited. Vaccine (2008) 26 Suppl 4:D59–66. doi: 10.1016/j.vaccine.2008.07.025
100. Hansel C, Jendrossek V, Klein D. Cellular senescence in the lung: The central role of senescent epithelial cells. Int J Mol Sci (2020) 21(9):3279. doi: 10.3390/ijms21093279
101. Lee S, Yu Y, Trimpert J, Benthani F, Mairhofer M, Richter-Pechanska P, et al. Virus-induced senescence is a driver and therapeutic target in covid-19. Nature (2021) 599(7884):283–9. doi: 10.1038/s41586-021-03995-1
102. Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor ppar-Γ by the cytokine gm-csf is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol (2014) 15(11):1026–37. doi: 10.1038/ni.3005
103. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via gm-csf. J Exp Med (2013) 210(10):1977–92. doi: 10.1084/jem.20131199
104. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med (2017) 214(8):2387–404. doi: 10.1084/jem.20162152
105. Li F, Piattini F, Pohlmeier L, Feng Q, Rehrauer H, Kopf M. Monocyte-derived alveolar macrophages autonomously determine severe outcome of respiratory viral infection. Sci Immunol (2022) 7(73):eabj5761. doi: 10.1126/sciimmunol.abj5761
106. Ma C, Zhang N. Transforming growth factor-β signaling is constantly shaping memory T-cell population. Proc Natl Acad Sci USA (2015) 112(35):11013–7. doi: 10.1073/pnas.1510119112
107. Honda K, Yanai H, Takaoka A, Taniguchi T. Regulation of the type I ifn induction: A current view. Int Immunol (2005) 17(11):1367–78. doi: 10.1093/intimm/dxh318
108. Molony RD, Nguyen JT, Kong Y, Montgomery RR, Shaw AC, Iwasaki A. Aging impairs both primary and secondary rig-I signaling for interferon induction in human monocytes. Sci Signal (2017) 10(509):eaan2392. doi: 10.1126/scisignal.aan2392
109. Feng E, Balint E, Poznanski SM, Ashkar AA, Loeb M. Aging and interferons: Impacts on inflammation and viral disease outcomes. Cells (2021) 10(3):708. doi: 10.3390/cells10030708
110. Sant AJ, DiPiazza AT, Nayak JL, Rattan A, Richards KA. Cd4 T cells in protection from influenza virus: Viral antigen specificity and functional potential. Immunol Rev (2018) 284(1):91–105. doi: 10.1111/imr.12662
111. Lefebvre JS, Maue AC, Eaton SM, Lanthier PA, Tighe M, Haynes L. The aged microenvironment contributes to the age-related functional defects of Cd4 T cells in mice. Aging Cell (2012) 11(5):732–40. doi: 10.1111/j.1474-9726.2012.00836.x
112. Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med (2017) 23(9):1072–9. doi: 10.1038/nm.4385
113. Musi N, Valentine JM, Sickora KR, Baeuerle E, Thompson CS, Shen Q, et al. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell (2018) 17(6):e12840. doi: 10.1111/acel.12840
114. Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell (2016) 15(5):973–7. doi: 10.1111/acel.12458
115. Jiang J, Bennett AJ, Fisher E, Williams-Bey Y, Shen H, Murasko DM. Limited expansion of virus-specific Cd8 T cells in the aged environment. Mech Ageing Dev (2009) 130(11-12):713–21. doi: 10.1016/j.mad.2009.08.007
116. Williams-Bey Y, Jiang J, Murasko DM. Expansion of regulatory T cells in aged mice following influenza infection. Mech Ageing Dev (2011) 132(4):163–70. doi: 10.1016/j.mad.2011.03.001
117. Quinn KM, Fox A, Harland KL, Russ BE, Li J, Nguyen THO, et al. Age-related decline in primary Cd8 + t cell responses is associated with the development of senescence in virtual memory Cd8 + t cells. Cell Rep (2018) 23(12):3512–24. doi: 10.1016/j.celrep.2018.05.057
118. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell (2018) 174(5):1054–66. doi: 10.1016/j.cell.2018.07.017
119. Fonseca W, Lukacs NW, Elesela S, Malinczak C-A. Role of Ilc2 in viral-induced lung pathogenesis. Front Immunol (2021) 12:675169. doi: 10.3389/fimmu.2021.675169
120. Chang Y-J, Kim HY, Albacker LA, Baumgarth N, McKenzie ANJ, Smith DE, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol (2011) 12(7):631–8. doi: 10.1038/ni.2045
121. Hams E, Armstrong ME, Barlow JL, Saunders SP, Schwartz C, Cooke G, et al. Il-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc Natl Acad Sci (2014) 111(1):367–72. doi: 10.1073/pnas.1315854111
122. D'Souza SS, Shen X, Fung ITH, Ye L, Kuentzel M, Chittur SV, et al. Compartmentalized effects of aging on group 2 innate lymphoid cell development and function. Aging Cell (2019) 18(6):e13019. doi: 10.1111/acel.13019
123. Bartleson JM, Radenkovic D, Covarrubias AJ, Furman D, Winer DA, Verdin E. Sars-Cov-2, covid-19 and the ageing immune system. Nat Aging (2021) 1(9):769–82. doi: 10.1038/s43587-021-00114-7
124. Metzger DW, Sun K. Immune dysfunction and bacterial coinfections following influenza. J Immunol (2013) 191(5):2047–52. doi: 10.4049/jimmunol.1301152
125. Cross RW, Betancourt AM, Schur MJ, Norton EB, Roy D, Hartley T, et al. Impact of primary influenza infection on the immune response to secondary bacterial infection in aged mice. Influenza Other Respir Viruses (2011) 5(Suppl 1):198–201.
126. Pawelec G, Akbar A, Caruso C, Effros R, Grubeck-Loebenstein B, Wikby A. Is immunosenescence infectious? Trends Immunol (2004) 25(8):406–10. doi: 10.1016/j.it.2004.05.006
127. Arens R, Remmerswaal EB, Bosch JA, van Lier RA. 5(Th) international workshop on cmv and immunosenescence - a shadow of cytomegalovirus infection on immunological memory. Eur J Immunol (2015) 45(4):954–7. doi: 10.1002/eji.201570044
128. Derhovanessian E, Theeten H, Hähnel K, Van Damme P, Cools N, Pawelec G. Cytomegalovirus-associated accumulation of late-differentiated Cd4 T-cells correlates with poor humoral response to influenza vaccination. Vaccine (2013) 31(4):685–90. doi: 10.1016/j.vaccine.2012.11.041
129. Frasca D, Blomberg BB. Aging induces b cell defects and decreased antibody responses to influenza infection and vaccination. Immun Ageing (2020) 17(1):37. doi: 10.1186/s12979-020-00210-z
130. Trzonkowski P, Myśliwska J, Szmit E, Wieckiewicz J, Lukaszuk K, Brydak LB, et al. Association between cytomegalovirus infection, enhanced proinflammatory response and low level of anti-hemagglutinins during the anti-influenza vaccination–an impact of immunosenescence. Vaccine (2003) 21(25-26):3826–36. doi: 10.1016/s0264-410x(03)00309-8
131. Furman D, Jojic V, Sharma S, Shen-Orr SS, L. Angel CJ, Onengut-Gumuscu S, et al. Cytomegalovirus infection enhances the immune response to influenza. Sci Trans Med (2015) 7(281):281ra43–ra43. doi: 10.1126/scitranslmed.aaa2293
132. Cicin-Sain L, Brien JD, Uhrlaub JL, Drabig A, Marandu TF, Nikolich-Zugich J. Cytomegalovirus infection impairs immune responses and accentuates T-cell pool changes observed in mice with aging. PloS Pathog (2012) 8(8):e1002849. doi: 10.1371/journal.ppat.1002849
133. Marandu TF, Oduro JD, Borkner L, Dekhtiarenko I, Uhrlaub JL, Drabig A, et al. Immune protection against virus challenge in aging mice is not affected by latent herpesviral infections. J Virol (2015) 89(22):11715–7. doi: 10.1128/jvi.01989-15
134. Nguyen QD, Moodie EM, Forget MF, Desmarais P, Keezer MR, Wolfson C. Health heterogeneity in older adults: Exploration in the Canadian longitudinal study on aging. J Am Geriatrics Soc (2021) 69(3):678–87. doi: 10.1111/jgs.16919
135. Lefebvre JS, Lorenzo EC, Masters AR, Hopkins JW, Eaton SM, Smiley ST, et al. Vaccine efficacy and T helper cell differentiation change with aging. Oncotarget (2016) 7(23):33581–94. doi: 10.18632/oncotarget.9254
136. McElhaney JE, Verschoor CP, Andrew MK, Haynes L, Kuchel GA, Pawelec G. The immune response to influenza in older humans: Beyond immune senescence. Immun Ageing (2020) 17:10. doi: 10.1186/s12979-020-00181-1
137. McElhaney JE, Xie D, Hager WD, Barry MB, Wang Y, Kleppinger A, et al. T Cell responses are better correlates of vaccine protection in the elderly. J Immunol (2006) 176(10):6333–9. doi: 10.4049/jimmunol.176.10.6333
138. McElhaney JE, Ewen C, Zhou X, Kane KP, Xie D, Hager WD, et al. Granzyme b: Correlates with protection and enhanced ctl response to influenza vaccination in older adults. Vaccine (2009) 27(18):2418–25. doi: 10.1016/j.vaccine.2009.01.136
139. Haynes L, Eaton SM, Burns EM, Randall TD, Swain SL. Cd4 T cell memory derived from young naive cells functions well into old age, but memory generated from aged naive cells functions poorly. Proc Natl Acad Sci USA (2003) 100(25):15053–8. doi: 10.1073/pnas.2433717100
140. Goplen NP, Wu Y, Son YM, Li C, Wang Z, Cheon IS, et al. Tissue-resident Cd8+ T cells drive age-associated chronic lung sequelae after viral pneumonia. Sci Immunol (2020) 5(53):eabc4557. doi: 10.1126/sciimmunol.abc4557
141. Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, et al. The achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell (2015) 14(4):644–58. doi: 10.1111/acel.12344
142. Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine (2018) 36:18–28. doi: 10.1016/j.ebiom.2018.09.015
143. Chang J, Wang Y, Shao L, Laberge R-M, Demaria M, Campisi J, et al. Clearance of senescent cells by Abt263 rejuvenates aged hematopoietic stem cells in mice. Nat Med (2016) 22(1):78–83. doi: 10.1038/nm.4010
144. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med (2018) 24(8):1246–56. doi: 10.1038/s41591-018-0092-9
145. Lorenzo EC, Torrance BL, Keilich SR, Al-Naggar I, Harrison A, Xu M, et al. Senescence-induced changes in Cd4 T cell differentiation can be alleviated by treatment with senolytics. Aging Cell (2022) 21(1):e13525. doi: 10.1111/acel.13525
146. Camell CD, Yousefzadeh MJ, Zhu Y, Prata LGPL, Huggins MA, Pierson M, et al. Senolytics reduce coronavirus-related mortality in old mice. Science (2021) 373(6552):eabe4832. doi: 10.1126/science.abe4832
147. Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine (2019) 47:446–56. doi: 10.1016/j.ebiom.2019.08.069
148. Wang B, Wang L, Gasek NS, Zhou Y, Kim T, Guo C, et al. An inducible p21-cre mouse model to monitor and manipulate p21-Highly-Expressing senescent cells in vivo. Nat Aging (2021) 1(10):962–73. doi: 10.1038/s43587-021-00107-6
Keywords: senescence, influenza, lung, aging, immunity
Citation: Torrance BL and Haynes L (2022) Cellular senescence is a key mediator of lung aging and susceptibility to infection. Front. Immunol. 13:1006710. doi: 10.3389/fimmu.2022.1006710
Received: 29 July 2022; Accepted: 16 August 2022;
Published: 31 August 2022.
Edited by:
Priyadharshini Devarajan, University of Massachusetts Medical School, United StatesReviewed by:
Michelle Ratliff, East Carolina University, United StatesMladen Jergovic, University of Arizona, United States
Claire E Gustafson, Allen Institute for Immunology, United States
Copyright © 2022 Torrance and Haynes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Haynes, bGhheW5lc0B1Y2hjLmVkdQ==